 Melissa Bowerman1
Melissa Bowerman1 Thierry Vincent1,2
Thierry Vincent1,2 Frédérique Scamps1
Frédérique Scamps1 Florence E. Perrin1,3
Florence E. Perrin1,3 William Camu1,4
William Camu1,4 Cédric Raoul1*
Cédric Raoul1*- 1The Neuroscience Institute of Montpellier, INM, INSERM UMR1051, Saint Eloi Hospital, Montpellier, France
- 2Department of Immunology, Saint Eloi Hospital, Montpellier, France
- 3Integrative Biology of Neuroregeneration, Faculty of Science, University of Montpellier 2, Montpellier, France
- 4Department of Neurology, ALS Reference Center, Gui-de-Chauliac Hospital, Montpellier, France
Amyotrophic lateral sclerosis (ALS) is a fatal paralytic disorder characterized by the progressive and selective loss of both upper and lower motoneurons. The neurodegenerative process is accompanied by a sustained inflammation in the brain and spinal cord. The neuron-immune interaction, implicating resident microglia of the central nervous system and blood-derived immune cells, is highly dynamic over the course of the disease. Here, we discuss the timely controlled neuroprotective and neurotoxic cues that are provided by the immune environment of motoneurons and their potential therapeutic applications for ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the selective and progressive loss of upper and lower motoneurons, with both genetic and sporadic events contributing to the development of the pathological process (reviewed in Bento-Abreu et al., 2010). The term “neuroinflammation” has been attributed to the inflammatory response that occurs within the central nervous system (CNS) concomitantly to neurodegeneration (reviewed in Glass et al., 2010). Astrocytes, microglia, and immune cells are the key cellular modulators of neuroinflammation and have all been shown to actively participate in ALS pathogenesis (Glass et al., 2010; McCombe and Henderson, 2011; Philips and Robberecht, 2011). Importantly, recent reports have highlighted the presence of both neuroprotective and neurotoxic inflammatory cells in ALS animal models and patients that appear to be mainly dependent on the stage of disease progression. Seeing as reviews on the relationship between astrocytic activation and ALS are numerous, we will focus herein on the dynamic functional changes of microglia and immune cells that take place during ALS pathogenesis. A better understanding of these time-dependent modifications is of utmost importance for the development of ALS therapeutic strategies aimed at targeting the neuroinflammatory process.
A Role for Microglia in Neuroinflammation
Activation Profile in Human and Animal Models of ALS
Microglia, the resident immune cells of the CNS, constantly survey the environment and become activated upon alterations resulting from disease or injury eliciting a strong pro-inflammatory response (reviewed in Hanisch and Kettenmann, 2007). In ALS patients, reactive microglia are observed in the motor cortex, motor nuclei of the brainstem, the entire corticospinal tract, the spinal cord, and within the cerebrospinal fluid (CSF; Engelhardt and Appel, 1990; Kawamata et al., 1992; Banati et al., 1995). Given the relationship between astrocytes and microglia and the importance of astrocytosis in ALS (Davalos et al., 2005; Yamanaka et al., 2008), it has been hypothesized that microgliosis may also participate in ALS pathogenesis.
In rodent ALS models, microgliosis occurs in pre-symptomatic and symptomatic SOD1G93A mice (Hall et al., 1998; Alexianu et al., 2001; Petrik et al., 2007; Gerber et al., 2012) and at both onset and early-stage of the disease in SOD1G37R mice (Boillee et al., 2006). An in-depth in vivo characterization of microgliosis in SOD1G93A mice shows that microglia are highly reactive in pre-symptomatic stages while they lose their ability to monitor the environment as the disease progresses (Dibaj et al., 2011). Indeed, microglia isolated from either neonatal or early onset SOD1G93A mice display an activated M2 phenotype and enhance motoneuron survival while microglia isolated from either adult or end stage mice have a classically activated M1 phenotype and induce motoneuron death (Weydt et al., 2004; Liao et al., 2012). In the pre-symptomatic and symptomatic SOD1G93A rat model, microglia aggregates are detected in both the spinal cord and brainstem and display a degenerative and apoptotic phenotype at end stage (Fendrick et al., 2007; Graber et al., 2010). Moreover, microglia of pre-symptomatic SOD1H46R rats express the proliferating marker Ki67 and the phagocytic markers ED1 and major histocompatibility complex (MHC) class II (Sanagi et al., 2010; Bataveljic et al., 2011). These data suggest that microgliosis not only typifies ALS but that microglia function changes during disease progression, thus exerting differential effects on motoneurons.
A Role for Microglia in ALS Pathogenesis
A key finding supporting the contribution of microglia in ALS pathogenesis is the significant extension in lifespan and delay in disease progression when the mutant protein is specifically deleted from macrophages and microglial lineages in both SOD1G37R and SOD1G85R mice (Boillee et al., 2006; Wang et al., 2009). Similarly, bone marrow transplantation (resulting in donor-derived microglia) of SOD1G93A microglia into PU.1-/- mice (that lack CNS microglia at birth) did not induce neurodegeneration whereas wild-type donor-derived microglia transplantation into SOD1G93A; PU.1-/-mice improved survival (Beers et al., 2006).
However, phenotypical analysis of microglia in different regions of SOD1G93A spinal cord suggests that both neuroprotective and neurotoxic population of microglial cells may co-exist during the disease and that depletion of proliferative microglia does not prevent motoneuron degeneration (Gowing et al., 2008; Beers et al., 2011b). Together, these studies thus suggest that microglia participates, through a complex balance between neuroprotective and neurotoxic signals, to ALS disease progression.
Proposed Mechanisms of Microglial-Derived Neurotoxicity
Various misregulated pathways within ALS microglia have been identified that may influence motoneuron survival. Endoplasmic reticulum (ER) stress is a characteristic of ALS pathogenesis (reviewed in Lautenschlaeger et al., 2012). In microglia of both sporadic ALS patients and symptomatic SOD1G93A mice, there is an increased expression of C/EBP homologous protein (CHOP; Ito et al., 2009), a member of the apoptotic ER stress pathway (reviewed in Oyadomari and Mori, 2004). It remains unclear if it directly participates in microglial neurotoxicity but exposure of microglia to interferon gamma (IFNγ), which levels are increased in the spinal cord of ALS mice and patients (Aebischer et al., 2011; Aebischer et al., 2012), elicits inducible nitric oxide (NO) synthase (iNOS) expression. The subsequent production of NO can cause an ER stress response that involves CHOP (Kawahara et al., 2001). Interestingly, several SOD1 mouse models show initiation of a specific ER stress response accompanied by microglial activation (Saxena et al., 2009).
Activation of the ligand-dependent CD14 lipopolysaccharide (LPS) receptor located at the microglial surface (Lacroix et al., 1998) initiates a pro-inflammatory Toll-like receptors (TLRs) dependent cascade (Laflamme and Rivest, 2001; Laflamme et al., 2001). Importantly, neurotoxic microglia activation by extracellular SOD1G93A is mediated by the CD14-TLR2 pathway and induces a subsequent release of pro-inflammatory cytokines, including tumor necrosis factor alpha (TNFα) and interleukin (IL)-1β (Liu et al., 2009; Zhao et al., 2010). Moreover, microglia from sporadic ALS patients show an enhanced TLR2 immunoreactivity (Casula et al., 2011). Microglia may thus participate in motoneuron loss following the specific activation of the CD14-TLR pathway by secreted SOD1 mutant, therefore propagating pro-inflammatory stimuli.
The release of extracellular nucleoside di- and tri-phosphates, in particular ATP, by degenerating neurons can elicit microglia activation through the ionotropic P2X and metabotropic P2Y purinergic receptors which can subsequently elicit a pro-inflammatory response, chemotaxis, and phagocytosis (reviewed in Inoue, 2006; Bours et al., 2011). Notably, P2X is increased within spinal cord microglia of ALS patients (Yiangou et al., 2006). Embryonic microglia and neonatal primary microglial cultures from mutant SOD1 mice display an upregulation of P2X4, P2X7, and P2Y6 receptors (D’Ambrosi et al., 2009). Further, activation of P2X7 in SOD1G93A microglia leads to the production of significantly higher levels of TNFα, which has a neurotoxic effect on motoneuron cultures (Ugolini et al., 2003), and of cyclooxygenase-2 (COX-2), which produces the potent inflammatory mediators prostaglandins (D’Ambrosi et al., 2009).
Moreover, a reduced ATP hydrolysis activity in mutant SOD1 microglia, suggests a potentiation of a purinergic-mediated inflammation that can participate to the neuroinflammatory state of microglial cells. Since ATP induces an astrocytic neurotoxic phenotype through P2X7 receptor signaling (Gandelman et al., 2010), one can hypothesize that increased extracellular ATP in ALS, whether exacerbated by motoneurons and/or microglia contributes to the pathogenic microgliosis.
The Potential Influence of Microglia on Neuronal Excitability
There is presently few assessment of the influence of microglia on motoneuron electrophysiology. However, studies on peripheral nerve or spinal cord injuries show that microglia activation has prominent effects on neuronal inhibitory control and loss of inhibitory control is a contributing mechanism to the motoneuron hyperexcitability that typifies ALS pathogenesis in humans (Bae et al., 2013).
Loss of neuronal inhibitory control occurs by several means including decrease in gamma-aminobutyric acid (GABA)ergic interneurons combined with changes in the expression of the GABAA receptor messenger RNA subunit (Petri et al., 2003; Maekawa et al., 2004). GABAA and glycine receptors are chloride (Cl-) channels and the expression of cation-chloride co-transporter contributes to inhibitory effects of these Cl- currents (Blaesse et al., 2009). Indeed, the entry of Cl- following the opening of GABAA and glycine receptor-gated Cl- channels inhibits neuron excitability by hyperpolarizing membrane potential. Under physiological condition, low intracellular Cl- concentration [Cl-]i is maintained by the potassium (K+)-chloride co-transporter KCC2 that extrudes Cl- from mature neurons (Rivera et al., 1999). Stimulation of spinal microglia following peripheral nerve injury induces a decrease in KCC2 expression among dorsal horn nociceptive neurons (Coull et al., 2003). KCC2 decrease is induced by the brain-derived neurotrophic factor (BDNF) and this is consistent with the previous observation that BDNF can be produced by non-neuronal cells involved in immune responses, including T and B lymphocytes, monocytes, and microglia (Kerschensteiner et al., 1999; Coull et al., 2005). BDNF produces a depolarizing shift in the anion reversal potential of dorsal horn lamina I neurons due to an increase in [Cl-]i. This shift prompts an inversion of inhibitory GABA currents that contributes to neuropathic pain following nerve injury (Coull et al., 2005). Decrease in KCC2 expression is thus responsible for the excitatory effects of GABA on neurons. Microglia activation and BDNF secretion are mediated through ATP activation of microglial P2X receptors. As discussed earlier, P2X receptors might be involved in ALS pathology since a higher density of P2X7-immunoreactive microglial cells/macrophages are found in affected regions of spinal cords from ALS patients (Yiangou et al., 2006). Furthermore, levels of BDNF have been found to be increased in microglial cells isolated from ALS mice at the onset of disease and KCC2 is decreased in vulnerable motoneurons in SOD1G93A mice (Fuchs et al., 2010; Liao et al., 2012). Additionally, BDNF might play a role in the influence of microglia on motoneuron electric activity as suggested by work on spasticity. Spasticity is characterized by a velocity-dependent increase in muscle tone resulting from hyperexcitable stretch reflexes, spasms and hypersensitivity to normally innocuous sensory stimulations. Spasticity develops following spinal cord injury and is also regarded as an ALS clinical symptom (Rowland and Shneider, 2001). The main mechanism hypothesized to be responsible for spasticity is increased motoneuron excitability and increased synaptic inputs in response to muscle stretch due to reduced inhibitory mechanisms. Recently, it has been demonstrated that, following spinal cord injury, increased levels of BDNF mediated spasticity, due to post-transcriptional downregulation of KCC2 (Boulenguez et al., 2010). Together, these studies suggest that reactive microglia in ALS may exert an aberrant effect on the electrical activity of motoneurons and highlight the importance of furthering our understanding of this functional interaction.
Lastly, a hypothetical scenario relates to the defect in astrocytic glutamate transporter and the neurotoxic accumulation of the excitatory amino acid. It has been demonstrated that TNFα promotes glutamate release by activated microglia through the cystine/glutamate exchanger (Xc; Piani and Fontana, 1994). Though the implication of the Xc system in ALS has not yet been investigated, it may represent a potential mechanism of microglia-mediated excitotoxicity that warrants further study (Qin et al., 2006).
The Dual role of Neuroimmunity in Motoneuron Disease
Pathological Phenotype of the Immune System in ALS
In addition to astrocytes and microglia, blood-derived immune cells may also play synergistic and critical functions during disease progression. Presence of a systemic immune activation is suggested by abnormalities observed in the blood and the CSF of ALS patients such as increased numbers of circulating lymphocytes (CD4+ helper T cells, CD8+ cytotoxic T lymphocytes, CTL, and natural killer, NK cells), increased expression of MHC class II molecules on monocytes as well as higher levels of inflammatory chemokines and cytokines (regulated on activation normal T cell expressed and secreted, RANTES, monocyte chemotactic protein, MCP-1, IL-12, IL-15, IL-17, and IL-23; Zhang et al., 2005; Rentzos et al., 2007, 2010, 2012; McCombe and Henderson, 2011). Further, post-mortem studies of brain and spinal cord lesions from ALS patients show that the activation and proliferation of microglia is associated with an infiltration of activated macrophages, mast cells and T lymphocytes which are found in close proximity to degenerating tissues (Engelhardt et al., 1993; Graves et al., 2004; Lewis et al., 2012). An in-depth autopsy of six ALS patients reveals an enrichment of T-cell receptor Vβ2-positive T cells in the spinal cord and CSF, suggesting an antigen-driven T cell selection (Panzara et al., 1999). Finally, ALS patients with a more rapidly progressing pathology show decreased numbers of regulatory T lymphocytes (Tregs), suggesting that the number of Tregs is inversely correlated with disease progression (Beers et al., 2011a; Rentzos et al., 2012). Tregs secrete anti-inflammatory cytokines such as IL-4, IL-10 and transforming growth factor beta (TGF-β) and has been show to induce the production of the neurotrophic factors glial-derived neurotrophic factor (GDNF) and BDNF by astrocytes (Reynolds et al., 2007). Tregs are also able to dampen a T helper (Th)1 pro-inflammatory response and attenuate toxic microglial responses. Contribution of the innate immune system is also suggested by the presence of immunoglobulins and complement deposition as well as a significant increase of NK cells in the blood of ALS patients (Donnenfeld et al., 1984; Engelhardt and Appel, 1990; Rentzos et al., 2012). While these investigations of ALS samples and tissues do not assess the contributory role of the immune system to disease pathogenesis, they do highlight its active presence.
In support of what is observed in humans, ALS rodent models also display a particular immunological phenotype. Indeed, SOD1G93A mice have allowed the demonstration that the inflammatory cellular subtypes are phenotypicaly and functionally different depending upon the disease stage (Liao et al., 2012). During the initial stages, infiltrating CD4+ T cells are mainly Th2 (IL-4+) while there is a skew toward Th1 (IFNγ+) cells and CD8+ T cells (both IL-17A positive and negative) as the disease progresses (Fiala et al., 2010; Beers et al., 2011b). Alteration in inflammatory cell subtypes is associated with, and maybe driven by, differences in Tregs. Interestingly, early symptomatic SOD1G93A mice have an increased number of Tregs and a decreased proliferation of effectors T lymphocytes (Teffs), whereas a decreased numbers of Tregs and an increased proliferation of Teffs is found in end stage animals (Beers et al., 2011a; Zhao et al., 2012). The innate immune system is also affected in ALS rodents, displayed by the substantial increase of NKT cells firstly in the liver and then in the spinal cord of SOD1G93A mice (Chiu et al., 2008; Finkelstein et al., 2011).
Whether neuroinflammation is a cause or a consequence of motoneuron dysfunction is still debated. It is interesting to note that inflammation is not limited to the CNS but systemic with a correlation between disease evolution and levels of plasma LPS as well as the numbers of activated circulating monocytes and T lymphocytes (Zhang et al., 2005, 2009). A thymic dysfunction also parallels the neurodegenerative process in mutant SOD1 mice and ALS patients (Seksenyan et al., 2010). In the CNS of ALS patients, TAR DNA-binding protein 43 (TDP-43) displays an increased expression and interacts with nuclear factor kappa B (NF-κB) in glial and neuronal cells. LPS-activation of NF-κB in microglial cells expressing the TDP-43 mutant is associated with the production of pro-inflammatory cytokines, including TNFα, IL-1β, IL-6, and IFNγ (Swarup et al., 2011). NF-κB, is also an important intermediate of the TLR signaling pathway that contribute to the initiation of inflammatory responses (O’Connell et al., 2012). The central role of inflammation and NF-κB in ALS was recently confirmed by the description in familial ALS of mutations in the gene encoding optineurin, a negative regulator of TNF-induced NF-κB activation (Maruyama et al., 2010).
Additional regulators of the neuroinflammatory response are the microRNAs (miRNA), an abundant class of small, non-coding RNA that regulate gene expression in a wide range of biological processes (O’Connell et al., 2012). Recently, a dominantly inherited mutation in the heterogeneous nuclear ribonucleoprotein (hnRNP) A1 has been associated with familial ALS (Kim et al., 2013). hnRNPA1 is a RNA-binding protein involved in RNA metabolism, including the regulation of alternative pre-mRNA splicing, mRNA export, and stability as well as the processing of miRNA (Guil and Caceres, 2007). Interestingly, hnRNPA1 can directly interact with TDP-43 (Buratti et al., 2005), and TDP-43 was proposed to contribute to the post-translational processing of miRNA through interaction with the endonucleases, Drosha and Dicer (Kawahara and Mieda-Sato, 2012). The activity of Dicer, which processes miRNA precursors at the RNA-induced silencing complex (Wilson and Doudna, 2013), is required to maintain motoneuron functional integrity. Indeed, the conditional deletion of Dicer in vesicular acetylcholine transporter-expressing cells leads to motoneuron degeneration and denervation atrophy in mice (Haramati et al., 2010). Another intriguing link with the miRNA pathway in the neuro-immune interaction has been recently revealed by the demonstration that the neurotransmitter acetylcholine can inhibit the production of pro-inflammatory cytokines, TNFα and IL-6, through induction of miRNA-124 in macrophages (Sun et al., 2013). In addition, a subset of CD4+ T cells has been described to produce acetylcholine to modulate the inflammatory response taking part of the autonomic homeostatic reflexes (Rosas-Ballina et al., 2011). Regarding ALS pathogenesis, a dysfunction of the cholinergic circuit has been reported in the spinal cord of SOD1 mutant mice, early in the disease course (Casas et al., 2013). Moreover, the choline acetyltransferase mRNA is a target of TDP-43 (Polymenidou et al., 2011), and the decrease in cholinergic input in the neuroinflammatory context of Alzheimer’s disease was also shown to lead to the down regulation of hnRNPA1 (Berson et al., 2012). Despite the sequential events implicating miRNAs and the cholinergic signaling needs to be further explored, this evidence concurs toward the contribution of the neuro-immune interaction in the degenerative process.
The information from pre-clinical models and ALS patients suggests that systemic immune activation (innate and adaptive) might play a key role in ALS pathogenesis and may represent an interesting target for the development of novel treatments. However, a better understanding of the specific roles played by the different subtypes of immune cells is of utmost necessity. Indeed, accumulative evidence suggests that inflammatory cells mediate both protective and deleterious effects on motoneuron survival and that these functions vary during disease progression.
The Protective Function of the Immune Response in ALS
Protective immunity, a crucial homeostatic phenomenon in the repair of damaged tissues, results from both the clearance of debris and the effects of cytokines and growth factors delivered by inflammatory cells to the site of injury (Hohlfeld et al., 2000; Schwartz and Moalem, 2001). The neuroprotective ability of immune cells is also evident in ALS. Indeed, when SOD1G93A mice are bred with mice lacking functional T cells or CD4+ T cells, microglia skew toward an M1 inflammatory phenotype and disease progression accelerates, suggesting that CD4+ T cells provide neuroprotection by suppressing the activation of cytotoxic microglia. Accordingly, reconstitution of T cells following bone marrow transplantation of SOD1G93A mice lacking functional T and B cells prolonged their survival and suppressed the activation of M1 microglia (Beers et al., 2008). Further analysis showed that neuroprotection is mainly supported by CD4+CD25+Foxp3+ Tregs that secrete IL-4, thus promoting M2 protective microglia and IL-4 secreting Th2 cells, while inhibiting the neurotoxic Th1 response and IFNγ secretion. The passive transfer of Tregs into ALS mice lacking functional T cells results in lengthened disease duration and prolonged survival (Beers et al., 2011a). Accordingly, these neuroprotective Tregs are increased in the peripheral blood of ALS patients during early stages but their numbers decrease as the disease progression accelerates and are thus inversely correlated with disease progression rates (Beers et al., 2011a; Rentzos et al., 2012; Henkel et al., 2013). Furthermore, Foxp3 and CD25 expression is reduced in Tregs from rapidly progressing patients and are also inversely correlated with disease progression rates (Henkel et al., 2013). Co-culture experiments showed that Tregs suppress the expression of cytotoxic factors Nox2 and iNOS from SOD1G93A microglia through IL-4 secretion and inhibit the proliferation of SOD1G93A Teffs via the combined secretion of IL-4, IL-10, and TGF-β (Zhao et al., 2012). Hence, Tregs enhance the neuroprotective properties of the immune system during the stable disease phase while a switch from a neuroprotective Tregs/M2 to a deleterious Th1/M1 response characterizes disease progression. The key role of this balance between protective and deleterious immune responses in modulating clinical outcome is confirmed by the temporal and regional association between neuroinflammation and motoneuron injury in ALS mice (Beers et al., 2011b). Indeed, initial weakness in the hindlimbs is associated with a Th1 proinflammatory infiltrate in the lumbar spinal cord, while a protective Th2 immune response is observed in the cervical cord and may explain the delayed motor weakness in the forelimbs (Beers et al., 2011b). Therefore, the inflammatory infiltrate observed in ALS lesions appears not simply as a consequence of motoneuron degeneration but is actively involved in the neurodegenerative process. Tregs and Th2 lymphocytes assume the majority of the neuroprotective functions of the immune system and targeting their signaling pathways may be an attractive therapeutic strategy in ALS.
The Neurotoxic Function of the Immune Response in ALS
Cytotoxic T lymphocytes and NK cells are important effector cells of the immune system that eliminate aberrant cells, classically virus-infected cells, or tumorigenic cells (Zhang and Bevan, 2011; Kaur et al., 2012). Interestingly, at symptomatic stage, an increased number of CD8+ T and NK cells is observed in the blood and spinal cord of ALS patients (Calvo et al., 2010; Rentzos et al., 2012). Neurotoxic effects might be associated with a Th1-driven CTL pro-inflammatory immune response. Accordingly, mutant SOD1 Th1 lymphocytes proliferate to a greater extend and produce more IFNγ during the rapidly progressing phase than Th1 lymphocytes isolated during the slowly progressing phase.
Different death pathways induced by CD8+ CTL lymphocytes could potentially lead to motoneuron death in ALS. CTL are antigen-specific effector cells that express the ligand for Fas (FasL) and most potential CTL targets express Fas at their surface. The activation of Fas (CD95) by its cognate ligand FasL commits cells to a death program through a caspase cascade (Peter et al., 2007). Interestingly, the activation of Fas triggers a death pathway in motoneurons that appeared restricted to this cell type (Raoul et al., 1999, 2002, 2006; Bernard-Marissal et al., 2012; Aebischer et al., 2013). Motoneurons expressing ALS-linked SOD1 mutations showed an increased susceptibility to Fas-mediated death through activation of a Fas/NO amplification loop (Raoul et al., 2002, 2006). Accordingly, mutant SOD1 mice with homozygous loss-of-function FasL mutation present a reduced loss of motoneurons and a prolonged life expectancy (Petri et al., 2006). It remains to be determined whether CTL contribute to Fas-induced motoneuron loss. Another cytotoxic mechanism of CTL-mediated killing of target cells is the perforin-granzyme system. Upon recognition of a target cell by CTL, cytotoxic granules containing perforin and granzyme are released in the extracellular space. Perforin is a pore forming protein allowing the entry in the target cells of granzyme serine proteases that subsequently induce caspase activation and cell death (van Domselaar and Bovenschen, 2011). It is noteworthy that increased levels of granzyme A and B isoforms are increased in the serum of ALS patients (Ilzecka, 2011). However, the functional significance of such an increase remains to be determined. IFNγ, which is produced by CTL cells, can exert both immunostimulatory and immunomodulatory effects during an immune response. IFNγ produced by mutant astrocytes and motoneurons can elicit a death program in motoneurons through the activation of the lymphotoxin beta receptor (LT-βR) by its ligand LIGHT (Aebischer et al., 2011, 2012). The genetic deletion of Light in SOD1G93A mice suggests that the LIGHT pathway contributes to the progression phase of the disease. Recently, the intracerebroventicular infusion of neutralizing anti-IFNγ antibody has been shown to delay the motor function decline in SOD1G93A mice, suggesting that IFNγ contributes to ALS pathogenesis (Otsmane et al., 2013). However, The precise contribution of IFNγ in the neuroinflammatory response remains to be investigated.
An infiltration of NK cells has been reported in the spinal cord of symptomatic ALS mice (Chiu et al., 2008). While the role of NK cells in ALS remains unknown, several hypothetical mechanisms can be raised about their pathogenic contribution. Indeed, activated NK cells inhibit neurite outgrowth of cerebellar neurons in a cell contact-dependent manner in vitro (Pool et al., 2012). In sensory neurons, IL-2-activated NK cells have a killing activity that requires the perforin-granzyme system (Backstrom et al., 2000). Further, the production of IFNγ by activated NK cells might directly trigger motoneuron death through the LIGHT/LT-βR pathway or potentiate a cytotoxic Th1/CTL response via the combined action of other NK-related cytokines such as IL-17 or IL-22 (Cella et al., 2010). NK cells thus represent an interesting branch of the immunopathology that should be further considered.
Several studies suggest that humoral immunity and immunoglobulins could also contribute to the disease. Autoantibodies to voltage-gated Ca2+ or K+ channels have been described in ALS patients, which induce specific motoneuron alterations both in vitro and in vivo after passive transfer in mice (Appel et al., 1991; Engelhardt et al., 1995; Demestre et al., 2005; Pagani et al., 2006; Nwosu et al., 2010). Abnormal levels of anti-Fas antibodies, able to induce neuronal apoptosis in vitro, have been detected in the serum of patients with ALS (Yi et al., 2000; Sengun and Appel, 2003). C5a and other complement activation products released after activation of the classical complement pathway by antibodies are elevated in the CSF and spinal cord of ALS mice and patients and specific inhibition of C5a receptor ameliorates disease in SOD1G93A mice (Woodruff et al., 2008; Heurich et al., 2011). Thus, both the innate and adaptive immune system appears to have deleterious consequences on the survival and maintenance of motoneurons in ALS (Figure 1).
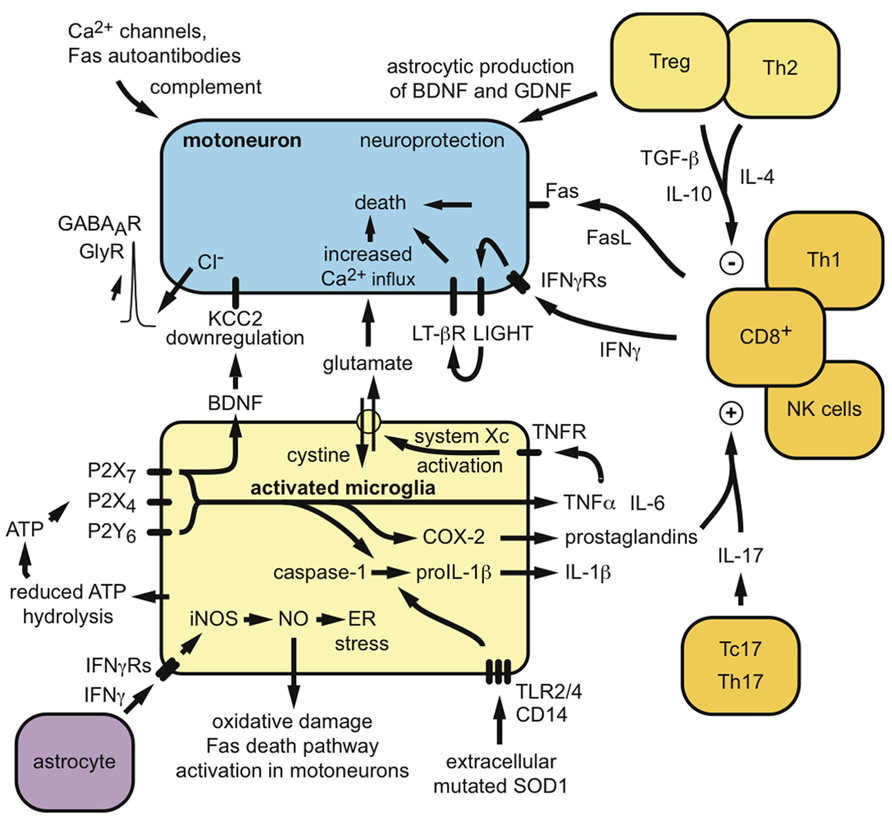
FIGURE 1. Potential mechanisms by which peripheral and central immunity might contribute to the neurodegenerative process in ALS. Both neuroprotective and neurotoxic functions can be proposed for the involvement of microglia and lymphocytes in ALS pathogenesis.
Exploiting the Neuroprotective and Neurotoxic Properties of Neuroimmunity for the Development of Therapeutic Strategies
In light of the dynamic functional changes of microglia and immune cells discussed above, attempts to develop therapeutic strategies targeting neuroinflammation have only emphasized the importance of understanding the temporal neuroinflammatory events in ALS.
In pre-clinical mouse models, genetic deletion of the P2X7 receptor, which was previously described as being upregulated in ALS microglia (D’Ambrosi et al., 2009), resulted in increased motoneuron loss, increased microgliosis, and accelerated disease progression, thus suggesting an unanticipated protective role for the P2X7 receptor (Apolloni et al., 2013). Similarly, as mentioned earlier, genetic depletion of functional T cells or CD4+ cells in SOD1G93A mice lead to increased disease progression, decreased survival as well as promoted production of pro-inflammatory effectors (Beers et al., 2008). Finally, eliminating the expression of galectin-3, a multifunctional immunomodulator that is increased in ALS microglia (Norling et al., 2009), in SOD1G93A mice, also results in aberrant microgliosis and increased disease progression (Lerman et al., 2012). These alterations (P2X7, immune cells and galectin-3) were embryonically and permanently induced, implying that at a certain time-point during the development of the animal and the progression of the disease, these molecular and cellular components are necessary for alleviating certain ALS symptoms and pathological features.
At the clinical level, the failure of certain trials assessing the influence of drugs that directly or indirectly impact neuroinflammation may be due to inappropriate knowledge of the dynamic changes that occur within microglia and immune cells. Indeed, drastic immunosuppressive strategies such as cyclosporine, cyclophosphamide, intravenous immunoglobulin G treatment, and total lymphoid irradiation did not provide any significant benefits to ALS patients (Brown et al., 1986; Drachman et al., 1994; Gourie-Devi et al., 1997). Similarly, drugs used to target specific neuroinflammatory effectors that showed promising results in pre-clinical models such as celecoxib and pioglitazone (Drachman et al., 2002; Schutz et al., 2005), proved to be ineffective in improving motor functions and survival in ALS patients (Cudkowicz et al., 2006; Dupuis et al., 2012).
The progressive spreading, extension and diffusion of the neurodegenerative process that typically occurs in ALS patients may result from the concurrent progressive invasion of the CNS by glial cells and most importantly, the functional changes that take place within these cells. Importantly, an incomplete understanding of said changes could lead to undesired and unexpected results. Indeed, both minocycline and thalidomide (an analog of lenalidomide) revealed serious harmful effects in patients during a randomized placebo-controlled phase III trial and a single arm, open label phase II study, respectively (Gordon et al., 2007; Stommel et al., 2009).
As translational therapy targeting neuroinflammatory and immunomodulatory effectors is rapidly progressing, it has become clear that a step backward is presently required to better assess the temporal functional changes that occur within glial and immune cells in ALS pathogenesis. The cellular environment being composed of both neuroprotective and neurotoxic functions, specific therapeutic windows may dictate the choice of drugs and their pathogenic targets. Alternatively, a combinatory therapeutic approach may be more efficient at modulating the contributions of non-neuronal cells to ALS pathology. Thus, while neuroinflammation undoubtedly plays a role in ALS pathogenesis, therapeutic success will be reached in limiting the activation and amplification of toxic glial and immune cells whilst preserving the cellular subtypes that are beneficial to motoneuron survival.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our work is supported by grants from the Institute National de la Santé et de la Recherche Médicale (INSERM), Association Française Contre les Myopathies (AFM), Association Française Pour la Recherche Sur la SLA (ARSLA), Direction de L’Hospitalisation et de L’Organisation des Soins (DHOS), and the Thierry Latran foundation. Melissa Bowerman is a recipient of a long-term EMBO Marie Curie Fellowship. We apologize to authors whose work could not have been cited due to space limitations.
References
Aebischer, J., Benard-Marissal, N., Pettmann, B., and Raoul, C. (2013). Death receptors in the selective degeneration of motoneurons in amyotrophic lateral sclerosis. J. Neurodegener. Dis. 2013, 746845. doi: 10.1155/2013/746845
Aebischer, J., Cassina, P., Otsmane, B., Moumen, A., Seilhean, D., Meininger, V., et al. (2011). IFNgamma triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death Differ. 18, 754–768. doi: 10.1038/cdd.2010.143
Aebischer, J., Moumen, A., Sazdovitch, V., Seilhean, D., Meininger, V., and Raoul, C. (2012). Elevated levels of IFNgamma and LIGHT in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. Eur. J. Neurol. 19, 752–759. doi: 10.1111/j.1468-1331.2011.03623.x
Alexianu, M. E., Kozovska, M., and Appel, S. H. (2001). Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 57, 1282–1289. doi: 10.1212/WNL.57.7.1282
Apolloni, S., Amadio, S., Montilli, C., Volonte, C., and D’Ambrosi, N. (2013). Ablation of P2X7 receptor exacerbates gliosis and motoneuron death in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 4102–4116. doi: 10.1093/hmg/ddt259
Appel, S. H., Engelhardt, J. I., Garcia, J., and Stefani, E. (1991). Immunoglobulins from animal models of motor neuron disease and from human amyotrophic lateral sclerosis patients passively transfer physiological abnormalities to the neuromuscular junction. Proc. Natl. Acad. Sci. U.S.A. 88, 647–651. doi: 10.1073/pnas.88.2.647
Backstrom, E., Chambers, B. J., Kristensson, K., and Ljunggren, H. G. (2000). Direct NK cell-mediated lysis of syngenic dorsal root ganglia neurons in vitro. J. Immunol. 165, 4895–4900.
Bae, J. S., Simon, N. G., Menon, P., Vucic, S., and Kiernan, M. C. (2013). The puzzling case of hyperexcitability in amyotrophic lateral sclerosis. J. Clin. Neurol. 9, 65–74. doi: 10.3988/jcn.2013.9.2.65
Banati, R. B., Gehrmann, J., Kellner, M., and Holsboer, F. (1995). Antibodies against microglia/brain macrophages in the cerebrospinal fluid of a patient with acute amyotrophic lateral sclerosis and presenile dementia. Clin. Neuropathol. 14, 197–200.
Bataveljic, D., Stamenkovic, S., Bacic, G., and Andjus, P. R. (2011). Imaging cellular markers of neuroinflammation in the brain of the rat model of amyotrophic lateral sclerosis. Acta Physiol. Hung. 98, 27–31. doi: 10.1556/APhysiol.98.2011.1.4
Beers, D. R., Henkel, J. S., Xiao, Q., Zhao, W., Wang, J., Yen, A. A., et al. (2006). Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 103, 16021–16026. doi: 10.1073/pnas.0607423103
Beers, D. R., Henkel, J. S., Zhao, W., Wang, J., and Appel, S. H. (2008). CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. U.S.A. 105, 15558–15563. doi: 10.1073/pnas.0807419105
Beers, D. R., Henkel, J. S., Zhao, W., Wang, J., Huang, A., Wen, S., et al. (2011a). Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134, 1293–1314. doi: 10.1093/brain/awr074
Beers, D. R., Zhao, W., Liao, B., Kano, O., Wang, J., Huang, A., et al. (2011b). Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav. Immun. 25, 1025–1035. doi: 10.1016/j.bbi.2010.12.008
Bento-Abreu, A., Van Damme, P., Van Den Bosch, L., and Robberecht, W. (2010). The neurobiology of amyotrophic lateral sclerosis. Eur. J. Neurosci. 31, 2247–2265. doi: 10.1111/j.1460-9568.2010.07260.x
Bernard-Marissal, N., Moumen, A., Sunyach, C., Pellegrino, C., Dudley, K., Henderson, C. E., et al. (2012). Reduced calreticulin levels link endoplasmic reticulum stress and Fas-triggered cell death in motoneurons vulnerable to ALS. J. Neurosci. 32, 4901–4912. doi: 10.1523/JNEUROSCI.5431-11.2012
Berson, A., Barbash, S., Shaltiel, G., Goll, Y., Hanin, G., Greenberg, D. S., et al. (2012). Cholinergic-associated loss of hnRNP-A/B in Alzheimer’s disease impairs cortical splicing and cognitive function in mice. EMBO Mol. Med. 4, 730–742. doi: 10.1002/emmm.201100995
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838. doi: 10.1016/j.neuron.2009.03.003
Boillee, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Boulenguez, P., Liabeuf, S., Bos, R., Bras, H., Jean-Xavier, C., Brocard, C., et al. (2010). Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 16, 302–307. doi: 10.1038/nm.2107
Bours, M. J., Dagnelie, P. C., Giuliani, A. L., Wesselius, A., and Di Virgilio, F. (2011). P2 receptors and extracellular ATP: a novel homeostatic pathway in inflammation. Front. Biosci. 3:1443–1456. doi: 10.2741/235
Brown, R. H. Jr., Hauser, S. L., Harrington, H., and Weiner, H. L. (1986). Failure of immunosuppression with a ten- to 14-day course of high-dose intravenous cyclophosphamide to alter the progression of amyotrophic lateral sclerosis. Arch. Neurol. 43, 383–384. doi: 10.1001/archneur.1986.00520040063021
Buratti, E., Brindisi, A., Giombi, M., Tisminetzky, S., Ayala, Y. M., and Baralle, F. E. (2005). TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 280, 37572–37584. doi: 10.1074/jbc.M505557200
Calvo, A., Moglia, C., Balma, M., and Chio, A. (2010). Involvement of immune response in the pathogenesis of amyotrophic lateral sclerosis: a therapeutic opportunity? CNS Neurol. Disord. Drug Targets 9, 325–330. doi: 10.2174/187152710791292657
Casas, C., Herrando-Grabulosa, M., Manzano, R., Mancuso, R., Osta, R., and Navarro, X. (2013). Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav. 3, 145–158. doi: 10.1002/brb3.104
Casula, M., Iyer, A. M., Spliet, W. G., Anink, J. J., Steentjes, K., Sta, M., et al. (2011). Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience 179, 233–243. doi: 10.1016/j.neuroscience.2011.02.001
Cella, M., Otero, K., and Colonna, M. (2010). Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. U.S.A. 107, 10961–10966. doi: 10.1073/pnas.1005641107
Chiu, I. M., Chen, A., Zheng, Y., Kosaras, B., Tsiftsoglou, S. A., Vartanian, T. K., et al. (2008). T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. U.S.A. 105, 17913–17918. doi: 10.1073/pnas.0804610105
Coull, J. A., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., et al. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. doi: 10.1038/nature04223
Coull, J. A., Boudreau, D., Bachand, K., Prescott, S. A., Nault, F., Sik, A., et al. (2003). Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942. doi: 10.1038/nature01868
Cudkowicz, M. E., Shefner, J. M., Schoenfeld, D. A., Zhang, H., Andreasson, K. I., Rothstein, J. D., et al. (2006). Trial of celecoxib in amyotrophic lateral sclerosis. Ann. Neurol. 60, 22–31. doi: 10.1002/ana.20903
D’Ambrosi, N., Finocchi, P., Apolloni, S., Cozzolino, M., Ferri, A., Padovano, V., et al. (2009). The proinflammatory action of microglial P2 receptors is enhanced in SOD1 models for amyotrophic lateral sclerosis. J. Immunol. 183, 4648–4656. doi: 10.4049/jimmunol.0901212
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
Demestre, M., Pullen, A., Orrell, R. W., and Orth, M. (2005). ALS-IgG-induced selective motor neurone apoptosis in rat mixed primary spinal cord cultures. J. Neurochem. 94, 268–275. doi: 10.1111/j.1471-4159.2005.03184.x
Dibaj, P., Steffens, H., Zschuntzsch, J., Nadrigny, F., Schomburg, E. D., Kirchhoff, F., et al. (2011). In vivo imaging reveals distinct inflammatory activity of CNS microglia versus PNS macrophages in a mouse model for ALS. PLoS ONE 6:e17910. doi: 10.1371/journal.pone.0017910
Donnenfeld, H., Kascsak, R. J., and Bartfeld, H. (1984). Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J. Neuroimmunol. 6, 51–57. doi: 10.1016/0165-5728(84)90042-0
Drachman, D. B., Chaudhry, V., Cornblath, D., Kuncl, R. W., Pestronk, A., Clawson, L., et al. (1994). Trial of immunosuppression in amyotrophic lateral sclerosis using total lymphoid irradiation. Ann. Neurol. 35, 142–150. doi: 10.1002/ana.410350205
Drachman, D. B., Frank, K., Dykes-Hoberg, M., Teismann, P., Almer, G., Przedborski, S., et al. (2002). Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann. Neurol. 52, 771–778. doi: 10.1002/ana.10374
Dupuis, L., Dengler, R., Heneka, M. T., Meyer, T., Zierz, S., Kassubek, J., et al. (2012). A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS ONE 7:e37885. doi: 10.1371/journal.pone.0037885
Engelhardt, J. I., and Appel, S. H. (1990). IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch. Neurol. 47, 1210–1216. doi: 10.1001/archneur.1990.00530110068019
Engelhardt, J. I., Siklos, L., Komuves, L., Smith, R. G., and Appel, S. H. (1995). Antibodies to calcium channels from ALS patients passively transferred to mice selectively increase intracellular calcium and induce ultrastructural changes in motoneurons. Synapse 20, 185–199. doi: 10.1002/syn.890200302
Engelhardt, J. I., Tajti, J., and Appel, S. H. (1993). Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch. Neurol. 50, 30–36. doi: 10.1001/archneur.1993.00540010026013
Fendrick, S. E., Xue, Q. S., and Streit, W. J. (2007). Formation of multinucleated giant cells and microglial degeneration in rats expressing a mutant Cu/Zn superoxide dismutase gene. J. Neuroinflammation 4, 9. doi: 10.1186/1742-2094-4-9
Fiala, M., Chattopadhay, M., La Cava, A., Tse, E., Liu, G., Lourenco, E., et al. (2010). IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J. Neuroinflammation 7, 76. doi: 10.1186/1742-2094-7-76
Finkelstein, A., Kunis, G., Seksenyan, A., Ronen, A., Berkutzki, T., Azoulay, D., et al. (2011). Abnormal changes in NKT cells, the IGF-1 axis, and liver pathology in an animal model of ALS. PLoS ONE 6:e22374. doi: 10.1371/journal.pone.0022374
Fuchs, A., Ringer, C., Bilkei-Gorzo, A., Weihe, E., Roeper, J., and Schutz, B. (2010). Downregulation of the potassium chloride cotransporter KCC2 in vulnerable motoneurons in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 1057–1070. doi: 10.1097/NEN.0b013e3181f4dcef
Gandelman, M., Peluffo, H., Beckman, J. S., Cassina, P., and Barbeito, L. (2010). Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: implications for amyotrophic lateral sclerosis. J. Neuroinflammation 7, 33. doi: 10.1186/1742-2094-7-33
Gerber, Y. N., Sabourin, J. C., Rabano, M., Vivanco, M., and Perrin, F. E. (2012). Early functional deficit and microglial disturbances in a mouse model of amyotrophic lateral sclerosis. PLoS ONE 7:e36000. doi: 10.1371/journal.pone.0036000
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Gordon, P. H., Moore, D. H., Miller, R. G., Florence, J. M., Verheijde, J. L., Doorish, C., et al. (2007). Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 6, 1045–1053. doi: 10.1016/S1474-4422(07)70270-3
Gourie-Devi, M., Nalini, A., and Subbakrishna, D. K. (1997). Temporary amelioration of symptoms with intravenous cyclophosphamide in amyotrophic lateral sclerosis. J. Neurol. Sci. 150, 167–172. doi: 10.1016/S0022-510X(97)00083-X
Gowing, G., Philips, T., Van Wijmeersch, B., Audet, J. N., Dewil, M., Van Den Bosch, L., et al. (2008). Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J. Neurosci. 28, 10234–10244. doi: 10.1523/JNEUROSCI.3494-08.2008
Graber, D. J., Hickey, W. F., and Harris, B. T. (2010). Progressive changes in microglia and macrophages in spinal cord and peripheral nerve in the transgenic rat model of amyotrophic lateral sclerosis. J. Neuroinflammation 7, 8. doi: 10.1186/1742-2094-7-8
Graves, M. C., Fiala, M., Dinglasan, L. A., Liu, N. Q., Sayre, J., Chiappelli, F., et al. (2004). Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 5, 213–219. doi: 10.1080/14660820410020286
Guil, S., and Caceres, J. F. (2007). The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat. Struct. Mol. Biol. 14, 591–596. doi: 10.1038/nsmb1250
Hall, E. D., Oostveen, J. A., and Gurney, M. E. (1998). Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 23, 249–256. doi: 10.1002/(SICI)1098-1136(199807)23:3<249::AID-GLIA7>3.0.CO;2-#
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Haramati, S., Chapnik, E., Sztainberg, Y., Eilam, R., Zwang, R., Gershoni, N., et al. (2010). miRNA malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. U.S.A. 107, 13111–13116. doi: 10.1073/pnas.1006151107
Henkel, J. S., Beers, D. R., Wen, S., Rivera, A. L., Toennis, K. M., Appel, J. E., et al. (2013). Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 5, 64–79. doi: 10.1002/emmm.201201544
Heurich, B., El Idrissi, N. B., Donev, R. M., Petri, S., Claus, P., Neal, J., et al. (2011). Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J. Neuroimmunol. 235, 104–109. doi: 10.1016/j.jneuroim.2011.03.011
Hohlfeld, R., Kerschensteiner, M., Stadelmann, C., Lassmann, H., and Wekerle, H. (2000). The neuroprotective effect of inflammation: implications for the therapy of multiple sclerosis. J. Neuroimmunol. 107, 161–166. doi: 10.1016/S0165-5728(00)00233-2
Ilzecka, J. (2011). Granzymes A and B levels in serum of patients with amyotrophic lateral sclerosis. Clin. Biochem. 44, 650–653. doi: 10.1016/j.clinbiochem.2011.02.006
Inoue, K. (2006). The function of microglia through purinergic receptors: neuropathic pain and cytokine release. Pharmacol. Ther. 109, 210–226. doi: 10.1016/j.pharmthera.2005.07.001
Ito, Y., Yamada, M., Tanaka, H., Aida, K., Tsuruma, K., Shimazawa, M., et al. (2009). Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 36, 470–476. doi: 10.1016/j.nbd.2009.08.013
Kaur, G., Trowsdale, J., and Fugger, L. (2012). Natural killer cells and their receptors in multiple sclerosis. Brain 136(Pt 9), 2657–2676. doi: 10.1093/brain/aws159
Kawamata, T., Akiyama, H., Yamada, T., and McGeer, P. L. (1992). Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 140, 691–707.
Kawahara, Y., and Mieda-Sato, A. (2012). TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. U.S.A. 109, 3347–3352. doi: 10.1073/pnas.1112427109
Kawahara, K., Oyadomari, S., Gotoh, T., Kohsaka, S., Nakayama, H., and Mori, M. (2001). Induction of CHOP and apoptosis by nitric oxide in p53-deficient microglial cells. FEBS Lett. 506, 135–139. doi: 10.1016/S0014-5793(01)02898-8
Kerschensteiner, M., Gallmeier, E., Behrens, L., Leal, V. V., Misgeld, T., Klinkert, W. E., et al. (1999). Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J. Exp. Med. 189, 865–870. doi: 10.1084/jem.189.5.865
Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Lacroix, S., Feinstein, D., and Rivest, S. (1998). The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol. 8, 625–640. doi: 10.1111/j.1750-3639.1998.tb00189.x
Laflamme, N., and Rivest, S. (2001). Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 15, 155–163. doi: 10.1096/fj.00-0339com
Laflamme, N., Soucy, G., and Rivest, S. (2001). Circulating cell wall components derived from gram-negative, not gram-positive, bacteria cause a profound induction of the gene-encoding Toll-like receptor 2 in the CNS. J. Neurochem. 79, 648–657. doi: 10.1046/j.1471-4159.2001.00603.x
Lautenschlaeger, J., Prell, T., and Grosskreutz, J. (2012). Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 166–177. doi: 10.3109/17482968.2011.641569
Lerman, B. J., Hoffman, E. P., Sutherland, M. L., Bouri, K., Hsu, D. K., Liu, F. T., et al. (2012). Deletion of galectin-3 exacerbates microglial activation and accelerates disease progression and demise in a SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Brain Behav. 2, 563–575. doi: 10.1002/brb3.75
Lewis, C. A., Manning, J., Rossi, F., and Krieger, C. (2012). The neuroinflammatory response in ALS: the roles of microglia and T cells. Neurol. Res. Int. 2012, 803701. doi: 10.1155/2012/803701
Liao, B., Zhao, W., Beers, D. R., Henkel, J. S., and Appel, S. H. (2012). Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 237, 147–152. doi: 10.1016/j.expneurol.2012.06.011
Liu, Y., Hao, W., Dawson, A., Liu, S., and Fassbender, K. (2009). Expression of amyotrophic lateral sclerosis-linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. J. Biol. Chem. 284, 3691–3699. doi: 10.1074/jbc.M804446200
Maekawa, S., Al-Sarraj, S., Kibble, M., Landau, S., Parnavelas, J., Cotter, D., et al. (2004). Cortical selective vulnerability in motor neuron disease: a morphometric study. Brain 127, 1237–1251. doi: 10.1093/brain/awh132
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
McCombe, P. A., and Henderson, R. D. (2011). The role of immune and inflammatory mechanisms in ALS. Curr. Mol. Med. 11, 246–254. doi: 10.2174/156652411795243450
Norling, L. V., Perretti, M., and Cooper, D. (2009). Endogenous galectins and the control of the host inflammatory response. J. Endocrinol. 201, 169–184. doi: 10.1677/JOE-08-0512
Nwosu, V. K., Royer, J. A., and Stickler, D. E. (2010). Voltage gated potassium channel antibodies in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 11, 392–394. doi: 10.3109/17482960903452283
O’Connell, R. M., Rao, D. S., and Baltimore, D. (2012). microRNA regulation of inflammatory responses. Annu. Rev. Immunol. 30, 295–312. doi: 10.1146/annurev-immunol-020711-075013
Otsmane, B., Aebischer, J., Moumen, A., and Raoul, C. (2013). Cerebrospinal fluid-targeted delivery of neutralizing anti-IFNgamma antibody delays motor decline in an ALS mouse model. Neuroreport doi: 10.1097/WNR.0000000000000043 [Epub ahead of print].
Oyadomari, S., and Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389. doi: 10.1038/sj.cdd.4401373
Pagani, M. R., Reisin, R. C., and Uchitel, O. D. (2006). Calcium signaling pathways mediating synaptic potentiation triggered by amyotrophic lateral sclerosis IgG in motor nerve terminals. J. Neurosci. 26, 2661–2672. doi: 10.1523/JNEUROSCI.4394-05.2006
Panzara, M. A., Gussoni, E., Begovich, A. B., Murray, R. S., Zang, Y. Q., Appel, S. H., et al. (1999). T cell receptor BV gene rearrangements in the spinal cords and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 6, 392–405. doi: 10.1006/nbdi.1999.0252
Peter, M. E., Budd, R. C., Desbarats, J., Hedrick, S. M., Hueber, A. O., Newell, M. K., et al. (2007). The CD95 receptor: apoptosis revisited. Cell 129, 447–450. doi: 10.1016/j.cell.2007.04.031
Petri, S., Kiaei, M., Wille, E., Calingasan, N. Y., and Flint Beal, M. (2006). Loss of Fas ligand-function improves survival in G93A-transgenic ALS mice. J. Neurol. Sci. 251, 44–49. doi: 10.1016/j.jns.2006.08.013
Petri, S., Krampfl, K., Hashemi, F., Grothe, C., Hori, A., Dengler, R., et al. (2003). Distribution of GABAA receptor mRNA in the motor cortex of ALS patients. J. Neuropathol. Exp. Neurol. 62, 1041–1051.
Petrik, M. S., Wilson, J. M., Grant, S. C., Blackband, S. J., Tabata, R. C., Shan, X., et al. (2007). Magnetic resonance microscopy and immunohistochemistry of the CNS of the mutant SOD murine model of ALS reveals widespread neural deficits. Neuromol. Med. 9, 216–229. doi: 10.1007/s12017-007-8002-1
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi: 10.1016/S1474-4422(11)70015-1
Piani, D., and Fontana, A. (1994). Involvement of the cystine transport system xc-in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J. Immunol. 152, 3578–3585.
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
Pool, M., Rambaldi, I., Darlington, P. J., Wright, M. C., Fournier, A. E., and Bar-or, A. (2012). Neurite outgrowth is differentially impacted by distinct immune cell subsets. Mol. Cell. Neurosci. 49, 68–76. doi: 10.1016/j.mcn.2011.09.004
Qin, S., Colin, C., Hinners, I., Gervais, A., Cheret, C., and Mallat, M. (2006). System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1–40. J. Neurosci. 26, 3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006
Raoul, C., Buhler, E., Sadeghi, C., Jacquier, A., Aebischer, P., Pettmann, B., et al. (2006). Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proc. Natl. Acad. Sci. U.S.A. 103, 6007–6012. doi: 10.1073/pnas.0508774103
Raoul, C., Estevez, A. G., Nishimune, H., Cleveland, D. W., Delapeyriere, O., Henderson, C. E., et al. (2002). Motoneuron death triggered by a specific pathway downstream of Fas. Potentiation by ALS-linked SOD1 mutations. Neuron 35, 1067–1083. doi: 10.1016/S0896-6273(02)00905-4
Raoul, C., Henderson, C. E., and Pettmann, B. (1999). Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J. Cell Biol. 147, 1049–1062. doi: 10.1083/jcb.147.5.1049
Rentzos, M., Evangelopoulos, E., Sereti, E., Zouvelou, V., Marmara, S., Alexakis, T., et al. (2012). Alterations of T cell subsets in ALS: a systemic immune activation? Acta Neurol. Scand. 125, 260–264. doi: 10.1111/j.1600-0404.2011.01528.x
Rentzos, M., Nikolaou, C., Rombos, A., Boufidou, F., Zoga, M., Dimitrakopoulos, A., et al. (2007). RANTES levels are elevated in serum and cerebrospinal fluid in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 8, 283–287. doi: 10.1080/17482960701419232
Rentzos, M., Rombos, A., Nikolaou, C., Zoga, M., Zouvelou, V., Dimitrakopoulos, A., et al. (2010). Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation? Acta Neurol. Scand. 122, 425–429. doi: 10.1111/j.1600-0404.2010.01333.x
Reynolds, A. D., Banerjee, R., Liu, J., Gendelman, H. E., and Mosley, R. L. (2007). Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J. Leukoc. Biol. 82, 1083–1094. doi: 10.1189/jlb.0507296
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Rosas-Ballina, M., Olofsson, P. S., Ochani, M., Valdes-Ferrer, S. I., Levine, Y. A., Reardon, C., et al. (2011). Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101. doi: 10.1126/science.1209985
Rowland, L. P., and Shneider, N. A. (2001). Amyotrophic lateral sclerosis. N. Engl. J. Med. 344, 1688–1700. doi: 10.1056/NEJM200105313442207
Sanagi, T., Yuasa, S., Nakamura, Y., Suzuki, E., Aoki, M., Warita, H., et al. (2010). Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a presymptomatic transgenic rat model of amyotrophic lateral sclerosis. J. Neurosci. Res. 88, 2736–2746. doi: 10.1002/jnr.22424.
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Schutz, B., Reimann, J., Dumitrescu-Ozimek, L., Kappes-Horn, K., Landreth, G. E., Schurmann, B., et al. (2005). The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J. Neurosci. 25, 7805–7812. doi: 10.1523/JNEUROSCI.2038-05.2005
Schwartz, M., and Moalem, G. (2001). Beneficial immune activity after CNS injury: prospects for vaccination. J. Neuroimmunol. 113, 185–192. doi: 10.1016/S0165-5728(00)00447-1
Seksenyan, A., Ron-Harel, N., Azoulay, D., Cahalon, L., Cardon, M., Rogeri, P., et al. (2010). Thymic involution, a co-morbidity factor in amyotrophic lateral sclerosis. J. Cell Mol. Med. 14, 2470–2482. doi: 10.1111/j.1582-4934.2009.00863.x
Sengun, I. S., and Appel, S. H. (2003). Serum anti-Fas antibody levels in amyotrophic lateral sclerosis. J. Neuroimmunol. 142, 137–140. doi: 10.1016/S0165-5728(03)00263-7
Stommel, E. W., Cohen, J. A., Fadul, C. E., Cogbill, C. H., Graber, D. J., Kingman, L., et al. (2009). Efficacy of thalidomide for the treatment of amyotrophic lateral sclerosis: a phase II open label clinical trial. Amyotroph. Lateral Scler. 10, 393–404. doi: 10.3109/17482960802709416
Sun, Y., Li, Q., Gui, H., Xu, D. P., Yang, Y. L., Su, D. F., et al. (2013). MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. doi: 10.1038/cr.2013.116 [Epub ahead of print].
Swarup, V., Phaneuf, D., Dupre, N., Petri, S., Strong, M., Kriz, J., et al. (2011). Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J. Exp. Med. 208, 2429–2447. doi: 10.1084/jem.20111313
Ugolini, G., Raoul, C., Ferri, A., Haenggeli, C., Yamamoto, Y., Salaun, D., et al. (2003). Fas/tumor necrosis factor receptor death signaling is required for axotomy-induced death of motoneurons in vivo. J. Neurosci. 23, 8526–8531.
van Domselaar, R., and Bovenschen, N. (2011). Cell death-independent functions of granzymes: hit viruses where it hurts. Rev. Med. Virol. doi: 10.1002/rmv.697 [Epub ahead of print].
Wang, L., Sharma, K., Grisotti, G., and Roos, R. P. (2009). The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol. Dis. 35, 234–240. doi: 10.1016/j.nbd.2009.05.002
Weydt, P., Yuen, E. C., Ransom, B. R., and Moller, T. (2004). Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia 48, 179–182. doi: 10.1002/glia.20062
Wilson, R. C., and Doudna, J. A. (2013). Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 42, 217–239. doi: 10.1146/annurev-biophys-083012-130404
Woodruff, T. M., Costantini, K. J., Crane, J. W., Atkin, J. D., Monk, P. N., Taylor, S. M., et al. (2008). The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J. Immunol. 181, 8727–8734.
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253. doi: 10.1038/nn2047
Yi, F. H., Lautrette, C., Vermot-Desroches, C., Bordessoule, D., Couratier, P., Wijdenes, J., et al. (2000). In vitro induction of neuronal apoptosis by anti-Fas antibody-containing sera from amyotrophic lateral sclerosis patients. J. Neuroimmunol. 109, 211–220. doi: 10.1016/S0165-5728(00)00288-5
Yiangou, Y., Facer, P., Durrenberger, P., Chessell, I. P., Naylor, A., Bountra, C., et al. (2006). COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 6:12. doi: 10.1186/1471-2377-6-12
Zhang, N., and Bevan, M. J. (2011). CD8(+) T cells: foot soldiers of the immune system. Immunity 35, 161–168. doi: 10.1016/j.immuni.2011.07.010
Zhang, R., Gascon, R., Miller, R. G., Gelinas, D. F., Mass, J., Hadlock, K., et al. (2005). Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 159, 215–224. doi: 10.1016/j.jneuroim.2004.10.009
Zhang, R., Miller, R. G., Gascon, R., Champion, S., Katz, J., Lancero, M., et al. (2009). Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 206, 121–124. doi: 10.1016/j.jneuroim.2008.09.017
Zhao, W., Beers, D. R., Henkel, J. S., Zhang, W., Urushitani, M., Julien, J. P., et al. (2010). Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 58, 231–243. doi: 10.1002/glia.20919
Keywords: inflammation, microglia, lymphocytes, astrocytes, hyperexcitability, cytokine, therapy
Citation: Bowerman M, Vincent T, Scamps F, Perrin FE, Camu W and Raoul C (2013) Neuroimmunity dynamics and the development of therapeutic strategies for amyotrophic lateral sclerosis. Front. Cell. Neurosci. 7:214. doi: 10.3389/fncel.2013.00214
Received: 09 August 2013; Paper pending published: 10 September 2013;
Accepted: 28 October 2013; Published online: 19 November 2013.
Edited by:
Ricardo Tapia, Instituto de Fisiologïa Celular, Universidad Nacional Autónoma de México, MexicoReviewed by:
Hermona Soreq, The Hebrew University of Jerusalem, IsraelPamela McCombe, The University of Queensland, Australia
Copyright © 2013 Bowerman, Vincent, Scamps, Perrin, Camu and Raoul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cédric Raoul, The Neuroscience Institute of Montpellier, INM, INSERM UMR1051, Saint Eloi Hospital, 80 rue Augustin Fliche, 34091 Montpellier, France e-mail:Y2VkcmljLnJhb3VsQGluc2VybS5mcg==