 Christian A. Hübner
Christian A. Hübner Knut Holthoff
Knut Holthoff- 1Institute of Human Genetics, Jena University Hospital, Friedrich Schiller University Jena, Jena, Germany
- 2Hans Berger Department of Neurology, Jena University Hospital, Friedrich Schiller University Jena, Jena, Germany
Whereas activation of GABAA receptors by GABA usually results in a hyperpolarizing influx of chloride into the neuron, the reversed chloride driving force in the immature nervous system results in a depolarizing efflux of chloride. This GABAergic depolarization is deemed to be important for the maturation of the neuronal network. The concept of a developmental GABA switch has mainly been derived from in vitro experiments and reliable in vivo evidence is still missing. As GABAA receptors are permeable for both chloride and bicarbonate, the net effect of GABA also critically depends on the distribution of bicarbonate. Whereas chloride can either mediate depolarizing or hyperpolarizing currents, bicarbonate invariably mediates a depolarizing current under physiological conditions. Intracellular bicarbonate is quickly replenished by cytosolic carbonic anhydrases. Intracellular bicarbonate levels also depend on different bicarbonate transporters expressed by neurons. The expression of these proteins is not only developmentally regulated but also differs between cell types and even subcellular regions. In this review we will summarize current knowledge about the role of some of these transporters for brain development and brain function.
Introduction
GABA signaling has a wide spectrum of functions in individual neurons and neuronal networks in the brain. It is well known that in the mature brain, GABA acts as the main inhibitory transmitter due to activation of hyperpolarizing chloride currents through GABAA receptors (Farrant and Kaila, 2007). In contrast, during early brain development, GABAergic transmission is assumed to provide the main excitatory drive in neuronal networks, at a time when glutamatergic synaptic contacts are less frequent than GABAergic synapses (Ben-Ari et al., 1989). Although this functional switch from excitatory to inhibitory GABA action during brain development has been observed in a wide range of preparations and different animal species, most of the experimental evidence relies on in vitro studies (comprehensively reviewed by Ben-Ari et al., 2007). They were performed using several electrophysiological means like intracellular recordings (Mueller et al., 1984; Luhmann and Prince, 1991) and less invasive techniques including perforated patch (Owens et al., 1996; Yamada et al., 2004) and cell-attached measurements (Wang et al., 2003; Rheims et al., 2008; Kirmse et al., 2010). Consistently, several groups reported intracellular calcium increases in immature neurons upon GABA application, most likely due to depolarization mediated activation of voltage-gated calcium channels (Yuste and Katz, 1991; Owens et al., 1996; Yamada et al., 2004; Kirmse and Kirischuk, 2006; Kirmse et al., 2010). However, in vivo evidence for depolarizing GABA action in immature neuronal networks is rare (Brustein et al., 2003) and often indirect (Sipila et al., 2006). Metabotropic GABAB-receptors are coupled to calcium or potassium channels, and cyclic AMP signaling. Although there is quite recent evidence that the non-hyperpolarizing activation of GABAB-receptors during development promotes neuronal migration and morphological maturation (Bony et al., 2013), this review will focus on GABAA-receptor signaling and how this relates to anion-transport.
The functional relevance of GABAA-receptor activation for activity patterns in immature neuronal networks has been investigated in different model systems in vitro and in vivo. In the immature hippocampus, it is widely accepted that GABAergic excitation drives the typical spontaneous network activity known as giant depolarization potentials or GDPs (Ben-Ari et al., 1989; Bonifazi et al., 2009). On the other hand in neocortex, glutamatergic excitation was shown to be dominating in the early generation of network activity like early network oscillations or ENOs (Garaschuk et al., 2000) and spindle-bursts (Minlebaev et al., 2007). However, more recent results suggest that GABAA receptor activation also supports the generation of early neocortical network activity (Allene et al., 2008). It is worth noting that depolarizing GABAA-receptor activation not necessarily needs to be excitatory (Morita et al., 2005, 2006). The GABA-induced increase in membrane conductance can also cause a so-called shunting inhibition, because according to Ohm’s law, the drop in membrane resistance would decrease the voltage change caused by a certain depolarizing current (e.g., a glutamatergic synaptic input). The inhibitory effect of shunting does not depend on the polarity of a GABA-induced membrane potential change and therefore on the chloride and bicarbonate reversal potentials, but is solely due to the GABA-induced drop in input resistance. Nevertheless, the paradoxical situation could happen that a depolarizing GABAA-receptor activation leads to an inhibitory restriction of network activity (Minlebaev et al., 2007).
Although many initial steps in early neuronal network development are genetically determined, there is a large body of evidence, that the proper functional maturation of cortical neuronal circuits is highly activity-dependent (Katz and Shatz, 1996). However, to what extent the spontaneously occurring network activity, partly driven by GABAA-receptor activation, contributes to the functional maturation in the pre-sensory period of the brain is still unclear. Undoubtedly, a fine tuned balance between excitation and inhibition at any stage of development is essential for providing a proper function of neuronal networks. In this context, the potentially depolarizing mode of GABAA-receptor activation during early development has been considered to contribute to the higher liability to pathological events like epileptic seizures during childhood (see Kirmse et al., 2011 for review). Later in development, the increasing GABAergic inhibition governs the on- and offset of the so-called critical period in the visual system, which is crucial for the activity-dependent functional refinement of the participating cortical circuits (Hensch et al., 1998; Fagiolini and Hensch, 2000). Therefore, GABAA-receptor activation plays a pivotal role at various developmental stages for the maturation, refinement and proper function of neuronal networks.
The ionotropic GABAA receptors are pentamers of 19 different subunits, which are grouped into eight different families according to sequence homology (α1–6, β1–3, γ1–3, δ, ε, θ, π, ρ1–3; Farrant and Kaila, 2007). Although the different receptor assemblies have different properties and different distributions, only chloride and bicarbonate are conducted under physiological conditions (Bormann et al., 1987). It is assumed that the relative bicarbonate/chloride permeability of ionotropic GABAA receptors ranges between 0.18 and 0.6 (Fatima-Shad and Barry, 1993). Because of a variety of different ion transporters within the plasma membrane, which mediate acid extrusion either by extrusion of H+ or by accumulation of bicarbonate, the bicarbonate equilibrium potential is much more depolarized (around -10 mV) than the resting membrane potential (Roos and Boron, 1981). Hence bicarbonate can only mediate a depolarizing current under normal conditions. Quite in contrast, the equilibrium potential for chloride is close to the resting membrane potential. Hence chloride can mediate both hyperpolarizing and depolarizing currents depending on the existing gradient, which is regulated during development. However, a depolarizing action of GABA does not exclude an inhibitory action but may result in shunting inhibition as outlined above.
Electrophysiological Methods to Quantify GABA Function
Before fluorescent indicators have been available, ion-selective micro-electrodes were the gold standard for the measurement of chloride activity and pH in the intracellular compartment (Walker, 1971; Thomas, 1974; Ammann et al., 1981). Because of the invasive nature of this method – the cellular membrane needs to be impaled by a double-barreled sharp micro-electrode – it was impossible to determine the ion activity of interest quantitatively without changing it at the same time due to the measurement. Leakage currents at the site of impalement and intracellular perfusion by the solution of the reference channel are only two possible sources of measurement errors. Nevertheless, this method provided first important insights into the ionic mechanisms of pH regulation or chloride homoeostasis (Thomas, 1977; Vaughan-Jones, 1979).
In order to keep the ionic composition of the intracellular compartment unchanged, most other electrophysiological means are based on cell-attached patch clamp recordings. In the following, we describe several methods which have been developed to determine membrane potentials and membrane currents without disrupting the plasma membrane. To provide electrical access to the intracellular compartment without interference with the intracellular milieu, a technique called perforated patch clamp was developed, originally using ATP in the pipette solution as a membrane permeabilizing agent (Lindau and Fernandez, 1986). In subsequent modifications of this method, different ionophores were added to the pipette solution, which, during the experiment, incorporate into the membrane patch under the pipette tip. Ionophores are lipid-soluble molecules which form hydrophilic pores in the cell membrane and mediate electrical access to the intracellular compartment without destroying the barrier function of the membrane patch for the ion of interest. In early studies, nystatin and amphotericin B were used to achieve low resistance electrical access (Horn and Marty, 1988; Rae et al., 1991). However, as a major drawback these substances also lead to chloride redistribution. Subsequently, gramicidin D, a mixture of different antibiotics, was added because it is only permeable for monovalent cations and uncharged low molecular substances but impermeable for chloride, leaving its concentration gradient over the cell membrane intact (Ebihara et al., 1995; Kyrozis and Reichling, 1995). However, indirect changes in intracellular chloride concentration are conceivable because it is mainly regulated by cation/chloride co-transporters. Several groups have applied this method successfully to determine e.g., the chloride equilibrium potential in hippocampal cells during brain development (Mohajerani and Cherubini, 2005; Sipila et al., 2006; Tyzio et al., 2007; Pfeffer et al., 2009).
As mentioned previously, the chloride concentration gradient is not the only determinant governing GABA-induced membrane potential changes, because GABAA-receptor channels are also permeable for other anions like bicarbonate. In order to quantify the GABA-reversal potential directly, Tyzio et al. (2003), 2006 developed a non-invasive method to measure the resting membrane potential and the GABA reversal potential at the same cell, using single N-methyl-D-aspartate and GABA channel recordings. The combination of both experimental approaches at the same cells provides the driving force for GABA-induced currents and the resting membrane potential in absolute numbers and, thereby, the GABA equilibrium potential. Although this is up to now the most reliable method to quantify these parameters, it is hardly applicable to complex preparations like in vivo recordings. If only the non-invasive quantification of the membrane potential change due to GABA-receptor activation is of interest, an alternative method described by Verheugen et al. (1999) can be applied (Kirmse et al., 2010). Assuming symmetrical potassium concentrations, this method uses the fact, that the reversal potential of voltage-dependent potassium currents in the cell-attached configuration represents a good estimate of the cell membrane potential (Verheugen et al., 1999). However, potential changes in intracellular potassium concentration would flaw the correct membrane potential quantification. Because a voltage-ramp protocol has to be applied for every time point of interest, the time resolution of this method is rather slow and phasic membrane potential changes are difficult to catch. In these cases, applying a current-clamp recording protocol in the cell-attached configuration might be beneficial, because it can provide a good estimate of the polarity of an induced membrane potential change at high time resolution (Perkins and Wong, 1996; Mason et al., 2005).
In summary, various non-invasive electrophysiological methods provide valuable estimates of GABA equilibrium potentials under different conditions in vitro and in vivo and enable the measurement of relative or absolute cell membrane potential changes without disturbing the intracellular milieu. In concert with complementary optical methods for quantification of intracellular chloride concentration and pH, they draw a detailed image of GABA-mediated physiological processes.
Optical Methods to Quantify GABA Function
A big step forward was the development of fluorescent indicator dyes which enabled the optical measurement of intracellular pH and chloride concentrations (Rink et al., 1982; Illsley and Verkman, 1987). The initially used small molecular fluorescent chloride indicator dyes were quinoline derivatives which change their fluorescent intensity upon changes in chloride concentration by a mechanism called collision quenching (Chen et al., 1988). With increasing chloride concentration the probability of a collision between a chloride ion and an indicator molecule increases and therefore, its fluorescence intensity decreases by quenching. A notably feature of this mechanism is, that different from the popular calcium indicator dyes, which change their fluorescence intensity upon binding to calcium, these chloride indicator dyes do not introduce any exogenous buffer capacity to the intracellular milieu, because no binding to the ion of interest takes place. Nevertheless, their excitation spectra in the ultraviolet range give rise to strong bleaching and photodynamic damage (Inglefield and Schwartz-Bloom, 1997). However, the combination of these dyes with two-photon imaging is able to reduce both side effects significantly (Marandi et al., 2002). Several years earlier, the measurement of intracellular pH has been revolutionized by the invention of BCECF, a fluorescence indicator derived from fluorescein, by Roger Tsien and coworkers (Rink et al., 1982). The absorption spectrum of BCECF is shifted depending on changes in pH and by applying ratiometric excitation the indicator can be calibrated to absolute pH units (Graber et al., 1986; Bright et al., 1987).
The unspecific loading of the exogenously applied fluorescent indicator dyes prevents a cell-specific labeling. Therefore, much effort was invested to develop genetically determined chloride indicator dyes (see Bregestovski et al., 2009 for review). Starting point of this development was the chloride binding property of the yellow fluorescent protein (YFP) a derivative of the green fluorescent protein (GFP). Because the sensitivity of wild-type YFP to chloride is low, many random chloride binding site mutations of YFP were tested and analyzed for improved sensitivity (Galietta et al., 2001). Besides the possibility of cell-specific expression of the chloride indicator, YFP-based indicator dyes have additional advantages. Different from quinolone-derived dyes, the optimal excitation wavelength is located in the visible range, providing less bleaching and photodynamic damage during the experiments. In addition, leakage during the measurements is less pronounced due to their relative large molecular weight of about 27 kDa (Bregestovski et al., 2009). Finally and different from calcium measurements, intracellular indicator concentrations are orders of magnitude smaller than that of the ion of interest, therefore exogenous buffering of chloride is negligible. On the other hand, there are also some disadvantages of YFP-based indicator dyes. Keeping in mind that changes in intracellular chloride concentration are often accompanied by changes in pH, the significant pH-sensitivity of many YFP derivatives is the most serious one. The only way to circumvent this restriction is the independent monitoring of pH changes and subsequent data correction. Another problem of YFP-based chloride indicator dyes are their rather slow kinetics or poor sensitivity, which either limits the detection of fast chloride concentration changes or leads to poor resolution at physiological chloride levels (Galietta et al., 2001). Originally, YFP-based indicator dyes were not able to report absolute levels of chloride concentration, because they lack an isosbestic point, at which they are insensitive to chloride concentration changes. Because the absolute measurement of intracellular chloride concentrations is imperative to determine the chloride equilibrium potential or the driving force for chloride, Kuner and Augustine (2000) developed a ratiometric chloride indicator named Clomeleon. Clomeleon uses the chloride-dependent interaction of two fluorophores (cyan fluorescent protein (CFP) as donor and a variant of YFP called topas fluorescent protein (TFP) as acceptor) by Förster energy transfer (FRET). Upon chloride binding to TFP, the efficiency of FRET between CFP and TFP declines. As a consequence, the ratio of TFP and CFP fluorescence emission drops with increasing chloride concentrations. Because the emission spectrum comprises an isosbestic point, calibration to absolute chloride levels is possible (Kuner and Augustine, 2000). Unfortunately, the sensitivity of Clomeleon with an IC50 of more than 160 mM is rather low and, at physiological levels, makes reliable measurements of absolute intracellular chloride concentration very difficult (Kuner and Augustine, 2000). Following genetic engineering of the YFP chloride binding site yielded a higher sensitivity of the resulting indicator called Cl-sensor with an IC50 around 30 mM, much closer to physiological intracellular chloride concentrations (Markova et al., 2008). However, Clomeleon and Cl-sensor have slow response kinetics and share the pH sensitivity of all YFP-based chloride indicators (Bregestovski et al., 2009).
To overcome the main drawbacks of Clomeleon and Cl-sensor, a new ratiometric but non-FRET-based sensor was developed (Arosio et al., 2010). This new indicator, called ClopHensor, is suitable for the simultaneous quantification of intracellular pH and chloride concentration. Therefore, a variant of enhanced GFP (E2GFP) with pH sensitivity and sensitivity to chloride comparable to Cl-sensor was fused with a pH- and chloride-insensitive monomeric DsRed. The E2GFP part of the fused protein allows chloride-independent ratiometric quantification of pH by exciting it subsequently at 458 and 488 nm. The ratiometric measurement of chloride concentration by alternative exciting E2GFP at 458 nm and the chloride-insensitive DsRed at 543 nm requires in addition the calibration at different pH values. However a simultaneous quantification of intracellular pH is now possible, ClopHensor still suffers from rather low sensitivity to chloride with an IC50 around 40 mM (Arosio et al., 2010; Mukhtarov et al., 2013). A more recent variant of ClopHensor exhibits a higher sensitivity with an IC50 of 20 mM, but at the expense of a significant lower dynamic range (Mukhtarov et al., 2013). In summary, the development of the ratiometric indicator ClopHensor provides a most valuable means for the simultaneous quantification of pH and chloride concentration, but variants with higher sensitivity to chloride are desired to increase the quantification accuracy at physiological concentration levels.
Ion Transporters Involved in the Regulation of Neuronal Chloride and Bicarbonate Levels
The role of cation-chloride co-transporters (Figure 1) in the regulation of the intraneuronal chloride concentration has been extensively studied and follows a well-defined developmental sequence with a high chloride concentration in immature neurons due to neuronal chloride accumulation. Chloride accumulation largely depends on the action of the Na+/K+/2Cl- co-transporter NKCC1 (Yamada et al., 2004; Sipila et al., 2006; Achilles et al., 2007; Blaesse et al., 2009; Pfeffer et al., 2009). But other mechanisms to accumulate chloride exist and maintain GABA depolarizing even in the absence of NKCC1 (Pfeffer et al., 2009). One candidate is the anion-exchanger AE3, which normally accumulates chloride in exchange for intracellular bicarbonate and thereby raises intracellular chloride levels. The so-called GABA switch from excitatory to inhibitory is brought about by the incipient expression of the cation-chloride co-transporter KCC2 (Rivera et al., 1999; Hübner et al., 2001; Stein et al., 2004), which extrudes chloride out of the cell. From our knockout studies other KCl co-transporters like KCC1 (Rust et al., 2007), KCC3 (Boettger et al., 2003; Seja et al., 2012), or KCC4 (Boettger et al., 2002) appear to be less important in the control of neuronal chloride levels.
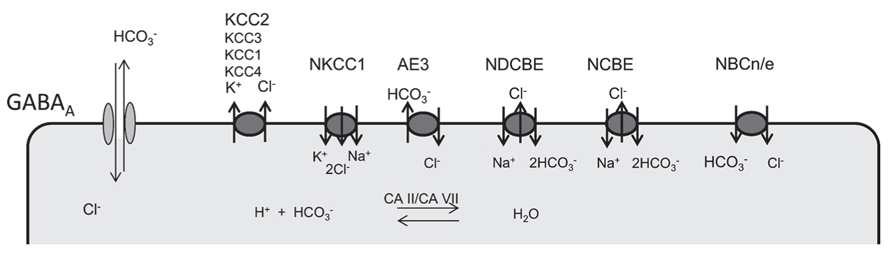
FIGURE 1. Ion transporters involved in the regulation of neuronal chloride and bicarbonate levels. GABA receptors are permeable for both chloride and bicarbonate. Several anion transporters are expressed in neurons and may thus also affect GABA signaling. Whereas NKCC1 is the main chloride accumulating transporter in most neurons, KCC2 is the most important chloride extruder. Anion-transporters of the SLC4A family of bicarbonate transporters can be sub-classified in Na+ dependent (NDCBE, NCBE) and Na+ independent anion exchangers (AE1,2,3). According to the stoichiometry Na+-coupled bicarbonate co-transports can be either electroneutral (NBCn) or electrogenic (NBCe). Although carbonic anhydrases cannot change the existing bicarbonate gradients, they promote anion-transport by members of the SlC4A family and replenish bicarbonate levels.
Several transporters either exchange bicarbonate and chloride or couple the transport of bicarbonate to sodium (Figure 1). For bicarbonate transporters that also transport chloride, the net effect for GABAA receptor signaling is difficult to predict. It is evident that changes of bicarbonate levels do not only directly affect the currents mediated by GABAA receptors, but are also tightly linked to alterations of the pH both within the cells and in the extracellular space, which can have a multitude of different effects. In neurons with a high intracellular chloride concentration as in the immature nervous system, however, the effect of bicarbonate on the GABAA reversal potential is quite low according to the Goldman equation (Farrant and Kaila, 2007).
Anion Exchangers
Whereas there are numerous reviews addressing the role of cation-chloride co-transporters for GABA transmission (Blaesse et al., 2009), the role of bicarbonate and hence the role of neuronal mechanisms to control intracellular bicarbonate levels are less acknowledged. Bicarbonate transport is mediated by members of the SLC4A or the SLC26A family of proteins. As members of the SLC26A family appear to play a minor role for neurons (Dorwart et al., 2008; Majumdar and Bevensee, 2010), we restrict our review to some selected members of the SLC4A family with known relevance for neuronal function and refer to some other more complete reviews (Romero et al., 2004; Dorwart et al., 2008; Majumdar and Bevensee, 2010). The SLC4 family can be subdivided into four main branches: the sodium independent anion-exchangers (AE1, AE2, and AE3) recently reviewed in Alper (2009), and sodium-coupled bicarbonate transporters recently reviewed in Majumdar and Bevensee (2010; Figure 2). The role of AE4 is still unclear: although originally cloned as a sodium-independent anion-exchanger, there is evidence that it rather serves as a Na+/HCO3- co-transporter (Parker et al., 2002). It localizes highly specific to the basolateral membrane of mouse type B intercalated cells and is involved in chloride recovery by these cells (Chambrey et al., 2013). Na+/HCO3- co-transporters can be either electroneutral (NBCn1 and NBCn2) or electrogenic (NBCe1 and NBCe2), whereas the sodium-dependent anion-exchangers (NDAE or NDCBE and NCBE) are electroneutral. SLC4A11 differs from the other family members because it rather mediates borate transport and is hence termed as BTR1 (Park et al., 2004). In the following we will focus on AE3, NCBE, and NDCBE which are strongly expressed in the brain.
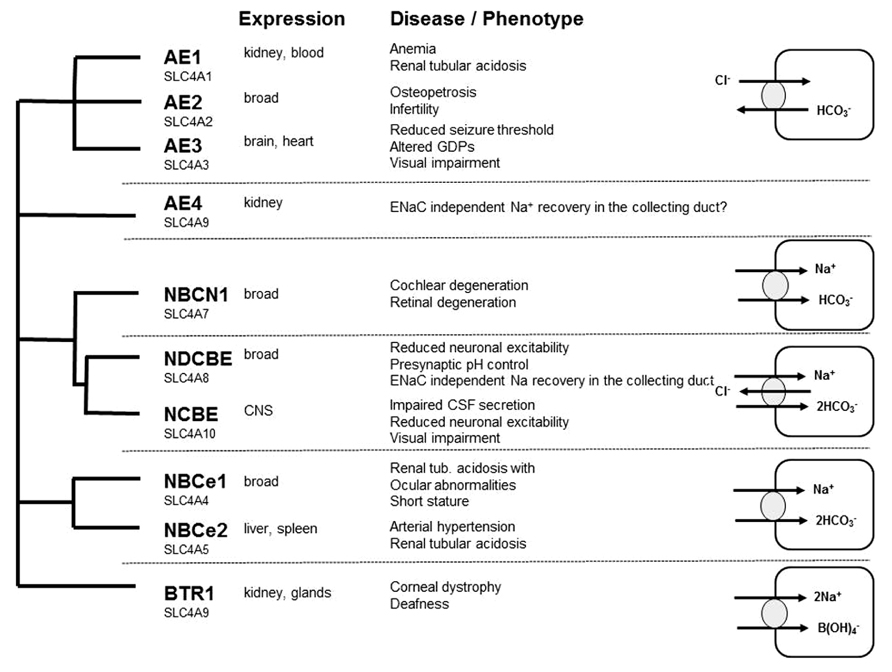
FIGURE 2. Overview of the SLC4A family of bicarbonate transporter: expression, loss of function phenotypes, and transport characteristics.
AE3
In nervous tissue, the AE3 transporter has been localized to neurons (Kopito et al., 1989; Raley-Susman et al., 1993), and to Muller cells and horizontal neurons within the retina (Kobayashi et al., 1994). In many neurons, anion-exchange is thought to be mainly mediated by AE3 (Kopito et al., 1989; Hentschke et al., 2006; Romero et al., 2013). In contrast, AE1 plays an important role for bicarbonate recovery of intercalated cells in the kidney and for red blood cells, where it is one of the most abundant proteins of the plasma membrane (band3). Accordingly, mutations in AE1 can cause renal tubular acidosis and/or hemolytic anemia (Alper, 2009). AE2 is the most widely expressed anion-exchanger, which localizes to the basolateral side in most epithelial cells (Romero et al., 2013). Like AE1 it appears to play a minor role for the control of the neuronal pH. The SLC4A3 gene encoding AE3 employs two different promoters to generate the brain and the cardiac variant, the latter having a shorter amino-terminal amino acid sequence. Because of its broad neuronal expression, the brain variant of AE3 is also often referred to as the neuronal anion-exchanger. Transcripts were already detected at early developmental stages of murine brain development starting around E11 (Hentschke et al., 2006). Because of its early expression and its predicted role to raise the intracellular chloride concentration above the electrochemical equilibrium, AE3 may also contribute to early GABAergic excitation. In particular, it has been hypothesized that AE3 is responsible for chloride accumulation in lateral superior olivary neurons at P0–P3 (Becker et al., 2003). During this time window, these neurons express AE3 but not NKCC1 and depolarize in response to glycine (Balakrishnan et al., 2003). Surprisingly, however, anion-exchange was nearly absent from cultured fetal neurons, although AE3 mRNA was found in both fetal and adult hippocampal neurons (Raley-Susman et al., 1993).
Interestingly, AE3 expression levels in cultured hippocampal neurons from rat increased during long-term exposure to ammonia and caused an ammonia induced increase of the intracellular chloride concentration (Irie et al., 1998), thus supporting a role of AE3 for the regulation of the intraneuronal chloride concentration. At the protein level, a clear band corresponding to AE3, which was absent from knockout tissues, was detected in murine P1 brain lysates with increasing signal intensities at P5 and P15 (Pfeffer et al., 2009). Unfortunately, the subcellular localization of AE3 in the brain is still unclear, because no antibody has been reported that reliably detects endogenous AE3 in brain sections. The GABA reversal potential and GABA-evoked Ca2+ responses of CA1 neurons of AE3 knockout mice did not differ between AE3 knockout and WT mice at P1 (Pfeffer et al., 2009), suggesting that in this type of neuron at this time point chloride accumulation by AE3 may be marginal compared to NKCC1. Nevertheless, this may change with increasing expression levels of AE3 during brain maturation. Supporting that AE3 modulates GABAergic transmission, similar to NKCC1 knockout mice GDPs, which largely depend on a depolarizing action of GABA (Leinekugel et al., 1997; Ben-Ari et al., 2007), were reduced in terms of frequency and amplitudes at postnatal day 5 in AE3 knockout mice (Figure 3; Pfeffer et al., 2009), but these changes may also be related to changes in neuronal pH homeostasis. Although the intraneuronal pH at steady-state conditions in principal neurons of the adult mouse hippocampus did not differ between genotypes, the recovery from an alkaline load was drastically reduced in neurons devoid of AE3 (Hentschke et al., 2006). Hence, the role of AE3 for chloride accumulation in hippocampal neurons should be re-addressed at later developmental stages and in different types of neurons. Indeed, in spinal cord motoneurons chloride accumulation was in part bicarbonate-dependent and sensitive to anion-exchange blockers (Gonzalez-Islas et al., 2009). These findings are in accordance with a previous report on GABA currents in embryonic motoneurons, which were dampened by bumetanide and removal of extracellular bicarbonate (Kulik et al., 2000). It has been estimated that NKCC1 is responsible for approximately two-thirds of the steady-state chloride accumulation, whereas AE3 for the remaining third (Gonzalez-Islas et al., 2009). NKCC1 and AE3 may thus have distinct functions in the recovery of chloride levels following chloride depletion in embryonic motoneurons.
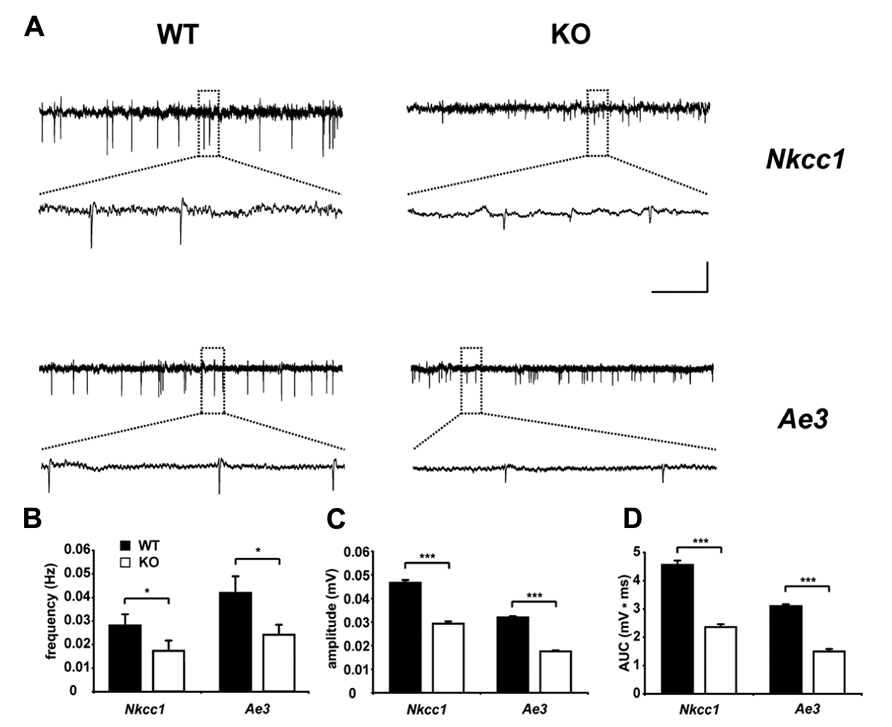
FIGURE 3. Reduced spontaneous electrical activity in hippocampal slices of NKCC1 and AE3 knockout mice. (A) Representative extracellular recordings from the stratum pyramidale (CA3) of postnatal day 5 NKCC1 and AE3 WT and KO slices. The framed parts are shown as enlargements below the original trace. Calibration: horizontal, 2 min (original trace), 7.5 s (enlargement); vertical, 0.04 mV. Quantification of frequency (B), amplitude (C), area under curve (AUC), and (D) of single spontaneous electrical events. The asterisks indicate significant difference (*p < 0.05, ***p < 0.001, t-test). Error bars indicate SEM. Reprinted from Pfeffer et al. (2009).
Overall, no obvious behavioral or morphological alterations of the brain of AE3 knockout mice have been reported (Hentschke et al., 2006; Alvarez et al., 2007). Notably, the seizure threshold in response to various proconvulsive agents was significantly reduced upon disruption of AE3 (Hentschke et al., 2006). This observation supports a previous report that a susceptibility locus for common idiopathic generalized epilepsy maps to chromosomal region 2q36 (Sander et al., 2002), which also includes SLC4A3. Indeed, in a subsequent study a common polymorphism within the coding sequence of SLC4A3, which entails the amino acid exchange Ala867Asp, was associated with an increased risk to develop idiopathic generalized epilepsy (Sander et al., 2002). Moreover, the Ala867Asp variant had a significantly reduced anion-exchange activity compared to wild-type in a heterologous expression system, whereas differences in expression levels or protein trafficking to the plasma membrane were excluded (Vilas et al., 2009). Nevertheless, it is still unclear whether the above-mentioned polymorphism 867Asp itself confers the increased risk for epileptic seizures or another gene in close proximity of SLC4A3 is involved.
Inner retina defects with late onset photoreceptor degeneration with optic nerve and retinal vessel anomalies, which resulted in reduction of the b-wave in electroretinograms, were noted in an independent AE3 knockout mouse (Alvarez et al., 2007). In the retina, the brain variant of AE3 localized to Müller cells, whereas the cardiac variant was detected in horizontal cells. Immuno-labeling of astrocytes showed that inner retina vessels were wrapped by dense astrocytic processes at 8 months of age in AE3 knockout mice. Moreover, inner retina blood vessels formed sporadic loops in the knockout, a finding which was not observed in wild-type mice. Immunoblotting analysis revealed that the Na+/HCO3- co-transporter (NBC1), and carbonic anhydrases (CAs) II and CA XIV protein expression were elevated in AE3 knockout mice mouse retinas, suggesting a partial compensation for loss of AE3. Anion-exchange activity mediated by AE3 is promoted by the action of extracellular CAs (Svichar et al., 2009). AE3 associates with the CAs and forms a bicarbonate transport metabolon to maximize bicarbonate fluxes across the plasma membrane (Casey et al., 2009).
Sodium-Coupled Anion Exchangers
Historically, sodium-dependent anion-exchange (NDAE) which extrudes chloride from cells was the first acid–base transport mechanism described to play a role in the control of intracellular pH (Boron and De Weer, 1976). A cDNA encoding a protein that mediates NDAE (also referred to as NDCBE) was initially cloned from Drosophila (Romero et al., 2000). The mRNA was expressed throughout Drosophila development with a prominent signal in the central nervous system and its disruption resulted in a lethal phenotype in Drosophila. A related cDNA coding for another protein mediating NCBE was cloned from a mouse insulinoma cell line (Wang et al., 2000). This initial transport characterization was subsequently confirmed for rat (Giffard et al., 2003; Damkier et al., 2010), whereas the human cDNA was rather characterized as an electroneutral Na+/HCO3- cotransporter (NBCn2) with chloride self-exchange activity (Parker et al., 2008b). Some of the controversy may be explained by the different expression systems used in the different studies like mammalian cells and Xenopus oocytes, temperature, and composition of solutions, the transfection/injection efficiency or molecular tagging of the transport proteins.
Sodium-coupled anion exchange is activated by intracellular acidification (Schwiening and Boron, 1994), suggesting that regulation of the chloride gradient by NDAEs may be closely linked to the regulation of cellular pH. As prolonged neuronal activity can cause neuronal acidification by efflux of bicarbonate through GABAA receptors (Kaila and Voipio, 1987), sodium-coupled anion exchange may help to maintain a hyperpolarizing chloride reversal potential and thus promote the inhibitory action of GABA. Thus activation of sodium-coupled anion exchange by acidosis may also contribute to seizure termination by promoting a more negative chloride reversal potential and thus promoting the inhibitory effects of GABA.
NDCBE. Several transcript variants have been reported for human and murine NDCBE. The functional comparison of the NDCBE variants expressed in Xenopus oocytes demonstrated that the variants with a shorter C-terminus had a reduced functional expression, whereas the different amino termini did not affect the basal functional expression of NDCBE (Parker et al., 2008a). NDCBE is encoded by SLC4A8 and is broadly expressed in different tissues including brain (Romero et al., 2004). A down-regulation of NDCBE protein expression was shown in different brain regions after chronic hypoxia with a different profile in neonates and adult mice (Chen et al., 2008a). Immunoreactivity for NDCBE was detected in different brain regions with no overlap to astrocyte markers (Chen et al., 2008b). This was also confirmed in a knockout controlled study with an independent polyclconal antibody against NDCBE. Moreover, this study reported that NDCBE localization overlapped with markers of presynaptic glutamatergic but not GABAergic nerve terminals (Sinning et al., 2011). From Western analysis of different brain lysate subfractions and immunogold electron microscopy studies on isolated synaptosomes, it was further concluded that NDCBE is enriched in presynaptic nerve endings of excitatory neurons. The localization in presynaptic glutamatergic terminals was also shown in an independent study, but in contrast to the previous report the latter study also detected NDCBE in terminals of parvalbumin-positive GABAergic cells (Burette et al., 2012). Hence, the authors speculated that NDCBE may play a role as a regulator of GABAergic neurotransmission.
Confirming the important role of NDCBE for pH regulation in neurons, its disruption caused a sustained decrease of the steady-state pH of cultured hippocampal neurons (Sinning et al., 2011). In accordance with the observation that NDCBE co-localizes with presynaptic glutamatergic nerve terminals, the frequency of miniature excitatory postsynaptic currents (mEPSCs) was drastically reduced in a pH-dependent manner in hippocampal neurons of mice devoid of NDCBE, whereas miniature inhibitory postsynaptic currents (mIPSCs) were unchanged. Importantly, the effect on mEPSCs could be at least in part restored by shifting the pH, strongly arguing against a structural defect (Sinning et al., 2011). Its role during early brain maturation, however, has not been studied.
Whether NDCBE also significantly contributes to the control of the intraneuronal chloride concentration in some neurons is still unclear. It was observed that dopaminergic neurons in the rat substantia nigra do not express KCC2, but still exhibit inhibitory responses to GABA that are dependent upon the presence of extracellular bicarbonate (Gulacsi et al., 2003). As the GABA reversal potential was significantly less negative in bicarbonate-free buffer in dopaminergic neurons, a sodium-dependent anion might substitute KCC2 in this type of neuron. This assumption was also supported by the demonstration that complex-spike activity in some auditory interneurons results in a pH-dependent negative shift of the glycine reversal potential, and it was suggested that sodium-coupled anion exchange via Slc4a8 may account for the reduction of intracellular chloride (Kim and Trussell, 2009).
In Caenorhabditis elegans sodium-coupled anion exchange is mediated by ABTS-1. While animals lacking ABTS-1 or KCC2 displayed only mild behavioral defects, disruption of both chloride extruders resulted in a paralytic phenotype (Bellemer et al., 2011). Although direct electrophysiological data were not provided, the authors speculated that the disruption of both transporters results in a reversal of chloride fluxes through GABAA receptors thus rather exciting than inhibiting cells. Moreover, neuronal expression of both transporters was up-regulated during neuronal differentiation and ABTS-1 expression was increased in mutants devoid of KCC2, suggesting that both transporters are important to control the cellular chloride gradient.
NCBE. For NCBE two different splice variants have been identified with different expression profiles (Giffard et al., 2003). The variant missing a 39-bp insert at the 3′ end is predicted to result in a protein with a C-terminal PDZ motif (Giffard et al., 2003). How this might relate to function has not been studied. Our expression analysis in the developing mouse brain with a probe detecting both transcript variants revealed a broad neuronal expression pattern and a particularly strong labeling of the choroid plexus (Hübner et al., 2004). At the protein level, NCBE localized to the basolateral membrane of choroid plexus epithelial cells (Jacobs et al., 2008). There, NCBE serves as a basolateral sodium entry pathway. According to this model, its disruption is predicted to impair cerebrospinal fluid secretion, which is supported by the finding that mice with a targeted disruption of NCBE display a collapse of their brain ventricles (Jacobs et al., 2008). Immunohistological studies revealed that the NCBE protein mainly localized to dendrites and somata of principal neurons, but not to axons or astrocytes (Chen et al., 2008b; Jacobs et al., 2008). There was also a considerable overlap between GABAergic interneurons as identified by the co-localization of GAD and NCBE (Jacobs et al., 2008). However, to which extent NCBE can be detected in different interneuron subtypes, still remains to be addressed. Notably, the ultrastructural analysis also localized NCBE preferentially to dendrites and spines both in the hippocampus as well as in the cerebellum (Jacobs et al., 2008).
Although there was no difference in the steady-state pH of principal neurons of the CA1 hippocampal region of NCBE knockout mice (Jacobs et al., 2008), the recovery to an acid load was delayed. How this affects network excitability was studied in the 4-aminopyridine model of interictal discharges in acute brain slices. The frequency of the interictal-like events at baseline levels did not differ between genotypes, however, the decreased frequency upon a propionate pulse was prolonged in the knockout. In accordance, knockout mice had an increased seizure threshold in response to different seizure inducing agents including pentylenetetrazole or pilocarpine. Quite in contrast to the mouse findings, in some patients with epilepsy larger heterozygous genomic deletions involving SLC4A10 were described (McMilin et al., 1998), however, the genetic evidence that the epilepsy phenotype is directly linked to the heterozygous loss of NCBE or rather to some other genes within this chromosomal region is obscure. Because of the different sites of NCBE expression both in excitatory and inhibitory neurons as well as in the choroid plexus different effects may add up in the total knockout. Thus, the exact role of NCBE for network excitability and synaptic transmission still needs to be addressed in more specific mouse models.
NCBE is also strongly expressed within the retina, where it localizes to ON and OFF bipolar cell axon terminals and to dendrites of OFF bipolar cells, where it co-localized with the main neuronal chloride extruder KCC2 (Hilgen et al., 2012). NCBE was also expressed in starburst amacrine cells, but was absent from neurons known to depolarize in response to GABA, like horizontal cells. These data suggest that NCBE may indeed contribute to the regulation of intracellular chloride and bicarbonate concentration in retinal neurons. Supporting this assumption, knockout mice displayed a decreased visual acuity and contrast sensitivity in behavioral experiments and smaller b-wave amplitudes and longer latencies in electroretinograms (Hilgen et al., 2012).
Carbonic Anhydrases
In the mature rat hippocampus, intense GABAA receptor activation causes neuronal excitation which is strictly dependent on the presence of bicarbonate and suppressed by membrane-permeant inhibitors of CA activity (Staley et al., 1995; Kaila et al., 1997; Fujiwara-Tsukamoto et al., 2007). Fifteen members of the CA family have been identified which differ in tissue distribution and subcellular localization. At least 13 family members catalyze the reversible hydration of CO2 to form bicarbonate and H+, accelerating this spontaneous reaction several thousand-fold. Thereby CAs influence the kinetics and amplitudes of pH transients in distinct intra- and extracellular compartments (Chesler, 2003; Casey et al., 2009) and can affect proton-sensitive membrane proteins involved in neuronal signaling such as GABAA receptors, NMDA receptors, and many more. CA also associate with anion exchangers to form bicarbonate transport metabolons, which enhance bicarbonate fluxes across the plasma membrane (McMurtrie et al., 2004). By forming isoform-specific metabolons with distinct acid–base transporters intracellular neuronal CAs may contribute to developmentally and spatially distinct pHi microdomains. In the brain, extracellular space CA activity is due mainly to isoforms CA IV and CA XIV, which both play important roles in the regulation of intracellular pH in hippocampal neurons by facilitating AE3-mediated Cl-/HCO3- exchange (Casey et al., 2009; Svichar et al., 2009).
CA II and CA VII are the only cytosolic isoforms present in both somata and dendrites of mature hippocampal CA1 pyramidal neurons. The functional expression of CA VII in mouse brain starts around postnatal day 10 (P10) and that of CA II around P20 and coincides with the appearance of bicarbonate-dependent high frequency stimulation (HFS)-induced tonic GABAergic excitation (Ruusuvuori et al., 2004, 2013). Synchronous neuronal activity in the form of GDPs, however, starts much earlier with an onset at approximately P0 (Ben-Ari et al., 1989) and is largely independent from the presence of bicarbonate (Ruusuvuori et al., 2004). These GDPs disappear with the on-going expression of the chloride extruding K+–Cl- cotransporter KCC2 which is up-regulated from P0 to P12 thus rendering GABAA responses hyperpolarizing (Rivera et al., 1999; Hübner et al., 2001; Stein et al., 2004). To study the role for cytoplasmic neuronal CAs for bicarbonate-dependent GABAergic depolarization, we recently established a CA VII knockout mouse model. Remarkably, CA VII knockout mice have a normal life span and show no gross behavioral abnormalities. At P13–14, when CA II is not yet expressed, CA VII KO mice show a complete absence of electrographic seizures (Ruusuvuori et al., 2013). These results point to a crucial role for the developmental expression of intrapyramidal CAs in shaping integrative functions, long-term plasticity and susceptibility to epileptogenesis and put intraneuronal CA in a key position in GABAergic excitation (Kaila et al., 1997; Ruusuvuori et al., 2004). Moreover, these observations give important insights into the antiepileptic actions of CA inhibitors.
Conclusion
There is ample evidence that brain development and brain function critically depends on anion gradients. Whereas chloride has been in the focus of the neuroscientific community, much less is known about bicarbonate. With the development of several mouse models with targeted disruption of selected players of intraneuronal bicarbonate levels, some described in this review, first clues how bicarbonate contributes to proper brain function like the production of the cerebrospinal fluid, neuronal excitability, and synaptic transmission evolved. The role of these processes for brain development is mostly unknown but it emerges that bicarbonate transporters modulate GABAergic transmission already in the developing brain. It will be essential to assess whether this reflects secondary effects in response to changes in pH or whether these effects rather reflect alterations of the existing anion gradients. Bicarbonate definitely plays an essential role for the GABAergic excitation observed upon massive GABAergic stimulation. This process is massively enhanced by CAs, which quickly replenish intraneuronal bicarbonate from P18 onwards. To understand the complex interplay of the different proteins in time and space is an emerging challenge for the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
No financial, commercial, or otherwise relationships that might be perceived by the academic community as representing a potential conflict of interest exist. This work was supported by a grant from the Interdisciplinary Centre for Clinical Research Jena to Christian A. Hübner and Knut Holthoff, funds of the DFG and the BMBF (01GQ0923) to Christian A. Hübner and Knut Holthoff. We thank Knut Kirmse for helpful discussions.
References
Achilles, K., Okabe, A., Ikeda, M., Shimizu-Okabe, C., Yamada, J., Fukuda, A., et al. (2007). Kinetic properties of Cl uptake mediated by Na+-dependent K+–2Cl cotransport in immature rat neocortical neurons. J. Neurosci. 27, 8616–8627.doi: 10.1523/JNEUROSCI.5041-06.2007
Allene, C., Cattani, A., Ackman, J. B., Bonifazi, P., Aniksztejn, L., Ben-Ari, Y., et al. (2008). Sequential generation of two distinct synapse-driven network patterns in developing neocortex. J. Neurosci. 28, 12851–12863.doi: 10.1523/JNEUROSCI.3733-08.2008
Alper, S. L. (2009). Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J. Exp. Biol. 212, 1672–1683.doi: 10.1242/jeb.029454
Alvarez, B. V., Gilmour, G. S., Mema, S. C., Martin, B. T., Shull, G. E., Casey, J. R., et al. (2007). Blindness caused by deficiency in AE3 chloride/bicarbonate exchanger. PLoS ONE 2:e839. doi: 10.1371/journal.pone.0000839
Ammann, D., Lanter, F., Steiner, R. A., Schulthess, P., Shijo, Y., and Simon, W. (1981). Neutral carrier based hydrogen ion selective microelectrode for extra- and intracellular studies. Anal. Chem. 53, 2267–2269.doi: 10.1021/ac00237a031
Arosio, D., Ricci, F., Marchetti, L., Gualdani, R., Albertazzi, L., and Beltram, F. (2010). Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat. Methods 7, 516–518.doi: 10.1038/nmeth.1471
Balakrishnan, V., Becker, M., Lohrke, S., Nothwang, H. G., Guresir, E., and Friauf, E. (2003). Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J. Neurosci. 23, 4134–4145.
Becker, M., Nothwang, H. G., and Friauf, E. (2003). Differential expression pattern of chloride transporters NCC, NKCC2, KCC1, KCC3, KCC4, and AE3 in the developing rat auditory brainstem. Cell Tissue Res. 312, 155–165.
Bellemer, A., Hirata, T., Romero, M. F., and Koelle, M. R. (2011). Two types of chloride transporters are required for GABA(A) receptor-mediated inhibition in C. elegans. EMBO J. 30, 1852–1863.doi: 10.1038/emboj.2011.83
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J. L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325.
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284.doi: 10.1152/physrev.00017.2006
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838.doi: 10.1016/j.neuron.2009.03.003
Boettger, T., Hübner, C. A., Maier, H., Rust, M. B., Beck, F. X., and Jentsch, T. J. (2002). Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 416, 874–878.doi: 10.1038/416874a
Boettger, T., Rust, M. B., Maier, H., Seidenbecher, T., Schweizer, M., Keating, D. J., et al. (2003). Loss of K–Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 22, 5422–5434.doi: 10.1093/emboj/cdg519
Bonifazi, P., Goldin, M., Picardo, M. A., Jorquera, I., Cattani, A., Bianconi, G., et al. (2009). GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science 326, 1419–1424.doi: 10.1126/science.1175509
Bony, G., Szczurkowska, J., Tamagno, I., Shelly, M., Contestabile, A., and Cancedda, L. (2013). Non-hyperpolarizing GABAB receptor activation regulates neuronal migration and neurite growth and specification by cAMP/LKB1. Nat. Commun. 4, 1800.doi: 10.1038/ncomms2820
Bormann, J., Hamill, O. P., and Sakmann, B. (1987). Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J. Physiol. 385, 243–286.
Boron, W. F., and De Weer, P. (1976). Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. J. Gen. Physiol. 67, 91–112.doi: 10.1085/jgp.67.1.91
Bregestovski, P., Waseem, T., and Mukhtarov, M. (2009). Genetically encoded optical sensors for monitoring of intracellular chloride and chloride-selective channel activity. Front. Mol. Neurosci. 2:15. doi: 10.3389/neuro.02.015.2009
Bright, G. R., Fisher, G. W., Rogowska, J., and Taylor, D. L. (1987). Fluorescence ratio imaging microscopy: temporal and spatial measurements of cytoplasmic pH. J. Cell Biol. 104, 1019–1033.doi: 10.1083/jcb.104.4.1019
Brustein, E., Marandi, N., Kovalchuk, Y., Drapeau, P., and Konnerth, A. (2003). “In vivo” monitoring of neuronal network activity in zebrafish by two-photon Ca2+ imaging. Pflugers Arch. 446, 766–773.doi: 10.1007/s00424-003-1138-4
Burette, A. C., Weinberg, R. J., Sassani, P., Abuladze, N., Kao, L., and Kurtz, I. (2012). The sodium-driven chloride/bicarbonate exchanger in presynaptic terminals. J. Comp. Neurol. 520, 1481–1492.doi: 10.1002/cne.22806
Casey, J. R., Sly, W. S., Shah, G. N., and Alvarez, B. V. (2009). Bicarbonate homeostasis in excitable tissues: role of AE3 Cl-/HCO3- exchanger and carbonic anhydrase XIV interaction. Am. J. Physiol. Cell Physiol. 297, C1091–C1102.doi: 10.1152/ajpcell.00177.2009
Chambrey, R., Kurth, I., Peti-Peterdi, J., Houillier, P., Purkerson, J. M., Leviel, F., et al. (2013). Renal intercalated cells are rather energized by a proton than a sodium pump. Proc. Natl. Acad. Sci. U.S.A. 110, 7928–7933.doi: 10.1073/pnas.1221496110
Chen, L. M., Haddad, G. G., and Boron, W. F. (2008a). Effects of chronic continuous hypoxia on the expression of SLC4A8 (NDCBE) in neonatal versus adult mouse brain. Brain Res. 1238, 85–92.doi: 10.1016/j.brainres.2008.08.033
Chen, L. M., Kelly, M. L., Parker, M. D., Bouyer, P., Gill, H. S., Felie, J. M., et al. (2008b). Expression and localization of Na-driven Cl-/HCO3- exchanger (SLC4A8) in rodent CNS. Neuroscience 153, 162–174.doi: 10.1016/j.neuroscience.2008.02.018
Chen, P. Y., Illsley, N. P., and Verkman, A. S. (1988). Renal brush-border chloride transport mechanisms characterized using a fluorescent indicator. Am. J. Physiol. 254, F114–F120.
Damkier, H. H., Aalkjaer, C., and Praetorius, J. (2010). Na+-dependent HCO3- import by the slc4a10 gene product involves Cl- export. J. Biol. Chem. 285, 26998–27007.doi: 10.1074/jbc.M110.108712
Dorwart, M. R., Shcheynikov, N., Yang, D., and Muallem, S. (2008). The solute carrier 26 family of proteins in epithelial ion transport. Physiology (Bethesda) 23, 104–114.doi: 10.1152/physiol.00037.2007
Ebihara, S., Shirato, K., Harata, N., and Akaike, N. (1995). Gramicidin-perforated patch recording: GABA response in mammalian neurones with intact intracellular chloride. J. Physiol. 484 (Pt 1), 77–86.
Fagiolini, M., and Hensch, T. K. (2000). Inhibitory threshold for critical-period activation in primary visual cortex. Nature 404, 183–186.doi: 10.1038/35004582
Farrant, M., and Kaila, K. (2007). The cellular, molecular and ionic basis of GABAA receptor signalling. Prog. Brain Res. 160, 59–87.doi: 10.1016/S0079-6123(06)60005-8
Fatima-Shad, K., and Barry, P. H. (1993). Anion permeation in GABA- and glycine-gated channels of mammalian cultured hippocampal neurons. Proc. Biol. Sci. 253, 69–75.doi: 10.1098/rspb.1993.0083
Fujiwara-Tsukamoto, Y., Isomura, Y., Imanishi, M., Fukai, T., and Takada, M. (2007). Distinct types of ionic modulation of GABA actions in pyramidal cells and interneurons during electrical induction of hippocampal seizure-like network activity. Eur. J. Neurosci. 25, 2713–2725.doi: 10.1111/j.1460-9568.2007.05543.x
Galietta, L. J., Haggie, P. M., and Verkman, A. S. (2001). Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 499, 220–224.doi: 10.1016/S0014-5793(01)02561-3
Garaschuk, O., Linn, J., Eilers, J., and Konnerth, A. (2000). Large-scale oscillatory calcium waves in the immature cortex. Nat. Neurosci. 3, 452–459.doi: 10.1038/74823
Giffard, R. G., Lee, Y. S., Ouyang, Y. B., Murphy, S. L., and Monyer, H. (2003). Two variants of the rat brain sodium-driven chloride bicarbonate exchanger (NCBE): developmental expression and addition of a PDZ motif. Eur. J. Neurosci. 18, 2935–2945.doi: 10.1046/j.1460-9568.2003.03053.x
Gonzalez-Islas, C., Chub, N., and Wenner, P. (2009). NKCC1 and AE3 appear to accumulate chloride in embryonic motoneurons. J. Neurophysiol. 101, 507–518.doi: 10.1152/jn.90986.2008
Graber, M. L., Dilillo, D. C., Friedman, B. L., and Pastoriza-Munoz, E. (1986). Characteristics of fluoroprobes for measuring intracellular pH. Anal. Biochem. 156, 202–212.doi: 10.1016/0003-2697(86)90174-0
Gulacsi, A., Lee, C. R., Sik, A., Viitanen, T., Kaila, K., Tepper, J. M., et al. (2003). Cell type-specific differences in chloride-regulatory mechanisms and GABAA receptor-mediated inhibition in rat substantia nigra. J. Neurosci. 23, 8237–8246.
Hensch, T. K., Fagiolini, M., Mataga, N., Stryker, M. P., Baekkeskov, S., and Kash, S. F. (1998). Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science 282, 1504–1508.doi: 10.1126/science.282.5393.1504
Hentschke, M., Wiemann, M., Hentschke, S., Kurth, I., Hermans-Borgmeyer, I., Seidenbecher, T., et al. (2006). Mice with a targeted disruption of the Cl-/HCO3- exchanger AE3 display a reduced seizure threshold. Mol. Cell. Biol. 26, 182–191.doi: 10.1128/MCB.26.1.182-191.2006
Hilgen, G., Huebner, A. K., Tanimoto, N., Sothilingam, V., Seide, C., Garrido, M. G., et al. (2012). Lack of the sodium-driven chloride bicarbonate exchanger NCBE impairs visual function in the mouse retina. PLoS ONE 7:e46155. doi: 10.1371/journal.pone.0046155
Horn, R., and Marty, A. (1988). Muscarinic activation of ionic currents measured by a new whole-cell recording method. J. Gen. Physiol. 92, 145–159.doi: 10.1085/jgp.92.2.145
Hübner, C. A., Hentschke, M., Jacobs, S., and Hermans-Borgmeyer, I. (2004). Expression of the sodium-driven chloride bicarbonate exchanger NCBE during prenatal mouse development. Gene Expr. Patterns 5, 219–223.doi: 10.1016/j.modgep.2004.08.002
Hübner, C. A., Stein, V., Hermans-Borgmeyer, I., Meyer, T., Ballanyi, K., and Jentsch, T. J. (2001). Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30, 515–524.doi: 10.1016/S0896-6273(01)00297-5
Illsley, N. P., and Verkman, A. S. (1987). Membrane chloride transport measured using a chloride-sensitive fluorescent probe. Biochemistry 26, 1215–1219.doi: 10.1021/bi00379a002
Inglefield, J. R., and Schwartz-Bloom, R. D. (1997). Confocal imaging of intracellular chloride in living brain slices: measurement of GABAA receptor activity. J. Neurosci. Methods 75, 127–135.doi: 10.1016/S0165-0270(97)00054-X
Irie, T., Hara, M., Yasukura, T., Minamino, M., Omori, K., Matsuda, H., et al. (1998). Chloride concentration in cultured hippocampal neurons increases during long-term exposure to ammonia through enhanced expression of an anion exchanger. Brain Res. 806, 246–256.doi: 10.1016/S0006-8993(98)00700-8
Jacobs, S., Ruusuvuori, E., Sipila, S. T., Haapanen, A., Damkier, H. H., Kurth, I., et al. (2008). Mice with targeted Slc4a10 gene disruption have small brain ventricles and show reduced neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 105, 311–316.doi: 10.1073/pnas.0705487105
Kaila, K., Lamsa, K., Smirnov, S., Taira, T., and Voipio, J. (1997). Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J. Neurosci. 17, 7662–7672.
Kaila, K., and Voipio, J. (1987). Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature 330, 163–165.doi: 10.1038/330163a0
Katz, L. C., and Shatz, C. J. (1996). Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138.doi: 10.1126/science.274.5290.1133
Kim, Y., and Trussell, L. O. (2009). Negative shift in the glycine reversal potential mediated by a Ca2+- and pH-dependent mechanism in interneurons. J. Neurosci. 29, 11495–11510.doi: 10.1523/JNEUROSCI.1086-09.2009
Kirmse, K., and Kirischuk, S. (2006). Ambient GABA constrains the strength of GABAergic synapses at Cajal–Retzius cells in the developing visual cortex. J. Neurosci. 26, 4216–4227.doi: 10.1523/JNEUROSCI.0589-06.2006
Kirmse, K., Witte, O. W., and Holthoff, K. (2010). GABA depolarizes immature neocortical neurons in the presence of the ketone body ss-hydroxybutyrate. J. Neurosci. 30, 16002–16007.doi: 10.1523/JNEUROSCI.2534-10.2010
Kirmse, K., Witte, O. W., and Holthoff, K. (2011). GABAergic depolarization during early cortical development and implications for anticonvulsive therapy in neonates. Epilepsia 52, 1532–1543.doi: 10.1111/j.1528-1167.2011.03128.x
Kobayashi, S., Morgans, C. W., Casey, J. R., and Kopito, R. R. (1994). AE3 anion exchanger isoforms in the vertebrate retina: developmental regulation and differential expression in neurons and glia. J. Neurosci. 14, 6266–6279.
Kopito, R. R., Lee, B. S., Simmons, D. M., Lindsey, A. E., Morgans, C. W., and Schneider, K. (1989). Regulation of intracellular pH by a neuronal homolog of the erythrocyte anion exchanger. Cell 59, 927–937.doi: 10.1016/0092-8674(89)90615-6
Kulik, A., Nishimaru, H., and Ballanyi, K. (2000). Role of bicarbonate and chloride in GABA- and glycine-induced depolarization and [Ca2+]i rise in fetal rat motoneurons in situ. J. Neurosci. 20, 7905–7913.
Kuner, T., and Augustine, G. J. (2000). A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron 27, 447–459.doi: 10.1016/S0896-6273(00)00056-8
Kyrozis, A., and Reichling, D. B. (1995). Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J. Neurosci. Methods 57, 27–35.doi: 10.1016/0165-0270(94)00116-X
Leinekugel, X., Medina, I., Khalilov, I., Ben-Ari, Y., and Khazipov, R. (1997). Ca2+ oscillations mediated by the synergistic excitatory actions of GABA(A) and NMDA receptors in the neonatal hippocampus. Neuron 18, 243–255.doi: 10.1016/S0896-6273(00)80265-2
Lindau, M., and Fernandez, J. M. (1986). IgE-mediated degranulation of mast cells does not require opening of ion channels. Nature 319, 150–153.doi: 10.1038/319150a0
Luhmann, H. J., and Prince, D. A. (1991). Postnatal maturation of the GABAergic system in rat neocortex. J. Neurophysiol. 65, 247–263.
Majumdar, D., and Bevensee, M. O. (2010). Na-coupled bicarbonate transporters of the solute carrier 4 family in the nervous system: function, localization, and relevance to neurologic function. Neuroscience 171, 951–972.doi: 10.1016/j.neuroscience.2010.09.037
Marandi, N., Konnerth, A., and Garaschuk, O. (2002). Two-photon chloride imaging in neurons of brain slices. Pflugers Arch. 445, 357–365.doi: 10.1007/s00424-002-0933-7
Markova, O., Mukhtarov, M., Real, E., Jacob, Y., and Bregestovski, P. (2008). Genetically encoded chloride indicator with improved sensitivity. J. Neurosci. Methods 170, 67–76.doi: 10.1016/j.jneumeth.2007.12.016
Mason, M. J., Simpson, A. K., Mahaut-Smith, M. P., and Robinson, H. P. (2005). The interpretation of current-clamp recordings in the cell-attached patch-clamp configuration. Biophys. J. 88, 739–750.doi: 10.1529/biophysj.104.049866
McMilin, K. D., Reiss, J. A., Brown, M. G., Black, M. H., Buckmaster, D. A., Durum, C. T., et al. (1998). Clinical outcomes of four patients with microdeletion in the long arm of chromosome 2. Am. J. Med. Genet. 78, 36–43.doi: 10.1002/(SICI)1096-8628(19980616)78:1<36::AID-AJMG8>3.0.CO;2-J
McMurtrie, H. L., Cleary, H. J., Alvarez, B. V., Loiselle, F. B., Sterling, D., Morgan, P. E., et al. (2004). The bicarbonate transport metabolon. J. Enzyme Inhib. Med. Chem. 19, 231–236.doi: 10.1080/14756360410001704443
Minlebaev, M., Ben-Ari, Y., and Khazipov, R. (2007). Network mechanisms of spindle-burst oscillations in the neonatal rat barrel cortex in vivo. J. Neurophysiol. 97, 692–700.doi: 10.1152/jn.00759.2006
Mohajerani, M. H., and Cherubini, E. (2005). Spontaneous recurrent network activity in organotypic rat hippocampal slices. Eur. J. Neurosci. 22, 107–118.doi: 10.1111/j.1460-9568.2005.04198.x
Morita, K., Tsumoto, K., and Aihara, K. (2005). Possible effects of depolarizing GABAA conductance on the neuronal input-output relationship: a modeling study. J. Neurophysiol. 93, 3504–3523.doi: 10.1152/jn.00988.2004
Morita, K., Tsumoto, K., and Aihara, K. (2006). Bidirectional modulation of neuronal responses by depolarizing GABAergic inputs. Biophys. J. 90, 1925–1938.doi: 10.1529/biophysj.105.063164
Mueller, A. L., Taube, J. S., and Schwartzkroin, P. A. (1984). Development of hyperpolarizing inhibitory postsynaptic potentials and hyperpolarizing response to gamma-aminobutyric acid in rabbit hippocampus studied in vitro. J. Neurosci. 4, 860–867.
Mukhtarov, M., Liguori, L., Waseem, T., Rocca, F., Buldakova, S., Arosio, D., et al. (2013). Calibration and functional analysis of three genetically encoded Cl-/pH sensors. Front. Mol. Neurosci. 6:9. doi: 10.3389/fnmol.2013.00009
Owens, D. F., Boyce, L. H., Davis, M. B., and Kriegstein, A. R. (1996). Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J. Neurosci. 16, 6414–6423.
Park, M., Li, Q., Shcheynikov, N., Zeng, W., and Muallem, S. (2004). NaBC1 is a ubiquitous electrogenic Na+-coupled borate transporter essential for cellular boron homeostasis and cell growth and proliferation. Mol. Cell. 16, 331–341.doi: 10.1016/j.molcel.2004.09.030
Parker, M. D., Boron W. F., and Tanner, M. J. A. (2002). Characterization of human ‘AE4’ as an electroneutral, sodium-dependent bicarbonate transporter. FASEB J. 16, A796.
Parker, M. D., Bouyer, P., Daly, C. M., and Boron, W. F. (2008a). Cloning and characterization of novel human SLC4A8 gene products encoding Na+-driven Cl-/HCO3- exchanger variants NDCBE-A, -C, and -D. Physiol. Genomics 34, 265–276.doi: 10.1152/physiolgenomics.90259.2008
Parker, M. D., Musa-Aziz, R., Rojas, J. D., Choi, I., Daly, C. M., and Boron, W. F. (2008b). Characterization of human SLC4A10 as an electroneutral Na/HCO3 cotransporter (NBCn2) with Cl- self-exchange activity. J. Biol. Chem. 283, 12777–12788.doi: 10.1074/jbc.M707829200
Perkins, K. L., and Wong, R. K. (1996). Ionic basis of the postsynaptic depolarizing GABA response in hippocampal pyramidal cells. J. Neurophysiol. 76, 3886–3894.
Pfeffer, C. K., Stein, V., Keating, D. J., Maier, H., Rinke, I., Rudhard, Y., et al. (2009). NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal development. J. Neurosci. 29, 3419–3430.doi: 10.1523/JNEUROSCI.1377-08.2009
Rae, J., Cooper, K., Gates, P., and Watsky, M. (1991). Low access resistance perforated patch recordings using amphotericin B. J. Neurosci. Methods 37, 15–26.doi: 10.1016/0165-0270(91)90017-T
Raley-Susman, K. M., Sapolsky, R. M., and Kopito, R. R. (1993). Cl-/HCO3- exchange function differs in adult and fetal rat hippocampal neurons. Brain Res. 614, 308–314.doi: 10.1016/0006-8993(93)91049-X
Rheims, S., Minlebaev, M., Ivanov, A., Represa, A., Khazipov, R., Holmes, G. L., et al. (2008). Excitatory GABA in rodent developing neocortex in vitro. J. Neurophysiol. 100, 609–619.doi: 10.1152/jn.90402.2008
Rink, T. J., Tsien, R. Y., and Pozzan, T. (1982). Cytoplasmic pH and free Mg2+ in lymphocytes. J. Cell Biol. 95, 189–196.doi: 10.1083/jcb.95.1.189
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255.doi: 10.1038/16697
Romero, M. F., Chen, A. P., Parker, M. D., and Boron, W. F. (2013). The SLC4 family of bicarbonate (HCO3-) transporters. Mol. Aspects Med. 34, 159–182.doi: 10.1016/j.mam.2012.10.008
Romero, M. F., Fulton, C. M., and Boron, W. F. (2004). The SLC4 family of HCO3- transporters. Pflugers Arch. 447, 495–509.doi: 10.1007/s00424-003-1180-2
Romero, M. F., Henry, D., Nelson, S., Harte, P. J., Dillon, A. K., and Sciortino, C. M. (2000). Cloning and characterization of a Na+-driven anion exchanger (NDAE1). A new bicarbonate transporter. J. Biol. Chem. 275, 24552–24559.doi: 10.1074/jbc.M003476200
Rust, M. B., Alper, S. L., Rudhard, Y., Shmukler, B. E., Vicente, R., Brugnara, C., et al. (2007). Disruption of erythroid K–Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J. Clin. Invest. 117, 1708–1717.doi: 10.1172/JCI30630
Ruusuvuori, E., Huebner, A. K., Kirilkin, I., Yukin, A., Blaesse, P., Helmy, M., et al. (2013). Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. EMBO J. 32, 2275–2286.doi: 10.1038/emboj.2013.160
Ruusuvuori, E., Li, H., Huttu, K., Palva, J. M., Smirnov, S., Rivera, C., et al. (2004). Carbonic anhydrase isoform VII acts as a molecular switch in the development of synchronous gamma-frequency firing of hippocampal CA1 pyramidal cells. J. Neurosci. 24, 2699–2707.doi: 10.1523/JNEUROSCI.5176-03.2004
Sander, T., Toliat, M. R., Heils, A., Leschik, G., Becker, C., Ruschendorf, F., et al. (2002). Association of the 867Asp variant of the human anion exchanger 3 gene with common subtypes of idiopathic generalized epilepsy. Epilepsy Res. 51, 249–255.doi: 10.1016/S0920-1211(02)00152-3
Schwiening, C. J., and Boron, W. F. (1994). Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na+-dependent Cl–HCO3- exchange. J. Physiol. 475, 59–67.
Seja, P., Schonewille, M., Spitzmaul, G., Badura, A., Klein, I., Rudhard, Y., et al. (2012). Raising cytosolic Cl- in cerebellar granule cells affects their excitability and vestibulo-ocular learning. EMBO J. 31, 1217–1230.doi: 10.1038/emboj.2011.488
Sinning, A., Liebmann, L., Kougioumtzes, A., Westermann, M., Bruehl, C., and Hübner, C. A. (2011). Synaptic glutamate release is modulated by the Na+-driven Cl-/HCO3- exchanger Slc4a8. J. Neurosci. 31, 7300–7311.doi: 10.1523/JNEUROSCI.0269-11.2011
Sipila, S. T., Schuchmann, S., Voipio, J., Yamada, J., and Kaila, K. (2006). The cation-chloride cotransporter NKCC1 promotes sharp waves in the neonatal rat hippocampus. J. Physiol. 573, 765–773.doi: 10.1113/jphysiol.2006.107086
Staley, K. J., Soldo, B. L., and Proctor, W. R. (1995). Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 269, 977–981.doi: 10.1126/science.7638623
Stein, V., Hermans-Borgmeyer, I., Jentsch, T. J., and Hübner, C. A. (2004). Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J. Comp. Neurol. 468, 57–64.doi: 10.1002/cne.10983
Svichar, N., Waheed, A., Sly, W. S., Hennings, J. C., Hübner, C. A., and Chesler, M. (2009). Carbonic anhydrases CA4 and CA14 both enhance AE3-mediated Cl-/HCO3- exchange in hippocampal neurons. J. Neurosci. 29, 3252–3258.doi: 10.1523/JNEUROSCI.0036-09.2009
Thomas, R. C. (1974). Intracellular pH of snail neurones measured with a new pH-sensitive glass mirco-electrode. J. Physiol. 238, 159–180.
Thomas, R. C. (1977). The role of bicarbonate, chloride and sodium ions in the regulation of intracellular pH in snail neurones. J. Physiol. 273, 317–338.
Tyzio, R., Cossart, R., Khalilov, I., Minlebaev, M., Hübner, C. A., Represa, A., et al. (2006). Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314, 1788–1792.doi: 10.1126/science.1133212
Tyzio, R., Ivanov, A., Bernard, C., Holmes, G. L., Ben-Ari, Y., and Khazipov, R. (2003). Membrane potential of CA3 hippocampal pyramidal cells during postnatal development. J. Neurophysiol. 90, 2964–2972.doi: 10.1152/jn.00172.2003
Tyzio, R., Holmes, G. L., Ben-Ari, Y., and Khazipov, R. (2007). Timing of the developmental switch in GABA(A) mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia 48(Suppl. 5), 96–105.doi: 10.1111/j.1528-1167.2007.01295.x
Vaughan-Jones, R. D. (1979). Non-passive chloride distribution in mammalian heart muscle: micro-electrode measurement of the intracellular chloride activity. J. Physiol. 295, 83–109.
Verheugen, J. A., Fricker, D., and Miles, R. (1999). Noninvasive measurements of the membrane potential and GABAergic action in hippocampal interneurons. J. Neurosci. 19, 2546–2555.
Vilas, G. L., Johnson, D. E., Freund, P., and Casey, J. R. (2009). Characterization of an epilepsy-associated variant of the human Cl-/HCO3- exchanger AE3. Am. J. Physiol. Cell Physiol. 297, C526–C536.doi: 10.1152/ajpcell.00572.2008
Walker, J. L. (1971). Ion specific liquid ion exchanger microelectrodes. Anal. Chem. 43, 89A–93A.doi: 10.1021/ac60298a039
Wang, C. Z., Yano, H., Nagashima, K., and Seino, S. (2000). The Na+-driven Cl-/HCO3- exchanger. Cloning, tissue distribution, and functional characterization. J. Biol. Chem. 275, 35486–35490.doi: 10.1074/jbc.C000456200
Wang, D. D., Krueger, D. D., and Bordey, A. (2003). GABA depolarizes neuronal progenitors of the postnatal subventricular zone via GABAA receptor activation. J. Physiol. 550, 785–800.doi: 10.1113/jphysiol.2003.042572
Yamada, J., Okabe, A., Toyoda, H., Kilb, W., Luhmann, H. J., and Fukuda, A. (2004). Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 557, 829–841.doi: 10.1113/jphysiol.2004.062471
Keywords: GABA, pH, chloride, bicarbonate, ion transporter
Citation: Hübner CA and Holthoff K (2013) Anion transport and GABA signaling. Front. Cell. Neurosci. 7:177. doi: 10.3389/fncel.2013.00177
Received: 25 July 2013; Accepted: 21 September 2013;
Published online: 24 October 2013.
Edited by:
Laura Cancedda, Istituto Italiano di Tecnologia, ItalyReviewed by:
Alexander Dityatev, German Center for Neurodegenerative Diseases, GermanyYehezkel Ben-Ari, Institut National de la Santé et de la Recherche Médicale, France
Copyright © 2013 Hübner and Holthoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian A. Hübner, Institute of Human Genetics, Jena University Hospital, Friedrich Schiller University Jena, Kollegiengasse 10, 07743 Jena, Germany e-mail:Y2hyaXN0aWFuLmh1ZWJuZXJAbWVkLnVuaS1qZW5hLmRl; Knut Holthoff, Hans Berger Department of Neurology, Jena University Hospital, Friedrich Schiller University Jena, Erlanger Allee 101, 07747 Jena, Germany e-mail:a251dC5ob2x0aG9mZkBtZWQudW5pLWplbmEuZGU=