 Eveliina Pollari
Eveliina Pollari Gundars Goldsteins
Gundars Goldsteins Geneviève Bart
Geneviève Bart Jari Koistinaho
Jari Koistinaho Rashid Giniatullin
Rashid Giniatullin- 1Molecular Brain Research Laboratory, Department of Neurobiology, A. I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, Kuopio, Finland
- 2Experimental Neurology - Laboratory of Neurobiology, Department of Neurosciences, Vesalius Research Center, KULeuven – University of Leuven, Leuven, Belgium
- 3Cell Biology Laboratory, Department of Neurobiology, A. I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, Kuopio, Finland
- 4Laboratory of Neurobiology, Department of Physiology, Kazan Federal University, Kazan, Russia
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive loss of motoneurons and degradation of the neuromuscular junctions (NMJ). Consistent with the dying-back hypothesis of motoneuron degeneration the decline in synaptic function initiates from the presynaptic terminals in ALS. Oxidative stress is a major contributory factor to ALS pathology and affects the presynaptic transmitter releasing machinery. Indeed, in ALS mouse models nerve terminals are sensitive to reactive oxygen species (ROS) suggesting that oxidative stress, along with compromised mitochondria and increased intracellular Ca2+ amplifies the presynaptic decline in NMJ. This initial dysfunction is followed by a neurodegeneration induced by inflammatory agents and loss of trophic support. To develop effective therapeutic approaches against ALS, it is important to identify the mechanisms underlying the initial pathological events. Given the role of oxidative stress in ALS, targeted antioxidant treatments could be a promising therapeutic approach. However, the complex nature of ALS and failure of monotherapies suggest that an antioxidant therapy should be accompanied by anti-inflammatory interventions to enhance the restoration of the redox balance.
NMJ as a Vulnerable Target of ALS (Dying Back Hypothesis)
Temporal analysis of axon and neuromuscular junction (NMJ) degeneration in sporadic ALS (sALS) and mouse mutant SOD1 (mSOD1) cases indicate that motoneuron pathology begins distally from the synaptic area (Figure 1) markedly earlier than clinical symptoms and proceeds towards soma in a retrograde dying back manner (Fischer et al., 2004; Rocha et al., 2013). Impaired axonal transport, Ca2+ imbalance and mitochondria dysfunction drive the axonal degeneration, and eventually lead to dying of the neuron (Fischer-Hayes et al., 2013).
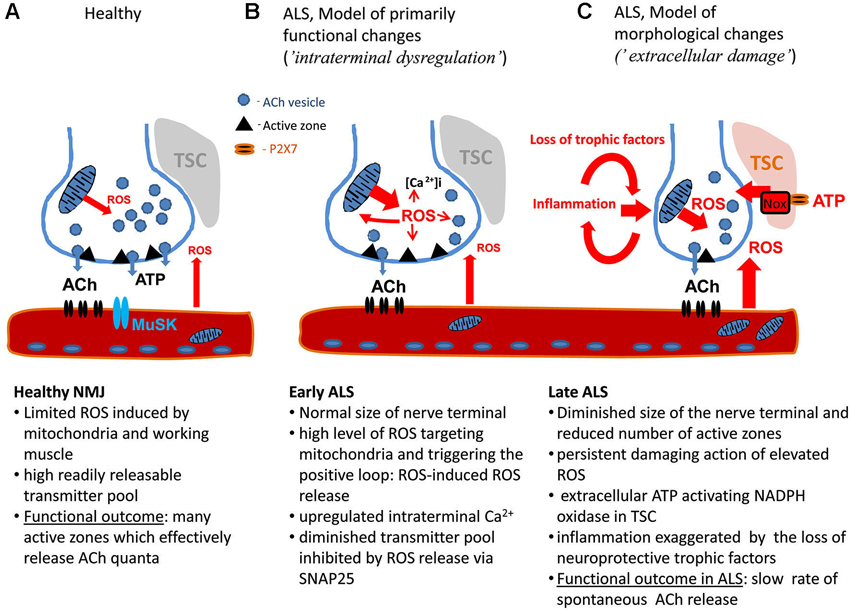
Figure 1. The model of motor nerve terminal dysregulation in ALS. (A) Healthy NMJ. (B) Pathological changes in NMJ during early stage of ALS. (C) Pathological changes in NMJ during late stage of ALS. ACh: Acetyl choline, MuSK: muscle-specific kinase, NMJ: Neuromuscular junction, ROS: Reactive oxygen species, TSC: Terminal Schwann Cells.
Figure 1 shows the principal structure of the NMJ including the presynaptic machinery restricted to active zones (AZ) releasing acetyl choline (ACh) in quantal manner and postsynaptic structures consisting of densely packed ACh receptors linked to the muscle-specific kinase (MuSK), agrin and other molecules involved in NMJ maturation and maintenance (reviewed in Shi et al., 2012). Thus, the dysfunction of the neuromuscular transmission can originate from the presynaptic site or from disorganized postsynaptic density. Notably, the motor nerve terminals are covered by the Terminal Schwann Cells (TSC) which can contribute to ALS progression.
In mSOD1 mice many motor terminals of the diaphragm muscle show a number of dysfunctional changes in the early disease stage (Naumenko et al., 2011). Muscle fibers are proposed to initiate the early changes leading to ALS progression (Wong and Martin, 2010). However, our results indicate that in the NMJ of ALS mice the presynaptic machinery is affected first (Naumenko et al., 2011). There is a noticeable variation in the probability of transmitter release between synapses, suggesting different degeneration rates of synapses. At the early symptomatic phase, only a few synapses have compromised function. Presumably, early in the disease course, the proportion of damaged synapses is low allowing compensation of the lost function by the healthy ones. Interestingly, during the pre-symptomatic stage enhanced neuromuscular transmission can be observed before the occurrence of the marked decline during the symptomatic phase, possibly due to compensatory mechanisms against the initial degeneration (Rocha et al., 2013).
Indeed, while some axon branches degenerate in ALS, others show sprouting thus compensating for lost synapses (Schaefer et al., 2005). Supporting the regenerating axons provides a therapeutic opportunity for maintaining innervation. However, as the disease progresses the proportion of damaged synapses increases and the sparse functional synapses cannot mediate synaptic transmission anymore. In mouse models of ALS axons of fast-fatiguable motoneurons are affected synchronously in hindlimbs, long before symptoms appear, whereas axons of slow motoneurons are more resistant. Thus it is possible that ALS involves predictable, selective vulnerability patterns of NMJs by physiological subtypes of axons, where NMJs of the resistant axons partially assume compensatory functions (Pun et al., 2006; Dibaj et al., 2011).
In some mSOD1 mouse models, oxidative stress appears to originate from distal muscles before the disease onset (Kraft et al., 2007). Reactive oxygen species (ROS) affect synaptic transmission by inhibiting transmitter release. Increasing ROS levels further inhibit NMJ function in spite of already elevated level of oxidative stress (Naumenko et al., 2011). This suggests that oxidative damage could start in peripheral tissues and proceed retrogradely to neurons. Skeletal muscle targeted expression of mSOD1 provokes motor deficits, but at a rather late age and without evident effect on the life expectancy (Wong and Martin, 2010). In this particular model, the muscle pathology is accompanied by NMJ abnormalities and distal motoneuron axonopathy. Initiation of the motoneuron degeneration by muscle cells supports the hypothesis of dying-back pathogenesis where the neurodegeneration starts from deficits in muscle and NMJs and proceeds from distal axons towards soma leading to apoptosis of motoneurons (Fischer et al., 2004; Dupuis and Loeffler, 2009).
Presynaptic Part of the NMJ as the Main Sensitive Part Reacting to Oxidative Stress
Measurements from the diaphragm muscle of G93A-SOD1 mice have revealed a dramatic reduction in the frequency of miniature end-plate potentials (MEPPs) during the early symptomatic stage (Naumenko et al., 2011). Remarkably, no changes in the amplitude of MEPPs were observed, indicating purely presynaptic decline in the synaptic transmission. The amplitude of single evoked EPPs remained unchanged suggesting vulnerability of spontaneous quantal transmitter release from nerve terminals.
This phenotype (selective depression of MEPPs with little affected EPPs) largely resembles the inhibitory action of ROS on transmitter release at the frog NMJ: exogenous H2O2 elicits a strong inhibition of spontaneous release with limited effect on EPPs (Giniatullin and Giniatullin, 2003). NMJ impairment in ALS could therefore be produced by mechanisms similar to those, which affect synapses damaged by oxidative stress. Recent studies revealed distinct mechanisms underlying spontaneous versus evoked transmitter release (Maximov et al., 2007; Pang et al., 2011; Melom et al., 2013). Soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) protein, Snap25, was identified as the main targets of ROS at the presynaptic level (Giniatullin et al., 2006) and could be one of the targets of ROS inhibiting transmitter release (Figure 1B).
Apart from ROS, another interesting candidate contributing to the damage of NMJ in ALS is extracellular ATP. ATP, the major co-transmitter of ACh at the NMJ (Redman and Silinsky, 1994), can produce a strong inhibitory action on transmitter release (Giniatullin and Sokolova, 1998) via ROS induction (Giniatullin et al., 2005; Sciancalepore et al., 2012). This mechanism could contribute to the motor nerve terminal dysregulation in ALS (Figure 1B) or, when applied persistently, to ATP-driven neurodegeneration of NMJ (Figure 1C). This view is consistent with recent data showing that extracellular ATP, operating via cytotoxic P2X7 receptors could largely regulate immune function and inflammatory responses (Volonté et al., 2012). Notably, TSC also express P2X7 receptor (Grafe et al., 1999; Colomar et al., 2003; Nobbio et al., 2009). Whereas accumulating evidence suggest that Schwann cells can contribute to ALS (De Winter et al., 2006; Gorlewicz et al., 2009; Lobsiger et al., 2009; Chen et al., 2010), the role of myelinating versus non-myelinating TSCs in ALS however requires, further studies (Turner et al., 2010).
We propose that the early damage to the NMJ in ALS is due to intraterminal dysregulation of nerve terminals without essential changes in their morphology (Figure 1B). Underlying mechanisms probably include dysfunctional mitochondria, intracellular Ca2+ and ROS. Elevated intraterminal Ca2+ can eventually support enhanced Ca2+-dependent evoked release during early stage of ALS (Rocha et al., 2013). The other model (Figure 1C), applicable to the later stage of ALS, suggests that the main damage results from the accumulation of toxic ROS, inflammatory factors, including glial transmitters from local Schwann cells and invasive immune cells, and absence of neuroprotective trophic factors. However, these two scenarios most likely co-exist within the same muscle during ALS progression providing a heterogeneous picture of morphological and functional changes (Rocha et al., 2013) and resulting in the pitfalls of the monotherapy in this disease.
Axonal Transport, Presynaptic Mitochondria and ROS-Induced ROS Release
Correct spatial distribution of mitochondria within a cell is an instrumental prerequisite for normal physiology. In neurons, mitochondria are subjected to both anterograde and retrograde axonal transport, which in case of motoneurons covers substantial distances. The transport of mitochondria in axons is driven along microtubules by kinesin and dynein motors (Pilling et al., 2006).
Accumulation of mitochondria at presynaptic nerve terminals of motoneurons is thought to support synaptic function through ATP production and partially take part in Ca2+ buffering during neurotransmission (Figure 1; Chouhan et al., 2010). Mitochondria are connected to the presynaptic membrane by a complex cytoskeletal superstructure, which is connected with nerve terminal filamentous linkages between synaptic vesicles, providing polarized organization for mitochondrial crista structures (Perkins et al., 2010). Defects in neuronal mitochondrial morphology and axonal transport have been demonstrated in primary neuronal cultures from ALS model animals (De Vos et al., 2007; Magrané et al., 2012). Importantly, these abnormalities are also observed in vivo in both SOD1 and TDP43 ALS mouse models, indicating that they are common denominators of different genetic forms of ALS (Magrané et al., 2014).
The high order of mitochondrial organization at presynaptic nerve terminals implies their participation in coordinated responses to various stimuli. One of the fundamental oxidative stress responses in mitochondria is mitochondrial permeability transition (MPT) pore opening, followed by sudden collapse of membrane potential and burst of ROS production, which might contribute to the spreading of MPT in bystanding mitochondria, and lead to the effect known as ROS-induced ROS release (Zorov et al., 2000). The latter can contribute to the functional dysregulation within the nerve terminal during the early stage of ALS (Figure 1B). Our studies have demonstrated that SOD1 activity is increased in ALS animal spinal cord mitochondria, and causes elevated hydroperoxide production (Ahtoniemi et al., 2008; Goldsteins et al., 2008). Augmented hydroperoxide flux from presynaptic mitochondria might contribute not only to reduced probability of quatal ACh release but also to the desynchronization of neurotransmitter release at NMJ (Tsentsevitsky et al., 2013) which would additionally diminish synaptic efficacy (Figure 1C). Apart from presynaptic location, muscle mitochondria and activity of NADPH oxidase in TSCs can serve as an additional source of ROS (Figure 1).
Neuroinflammation, Immune Cells and Oxidative Stress in Spinal Cord in ALS
Oxidative stress, such as free radical damage and abnormal free radical metabolism, is evident in sALS and fALS patients (Shaw et al., 1995; Ferrante et al., 1997; Smith et al., 1998; Chang et al., 2008). The aberrant activity of mSOD1 leads to oxidative damage (Wiedau-Pazos et al., 1996; Crow et al., 1997) and other ALS-linked proteins, such as mutant TDP-43, promote oxidative stress in a motoneuron cell line (Duan et al., 2010). Excitotoxicity and oxidative stress caused by astrocytes arises from aberrant glutamate receptor function which leads to misregulated glutamate homeostasis (Rothstein et al., 1992). Oxidative stress promotes tissue damage by exacerbating and interacting with other pathological events that promote motoneuron degeneration.
Inflammation, which is an additional source of ROS, is evident in ALS patients and mSOD1 mice; microglia are activated and proliferating whereas the T cells and dendritic cells infiltrate into the spinal cord (Engelhardt et al., 1993; Henkel et al., 2004, 2006). Moreover, there is marked increase in pro-inflammatory cytokines and enzymes, such as interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), IL-8, and cyclooxygenase-2 (Cox-2) (Sekizawa et al., 1998; Almer et al., 2001, 2002; Elliott, 2001; Hensley et al., 2002; Kuhle et al., 2009). Astrocytes expressing mSOD1 are also prone to exhibit an activated pro-inflammatory state (Hensley et al., 2006; Di Giorgio et al., 2008; Marchetto et al., 2008). Activated pro-inflammatory M1 microglia cause ROS and glutamate excitotoxicity induced motoneuron injury and death (Zhao et al., 2004). MSOD1 induced oligodendrocyte dysfunction drives demyelination in the spinal cord and accelerates motoneuron degeneration (Kang et al., 2013).
Immune responses are also activated in peripheral tissues of ALS patients (Mantovani et al., 2009). Regulatory T (Treg) cells lower neuroinflammation through microglia by inducing secretion of anti-inflammatory cytokines IL-10 and transforming growth factor-β (TGF-β; Kipnis et al., 2004; Mantovani et al., 2009). In ALS patients, elevated levels of Treg cells and CD4 T cells in blood correlate with slow disease progression (Beers et al., 2011). T cell infiltration in the spinal cord in mSOD1 mice is amplified during the presymptomatic stage and the number of T cells in the spinal cord increases as the disease progresses (Beers et al., 2008; Chiu et al., 2008). The spinal cord T cell population is mainly composed of helper CD4 cells. The proportion of cytotoxic CD8 becomes prominent at the end-stage. This supports the assumption that during the early stages of the disease, there is a systemic combat to maintain neuroprotective responses, but as the disease aggravates, the immune response shifts towards cytotoxic.
Macrophages infiltrate ventral spinal roots, peripheral motor nerves and skeletal muscles in ALS mouse models (Chiu et al., 2009; Graber et al., 2010). The role of macrophages in affected tissues in ALS mice appears to be the phagocytic removal of debris from axonal degeneration. Thus, activated macrophages could contribute to ROS production in axons and muscle in ALS and along with other inflammatory agents participate in triggering of sprouting in nerve terminals. However, in ALS mice the majority of activated macrophages accumulated within fascicles of motoneurons in the peripheral tissues and were only rarely found adjacent to end-plate of NMJs. It is therefore unlikely, that macrophages directly contribute to oxidative damage of NMJs in ALS.
Interestingly, in ALS motoneurons in the brainstem oculomotor nuclei and Onuf’s nucleus in the sacral spinal cord are preserved and selective vulnerability seems to be related to oxidative stress. Reduced capability to maintain calcium homeostasis and disturbed mitochondrial function predispose specific motoneurons to degeneration in ALS (Vanselow and Keller, 2000; Jaiswal and Keller, 2009). In addition, the most vulnerable motoneurons are more prone to endoplasmic reticulum stress and exhibit increased susceptibility to excitotoxicity (Saxena et al., 2009; Brockington et al., 2013).
Gender Dependence of ALS and Oxidative Stress
ALS affects men more than women, with earlier age of onset for men as well a tendency for spinal initiation of the disease whereas in women it is more commonly bulbar (McComb and Henderson, 2010). Most of these features were also observed in mSOD1 animals (Veldink et al., 2003; Suzuki et al., 2007). Gender specific differences are also detected at the synapses: Synaptic vesicle release being more frequent in females’ end-plate with impairment only observed in males (Naumenko et al., 2011). Specific interneurons control motoneuron excitability via specialized cholinergic synaptic boutons: C-boutons. In ALS there is no gender difference in the number of C-boutons, but their size is bigger in male mSOD1 mice (Herron and Miles, 2012).
The most obvious explanation for gender differences is a protective effect of estrogen. ROS damage in muscle is limited in young women. Even aging women have significantly less lipid peroxidation, protein carbonylation and mitochondrial DNA damage than men (Pansarasa et al., 2000). Several estrogen-controlled pathways might protect females against fast neuromuscular degeneration in ALS, for instance estrogen-mediated cyclophilin D prevention of mitochondrial calcium overload (Kim et al., 2012). However, experiments with ovariectomized mSOD1 mice or rats with and without supplemental 17β-estradiol do not support the idea that estrogen could explain the gender differences in ALS (Choi et al., 2008; Hayes-Punzo et al., 2012).
Additional evidence for gender differences in the ROS balance are coming to light, for instance, lower blood level of uric acid (UA) were observed in ALS patients (Keizman et al., 2009). UA, a scavenger of NO radical and superoxide, reduces damage to cells, by preventing protein nitration on tyrosine residues by peroxynitrites, and higher level of UA in blood increases likelihood of longer survival in men (Paganoni et al., 2012).
Gender differences are also striking in the response to treatments. Examples of therapeutic approaches with gender bias are specific inhibition of spinal cord microglial P2X7, which appears to increase the duration of life without affecting the age of symptom onset in male (Cervetto et al., 2013) and G-CSF treatment which delays disease progression in male mSOD1 mice (Pitzer et al., 2008; Naumenko et al., 2011; Pollari et al., 2011).
Promising Therapeutic Approaches and Challenges of the Antioxidant Therapy in ALS
Several molecules with antioxidant capabilities have failed in clinics after showing promise in animal models (Gordon, 2013; Musarò, 2013; Pandya et al., 2013; Sreedharan and Brown, 2013). Still, riluzole is the only approved drug that delays the progression of ALS.
The translational failures in ALS can be explained by the same arguments as in other neurodegenerative diseases: (a) animal models represent only a fraction of genetic variations among ALS patients and do not model well sALS; (b) the preclinical studies are characterized by inadequate randomization, blinding, statistical power, control cohorts and consideration of comorbidities; and (c) optimal dosing and administration route in clinics are unknown, flaws in patient stratification or identification of proper patients, and insufficient samples size (Ubogu, 2012; Planas, 2013).
Perhaps the most important reason for the translation block is that by the time of diagnosis, ALS has already progressed too far, making prevention of further deterioration challenging. The late diagnosis allows multiple disease mechanisms to accelerate their contributions to motoneuron death overriding the regenerative mechanisms and preconditions. Also, nerve terminal retraction, axonal degeneration and eventual neuronal death may take weeks or months before their deteriorating effects become clinically noticeable (Coleman and Freeman, 2010; Sreedharan and Brown, 2013). The late time of diagnosis is an especially important concern for protection of NMJ functions, as NMJ degeneration is among the earliest pathological alterations in ALS.
During the presymptomatic phase of the ALS, oxidative stress may be triggered by increased production of superoxide and nitric oxide in neurons, central and peripheral glia and even in muscle cells. Also, levels of a major intracellular antixodiant, glutathione, reduce early in ALS tissues. At the same time, blood-spinal cord barrier appears to become leaky and infiltration of inflammatory cells into the spinal cord, motor nerves and muscles contributes to oxidative stress present prior to the disease onset (Henkel et al., 2004; Chang et al., 2008; Chiu et al., 2009; Chen et al., 2010; Halter et al., 2010; Dibaj et al., 2011; Drechsel et al., 2012; Winkler et al., 2014). In fact, several findings favor the idea of linking ALS therapy to the oxidative stress-related degeneration of NMJ. First, normal SOD1 activity is required for maintenance of NMJ function in aged rodents (Sakellariou et al., 2014) and, in a zebrafish model expressing mSOD1 in physiological levels, NMJ has increased susceptibility to oxidative stress showing early morphological alterations (Da Costa et al., 2014). Second, even though pathological changes in synapses and axons occur early during the ALS pathogenesis, these self-destructive mechanisms could be delayed by correcting molecular environment (Sreedharan and Brown, 2013). Third, the NOX-mediated increase in superoxide production takes place in neural cells in mSOD1 models of ALS (Harraz et al., 2008; Jaronen et al., 2013). Even though is it not known whether NOX is expressed in Schwann cells around NMJs, NOX could well contribute to oxidative deterioration of NMJ in ALS, as muscle cells express various isoforms of NOX. While it is unclear whether ALS-linked mutations or conditions in sALS could trigger activation of NOX in skeletal muscles, inhibitors of NOX activation are known to provide protection in ALS models. Considering that NOX activation pathway is a readily druggable target, the role of NOX in NMJ degeneration in ALS models is worth exploring (Sullivan-Gunn and Lewandowski, 2013). Finally, it is of interest that Vitamin D, an essential dietary vitamin with multiple physiological functions, has been demonstrated to influence several aspect of ALS pathology, including skeletal muscle strength and oxidative stress (Gianforcaro and Hamadeh, 2014).
Overall, recent research on NMJ, oxidative stress and inflammation in ALS models warrant further preclinical investigation of the possibility of developing an ALS therapy by targeting the signaling pathways of NMJ dysfunction, provided that early diagnosis of ALS and biomarkers for NMJ dysfunctions become available (Figure 1). While keeping in mind the previous failures in clinical trials for ALS, it is evident that multiple mechanisms, including oxidative stress in the center, contribute to ALS pathogenesis. The concept of combination therapy is not novel in the field of neurodegenerative diseases, but it is still a valid approach once most of the key targets of the disease mechanisms, including oxidative stress become identified.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by Photonic program from the Finnish Academy (ROSim Grant 135179). Rashid Giniatullin was supported by the Program of Competitiveness of Kazan University.
References
Ahtoniemi, T., Jaronen, M., Keksa-Goldsteine, V., Goldsteins, G., and Koistinaho, J. (2008). Mutant SOD1 from spinal cord of G93A rats is destabilized and binds to inner mitochondrial membrane. Neurobiol. Dis. 32, 479–485. doi: 10.1016/j.nbd.2008.08.010
Almer, G., Guégan, C., Teismann, P., Naini, A., Rosoklija, G., Hays, A. P., et al. (2001). Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann. Neurol. 49, 176–185. doi: 10.1002/1531-8249(20010201)49:2<176::aid-ana37>3.3.co;2-o
Almer, G., Teismann, P., Stevic, Z., Halaschek-Wiener, J., Deecke, L., Kostic, V., et al. (2002). Increased levels of the pro-inflammatory prostaglandin PGE2 in CSF from ALS patients. Neurology 58, 1277–1279. doi: 10.1212/wnl.58.8.1277
Beers, D. R., Henkel, J. S., Zhao, W., Wang, J., and Appel, S. H. (2008). CD4+ T cells support glial neuroprotection, slow disease progression and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. U S A 105, 15558–15563. doi: 10.1073/pnas.0807419105
Beers, D. R., Henkel, J. S., Zhao, W., Wang, J., Huang, A., Wen, S., et al. (2011). Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134, 1293–1314. doi: 10.1093/brain/awr074
Brockington, A., Nin, G. K., Heath, P. R., Wood, E., Kirby, J., Fusi, N., et al. (2013). Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 125, 95–109. doi: 10.1007/s00401-012-1058-5
Cervetto, C., Frattaroli, D., Maura, G., and Marcoli, M. (2013). Motor neuron dysfunction in a mouse model of ALS: gender-dependent effect of P2X7 antagonism. Toxicology 311, 69–77. doi: 10.1016/j.tox.2013.04.004
Chang, Y., Kong, Q., Shan, X., Tian, G., Ilieva, H., Cleveland, D. W., et al. (2008). Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One 3:2849. doi: 10.1371/journal.pone.0002849
Chen, K., Northington, F. J., and Martin, L. J. (2010). Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct. Funct. 214, 219–234. doi: 10.1007/s00429-009-0226-4
Chiu, I. M., Chen, A., Zheng, Y., Kosaras, B., Tsiftsoglou, S. A., Vartanian, T. K., et al. (2008). T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. U S A 105, 17913–17918. doi: 10.1073/pnas.0804610105
Chiu, I. M., Phatnani, H., Kuligowski, M., Tapia, J. C., Carrasco, M. A., Zhang, M., et al. (2009). Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc. Natl. Acad. Sci. U S A 106, 20960–20965. doi: 10.1073/pnas.0911405106
Choi, C. I., Lee, Y. D., Gwag, B. J., Cho, S. I., Kim, S. S., and Suh-Kim, H. (2008). Effects of estrogen on lifespan and motor functions in female hSOD1 G93A transgenic mice. J. Neurol. Sci. 268, 40–47. doi: 10.1016/j.jns.2007.10.024
Chouhan, A., Zhang, J., Zinsmaier, K., and Macleod, G. (2010). Presynaptic mitochondria in functionally different motor neurons exhibit similar affinities for Ca2+ but exert little influence as Ca2+ buffers at nerve firing rates in situ. J. Neurosci. 30, 1869–1881. doi: 10.1523/jneurosci.4701-09.2010
Coleman, M. P., and Freeman, M. R. (2010). Wallerian degeneration, wld(s) and nmnat. Annu. Rev. Neurosci. 33, 245–267. doi: 10.1146/annurev-neuro-060909-153248
Colomar, A., Marty, V., Médina, C., Combe, C., Parnet, P., and Amédée, T. (2003). Maturation and release of interleukin-1beta by lipopolysaccharide-primed mouse Schwann cells require the stimulation of P2X7 receptors. J. Biol. Chem. 278, 30732–30740. doi: 10.1074/jbc.m304534200
Crow, J. P., Ye, Y. Z., Strong, M., Kirk, M., Barnes, S., and Beckman, J. S. (1997). Superoxide dismutase catalyzes nitration of tyrosines by peroxynitrite in the rod and head domains of neurofilament-L. J. Neurochem. 69, 1945–1953. doi: 10.1046/j.1471-4159.1997.69051945.x
Da Costa, M. M., Allen, C. E., Higginbottom, A., Ramesh, T., Shaw, P. J., and McDermott, C. J. (2014). A new zebrafish model produced by TILLING of SOD1-related amyotrophic lateral sclerosis replicates key features of the disease and represents a tool for in vivo therapeutic screening. Dis. Model Mech. 7, 73–81. doi: 10.1242/dmm.012013
De Vos, K. J., Chapman, A. L., Tennant, M. E., Manser, C., Tudor, E. L., Lau, K., et al. (2007). Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 16, 2720–2728. doi: 10.1093/hmg/ddm226
De Winter, F., Vo, T., Stam, F. J., Wisman, L. A., Bär, P. R., Niclou, S. P., et al. (2006). The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol. Cell. Neurosci. 32, 102–117. doi: 10.1016/j.mcn.2006.03.002
Di Giorgio, F. P., Boulting, G. L., Bobrowicz, S., and Eggan, K. C. (2008). Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an als-causing mutation. Cell Stem Cell 3, 637–648. doi: 10.1016/j.stem.2008.09.017
Dibaj, P., Steffens, H., Zschüntzsch, J., Nadrigny, F., Schomburg, E. D., Kirchhoff, F., et al. (2011). In Vivo imaging reveals distinct inflammatory activity of CNS microglia versus PNS macrophages in a mouse model for ALS. PLoS One 6:e17910. doi: 10.1371/journal.pone.0017910
Drechsel, D. A., Estévez, A. G., Barbeito, L., and Beckman, J. S. (2012). Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotox. Res. 22, 251–264. doi: 10.1007/s12640-012-9322-y
Duan, W., Li, X., Shi, J., Guo, Y., Li, Z., and Li, C. (2010). Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 169, 1621–1629. doi: 10.1016/j.neuroscience.2010.06.018
Dupuis, L., and Loeffler, J. (2009). Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models. Curr. Opin. Pharmacol. 9, 341–346. doi: 10.1016/j.coph.2009.03.007
Elliott, J. L. (2001). Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Brain Res. Mol. Brain Res. 95, 172–178. doi: 10.1016/s0169-328x(01)00242-x
Engelhardt, J. I., Tajti, J., and Appel, S. H. (1993). Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch. Neurol. 50, 30–36. doi: 10.1001/archneur.1993.00540010026013
Ferrante, R. J., Browne, S. E., Shinobu, L. A., Bowling, A. C., Baik, M. J., MacGarvey, U., et al. (1997). Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 69, 2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Fischer-Hayes, L. R., Brotherton, T., and Glass, J. D. (2013). Axonal degeneration in the peripheral nervous system: implications for the pathogenesis of amyotrophic lateral sclerosis. Exp. Neurol. 246, 6–13. doi: 10.1016/j.expneurol.2013.05.001
Gianforcaro, A., and Hamadeh, M. J. (2014). Vitamin D as a potential therapy in amyotrophic lateral sclerosis. CNS Neurosci. Ther. 20, 101–111. doi: 10.1111/cns.12204
Giniatullin, A., Grishin, S., Sharifullina, E., Petrov, A., Zefirov, A., and Giniatullin, R. (2005). Reactive oxygen species contribute to the presynaptic action of extracellular ATP at the frog neuromuscular junction. J. Physiol. 565, 229–242. doi: 10.1113/jphysiol.2005.084186
Giniatullin, A. R., and Giniatullin, R. A. (2003). Dual action of hydrogen peroxide on synaptic transmission at the frog neuromuscular junction. J. Physiol. 552, 283–293. doi: 10.1113/jphysiol.2003.050690
Giniatullin, A. R., Darios, F., Shakirzyanova, A., Davletov, B., and Giniatullin, R. (2006). SNAP25 is a pre-synaptic target for the depressant action of reactive oxygen species on transmitter release. J. Neurochem. 98, 1789–1797. doi: 10.1111/j.1471-4159.2006.03997.x
Giniatullin, R., and Sokolova, E. (1998). ATP and adenosine inhibit transmitter release at the frog neuromuscular junction through distinct presynaptic receptors. Br. J. Pharmacol. 124, 839–844. doi: 10.1038/sj.bjp.0701881
Goldsteins, G., Keksa-Goldsteine, V., Ahtoniemi, T., Jaronen, M., Arens, E., Akerman, K., et al. (2008). Deleterious role of superoxide dismutase in the mitochondrial intermembrane space. J. Biol. Chem. 283, 8446–8452. doi: 10.1074/jbc.m706111200
Gordon, P. H. (2013). Amyotrophic lateral sclerosis: an update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis. 4, 295–310. doi: 10.14336/ad.2013.0400295
Gorlewicz, A., Wlodarczyk, J., Wilczek, E., Gawlak, M., Cabaj, A., Majczynski, H., et al. (2009). CD44 is expressed in non-myelinating Schwann cells of the adult rat and may play a role in neurodegeneration-induced glial plasticity at the neuromuscular junction. Neurobiol. Dis. 34, 245–258. doi: 10.1016/j.nbd.2009.01.011
Graber, D. J., Hickey, W. F., and Harris, B. T. (2010). Progressive changes in microglia and macrophages in spinal cord and peripheral nerve in the transgenic rat model of amyotrophic lateral sclerosis. J. Neuroinflammation 7:8. doi: 10.1186/1742-2094-7-8
Grafe, P., Mayer, C., Takigawa, T., Kamleiter, M., and Sanchez-Brandelik, R. (1999). Confocal calcium imaging reveals an ionotropic P2 nucleotide receptor in the paranodal membrane of rat Schwann cells. J. Physiol. 515, 377–383. doi: 10.1111/j.1469-7793.1999.377ac.x
Halter, B., Gonzalez de Aguilar, J. L., Rene, F., Petri, S., Fricker, B., Echaniz-Laguna, A., et al. (2010). Oxidative stress in skeletal muscle stimulates early expression of Rad in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 48, 915–923. doi: 10.1016/j.freeradbiomed.2010.01.014
Harraz, M. M., Marden, J. J., Zhou, W., Zhang, Y., Williams, A., Sharov, V. S., et al. (2008). SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Invest. 118, 659–670. doi: 10.1172/JCI34060
Hayes-Punzo, A., Mulcrone, P., Meyer, M., McHugh, J., Svendsen, C. N., and Suzuki, M. (2012). Gonadectomy and dehydroepiandrosterone (DHEA) do not modulate disease progression in the G93A mutant SOD1 rat model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 311–314. doi: 10.3109/17482968.2012.654393
Henkel, J. S., Beers, D. R., Siklós, L., and Appel, S. H. (2006). The chemokine MCP-1 and the dendritic and myeloid cells it attracts are increased in the mSOD1 mouse model of ALS. Mol. Cell. Neurosci. 31, 427–437. doi: 10.1016/j.mcn.2005.10.016
Henkel, J. S., Engelhardt, J. I., Siklós, L., Simpson, E. P., Kim, S. H., Pan, T., et al. (2004). Presence of dendritic cells, MCP-1 and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 55, 221–235. doi: 10.1002/ana.10805
Hensley, K., Abdel-Moaty, H., Hunter, J., Mhatre, M., Mou, S., Nguyen, K., et al. (2006). Primary glia expressing the G93A-SOD1 mutation present a neuroinflammatory phenotype and provide a cellular system for studies of glial inflammation. J. Neuroinflammation 3:2. doi: 10.1186/1742-2094-3-2
Hensley, K., Floyd, R. A., Gordon, B., Mou, S., Pye, Q. N., Stewart, C., et al. (2002). Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J. Neurochem. 82, 365–374. doi: 10.1046/j.1471-4159.2002.00968.x
Herron, L. R., and Miles, G. B. (2012). Gender-specific perturbations in modulatory inputs to motoneurons in a mouse model of amyotrophic lateral sclerosis. Neuroscience 226, 313–323. doi: 10.1016/j.neuroscience.2012.09.031
Jaiswal, M. K., and Keller, B. U. (2009). Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 75, 478–489. doi: 10.1124/mol.108.050831
Jaronen, M., Vehviläinen, P., Malm, T., Keksa-Goldsteine, V., Pollari, E., Valonen, P., et al. (2013). Protein disulfide isomerase in ALS mouse glia links protein misfolding with NADPH oxidase-catalyzed superoxide production. Hum. Mol. Genet. 22, 646–655. doi: 10.1093/hmg/dds472
Kang, S. H., Li, Y., Fukaya, M., Lorenzini, I., Cleveland, D. W., Ostrow, L. W., et al. (2013). Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 16, 571–579. doi: 10.1038/nn.3357
Keizman, D., Ish-Shalom, M., Berliner, S., Maimon, N., Vered, Y., Artamonov, I., et al. (2009). Low uric acid levels in serum of patients with ALS: further evidence for oxidative stress? J. Neurol. Sci. 285, 95–99. doi: 10.1016/j.jns.2009.06.002
Kim, H. J., Magranè, J., Starkov, A. A., and Manfredi, G. (2012). The mitochondrial calcium regulator cyclophilin D is an essential component of oestrogen-mediated neuroprotection in amyotrophic lateral sclerosis. Brain 135, 2865–2874. doi: 10.1093/brain/aws208
Kipnis, J., Avidan, H., Caspi, R. R., and Schwartz, M. (2004). Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc. Natl. Acad. Sci. U S A 101, 14663–14669. doi: 10.1073/pnas.0404842101
Kraft, A. D., Resch, J. M., Johnson, D. A., and Johnson, J. A. (2007). Activation of the Nrf2–ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp. Neurol. 207, 107–117. doi: 10.1016/j.expneurol.2007.05.026
Kuhle, J., Lindberg, R. L. P., Regeniter, A., Mehling, M., Steck, A. J., Kappos, L., et al. (2009). Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur. J. Neurol. 16, 771–774. doi: 10.1111/j.1468-1331.2009.02560.x
Lobsiger, C. S., Boillee, S., McAlonis-Downes, M., Khan, A. M., Feltri, M. L., Yamanaka, K., et al. (2009). Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. U S A 106, 4465–4470. doi: 10.1073/pnas.0813339106
Magrané, J., Cortez, C., Gan, W.-B., and Manfredi, G. (2014). Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 23, 1413–1424. doi: 10.1093/hmg/ddt528
Magrané, J., Sahawneh, M. A., Przedborski, S., Estévez, Á. G., and Manfredi, G. (2012). Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J. Neurosci. 32, 229–242. doi: 10.1523/jneurosci.1233-11.2012
Mantovani, S., Garbelli, S., Pasini, A., Alimonti, D., Perotti, C., Melazzini, M., et al. (2009). Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J. Neuroimmunol. 210, 73–79. doi: 10.1016/j.jneuroim.2009.02.012
Marchetto, M. C. N., Muotri, A. R., Mu, Y., Smith, A. M., Cezar, G. G., and Gage, F. H. (2008). Non-cell-autonomous effect of human SOD1G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3, 649–657. doi: 10.1016/j.stem.2008.10.001
Maximov, A., Shin, O. H., Liu, X., and Sudhof, T. C. (2007). Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. J. Cell Biol. 176, 113–124. doi: 10.1083/jcb.200607021
McComb, P. A., and Henderson, R. D. (2010). Effects of gender in amyotrophic lateral sclerosis. Gend. Med. 7, 557–570. doi: 10.1016/j.genm.2010.11.010
Melom, J. E., Akbergenova, Y., Gavornik, J. P., and Littleton, J. T. (2013). Spontaneous and evoked release are independently regulated at individual active zones. J. Neurosci. 33, 17253–17263. doi: 10.1523/jneurosci.3334-13.2013
Musarò, A. (2013). Understanding ALS: new therapeutic approaches. FEBS J. 280, 4315–4322. doi: 10.1111/febs.12087
Naumenko, N., Pollari, E., Kurronen, A., Giniatullina, R., Shakirzyanova, A., Magga, J., et al. (2011). Gender-specific mechanism of synaptic impairment and its prevention by GCSF in a mouse model of ALS. Front. Cell. Neurosci. 5:26. doi: 10.3389/fncel.2011.00026
Nobbio, L., Sturla, L., Fiorese, F., Usai, C., Basile, G., Moreschi, I., et al. (2009). P2X7-mediated increased intracellular calcium causes functional derangement in Schwann cells from rats with CMT1A neuropathy. J. Biol. Chem. 284, 23146–23158. doi: 10.1074/jbc.m109.027128
Paganoni, S., Zhang, M., Quiroz Zárate, A., Jaffa, M., Yu, H., Cudkowicz, M. E., et al. (2012). Uric acid levels predict survival in men with amyotrophic lateral sclerosis. J. Neurol. 259, 1923–1928. doi: 10.1007/s00415-012-6440-7
Pandya, R. S., Zhu, H., Li, W., Bowser, R., Friedlander, R. M., and Wang, X. (2013). Therapeutic neuroprotective agents for amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 70, 4729–4745. doi: 10.1007/s00018-013-1415-0
Pang, Z. P., Bacaj, T., Yang, X., Zhou, P., Xu, W., and Sudhof, T. C. (2011). Doc2 supports spontaneous synaptic transmission by a Ca2+-independent mechanism. Neuron 70, 244–251. doi: 10.1016/j.neuron.2011.03.011
Pansarasa, O., Castagna, L., Colombi, B., Vecchiet, J., Felzani, G., and Marzatico, F. (2000). Age and sex differences in human skeletal muscle: role of reactive oxygen species. Free Radic. Res. 33, 287–293. doi: 10.1080/10715760000301451
Perkins, G. A., Tjong, J., Brown, J. M., Poquiz, P. H., Scott, R. T., Kolson, D. R., et al. (2010). The micro-architecture of mitochondria at active zones: electron tomography reveals novel anchoring scaffolds and cristae structured for high-rate metabolism. J. Neurosci. 30, 1015–1026. doi: 10.1523/jneurosci.1517-09.2010
Pilling, A. D., Horiuchi, D., Lively, C. M., and Saxton, W. M. (2006). Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in drosophila motor axons. Mol. Biol. Cell 17, 2057–2068. doi: 10.1091/mbc.e05-06-0526
Pitzer, C., Krüger, C., Plaas, C., Kirsch, F., Dittgen, T., Müller, R., et al. (2008). Granulocyte-colony stimulating factor improves outcome in a mouse model of amyotrophic lateral sclerosis. Brain 131, 3335–3347. doi: 10.1093/brain/awn243
Planas, A. M. (2013). Advances in stroke: translational medicine 2012. Stroke 44, 318–319. doi: 10.1161/strokeaha.111.000495
Pollari, E., Savchenko, E., Jaronen, M., Kanninen, K., Malm, T., Wojciechowski, S., et al. (2011). Granulocyte colony stimulating factor attenuates inflammation in a mouse model of amyotrophic lateral sclerosis. J. Neuroinflammation 8:74. doi: 10.1186/1742-2094-8-74
Pun, S., Santos, A. F., Saxena, S., Xu, L., and Caroni, P. (2006). Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 9, 408–419. doi: 10.1038/nn1653
Redman, R. S., and Silinsky, E. M. (1994). ATP released together with acetylcholine as the mediator of neuromuscular depression at frog motor nerve endings. J. Physiol. 477, 117–127.
Rocha, M. C., Pousinha, P. A., Correia, A. M., Sebastião, A. M., and Ribeiro, J. A. (2013). Early changes of neuromuscular transmission in the SOD1(G93A) mice model of als start long before motor symptoms onset. PLoS One 8:e73846. doi: 10.1371/journal.pone.0073846
Rothstein, J. D., Martin, L. J., and Kuncl, R. W. (1992). Decreased glutamate transport by the brain and spinal-cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 326, 1464–1468. doi: 10.1056/nejm199205283262204
Sakellariou, G. K., Davis, C. S., Shi, Y., Ivannikov, M. V., Zhang, Y., Vasilaki, A., et al. (2014). Neuron-specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD-knockout mice. FASEB J. 28, 1666–1681. doi: 10.1096/fj.13-240390
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Schaefer, A. M., Sanes, J. R., and Lichtman, J. W. (2005). A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 490, 209–219. doi: 10.1002/cne.20620
Sciancalepore, M., Luin, E., Parato, G., Ren, E., Giniatullin, R., Fabbretti, E., et al. (2012). Reactive oxygen species contribute to the promotion of the ATP-mediated proliferation of mouse skeletal myoblasts. Free Radic. Biol. Med. 53, 1392–1398. doi: 10.1016/j.freeradbiomed.2012.08.002
Sekizawa, T., Openshaw, H., Ohbo, K., Sugamura, K., Itoyama, Y., and Niland, J. C. (1998). Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases. J. Neurol. Sci. 154, 194–199. doi: 10.1016/s0022-510x(97)00228-1
Shaw, P. J., Ince, P. G., Falkous, G., and Mantle, D. (1995). Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann. Neurol. 38, 691–695. doi: 10.1002/ana.410380424
Shi, L., Fu, A. K., and Ip, N. Y. (2012). Molecular mechanisms underlying maturation and maintenance of the vertebrate neuromuscular junction. Trends Neurosci. 35, 441–453. doi: 10.1016/j.tins.2012.04.005
Smith, R. G., Henry, Y. K., Mattson, M. P., and Appel, S. H. (1998). Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 44, 696–699. doi: 10.1002/ana.410440419
Sreedharan, J., and Brown, R. H. Jr. (2013). Amyotrophic lateral sclerosis: problems and prospects. Ann. Neurol. 74, 309–316. doi: 10.1002/ana.24012
Sullivan-Gunn, M. J., and Lewandowski, P. A. (2013). Elevated hydrogen peroxide and decreased catalase and glutathione peroxidase protection are associated with aging sarcopenia. BMC Geriatr. 13:104. doi: 10.1186/1471-2318-13-104
Suzuki, M., Tork, C., Shelley, B., McHugh, J., Wallace, K., Klein, S. M., et al. (2007). Sexual dimorphism in disease onset and progression of a rat model of ALS. Amyotroph. Lateral Scler. 8, 20–25. doi: 10.1080/17482960600982447
Tsentsevitsky, A., Kovyazina, I., Nikolsky, E., Bukharaeva, E., and Giniatullin, R. (2013). Redox-sensitive synchronizing action of adenosine on transmitter release at the neuromuscular junction. Neuroscience 248, 699–707. doi: 10.1016/j.neuroscience.2013.05.065
Turner, B. J., Ackerley, S., Davies, K. E., and Talbot, K. (2010). Dismutase-competent SOD1 mutant accumulation in myelinating Schwann cells is not detrimental to normal or transgenic ALS model mice. Hum. Mol. Genet. 19, 815–824. doi: 10.1093/hmg/ddp550
Ubogu, E. E. (2012). Translational strategies in peripheral neuroinflammation and neurovascular repair. Transl. Neurosci. 3, 373–383. doi: 10.2478/s13380-012-0039-4
Vanselow, B. K., and Keller, B. U. (2000). Calcium dynamics and buffering in oculomotor neurones from mouse that are particularly resistant during amyotrophic lateral sclerosis (ALS)-related motoneurone disease. J. Physiol. 525, 433–445. doi: 10.1111/j.1469-7793.2000.t01-1-00433.x
Veldink, J. H., Bär, P. R., Joosten, E. A., Otten, M., Wokke, J. H., and van den Berg, L. H. (2003). Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul. Disord. 13, 737–743. doi: 10.1016/s0960-8966(03)00104-4
Volonté, C., Apolloni, S., Skaper, S. D., and Burnstock, G. (2012). P2X7 receptors: channels, pores and more. CNS Neurol. Disord. Drug Targets 11, 705–721. doi: 10.2174/187152712803581137
Wiedau-Pazos, M., Goto, J. J., Rabizadeh, S., Gralla, E. B., Roe, J. A., Lee, M. K., et al. (1996). Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science 271, 515–518. doi: 10.1126/science.271.5248.515
Winkler, E. A., Sengillo, J. D., Sagare, A. P., Zhao, Z., Ma, Q., Zuniga, E., et al. (2014). Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. U S A 111, E1035–E1042. doi: 10.1073/pnas.1401595111
Wong, M., and Martin, L. J. (2010). Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 19, 2284–2302. doi: 10.1093/hmg/ddq106
Zhao, W., Xie, W., Le, W., Beers, D. R., He, Y., Henkel, J. S., et al. (2004). Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J. Neuropathol. Exp. Neurol. 63, 964–977.
Keywords: ALS, neuromuscular junction, ROS, oxidative stress, neurodegeneration
Citation: Pollari E, Goldsteins G, Bart G, Koistinaho J and Giniatullin R (2014) The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8:131. doi: 10.3389/fncel.2014.00131
Received: 28 February 2014; Accepted: 27 April 2014;
Published online: 13 May 2014.
Edited by:
Manoj Kumar Jaiswal, Center for Neuroscience and Regenerative Medicine, USAReviewed by:
David Marcinek, University of Washington, USADan Lindholm, Helsinki university, Finland
Andreas H. Kottmann, The Sophie Davis School of Biomedical Education, City University of New York, USA
Copyright © 2014 Pollari, Goldsteins, Bart, Koistinaho and Giniatullin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eveliina Pollari, Molecular Brain Research Laboratory, Department of Neurobiology, A. I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, Yliopistonranta 1, P. O. Box 1627, Kuopio 70211, Finland e-mail:RXZlbGlpbmEucG9sbGFyaUB1ZWYuZmk=