 Nadine Ehmann1
Nadine Ehmann1 Markus Sauer
Markus Sauer Robert J. Kittel
Robert J. Kittel- 1Department of Neurophysiology, Institute of Physiology, University of Würzburg, Würzburg, Germany
- 2Department of Biotechnology and Biophysics, University of Würzburg, Würzburg, Germany
Brain function relies on accurate information transfer at chemical synapses. At the presynaptic active zone (AZ) a variety of specialized proteins are assembled to complex architectures, which set the basis for speed, precision and plasticity of synaptic transmission. Calcium channels are pivotal for the initiation of excitation-secretion coupling and, correspondingly, capture a central position at the AZ. Combining quantitative functional studies with modeling approaches has provided predictions of channel properties, numbers and even positions on the nanometer scale. However, elucidating the nanoscopic organization of the surrounding protein network requires direct ultrastructural access. Without this information, knowledge of molecular synaptic structure-function relationships remains incomplete. Recently, super-resolution microscopy (SRM) techniques have begun to enter the neurosciences. These approaches combine high spatial resolution with the molecular specificity of fluorescence microscopy. Here, we discuss how SRM can be used to obtain information on the organization of AZ proteins.
Introduction
At chemical synapses, neurotransmitter release takes place at presynaptic active zones (AZs). Morphologically, AZs can be identified via their electron-dense cytomatrix—an intricate network of specialized proteins precisely organized to execute and modulate exocytosis (Zhai and Bellen, 2004; Jahn and Fasshauer, 2012; Südhof, 2012). Structure and function of AZs display varying degrees of diversity between different neuron types, between synapses of the same neuron innervating different follower cells and even between individual synapses formed by the same partner cells (Rozov et al., 2001; Atwood and Karunanithi, 2002; Zhai and Bellen, 2004; Peled and Isacoff, 2011; Ehmann et al., 2014; Paul et al., submitted). Moreover, the functional properties and the molecular composition of AZs are dynamic and can be modified in an activity-dependent manner (e.g., Wojtowicz et al., 1994; Castillo et al., 2002; Matz et al., 2010; Weyhersmüller et al., 2011). Moving from correlation to causality to clarify how different molecular architectures of AZs give rise to specific physiological properties remains a major challenge.
As an AZ contains a multitude of densely packed proteins in a small sub-cellular compartment (around 200–400 nm diameter at a central synapse; Siksou et al., 2007) diffraction-limited light microscopy delivers only very coarse structural information. Hence, morphological investigations of the fine structure and the molecular organization of AZs have mainly been restricted to electron microscopy (EM). Recently, the development of super-resolution microscopy (SRM) techniques has provided means to bypass the diffraction barrier of ~300 nm in lateral dimensions (Abbe, 1873) and to bridge the gap between conventional light microscopy and EM (for detailed recent overviews, see Hell, 2009; Patterson et al., 2010; Schermelleh et al., 2010; Galbraith and Galbraith, 2011; Sauer, 2013). These emerging technologies offer promising new options for studying nanoscopic sub-cellular structures.
Recent work has reviewed the molecular composition of AZs (Owald and Sigrist, 2009; Jahn and Fasshauer, 2012; Südhof, 2012). This perspective will focus on excitation-secretion coupling, i.e., the transduction of an electrical signal into Calcium (Ca2+)-dependent neurotransmitter release (Schneggenburger and Neher, 2005; Wojcik and Brose, 2007). We will summarize current information on functional determinants of the AZ and explore how the search for structural correlates can be supported by SRM to improve our mechanistic understanding of neurotransmission.
Microscopy
Fluorescence microscopy is the method of choice for visualizing biomolecules in fixed and living cells as it enables their selective and specific detection with a high signal-to-background ratio (Lichtman and Conchello, 2005). However, while light micro-scopy is ideally suited to investigate macroscopic arrangements, it fails to uncover organizational principles at the molecular scale due to its limited spatial resolution.
EM provides substantially increased resolution, though its application is restricted to lifeless, fixed and embedded biological samples. EM studies have been instrumental in recognizing the large morphological diversity of AZs (Zhai and Bellen, 2004) and in identifying repetitive structural elements within individual, chemically fixed AZs (Pfenninger et al., 1972; Phillips et al., 2001). Moreover, alternative tissue preparation and fixation techniques have enabled analyses of filamentous AZ structures and their associated synaptic vesicles in various organisms (Landis et al., 1988; Siksou et al., 2007; Jiao et al., 2010; Wichmann and Sigrist, 2010; Fernández-Busnadiego et al., 2013). The resolving power of EM is exemplified by a classical tomographic study at the frog neuromuscular junction (NMJ). The results revealed an intricate fine structure of the AZ, which establishes a regular and precisely organized arrangement of synaptic vesicles relative to Ca2+ channels at release sites (Harlow et al., 2001). As more substructural details are uncovered (Szule et al., 2012), knowledge of the underlying protein species becomes increasingly desirable. Immunogold labeling provides a means to locate specific proteins in electron micrographs with nanometer resolution and has been used to examine the topology of AZs (e.g., Limbach et al., 2011). However, specific labeling with antibody-coupled gold particles is inefficient and a compromise must be made between optimal tissue preservation and structural resolution. Consequently, the ideal microscope should combine the minimal invasiveness and efficient specific labeling possibilities of optical microscopy with the high spatial resolution of EM. Technologies that merge these features, at least to a certain extent, are collectively termed SRM. These include structured illumination microscopy (SIM), stimulated emission depletion (STED) and single-molecule based localization microscopy methods, such as photo-activated localization microscopy (PALM) and direct stochastic optical reconstruction microscopy (dSTORM). The techniques can be subdivided based on their principle of bypassing the diffraction barrier: deterministic approaches, such as STED, use a phase mask to define the coordinates of fluorescence emission predefined in space by the zero-node, whereas PALM and dSTORM use stochastic activation of individual fluorophores and precise position determination (localization).
SIM relies on patterned illumination of the specimen with a high spatial frequency in various orientations providing a lateral resolution of approximately 100 nm (Heintzmann and Cremer, 1999; Gustafsson, 2000). Fortunately, SIM does not depend on any specific fluorophore properties, such as high photostability or particular transitions between orthogonal states, and can therefore be generally applied. A further modification of SIM, known as SSIM (saturated-SIM) exhibits higher spatial resolution but requires photostable samples (Gustafsson, 2005). As SIM enables multicolor 3D-imaging with standard fluorescent dyes, it has attracted considerable interest among biologists (Maglione and Sigrist, 2013).
In STED microscopy, the lateral resolution is improved by decreasing the size of the excitation point-spread-function (PSF) by stimulated emission of fluorophores at the rim of the PSF (Hell and Wichmann, 1994). Since the resolution enhancement in STED microscopy scales with the intensity of the depletion beam (Hell, 2007), only very photostable fluorophores allow spatial resolutions in the 30–50 nm range (Hell, 2007; Meyer et al., 2008). Nevertheless, STED has also been used for live-cell super-resolution imaging albeit at lower resolution (Nägerl et al., 2008; Tønnesen et al., 2014).
Single-molecule based localization microscopy techniques such as PALM, STORM and dSTORM rely on stochastic photoactivation, photoconversion, or photoswitching of fluorophores, such that only a small subset emits photons at any given time. By fitting a 2D Gaussian function to the PSF of individual, spatially isolated emitters, their positions can be precisely localized and used to reconstruct a super-resolved image, as long as all fluorophores determining the structure of interest have been detected and localized at least once during acquisition (Betzig et al., 2006; Hess et al., 2006; Rust et al., 2006; Heilemann et al., 2008). Localization microscopy methods differ in their use of fluorescent probes: PALM is conducted with genetically expressed photoactivatable fluorescent proteins (Betzig et al., 2006; Hess et al., 2006), STORM requires photoswitchable dye pairs (Rust et al., 2006) and dSTORM takes advantage of the reversible photoswitching of standard organic fluorophores in thiol-containing aqueous buffer (Heilemann et al., 2008; van de Linde et al., 2011). Since localization microscopy exhibits explicit single-molecule sensitivity, all approaches can deliver quantitative information on molecular distributions and even have the potential to report absolute numbers of proteins present in sub-cellular compartments (Sauer, 2013). These features provide insight into biological systems at a molecular level and can yield direct experimental feedback for modeling the complexity of biological interactions.
Functional Parameters of the AZ
Derived from the quantal hypothesis (Del Castillo and Katz, 1954) it is understood that synaptic strength, i.e., the amplitude of an excitatory postsynaptic current (EPSC), can be described by the product of three basic parameters: N, the number of fusion competent synaptic vesicles also termed readily-releasable vesicles (RRVs), p, their probability of exocytosis and q, usually taken to reflect postsynaptic sensitivity (Equation 1). This conceptual framework plays an important role in explaining synaptic function and plasticity (Zucker and Regehr, 2002), and identifies N and p as major functional determinants of the presynapse.
The parameter N can be estimated by electrophysiological means, such as high-frequency electrical stimulation or fluctuation analysis of synaptic responses (Clements and Silver, 2000). Results obtained by either approach must, however, be interpreted carefully, as additional factors complicate the analysis (Sakaba et al., 2002; Hallermann et al., 2010a). For example, asynchronous release, the kinetics of vesicle pool refilling (Hosoi et al., 2007) and postsynaptic contributions, such as receptor desensitization and saturation (Scheuss et al., 2002), can influence approximations of N. Hypertonic sucrose stimulation can be used as another technique to approximate N (Fatt and Katz, 1952; Rosenmund and Stevens, 1996). However, being independent of Ca2+-triggered fusion, it remains uncertain whether hypertonically released vesicles are generally also readily released under physiological conditions (Moulder and Mennerick, 2005).
Alternatively, N can be defined as the number of release sites, in which case p denotes the probability that a vesicle will fuse at a given release site (Schneggenburger et al., 2002). Nerve terminals vary greatly in size and correspondingly contain between one (e.g., at certain cortical synapses; Xu-Friedman et al., 2001) and many hundreds of AZs (e.g., at the Calyx of Held; Sätzler et al., 2002). It is therefore helpful to view the AZ as a fundamental unit of presynaptic function (Alabi and Tsien, 2012). That said, morphology and function of AZs are highly heterogeneous (Zhai and Bellen, 2004), also varying within one and the same neuron (Atwood and Karunanithi, 2002; Peled and Isacoff, 2011; Ehmann et al., 2014). Correspondingly, functional estimates of p at central mammalian synapses have reported both AZs operating with uniquantal release and AZs capable of multivesicular release (Tong and Jahr, 1994; Auger et al., 1998; Silver et al., 2003). To date, this next level of AZ organization has been difficult to study as specific molecular markers or structural correlates of release sites remain uncertain.
Functional estimates of p can be obtained with several methods that provide relative or absolute values. These include electro-physiology-based approaches such as paired-pulse stimulation or fluctuation analysis (Clements and Silver, 2000; Sakaba et al., 2002; Zucker and Regehr, 2002) and dynamic optical readouts of exocytosis or postsynaptic activation (Branco and Staras, 2009; Zhang et al., 2009; Peled and Isacoff, 2011; Marvin et al., 2013). Since p is highly Ca2+-dependent, its value for a given synaptic vesicle will be strongly influenced by the vesicle’s position relative to voltage-gated Ca2+ channels at the AZ (Neher, 1998; Eggermann et al., 2012).
Ca2+ channels are essential components of the macro-molecular exocytosis machinery. Their opening elicits Ca2+ influx, which serves as the fusion trigger for nearby vesicles. Early computational and functional studies introduced the concept of “microdomains” to describe transient, local regions of high Ca2+ concentration (Chad and Eckert, 1984; Llinás et al., 1992). Such microdomains possess complex spatial distributions of Ca2+ elevation, which are controlled by Ca2+ diffusion, Ca2+ buffering and the geometric arrangement of Ca2+ channels in the AZ membrane (Neher, 1998). Due to their major functional significance for synaptic transmission, detailed understanding of Ca2+ signals and the arrangement of synaptic vesicles relative to local domains is important. Using electrophysiology, modeling, Ca2+ imaging and Ca2+ uncaging, considerable quantitative information on excitation-secretion coupling has been obtained at the Calyx of Held, a large glutamatergic synapse in the mammalian auditory brainstem (Bollmann et al., 2000; Schneggenburger and Neher, 2005; Sun et al., 2007). At calyceal terminals, electrophysiology has even delivered direct functional readouts (Stanley, 1993) and estimates of AZ Ca2+ channel numbers (Sheng et al., 2012). Application of synthetic Ca2+ chelators with different binding rates [BAPTA (1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid) and EGTA (ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid)] can differentiate between very tight (“nanodomain”, <100 nm) and larger distance (“microdomain”, >100 nm) coupling regimes of synaptic vesicles and Ca2+ channels (Eggermann et al., 2012). By combining data from such investigations, the vesicle-Ca2+ channel topography has now been modeled at several mammalian central AZs (Meinrenken et al., 2002; Schmidt et al., 2013; Vyleta and Jonas, 2014). While it would be desirable to study the ultrastructural organization underlying coupling modes directly, information on the exact arrangement of Ca2+ channels derived from EM is sparse (Feeney et al., 1998; Holderith et al., 2012; Indriati et al., 2013). Conventional light microscopy, in turn, cannot measure the physical distance between channels and vesicles or resolve whether the Ca2+ signal is shaped by a single channel (Augustine et al., 1991; Stanley, 1993) or the superposition of multiple channels (Borst and Sakmann, 1996).
There appears to be no general map of synaptic vesicle and Ca2+ channel arrangements at the AZ. In fact, vesicle-channel coupling may differ significantly at AZs belonging to the same neuron (Rozov et al., 2001) and at single presynaptic terminals over time (Fedchyshyn and Wang, 2005; Erazo-Fischer et al., 2007; Wong et al., 2013). Before a synaptic vesicle becomes fusion competent, the release machinery must build up a primed state (Wojcik and Brose, 2007). In addition to such “molecular priming”, evidence also suggests that “positional priming”, i.e., moving primed vesicles closer to Ca2+ channels, can contribute to a heterogeneous p of RRVs (Neher and Sakaba, 2008). However, information on spatial relationships of AZ molecules in these distinct states has not yet been collected. Importantly, proteins which influence AZ function and plasticity by tightening vesicle-Ca2+ channel coupling have been identified in fly and mouse (Kittel et al., 2006; Yang et al., 2010). Investigating the organization of such key components relative to other AZ constituents should help to improve our mechanistic understanding of AZ structure-function relationships.
SRM of the AZ
Quantitative information on functional determinants of the AZ has mainly been derived from large, electrophysiologically accessible presynaptic terminals, such as the Calyx of Held (Forsythe, 1994; Meinrenken et al., 2002; Neher and Sakaba, 2008). While sophisticated electrophysiology has extended direct studies of transmitter release to smaller terminals (see e.g., Hallermann et al., 2003; Rancz et al., 2007; Bucurenciu et al., 2008), there remains an obvious demand for correlative structural information.
Here, SRM techniques can be expected to make a significant contribution. Several SRM studies, mostly conducted in cell culture, have provided indirect information on AZ function by analyzing the vesicle cycle. In one of its first biological applications, STED microscopy showed that the vesicular Ca2+ sensor Synaptotagmin remains clustered in isolated patches following exocytosis in cultured neurons (Willig et al., 2006). Subsequent work introduced live cell STED imaging to visualize synaptic vesicle movement between and within presynaptic boutons (Westphal et al., 2008), while multicolor imaging has been used to differentiate molecularly-defined synaptic vesicle pools at calyceal synapses in rat brain tissue (Kempf et al., 2013). Focussing on Syntaxin as a component of the vesicle fusion machinery, two independently conducted investigations using STED and dSTORM provided detailed information on its arrangement in clusters at the plasma membrane of PC12 cells (Sieber et al., 2007; Bar-On et al., 2012). Moreover, 3-D applications of STORM and PALM have been utilized to investigate vesicle endocytosis by Clathrin nanostructures in cultured cell lines (Jones et al., 2011; Sochacki et al., 2012).
Analysis of the AZ nanoarchitecture in tissue was first carried out with SRM by using STED at the Drosophila NMJ. Beginning with the identification of Bruchpilot (Brp) as a major component of the AZ cytomatrix (Kittel et al., 2006; Wagh et al., 2006), subsequent work described the polarized, elongated orientation of this large filamentous protein and resolved the organization of further AZ components, such as Ca2+ channels, Syd-1, Liprin-α and RIM binding protein (RBP) relative to the Brp hub (Fouquet et al., 2009; Owald et al., 2010; Liu et al., 2011). This has generated an increasingly detailed picture of the protein scaffold at Drosophila AZs (Maglione and Sigrist, 2013), which is currently being extended by photobleaching microscopy techniques (PiMP, photo-bleaching microscopy with nonlinear processing; Khuong et al., 2013) and SRM via dSTORM (Figure 1; Ehmann et al., 2014, Paul et al., submitted). In a separate effort, STORM was used to measure the axial positions of the AZ-specific proteins RIM1, Piccolo and Bassoon at synapses in mouse brain tissue (Dani et al., 2010). It is of obvious interest to compare such AZ topographies from different synapses, to identify conserved and specialized principles of organization and to test whether these are causally linked to functional diversity.
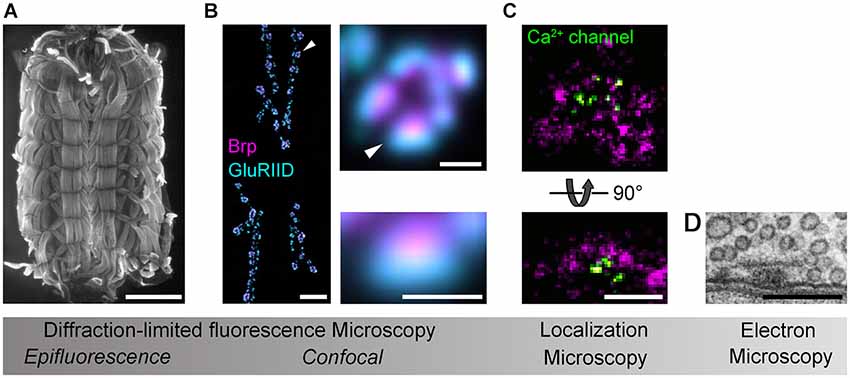
Figure 1. Imaging Drosophila neuromuscular AZs. Gradual increases in spatial resolution show (A) a Drosophila larval preparation imaged with epifluorescence microscopy (phalloidin staining); (B) a confocal image of the glutamatergic neuromuscular junction (left panel), a single bouton (upper panel) and an individual synapse (lower panel) stained against the AZ protein Brp (magenta) and the postsynaptic glutamate receptor subunit GluRIID (cyan; arrowheads indicate enlarged regions); (C) dSTORM images of AZs stained against Brp (C-terminal epitope, magenta) and Ca2+ channels (nanobody recognizing a GFP-tagged α1-subunit, CacGFP; Kawasaki et al., 2004) viewed en face (optical axis perpendicular to AZ membrane, upper panel) and from the side (optical axis parallel to AZ membrane, lower panel; cf. D); (D) an electron micrograph of the AZ cytomatrix and opposed pre- and postsynaptic membranes. Electron micrograph kindly provided by C. Wichmann and S.J. Sigrist. Scale bars: (A) 1 mm; (B) 10µm (NMJ), 1µm (bouton), 500 nm (synapse); (C,D) 200 nm.
Extending beyond descriptive ultrastructural studies, microscopy can contribute to identifying structural correlates of synaptic function (Wojtowicz et al., 1994). Considering their fundamental impact on neurotransmission there has thus been a long standing motivation to resolve the nanoscopic organization of Ca2+ channels at the AZ. However, to date little direct information has been collected on their ultrastructural distribution (Haydon et al., 1994; Feeney et al., 1998; Holderith et al., 2012; Indriati et al., 2013). Notably, a recent study at hippocampal neurons elegantly combined Ca2+ imaging with EM to estimate the number of Ca2+ channels contributing to one microdomain and to identify a close correlation between the number of docked vesicles, AZ area and p (Holderith et al., 2012). Combining STED with molecular manipulations and electrophysiology has identified functional roles of the AZ proteins Brp and RBP in the recruitment and spatial arrangement of Ca2+ channels to promote p at the Drosophila AZ (Kittel et al., 2006; Hallermann et al., 2010b; Liu et al., 2011). Moreover, dynamic reorganizations of Brp accompany rapid AZ strengthening and increase the number of release sites during homeostatic synaptic plasticity (Weyhersmüller et al., 2011). Similarly, studies at mammalian hair cell synapses have demonstrated a role of the AZ protein Bassoon, functionally related to Brp (Hallermann and Silver, 2013), in shaping Ca2+ channel arrangement and establishing release sites (Frank et al., 2010).
Despite the high spatial resolution provided by SRM, estimates of protein abundance are mainly obtained from fluorescence intensity measurements and therefore deliver only relative values. However, quantitative information on endogenous protein copies, in addition to their spatial organization, is required for a comprehensive mechanistic understanding of AZ structure-function relationships. While stepwise photobleaching can be used to count low protein numbers (Ulbrich and Isacoff, 2007) the densely packed protein assembly at the AZ requires alternative methods. Several recent reports have addressed this issue.
Wilhelm et al. combined quantitative biochemistry with EM and STED to estimate average protein copies and to localize these to specific sub-cellular regions of biochemically isolated presynaptic terminals (Wilhelm et al., 2014). This approach has delivered a wealth of quantitative information on presynaptic proteins. However, it does not connect structural features with functional properties at the single synapse level.
Since localization microscopy is an explicit single-molecule imaging technique, it can be used to obtain quantitative information on both the spatial distribution and the copy number of labeled proteins in situ, as long as antibody binding features (e.g., in dSTORM) or fluorescent protein expression and folding properties (as e.g., in PALM) are taken into account. By engaging dSTORM, this principle was recently utilized to study the nanoscopic arrangement of endogenous Brp proteins at AZs in tissue (Ehmann et al., 2014). The results provided an estimate of the number of Brp copies per AZ and were correlated with electrophysiological features to offer an interpretation of how the protein’s organization is linked to AZ function.
These current developments open up new perspectives for clarifying how functional properties are encoded in the protein architecture of AZs. Logical next steps could include searching for molecular determinants of vesicle release sites and quantitative ultrastructural studies of Ca2+ channel-vesicle topographies.
Outlook
Despite a gradually emerging comprehensive protein catalog, we still lack basic information describing how the nanoscopic organization of proteins at the AZ gives rise to neurotransmission. Arguably, this is due to the diffraction-limited resolution of conventional light microscopy, which has hindered access to the spatial nanodomain in a physiologically relevant experimental setting.
Several SRM techniques now exist that have the capacity to localize proteins on the nanometer scale and to resolve components of macromolecular assemblies in their native environment. In this context, we believe that localization microscopy is of particular value, as it can be used to provide direct access to molecular coordinates and to count endogenous protein epitopes (Specht et al., 2013; Andreska et al., 2014; Ehmann et al., 2014). We expect that combining such quantitative information on protein organization with results from electrophysiology will contribute to a better understanding of the molecular mechanisms controlling AZ function. In addition, other correlative approaches, such as pairing SRM with biochemistry (Wilhelm et al., 2014), EM (Watanabe et al., 2011; Löschberger et al., 2014) and array tomography (Nanguneri et al., 2012) hold great promise for uncovering multiprotein architectures.
Harnessing the full potential of SRM will require expanding the repertoire of robust test samples and introducing optimized analytical tools (Bar-On et al., 2012). Likewise, small fluorescent probes with both efficient and specific binding properties will have to be developed to allow for simultaneous visualization of multiple targets in their native settings (Sauer, 2013). As already common practice in EM, users of SRM have to accept that fluorophores, labeling protocols and sample preparations need to be optimized for each new target molecule under investigation.
Dynamic, live-cell SRM remains challenging. As a rule of thumb, spatial resolution always comes at the cost of temporal resolution. Therefore, imaging complex structures, such as the cytoskeleton of a whole cell, requires several minutes acquisition time at a lateral resolution of about 20 nm. This clearly limits the obtainable dynamic information. In contrast, modified SIM can easily resolve the movement of microtubules in entire living cells, albeit at lower spatial resolution (Chen et al., 2014). Hence, future efforts will have to optimize the trade-off between imaging area, temporal information and spatial resolution in order to monitor dynamic protein re-arrangements at the AZ directly. In principle, fluorescent protein-based SRM techniques offer the possibility of in vivo imaging in fully intact organisms. However, the feasibility of such applications must take into account light scattering and aberration in biological tissue, less amenable photophysical properties of fluorescent proteins compared with organic fluorophores and possible physiological alterations induced by recombinant protein expression (Sauer, 2013).
Despite its capacity to resolve multiprotein structures, so far relatively few studies have engaged SRM to study synaptic AZs. We anticipate that this situation will change as SRM techniques become increasingly available and affordable (Holm et al., 2014). Progress in efficient and stoichiometric labeling of endogenous proteins, together with the development of sample preparations that accurately preserve the molecular details of interest, will further advance SRM to shed light on the AZ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank M. Heckmann and T. Langenhan for discussions, C. Wichmann and S. J. Sigrist for providing the electron micrograph in figure panel D and L. Pließ for technical support. This work was supported by grants from the German Research foundation (KI 1460/1-1 and SFB 1047/A05 to Robert J. Kittel), a fellowship from the GSLS, University of Würzburg (Nadine Ehmann), the Biophotonics Initiative of the German Ministry of Research and Education (BMBF/Grants #13N11019 and #13N12507 to Markus Sauer), and the German-Israeli Foundation (GIF/Grant #1125-145.1/2010 to Markus Sauer).
References
Abbe, E. (1873). Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch. Mikroskopische Anat. 9, 456–468. doi: 10.1007/bf02956176
Alabi, A. A., and Tsien, R. W. (2012). Synaptic vesicle pools and dynamics. Cold Spring Harb. Perspect. Biol. 4:a013680. doi: 10.1101/cshperspect.a013680
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andreska, T., Aufmkolk, S., Sauer, M., and Blum, R. (2014). High abundance of BDNF within glutamatergic presynapses of cultured hippocampal neurons. Front. Cell. Neurosci. 8:107. doi: 10.3389/fncel.2014.00107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Atwood, H. L., and Karunanithi, S. (2002). Diversification of synaptic strength: presynaptic elements. Nat. Rev. Neurosci. 3, 497–516. doi: 10.1038/nrn876
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Auger, C., Kondo, S., and Marty, A. (1998). Multivesicular release at single functional synaptic sites in cerebellar stellate and basket cells. J. Neurosci. 18, 4532–4547.
Augustine, G. J., Adler, E. M., and Charlton, M. P. (1991). The calcium signal for transmitter secretion from presynaptic nerve terminals. Ann. N Y Acad. Sci. 635, 365–381. doi: 10.1111/j.1749-6632.1991.tb36505.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bar-On, D., Wolter, S., van de Linde, S., Heilemann, M., Nudelman, G., Nachliel, E., et al. (2012). Super-resolution imaging reveals the internal architecture of nano-sized syntaxin clusters. J. Biol. Chem. 287, 27158–27167. doi: 10.1074/jbc.M112.353250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., et al. (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645. doi: 10.1126/science.1127344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bollmann, J. H., Sakmann, B., and Borst, J. G. (2000). Calcium sensitivity of glutamate release in a calyx-type terminal. Science 289, 953–957. doi: 10.1126/science.289.5481.953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Borst, J. G., and Sakmann, B. (1996). Calcium influx and transmitter release in a fast CNS synapse. Nature 383, 431–434. doi: 10.1038/383431a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Branco, T., and Staras, K. (2009). The probability of neurotransmitter release: variability and feedback control at single synapses. Nat. Rev. Neurosci. 10, 373–383. doi: 10.1038/nrn2634
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bucurenciu, I., Kulik, A., Schwaller, B., Frotscher, M., and Jonas, P. (2008). Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron 57, 536–545. doi: 10.1016/j.neuron.2007.12.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castillo, P. E., Schoch, S., Schmitz, F., Südhof, T. C., and Malenka, R. C. (2002). RIM1α is required for presynaptic long-term potentiation. Nature 415, 327–330. doi: 10.1038/415327a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chad, J. E., and Eckert, R. (1984). Calcium domains associated with individual channels can account for anomalous voltage relations of CA-dependent responses. Biophys. J. 45, 993–999. doi: 10.1016/s0006-3495(84)84244-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, B.-C., Legant, W. R., Wang, K., Shao, L., Milkie, D. E., Davidson, M. W., et al. (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 346:1257998. doi: 10.1126/science.1257998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clements, J. D., and Silver, R. A. (2000). Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 23, 105–113. doi: 10.1016/s0166-2236(99)01520-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dani, A., Huang, B., Bergan, J., Dulac, C., and Zhuang, X. (2010). Superresolution imaging of chemical synapses in the brain. Neuron 68, 843–856. doi: 10.1016/j.neuron.2010.11.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Castillo, J., and Katz, B. (1954). Quantal components of the end-plate potential. J. Physiol. 124, 560–573. doi: 10.1113/jphysiol.1954.sp005129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eggermann, E., Bucurenciu, I., Goswami, S. P., and Jonas, P. (2012). Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci. 13, 7–21. doi: 10.1038/nrn3125
Ehmann, N., van de Linde, S., Alon, A., Ljaschenko, D., Keung, X. Z., Holm, T., et al. (2014). Quantitative super-resolution imaging of Bruchpilot distinguishes active zone states. Nat. Commun. 5:4650. doi: 10.1038/ncomms5650
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Erazo-Fischer, E., Striessnig, J., and Taschenberger, H. (2007). The role of physiological afferent nerve activity during in vivo maturation of the calyx of held synapse. J. Neurosci. 27, 1725–1737. doi: 10.1523/jneurosci.4116-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fatt, P., and Katz, B. (1952). Spontaneous subthreshold activity at motor nerve endings. J. Physiol. 117, 109–128.
Fedchyshyn, M. J., and Wang, L.-Y. (2005). Developmental transformation of the release modality at the calyx of held synapse. J. Neurosci. 25, 4131–4140. doi: 10.1523/jneurosci.0350-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feeney, C. J., Karunanithi, S., Pearce, J., Govind, C. K., and Atwood, H. L. (1998). Motor nerve terminals on abdominal muscles in larval flesh flies, Sarcophaga bullata: comparisons with Drosophila. J. Comp. Neurol. 402, 197–209. doi: 10.1002/(sici)1096-9861(19981214)402:2<197::aid-cne5>3.3.co;2-h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernández-Busnadiego, R., Asano, S., Oprisoreanu, A.-M., Sakata, E., Doengi, M., Kochovski, Z., et al. (2013). Cryo-electron tomography reveals a critical role of RIM1α in synaptic vesicle tethering. J. Cell Biol. 201, 725–740. doi: 10.1083/jcb.201206063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forsythe, I. D. (1994). Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. J. Physiol. 479, 381–387. doi: 10.1113/jphysiol.1994.sp020303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fouquet, W., Owald, D., Wichmann, C., Mertel, S., Depner, H., Dyba, M., et al. (2009). Maturation of active zone assembly by Drosophila Bruchpilot. J. Cell Biol. 186, 129–145. doi: 10.1083/jcb.200812150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frank, T., Rutherford, M. A., Strenzke, N., Neef, A., Pangršič, T., Khimich, D., et al. (2010). Bassoon and the synaptic ribbon organize Ca2+ channels and vesicles to add release sites and promote refilling. Neuron 68, 724–738. doi: 10.1016/j.neuron.2010.10.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galbraith, C. G., and Galbraith, J. A. (2011). Super-resolution microscopy at a glance. J. Cell Sci. 124, 1607–1611. doi: 10.1242/jcs.080085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gustafsson, M. G. L. (2000). Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 198, 82–87. doi: 10.1046/j.1365-2818.2000.00710.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gustafsson, M. G. L. (2005). Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. U S A 102, 13081–13086. doi: 10.1073/pnas.0406877102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hallermann, S., Heckmann, M., and Kittel, R. J. (2010a). Mechanisms of short-term plasticity at neuromuscular active zones of Drosophila. HFSP J. 4, 72–84. doi: 10.2976/1.3338710
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hallermann, S., Kittel, R. J., Wichmann, C., Weyhersmüller, A., Fouquet, W., Mertel, S., et al. (2010b). Naked dense bodies provoke depression. J. Neurosci. 30, 14340–14345. doi: 10.1523/JNEUROSCI.2495-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hallermann, S., Pawlu, C., Jonas, P., and Heckmann, M. (2003). A large pool of releasable vesicles in a cortical glutamatergic synapse. Proc. Natl. Acad. Sci. U S A 100, 8975–8980. doi: 10.1073/pnas.1432836100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hallermann, S., and Silver, R. A. (2013). Sustaining rapid vesicular release at active zones: potential roles for vesicle tethering. Trends Neurosci. 36, 185–194. doi: 10.1016/j.tins.2012.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harlow, M. L., Ress, D., Stoschek, A., Marshall, R. M., and McMahan, U. J. (2001). The architecture of active zone material at the frog’s neuromuscular junction. Nature 409, 479–484. doi: 10.1038/35054000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haydon, P. G., Henderson, E., and Stanley, E. F. (1994). Localization of individual calcium channels at the release face of a presynaptic nerve terminal. Neuron 13, 1275–1280. doi: 10.1016/0896-6273(94)90414-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heilemann, M., van de Linde, S., Schüttpelz, M., Kasper, R., Seefeldt, B., Mukherjee, A., et al. (2008). Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed Engl. 47, 6172–6176. doi: 10.1002/anie.200802376
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heintzmann, R., and Cremer, C. G. (1999). Laterally modulated excitation microscopy: improvement of resolution by using a diffraction grating. Proc. SPIE 3568, 185–196. doi: 10.1117/12.336833
Hell, S. W. (2007). Far-field optical nanoscopy. Science 316, 1153–1158. doi: 10.1126/science.1137395
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hell, S. W. (2009). Microscopy and its focal switch. Nat. Methods 6, 24–32. doi: 10.1038/nmeth.1291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hell, S. W., and Wichmann, J. (1994). Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782. doi: 10.1364/ol.19.000780
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hess, S. T., Girirajan, T. P. K., and Mason, M. D. (2006). Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 91, 4258–4272. doi: 10.1529/biophysj.106.091116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holderith, N., Lorincz, A., Katona, G., Rózsa, B., Kulik, A., Watanabe, M., et al. (2012). Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nat. Neurosci. 15, 988–997. doi: 10.1038/nn.3137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holm, T., Klein, T., Löschberger, A., Klamp, T., Wiebusch, G., van de Linde, S., et al. (2014). A blueprint for cost-efficient localization microscopy. Chemphyschem 15, 651–654. doi: 10.1002/cphc.201300739
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hosoi, N., Sakaba, T., and Neher, E. (2007). Quantitative analysis of calcium-dependent vesicle recruitment and its functional role at the calyx of held synapse. J. Neurosci. 27, 14286–14298. doi: 10.1523/jneurosci.4122-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Indriati, D. W., Kamasawa, N., Matsui, K., Meredith, A. L., Watanabe, M., and Shigemoto, R. (2013). Quantitative localization of Cav2.1 (P/Q-type) voltage-dependent calcium channels in Purkinje cells: somatodendritic gradient and distinct somatic coclustering with calcium-activated potassium channels. J. Neurosci. 33, 3668–3678. doi: 10.1523/JNEUROSCI.2921-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jahn, R., and Fasshauer, D. (2012). Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207. doi: 10.1038/nature11320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiao, W., Masich, S., Franzén, O., and Shupliakov, O. (2010). Two pools of vesicles associated with the presynaptic cytosolic projection in Drosophila neuromuscular junctions. J. Struct. Biol. 172, 389–394. doi: 10.1016/j.jsb.2010.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, S. A., Shim, S.-H., He, J., and Zhuang, X. (2011). Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods 8, 499–508. doi: 10.1038/nmeth.1605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawasaki, F., Zou, B., Xu, X., and Ordway, R. W. (2004). Active zone localization of presynaptic calcium channels encoded by the cacophony locus of Drosophila. J. Neurosci. 24, 282–285. doi: 10.1523/jneurosci.3553-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kempf, C., Staudt, T., Bingen, P., Horstmann, H., Engelhardt, J., Hell, S. W., et al. (2013). Tissue multicolor STED nanoscopy of presynaptic proteins in the calyx of held. PLoS One 8:e62893. doi: 10.1371/journal.pone.0062893
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khuong, T. M., Habets, R. L. P., Kuenen, S., Witkowska, A., Kasprowicz, J., Swerts, J., et al. (2013). Synaptic PI(3,4,5)P3 is required for Syntaxin1A clustering and neurotransmitter release. Neuron 77, 1097–1108. doi: 10.1016/j.neuron.2013.01.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kittel, R. J., Wichmann, C., Rasse, T. M., Fouquet, W., Schmidt, M., Schmid, A., et al. (2006). Bruchpilot promotes active zone assembly, Ca2+ channel clustering and vesicle release. Science 312, 1051–1054. doi: 10.1126/science.1126308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Landis, D. M., Hall, A. K., Weinstein, L. A., and Reese, T. S. (1988). The organization of cytoplasm at the presynaptic active zone of a central nervous system synapse. Neuron 1, 201–209. doi: 10.1016/0896-6273(88)90140-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lichtman, J. W., and Conchello, J.-A. (2005). Fluorescence microscopy. Nat. Methods 2, 910–919. doi: 10.1038/nmeth817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Limbach, C., Laue, M. M., Wang, X., Hu, B., Thiede, N., Hultqvist, G., et al. (2011). Molecular in situ topology of Aczonin/Piccolo and associated proteins at the mammalian neurotransmitter release site. Proc. Natl. Acad. Sci. U S A 108, E392–E401. doi: 10.1073/pnas.1101707108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, K. S. Y., Siebert, M., Mertel, S., Knoche, E., Wegener, S., Wichmann, C., et al. (2011). RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science 334, 1565–1569. doi: 10.1126/science.1212991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Llinás, R., Sugimori, M., and Silver, R. B. (1992). Microdomains of high calcium concentration in a presynaptic terminal. Science 256, 677–679. doi: 10.1126/science.1350109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Löschberger, A., Franke, C., Krohne, G., van de Linde, S., and Sauer, M. (2014). Correlative super-resolution fluorescence and electron microscopy of the nuclear pore complex with molecular resolution. J. Cell Sci. 127, 4351–4355. doi: 10.1242/jcs.156620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maglione, M., and Sigrist, S. J. (2013). Seeing the forest tree by tree: super-resolution light microscopy meets the neurosciences. Nat. Neurosci. 16, 790–797. doi: 10.1038/nn.3403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marvin, J. S., Borghuis, B. G., Tian, L., Cichon, J., Harnett, M. T., Akerboom, J., et al. (2013). An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 10, 162–170. doi: 10.1038/nmeth.2333
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matz, J., Gilyan, A., Kolar, A., McCarvill, T., and Krueger, S. R. (2010). Rapid structural alterations of the active zone lead to sustained changes in neurotransmitter release. Proc. Natl. Acad. Sci. U S A 107, 8836–8841. doi: 10.1073/pnas.0906087107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meinrenken, C. J., Borst, J. G. G., and Sakmann, B. (2002). Calcium secretion coupling at calyx of held governed by nonuniform channel-vesicle topography. J. Neurosci. 22, 1648–1667.
Meyer, L., Wildanger, D., Medda, R., Punge, A., Rizzoli, S. O., Donnert, G., et al. (2008). Dual-color STED microscopy at 30-nm focal-plane resolution. Small 4, 1095–1100. doi: 10.1002/smll.200800055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moulder, K. L., and Mennerick, S. (2005). Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J. Neurosci. 25, 3842–3850. doi: 10.1523/jneurosci.5231-04.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nägerl, U. V., Willig, K. I., Hein, B., Hell, S. W., and Bonhoeffer, T. (2008). Live-cell imaging of dendritic spines by STED microscopy. Proc. Natl. Acad. Sci. U S A 105, 18982–18987. doi: 10.1073/pnas.0810028105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nanguneri, S., Flottmann, B., Horstmann, H., Heilemann, M., and Kuner, T. (2012). Three-dimensional, tomographic super-resolution fluorescence imaging of serially sectioned thick samples. PLoS One 7:e38098. doi: 10.1371/journal.pone.0038098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neher, E. (1998). Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20, 389–399. doi: 10.1016/s0896-6273(00)80983-6
Neher, E., and Sakaba, T. (2008). Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59, 861–872. doi: 10.1016/j.neuron.2008.08.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Owald, D., Fouquet, W., Schmidt, M., Wichmann, C., Mertel, S., Depner, H., et al. (2010). A Syd-1 homologue regulates pre- and postsynaptic maturation in Drosophila. J. Cell Biol. 188, 565–579. doi: 10.1083/jcb.200908055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Owald, D., and Sigrist, S. J. (2009). Assembling the presynaptic active zone. Curr. Opin. Neurobiol. 19, 311–318. doi: 10.1016/j.conb.2009.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patterson, G., Davidson, M., Manley, S., and Lippincott-Schwartz, J. (2010). Superresolution imaging using single-molecule localization. Annu. Rev. Phys. Chem. 61, 345–367. doi: 10.1146/annurev.physchem.012809.103444
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peled, E. S., and Isacoff, E. Y. (2011). Optical quantal analysis of synaptic transmission in wild-type and rab3-mutant Drosophila motor axons. Nat. Neurosci. 14, 519–526. doi: 10.1038/nn.2767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfenninger, K., Akert, K., Moor, H., and Sandri, C. (1972). The fine structure of freeze-fractured presynaptic membranes. J. Neurocytol. 1, 129–149. doi: 10.1007/bf01099180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Phillips, G. R., Huang, J. K., Wang, Y., Tanaka, H., Shapiro, L., Zhang, W., et al. (2001). The presynaptic particle web: ultrastructure, composition, dissolution and reconstitution. Neuron 32, 63–77. doi: 10.1016/S0896-6273(01)00450-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rancz, E. A., Ishikawa, T., Duguid, I., Chadderton, P., Mahon, S., and Häusser, M. (2007). High-fidelity transmission of sensory information by single cerebellar mossy fibre boutons. Nature 450, 1245–1248. doi: 10.1038/nature05995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosenmund, C., and Stevens, C. F. (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16, 1197–1207. doi: 10.1016/s0896-6273(00)80146-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rozov, A., Burnashev, N., Sakmann, B., and Neher, E. (2001). Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J. Physiol. 531, 807–826. doi: 10.1111/j.1469-7793.2001.0807h.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rust, M. J., Bates, M., and Zhuang, X. (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795. doi: 10.1038/nmeth929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakaba, T., Schneggenburger, R., and Neher, E. (2002). Estimation of quantal parameters at the calyx of held synapse. Neurosci. Res. 44, 343–356. doi: 10.1016/s0168-0102(02)00174-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sätzler, K., Söhl, L. F., Bollmann, J. H., Borst, J. G. G., Frotscher, M., Sakmann, B., et al. (2002). Three-dimensional reconstruction of a calyx of held and its postsynaptic principal neuron in the medial nucleus of the trapezoid body. J. Neurosci. 22, 10567–10579.
Sauer, M. (2013). Localization microscopy coming of age: from concepts to biological impact. J. Cell Sci. 126, 3505–3513. doi: 10.1242/jcs.123612
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schermelleh, L., Heintzmann, R., and Leonhardt, H. (2010). A guide to super-resolution fluorescence microscopy. J. Cell Biol. 190, 165–175. doi: 10.1083/jcb.201002018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scheuss, V., Schneggenburger, R., and Neher, E. (2002). Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of held synapse. J. Neurosci. 22, 728–739.
Schmidt, H., Brachtendorf, S., Arendt, O., Hallermann, S., Ishiyama, S., Bornschein, G., et al. (2013). Nanodomain coupling at an excitatory cortical synapse. Curr. Biol. 23, 244–249. doi: 10.1016/j.cub.2012.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneggenburger, R., and Neher, E. (2005). Presynaptic calcium and control of vesicle fusion. Curr. Opin. Neurobiol. 15, 266–274. doi: 10.1016/j.conb.2005.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneggenburger, R., Sakaba, T., and Neher, E. (2002). Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 25, 206–212. doi: 10.1016/s0166-2236(02)02139-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sheng, J., He, L., Zheng, H., Xue, L., Luo, F., Shin, W., et al. (2012). Calcium-channel number critically influences synaptic strength and plasticity at the active zone. Nat. Neurosci. 15, 998–1006. doi: 10.1038/nn.3129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sieber, J. J., Willig, K. I., Kutzner, C., Gerding-Reimers, C., Harke, B., Donnert, G., et al. (2007). Anatomy and dynamics of a supramolecular membrane protein cluster. Science 317, 1072–1076. doi: 10.1126/science.1141727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Siksou, L., Rostaing, P., Lechaire, J.-P., Boudier, T., Ohtsuka, T., Fejtová, A., et al. (2007). Three-dimensional architecture of presynaptic terminal cytomatrix. J. Neurosci. 27, 6868–6877. doi: 10.1523/jneurosci.1773-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Silver, R. A., Lubke, J., Sakmann, B., and Feldmeyer, D. (2003). High-probability uniquantal transmission at excitatory synapses in barrel cortex. Science 302, 1981–1984. doi: 10.1126/science.1087160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sochacki, K. A., Larson, B. T., Sengupta, D. C., Daniels, M. P., Shtengel, G., Hess, H. F., et al. (2012). Imaging the post-fusion release and capture of a vesicle membrane protein. Nat. Commun. 3:1154. doi: 10.1038/ncomms2158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Specht, C. G., Izeddin, I., Rodriguez, P. C., El Beheiry, M., Rostaing, P., Darzacq, X., et al. (2013). Quantitative nanoscopy of inhibitory synapses: counting gephyrin molecules and receptor binding sites. Neuron 79, 308–321. doi: 10.1016/j.neuron.2013.05.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stanley, E. F. (1993). Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron 11, 1007–1011. doi: 10.1016/0896-6273(93)90214-c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Südhof, T. C. (2012). The presynaptic active zone. Neuron 75, 11–25. doi: 10.1016/j.neuron.2012.06.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, J., Pang, Z. P., Qin, D., Fahim, A. T., Adachi, R., and Südhof, T. C. (2007). A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450, 676–682. doi: 10.1038/nature06308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szule, J. A., Harlow, M. L., Jung, J. H., De-Miguel, F. F., Marshall, R. M., and McMahan, U. J. (2012). Regulation of synaptic vesicle docking by different classes of macromolecules in active zone material. PLoS One 7:e33333. doi: 10.1371/journal.pone.0033333
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tong, G., and Jahr, C. E. (1994). Multivesicular release from excitatory synapses of cultered hippocampal neurons. Neuron 12, 51–59. doi: 10.1016/0896-6273(94)90151-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tønnesen, J., Katona, G., Rózsa, B., and Nägerl, U. V. (2014). Spine neck plasticity regulates compartmentalization of synapses. Nat. Neurosci. 17, 678–685. doi: 10.1038/nn.3682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ulbrich, M. H., and Isacoff, E. Y. (2007). Subunit counting in membrane-bound proteins. Nat. Methods 4, 319–321. doi: 10.1038/nmeth1024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van de Linde, S., Löschberger, A., Klein, T., Heidbreder, M., Wolter, S., Heilemann, M., et al. (2011). Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 6, 991–1009. doi: 10.1038/nprot.2011.336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vyleta, N. P., and Jonas, P. (2014). Loose coupling between Ca2+ channels and release sensors at a plastic hippocampal synapse. Science 343, 665–670. doi: 10.1126/science.1244811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagh, D. A., Rasse, T. M., Asan, E., Hofbauer, A., Schwenkert, I., Dürrbeck, H., et al. (2006). Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron 49, 833–844. doi: 10.1016/j.neuron.2006.02.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watanabe, S., Punge, A., Hollopeter, G., Willig, K. I., Hobson, R. J., Davis, M. W., et al. (2011). Protein localization in electron micrographs using fluorescence nanoscopy. Nat. Methods 8, 80–84. doi: 10.1038/nmeth.1537
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westphal, V., Rizzoli, S. O., Lauterbach, M. A., Kamin, D., Jahn, R., and Hell, S. W. (2008). Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science 320, 246–249. doi: 10.1126/science.1154228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weyhersmüller, A., Hallermann, S., Wagner, N., and Eilers, J. (2011). Rapid active zone remodeling during synaptic plasticity. J. Neurosci. 31, 6041–6052. doi: 10.1523/JNEUROSCI.6698-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wichmann, C., and Sigrist, S. J. (2010). The active zone T-bar–a plasticity module? J. Neurogenet. 24, 133–145. doi: 10.3109/01677063.2010.489626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wilhelm, B. G., Mandad, S., Truckenbrodt, S., Kröhnert, K., Schäfer, C., Rammner, B., et al. (2014). Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344, 1023–1028. doi: 10.1126/science.1252884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Willig, K. I., Rizzoli, S. O., Westphal, V., Jahn, R., and Hell, S. W. (2006). STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature 440, 935–939. doi: 10.1038/nature04592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wojcik, S. M., and Brose, N. (2007). Regulation of membrane fusion in synaptic excitation-secretion coupling: speed and accuracy matter. Neuron 55, 11–24. doi: 10.1016/j.neuron.2007.06.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wojtowicz, J. M., Marin, L., and Atwood, H. L. (1994). Activity-induced changes in synaptic release sites at the crayfish neuromuscular junction. J. Neurosci. 14, 3688–3703.
Wong, A. B., Jing, Z., Rutherford, M. A., Frank, T., Strenzke, N., and Moser, T. (2013). Concurrent maturation of inner hair cell synaptic Ca2+ influx and auditory nerve spontaneous activity around hearing onset in mice. J. Neurosci. 33, 10661–10666. doi: 10.1523/JNEUROSCI.1215-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu-Friedman, M. A., Harris, K. M., and Regehr, W. G. (2001). Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J. Neurosci. 21, 6666–6672.
Yang, X., Kaeser-Woo, Y. J., Pang, Z. P., Xu, W., and Südhof, T. C. (2010). Complexin clamps asynchronous release by blocking a secondary Ca2+ sensor via its accessory α helix. Neuron 68, 907–920. doi: 10.1016/j.neuron.2010.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhai, R. G., and Bellen, H. J. (2004). The architecture of the active zone in the presynaptic nerve terminal. Physiology (Bethesda) 19, 262–270. doi: 10.1152/physiol.00014.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Q., Li, Y., and Tsien, R. (2009). The dynamic control of kiss-and-run and vesicular reuse probed with single nanoparticles. Science 323, 1448–1453. doi: 10.1126/science.1167373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zucker, R. S., and Regehr, W. G. (2002). Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405. doi: 10.1146/annurev.physiol.64.092501.114547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: active zone, super-resolution microscopy, excitation-secretion coupling, structure-function relationships, Ca2+ channels
Citation: Ehmann N, Sauer M and Kittel RJ (2015) Super-resolution microscopy of the synaptic active zone. Front. Cell. Neurosci. 9:7. doi: 10.3389/fncel.2015.00007
Received: 30 September 2014; Accepted: 07 January 2015;
Published online: 30 January 2015.
Edited by:
Hartmut Schmidt, University of Leipzig, GermanyReviewed by:
Ania K. Majewska, University of Rochester, USAValentin Nägerl, University of Bordeaux, France
Copyright © 2015 Ehmann, Sauer and Kittel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Sauer, Department of Biotechnology and Biophysics, University of Würzburg, Am Hubland, 97074 Würzburg, Germany e-mail:bS5zYXVlckB1bmktd3VlcnpidXJnLmRl;
Robert J. Kittel, Department of Neurophysiology, Institute of Physiology, University of Würzburg, Röntgenring 9, 97070 Würzburg, Germany e-mail:cm9iZXJ0LmtpdHRlbEB1bmktd3VlcnpidXJnLmRl