Abstract
Amyotrophic lateral sclerosis (ALS) is the most common and most aggressive form of adult motor neuron (MN) degeneration. The cause of the disease is still unknown, but some protein mutations have been linked to the pathological process. Loss of upper and lower MNs results in progressive muscle paralysis and ultimately death due to respiratory failure. Although initially thought to derive from the selective loss of MNs, the pathogenic concept of non-cell-autonomous disease has come to the forefront for the contribution of glial cells in ALS, in particular microglia. Recent studies suggest that microglia may have a protective effect on MN in an early stage. Conversely, activated microglia contribute and enhance MN death by secreting neurotoxic factors, and impaired microglial function at the end-stage may instead accelerate disease progression. However, the nature of microglial–neuronal interactions that lead to MN degeneration remains elusive. We review the contribution of the neurodegenerative network in ALS pathology, with a special focus on each glial cell type from data obtained in the transgenic SOD1G93A rodents, the most widely used model. We further discuss the diverse roles of neuroinflammation and microglia phenotypes in the modulation of ALS pathology. We provide information on the processes associated with dysfunctional cell–cell communication and summarize findings on pathological cross-talk between neurons and astroglia, and neurons and microglia, as well as on the spread of pathogenic factors. We also highlight the relevance of neurovascular disruption and exosome trafficking to ALS pathology. The harmful and beneficial influences of NG2 cells, oligodendrocytes and Schwann cells will be discussed as well. Insights into the complex intercellular perturbations underlying ALS, including target identification, will enhance our efforts to develop effective therapeutic approaches for preventing or reversing symptomatic progression of this devastating disease.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a non-cell-autonomous disease targeting motor neurons (MNs) and neighboring glia, with microgliosis directly contributing to neurodegeneration (Beers et al., 2006; Lobsiger and Cleveland, 2007; Yamanaka et al., 2008a). Indeed, the neurodegenerative process in ALS was shown to be accompanied by a sustained inflammation in the brain and spinal cord (SC) (Bowerman et al., 2013). Recently, microglia were suggested to be implicated in ALS initiation (Gerber et al., 2012), as well as to lose their surveillance capacity by switching from an activated to a neurodegenerative phenotype as the disease progresses (Weydt et al., 2004; Dibaj et al., 2011). Therefore, a better therapeutic strategy should envisage the recovery of healthy microglia from those transformed cells near the most affected MNs in the SC. In such way we may preserve the passage of toxic mediators to environmental cells, and maintain the homeostatic conditions.
For the vast majority of patients with ALS, the etiology of the disorder is unknown. Actually, only some ALS cases (less than 10%) have been linked to mutations in a number of genes, including in the enzyme Cu, Zn superoxide dismutase 1 (SOD1), TAR DNA binding protein (TDP-43), fused in sarcoma (FUS), optineurin (OPTN), valosin-containing protein (VCP), ubiquilin 2 (UBQLN2), profilin 1 (PFN1), and chromosome 9 open reading frame 72 (C9ORF72) repeat expansions (Tovar et al., 2009a; DeJesus-Hernandez et al., 2011; Ince et al., 2011; Renton et al., 2011, 2014; Bertolin et al., 2013). Interestingly, both the non-genetic and the genetic forms of ALS are suggested to have common pathogenic mechanisms (Lilo et al., 2013), as well as similar clinical courses and dysfunctional features, such as the abnormal accumulation of neurofilaments in degenerating MNs (Julien, 2001). Actually, cytoplasmic aggregation of nuclear TDP-43 and FUS in the degenerating neurons and glia of ALS patients, and release of the accumulated cytoplasmic mutant SOD1 (mSOD1) to the extracellular space that can be taken up by other cells, are common features (Li et al., 2013; Ogawa and Furukawa, 2014). The identification of C9ORF72 repeat expansions in patients with ALS but without a family history of ALS challenged the division between genetic (familial) and non-genetic (sporadic) cases (Turner et al., 2013). As indicated by Kiernan (2014), the true substrate of ALS may reside in a pathogenic signature of nuclear protein mishandling.
There are several in vitro and in vivo models of MN degeneration. In vitro experimental models include SC cultures, NSC-34 cell line expressing the mSOD1 and organotypic cultures, while the axotomy-induced MN death, the naturally occurring ALS models, and the transgenic models are the most commonly used in vivo models (Elliott, 1999; Tovar et al., 2009a). Among the various transgenic models used in the study of ALS pathogenesis (Weydt et al., 2004; Kato, 2008), the transgenic rodent overexpressing mSOD1, in particular the SOD1G93A strain, is the most utilized and characterized. Transgenic mice containing other mSOD1 genes (G85R, G37R, D90A, or G93A missense mutations or truncated SOD1) and the related mutant (G86R) mouse have also shown progressive neurodegeneration of the motor system and resemblance to ALS (for review, see Van Den Bosch, 2011). Distinctive injurious effects between SOD1G93A and SOD1H46R on two different genetic backgrounds were recently recognized (Pan et al., 2012). Additionally developed models are based on TDP-43 (Wegorzewska et al., 2009; Liu et al., 2013; Yang et al., 2014) and FUS mutations (Verbeeck et al., 2012), but none of these models is currently used to study the pathogenesis of ALS and to test new drugs. Thus, the human mSOD1 murine model is the most widely used in the evaluation of the involved molecular targets, biomarkers and novel drugs/treatments for ALS. Apart from developing loss of MNs and symptoms that resemble human ALS by mSOD1, the model evidences molecular links between genetic and non-genetic cases of ALS (Andjus et al., 2009; Synofzik et al., 2010). To note, that non-genetic perturbations of the wild-type (wt) SOD1 protein may lead to SOD1 misfolding with a conformation much similar to genetic SOD1 variants (Cereda et al., 2006). Therefore, in this review we will summarize the most recent developments obtained in the SOD1G93A transgenic model to give consistency and cohesion between the data disclosed, and because we admit that common factors and pathways are shared in both genetic and non-genetic derived ALS cases, in particular changes in microglia performance and in neuron–glia communication.
It was initially considered that the selective death of MNs expressing the mutant protein was the player in the disease onset. However, non-cell-autonomous processes associated with mSOD1 in glial cells are believed to be implicated not only in disease progression and extent, but also to be related with the onset and early stage of the disease, thus underlying MN dysfunction and loss. Indeed, healthy glia evidenced to delay the progression of the disease (Boillée et al., 2006b; Yamanaka et al., 2008b) and the replacement of mSOD1 microglia by wt microglia slowed disease progression and prolonged mice survival (Lee et al., 2012). This finding is in line with previous studies showing that mSOD1 in microglia leads to the disease (Clement et al., 2003) and that the reduction of the mutant levels in the cells slows ALS progression (Boillée et al., 2006b). Indeed, damage to MNs by neighboring cells expressing mSOD1 seems to be required for MN degeneration (Pramatarova et al., 2001; Lino et al., 2002). Accumulating knowledge on the active participation of different microglia phenotypes in ALS was recently obtained when microglia were isolated from SOD1G93A rats at presymptomatic, symptom onset and end-stage periods (Nikodemova et al., 2013). Microglia were shown to be regionally different and to evidence a heterogeneity of phenotypes with the disease progression. Thus, it will be interesting to investigate the influence of changes caused by aging in the performance of the mutated microglia, namely on the interconnectivity with neurons and other glial cells. Moreover, our preliminary data indicate a decreased phagocytic and migration ability of healthy microglia by the soluble factors (SFs) released by NSC-34 cells expressing mSOD1, thus causing the loss of important microglia properties (Cunha, 2012). In addition, when this microglia was co-cultured with the mMNs we observed a reduction of the activation of matrix metalloproteinases (MMP)-2 and -9 in the extracellular media after cultivation for 4 days in vitro (DIV) (Barbosa, 2013), evidencing the beneficial effect of the healthy microglia in decreasing mMN stress.
Finally, we provide an outlook on the extent to which a diverse cellular environment may determine different pathological windows along disease progression opening new opportunities to explore distinct therapeutic approaches to either trigger a less reactive microglia phenotype at the early-onset, or a recovery of microglia dynamics at late-stage ALS. It will be also summarized recent advances in regenerative medicine technologies with potential to reverse or halt ALS progression by slowing MN death.
NEURODEGENERATIVE NETWORKING IN ALS
Neuronal homeostasis and survival were shown to be compromised in ALS due to multiple aberrant biological processes and to deregulated communication between neurons and glial cells in the brain and in SC by the disease. ALS, once considered a MN disease, is now known to have multiple influences and regarded as a multi-cellular/multi-systemic disease (Nardo et al., 2011). In fact, MN death seems to be driven by a convergence of damaging mechanisms, including glial cell pathology and inflammatory conditions, such as microglial activation or the invasion of lymphocytes, and calcium dysregulation (Grosskreutz et al., 2010; Beers et al., 2011b). It can be also determined by excitotoxicity due to the selective loss of the astrocytic glutamate transporter GLT-1, and consequent accumulation of extracellular glutamate (Rothstein, 2009). Our late results with astrocytes isolated from SOD1G93A mice and cultivated for 13 DIV suggest that both GLT-1 and glutamate aspartate transporter (GLAST) are compromised (personal communication) and corroborate other findings indicating that expression of such transporters are less potently activated by lipopolysaccharide (LPS) in astrocytes from the mSOD1 model than in those from the wt mice (Benkler et al., 2013). Other contributing processes include from neurovascular changes and compromised barriers of the central nervous system (CNS), to dysfunctional communication between neurons, abnormal neuron–glia interactions, and microglia and astroglia loss-of-function.
In fact, for the optimal functioning of the CNS, i.e., brain and SC, accounts the constant immune surveillance promoted by cells such as microglia, and the blood–brain barrier (BBB), the blood–SC barrier (BSCB), and the blood–cerebrospinal fluid (BCSF) barrier that uniquely shield CNS from potential mediators of infection and damage. Recently, brain pericytes, as important components of the neurovascular unit in the CNS, have shown to have pleiotropic and regulatory activities in brain vessel function and homeostasis, blood flow and barrier function (Lange et al., 2012). Astrocytes also participate in maintaining homeostasis and supporting neuronal function. In addition to microglia, neurons have lately been indicated to intervene in immune responses through control of T cells infiltration into the CNS and glial cell immunoreactivity (Czirr and Wyss-Coray, 2012; Tian et al., 2012; Liblau et al., 2013). This integrative network sustains ionic, energetic, and redox homeostasis for proper function.
Moreover, ALS progression was suggested to involve cell-to-cell transmission of mSOD1 aggregates involving MNs, microglia and astrocytes, similarly to prion disease (Chia et al., 2010; Munch et al., 2011). Interestingly, the transcellular spread of SOD1 aggregates evidenced to not require cell-to-cell contacts but to depend from their fragmentation and extracellular release. However, additional studies should demonstrate that similar findings also occur in vivo. Intriguingly, while extracellular mSOD1G93A has shown to not have direct toxic effects on MNs, it morphologically and functionally activates microglia, supporting the non-cell-autonomous nature of MN toxicity in ALS (Zhao et al., 2010). Indeed, it was shown that this extracellular mSOD1 can be endocytosed into microglia, determining the activation of caspase-1 and the up-regulation of interleukin (IL)-1β (Zhao et al., 2013).
Another interesting concept is that astrocyte and microglia activation, which is regulated by a variety of signaling pathways, should not be considered merely as pernicious for CNS homeostasis, once it promotes metabolic support, wound healing and repair. In fact, the production of cytokines and chemokines initiate and coordinate diverse cellular and intercellular actions. Although neurons and glial cells of the CNS express receptors for cytokines and chemokines, the biological consequence of receptor activation is not fully understood. In contrast, it must be considered as part of the pathological processes the excessive or deregulated signaling pathways leading to microglia and astrocyte abnormalities that culminate in abnormal CNS function.
Thus, progressive neurodegeneration of MNs in ALS may result from a combination of intrinsic MN vulnerability to mSOD1 aggregates and of non-cell-autonomous toxicity derived from neighboring cells (Tables 1 and 2). Therefore, pathological changes in ALS indicate a broken homeostasis in the CNS. We will focus on how CNS homeostasis is lost in ALS and in what way BBB and neural networks dysregulation contribute to neurodegeneration in ALS.
Table 1
| Changes in MN signaling by ALS | ALS stages | Reference |
|---|---|---|
| Elevation of matrix metalloproteinase (MMP)-9 | Pre-symptomatic phase | Soon et al. (2010), Kaplan et al. (2014) |
| Impaired antioxidative Keap1/Nrf2/ARE system | Along disease progression | Mimoto et al. (2012) |
| Oxidative and nitrosative stress | Early phase | Drechsel et al. (2012) |
| Glutamate excitotoxicity | Onset and progression | Spalloni et al. (2013) |
| Release of ATP | Along disease progression | Glass et al. (2010) |
| Protein misfolding, aggregation, and accumulation | Onset and progression | Julien (2001), Li et al. (2013) |
| Changes in fractalkine (CX3CL1), CD200, and CCL21 | Not clarified | – |
| Decreased high-mobility group box protein 1 (HMGB1) cellular expression | Advanced phase | Lo Coco et al. (2007), Fang et al. (2012) |
| Release of neuregulin-1 (NRG1) | Along disease progression | Song et al. (2012) |
| Up-regulation of major histocompatibility complex (MHC) class I and β2-microglobulin mRNAs | Along disease progression | Staats et al. (2013) |
Functional alterations of motor neurons (MNs) in amyotrophic lateral sclerosis (ALS): candidate molecular targets.
Table 2
| Mutant SOD1 cells | Loss of supportive functions | Contribution to ALS disease and MN death | Reference |
|---|---|---|---|
| Astrocytes | Deficient astrocyte-specific glutamate transporter EAAT2 (GLT-1) | Increase in the excitatory amino acid glutamate | Valori et al. (2014) |
| Increased release of D-serine | co-activator of the N-methyl-D-aspartate (NMDA) receptors, exacerbating glutamate toxicity on MNs | Valori et al. (2014) | |
| Mitochondrial dysfunction | Increased production of reactive oxygen species (ROS) | Valori et al. (2014) | |
| Release of interferon-γ and transforming growth factor-β (TGF-β) | Increased neuroinflammation | Valori et al. (2014) | |
| Ubiquitin- and active caspase-3-immunopositive | Degenerating astrocytes at the pre-symptomatic stage when MNs show axonal damage but are still alive | Valori et al. (2014) | |
| Increased nerve growth factor (NGF) and NO production | MN apoptosis | Pehar et al. (2004) | |
| Astrocytes (aberrant) | Increased S100B and connexin-43 (Cx-43) | Decreased MN survival | Diaz-Amarilla et al. (2011) |
| Microglia (spinal cord – early stage) | Recruitment of peripheral monocytes to the CNS | Neuronal viability impairment | Butovsky et al. (2012) |
| Microglia (spinal cord – end stage) | Decreased expression of M1 and M2 markers | Decreased reactivity to stimuli | Nikodemova et al. (2013) |
| Microglia (M2) – early stage | High levels of anti-inflammatory cytokines and neurotrophins | Enhancement of MN survival (neuroprotection) at ALS early stage | Zhao et al. (2013) |
| Microglia (M1) – progressive stage | Increased release of reactive oxygen species (ROS), tumor necrosis factor-α (TNF-α) and interleukin (IL)-1β | Toxicity to MN (death) in the late rapid phase of ALS | Zhao et al. (2013) |
| Dystrophic microglia – end stage | Decreased migration and phagocytosis by aging (not yet confirmed in ALS) | Neuronal degeneration by failure of the senescent microglia response to stimuli | Luo and Chen (2012) |
| Oligodendrocytes | Loss of the monocarboxylate transporter 1 (MCT1) | Decreased delivery of the metabolic substrate lactate to MNs and axonal sufferance | Philips et al. (2013) |
| NG2+ cells | Increased proliferation rate and degeneration of early-born oligodendrocytes | Gray matter demyelination | Kang et al. (2013) |
| Schwann cells | Signs of distress at the asymptomatic stage | Not known | Valori et al. (2014) |
Glial impairment and deregulated glia–motor neuron (MN) interconnectivity in amyotrophic lateral sclerosis (ALS).
MOTOR NEURON DYSFUNCTION
A certain number of cases are linked to mutations in SOD1 and candidate mechanisms are the formation of protein aggregates and pro-oxidant effects. Therefore, most of the experiments are generally conducted in the MN-like hybridoma cell line NSC-34 expressing human mSOD1 (hSOD1G93A) (Tovar et al., 2009a) or in the transgenic mice generated by Gurney et al. (1994) that also over-express hSOD1G93A (Riboldi et al., 2011).
SOD1 aggregates are observed in both genetic and non-genetic ALS cases, but their contribution to MN toxicity remains to be established, although deregulation of Golgi, endoplasmic reticulum (ER), and mitochondria, together with axonal transport defects have been indicated (for review, see Boillée et al., 2006a; Rothstein, 2009). Interestingly, when working with the NSC-34/hSOD1G93A cells we found that one of the most sensitive indicators of SOD1 accumulation and neuronal dysfunction was the elevation of MMP-9, but not of MMP-2 that remained unchanged (Cunha, 2012; Vaz et al., 2014). Elevation of the MMP-9 levels was previously observed in the SC of ALS mice from pre-symptomatic phase, predominantly in MNs but also in glia (Soon et al., 2010). These authors suggest that circulating MMP-9 is associated with the disease onset and is mainly derived from degenerating SC MNs and circulating cells. Its inhibition was shown to enhance animal survival in more than 30% (for review, see Riboldi et al., 2011). It was also found in the CNS, muscles, plasma, and skin of patients. Major inducers are reactive oxygen species (ROS) and cytokines released by microglia. Once MMP-9 is indicated to promote the regeneration of the injured neuron, it may be hypothesized that its increase results from a MN response to the pathology. However, it was recently claimed that MMP-9 is indeed a determinant of the selective neurodegeneration (Kaplan et al., 2014). The Authors demonstrated that MMP-9 is only expressed by the selectively vulnerable fast MNs and can originate ER stress and axonal dye-back. This finding provides a basis for considering MMP-9 as a candidate target for novel therapeutic approaches to ALS.
Oxidative damage at the level of proteins, lipids, and DNA was also observed in the transgenic hSOD1 mice models (Liu et al., 1999; Casoni et al., 2005; Poon et al., 2005; Barbosa, 2013) where mSOD1 revealed to be the most severely oxidized protein mouse (Andrus et al., 1998). Also accounting to MN disturbance is the failure of the Keap1/Nrf2/ARE system in regulating stress proteins, as evidenced along disease progression in MNs from the SC of hSOD1G93A mice (Mimoto et al., 2012). In addition, failure in MN autophagy was revealed as critical in the pathogenesis and progression of ALS (for review, see Pasquali et al., 2009; Chen et al., 2012; Otomo et al., 2012).
Currently, there is no cure for the ALS disease, although Riluzole, the only drug approved by the U.S. Food and Drug Administration (FDA), was shown to prolong median patient survival by 2–3 months and to be more effective when administered at an early stage of the disease (Zoccolella et al., 2007; Lee et al., 2013a). More potent therapeutic strategies may derive from better target identification and clarification of signaling mechanisms able to be modulated and from the use of cell-based therapies, such as the administration of mononuclear cells from human umbilical cord blood (Garbuzova-Davis et al., 2012) and of mesenchymal stromal (stem) cells (Uccelli et al., 2012), as referred to in Section “Challenges to Nerve Regeneration in ALS.” Bellow we introduce what is known about the most prominent MN membrane proteins and SFs suggested to be implicated in ALS pathology, but still deserving to be more explored.
Perturbations in glutamate handling
Glutamate excitotoxicity is one primarily cause of neuronal death by necrosis and apoptosis and a critical player in ALS onset and progression. Indeed the blockade of the glutamate transporter in a SC organotypic slice model from SOD1G93A rats has shown to result in an increased survival of MNs (Yin and Weiss, 2012). Glutamate, by activating the glutamatergic ionotropic receptor N-methyl-D-aspartate (NMDAR), triggers the influx of Ca2+ into neurons, increasing its intracellular levels (for review, see Spalloni et al., 2013). In these conditions, changes in mitochondria dynamic properties are produced leading to excessive oxidative phosphorylation and increased generation of ROS and reactive nitrogen species (RNS) that culminate in apoptosis. In parallel, the NMDAR excitotoxicity also results in the release of Ca2+ from the ER. In addition, it was shown that ER stress resulting from the accumulation of aggregated mSOD1 and dysfunction of the unfolded protein response (UPR) activation contributes to the apoptotic signaling cascade in ALS (for review, see Sofroniew, 2009).
Besides the synaptic glutamate pool directly implicated in the excitatory neurotransmission it has been lately additionally considered the extra-synaptic glutamate pool that influences cell communication. This pool, mainly derived from astrocyte and microglia release, is greatly increased by pathological stimuli (for review, see Rodriguez et al., 2013). Besides being implicated in many physiological conditions glutamate contribution to neuron–glia alterations of homeostasis and to the pathophysiology of neurodegenerative diseases, such as ALS, needs to be explored in the near future, due to controversial results. In fact, although linkage between glutamate and neuroinflammation was suggested to have a role in potentiating MN death in a model mimicking ALS disease (Tolosa et al., 2011), other Authors have demonstrated that glutamate by itself was not able to induce MN damage in ALS (Tovar et al., 2009b). Indeed, it has been questioned the involvement of glutamate in ALS-induced MN death (Le Verche et al., 2011) and the cytotoxicity of the cerebrospinal fluid (CSF) from patients with ALS evidenced to not be related to glutamate (Gomez-Pinedo et al., 2013).
Membrane-bound and soluble fractalkine (CX3CL1)
Fractalkine (FKN) mRNA is dominantly expressed in neuronal cells, particularly in those at the cortex, hippocampus, caudate putamen, thalamus, and olfactory bulb (Harrison et al., 1998). FKN mRNA was also detected in unstimulated astrocytes (more) and microglia (less) (Mizuno et al., 2003). Interestingly, FKN immunoreactivity and mRNA was observed also in the rat SC and dorsal root ganglia neurons, but not in glia, and levels were not enhanced by neuropathic conditions (Verge et al., 2004). FKN, or CX3CL1, exists as a membrane-bound and soluble protein (sFKN) allowing both adhesive and chemoattractive properties (Bazan et al., 1997). In fact, the sFKN has potent chemoattractant activity, recruiting CXCR1-expressing T cells, monocytes, and microglia to the injured neurons, as well as in regulating the phagocytic capacity of microglia (Cardona et al., 2006). sFKN was shown to increase upon stimulation of glutamate (Chapman et al., 2000) and to induce proliferation of human microglia (Hatori et al., 2002). The constitutive expression of FKN and its receptor CX3CR1 in microglia indicates its involvement in fundamental processes of communication between neurons and microglia (Harrison et al., 1998).
Under stimulation, such as an excitotoxic stimulus (Chapman et al., 2000), the membrane-bound form of FKN is rapidly cleaved from cultured neurons and significantly reduces neuronal NMDA-induced apoptosis (Deiva et al., 2004). Indeed attenuated glutamate-induced neuronal cell death was observed after treatment of primary neuron–microglia co-cultures with sFKN (Noda et al., 2011). Proteolytic cleavage of CX3CL1 is mediated by action of metalloproteinase ADAM10 (Gough et al., 2004), ADAM17 (Garton et al., 2001; Tsou et al., 2001), cathepsin S (CatS) (Clark et al., 2009), and MMP-2 (Bourd-Boittin et al., 2009). Thus, cleavage of FKN may be prevented by the inhibition of MMPs (Chapman et al., 2000). The source of CatS is the activated microglial cells that upon stimulation with LPS secrete sFKN to the extracellular media (Clark et al., 2009). Similarly, stromal cell-derived factor-1 (SDF-1) was indicated to stimulate the expression of ADAM17 and to increase sFKN, while up-regulating FKN expression (Cook et al., 2010).
In addition, FKN has a neuroprotective function by inhibiting the nitric oxide (NO) production and the expression of inducible NO synthase (iNOS) mRNA in activated microglia (Mizuno et al., 2003). Interestingly, it was observed that FKN expression was reduced in the brain of aged rats probably accounting for the increase in microglial activation in such condition. In fact, treatment with FKN has attenuated the age-related increase in microglial activation (Lyons et al., 2009).
Taken together, FKN seems to have both intrinsic and anti-inflammatory properties in the CNS and to act by interfering with toxic microglial–neuron interactions (Suzuki et al., 2011). Whether FKN may have a role in the development of ALS is still not known.
High-mobility group box 1 protein
High-mobility group box 1 (HMGB1) protein, also known as amphoterin, is an inflammatory factor that can be released by astrocytes, microglia, and neurons, mainly when cells are dying (for review, see Fang et al., 2012). HMGB1 has dual activities depending on whether is alone (probably promoting inflammation resolution and tissue regeneration) or forming complexes with several proinflammatory mediators (potentiating inflammation and promoting innate immune cell activation) (Bianchi, 2009). Nuclear HMGB1 regulates transcription of different sets of genes, including proinflammatory genes (Bianchi and Manfredi, 2009; Park et al., 2009; Wong, 2013). Beneficial effects were observed in early CNS development but increased levels of HMGB1 were shown to be correlated with apoptosis and degeneration of neurons (Liu et al., 2009; Kawabata et al., 2010). When neurons are injured, secretion of HMBG1 activates microglia through receptor for advanced glycation end products (RAGE), Toll-like receptors (TLRs) 2, 4, and 9, as well as Mac1 receptors (Kim et al., 2006a; Park et al., 2006; Neusch et al., 2007), as depicted in Figure 1. Release of inflammatory mediators by activated microglia further induces neuronal necrosis. HMGB1 was indicated to decrease in neurons and to increase in astrocytes with aging (Enokido et al., 2008). When expression and localization of HMBG1 was evaluated in the lumbar SC of SOD1G93A transgenic mice, although intense reactivity was found, no differences were obtained between controls and the SOD1 mice (Lo Coco et al., 2007). However, since HMBG1 was identified in the cytoplasm of astrocytes and microglia in SC samples from ALS patients (Casula et al., 2011) it may trigger TLR signaling pathways. The finding was observed at the late ALS phase and, thus, it will be important to follow the TLR/RAGE cascade in animal models at different stages of the disease. In contrast, a progressive reduction of HMGB1 immunopositive MNs was found at advanced stages and may reflect the loss of MNs, reduced synthesis or enhanced released of the cytokine (Lo Coco et al., 2007). Additional studies are needed to investigate the causative hypothesis indicated for the decreased HMGB1 immunoreactivity in MNs, inasmuch since it can also have beneficial effects on neuroregeneration (Fang et al., 2012).
FIGURE 1
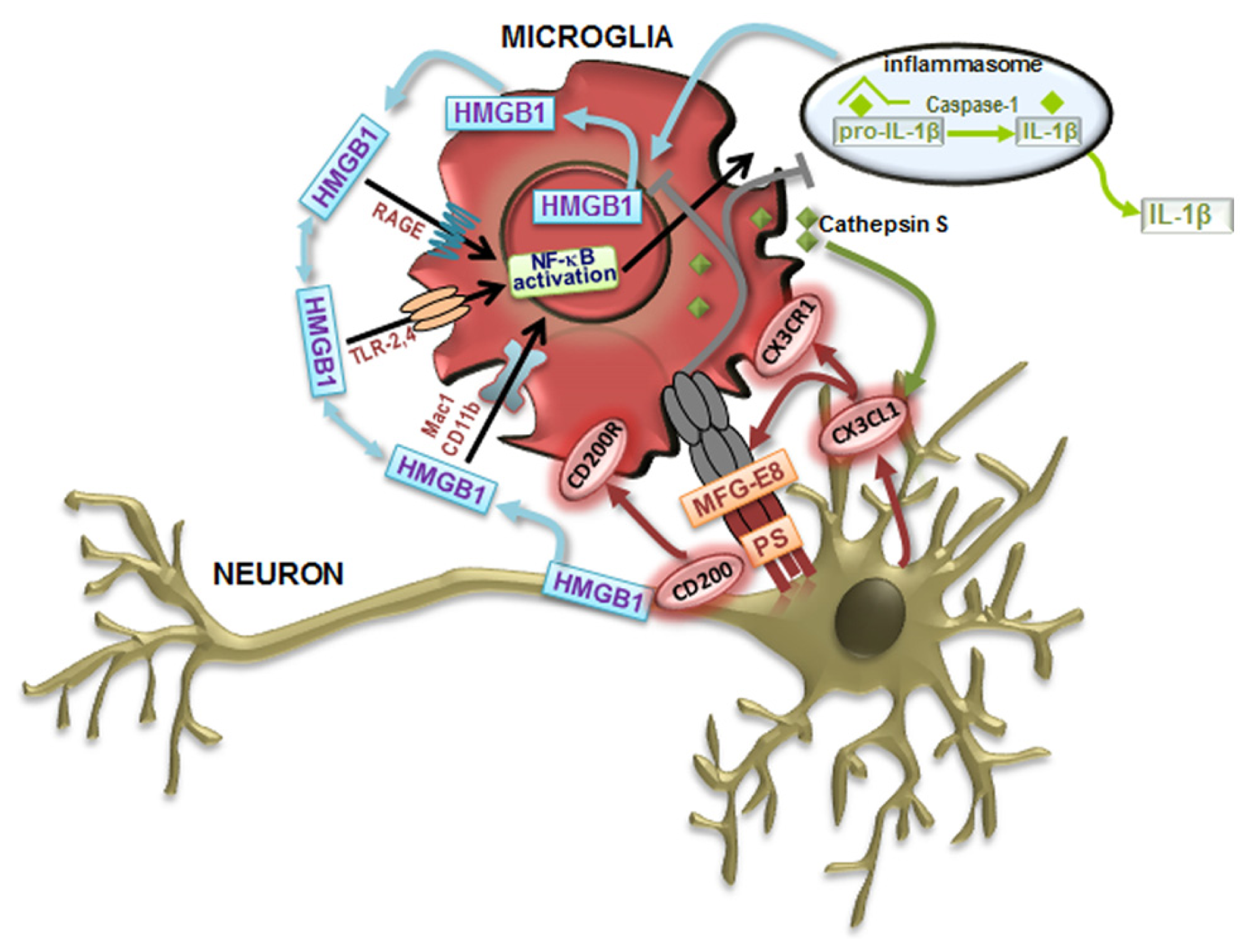
Neuron–microglia communication signaling pathways that modulate microglia cell phenotypes. Toll-like receptor (TLR) signaling contributes to classically activated microglia (M1) in response to damage-associated molecular patterns (DAMPs). Following recognition of DAMPs, TLRs activate downstream signaling cascades, activate nuclear factor-κB (NF-κB) inducing the transcription of inflammatory mediators associated with the M1-like microglial cell phenotype, such as the proinflammatory cytokines interleukin (IL)-1β and tumor necrosis factor (TNF)-α. The production of IL-1β can be achieved by the inflammasome activation by DAMPs, leading to caspase-1 activation that orchestrates the cleavage of pro-IL-1β to form active IL-1β, which leaves and binds to the IL-1 receptor, resulting in inflammation. Following activation, microglial cells augment the immune response by releasing metalloproteinases (MMPs) and increasing proinflammatory cytokines. High-mobility group box 1 (HMGB1) is an alarmin that signals cell damage in response to injury and are associated to all the described signaling events. HMGB1 released by activated microglia and damaged neurons concur for a vicious cycle mediating chronic, progressive neurodegeneration associated with neuroinflammation. HMGB1 can interact with receptors that include RAGE (receptor for advanced glycation end products), TLR-2, TLR-4, Mac-1 also known as CD11b, and possibly others. Alternatively activated microglia (M2) functions include phagocytosis. Milk fat globule factor-E8 (MFG-E8) produced by microglia recognizes phosphatidylserine (PS) as “eat me” signals expressed on the surface of apoptotic neurons, triggering a signaling cascade that stimulates phagocytosis to engulf the dying cell. In order to maintain a quiescent microglia phenotype (M0) under steady-state conditions, neurons suppress the activation of microglia through cell–cell contact (CD200–CD200R) and by the release of the chemokine ligand 1 (CX3CL1) mediated by the cathepsin S that binds to its receptor CX3CR1 on microglia.
CCL21
CCL21 was shown to be implicated in signaling neuronal injury to microglia through the receptor CXCR3. CCL21 expression was demonstrated to increase in cortical neurons, in vitro, 2 h after excitotoxic stimulus (de Jong et al., 2005). Intriguingly, these authors demonstrated its location within vesicles that are transported along the neuronal process till presynaptic structures. This chemokine is considered a chemotactic agent important to drive microglia to the site of lesion. Astrocytes were also indicated to have the receptor CXCR3, but unless high levels of CCL21 are produced, no relevant changes can be observed, indicating separate functions from microglia (van Weering et al., 2010). CCL21 expression in CNS revealed to induce a massive brain inflammation, but not lymphocytic infiltration in transgenic mice expressing the chemokine (Chen et al., 2002). Interestingly, CCL21/CXCR3 signaling axis was never explored in ALS.
CD200
The membrane glycoprotein CD200 is expressed in neurons and in endothelial cells and its receptor CD200R is restricted to cells of myeloid origin including macrophages and microglia. CD200 was also evidenced to be induced by kainic acid in microglia (Yi et al., 2012) and the authors have suggested that microglia are maintained in an activated state with autocrine signaling by interactions between microglial CD200 and CD200R, as well as in a surveillant/quiescent state by interactions between neuronal CD200 and microglial CD200R. In addition, they also demonstrated that IL-4 leads to an increased expression of CD200 what may be the mechanism down-regulating microglia activation whenever IL-4 is produced. In conclusion, deficits in CD200–CD200R system exacerbate microglia activation and the release of proinflammatory cytokines (for review, see Jurgens and Johnson, 2012). Curiously, multivariate analyses of gene expression have previously identified alterations on the CD200R expression in presymptomatic SOD1 mice model (Chen et al., 2004). Thus, more studies on the role of CD200/CD200R in ALS are needed to evaluate whether its disruption is implicated in ALS.
Neuregulins
Neuregulins have been showing to be implicated in a wide range of neurological and psychiatric disorders including multiple sclerosis, schizophrenia, and AD. Neuregulin precursors are expressed predominantly in cortical neurons, but also accumulate at the surface of white matter astrocytes. CSF neuregulin was found reduced in ALS and increased in AD (Pankonin et al., 2009). Neuregulin-1 (NRG1) is a growth and differentiation factor that binds to erbB receptors in microglia. NRG1–erbB signaling is activated after peripheral nerve injury and contributes to microgliosis and neuropathic pain (Calvo et al., 2010). Recently, it was suggested that NRG1 released from damaged neurons and other cells in the SC triggers microglial activation leading to progressive MN degeneration in ALS (Song et al., 2012). Indeed, it was observed that in the early stage of ALS there was activation of NRG1 receptors on microglia. Later on, a reduced membrane-bound NRG1 and an increased mRNA expression were noticed in the SC. More studies are, however, needed to sustain the NRG1 contribution to ALS pathogenesis.
β2-Microglobulin
Major histocompatibility complex (MHC) class I and class II molecules present fragments of peptide antigens to CD8+ or CD4+ T cells, respectively, and cells lacking such molecules are unable to interact with immunocompetent T cells (Parnes, 1989). Presentation of antigenic peptides to CD8+ cells is mediated by β2-microglobulin, which is non-covalently bound to MHC class I. Despite being recently observed during hippocampal neurodevelopment (Leinster et al., 2013), adult neurons do not constitutively express MHC class I (Arthur-Farraj et al., 2012). In contrast, spinal MNs were shown to display a high constitutive expression of both MHC class I and β2-microglobulin mRNAs (Thams et al., 2009). A latest paper describes a strong up-regulation of β2-microglobulin in MNs during disease progression in the SOD1G93A mice model, which revealed to be important for the ALS mouse survival (Staats et al., 2013). Therefore, β2-microglobulin target driven therapies may be of help in strategies counteracting ALS.
GLIAL CELL RESPONSES
It is believed that non-neuronal cells expressing mSOD1 may secrete toxic mediators or fail to secrete trophic factors, or both, resulting in either reduced function or survival of MNs. Indeed, several SFs were shown to drive axonal degeneration and regional-specific microglia activation expressing SODG93A was suggested to be implicated (Kim et al., 2006b). This calls our attention for the existence of multiple factors, and MNs and glial cells interplay in contributing to ALS onset and progression. Microglia activation and dysfunction have lately been related with the onset, but mostly with the progression, of several neurodegenerative diseases. Particularly, in ALS, it has been suggested that besides MNs, also glia and muscles are implicated in the disease.
Prominent neuroinflammation is a pathological hallmark in human ALS and mouse models of the disease. Gliosis and accumulation of large numbers of activated microglia and astrocytes can be observed in CNS and in SC areas. Active contributions of glial cells in ALS pathology have recently been reviewed (Lasiene and Yamanaka, 2011; Zhao et al., 2013). Conflicting results were published on whether the expression of mSOD1 in astrocytes and microglia really contributes to the progression of ALS disease. Some argues against (Yoshii et al., 2011) and others in favor (for review, see Boillée et al., 2006b). Overall, it will be important to identify the molecules released by glia, as well as the ones secreted by dysfunctional neurons targeting microglia or even acting retrogradely.
Toll-like receptor activation
Glial cells (astrocytes, microglia, oligodendrocytes, and Schwann cells), as well as neurons are known to express different members of the TLR family (Lee et al., 2013b). Increasing evidence indicates that, in the absence of pathogens, TLR signaling can be activated by molecules released by the injured tissue, namely the HMGB1 protein, one of the damage-associated molecular patterns (DAMPs) molecules (Bianchi and Manfredi, 2009). Recent findings have underlined the activation of TLRs and RAGE signaling pathways in ALS (Casula et al., 2011). TLR2, TLR4, and RAGE expression was increased in reactive glial cells in both gray (ventral horn) and white matter in SC. TLR2 was predominantly detected in cells of the microglia/macrophage lineage, whereas TLR4 and RAGE were strongly expressed in astrocytes. Activated macrophages (He et al., 2012) and microglia (personal communication) release HMBG1 that promotes the secretion of IL-1β and IL-18 further inducing the necrosis of neighboring neurons and the amount of extracellular HMBG1, which binds to microglial Mac1 leading to the activation of nuclear factor-κB (NF-κB) pathway (Gao et al., 2011) and inflammasome (Lu et al., 2013), forming a vicious circle that sustains progressive neurodegeneration (Figure 1). Recent data have demonstrated that NF-κB activation in wt microglia causes MN death providing a therapeutic target for ALS (Frakes et al., 2014).
When evaluating whether extracellular mSOD1 caused a direct or indirect injury to MNs, it was observed that neurodegeneration was mediated by microglia and concerted activation of CD14/TLR pathway involving both TLR2 and TLR4 (Zhao et al., 2010). In addition, granulocyte macrophage-colony stimulating factor (GM-CSF), a pleiotropic cytokine predominantly released by astrocytes that up-regulates TLR4 and CD14 expression in microglia, may function to exacerbate TLR signaling in ALS disease (Parajuli et al., 2012). Indeed, when GM-CSF was blocked it was observed a delayed onset and increased life span in the ALS mice (Turner and Talbot, 2008). The activation of these pathways may contribute to the progression of inflammation and to a further injury to MNs.
NG2 Cells, oligodendrocytes, and Schwann cells
Oligodendrocytes in CNS, and Schwann cells in the peripheral nervous system (PNS), are responsible for the myelin sheaths surrounding neurons which provide electrical insulation essential for rapid signal conduction. Schwann cells also participate in the clearance of debris and in guiding the axon after neuron damage (Ilieva et al., 2009). In the SC injury, reactive gliosis emerges in the lesion accompanied by the up-regulation of chondroitin sulfate proteoglycans (CSPGs) in oligodendrocytes and Schwann cells.
Considering the regenerative capacity of NG2 cells, one of the first cells responding to any alteration in CNS environment, it will be important to clarify their role in ALS since they can be mobilized to originate cells or to release factors. Nevertheless, NG2 cells were shown to remain committed to an oligodendrocyte lineage in both controls and mutant ALS mice, indicating their scarce participation (Kang et al., 2010). However, increased proliferation rate of NG2 revealed to mediate an elevated number of early-born oligodendrocytes that degenerate, resulting in gray matter demyelination in ALS mice and human CNS (Kang et al., 2013).
Although only a few studies have examined whether oligodendrocytes or myelin sheaths have a role in ALS, the myelin abnormalities consisting in loss of compact myelin and lamellae detachment in the SC of pre-symptomatic SOD1 transgenic rats and aggravated at symptomatic stages (Lasiene and Yamanaka, 2011) suggest that it may be an interesting target for further study. Biochemical examinations of the myelin structural components revealed a decrease in the phospholipid content along disease progression, as well as in cholesterol (already in the early presymptomatic stage) and cerebrosides (in the paralyzed animals) (Niebroj-Dobosz et al., 2007). Degeneration of oligodendrocytes was observed in human patients and mice models of ALS prior to the disease (Kim et al., 2013). Cells that were lost were replaced by newly differentiated oligodendrocyte precursor cells, which, however, evidenced reduced myelin basic protein. This finding is suggestive that they may contribute to MN degeneration in ALS (Philips et al., 2013). Moreover, these cells evidenced a loss of the monocarboxylate transporter 1 (MCT1), thus compromising the supply of lactate to MNs (Table 2).
Little is known about Schwann cell involvement in ALS pathology. Schwann cells are located near the MN axons and are known to bridge between denervated and reinnervated endplates, and to guide axonal sprouts (Magrane et al., 2009). Expression of mSOD1 in perisynaptic Schwann cells was suggested to interfere with the trophic maintenance of normal or regenerating motor axons (Inoue et al., 2003). In vivo evidence suggests that glial fibrillary acidic protein (GFAP) up-regulation in the stressed/proliferating Schwann cells may be the underlying pathological events (Keller et al., 2009). Conversely, different results were obtained in SOD1G73R mice where elimination of mutant SOD1G37R from Schwann cells failed to slow disease progression (Lobsiger et al., 2009). Nevertheless, Wang et al. (2012) found that knockdown of mSOD1 in Schwann cells of SODG85R transgenic mice delayed disease onset and extended survival indicating that SOD1G85R expression is neurotoxic. These results imply that diverse mutations confer different outcomes to cell toxicity and, in the case of Schwann cells, oxidative damage seems to be an important feature in the context of ALS.
Astrocyte reactivity
Astrocytes are a potential source of both pro- and anti-inflammatory cytokines and are ideally placed in close proximity to BBB and BSCB, thus translating signals from the periphery to the CNS. Although not being immune cells, they can also contribute to the immune response. Astroglial activation, or astrogliosis, is characterized by hyperplasia, hypertrophy of cell bodies and cytoplasmic processes, up-regulation of intermediate filament proteins, namely GFAP and vimentin, mediating a histologically apparent glial scar at the lesion site in the damaged SC (Sofroniew, 2009). Reactive astrogliosis in ALS was first revealed in 1990s by increased GFAP staining in the subcortical white matter (Kushner et al., 1991), and later on similarly observed in the ventral and dorsal horns of the SC, as well as in the transition between gray matter and anterior and lateral funiculi, where the dystrophy of neuritis exists (Schiffer et al., 1996). SC astrocytes were shown to assume a neurotoxic phenotype in response to extracellular ATP. In SOD1G93A astrocytes this activation is mediated through P2X7 receptor signaling (Gandelman et al., 2010). Reactive astrocytes surround degenerating MNs in patients and transgenic animal models of ALS, and in particular those localized in the ventral SC of SOD1G93A mice, are a source of nerve growth factor (NGF) and NO, which are both required for MN apoptosis (Table 2) (Pehar et al., 2004). Interestingly, the NO-induced MN death was shown to be mediated by astrocytes expressing SOD1 and TDP-43 mutations (Rojas et al., 2014). Ferraiuolo et al. (2011) have shown that the increase in total NGF is due to the secreted pro-NGF fraction before cleavage to the mature form, achieving a twofold increased ratio in the SOD1G93 mice as compared to control conditions. Such finding may derive from the increased ALS-associated MMP-9 (Niebroj-Dobosz et al., 2010), one of the enzymes that degrade mature NGF in the extracellular space. In this scenario, neuroprotection of MNs could be achieved by promoting pro-NGF cleavage.
Recently, it was demonstrated that loss of GFAP did not affect disease onset and marginally shorten the SOD1H46R mice survival, indicating that GFAP only plays modulatory effects (Yoshii et al., 2011), at least in this mice. On the contrary, astrogliosis was observed in the SOD1G93A mice in either symptomatic or presymptomatic phase, preceding microglial activation (Yang et al., 2011). Indeed, depending on the ALS animal model used, astrocyte activation was observed to occur earlier or later and to be rather complex, increasing and decreasing in waves at different times throughout the disease (for review, see Evans et al., 2013). Despite no differences in the astrocyte number, increased GFAP labeling in the ventral horn of lumbar SC from SOD1G93A mice was observed before MN loss (Gerber et al., 2012). Astrocytes derived from such mice evidenced to uptake glutamate less efficiently and showed a reduced trophic response to activation, deficiently protecting MNs (Benkler et al., 2013). Attesting this low astrocyte efficiency, the mainly expressed astroglial S100B protein was found to be decreased in CSF (Sussmuth et al., 2003) and serum (Otto et al., 1998) from ALS patients. Interestingly, exposure of primary cultures of astrocytes from ALS patients to CSF evidenced to enhance GFAP and S100B expression (Shobha et al., 2010). Similarly to neurons, astrocytes have demonstrated constitutive and regulated expression of FKN, which may control the migration and function of the microglia (Sunnemark et al., 2005). Additional contribution of astrocytes to MN death involves the release of ROS mediated by mitochondrial dysfunction and of D-serine that is a co-activator of the NMDARs, thus exacerbating glutamate excitotoxicity, among other factors as indicated in Table 2 and reviewed in Valori et al. (2014).
A recent publication has characterized a specific astrocytic phenotype (aberrant astrocytes) obtained from primary SC cultures of SOD1G93A symptomatic rats (Diaz-Amarilla et al., 2011). These aberrant astrocytes isolated on the basis of marked proliferative capacity and lack of replicative senescence, lack GLT-1 and the NG2 marker and were shown to release toxic factors accounting for a MN hostile environment. This specific astrocyte phenotype expressing increased S100B and connexin-43 (Cx-43) is abundant in the symptomatic phase of the disease and seems to be located close to MNs, representing a new potential target for delaying ALS progression.
Data in postmortem tissues of ALS patients revealed changes in the morphology of astrocytes together with elevated GFAP and aldehyde dehydrogenase family 1, member L1 (ALDH1L1) (Philips and Robberecht, 2011), corroborating findings in animal models of ALS. Indeed, mSOD1 gene excision from microglia and selective reduction in astrocytes significantly slowed disease progression (Yamanaka et al., 2008b). However, there is still some controversy on whether astrogliosis is detrimental or beneficial, what surely will depend from spectrum intensity and toxicity potential of the aberrant phenotype.
Microglia activation
Microglia are the immune resident cells of the CNS with both a supporting role to neurons and astrocytes, and immunological properties with either neuroprotective or neurotoxic potential. Though microglia activation was indicated to precede astrocyte reactivity (Alexianu et al., 2001), reduced neuroprotective behavior of mSOD1 microglia in a resting-state (Sargsyan et al., 2011), and at the disease end-stage (Nikodemova et al., 2013) was documented. If confirmed, boosting of microglia with stimulatory factors may reveal to be clinically useful.
In a recent paper, Roberts et al. (2013) verified that incubation of microglia with aggregated SOD1 first drives its location at the membrane level, and later within the cell. Moreover, supernatants of these SOD1 activated microglia caused a significant decrease in MN viability, which was not related with tumor necrosis factor-α (TNF-α) secretion, NO, or superoxide anion radical. The toxic factors involved are currently unknown, although it was shown that expression of mSOD1 increases TNF-α secretion (Liu et al., 2009), which may induce neurotoxicity by increasing the glutamate release by microglia in an autocrine manner (Takeuchi et al., 2006). Therefore, a very early stage of the disease may rely in this link between protein aggregation and microglial activation. Indeed, neurotoxic potential of mutant microglia in MN degeneration in ALS is a very well documented issue: (1) mSOD1 acting on microglia is required to cause the disease (Clement et al., 2003); (2) limiting mutant damage to microglia slows progression (Boillée et al., 2006b); replacement of mSOD1 microglia for wt microglia delays disease and prolongs mice survival (Lee et al., 2012).
Microgliosis at sites of MN injury is a neuropathological hallmark of ALS and recent therapeutic interventions are looking into factors capable of skewing microglia neurotoxic potential into a neuroprotective phenotype. However, a trophic role for activated microglia has been suggested at early stages of the disease (for review, see Lewis et al., 2012). In the SOD1G93A mice, it was observed a decrease in microglia number in the entire SC at the pre-symptomatic age (Gerber et al., 2012). The authors identified two diverse microglia subpopulations (low and high Iba1 expression cells), suggesting a distinct microglia involvement at pre- and early-symptomatic ALS stages. Interestingly, microglia heterogeneity observed between cervical and lumbar SC regions in ALS mice may derive from different environmental specificities determined by local MN/astrocyte/lymphocyte disposition and activation (Beers et al., 2011b). While cortical microglia appear unaffected by the disease, additional studies evidenced an increased microglial number in the lumbar SC at symptom onset, and neither the typical inflammatory nor the anti-inflammatory phenotypes were identified at end-stage (Nikodemova et al., 2013). In addition, elevated TNF-α gene expression and immunoreactivity were observed in lumbar SC of mSOD1 mice and related with invading microglia (Yoshihara et al., 2002). Early microglia activation in ALS may be further explored in vivo by positron emission tomography (PET). The possibilities of PET suggest its valuable contribution to monitor the progression of the disease and the efficacy of the therapy in use (Corcia et al., 2012).
Context-dependent neuroprotective and neurotoxic properties . An important neuroprotective role of microglia is phagocytosis. Following signaling by neuronal FKN, milk-fat globule EGF factor-8 protein (MFG-E8, SED1) is up-regulated and serve as a bridge via specific integrins between apoptotic neurons and microglia (Figure 1) (Leonardi-Essmann et al., 2005). The authors hypothesize that MFG-E8 is assembled on the surface of exosomes and apoptotic neurons so microglia can recognize their target cells. MFG-E8 seems to be essential for microglia engulfment and removal of the dying neurons. An interesting concept is that inflammatory microglia can also phagocyte viable neurons (Fricker et al., 2012a) through MFG-E8 mediation (Fricker et al., 2012b) accounting for a reduced number of neurons. Phagocytic ability may, however, be lost if the cell is continuously stressed by a neurotoxic stimulus.
Our recent data with the neurotoxic unconjugated bilirubin have evidenced that following the phagocytic ability at eliminating cell debris microglia changes to a more inflammatory phenotype (Silva et al., 2010) and even to a senescent-like cell morphology and death if the duration of exposure is prolonged. However, microglia behavior in the presence of unconjugated bilirubin is modulated by the presence of both astrocytes and neurons (Silva et al., 2011). Also to consider is that during the inflammatory phenotype, microglia may intervene in the glutamate homeostasis, but may also contribute to neurite degeneration through the release of NO (Silva et al., 2012). Interestingly, microglia phagocytic features were shown to occur early in ALS disease. Microglia were revealed to aggregate, proliferate, and phagocyte in the lumbar SC of pre-symptomatic mutant SOD1H46R transgenic mice (Sanagi et al., 2010). However, in other studies microglia have shown to contribute to MN death (Dibaj et al., 2011; Brettschneider et al., 2012) and to decrease in number within disease progression (Butovsky et al., 2012), thus contributing to the disease propagation. By using in vivo imaging by two-photon laser-scanning microscopy and axonal transection, it was observed different phases of microglia-mediated inflammation in the ALS mice model. Indeed, a first phase (preclinical) with highly reactive microglia was followed by another (clinical stage) with morphologically transformed microglia presenting reduced surveillance activity and reactivity (Dibaj et al., 2011). Regional, temporal, and immune environmental differences may contribute to changes in microglia phenotypes and response heterogeneity, thus requiring differentiated immunomodulatory or even combinatory therapeutic approaches along ALS disease progression.
Microglia phenotypes . Activation of microglia may be observed through the up-regulation of CD11b, Iba1, and CD68 markers. Primary microglia differ from other blood macrophages in the expression of CD11b/CD45low/high and CD68low/high (for review, see Hinze and Stolzing, 2011), although not specific for microglia in a pathologic brain (Matsumoto et al., 2007). These cells show a round morphology with an enlarged cell body and smaller and thicker processes in the resting/quiescent state (M0; Figure 2). When activated in response to an insult or injury, microglia are capable of acquiring diverse and complex phenotypes, allowing them to participate in the cytotoxic response, immune regulation, and injury resolution. Microglia may then favor the entrance of inflammatory T cells with which the cells seem to interact. Recent work have classified and characterized M1 and M2 phenotypes (Table 2) (for review, see Evans et al., 2013). The first is cytotoxic, characterized by the release of proinflammatory cytokines and influenced by T helper cell type 1 (Th1) that release GM-CSF and interferon-γ (IFN-γ), triggering M1 proliferation. Cytotoxic M1 markers include IL-1β, IL-6, TNF-α, iNOS, COX2, and CX3CL1, and known inducers are the TLR4 agonist LPS and IFN-γ (Chhor et al., 2013).
FIGURE 2
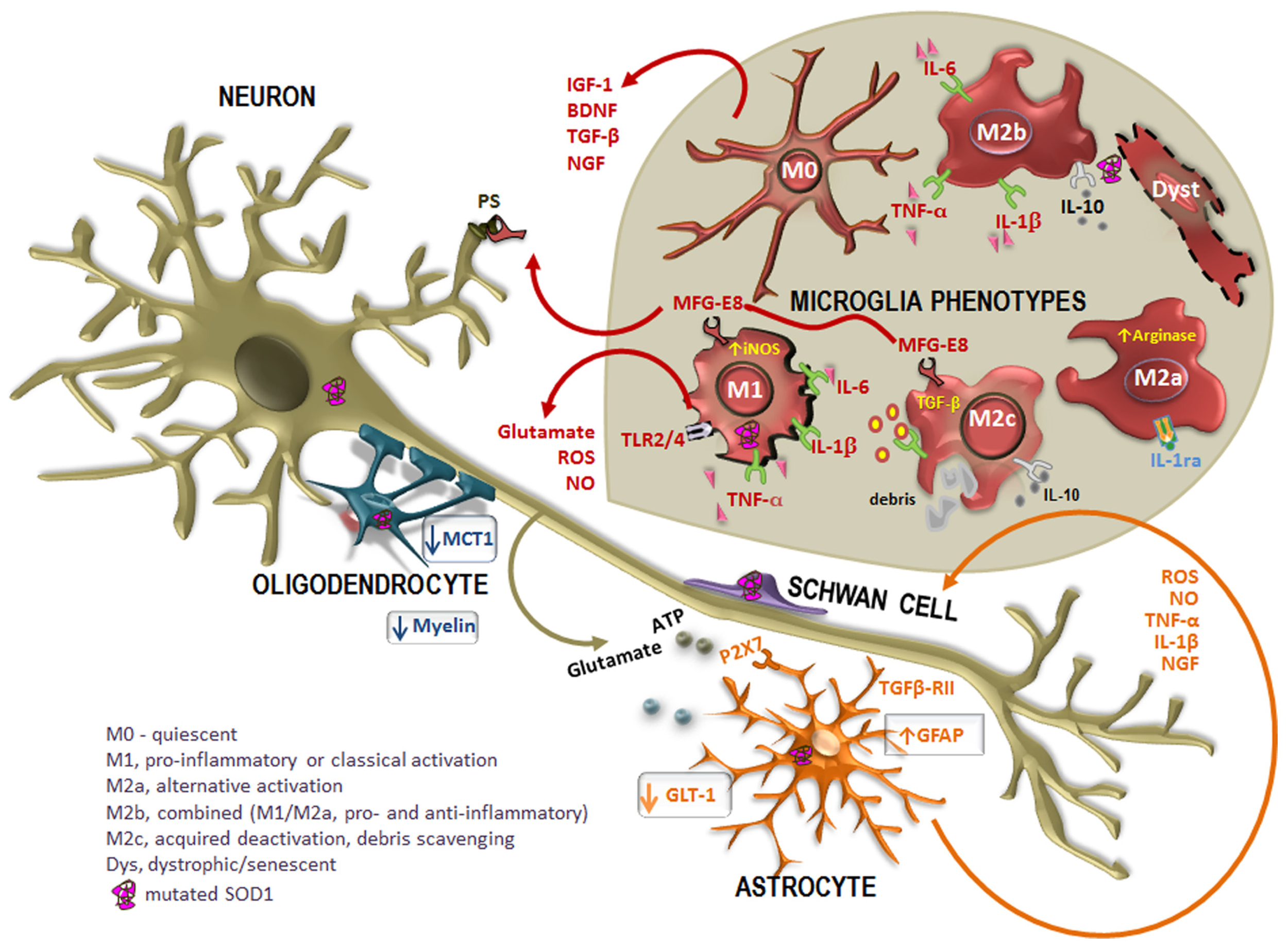
Altered cross-talk between glial cells and motor neurons (MNs) in ALS disease. Many studies report the intervention of mutated superoxide dismutase-1 (SOD1)-expressing non-neuronal cells in the pathogenesis of the disease. In case of injury, astrocytes become activated in a process called astrogliosis, characterized by the up-regulation of intermediate filament glial fibrillary acidic protein (GFAP) and increased release of toxic products into the extracellular media, such as pro-inflammatory cytokines [tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β)] oxidative stressors [reactive oxygen species (ROS), nitric oxide (NO)], as well as glutamate and ATP mediated by the receptor P2X7. In addition, astrocytes also evidence a reduced expression of the glutamate transporter GLT-1, which additionally contribute to neuronal excitotoxicity. However, reactive astrocytes up-regulate nerve growth factor (NGF), which can modulate neuronal survival. Disorganization and destruction of the myelin sheath with a progressive loss of phospholipids and cholesterol is also observed. Moreover, oligodendrocytes also evidence a loss of the of the monocarboxylate transporter 1 (MCT1), thus compromising the energy supply to MNs. On the other hand, increased mutated SOD1 expression in Schwann cells may have intricate ways contributing to slow ALS progression. When mutated SOD1 accumulates within microglia, there is a pattern of activation. Microglia acquire different activation phenotypes (M0, M1, M2a, M2b, M2c, and dystrophic/senescent) and, consequently, produce a diversity of substances that may be either beneficial [insulin-like growth factor 1 (IGF-1), transforming growth factor-β (TGF-β), brain-derived neurotrophic factor (BDNF), and nerve growth factor (NGF)] or toxic (glutamate, ROS, NO) to other cells. Phagocytosis is mediated by the release of milk factor globule-8 (MFG-E8) from microglia M1 and M2c phenotypes that recognizes phosphatidylserine (PS) in apoptotic neurons. Reliable markers for M2a are high IL-1 receptor antagonist (IL-1Ra) and high arginase, for M2b are IL-10, TNF-α, IL-6, and IL-1β, for M2c are TGF-β and IL-10 and for M1 are TNF-α, IL-6, and IL-1β, increased inducible nitric oxide synthase (iNOS) and toll-like receptors (TLR)2 and 4.
The M2, promoted by the cytokines IL-4 and IL-13 released by Th2, contribute to neuroprotection once they also secrete anti-inflammatory cytokines, such as IL-4 and IL-10, and growth factors such as insulin-like growth factor 1 (IGF-1). Indeed, IL-4 was shown to protect MNs from the injury produced by LPS-activated microglia (Zhao et al., 2006). Thus, M2 polarization may be desirable, although excessive or prolonged M2 polarization may become prejudicial in allowing unwanted fibrotic responses and scarring, not facilitating axonal growth. However, there are three M2 phenotypes: the M2a or alternate activation repair/regeneration/remodeling phenotype, the M2b immunoregulatory and the M2c acquired-deactivating (Figure 2). Reliable markers for M2a stimulated by IL-4 and IL-13 are high IL-1 receptor antagonist (IL-1Ra) and high arginase (Arg1). M2b is stimulated by immune complexes, TLR agonists and IL-1R ligands. Useful markers are IL-1Ra and SOCS3. Characterization of M2c stimulated by IL-10, transforming growth factor-β (TGF-β) and glucocorticoids is obtained through the increased levels of anti-inflammatory cytokines (IL-10, TGF-β), low levels of pro-inflammatory cytokines and enhanced IL-4Rα, Arg1, SOCS3, and CD206 (David and Kroner, 2011; Wilcock, 2012; Chhor et al., 2013). While LPS, IL-1β, TNF-α, and IFN-γ lead to cytotoxic M1 and immunomodulatory M2b activation states, IL-4 mainly triggers M2a phenotype (Chhor et al., 2013).
Therefore, balance between M1 and M2 phenotypes may be a desirable therapeutic goal. Interestingly, mSOD1 microglia isolated from ALS mice at disease onset showed higher levels of Ym1, CD163, and brain-derived neurotrophic factor (BDNF) (M2 markers) and lower levels of Nox2 mRNA (M1 marker) as compared to the end-stage disease (Liao et al., 2012). Interestingly, when co-cultured with wt MNs the first microglia phenotype exerted neuroprotection while the M1 phenotype was neurotoxic, supporting the pathoprogression-related changes in microglia. We may probably consider that microglia display the M2 phenotype at an early stage of the disease switching to the M1 phenotype during the late rapid phase (reviewed in Zhao et al., 2013). However, we should also consider that microglia may become functionally impaired at the end-stage, as recently observed in the SOD1G93A SC microglia (Nikodemova et al., 2013). Different roles of microglia depending on the neurological disease context was recently suggested by Chiu et al. (2013) based on the observation by FACS-transcriptome comparisons that SOD1G93A microglia show a unique phenotype that differs from M1 or M2 macrophages, and from activation with LPS. Robust up-regulation of MMP-12, IGF-1 and osteopontin were pointed as hallmarks. Transcriptomic technology has been carried out to examine the gene expression changes of ALS tissue as compared to controls (reviewed in Heath et al., 2013) and in the future may provide insights into the microglia phenotype profiling prior to disease onset and along ALS progression to assist in the development of new treatments for ALS at different stages.
Inflammatory microRNA profiling . MicroRNAs (miRNAs), small, non-coding RNAs, have been recently pointed to mediate cell-to-cell communication (Xu et al., 2013). Inflammatory phenotypes are miR-155, miR-21, miR-146a/b, and miR-124 (Quinn and O’Neill, 2011). Recent studies demonstrated the miR-124 involvement in promoting microglia quiescence by skewing their polarization from an M1 to an M2 phenotype (Willemen et al., 2012) and that miR-155 together with miR-124 are likely to be directly related to M1 and M2 phenotypes, respectively (Cardoso et al., 2012; Ponomarev et al., 2013). Up-regulation of miR-155 is induced by TNF-α and Il-1β (Pottier et al., 2009), as well as by HMGB1 through the activation of the TLR2/MyD88/miR-155 pathway (Wen et al., 2013), while it is down-regulated by TGF-β (Pottier et al., 2009). Increase of miR-146a expression was shown to occur in the aged mice (Rodier and Campisi, 2011), as well as after LPS stimulation (Jiang et al., 2012), and is related with the microglia phagocytic potential (Saba et al., 2012). Moreover, up-regulation of miR-21 in murine models of SC injury was pointed as a modulator of the pro-reactive effects of inflammatory signaling cascades (Nieto-Diaz et al., 2014). Dysregulation of miRNAs expression was lately found in the SC of ALS patients and in microglia isolated from the hSOD1G93A mice. Promising candidates that were found to be altered in patients were miR-146a*, miR-524-5p, and miR-582-3p (Campos-Melo et al., 2013), while those up-regulated in the mice model were miR-155, miR-146b, miR-22, miR-365, and miR-125b (Parisi et al., 2013), which need to be further investigated for their relevance as ALS biomarkers and therapeutic targets. Actually, it was recently discovered that inhibition of miR-155 prolongs survival in the mSOD1 mice (Koval et al., 2013).
Autotaxin . Autotaxin (ATX) is a secreted lysophospholipase D that converts lysophosphatidylcholine (LPC) to lysophosphatidic acid (LPA), a phospholipid growth factor that activates several transduction pathways, and is involved in migration, proliferation, and survival of various cells (Mori et al., 2011; Furukawa et al., 2013). LPA acts as an autocrine and/or paracrine signaling molecule mediating a broad range of intracellular signaling cascades, especially the RHOA pathway (reviewed in Willier et al., 2011). LPA receptors are expressed by astrocytes, oligodendrocytes, and microglia (Lorenzl et al., 2006; Shi et al., 2010a; He et al., 2013).
Data suggest that ATX levels increase within reactive astrocytes following neurotrauma (Savaskan et al., 2007), but other studies have obtained a down-regulation instead (Shi et al., 2010b). Most fascinating, it was observed that overexpression of ATX inhibits microglial activation and protects the cell against oxidative stress (Awada et al., 2012). Thus, reduced expression of ATX in microglia may contribute to a sustained neuroinflammation condition. Since ATX is also a motility driver that stimulates cell invasion (Sau et al., 2007; Hoelzinger et al., 2008; Magrane and Manfredi, 2009) it can be involved in microglia migration, a property not exploited till now. Although ATX is considered an inflammatory mediator we were not able to find studies demonstrating its relevance or not in ALS.
NEUROVASCULAR CHANGES
BBB and BSCB are dynamic and complex interfaces between the blood and the CNS. Endothelial cells and tight junctional complexes physically limit solute exchanges between the blood and the brain. These cells together with pericytes, astrocytes, neurons, microglia, and oligodendrocytes and the basement membrane form the neurovascular unit (for review, see Cardoso et al., 2010; Sá-Pereira et al., 2012). The interaction between all these components provides a sustainable environment for neural function while restricting permeability and transport. Alterations in barrier properties and dynamics were observed in transgenic SOD1 rats (Andjus et al., 2009; Nicaise et al., 2009). In particular, it was noticed a reduction of endothelial tight junctions (Zhong et al., 2008), together with the disruption of the neurovascular unit and up-regulation of MMP-9 (Miyazaki et al., 2011) prior to MN degeneration. MMP-9 activation was observed in blood vessel-like structures and microglia. Increased microvascular microglia, expressing CX3CR1 and weakly labeling Iba1, were detected in the SCs from the ALS mice model and suggested to have a bone marrow origin (Lewis et al., 2009). In addition, it was recently demonstrated that SC microglia in the mSOD1 mice promote the recruitment of inflammatory monocytes into the CNS well before the onset of the disease, triggering microglia apoptosis (Table 2) (Butovsky et al., 2012). Actually, the BSCB has been shown to be disrupted in different mSOD1 mice before MN degeneration, thus favoring blood monocytes infiltration (Barbeito et al., 2010). To that it may account the 54% reduction in pericytes at the BSCB level in patients with ALS (Winkler et al., 2013). In fact, some authors consider ALS as a neurovascular disease (Garbuzova-Davis et al., 2012) with evidence indicating impairment of all neurovascular unit components, including the BBB and the BSCB in both ALS patients and animal models. Disruption of BBB and BSCB was observed in the G93A mice by electron microscopy in places of MN degeneration at both early and late disease stages (Garbuzova-Davis et al., 2007). The damage of barriers besides allowing the entrance of T lymphocytes, such as CD4+ (helper/inducer) and CD8+ (cytotoxic) into the brain parenchyma, may also permit the entrance of harmful substances that will contribute to disrupt neuronal homeostasis and accelerate MN degeneration. The impact of microvascular damage in ALS pathology is a novel and promising research topic in that interactions with glia may determine neuroprotective or cytotoxic cell phenotypes with consequences on MN survival.
INFLAMMATORY COMPONENTS IN ALS AND PATHOLOGICAL CELL–CELL COMMUNICATION
Communication between neurons and microglia is essential for maintaining homeostasis in the CNS and altered cross-talk is implicated in the pathogenesis of ALS and other MN diseases (Appel et al., 2011). Dysregulated neuron–neuron signaling and neuron–microglia cross-talk in ALS, and other neurodegenerative diseases, may derive from: (i) secretion of toxic mediators or fail to secrete trophic factors, or both, resulting in reduced function and survival of MNs; (ii) SFs released by neurons and their action on microglia receptors; (iii) changes in direct cellular interactions.
Reduced expression of trophic factors such as BDNF, fibroblast growth factor 2 (FGF2), and IGF-1 was found in the SC of the newborn rat after intrathecal administration of CSF from ALS patients (Deepa et al., 2011). To note, that the release of mSOD1 was shown to trigger microgliosis and neuronal death (Urushitani et al., 2006) and increased levels of ROS/RNS, mainly of NO, were shown to play a critical role in the earliest stages of neuronal dysfunction in ALS (Drechsel et al., 2012). NO mainly produced by glia through the activation of iNOS can react with superoxide producing the potent oxidant peroxynitrite (Beckman et al., 1990), which mediates apoptosis or necrosis, depending on the concentration (Bonfoco et al., 1995). Peroxynitrite also induces toxic glial phenotypes further propagating oxidative damage and cellular dysfunction. Interestingly, the same concentrations of NO may either promote the survival of MNs healthy conditions or induce apoptosis and glia reactivity when in the presence of stressors. Up-regulation of neuronal NO synthase (nNOS) was shown to occur in MNs before iNOS in either pre-symptomatic ALS animal models or in patients and to be associated with MN loss (Moreno-Lopez et al., 2011; Drechsel et al., 2012).
Activated microglia and astrocytes amplify the initial damage to the MNs by activating AP-1 and NF-κB through production of proinflammatory cytokines and apoptosis-triggering molecules such as TNF-α and Fas ligand (FASL). TNF-α and IL-1β exert neurotoxic effects in vitro, but deletion of the individual genes seems to not affect the course of the disease. In addition, dying MNs release ATP that can further activate glia through the purinergic receptor P2X7 expressed by both microglia and astrocytes (Figure 2) (Gandelman et al., 2010; Glass et al., 2010). Classical microglia activation results in upregulation of MHC class II proteins that are involved in presentation of antigens to T lymphocytes. Microglia activation is mediated by the release of SFs and/or expression of surface receptors by neurons and astrocytes, such as CX3CL1, CD200, and CCL21. Microglia also express a diverse set of pattern recognition receptors (PRRs) for pathogen-associated molecular patterns (PAMPs) and endogenous ligands derived from injury that include TLRs and inflammasome. In addition, emerging evidence suggests that members of the nuclear receptor (NR) family of transcription factors, many of which are ligand-dependent, control the activation of microglia under physiological and pathological conditions (for review, see Saijo et al., 2013).
Neuroinflammation involves the activation and proliferation of microglia and the infiltration of T cells into the brain and SC. In these conditions, astrocytes and microglia release IL-1β, TNF-α, and IL-6 (Figure 2). Cytokines can then up-regulate oxidative stress by NO and superoxide (O2-) generation (for review, see Philips and Robberecht, 2011). Repair and limitation of the damage is promoted by the release of trophic and anti-inflammatory factors. Deleterious microglia M1 and benign M2 phenotypes are influenced by astrocytes and T cell subsets. In ALS, it is still currently unknown the precise function of microglia and astrocytes and how they mediate neuroinflammation and contribute to the pathology. Identification of the factors driving microglia phenotype and consequent functional changes, once known, may advance our knowledge on their role and on ways to modulate these cells. In addition, it will be important to clarify whether inflammation contributes to ALS pathogenesis or in opposite is a protective response, and if different individual immune responses to the disease are also implicated, so that immunomodulatory therapies can be pursued.
MICROGLIA–T CELL CROSS-TALK
Evidence for autoimmunity in ALS was proposed in 1990s (for review, see Appel et al., 1993). Inflammation is not a resultant from MN degeneration since it regulates the balance between neuroprotection and neurotoxicity (Henkel et al., 2009). Initially, the diverse populations and phenotypes of CD4+ T cells that cross-talk with microglia can slow disease progression, but later they may contribute to the acceleration of the disease. The inflammatory process involves infiltration of T cell subpopulations at sites of neuronal injury in the brain parenchyma. T cells may damage MNs by cell–cell contact or cytokine secretion through the activation of microglia (Holmoy, 2008). CD8+ cytotoxic T cells and natural killer T (NKT) cells were found significantly increased in patients with ALS, while regulatory T (Treg) cells were decreased (Rentzos et al., 2012). It was suggested that CD4+ T cells may trigger oxidative phosphorylation in microglia and CD8+ may stimulate phagocytosis accordingly to data obtained by FACS-transcriptome comparisons in cells isolated from the SC of SOD1G93A mice (Chiu et al., 2013).
The naive T cells, or Th0 cells, expand and differentiate into at least four functionally distinct subsets upon stimulation: Th1, Th2, Treg, and Th17 cells. Th1 cells secret IFN-γ and turn resting microglia into M1 phenotype; in contrast Th2 and Treg cells release IL-4 which induces activation of resting microglia into M2 phenotype (for review, see Lewis et al., 2012). Th1 and Th17 are CD4+ T cells that produce proinflammatory cytokines, causing damage while CD4+CD25high T lymphocytes (Treg) suppress Th1 cell effector function (Dittel, 2008).
Interestingly, it was demonstrated that mSOD1 mice lacking CD4+ T cells evidence a faster disease progression and decreased microglia reactivity and as so, future therapeutic interventions should consider the benefits that T cells may have (Beers et al., 2008; Chiu et al., 2008; Zhao et al., 2013). Compromise of Treg lymphocytes with disease progression diminishes the secretion of IL-4 and fails in suppressing the toxic properties of microglia (Beers et al., 2011a). Indeed, mSOD1 Treg suppress immune toxicity by inhibiting microglial activation, CD4+CD25- (T effector cells) proliferation, and the accompanying cytotoxicity, thus providing MN protection in ALS (Zhao et al., 2012). As disease progresses, the supportive Treg/Th2/M2 changes to an injurious Th1/M1 response triggering increased TNF-α secretion that was shown to induce the dysfunction of Tregs (Zhao et al., 2013). Therefore, elevation of Tregs in patients with ALS may trigger a longer life expectancy.
MN–ASTROCYTE CROSS-TALK
It was before considered that the mutant protein was the responsible for the disease onset in MNs with microglia and astrocytes only determining disease progression and extent. However, expression of mSOD1 in microglia and astrocytes is now being related with the disease onset and early stage disease, while healthy SOD1 in those cells was shown to delay ALS progression.
Several studies evidenced the demise of MNs in the presence of astrocytes harboring SOD1 mutations (Yamanaka et al., 2008b), attesting the non-cell-autonomous pathology in ALS, i.e., degeneration of MNs requires mSOD1 expression in other cells additionally to neurons. This finding was similarly obtained for astrocytes derived from ALS patients, which have shown to cause MN death (Haidet-Phillips et al., 2011). Deregulation between astrocytes and MNs communication seems to involve TGF-β signaling pathways (Phatnani et al., 2013) and miR-124a mediated regulation (Morel et al., 2013). Evidence for the role of miRNAs in MN diseases is substantiated by the relevance that the proteins TDP-43 and FUS/TLS, responsible for the processing of miRNAs, RNA maturation and splicing (Kiernan et al., 2011), have recently acquired in ALS (Lagier-Tourenne and Cleveland, 2009; Haramati et al., 2010). A major challenge would be to establish circulating miRNAs as particularly accessible biomarkers to monitor ALS.
Activated astrocytes (see Astrocyte Reactivity for more details) may also interfere with MNs function due to the reduced secretion of trophic factors such as BDNF, glial cell line-derived neurotrophic factor (GDNF) and vascular endothelial growth factor (VEGF), but this is not a clarified issue yet (for review, see Evans et al., 2013).
REGULATION OF ASTROCYTE–MICROGLIA INTERCONNECTIVITY ACTIVATION
Both in vitro and in vivo experiments have shown that astrocytes and microglia containing mSOD1 exert deleterious effects on MNs, by releasing proinflammatory factors (Phani et al., 2012). Microglia may respond earlier than astrocytes to injury and stress by first activating NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways, thus leading to a faster release of TNF-α and IL-1β (Brites, 2012). In that way one may believe that the activation of microglia precedes the reactivity of astrocytes and depends from the factors and cytokines that microglia release. Curiously, TNF-α and IL-1β released from activated microglia were reported to produce an inhibitory effect on Cx-43 expression, the main constitutive protein of gap junctions. Therefore, blockage of communication between astrocytes by the activated microglia can contribute to decrease the neuroprotective role of astrocytes (Meme et al., 2006). When considering the p25-mediated neuroinflammation, astrogliosis was shown to precede microglia activation, and apparently mediate the production of LPC, which as a chemoattractant for T cells may recruit peripheral cells into the brain (Sundaram et al., 2012). Recently, it was shown that astrocytes depend on functional microglia for response to LPS and to TLR2, 3, and 4 ligation. In their absence, astrocytes did not respond to TLR4 ligation and only weakly responded to TLR2 and 3 ligation (Holm et al., 2012). Thus, activation of astrocytes may be modulated by the proinflammatory cytokines released from microglia in an attempt to diminish the extent of excitotoxicity (Tilleux et al., 2007), but they can modulate microglia activation, as well (Welser and Milner, 2012). Which cells between microglia and astrocytes are first activated by the aggregated mSOD1 is a very controversial issue since different studies have obtained diverse profiles of glial activation in ALS models along disease progression. Some studies point out that microglia activation precede astrocyte reactivity (Alexianu et al., 2001). Others that astrogliosis is initiated in the symptomatic phase, while prominent microgliosis is only evident later at the moribund phase (Yang et al., 2011). However, since a close interaction/communication of both cells occurs in ALS, a better understanding of the benefits and risks of astrocyte and microglia activation in ALS will help to determine whether therapeutic strategies should envisage enhancing or impairing the actions of glial cells in ALS.
MN–MICROGLIA SIGNALING
Communication between MNs and microglia is essential for maintaining local homeostasis during both physiological and inflammatory conditions. Neuroprotective signaling from MN to microglia involve FKN and CD200, as previously mentioned. In addition, microglia “calming” effects during neuroinflammation are mediated through ATX (Awada et al., 2012). Insights into the complex intercellular perturbations and influence of alarming and calming factors underlying neurodegeneration will enhance our efforts toward target-driven directed therapeutic strategies in ALS.
CX3CR1 deficiency
The neuroprotective/neurotoxic role of CX3CL1/CX3CR1 signaling is still a matter of debate once it seems to depend upon the CNS insult (for review, see Desforges et al., 2012). Deletion of CX3CR1 in a transgenic model of ALS mice was shown to extend neuronal cell loss, suggesting that CX3CL1/CX3CR1 signaling limits microglial toxicity in ALS (Cardona et al., 2006). Interestingly, it was also shown that treatment with LPS down-regulated the expression of CX3CR1, thus suppressing the functional response to FKN (Boddeke et al., 1999) and potentiating LPS neurotoxic effects (Zujovic et al., 2000). Accordingly, in ALS, the disruption of CX3CL1/CX3CR1 signaling evidenced to promote neurodegeneration following LPS administration (Cardona et al., 2006).
Cathepsin S influence
Although it has been proposed that cathepsins compensate for each other because of their overlapping substrate specificities, there is increasing evidence that disturbance of the normal balance and extralysosomal localization of cathepsins contribute to age-related diseases (for review, see Nakanishi, 2003). CatS that is a lysosomal/endosomal cysteine protease that degrades extracellular matrix proteins, even at neutral pH, was shown to be increasingly expressed by microglia upon LPS surcharge (Petanceska et al., 1996). However, some authors indicate that a further challenge with ATP is required to observe CatS release (Clark and Malcangio, 2012). This protease is preferentially expressed by antigen-presenting cells, including microglia.
Microglial CatS seems to be responsible for the liberation of neuronal FKN (Figure 1), which via the specific receptor CX3CR1 in microglia activate p38 MAPK pathway leading to the release of mediators that interact with neurons (Clark et al., 2007). Several inhibitors have been tried to regulate the immune modulatory effects of CatS (Clark and Malcangio, 2012) and a shift from a Th1/Th17 type response to a Th2 type of response was obtained (Baugh et al., 2011).
The up-regulation of cathepsins appears as a general transcriptional event in ALS (Gonzalez de Aguilar et al., 2008; Offen et al., 2009; Boutahar et al., 2011). However, reference to CatS and microglia activation was not until now indicated in the context of ALS. Therefore, if CX3CL1/CX3CR1 demonstrates to have a role in any stage of ALS progression, inhibition of CatS may constitute a therapeutic approach for ALS.
Exosomes and disease spread
Exosomes are secretory vesicles deriving from late endosomes and multivesicular bodies that mediate neuron–glia communication, have 50–100 nm in size and carry specific protein and RNA cargo (Fitzner et al., 2011; Fruhbeis et al., 2012; El Andaloussi et al., 2013). Exosomes contain both miRNAs and mRNAs that may be delivered and be functional in another cell (Valadi et al., 2007). Selectivity for miRNA incorporation into exosomes is proposed based on the fact that some are exclusive in exosomes derived from immature dendritic cells while others only exist in those from mature dendritic cells (Stoorvogel, 2012). Indeed, exosomes and miRNAs have been found to participate in cellular senescence and contribute to aging (Xu and Tahara, 2013). In addition senescent cells produce high levels of exosomes thus interacting and inducing the senescence of neighboring cells. As so, exosomal miRNAs might become useful biomarkers of disease. Recently, it was suggested that exosomes may be important candidates to deliver siRNA (El Andaloussi et al., 2013) and specific drugs, based on the perceived advantages of nanoparticle size and non-cytotoxicity (Sun et al., 2013), thus constituting a therapeutic platform.
Microglia was shown to internalize oligodendroglial exosomes, thus participating in the degradation of oligodendroglial membrane (Fitzner et al., 2011). However, microglia also release exosomes (Potolicchio et al., 2005; Hooper et al., 2012), which generate IL-1β to the extracellular environment propagating inflammation (Turola et al., 2012). Increased exosome discharge by microglia was observed after stimulation with α-synuclein and such activated exosomes revealed an increased membrane content in TNF-α (Chang et al., 2013). Interestingly, it was shown that mouse MN-like NSC-34 cells overexpressing hSOD1G93A secrete SOD1 via exosomes probably accounting to cell–cell-mediated mutant toxicity in ALS pathogenesis (Gomes et al., 2007). Therefore, exosomes from microglia may spread pathogenic factors, such as SOD1 and promote inflammation while influencing neuronal survival (Chang et al., 2013). In line with this, a latest study reports that cell-to-cell transmission of SOD1 misfolding is mediated by two non-exclusive mechanisms: through the release of protein aggregates that are taken up by macropinocytosis or via exosomes secreted from living cells (Grad et al., 2014).
CHALLENGES TO NERVE REGENERATION IN ALS
Reconstruction of neural network implies the restoration of tissue architecture and cell functionality. Cell replacement-based repair strategies have been tested both in vitro and in vivo and a major challenge resides in maintaining the cell function when transplanted to a broken parenchyma homeostasis. Confront with excessive neuroinflammation and cell senescence may compromise the success of the strategy. In particular, reactive astrocytes exacerbate inflammation and by forming glial scars impede regenerating axons from traversing the lesions, while myelin debris prevent axon growth and microglia lose the ability to migrate, phagocyte or sustain inflammation from spread. Therefore, the rewiring of the CNS to foster a permissive environment for neuroregeneration is the key to a successful functional integration and repair (Xu et al., 2011; Kim et al., 2012).
CELL SENESCENCE
Redox changes within neurons (Olivieri et al., 2001), ER stress condition (Di Virgilio, 2000), and mitochondria dysfunction (Konnecke and Bechmann, 2013) are accelerated by aging and are emerging as common features relevant to the pathogenesis of neurological disorders, including ALS. Actually, the variation in SOD1 activity in aging ALS patients, when compared to younger ones, point to an increased oxidative misbalance vulnerability (Fiszman et al., 1999). Nevertheless, it was recently indicated that the ALS incidence decline in the elderly (Schoser and Blottner, 1999; Demestre et al., 2005), suggesting that the disease is not merely the result of aging. However, markers of senescence were found increased in satellite cells from ALS muscle biopsies suggesting a vulnerability to muscle atrophy (Gottschall and Deb, 1996). In addition, autophagic dysfunction and mitochondrial DNA damage in the CNS are prominently found in microglia with aging and may lead to a defective turnover of mitochondria and accumulation of hypergenerated ROS (reviewed in Nakanishi and Wu, 2009). Interestingly, dysfunctional and senescent microglia may even release compounds that inhibit neuronal autophagy (Alirezaei et al., 2008) and neurogenesis (reviewed in Wong, 2013). Given the important and necessary functions of microglia in the CNS homeostasis it is of major relevance to understand the multiple stage-related microglia phenotypes in ALS, including the increased vulnerability to a senescent cell with the disease progression, as observed in other neurodegenerative and age-related CNS disorders (Luo et al., 2010; Kaplan et al., 2014).
Microglia degeneration
Recent data obtained in the mSOD1 mouse model suggest a dominant neuroinflammatory response in the CNS, with a reactive microglia in preclinical stages that turns into an irresponsive cell during disease progression, and a degenerative process in the PNS (Dibaj et al., 2011). Earlier studies evidenced that mutated SOD1 microglia have an age-dependent cytotoxic potential which reveals upon a stimulatory effect (Weydt et al., 2004). Interestingly, extracellular SOD1G93A mediates the activation of CD14–TLR2 pathway with the consequent release of TNF-α and IL-1β, thus propagating the proinflammatory stimuli (Bowerman et al., 2013). However, since immunosuppressive strategies have not proven consistent efficacy (Appel et al., 1993, 2011; Baugh et al., 2011), one may believe that microglia function may change along disease progression with consequent differential effects on MNs. Late data evidenced that SC microglia proliferate and that TNF-α mRNA expression decreases during disease progression in SOD1G93A rats (Nikodemova et al., 2013). Indeed, microglia revealed to not be polarized to M1 or M2 phenotypes at any disease stage and CNS region evaluated. In addition, SC microglia evidenced to be irresponsive to experimental systemic inflammation at ALS end-stage. Diminished glial neuroprotection by senescent and/or dysfunctional microglia has been suggested to play a role in neurodegenerative diseases, mainly in the late stage (Streit, 2006). In such circumstances the aged microglia evidenced a “dystrophic” morphology with the loss of finely branched cytoplasmic processes, cytoplasmic beading/spheroid formation, and cytoplasmic fragmentation (cytorrhexis; Figure 2) (Streit et al., 2004). Such severe abnormalities in microglia, including cell fusion (multinucleated giant cells) at the symptomatic stage and cytorrhexis at the end stage are indicative of microglial aberrant activation and degeneration, respectively, and were observed in SOD1G93A transgenic rats (Fendrick et al., 2007). Although prevalent in older human subjects this dystrophic cells may also in rare instances be observed in the young brain (Luo and Chen, 2012). Pathogenic miRNAs, such as miR-155, miR-146a, and miR-124 in microglia may be associated with the acquisition of a senescent phenotype (Saba et al., 2012; Ponomarev et al., 2013). We recently observed that aged-cultured microglia exhibit lower phagocytic ability and higher expression levels of miR-146a than younger cells (Caldeira et al., 2013). Whether these findings are related with a dysfunctional microglia at ALS end-stages deserve to be further investigated.
Demyelination progression
Although new oligodendrocytes were observed in the SC of SOD1G93A mice, they do not fully maturate, resulting in progressive demyelination and accelerated disease (Liu et al., 2002). The authors also found similar myelination defects in postmortem samples taken from SC and motor cortex from ALS patients. Actually, there are many potent inhibitors of axonal regeneration in the injured CNS including myelin-associated proteins, fibrinogen, and axonal guidance molecules, where epidermal growth factor receptor (EGFR) and eukaryotic ribosome biogenesis protein 1 (Erb1) may have a special role in reducing the effects of multiple inhibitors of axonal regeneration (Arthur-Farraj et al., 2012; Leinster et al., 2013). However, the role of EGFR on the protection of MN synapses and survival extension in SOD1G93A mice is still a matter of debate; hence, it was reported that EGFR inhibitors failed to extend ALS mouse survival although influencing disease progression (Le Pichon et al., 2013).
Another important point to be considered is the decrease in the phagocytic clearance by the dysfunctional microglia that results in the accumulation of myelin debris, leading to oligodendrocyte differentiation arrest and decreased recruitment of oligodendrocyte precursor cells (Walter and Neumann, 2009). Activated microglia were also shown to attenuate the proliferation of the oligodendrocyte precursor cells, thus concurring for demyelination progression (Taylor et al., 2010), and reinforcing the recovery of healthy microglia as a potential therapeutic target in ALS. Indeed, studies on the global gene expression during demyelination and remyelination by microarray analysis reinforced that the primary function of microglia is the tolerance induction and support to regeneration (Olah et al., 2012).
CELL REPLACEMENT THERAPY
Cell replacement therapy has been suggested as a promising strategy for MN disease. The use of combined strategies to restore both the healthy state of MNs and glial cells, such as microglia, and their correct cross-talk to face persistent neurotoxic insults may even provide better benefits for this devastating disease.
Mesenchymal stem cells (MSCs) isolated from the bone marrow of ALS patients did not show morphological or functional differences from those obtained from donors and seem to be useful for cell-based therapy for ALS patients (Hadass et al., 2013). In a few well-monitored ALS patients the autologous transplantation of such MSCs into the SC evidenced to be well tolerated and to promote some clinical improvement (Min et al., 2012).
Another proposed strategy is the olfactory ensheathing cell (OEC) transplantation that although evidencing to slow the rate of ALS progression in a short period (Inoue et al., 2003) and to improve pulmonary function (Guegan et al., 2001) has been a matter of debate. Indeed, prominent glial and inflammatory reaction around the brain delivery track was observed in postmortem samples (Reyes et al., 2010).
Efficacy of transplantation-based astrocyte replacement was also evidenced as a promising therapy for slowing focal MN loss associated with ALS while also reducing microgliosis in the hSOD1G93A rodents (Lepore et al., 2008). Effects may derive from the release of growth factors that are decreased in ALS and already evidenced to increase mSOD1 mice survival (Kaspar et al., 2003; Park et al., 2009). However, limited efficacy was obtained in a later study (Lepore et al., 2011). The use of induced pluripotent stem cell (iPSC) technologies may allow in the future the autologous cell transplantation in ALS patients, including MNs (Papadeas and Maragakis, 2009).
More recently, depletion of microglia cells expressing mSOD1 with clodronate liposomes and subsequent transplantation with bone marrow cells (BMCs) expressing wtSOD1 was shown to trigger microglia replacement and to slow ALS disease progression in the SOD1G93A mice model (Lee et al., 2012). The method seems to afford better therapeutic effects than the one by Ohnishi et al. (2009) using BMCs transplantation since microglia renewal is better achieved by tissue-resident microglia rather than by BMCs (Davoust et al., 2008). Overall, the mechanisms and functional implications of microglia replacement require further elucidation, inasmuch because myeloid-derived infiltrating cells (monocyte-derived macrophages) revealed to be functionally distinct from the resident microglia assisting them (Jung and Schwartz, 2012; London et al., 2013). Clearly, additional research is required to address these issues and contribute to develop strategies able to stop, or at least delay, ALS progression.
CONCLUSION
Amyotrophic lateral sclerosis is a fatal neurodegenerative disorder with limited identified targets, biomarkers, and therapeutic options. Therefore, a better comprehension of the underlying molecular mechanisms is necessary to develop novel etiological therapeutic strategies. Multiple studies suggest that a complex pathological interplay between MNs and glial cells, involving neuroinflammation and microglia physiopathological changes drive the performance of these glial cells before the ALS onset and during disease progression till late and end stages. In this context, we here summarized the growing body of evidence supporting the key role of microglia in the deregulated motor-neuron interconnectivity and in the dreadful chain of events leading to MN degeneration in ALS. As such, insights into the complex intercellular perturbations underlying ALS disease and centered on microglia phenotypic changes, and associated detrimental functions, will help on our efforts to develop effective therapeutic approaches for recovering adequate microglia function in initiation and progression phases of ALS. In conclusion, the role of microglia in keeping brain homeostasis leads to consider that healthy microglia may be used to replace the senescent or irresponsive cell. In addition, dysfunctional microglia may be differently modulated, either directly or indirectly, to be transformed in a less reactive cell to challenges in excessive and chronic neuroinflammation, or alternatively rejuvenated to enhance their capacity to fight the insult and improve disease outcomes. Considerably more research is necessary to realize the feasibility and usefulness of such strategies before their potential in clinic can be realized.
Statements
Acknowledgments
This work was primarily supported by FEDER (COMPETE Programme) and by National grants from Fundação para a Ciência e a Tecnologia (FCT) through the Projects PTDC/SAU-FAR/118787/2010 and PEst-OE/SAU/UI4013/2011-2013 to Dora Brites. Ana R. Vaz was recipient of a post-doctoral fellowship from FCT (SFRH/BPD/76590/2011).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Alexianu M. E. Kozovska M. Appel S. H. (2001). Immune reactivity in a mouse model of familial ALS correlates with disease progression.Neurology571282–1289. 10.1212/WNL.57.7.1282
2
Alirezaei M. Kiosses W. B. Flynn C. T. Brady N. R. Fox H. S. (2008). Disruption of neuronal autophagy by infected microglia results in neurodegeneration.PLoS ONE3:e2906. 10.1371/journal.pone.0002906
3
Andjus P. R. Bataveljic D. Vanhoutte G. Mitrecic D. Pizzolante F. Djogo N. et al (2009). In vivo morphological changes in animal models of amyotrophic lateral sclerosis and Alzheimer’s-like disease: MRI approach.Anat. Rec. (Hoboken)2921882–1892. 10.1002/ar.20995
4
Andrus P. K. Fleck T. J. Gurney M. E. Hall E. D. (1998). Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis.J. Neurochem.712041–2048. 10.1046/j.1471-4159.1998.71052041.x
5
Appel S. H. Smith R. G. Engelhardt J. I. Stefani E. (1993). Evidence for autoimmunity in amyotrophic lateral sclerosis.J. Neurol. Sci.118169–174. 10.1016/0022-510X(93)90106-9
6
Appel S. H. Zhao W. Beers D. R. Henkel J. S. (2011). The microglial–motoneuron dialogue in ALS.Acta Myol.304–8.
7
Arthur-Farraj P. J. Latouche M. Wilton D. K. Quintes S. Chabrol E. Banerjee A. et al (2012). c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration.Neuron75633–647. 10.1016/j.neuron.2012.06.021
8
Awada R. Rondeau P. Gres S. Saulnier-Blache J. S. Lefebvre D’Hellencourt C. Bourdon E. (2012). Autotaxin protects microglial cells against oxidative stress.Free Radic. Biol. Med.52516–526. 10.1016/j.freeradbiomed.2011.11.014
9
Barbeito A. G. Mesci P. Boillee S. (2010). Motor neuron-immune interactions: the vicious circle of ALS.J. Neural. Transm.117981–1000. 10.1007/s00702-010-0429-0
10
Barbosa M. I. (2013). Dissecting Cross-talk Between Microglia and Motoneurons in ALS: Signalling Events and Soluble Factors. Master’s thesis, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Lisbon.
11
Baugh M. Black D. Westwood P. Kinghorn E. Mcgregor K. Bruin J. et al (2011). Therapeutic dosing of an orally active, selective cathepsin S inhibitor suppresses disease in models of autoimmunity.J. Autoimmun.36201–209. 10.1016/j.jaut.2011.01.003
12
Bazan J. F. Bacon K. B. Hardiman G. Wang W. Soo K. Rossi D. et al (1997). A new class of membrane-bound chemokine with a CX3C motif.Nature385640–644. 10.1038/385640a0
13
Beckman J. S. Beckman T. W. Chen J. Marshall P. A. Freeman B. A. (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide.Proc. Natl. Acad. Sci. U.S.A.871620–1624. 10.1073/pnas.87.4.1620
14
Beers D. R. Henkel J. S. Xiao Q. Zhao W. Wang J. Yen A. A. et al (2006). Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis.Proc. Natl. Acad. Sci. U.S.A.10316021–16026. 10.1073/pnas.0607423103
15
Beers D. R. Henkel J. S. Zhao W. Wang J. Appel S. H. (2008). CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS.Proc. Natl. Acad. Sci. U.S.A.10515558–15563. 10.1073/pnas.0807419105
16
Beers D. R. Henkel J. S. Zhao W. Wang J. Huang A. Wen S. et al (2011a). Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis.Brain1341293–1314. 10.1093/brain/awr074
17
Beers D. R. Zhao W. Liao B. Kano O. Wang J. Huang A. et al (2011b). Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice.Brain Behav. Immun.251025–1035. 10.1016/j.bbi.2010.12.008
18
Benkler C. Ben-Zur T. Barhum Y. Offen D. (2013). Altered astrocytic response to activation in SOD1(G93A) mice and its implications on amyotrophic lateral sclerosis pathogenesis.Glia61312–326. 10.1002/glia.22428
19
Bertolin C. D’Aascenzo C. Querin G. Gaiani A. Boaretto F. Salvoro C. et al (2013). Improving the knowledge of amyotrophic lateral sclerosis genetics: novel SOD1 and FUS variants.Neurobiol. Aging351212.e7–1212.e10. 10.1016/j.neurobiolaging.2013.10.093
20
Bianchi M. E. (2009). HMGB1 loves company.J. Leukoc. Biol.86573–576. 10.1189/jlb.1008585
21
Bianchi M. E. Manfredi A. A. (2009). Immunology. Dangers in and out.Science3231683–1684. 10.1126/science.1172794
22
Boddeke E. W. Meigel I. Frentzel S. Biber K. Renn L. Q. Gebicke-Harter P. (1999). Functional expression of the fractalkine (CX3C) receptor and its regulation by lipopolysaccharide in rat microglia.Eur. J. Pharmacol.374309–313. 10.1016/S0014-2999(99)00307-6
23
Boillée S. Vande Velde C. Cleveland D. W. (2006a). ALS: a disease of motor neurons and their nonneuronal neighbors.Neuron5239–59. 10.1016/j.neuron.2006.09.018
24
Boillée S. Yamanaka K. Lobsiger C. S. Copeland N. G. Jenkins N. A. Kassiotis G. et al (2006b). Onset and progression in inherited ALS determined by motor neurons and microglia.Science3121389–1392. 10.1126/science.1123511
25
Bonfoco E. Krainc D. Ankarcrona M. Nicotera P. Lipton S. A. (1995). Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures.Proc. Natl. Acad. Sci. U.S.A.927162–7166. 10.1073/pnas.92.16.7162
26
Bourd-Boittin K. Basset L. Bonnier D. L’helgoualc’h A. Samson M. Théret N. (2009). CX3CL1/fractalkine shedding by human hepatic stellate cells: contribution to chronic inflammation in the liver.J. Cell. Mol. Med.131526–1535. 10.1111/j.1582-4934.2009.00787.x
27
Boutahar N. Wierinckx A. Camdessanche J. P. Antoine J. C. Reynaud E. Lassabliere F. et al (2011). Differential effect of oxidative or excitotoxic stress on the transcriptional profile of amyotrophic lateral sclerosis-linked mutant SOD1 cultured neurons.J. Neurosci. Res.891439–1450. 10.1002/jnr.22672
28
Bowerman M. Vincent T. Scamps F. Perrin F. E. Camu W. Raoul C. (2013). Neuroimmunity dynamics and the development of therapeutic strategies for amyotrophic lateral sclerosis.Front. Cell. Neurosci.7:214. 10.3389/fncel.2013.00214
29
Brettschneider J. Toledo J. B. Van Deerlin V. M. Elman L. Mccluskey L. Lee V. M. et al (2012). Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis.PLoS ONE7:e39216. 10.1371/journal.pone.0039216
30
Brites D. (2012). The evolving landscape of neurotoxicity by unconjugated bilirubin: role of glial cells and inflammation.Front. Pharmacol.3:88. 10.3389/fphar.2012.00088
31
Butovsky O. Siddiqui S. Gabriely G. Lanser A. J. Dake B. Murugaiyan G. et al (2012). Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS.J. Clin. Invest.1223063–3087. 10.1172/JCI62636
32
Caldeira C. Frederico A. Vaz A. Fernandes A. Brites D. (2013). Age-related differences in microglia reactivity: relevance to Alzheimer’s disease.J. Neurochem. 125 (Suppl.1)185.
33
Calvo M. Zhu N. Tsantoulas C. Ma Z. Grist J. Loeb J. A. et al (2010). Neuregulin-ErbB signalling promotes microglial proliferation and chemotaxis contributing to microgliosis and pain after peripheral nerve injury.J. Neurosci.305437–5450. 10.1523/JNEUROSCI.5169-09.2010
34
Campos-Melo D. Droppelmann C. A. He Z. Volkening K. Strong M. J. (2013). Altered microRNA expression profile in Amyotrophic Lateral Sclerosis: a role in the regulation of NFL mRNA levels.Mol. Brain62610.1186/1756-6606-6-26
35
Cardona A. E. Pioro E. P. Sasse M. E. Kostenko V. Cardona S. M. Dijkstra I. M. et al (2006). Control of microglial neurotoxicity by the fractalkine receptor.Nat. Neurosci.9917–924. 10.1038/nn1715
36
Cardoso A. L. Guedes J. R. Pereira de Almeida L Pedroso De Lima M. C. (2012). miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production.Immunology13573–88. 10.1111/j.1365-2567.2011.03514.x
37
Cardoso F. L. Brites D. Brito M. A. (2010). Looking at the blood–brain barrier: molecular anatomy and possible investigation approaches.Brain Res. Rev.64328–363. 10.1016/j.brainresrev.2010.05.003
38
Casoni F. Basso M. Massignan T. Gianazza E. Cheroni C. Salmona M. et al (2005). Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: possible multifunctional role in the pathogenesis.J. Biol. Chem.28016295–16304. 10.1074/jbc.M413111200
39
Casula M. Iyer A. M. Spliet W. G. Anink J. J. Steentjes K. Sta M. et al (2011). Toll-like receptor signalling in amyotrophic lateral sclerosis spinal cord tissue.Neuroscience179233–243. 10.1016/j.neuroscience.2011.02.001
40
Cereda C. Cova E. Di Poto C. Galli A. Mazzini G. Corato M. et al (2006). Effect of nitric oxide on lymphocytes from sporadic amyotrophic lateral sclerosis patients: toxic or protective role?Neurol. Sci.27312–316. 10.1007/s10072-006-0702-z
41
Chang C. Lang H. Geng N. Wang J. Li N. Wang X. (2013). Exosomes of BV-2 cells induced by alpha-synuclein: important mediator of neurodegeneration in PD.Neurosci. Lett.548190–195. 10.1016/j.neulet.2013.06.009
42
Chapman G. A. Moores K. Harrison D. Campbell C. A. Stewart B. R. Strijbos P. J. (2000). Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage.J. Neurosci.20RC87.
43
Chen L. C. Smith A. Ben Y. Zukic B. Ignacio S. Moore D. et al (2004). Temporal gene expression patterns in G93A/SOD1 mouse.Amyotroph. Lateral Scler. Other Motor Neuron Disord.5164–171. 10.1080/14660820410017091
44
Chen S. Zhang X. Song L. Le W. (2012). Autophagy dysregulation in amyotrophic lateral sclerosis.Brain Pathol.22110–116. 10.1111/j.1750-3639.2011.00546.x
45
Chen S. C. Leach M. W. Chen Y. Cai X. Y. Sullivan L. Wiekowski M. et al (2002). Central nervous system inflammation and neurological disease in transgenic mice expressing the CC chemokine CCL21 in oligodendrocytes.J. Immunol.1681009–1017.
46
Chhor V. Le Charpentier T. Lebon S. Ore M. V. Celador I. L. Josserand J. et al (2013). Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro.Brain Behav. Immun.3270–85. 10.1016/j.bbi.2013.02.005
47
Chia R. Tattum M. H. Jones S. Collinge J. Fisher E. M. Jackson G. S. (2010). Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis.PLoS ONE5:e10627. 10.1371/journal.pone.0010627
48
Chiu I. M. Chen A. Zheng Y. Kosaras B. Tsiftsoglou S. A. Vartanian T. K. et al (2008). T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS.Proc. Natl. Acad. Sci. U.S.A.10517913–17918. 10.1073/pnas.0804610105
49
Chiu I. M. Morimoto E. T. Goodarzi H. Liao J. T. O’Keeffe S. Phatnani H. P. et al (2013). A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model.Cell Rep.4385–401. 10.1016/j.celrep.2013.06.018
50
Clark A. K. Malcangio M. (2012). Microglial signalling mechanisms: cathepsin S and fractalkine.Exp. Neurol.234283–292. 10.1016/j.expneurol.2011.09.012
51
Clark A. K. Yip P. K. Grist J. Gentry C. Staniland A. A. Marchand F. et al (2007). Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain.Proc. Natl. Acad. Sci. U.S.A.10410655–10660. 10.1073/pnas.0610811104
52
Clark A. K. Yip P. K. Malcangio M. (2009). The liberation of fractalkine in the dorsal horn requires microglial cathepsin S.J. Neurosci.296945–6954. 10.1523/JNEUROSCI.0828-09.2009
53
Clement A. M. Nguyen M. D. Roberts E. A. Garcia M. L. Boillee S. Rule M. et al (2003). Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice.Science302113–117. 10.1126/science.1086071
54
Cook A. Hippensteel R. Shimizu S. Nicolai J. Fatatis A. Meucci O. (2010). Interactions between chemokines: regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons.J. Biol. Chem.28510563–10571. 10.1074/jbc.M109.035477
55
Corcia P. Tauber C. Vercoullie J. Arlicot N. Prunier C. Praline J. et al (2012). Molecular imaging of microglial activation in amyotrophic lateral sclerosis.PLoS ONE7:e52941. 10.1371/journal.pone.0052941
56
Cunha C. (2012). Exploring Motor Neuron Degeneration in ALS – Prevention by Glycoursodeoxycholic Acid and Signalling to Microglia. Master’s thesis, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Lisbon.
57
Czirr E. Wyss-Coray T. (2012). The immunology of neurodegeneration.J. Clin. Invest.1221156–1163. 10.1172/JCI58656
58
David S. Kroner A. (2011). Repertoire of microglial and macrophage responses after spinal cord injury.Nat. Rev. Neurosci.12388–399. 10.1038/nrn3053
59
Davoust N. Vuaillat C. Androdias G. Nataf S. (2008). From bone marrow to microglia: barriers and avenues.Trends Immunol.29227–234. 10.1016/j.it.2008.01.010
60
Deepa P. Shahani N. Alladi P. A. Vijayalakshmi K. Sathyaprabha T. N. Nalini A. et al (2011). Down regulation of trophic factors in neonatal rat spinal cord after administration of cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients.J. Neural. Transm.118531–538. 10.1007/s00702-010-0520-6
61
Deiva K. Geeraerts T. Salim H. Leclerc P. Hery C. Hugel B. et al (2004). Fractalkine reduces N-methyl-D-aspartate-induced calcium flux and apoptosis in human neurons through extracellular signal-regulated kinase activation.Eur. J. Neurosci.203222–3232. 10.1111/j.1460-9568.2004.03800.x
62
DeJesus-Hernandez M. Mackenzie I. R. Boeve B. F. Boxer A. L. Baker M. Rutherford N. J. et al (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS.Neuron72245–256. 10.1016/j.neuron.2011.09.011
63
de Jong E. K. Dijkstra I. M. Hensens M. Brouwer N. Van Amerongen M. Liem R. S. et al (2005). Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion.J. Neurosci.257548–7557. 10.1523/JNEUROSCI.1019-05.2005
64
Demestre M. Parkin-Smith G. Petzold A. Pullen A. H. (2005). The pro and the active form of matrix metalloproteinase-9 is increased in serum of patients with amyotrophic lateral sclerosis.J. Neuroimmunol.159146–154. 10.1016/j.jneuroim.2004.09.015
65
Desforges N. M. Hebron M. L. Algarzae N. K. Lonskaya I. Moussa C. E. (2012). Fractalkine mediates communication between pathogenic proteins and microglia: implications of anti-inflammatory treatments in different stages of neurodegenerative diseases.Int. J. Alzheimers Dis.201234547210.1155/2012/345472
66
Diaz-Amarilla P. Olivera-Bravo S. Trias E. Cragnolini A. Martinez-Palma L. Cassina P. et al (2011). Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis.Proc. Natl. Acad. Sci. U.S.A.10818126–18131. 10.1073/pnas.1110689108
67
Dibaj P. Steffens H. Zschuntzsch J. Kirchhoff F. Schomburg E. D. Neusch C. (2011). In vivo imaging reveals rapid morphological reactions of astrocytes towards focal lesions in an ALS mouse model.Neurosci. Lett.497148–151. 10.1016/j.neulet.2011.04.049
68
Dittel B. N. (2008). CD4 T cells: balancing the coming and going of autoimmune-mediated inflammation in the CNS.Brain Behav. Immun.22421–430. 10.1016/j.bbi.2007.11.010
69
Di Virgilio F. (2000). Dr. Jekyll/Mr. Hyde: the dual role of extracellular ATP.J. Auton. Nerv. Syst.8159–63. 10.1016/S0165-1838(00)00114-4
70
Drechsel D. A. Estevez A. G. Barbeito L. Beckman J. S. (2012). Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS.Neurotox. Res.22251–264. 10.1007/s12640-012-9322-y
71
El Andaloussi S. Lakhal S. Mager I. Wood M. J. (2013). Exosomes for targeted siRNA delivery across biological barriers.Adv. Drug Deliv. Rev.65391–397. 10.1016/j.addr.2012.08.008
72
Elliott J. L. (1999). Experimental models of amyotrophic lateral sclerosis.Neurobiol. Dis.6310–320. 10.1006/nbdi.1999.0266
73
Enokido Y. Yoshitake A. Ito H. Okazawa H. (2008). Age-dependent change of HMGB1 and DNA double-strand break accumulation in mouse brain.Biochem. Biophys. Res. Commun.376128–133. 10.1016/j.bbrc.2008.08.108
74
Evans M. C. Couch Y. Sibson N. Turner M. R. (2013). Inflammation and neurovascular changes in amyotrophic lateral sclerosis.Mol. Cell. Neurosci.5334–41. 10.1016/j.mcn.2012.10.008
75
Fang P. Schachner M. Shen Y. Q. (2012). HMGB1 in development and diseases of the central nervous system.Mol. Neurobiol.45499–506. 10.1007/s12035-012-8264-y
76
Fendrick S. E. Xue Q. S. Streit W. J. (2007). Formation of multinucleated giant cells and microglial degeneration in rats expressing a mutant Cu/Zn superoxide dismutase gene.J. Neuroinflammation4910.1186/1742-2094-4-9
77
Ferraiuolo L. Higginbottom A. Heath P. R. Barber S. Greenald D. Kirby J. et al (2011). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis.Brain1342627–2641. 10.1093/brain/awr193
78
Fiszman M. L. Borodinsky L. N. Ricart K. C. Sanz O. P. Sica R. E. (1999). Cu/Zn superoxide dismutase activity at different ages in sporadic amyotrophic lateral sclerosis.J. Neurol. Sci.16234–37. 10.1016/S0022-510X(98)00272-X
79
Fitzner D. Schnaars M. Van Rossum D. Krishnamoorthy G. Dibaj P. Bakhti M. et al (2011). Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis.J. Cell Sci.124447–458. 10.1242/jcs.074088
80
Frakes A. E. Ferraiuolo L. Haidet-Phillips A. M. Schmelzer L. Braun L. Miranda C. J. et al (2014). Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis.Neuron811009–1023. 10.1016/j.neuron.2014.01.013
81
Fricker M. Oliva-Martin M. J. Brown G. C. (2012a). Primary phagocytosis of viable neurons by microglia activated with LPS or Abeta is dependent on calreticulin/LRP phagocytic signalling.J. Neuroinflammation919610.1186/1742-2094-9-196
82
Fricker M. Neher J. J. Zhao J. W. Thery C. Tolkovsky A. M. Brown G. C. (2012b). MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation.J. Neurosci.322657–2666. 10.1523/JNEUROSCI.4837-11.2012
83
Fruhbeis C. Frohlich D. Kramer-Albers E. M. (2012). Emerging roles of exosomes in neuron–glia communication.Front. Physiol.3:119. 10.3389/fphys.2012.00119
84
Furukawa Y. Kaneko K. Watanabe S. Yamanaka K. Nukina N. (2013). Intracellular seeded aggregation of mutant Cu,Zn-superoxide dismutase associated with amyotrophic lateral sclerosis.FEBS Lett.5872500–2505. 10.1016/j.febslet.2013.06.046
85
Gandelman M. Peluffo H. Beckman J. S. Cassina P. Barbeito L. (2010). Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: implications for amyotrophic lateral sclerosis.J. Neuroinflammation73310.1186/1742-2094-7-33
86
Gao H. M. Zhou H. Zhang F. Wilson B. C. Kam W. Hong J. S. (2011). HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration.J. Neurosci.311081–1092. 10.1523/JNEUROSCI.3732-10.2011
87
Garbuzova-Davis S. Haller E. Saporta S. Kolomey I. Nicosia S. V. Sanberg P. R. (2007). Ultrastructure of blood–brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS.Brain Res.1157126–137. 10.1016/j.brainres.2007.04.044
88
Garbuzova-Davis S. Rodrigues M. C. Mirtyl S. Turner S. Mitha S. Sodhi J. et al (2012). Multiple intravenous administrations of human umbilical cord blood cells benefit in a mouse model of ALS.PLoS ONE7:e31254. 10.1371/journal.pone.0031254
89
Garton K. J. Gough P. J. Blobel C. P. Murphy G. Greaves D. R. Dempsey P. J. et al (2001). Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1).J. Biol. Chem.27637993–38001. 10.1074/jbc.M106434200
90
Gerber Y. N. Sabourin J. C. Rabano M. Vivanco M. Perrin F. E. (2012). Early functional deficit and microglial disturbances in a mouse model of amyotrophic lateral sclerosis.PLoS ONE7:e36000. 10.1371/journal.pone.0036000
91
Glass C. K. Saijo K. Winner B. Marchetto M. C. Gage F. H. (2010). Mechanisms underlying inflammation in neurodegeneration.Cell140918–934. 10.1016/j.cell.2010.02.016
92
Gomes C. Keller S. Altevogt P. Costa J. (2007). Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis.Neurosci. Lett.42843–46. 10.1016/j.neulet.2007.09.024
93
Gomez-Pinedo U. Yanez M. Matias-Guiu J. Galan L. Guerrero-Sola A. Benito-Martin M. S. et al (2013). Cellular changes in motor neuron cell culture produced by cytotoxic cerebrospinal fluid from patients with amyotrophic lateral sclerosis.Neurologia.10.1016/j.nrl.2013.08.001[Epub ahead of print].
94
Gonzalez de Aguilar J. L. Niederhauser-Wiederkehr C. Halter B. De Tapia M. Di Scala F. Demougin P. et al (2008). Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model.Physiol. Genomics32207–218. 10.1152/physiolgenomics.00017.2007
95
Gottschall P. E. Deb S. (1996). Regulation of matrix metalloproteinase expressions in astrocytes, microglia and neurons.Neuroimmunomodulation369–75. 10.1159/000097229
96
Gough P. J. Garton K. J. Wille P. T. Rychlewski M. Dempsey P. J. Raines E. W. (2004). A disintegrin and metalloproteinase 10-mediated cleavage and shedding regulates the cell surface expression of CXC chemokine ligand 16.J. Immunol.1723678–3685. 10.4049/jimmunol.172.6.3678
97
Grad L. I. Yerbury J. J. Turner B. J. Guest W. C. Pokrishevsky E. O’neill M. A. et al (2014). Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc.Natl. Acad. Sci. U.S.A.1113620–3625. 10.1073/pnas.1312245111
98
Grosskreutz J. Van Den Bosch L. Keller B. U. (2010). Calcium dysregulation in amyotrophic lateral sclerosis.Cell Calcium47165–174. 10.1016/j.ceca.2009.12.002
99
Guegan C. Vila M. Rosoklija G. Hays A. P. Przedborski S. (2001). Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis.J. Neurosci.216569–6576.
100
Gurney M. E. Pu H. Chiu A. Y. Dal Canto M. C. Polchow C. Y. Alexander D. D. et al (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation.Science2641772–1775. 10.1126/science.8209258
101
Hadass O. Tomlinson B. N. Gooyit M. Chen S. Purdy J. J. Walker J. M. et al (2013). Selective inhibition of matrix metalloproteinase-9 attenuates secondary damage resulting from severe traumatic brain injury.PLoS ONE8:e76904. 10.1371/journal.pone.0076904
102
Haidet-Phillips A. M. Hester M. E. Miranda C. J. Meyer K. Braun L. Frakes A. et al (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons.Nat. Biotechnol.29824–828. 10.1038/nbt.1957
103
Haramati S. Chapnik E. Sztainberg Y. Eilam R. Zwang R. Gershoni N. et al (2010). miRNA malfunction causes spinal motor neuron disease.Proc. Natl. Acad. Sci. U.S.A.10713111–13116. 10.1073/pnas.1006151107
104
Harrison J. K. Jiang Y. Chen S. Xia Y. Maciejewski D. Mcnamara R. K. et al (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3BC1-expressing microglia.Proc. Natl. Acad. Sci. U.S.A.9510896–10901. 10.1073/pnas.95.18.10896
105
Hatori K. Nagai A. Heisel R. Ryu J. K. Kim S. U. (2002). Fractalkine and fractalkine receptors in human neurons and glial cells.J. Neurosci. Res.69418–426. 10.1002/jnr.10304
106
He Q. You H. Li X. M. Liu T. H. Wang P. Wang B. E. (2012). HMGB1 promotes the synthesis of pro-IL-1beta and pro-IL-18 by activation of p38 MAPK and NF-kappaB through receptors for advanced glycation end-products in macrophages.Asian Pac. J. Cancer Prev.131365–1370. 10.7314/APJCP.2012.13.4.1365
107
He X. Zhang L. Yao X. Hu J. Yu L. Jia H. et al (2013). Association studies of MMP-9 in Parkinson’s disease and amyotrophic lateral sclerosis.PLoS ONE8:e73777. 10.1371/journal.pone.0073777
108
Heath P. R. Kirby J. Shaw P. J. (2013). Investigating cell death mechanisms in amyotrophic lateral sclerosis using transcriptomics.Front. Cell. Neurosci.7:259. 10.3389/fncel.2013.00259
109
Henkel J. S. Beers D. R. Zhao W. Appel S. H. (2009). Microglia in ALS: the good, the bad, and the resting.J. Neuroimmune Pharmacol.4389–398. 10.1007/s11481-009-9171-5
110
Hinze A. Stolzing A. (2011). Differentiation of mouse bone marrow derived stem cells toward microglia-like cells.BMC Cell Biol.12:35. 10.1186/1471-2121-12-35
111
Hoelzinger D. B. Nakada M. Demuth T. Rosensteel T. Reavie L. B. Berens M. E. (2008). Autotaxin: a secreted autocrine/paracrine factor that promotes glioma invasion.J. Neurooncol.86297–309. 10.1007/s11060-007-9480-6
112
Holm T. H. Draeby D. Owens T. (2012). Microglia are required for astroglial Toll-like receptor 4 response and for optimal TLR2 and TLR3 response.Glia60630–638. 10.1002/glia.22296
113
Holmoy T. (2008). T cells in amyotrophic lateral sclerosis.Eur. J. Neurol.15360–366. 10.1111/j.1468-1331.2008.02065.x
114
Hooper C. Sainz-Fuertes R. Lynham S. Hye A. Killick R. Warley A. et al (2012). Wnt3a induces exosome secretion from primary cultured rat microglia.BMC Neurosci.13:144. 10.1186/1471-2202-13-144
115
Ilieva H. Polymenidou M. Cleveland D. W. (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond.J. Cell Biol.187761–772. 10.1083/jcb.200908164
116
Ince P. G. Highley J. R. Kirby J. Wharton S. B. Takahashi H. Strong M. J. et al (2011). Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology.Acta Neuropathol.122657–671. 10.1007/s00401-011-0913-0
117
Inoue H. Tsukita K. Iwasato T. Suzuki Y. Tomioka M. Tateno M. et al (2003). The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model.EMBO J.226665–6674. 10.1093/emboj/cdg634
118
Jiang M. Xiang Y. Wang D. Gao J. Liu D. Liu Y. et al (2012). Dysregulated expression of miR-146a contributes to age-related dysfunction of macrophages.Aging Cell1129–40. 10.1111/j.1474-9726.2011.00757.x
119
Julien J. P. (2001). Amyotrophic lateral sclerosis. unfolding the toxicity of the misfolded.Cell104581–591. 10.1016/S0092-8674(01)00244-6
120
Jung S. Schwartz M. (2012). Non-identical twins – microglia and monocyte-derived macrophages in acute injury and autoimmune inflammation.Front. Immunol.3:89. 10.3389/fimmu.2012.00089
121
Jurgens H. A. Johnson R. W. (2012). Dysregulated neuronal-microglial cross-talk during aging, stress and inflammation.Exp. Neurol.23340–48. 10.1016/j.expneurol.2010.11.014
122
Kang S. H. Fukaya M. Yang J. K. Rothstein J. D. Bergles D. E. (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration.Neuron68668–681. 10.1016/j.neuron.2010.09.009
123
Kang S. H. Li Y. Fukaya M. Lorenzini I. Cleveland D. W. Ostrow L. W. et al (2013). Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis.Nat. Neurosci.16571–579. 10.1038/nn.3357
124
Kaplan A. Spiller K. J. Towne C. Kanning K. C. Choe G. T. Geber A. et al (2014). Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration.Neuron81333–348. 10.1016/j.neuron.2013.12.009
125
Kaspar B. K. Llado J. Sherkat N. Rothstein J. D. Gage F. H. (2003). Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model.Science301839–842. 10.1126/science.1086137
126
Kato S. (2008). Amyotrophic lateral sclerosis models and human neuropathology: similarities and differences.Acta Neuropathol.11597–114. 10.1007/s00401-007-0308-4
127
Kawabata H. Setoguchi T. Yone K. Souda M. Yoshida H. Kawahara K. et al (2010). High mobility group box 1 is upregulated after spinal cord injury and is associated with neuronal cell apoptosis.Spine (Phila Pa 1976)351109–1115. 10.1097/BRS.0b013e3181bd14b6
128
Keller A. F. Gravel M. Kriz J. (2009). Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells.Glia571130–1142. 10.1002/glia.20836
129
Kiernan M. C. (2014). ALS and neuromuscular disease: in search of the Holy Grail.Lancet Neurol.1313–14. 10.1016/S1474-4422(13)70226-6
130
Kiernan M. C. Vucic S. Cheah B. C. Turner M. R. Eisen A. Hardiman O. et al (2011). Amyotrophic lateral sclerosis.Lancet377942–955. 10.1016/S0140-6736(10)61156-7
131
Kim H. Cooke M. J. Shoichet M. S. (2012). Creating permissive microenvironments for stem cell transplantation into the central nervous system.Trends Biotechnol.3055–63. 10.1016/j.tibtech.2011.07.002
132
Kim J. B. Sig Choi J. Yu Y. M. Nam K. Piao C. S. Kim S. W. et al (2006a). HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain.J. Neurosci.266413–6421. 10.1523/JNEUROSCI.3815-05.2006
133
Kim Y. S. Martinez T. Deshpande D. M. Drummond J. Provost-Javier K. Williams A. et al (2006b). Correction of humoral derangements from mutant superoxide dismutase 1 spinal cord.Ann. Neurol.60716–728. 10.1002/ana.21034
134
Kim J. M. Noh E. M. Kwon K. B. Kim J. S. You Y. O. Hwang J. K. et al (2013). Suppression of TPA-induced tumor cell invasion by sulfuretin via inhibition of NF-kappaB-dependent MMP-9 expression.Oncol. Rep.291231–1237. 10.3892/or.2012.2218
135
Konnecke H. Bechmann I. (2013). The role of microglia and matrix metalloproteinases involvement in neuroinflammation and gliomas.Clin. Dev. Immunol.201391410410.1155/2013/914104
136
Koval E. D. Shaner C. Zhang P. Du Maine X. Fischer K. Tay J. et al (2013). Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice.Hum. Mol. Genet.224127–4135. 10.1093/hmg/ddt261
137
Kushner P. D. Stephenson D. T. Wright S. (1991). Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain.J. Neuropathol. Exp. Neurol.50263–277. 10.1097/00005072-199105000-00008
138
Lagier-Tourenne C. Cleveland D. W. (2009). Rethinking ALS: the FUS about TDP-43.Cell1361001–1004. 10.1016/j.cell.2009.03.006
139
Lange S. Trost A. Tempfer H. Bauer H. C. Bauer H. Rohde E. et al (2012). Brain pericyte plasticity as a potential drug target in CNS repair.Drug Discov. Today18456–46310.1016/j.drudis.2012.12.007
140
Lasiene J. Yamanaka K. (2011). Glial cells in amyotrophic lateral sclerosis.Neurol. Res. Int.201171898710.1155/2011/718987
141
Lee C. T. Chiu Y. W. Wang K. C. Hwang C. S. Lin K. H. Lee I. T. et al (2013a). Riluzole and prognostic factors in amyotrophic lateral sclerosis long-term and short-term survival: a population-based study of 1149 cases in Taiwan.J. Epidemiol.2335–40. 10.2188/jea.JE20120119
142
Lee H. Lee S. Cho I. H. Lee S. J. (2013b). Toll-like receptors: sensor molecules for detecting damage to the nervous system.Curr. Protein Pept. Sci.1433–42. 10.2174/1389203711314010006
143
Lee J. C. Seong J. Kim S. H. Lee S. J. Cho Y. J. An J. et al (2012). Replacement of microglial cells using clodronate liposome and bone marrow transplantation in the central nervous system of SOD1(G93A) transgenic mice as an in vivo model of amyotrophic lateral sclerosis.Biochem. Biophys. Res. Commun.418359–365. 10.1016/j.bbrc.2012.01.026
144
Leinster V. H. Joy M. T. Vuononvirta R. E. Bolsover S. R. Anderson P. N. (2013). ErbB1 epidermal growth factor receptor is a valid target for reducing the effects of multiple inhibitors of axonal regeneration.Exp. Neurol.23982–90. 10.1016/j.expneurol.2012.09.007
145
Leonardi-Essmann F. Emig M. Kitamura Y. Spanagel R. Gebicke-Haerter P. J. (2005). Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia.J. Neuroimmunol.16092–101. 10.1016/j.jneuroim.2004.11.012
146
Le Pichon C. E. Dominguez S. L. Solanoy H. Ngu H. Lewin-Koh N. Chen M. et al (2013). EGFR inhibitor erlotinib delays disease progression but does not extend survival in the SOD1 mouse model of ALS.PLoS ONE8:e62342. 10.1371/journal.pone.0062342
147
Lepore A. C. O’Donnell J. Kim A. S. Williams T. Tuteja A. Rao M. S. et al (2011). Human glial-restricted progenitor transplantation into cervical spinal cord of the SOD1 mouse model of ALS.PLoS ONE6:e25968. 10.1371/journal.pone.0025968
148
Lepore A. C. Rauck B. Dejea C. Pardo A. C. Rao M. S. Rothstein J. D. et al (2008). Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease.Nat. Neurosci.111294–1301. 10.1038/nn.2210
149
Le Verche V. Ikiz B. Jacquier A. Przedborski S. Re D. B. (2011). Glutamate pathway implication in amyotrophic lateral sclerosis: what is the signal in the noise?J. Receptor Ligand Channel Res.41–22. 10.2147/JRLCR.S6504
150
Lewis C. A. Manning J. Rossi F. Krieger C. (2012). The neuroinflammatory response in ALS: the roles of microglia and T cells.Neurol. Res. Int.201280370110.1155/2012/803701
151
Lewis C. A. Solomon J. N. Rossi F. M. Krieger C. (2009). Bone marrow-derived cells in the central nervous system of a mouse model of amyotrophic lateral sclerosis are associated with blood vessels and express CX(3)BC1.Glia571410–1419. 10.1002/glia.20859
152
Li Y. R. King O. D. Shorter J. Gitler A. D. (2013). Stress granules as crucibles of ALS pathogenesis.J. Cell Biol.201361–372. 10.1083/jcb.201302044
153
Liao B. Zhao W. Beers D. R. Henkel J. S. Appel S. H. (2012). Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS.Exp. Neurol.237147–152. 10.1016/j.expneurol.2012.06.011
154
Liblau R. S. Gonzalez-Dunia D. Wiendl H. Zipp F. (2013). Neurons as targets for T cells in the nervous system.Trends Neurosci.36315–324. 10.1016/j.tins.2013.01.008
155
Lilo E. Wald-Altman S. Solmesky L. J. Ben Yaakov K. Gershoni-Emek N. Bulvik S. et al (2013). Characterization of human sporadic ALS biomarkers in the familial ALS transgenic mSOD1(G93A) mouse model.Hum. Mol. Genet.224720–4725. 10.1093/hmg/ddt325
156
Lino M. M. Schneider C. Caroni P. (2002). Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease.J. Neurosci.224825–4832.
157
Liu D. Wen J. Liu J. Li L. (1999). The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids.FASEB J.132318–2328.
158
Liu R. Li B. Flanagan S. W. Oberley L. W. Gozal D. Qiu M. (2002). Increased mitochondrial antioxidative activity or decreased oxygen free radical propagation prevent mutant SOD1-mediated motor neuron cell death and increase amyotrophic lateral sclerosis-like transgenic mouse survival.J. Neurochem.80488–500. 10.1046/j.0022-3042.2001.00720.x
159
Liu Y. Hao W. Dawson A. Liu S. Fassbender K. (2009). Expression of amyotrophic lateral sclerosis-linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2.J. Biol. Chem.2843691–3699. 10.1074/jbc.M804446200
160
Liu Y. C. Chiang P. M. Tsai K. J. (2013). Disease animal models of TDP-43 proteinopathy and their pre-clinical applications.Int. J. Mol. Sci.1420079–20111. 10.3390/ijms141020079
161
Lobsiger C. S. Boillee S. Mcalonis-Downes M. Khan A. M. Feltri M. L. Yamanaka K. et al (2009). Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice.Proc. Natl. Acad. Sci. U.S.A.1064465–4470. 10.1073/pnas.0813339106
162
Lobsiger C. S. Cleveland D. W. (2007). Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease.Nat. Neurosci.101355–1360. 10.1038/nn1988
163
Lo Coco D. Veglianese P. Allievi E. Bendotti C. (2007). Distribution and cellular localization of high mobility group box protein 1 (HMGB1) in the spinal cord of a transgenic mouse model of ALS.Neurosci. Lett.41273–77. 10.1016/j.neulet.2006.10.063
164
London A. Cohen M. Schwartz M. (2013). Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair.Front. Cell. Neurosci.7:34. 10.3389/fncel.2013.00034
165
Lorenzl S. Narr S. Angele B. Krell H. W. Gregorio J. Kiaei M. et al (2006). The matrix metalloproteinases inhibitor Ro 28-2653 [correction of Ro 26-2853] extends survival in transgenic ALS mice.Exp. Neurol.200166–171. 10.1016/j.expneurol.2006.01.026
166
Lu B. Wang H. Andersson U. Tracey K. J. (2013). Regulation of HMGB1 release by inflammasomes.Protein Cell4163–167. 10.1007/s13238-012-2118-2
167
Luo X. G. Chen S. D. (2012). The changing phenotype of microglia from homeostasis to disease.Transl. Neurodegener.1910.1186/2047-9158-1-9
168
Luo X. G. Ding J. Q. Chen S. D. (2010). Microglia in the aging brain: relevance to neurodegeneration.Mol. Neurodegener.51210.1186/1750-1326-5-12
169
Lyons A. Lynch A. M. Downer E. J. Hanley R. O’Sullivan J. B. Smith A. et al (2009). Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attenuates microglial activation in vivo and in vitro.J. Neurochem.1101547–1556. 10.1111/j.1471-4159.2009.06253.x
170
Magrane J. Hervias I. Henning M. S. Damiano M. Kawamata H. Manfredi G. (2009). Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities.Hum. Mol. Genet.184552–4564. 10.1093/hmg/ddp421
171
Magrane J. Manfredi G. (2009). Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis.Antioxid. Redox Signal.111615–1626. 10.1089/ARS.2009.2604
172
Matsumoto H. Kumon Y. Watanabe H. Ohnishi T. Shudou M. Ii C. et al (2007). Antibodies to CD11b, CD68, and lectin label neutrophils rather than microglia in traumatic and ischemic brain lesions.J. Neurosci. Res.85994–1009. 10.1002/jnr.21198
173
Meme W. Calvo C. F. Froger N. Ezan P. Amigou E. Koulakoff A. et al (2006). Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: potentiation by beta-amyloid.FASEB J.20494–496. 10.1096/fj.05-4297fje
174
Mimoto T. Miyazaki K. Morimoto N. Kurata T. Satoh K. Ikeda Y. et al (2012). Impaired antioxidative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice.Brain Res.1446109–118. 10.1016/j.brainres.2011.12.064
175
Min J. H. Hong Y. H. Sung J. J. Kim S. M. Lee J. B. Lee K. W. (2012). Oral solubilized ursodeoxycholic acid therapy in amyotrophic lateral sclerosis: a randomized cross-over trial.J. Korean Med. Sci.27200–206. 10.3346/jkms.2012.27.2.200
176
Miyazaki K. Ohta Y. Nagai M. Morimoto N. Kurata T. Takehisa Y. et al (2011). Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis.J. Neurosci. Res.89718–728. 10.1002/jnr.22594
177
Mizuno T. Kawanokuchi J. Numata K. Suzumura A. (2003). Production and neuroprotective functions of fractalkine in the central nervous system.Brain Res.97965–70. 10.1016/S0006-8993(03)02867-1
178
Morel L. Regan M. Higashimori H. Ng S. K. Esau C. Vidensky S. et al (2013). Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1.J. Biol. Chem.2887105–7116. 10.1074/jbc.M112.410944
179
Moreno-Lopez B. Sunico C. R. Gonzalez-Forero D. (2011). NO orchestrates the loss of synaptic boutons from adult “sick” motoneurons: modeling a molecular mechanism.Mol. Neurobiol.4341–66. 10.1007/s12035-010-8159-8
180
Mori A. Yamashita S. Uchino K. Suga T. Ikeda T. Takamatsu K. et al (2011). Derlin-1 overexpression ameliorates mutant SOD1-induced endoplasmic reticulum stress by reducing mutant SOD1 accumulation.Neurochem. Int.58344–353. 10.1016/j.neuint.2010.12.010
181
Munch C. O’Brien J. Bertolotti A. (2011). Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells.Proc. Natl. Acad. Sci. U.S.A.1083548–3553. 10.1073/pnas.1017275108
182
Nakanishi H. (2003). Neuronal and microglial cathepsins in aging and age-related diseases.Ageing Res. Rev.2367–381. 10.1016/S1568-1637(03)00027-8
183
Nakanishi H. Wu Z. (2009). Microglia-aging: roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging.Behav. Brain Res.2011–7. 10.1016/j.bbr.2009.02.001
184
Nardo G. Pozzi S. Pignataro M. Lauranzano E. Spano G. Garbelli S. et al (2011). Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells.PLoS ONE6:e25545. 10.1371/journal.pone.0025545
185
Neusch C. Bahr M. Schneider-Gold C. (2007). Glia cells in amyotrophic lateral sclerosis: new clues to understanding an old disease?Muscle Nerve35712–724. 10.1002/mus.20768
186
Nicaise C. Mitrecic D. Demetter P. De Decker R. Authelet M. Boom A. et al (2009). Impaired blood–brain and blood–spinal cord barriers in mutant SOD1-linked ALS rat.Brain Res.1301152–162. 10.1016/j.brainres.2009.09.018
187
Niebroj-Dobosz I. Janik P. Sokolowska B. Kwiecinski H. (2010). Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis.Eur. J. Neurol.17226–231. 10.1111/j.1468-1331.2009.02775.x
188
Niebroj-Dobosz I. Rafalowska J. Fidzianska A. Gadamski R. Grieb P. (2007). Myelin composition of spinal cord in a model of amyotrophic lateral sclerosis (ALS) in SOD1G93A transgenic rats.Folia Neuropathol.45236–241.
189
Nieto-Diaz M. Esteban F. J. Reigada D. Munoz-Galdeano T. Yunta M. Caballero-Lopez M. et al (2014). MicroRNA dysregulation in spinal cord injury: causes, consequences and therapeutics.Front. Cell Neurosci.85310.3389/fncel.2014.00053
190
Nikodemova M. Small A. L. Smith S. M. Mitchell G. S. Watters J. J. (2013). Spinal but not cortical microglia acquire an atypical phenotype with high VEGF, galectin-3 and osteopontin, and blunted inflammatory responses in ALS rats.Neurobiol. Dis.10.1016/j.nbd.2013.11.009[Epub ahead of print].
191
Noda M. Doi Y. Liang J. Kawanokuchi J. Sonobe Y. Takeuchi H. et al (2011). Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression.J. Biol. Chem.2862308–2319. 10.1074/jbc.M110.169839
192
Offen D. Barhum Y. Melamed E. Embacher N. Schindler C. Ransmayr G. (2009). Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant.J. Mol. Neurosci.3885–93. 10.1007/s12031-007-9004-z
193
Ogawa M. Furukawa Y. (2014). A seeded propagation of Cu, Zn-superoxide dismutase aggregates in amyotrophic lateral sclerosis.Front. Cell. Neurosci.8:83. 10.3389/fncel.2014.00083
194
Ohnishi S. Ito H. Suzuki Y. Adachi Y. Wate R. Zhang J. et al (2009). Intra-bone marrow-bone marrow transplantation slows disease progression and prolongs survival in G93A mutant SOD1 transgenic mice, an animal model mouse for amyotrophic lateral sclerosis.Brain Res.1296216–224. 10.1016/j.brainres.2009.08.012
195
Olah M. Amor S. Brouwer N. Vinet J. Eggen B. Biber K. et al (2012). Identification of a microglia phenotype supportive of remyelination.Glia60306–321. 10.1002/glia.21266
196
Olivieri G. Baysang G. Meier F. Muller-Spahn F. Stahelin H. B. Brockhaus M. et al (2001). N-acetyl-L-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: effects on beta-amyloid secretion and tau phosphorylation.J. Neurochem.76224–233. 10.1046/j.1471-4159.2001.00090.x
197
Otomo A. Pan L. Hadano S. (2012). Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases.Neurol. Res. Int.201249842810.1155/2012/498428
198
Otto M. Bahn E. Wiltfang J. Boekhoff I. Beuche W. (1998). Decrease of S100 beta protein in serum of patients with amyotrophic lateral sclerosis.Neurosci. Lett.240171–173. 10.1016/S0304-3940(97)00947-6
199
Pan L. Yoshii Y. Otomo A. Ogawa H. Iwasaki Y. Shang H. F. et al (2012). Different human copper-zinc superoxide dismutase mutants, SOD1G93A and SOD1H46R, exert distinct harmful effects on gross phenotype in mice.PLoS ONE7:e33409. 10.1371/journal.pone.0033409
200
Pankonin M. S. Sohi J. Kamholz J. Loeb J. A. (2009). Differential distribution of neuregulin in human brain and spinal fluid.Brain Res.12581–11. 10.1016/j.brainres.2008.12.047
201
Papadeas S. T. Maragakis N. J. (2009). Advances in stem cell research for amyotrophic lateral sclerosis.Curr. Opin. Biotechnol.20545–551. 10.1016/j.copbio.2009.09.003
202
Parajuli B. Sonobe Y. Kawanokuchi J. Doi Y. Noda M. Takeuchi H. et al (2012). GM-CSF increases LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia.J. Neuroinflammation926810.1186/1742-2094-9-268
203
Parisi C. Arisi I. D’Ambrosi N. Storti A. E. Brandi R. D’Onofrio M. et al (2013). Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation.Cell Death Dis.4e95910.1038/cddis.2013.491
204
Park J. S. Gamboni-Robertson F. He Q. Svetkauskaite D. Kim J. Y. Strassheim D. et al (2006). High mobility group box 1 protein interacts with multiple Toll-like receptors.Am. J. Physiol. Cell Physiol.290C917–C924. 10.1152/ajpcell.00401.2005
205
Park S. Kim H. T. Yun S. Kim I. S. Lee J. Lee I. S. et al (2009). Growth factor-expressing human neural progenitor cell grafts protect motor neurons but do not ameliorate motor performance and survival in ALS mice.Exp. Mol. Med.41487–500. 10.3858/emm.2009.41.7.054
206
Parnes J. R. (1989). Molecular biology and function of CD4 and CD8.Adv. Immunol.44265–311. 10.1016/S0065-2776(08)60644-6
207
Pasquali L. Longone P. Isidoro C. Ruggieri S. Paparelli A. Fornai F. (2009). Autophagy, lithium, and amyotrophic lateral sclerosis.Muscle Nerve40173–194. 10.1002/mus.21423
208
Pehar M. Cassina P. Vargas M. R. Castellanos R. Viera L. Beckman J. S. et al (2004). Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis.J. Neurochem.89464–473. 10.1111/j.1471-4159.2004.02357.x
209
Petanceska S. Canoll P. Devi L. A. (1996). Expression of rat cathepsin S in phagocytic cells.J. Biol. Chem.2714403–4409. 10.1074/jbc.271.8.4403
210
Phani S. Re D. B. Przedborski S. (2012). The role of the innate immune system in ALS.Front. Pharmacol.3:150. 10.3389/fphar.2012.00150
211
Phatnani H. P. Guarnieri P. Friedman B. A. Carrasco M. A. Muratet M. O’Keeffe S. et al (2013). Intricate interplay between astrocytes and motor neurons in ALS.Proc. Natl. Acad. Sci. U.S.A.110E756–E765. 10.1073/pnas.1222361110
212
Philips T. Bento-Abreu A. Nonneman A. Haeck W. Staats K. Geelen V. et al (2013). Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis.Brain136471–482. 10.1093/brain/aws339
213
Philips T. Robberecht W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease.Lancet Neurol.10253–263. 10.1016/S1474-4422(11)70015-1
214
Ponomarev E. D. Veremeyko T. Weiner H. L. (2013). MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS.Glia6191–103. 10.1002/glia.22363
215
Poon H. F. Hensley K. Thongboonkerd V. Merchant M. L. Lynn B. C. Pierce W. M. et al (2005). Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice – a model of familial amyotrophic lateral sclerosis.Free Radic. Biol. Med.39453–462. 10.1016/j.freeradbiomed.2005.03.030
216
Potolicchio I. Carven G. J. Xu X. Stipp C. Riese R. J. Stern L. J. et al (2005). Proteomic analysis of microglia-derived exosomes: metabolic role of the aminopeptidase CD13 in neuropeptide catabolism.J. Immunol.1752237–2243.
217
Pottier N. Maurin T. Chevalier B. Puissegur M. P. Lebrigand K. Robbe-Sermesant K. et al (2009). Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: implication in epithelial-mesenchymal interactions.PLoS ONE4:e6718. 10.1371/journal.pone.0006718
218
Pramatarova A. Laganiere J. Roussel J. Brisebois K. Rouleau G. A. (2001). Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment.J. Neurosci.213369–3374. 10.1523/JNEUROSCI.5258-07.2008
219
Quinn S. R O’Neill L. A. (2011). A trio of microRNAs that control Toll-like receptor signalling.Int. Immunol.23421–425. 10.1093/intimm/dxr034
220
Renton A. E. Chio A. Traynor B. J. (2014). State of play in amyotrophic lateral sclerosis genetics.Nat. Neurosci.1717–23. 10.1038/nn.3584
221
Renton A. E. Majounie E. Waite A. Simon-Sanchez J. Rollinson S. Gibbs J. R. et al (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD.Neuron72257–268. 10.1016/j.neuron.2011.09.010
222
Rentzos M. Evangelopoulos E. Sereti E. Zouvelou V. Marmara S. Alexakis T. et al (2012). Alterations of T cell subsets in ALS: a systemic immune activation?Acta Neurol. Scand.125260–264. 10.1111/j.1600-0404.2011.01528.x
223
Reyes N. A. Fisher J. K. Austgen K. Vandenberg S. Huang E. J. Oakes S. A. (2010). Blocking the mitochondrial apoptotic pathway preserves motor neuron viability and function in a mouse model of amyotrophic lateral sclerosis.J. Clin. Invest.1203673–3679. 10.1172/JCI42986
224
Riboldi G. Nizzardo M. Simone C. Falcone M. Bresolin N. Comi G. P. et al (2011). ALS genetic modifiers that increase survival of SOD1 mice and are suitable for therapeutic development.Prog. Neurobiol.95133–148. 10.1016/j.pneurobio.2011.07.009
225
Roberts K. Zeineddine R. Corcoran L. Li W. Campbell I. L. Yerbury J. J. (2013). Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype.Glia61409–419. 10.1002/glia.22444
226
Rodier F. Campisi J. (2011). Four faces of cellular senescence.J. Cell Biol.192547–556. 10.1083/jcb.201009094
227
Rodriguez M. Sabate M. Rodriguez-Sabate C. Morales I. (2013). The role of non-synaptic extracellular glutamate.Brain Res. Bull.9317–26. 10.1016/j.brainresbull.2012.09.018
228
Rojas F. Cortes N. Abarzua S. Dyrda A Van Zundert B. (2014). Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress.Front. Cell. Neurosci.8:24. 10.3389/fncel.2014.00024
229
Rothstein J. D. (2009). Current hypotheses for the underlying biology of amyotrophic lateral sclerosis.Ann. Neurol. 65 (Suppl.1)S3–S9. 10.1002/ana.21543
230
Saba R. Gushue S. Huzarewich R. L. Manguiat K. Medina S. Robertson C. et al (2012). MicroRNA 146a (miR-146a) is over-expressed during prion disease and modulates the innate immune response and the microglial activation state.PLoS ONE7:e30832. 10.1371/journal.pone.0030832
231
Saijo K. Crotti A. Glass C. K. (2013). Regulation of microglia activation and deactivation by nuclear receptors.Glia61104–111. 10.1002/glia.22423
232
Sanagi T. Yuasa S. Nakamura Y. Suzuki E. Aoki M. Warita H. et al (2010). Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a presymptomatic transgenic rat model of amyotrophic lateral sclerosis.J. Neurosci. Res.882736–2746. 10.1002/jnr.22424
233
Sá-Pereira I. Brites D. Brito M. A. (2012). Neurovascular unit: a focus on pericytes.Mol. Neurobiol.45327–347. 10.1007/s12035-012-8244-2
234
Sargsyan S. A. Blackburn D. J. Barber S. C. Grosskreutz J. De Vos K. J. Monk P. N. et al (2011). A comparison of in vitro properties of resting SOD1 transgenic microglia reveals evidence of reduced neuroprotective function.BMC Neurosci.12:91. 10.1186/1471-2202-12-91
235
Sau D. De Biasi S. Vitellaro-Zuccarello L. Riso P. Guarnieri S. Porrini M. et al (2007). Mutation of SOD1 in ALS: a gain of a loss of function.Hum. Mol. Genet.161604–1618. 10.1093/hmg/ddm110
236
Savaskan N. E. Rocha L. Kotter M. R. Baer A. Lubec G. Van Meeteren L. A. et al (2007). Autotaxin (NPP-2) in the brain: cell type-specific expression and regulation during development and after neurotrauma.Cell. Mol. Life Sci.64230–243. 10.1007/s00018-006-6412-0
237
Schiffer D. Cordera S. Cavalla P. Migheli A. (1996). Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis.J. Neurol. Sci.139(Suppl.)27–33. 10.1016/0022-510X(96)00073-1
238
Schoser B. G. Blottner D. (1999). Matrix metalloproteinases MMP-2, MMP-7 and MMP-9 in denervated human muscle.Neuroreport102795–2797. 10.1097/00001756-199909090-00018
239
Shi P. Gal J. Kwinter D. M. Liu X. Zhu H. (2010a). Mitochondrial dysfunction in amyotrophic lateral sclerosis.Biochim. Biophys. Acta180245–51. 10.1016/j.bbadis.2009.08.012
240
Shi P. Wei Y. Zhang J. Gal J. Zhu H. (2010b). Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis.J. Alzheimers. Dis. 20(Suppl.2)S311–S324. 10.3233/JAD-2010-100366
241
Shobha K. Alladi P. A. Nalini A. Sathyaprabha T. N. Raju T. R. (2010). Exposure to CSF from sporadic amyotrophic lateral sclerosis patients induces morphological transformation of astroglia and enhances GFAP and S100beta expression.Neurosci. Lett.47356–61. 10.1016/j.neulet.2010.02.022
242
Silva S. L. Osório C. Vaz A. R. Barateiro A. Falcão A. S. Silva R. F. et al (2011). Dynamics of neuron–glia interplay upon exposure to unconjugated bilirubin.J. Neurochem.117412–424. 10.1111/j.1471-4159.2011.07200.x
243
Silva S. L. Vaz A. R. Barateiro A. Falcão A. S. Fernandes A. Brito M. A. et al (2010). Features of bilirubin-induced reactive microglia: from phagocytosis to inflammation.Neurobiol. Dis.40663–675. 10.1016/j.nbd.2010.08.010
244
Silva S. L. Vaz A. R. Diogenes M. J. Van Rooijen N. Sebastiao A. M. Fernandes A. et al (2012). Neuritic growth impairment and cell death by unconjugated bilirubin is mediated by NO and glutamate, modulated by microglia, and prevented by glycoursodeoxycholic acid and interleukin-10.Neuropharmacology622397–2407. 10.1016/j.neuropharm.2012.02.002
245
Sofroniew M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation.Trends Neurosci.32638–647. 10.1016/j.tins.2009.08.002
246
Song F. Chiang P. Wang J. Ravits J. Loeb J. A. (2012). Aberrant neuregulin 1 signalling in amyotrophic lateral sclerosis.J. Neuropathol. Exp. Neurol.71104–115. 10.1097/NEN.0b013e3182423c43
247
Soon C. P. Crouch P. J. Turner B. J. Mclean C. A. Laughton K. M. Atkin J. D. et al (2010). Serum matrix metalloproteinase-9 activity is dysregulated with disease progression in the mutant SOD1 transgenic mice.Neuromuscul. Disord.20260–266. 10.1016/j.nmd.2009.11.015
248
Spalloni A. Nutini M. Longone P. (2013). Role of the N-methyl-D-aspartate receptors complex in amyotrophic lateral sclerosis.Biochim. Biophys. Acta1832312–322. 10.1016/j.bbadis.2012.11.013
249
Staats K. A. Schonefeldt S. Van Rillaer M. Van Hoecke A. Van Damme P. Robberecht W. et al (2013). Beta-2 microglobulin is important for disease progression in a murine model for amyotrophic lateral sclerosis.Front. Cell. Neurosci.7:249. 10.3389/fncel.2013.00249
250
Stoorvogel W. (2012). Functional transfer of microRNA by exosomes.Blood119646–648. 10.1182/blood-2011-11-389478
251
Streit W. J. (2006). Microglial senescence: does the brain’s immune system have an expiration date?Trends Neurosci.29506–510. 10.1016/j.tins.2006.07.001
252
Streit W. J. Sammons N. W. Kuhns A. J. Sparks D. L. (2004). Dystrophic microglia in the aging human brain.Glia45208–212. 10.1002/glia.10319
253
Sun D. Zhuang X. Zhang S. Deng Z. B. Grizzle W. Miller D. et al (2013). Exosomes are endogenous nanoparticles that can deliver biological information between cells.Adv. Drug Deliv. Rev.65342–347. 10.1016/j.addr.2012.07.002
254
Sundaram J. R. Chan E. S. Poore C. P. Pareek T. K. Cheong W. F. Shui G. et al (2012). Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration.J. Neurosci.321020–1034. 10.1523/JNEUROSCI.5177-11.2012
255
Sunnemark D. Eltayeb S. Nilsson M. Wallstrom E. Lassmann H. Olsson T. et al (2005). CX3CL1 (fractalkine) and CX3CR1 expression in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis: kinetics and cellular origin.J. Neuroinflammation21710.1186/1742-2094-2-17
256
Sussmuth S. D. Tumani H. Ecker D. Ludolph A. C. (2003). Amyotrophic lateral sclerosis: disease stage related changes of tau protein and S100 beta in cerebrospinal fluid and creatine kinase in serum.Neurosci. Lett.35357–60. 10.1016/j.neulet.2003.09.018
257
Suzuki M. El-Hage N. Zou S. Hahn Y. K. Sorrell M. E. Sturgill J. L. et al (2011). Fractalkine/CX3CL1 protects striatal neurons from synergistic morphine and HIV-1 Tat-induced dendritic losses and death.Mol. Neurodegener.67810.1186/1750-1326-6-78
258
Synofzik M. Fernandez-Santiago R. Maetzler W. Schols L. Andersen P. M. (2010). The human G93A SOD1 phenotype closely resembles sporadic amyotrophic lateral sclerosis.J. Neurol. Neurosurg. Psychiatry81764–767. 10.1136/jnnp.2009.181719
259
Takeuchi H. Jin S. Wang J. Zhang G. Kawanokuchi J. Kuno R. et al (2006). Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner.J. Biol. Chem.28121362–21368. 10.1074/jbc.M600504200
260
Taylor D. L. Pirianov G. Holland S. Mcginnity C. J. Norman A. L. Reali C. et al (2010). Attenuation of proliferation in oligodendrocyte precursor cells by activated microglia.J. Neurosci. Res.881632–1644. 10.1002/jnr.22335
261
Thams S. Brodin P. Plantman S. Saxelin R. Karre K. Cullheim S. (2009). Classical major histocompatibility complex class I molecules in motoneurons: new actors at the neuromuscular junction.J. Neurosci.2913503–13515. 10.1523/JNEUROSCI.0981-09.2009
262
Tian L. Ma L. Kaarela T. Li Z. (2012). Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases.J. Neuroinflammation915510.1186/1742-2094-9-155
263
Tilleux S. Berger J. Hermans E. (2007). Induction of astrogliosis by activated microglia is associated with a down-regulation of metabotropic glutamate receptor 5.J. Neuroimmunol.18923–30. 10.1016/j.jneuroim.2007.06.011
264
Tolosa L. Caraballo-Miralles V. Olmos G. Llado J. (2011). TNF-alpha potentiates glutamate-induced spinal cord motoneuron death via NF-kappaB.Mol. Cell. Neurosci.46176–186. 10.1016/j.mcn.2010.09.001
265
Tovar Y. R. L. B. Santa-Cruz L. D. Tapia R. (2009a). Experimental models for the study of neurodegeneration in amyotrophic lateral sclerosis.Mol. Neurodegener.43110.1186/1750-1326-4-31
266
Tovar Y. R. L. B. Santa-Cruz L. D. Zepeda A. Tapia R. (2009b). Chronic elevation of extracellular glutamate due to transport blockade is innocuous for spinal motoneurons in vivo.Neurochem. Int.54186–191. 10.1016/j.neuint.2008.09.015
267
Tsou C. L. Haskell C. A. Charo I. F. (2001). Tumor necrosis factor-alpha-converting enzyme mediates the inducible cleavage of fractalkine.J. Biol. Chem.27644622–44626. 10.1074/jbc.M107327200
268
Turner B. J. Talbot K. (2008). Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS.Prog. Neurobiol.8594–134. 10.1016/j.pneurobio.2008.01.001
269
Turner M. R. Hardiman O. Benatar M. Brooks B. R. Chio A. De Carvalho M. et al (2013). Controversies and priorities in amyotrophic lateral sclerosis.Lancet Neurol.12310–322. 10.1016/S1474-4422(13)70036-X
270
Turola E. Furlan R. Bianco F. Matteoli M. Verderio C. (2012). Microglial microvesicle secretion and intercellular signalling.Front. Physiol.3:149. 10.3389/fphys.2012.00149
271
Uccelli A. Milanese M. Principato M. C. Morando S. Bonifacino T. Vergani L. et al (2012). Intravenous mesenchymal stem cells improve survival and motor function in experimental amyotrophic lateral sclerosis.Mol. Med.18794–804. 10.2119/molmed.2011.00498
272
Urushitani M. Sik A. Sakurai T. Nukina N. Takahashi R. Julien J. P. (2006). Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis.Nat. Neurosci.9108–118. 10.1038/nn1603
273
Valadi H. Ekstrom K. Bossios A. Sjostrand M. Lee J. J. Lotvall J. O. (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells.Nat. Cell Biol.9654–659. 10.1038/ncb1596
274
Valori C. F. Brambilla L. Martorana F. Rossi D. (2014). The multifaceted role of glial cells in amyotrophic lateral sclerosis.Cell. Mol. Life Sci.71287–297. 10.1007/s00018-013-1429-7
275
Van Den Bosch L. (2011). Genetic rodent models of amyotrophic lateral sclerosis.J. Biomed. Biotechnol.201134876510.1155/2011/348765
276
van Weering H. R. De Jong A. P. De Haas A. H. Biber K. P. Boddeke H. W. (2010). CCL21-induced calcium transients and proliferation in primary mouse astrocytes: CXCR3-dependent and independent responses.Brain Behav. Immun.24768–775. 10.1016/j.bbi.2009.04.007
277
Vaz A. R. Cunha C. Comes C. Fernandes A. Brites D. (2014). Glycoursodeoxycholic acid reduces matrix metalloproteinase-9 and caspase-9 activation in a cellular model of superoxide-dismutase-1 neurodegeneration.Mol. Neurobiol.10.1007/s12035-014-8731-8[Epub ahead of print].
278
Verbeeck C. Deng Q. Dejesus-Hernandez M. Taylor G. Ceballos-Diaz C. Kocerha J. et al (2012). Expression of fused in sarcoma mutations in mice recapitulates the neuropathology of FUS proteinopathies and provides insight into disease pathogenesis.Mol. Neurodegener.75310.1186/1750-1326-7-53
279
Verge G. M. Milligan E. D. Maier S. F. Watkins L. R. Naeve G. S. Foster A. C. (2004). Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions.Eur. J. Neurosci.201150–1160. 10.1111/j.1460-9568.2004.03593.x
280
Walter L. Neumann H. (2009). Role of microglia in neuronal degeneration and regeneration.Semin. Immunopathol.31513–525. 10.1007/s00281-009-0180-5
281
Wang L. Pytel P. Feltri M. L. Wrabetz L. Roos R. P. (2012). Selective knockdown of mutant SOD1 in Schwann cells ameliorates disease in G85R mutant SOD1 transgenic mice.Neurobiol. Dis.4852–57. 10.1016/j.nbd.2012.05.014
282
Wegorzewska I. Bell S. Cairns N. J. Miller T. M. Baloh R. H. (2009). TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration.Proc. Natl. Acad. Sci. U.S.A.10618809–18814. 10.1073/pnas.0908767106
283
Welser J. V. Milner R. (2012). Use of astrocyte–microglial cocultures to examine the regulatory influence of astrocytes on microglial activation.Methods Mol. Biol.814367–380. 10.1007/978-1-61779-452-0_24
284
Wen Z. Xu L. Chen X. Xu W. Yin Z. Gao X. et al (2013). Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway.J. Immunol.1905411–5422. 10.4049/jimmunol.1203301
285
Weydt P. Yuen E. C. Ransom B. R. Moller T. (2004). Increased cytotoxic potential of microglia from ALS-transgenic mice.Glia48179–182. 10.1002/glia.20062
286
Wilcock D. M. (2012). A changing perspective on the role of neuroinflammation in Alzheimer’s disease.Int. J. Alzheimers Dis.201249524310.1155/2012/495243
287
Willemen H. L. Huo X. J. Mao-Ying Q. L. Zijlstra J. Heijnen C. J. Kavelaars A. (2012). MicroRNA-124 as a novel treatment for persistent hyperalgesia.J. Neuroinflammation914310.1186/1742-2094-9-143
288
Willier S. Butt E. Richter G. H. Burdach S. Grunewald T. G. (2011). Defining the role of TRIP6 in cell physiology and cancer.Biol. Cell103573–591. 10.1042/BC20110077
289
Winkler E. A. Sengillo J. D. Sullivan J. S. Henkel J. S. Appel S. H. Zlokovic B. V. (2013). Blood–spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis.Acta Neuropathol.125111–120. 10.1007/s00401-012-1039-8
290
Wong W. T. (2013). Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation.Front. Cell. Neurosci.7:22. 10.3389/fncel.2013.00022
291
Xu D. Tahara H. (2013). The role of exosomes and microRNAs in senescence and aging.Adv. Drug Deliv. Rev.65368–375. 10.1016/j.addr.2012.07.010
292
Xu L. Yang B. F. Ai J. (2013). MicroRNA transport: a new way in cell communication.J. Cell. Physiol.2281713–1719. 10.1002/jcp.24344
293
Xu X. Warrington A. E. Bieber A. J. Rodriguez M. (2011). Enhancing CNS repair in neurological disease: challenges arising from neurodegeneration and rewiring of the network.CNS Drugs25555–573. 10.2165/11587830-000000000-00000
294
Yamanaka K. Boillee S. Roberts E. A. Garcia M. L. Mcalonis-Downes M. Mikse O. R. et al (2008a). Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice.Proc. Natl. Acad. Sci. U.S.A.1057594–7599. 10.1073/pnas.0802556105
295
Yamanaka K. Chun S. J. Boillee S. Fujimori-Tonou N. Yamashita H. Gutmann D. H. et al (2008b). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis.Nat. Neurosci.11251–253. 10.1038/nn2047
296
Yang C. Wang H. Qiao T. Yang B. Aliaga L. Qiu L. et al (2014). Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis.Proc. Natl. Acad. Sci. U.S.A.111E1121–E1129. 10.1073/pnas.1322641111
297
Yang W. W. Sidman R. L. Taksir T. V. Treleaven C. M. Fidler J. A. Cheng S. H. et al (2011). Relationship between neuropathology and disease progression in the SOD1(G93A) ALS mouse.Exp. Neurol.227287–295. 10.1016/j.expneurol.2010.11.019
298
Yi M. H. Zhang E. Kang J. W. Shin Y. N. Byun J. Y. Oh S. H. et al (2012). Expression of CD200 in alternative activation of microglia following an excitotoxic lesion in the mouse hippocampus.Brain Res.148190–96. 10.1016/j.brainres.2012.08.053
299
Yin H. Z. Weiss J. H. (2012). Marked synergism between mutant SOD1 and glutamate transport inhibition in the induction of motor neuronal degeneration in spinal cord slice cultures.Brain Res.1448153–162. 10.1016/j.brainres.2012.02.005
300
Yoshihara T. Ishigaki S. Yamamoto M. Liang Y. Niwa J. Takeuchi H. et al (2002). Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis.J. Neurochem.80158–167. 10.1046/j.0022-3042.2001.00683.x
301
Yoshii Y. Otomo A. Pan L. Ohtsuka M. Hadano S. (2011). Loss of glial fibrillary acidic protein marginally accelerates disease progression in a SOD1(H46R) transgenic mouse model of ALS.Neurosci. Res.70321–329. 10.1016/j.neures.2011.03.006
302
Zhao W. Beers D. R. Appel S. H. (2013). Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis.J. Neuroimmune Pharmacol.8888–899. 10.1007/s11481-013-9489-x
303
Zhao W. Beers D. R. Henkel J. S. Zhang W. Urushitani M. Julien J. P. et al (2010). Extracellular mutant SOD1 induces microglial-mediated motoneuron injury.Glia58231–243. 10.1002/glia.20919
304
Zhao W. Beers D. R. Liao B. Henkel J. S. Appel S. H. (2012). Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms.Neurobiol. Dis.48418–428. 10.1016/j.nbd.2012.07.008
305
Zhao W. Xie W. Xiao Q. Beers D. R. Appel S. H. (2006). Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia.J. Neurochem.991176–1187. 10.1111/j.1471-4159.2006.04172.x
306
Zhong Z. Deane R. Ali Z. Parisi M. Shapovalov Y. O’Banion M. K. et al (2008). ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration.Nat. Neurosci.11420–422. 10.1038/nn2073
307
Zoccolella S. Beghi E. Palagano G. Fraddosio A. Guerra V. Samarelli V. et al (2007). Riluzole and amyotrophic lateral sclerosis survival: a population-based study in southern Italy.Eur. J. Neurol.14262–268. 10.1111/j.1468-1331.2006.01575.x
308
Zujovic V. Benavides J. Vige X. Carter C. Taupin V. (2000). Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation.Glia29305–315. 10.1002/(SICI)1098-1136(20000215)29:4<305
Summary
Keywords
amyotrophic lateral sclerosis, microglia activation phenotypes, motor neuron, neuroinflammation, neurodegeneration, pathological cell–cell communication, SOD1G93A transgenic mouse/rat
Citation
Brites D and Vaz AR (2014) Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front. Cell. Neurosci. 8:117. doi: 10.3389/fncel.2014.00117
Received
28 February 2014
Accepted
10 April 2014
Published
22 May 2014
Volume
8 - 2014
Edited by
Raquel Ferreira, University of Southern California, USA
Reviewed by
Daniel Kaganovich, Hebrew University of Jerusalem, Israel; Cinzia Volonté, Consiglio Nazionale delle Ricerche, Italy
Copyright
© 2014 Brites and Vaz.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dora Brites, Research Institute for Medicines (iMed.ULisboa), Faculdade de Farmácia, Universidade de Lisboa, Avenida Professor Gama Pinto, 1649-003 Lisbon, Portugal e-mail: dbrites@ff.ul.pt
This article was submitted to the journal Frontiers in Cellular Neuroscience.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.