 Bastien Paré
Bastien Paré Lydia T. Deschênes
Lydia T. Deschênes Roxane Pouliot
Roxane Pouliot Nicolas Dupré
Nicolas Dupré Francois Gros-Louis
Francois Gros-Louis- 1Division of Regenerative Medicine, Laval University Experimental Organogenesis Research Center/LOEX, CHU de Québec Research Center – Enfant-Jésus Hospital, Québec, QC, Canada
- 2Department of Surgery, Faculty of Medicine, Laval University, Québec, QC, Canada
- 3Faculty of Pharmacy, Laval University, Québec, QC, Canada
- 4Neuroscience Division of the CHU de Québec, Department of Medicine of the Faculty of Medicine, Laval University, Québec, QC, Canada
The proteins secreted by a particular type of cell, the secretome, play important roles in the regulation of many physiological processes via paracrine/autocrine mechanisms, and they are of increasing interest to help understanding rare diseases and to identify potential biomarkers and therapeutic targets. To facilitate ongoing research involving secreted proteins, we revisited cell culture protocols and whole secreted protein enrichment protocols. A reliable method for culturing and precipitating secreted protein from patient-derived fibroblast conditioned-medium was established. The method is based on the optimization of cell confluency and incubation time conditions. The well-established carrier-based TCA-DOC protein precipitation method was consistently found to give higher protein recovery yield. According to our results, we therefore propose that protein enrichment should be performed by TCA-DOC precipitation method after 48 h at 95% of confluence in a serum-deprived culture medium. Given the importance of secreted proteins as a source to elucidate the pathogenesis of rare diseases, especially neurological disorders, this approach may help to discover novel candidate biomarkers with potential clinical significance.
Introduction
The secretome, a complex set of molecules secreted from living cells, represents a major class of proteins. It is considered to be encoded by ~10% of the human genome (Pavlou and Diamandis, 2010). It is well known that secreted proteins are associated with a broad range of physiological processes and play a key role in homeostasis, growth, differentiation, proteolysis, apoptosis, signaling, cell adhesion, and development of the extracellular matrix (ECM; Luo et al., 2012; Stastna and Van Eyk, 2012). Furthermore, the proteins secreted by particular types of cells are of increasing interest as a potential source of biomarker discoveries and therapeutic targets in diseases. Although the proteins released by cells into conditioned-media in vitro have been studied abundantly to better understand pathological conditions and mechanisms, the concept of the secretome has become an attractive and challenging proteomic technology in recent years.
There has been an increasing number of studies over the last decade targeting the secretome characterization, spanning a wide range of biological samples from microorganisms and lower animal models to human embryos and cell lines leading to the generation of a valuable secretome catalogs (Leroy et al., 2006; Klee, 2008; Katz-Jaffe et al., 2009; Brown et al., 2013; Gelman et al., 2013; Hartwig et al., 2013; Makridakis et al., 2013; Meissner et al., 2013). These efforts have been greatly facilitated by significant technological advances in the field of proteomics during the last two decades [improvement of proteomic platforms, mass spectrometry instrumentation and 2D-difference gel electrophoresis (2D-DIGE); Klose, 1975; O'Farrell, 1975; Beckett, 2012; Mukherjee and Mani, 2013]. The 2D-GE technique, introduced in 1976, enables protein separation according to their isoelectric point (pI) and molecular weight (Bensadoun and Weinstein, 1976; Peterson, 1977; Feist and Hummon, 2015). The 2D-DIGE technique addresses the problems encountered in the 2D-GE technique, which were a lack of reproducibility and quantification, and was introduced by Unlü et al. (Unlü et al., 1997). Using CyDyes (2, 3, and 5) for protein labeling, which are fluorescent dyes, the samples can be pooled and separated on one single 2D-PAGE (polyacrylamide gel electrophoresis) gel and then scanned by detecting each CyDye independently. The spots observed can be analyzed for expression levels and extracted from the gels for further analysis by mass spectrometry. The direct advantages of this technique is the use of an internal standard, generally a protein pool conjugated with the Cy2 fluorescent dye, and the multiplexing of the samples implying the simultaneous measurement of multiple analytes in a single analytical run and therefore increasing the power for statistical analysis (Minden, 2007). Along these lines, there has been a growing effort over the last few years to analyze fibroblast secretomes by proteomic approaches (secretomics), to allow the identification of molecules secreted by fibroblast cells and consequently gain insight into the mechanisms of immunomodulation, inflammation, angiogenesis, cell survival, or cell differentiation and recruitment in various health and diseases or conditions. Despite these advances, secretome analysis still faces some difficulties mainly related to sample collection and preparation.
The secretome is a challenging sample to analyze, mainly due to inherent difficulties with sample collection and preparation since secreted proteins are quite diluted and often present in μg to ηg. In addition, culture media often contains salts and other compounds such as phospholipids, lipids and polysaccharides, which interfere with most proteomics techniques. Thus, selective carrier-based precipitation of proteins becomes almost mandatory for proper subsequent secretomic analysis (Chevallet et al., 2007). Therefore, since the analysis of secreted proteins is challenging due to technical difficulties related to cell collection and preparation, the principal aim of this study was to determine the optimal time to harvest fibroblast conditioned-media and the best protein precipitation and conjugation methods for the subsequent secretomic screening experiments. A variety of methods have been previously described to extract diluted secreted proteins usually present in conditioned culture media in low concentration. These techniques present several drawbacks, such as coprecipitation of salts or poor yields at low protein concentrations. Samples should have a high protein concentration and be free of salt and other disturbing factors, such as ionic detergents, nucleic acids, and lipids that could interfere with downstream analysis. Protein concentration methods, such as absorption to hydrophobic resin, ultrafiltration, lyophilisation, and dialysis, are therefore not ideal for subsequent secrotomic analysis. From the data already present in the literature, we ruled out two carrier-based protein precipitation methods proven to be highly efficient to recover diluted proteins from culture media, trichloroacetic—sodium deoxycholate (TCA-DOC) and tetrahydrofuran—sodium lauroyl sarcosinate (TCA-NLS-THF) (Bensadoun and Weinstein, 1976; Chevallet et al., 2007; Feist and Hummon, 2015). In the current work, we found that the TCA precipitation protocol we described, enabling protein precipitation from large sample volumes, gave high yield protein recovery.
Materials and Methods
Sample Selection and Collection
All cases signed a consent form approved by our Institutional Ethics Committees prior to being enrolled in the study and were recruited on a voluntary basis (Ethical research board of the CHU de Québec. Protocol number: 2012-1316. For more information, please contact Z3VyZWNoZXJjaGVAY2h1cS5xYy5jYQ==). Skin biopsies were collected from affected and unaffected individuals. Two different pathologies and control individuals were targeted in order to fully validate our results. Besides collecting skin biopsies, blood samples were also collected and used for DNA extraction and patient's genotyping to confirm diagnosis when possible. In total, 6 Amyotrophic Lateral Sclerosis (ALS), 3 neurofibromatosis type 1 (NF1) cases, and 3 controls individuals were recruited for this study (Table 1). Fibroblasts were used in this study as they are known to be affected in different neurological conditions as ALS and NF1 (De Schepper et al., 2005; Maertens et al., 2007; Paré et al., 2015; García-Romero et al., 2016).
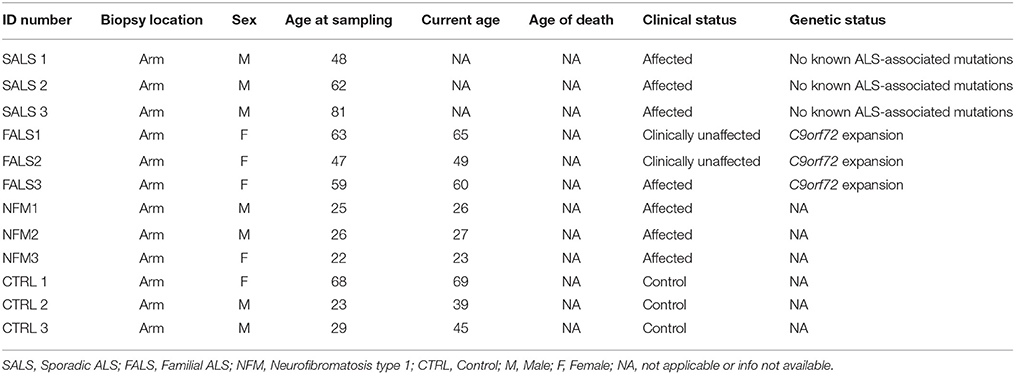
Table 1. Detailed clinical information of patients and controls enrolled in this study.
Skin Biopsies and Fibroblast Cell Extraction/Culture
For each participant, two skin biopsies were collected using a 6-mm diameter punch biopsy. All skin biopsies were taken from the same body area for each subject and fibroblasts were isolated as described before (Paré et al., 2015). Briefly, the skin biopsies were incubated with 0.05% thermolysin (Sigma, Oakville, QC, Canada), overnight at 4°C, in order to facilitate the mechanical separation of the epidermis from the dermis. Then, fibroblasts were isolated from the dermis after treatment with 0.2 IU/ml collagenase H (Roche, Missisauga, Ontario, Canada). Cultured fibroblasts, grown in DMEM (Dulbecco-Vogt modification of Eagle's medium; Invitrogen, Burlington, ON, Canada) supplemented with 10% Bovine Growth Serum (HyClone BGS; GE Healthcare Life Sciences, HyClone Laboratories, Logan, UT, USA), 100 IU/ml penicillin G (Sigma, Oakville, QC, Canada), and 25 μg/ml gentamicin (Schering, Pointe-Claire, QC, Canada) in 5% CO2 at 37°C, at passage two were seeded at a concentration of 2.5 × 105 cells on tissue culture dishes (75 cm2).
Fibroblast Induction and Protein Precipitation
Secreted proteins are often masked by high amounts of protein supplements in the culture medium. We have therefore developed a serum-deprived efficient method for the enrichment and analysis of the secretome of different fibroblast cell lines to overcome this difficulty. After three washes with 10 ml of phosphate-buffered saline (PBS) [0.8% (W/V) NaCl (BioBasic Inc., Markham, Ontario, Canada), 0.02% (W/V) KCl (BioBasic Inc., Markham, Ontario, Canada), 0.15% (W/V) Na2HPO4 (BioBasic Inc., Markham, Ontario, Canada), 0.02% (W/V) K2HPO4 (BioBasic Inc., Markham, Ontario, Canada)], the fibroblasts were deprived of serum [DMEM (Dulbecco-Vogt modification of Eagle's medium) (Invitrogen, Burlington, ON, Canada) supplemented with 100 IU/ml penicillin G (Sigma, Oakville, QC, Canada) and 25 μg/ml gentamicin (Schering, Pointe-Claire, QC, Canada) in 5% CO2 at 37°C]. This technique has been previously shown to induce secretion of proteins (Villarreal et al., 2013). Using this approach, previous work also demonstrated that short serum deprivation periods did not affect cellular viability from 48 h and inward (Del Galdo et al., 2010; Villarreal et al., 2013; Rogers et al., 2014). Fibroblast's supernatants were therefore collected at different confluence levels and at different collection times after serum deprivation; 24 h pre-confluence, 48 h pre-confluence, 24 h post-confluence, and 48-post confluence (please note that the term pre-confluence is defined as a confluence of 40% and post-confluence as a confluence of 95%). Four tissue culture dishes (75 cm2) were seeded for each condition and all the supernatants were collected. They were then centrifuged at 300 g for 10 min (4°C) and kept at −80°C until use. The proteins present in the patient-derived fibroblast conditioned-media were concentrated by the TCA-DOC and the TCA-NLS-THF precipitation methods as described in the Supplementary Data. The protein recovery yield for each culture condition was assessed via a Bradford assay (BioBasic Inc., Markham, Ontario, Canada).
TCA-DOC
The supernatants (40 ml) were thawed on ice in high-speed centrifuge tube. 1% (v/v) of a 2% sodium deoxycholate solution (Fisher Scientific, Ottawa, Ontario, Canada) was added to each tube and they were incubated on ice for 30 min after mixing. Trichloroacetic acid was then added to a final concentration of 7.5% (v/v) (BioBasic Inc., Markham, Ontario, Canada) and incubated again on ice for 60 min after mixing. Proteins were precipitated by centrifugation (15,000 g for 20 min at 4°C) and the supernatants were discarded. Twenty milliliters of 100% ice-cold (−20°C) acetone (EMD, Darmstadt, Germany) were added to the pellets, gently vortexed and kept at −20°C for 5 min. After centrifugation (15,000 g for 5 min at 4°C), the supernatants were discarded and 5 ml of 100% ice-cold (−20°C) acetone were added to the pellets. The pellets were again gently vortexed, kept at −20°C for 5 min, centrifuged (15,000 g for 5 min at 4°C), and the supernatant discarded. The pellets were air-dried in a chemical hood for 30 min, dissolved in 210 μl of Isoelectric Focusing (IEF) buffer, pH 8.5 [7M urea (BioBasic Inc., Markham, Ontario, Canada), 2M thiourea (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom), 30 mM TRIS (BioBasic Inc., Markham, Ontario, Canada), and 4% CHAPS (BioBasic Inc., Markham, Ontario, Canada)], vortexed, centrifuged (15,000 g for 10 min at room temperature), and kept at −80°C until use.
TCA-NLS-THF
The supernatants (40 ml) were thawed on ice in high-speed centrifuge tubes. 1% (v/v) of a 2% sodium layroyl sarcosine solution (Sigma-Aldrich, Oakville, Ontario, Canada) was added to the tubes and they were incubated on ice for 30 min after mixing. Trichloroacetic acid was added to a final concentration of 7.5% (v/v) (BioBasic Inc., Markham, Ontario, Canada) and the supernatants were incubated on ice for 60 min after mixing. Proteins were precipitated by centrifugation (15,000 g for 20 min at 4°C) and the supernatants were then discarded. 10% of the initial volume of ice-cold (−20°C) tetrahydrofuran (BioBasic Inc., Markham, Ontario, Canada) was added, vortexed until complete dissolution of the pellets and kept at −20°C for 5 min. After a centrifugation (15,000 g for 20 min at 4°C), the supernatants were discarded and 10% of the final volume of ice-cold (−20°C) tetrahydrofuran (BioBasic Inc., Markham, Ontario, Canada) was again added, vortexed until complete dissolution of the pellets and kept at −20°C for 5 min. After centrifugation (15,000 g for 20 min at 4°C), the supernatants were discarded and the pellets air-dried in a chemical hood for 30 min, dissolved in 210 μl of IEF buffer (pH 8.5), vortexed, centrifuged (15,000 g for 10 min at room temperature), and kept at −80°C until use.
Protein Conjugation
The proteins were conjugated with fluorescent cyanine dyes (CyDye) and resolved by SDS-PAGE and 2D-DIGE according to standard protocols. The CyDyes were synthesized by the Sherbrooke Molecular Imaging Center based on a protocol by Jung et al. (Jung et al., 2012). Briefly, the internal standard was a mix of equivalent fractions of all the samples used in this study, at a concentration higher than 25 μg, and conjugated with the CyDye Fluor Cy2 dye. The different CyDyes were diluted to a concentration of 400 pmol/μL in dimethylformamide (DMF; BioBasic Inc., Markham, Ontario, Canada). The samples and internal standards were adjusted to a pH of 8.5 with 2M NaOH and conjugated as follow: 30 μg of protein at 1 μg/μL were mixed with 1 μl of each CyDye (400 pmol/μl), vortexed, centrifuged (12,000 g for 30 s at room temperature), and kept on ice for 30 min in the dark. The reaction was stopped with 1 μl of 10 mM L-lysine (BioBasic Inc., Markham, Ontario, Canada), vortexed, centrifuged (12,000 g for 30 s at room temperature), incubated on ice for 10 min, and immediately used or kept at −80°C up to 3 months in the dark.
Electrophoresis
1D Electrophoresis
Gel electrophoresis of the conjugated proteins was performed on a 4% stacking gel and 14% acrylamide/bisacrylamide (37.5:1) resolving gel. Gels were 1.5 mm thick and casted following a specific protocol (Table 2). The migration was done using the Mini-PROTEAN® system (Biorad, Hercules, CA, USA) and performed at a constant rate of 60 mA, 2 h in 1X running buffer (Table 2). Gel readings were done using a Typhoon Trio+ (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom). The detection of CyDyes 3 and 5 was done at the optimal photomultiplier voltage (PMT) for each one of them, for each gel analyzed.
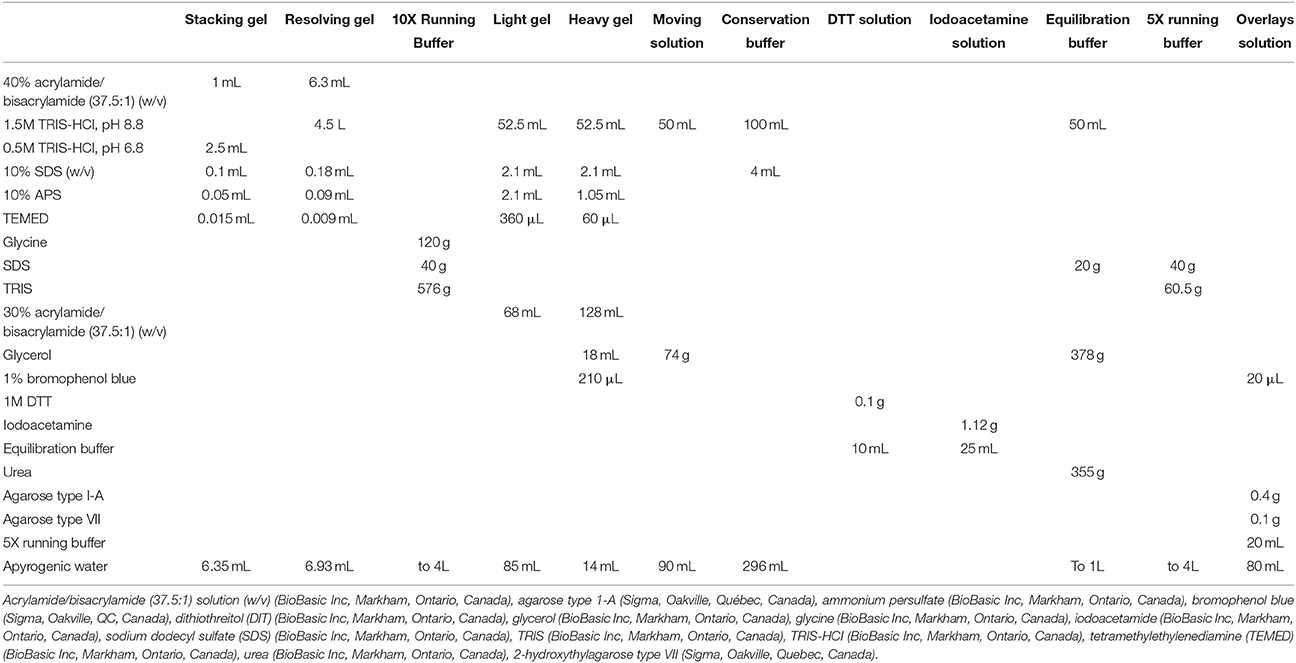
Table 2. Detailed protocol for gel and electrophoresis experiments.
2D Electrophoresis
The different CyDyes mixes were pooled together. Ninety microliters of the CyDye mix was added to 90 μl of a reduction solution [7M urea (BioBasic Inc., Markham, Ontario, Canada), 2M thiourea (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom), 4% (w/v) CHAPS (BioBasic Inc., Markham, Ontario, Canada), 0.5% (v/v) IPG (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom), 1 mM dithiothreitol (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom), 0.05% (v/v) bromophenol blue (Sigma, Oakville, QC, Canada)], vortexed and incubated for 15 min on ice in the dark. The sample was centrifuged (21,000 g for 30 s at room temperature) and the supernatant collected. The dyes were randomly conjugated to the different protein samples.
The first dimension of the 2D electrophoresis, the isoelectric focusing, was done on Immobiline DryStrip pH 3-11 NL, 24 cm (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) strips. They were rehydrated at room temperature overnight with 450 μl of a mix of the remaining reduced supernatant and rehydration solution [7M urea (BioBasic Inc., Markham, Ontario, Canada), 2M thiourea (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom), 4% CHAPS (BioBasic Inc., Markham, Ontario, Canada), 0.5% IPG (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom), 1.2% DeStreak rehydration solution (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom), 0.05% bromophenol blue (Sigma, Oakville, QC, Canada)]. Migration of the strips was done with the Ettan IPGphor 3 IEF system (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) following a specific protocol (Table 3). The strips were covered with Plus One DryStrip cover fluid (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) during the entire migration and kept at −80°C until use.
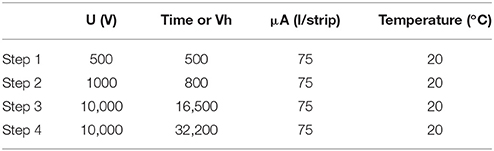
Table 3. Detailed migration protocol for the 2D gel electrophoresis.
The second dimension of the 2D electrophoresis was done on 10–18% gradient polyacrylamide gels. The gels (26 × 20 cm) were casted with the DALTsix gel caster and the DALTsix gradient maker (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom). The gels were cast following a specific recipe (Table 2) combining light and heavy acrylamide solutions, in order to generate a linear gradient between the two concentrations. The light gel represents the solution with the lowest percentage of total acrylamide (9.7%) while the heavy gel represents the solution with the highest (maximum) percentage of total acrylamide (17.8%). The gels were covered with 30% isopropyl alcohol (Laboratoire MAT, Québec, Québec, Canada) and polymerized for 3 h. The gels were then washed with apyrogenic water, covered with a conservation buffer (Table 2) and kept polymerizing for another 2 h.
The strips were thawed at room temperature and washed for 20 min under agitation in a DTT solution (Table 2). A second 20 min wash under agitation in an iodoacetamide solution (Table 2) was done and the strips were added on top of the gels. To maintain them in place, an overlays solution was added over the strips (Table 2).
The migration was done using the Ettan DALTsix large vertical system and performed at a constant rate of 0.5 W/gel for 1 h and then over night at a constant rate of 1.25 W/gel in 1X running buffer (Table 2). The whole system was kept at 20°C with a water-cooling system. Gels readings were done using a Typhoon Trio+ (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) as described above.
Visualization and Quantification of the In-Gel Fluorescent Signals
CyDye-labeled gels were visualized using a Typhoon Trio Plus laser scanner (GE Healthcare). Excitation and emission wavelengths were chosen according to manufacturer's recommendations (488, 532, and 633 nm laser beams; 520, 580, and 670 nm emission filters). Quantification of the fluorescent signal was done using ImageJ software. The average pixel saturation of the fluorescent signal from the Typhoon Image was calculated and normalized for background fluorescence.
Results
Comparison of Protein Extraction Methods and the Effect of the Confluence Level and Collection Time on the Protein Recovery Yield
Repeated experiments, using fibroblasts collected from two different neuronal pathologies (SALS/FALS and neurofibromatosis) and controls revealed that the TCA-DOC protein precipitation method was consistently superior to the TCA-NLS-THF method to recover proteins (Figure 1). A statistically significant difference between the two used protein precipitation methods was revealed by a grouped analysis. A 2-way ANOVA followed by a Bonferroni post-hoc test with a significance level of 0.05 was done using PRISM software (GraphPad Software, Inc., San Diego). The error bar represents the standard deviation. The TCA-DOC protein precipitation method was consistently giving higher recovery yield when compared to the TCA-NLS-THF method for each tested condition (Figure 1). No significant differences were observed in the recovered amount of secreted proteins between ALS (SALS/FALS), NF1, and controls fibroblasts. We then tested the influence of the confluence level as well as the collection time (24 and 48 h at 40 or 95% of confluence) on the total amount of secreted proteins recovered from each fibroblast conditioned-media. The 48 h post-confluency culture condition for both protein precipitation methods was proven to give the better recovery yield (Figure 1). However, the higher recovery yield was consistently obtained after a 48 h induction at 95% of cell confluence with the TCA-DOC (1045.2 μg/ml) protein precipitation method compared to the TCA-NLS-THF method (713.9 μg/ml).
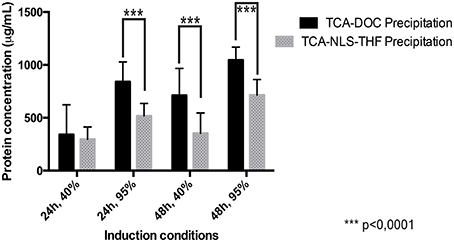
Figure 1. Protein recovery yield in function of precipitation methods, collection times, and confluence levels measured by the Bradford assay. The yield of protein recovered following the two tested protein precipitation methods (TCA-DOC and TCA-NLS-THF) and four different culture conditions was measured by the Bradford assay. The amount of recovered protein for all the tested conditions is given in μg/ml of conditioned-supernatant. The TCA-DOC method consistently gave better recovery yields independent of the induction conditions that were used. ***Corresponds to a P-value of < 0.0001.
Effect of Cyanine Dye Conjugation on the Recovered Protein
In order to determine if any of the protein precipitation methods interfered with cyanine dye conjugation, we performed a one dimension SDS-PAGE gel of secreted proteins precipitated from fibroblast conditioned-medium and conjugated with different commonly used CyDyes. As illustrated in Figure 2, both the TCA-DOC and TCA-NLS-THF methods have no significant effect on the conjugation with the CyDyes (3 and 5; Figure 2). A 2-way ANOVA followed by a Bonferroni post-hoc test with a significance level of 0.05 was done using PRISM software (GraphPad Software, Inc., San Diego). The error bar represents the standard deviation. The TCA-DOC method was therefore selected for downstream 2D-DIGE proteomic studies since it gives the higher and significant (P < 0.0001) recovery yield for all the induction conditions tested.
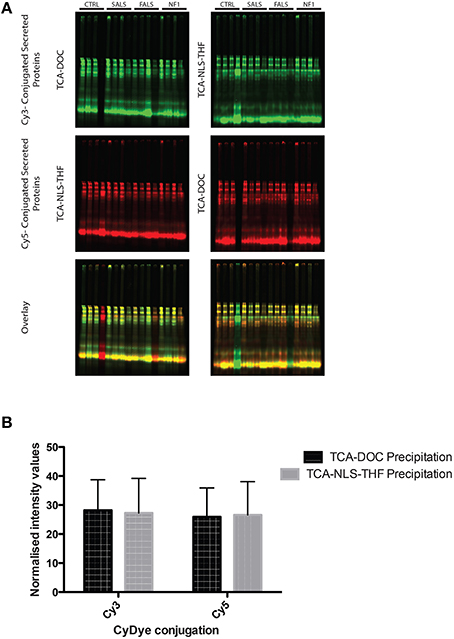
Figure 2. One dimension SDS-PAGE gel of secreted proteins precipitated from fibroblast conditioned-medium and conjugated with CyDyes. (A) Conjugation of CyDyes 3 and 5 to TCA-DOC and TCA-NLS-THF precipitated proteins. A 14% acrylamide/bisacrylamide (37.5:1) gel was used to resolve the CyDye conjugated supernatant protein. An internal standard was produced by conjugating protein from the TCA-DOC (Post-48 h) with both Cy3 (green) and Cy5 (red) dyes. One fibroblast cell line per tested condition for each of the studied pathology and control was tested and loaded in the gels in this order: pre-confluence/24 h, post-confluence/48 h, pre-confluence/48 h, post-confluence/48 h. Equal amount of proteins was loaded in all the gels. (B) Quantification of the fluorescent signal, detected with our laser-scanner Typhoon device, was done using ImageJ software. No statistical differences were noted for each of the tested condition, indicating that any of the protein precipitation method used in this study did not interfere with the CyDye conjugation.
Visualization of Whole Secreted Protein Resolved with 2D-DIGE
A pool of precipitated proteins collected from all the samples tested in this study, using the TCA-DOC method 48 h post-confluence and induction, were conjugated with each CyDyes (2, 3, and 5) and resolved with 2D-DIGE. The results show that each CyDyes bind at the same effectiveness to the proteins, giving an equal number of detected spots (502) for each of the conjugation (Figure 3) indicating that the choice of a specific CyDye does not affect or interfere with protein detection. Conjugation of the samples for each tested conditions was done in a randomized fashion between the two CyDyes used in the study (Cy3 and Cy5). The conjugated proteins mixture, precipitated from fibroblast conditioned-medium for two different conditions, was subjected to isoelectric focusing followed by a 10–18% SDS-PAGE gradient migration in the 2nd dimension. To determine the number of spots detected, the Delta 2D analysis software V4.2 (Decodon, Greifswald, Germany) was used. A data pool, comprised of all the gels and the CyDyes, was created. Quantification of the number of spots detected between different induction conditions and precipitation methods (TCA-DOC post-confluence, 24 h of incubation/TCA-DOC, post-confluence, 48 h of incubation/TCA-NLS-THF, post-confluence, 48 h of incubation) was also assessed. In line with our previous results showing that the TCA-DOC 48 h post-confluency was given better protein recovery yield, more spots were also detected using this method when compared to the two other conditions tested (Figure 4).
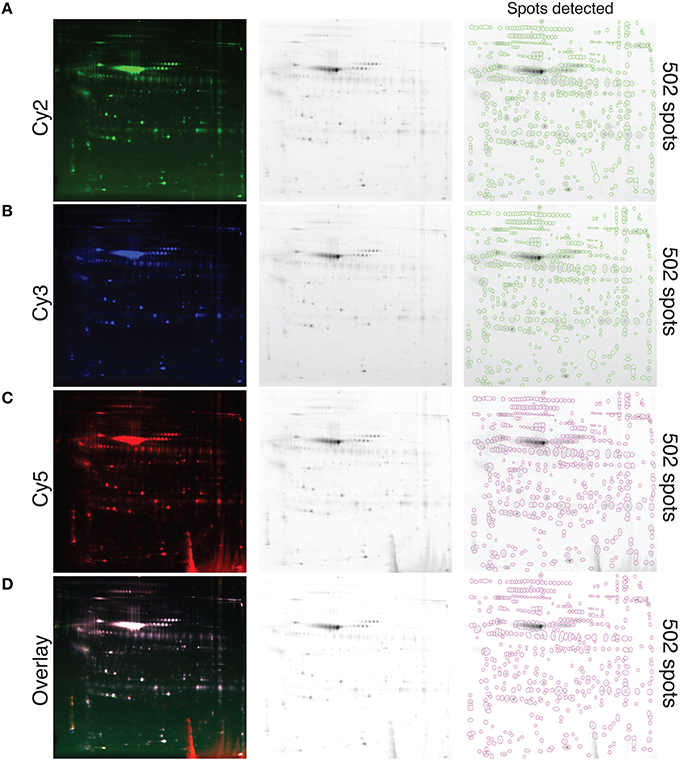
Figure 3. Visualization of whole secreted protein and quantification of the protein spots resolved with 2D-DIGE at the optimal induction condition. Conjugation of each CyDye, Cy2 (A), Cy3 (B), and Cy5 (C), to precipitated proteins extracted from fibroblast conditioned-medium via the TCA-DOC method and resolved by 2D-DIGE is shown. Please note that all technical replicates (disease and control) obtained for the 48 h post-confluence induction condition were pooled together prior CyDye conjugation and loaded on gel. (D) Represents a fusion image of the three different CyDyes. Each individual spot is circled. The number of detected spots (502) is indicated at the far right. Equal number of spots is detected for each of the tested CyDyes indicating that each of the specific tested CyDyes bond with the same effectiveness and therefore that the conjugation efficiency is independent of the CyDye fluors.

Figure 4. Visualization and quantification of the protein spots of different tested induction conditions and precipitation methods. Proteins were conjugated with the different CyDyes after extraction with the TCA-DOC, 24 h, post-confluence (Cy2) (A), TCA-NLS-THF, 48 h, post-confluence (Cy3) (B) and TCA-DOC, 48 h post-confluence (Cy5) (C) induction conditions and precipitation methods and resolved by 2D-DIGE from one control cell line. Each individual spot is circled and was automatically quantified by the software (far right). Note that the TCA-DOC 48 h post-confluence induction condition, which was also consistently given better protein recovery yield, gave the highest number of protein spots.
Discussion
The proteins secreted by a particular type of cell, the secretome, play important roles in the regulation of many physiological processes via paracrine/autocrine mechanisms, and they are of increasing interest as potential biomarkers and therapeutic targets in diseases (Pocsfalvi et al., 2011). The proteins released by cells into conditioned media in vitro have been studied to better understand pathological conditions and mechanisms in vivo (Pocsfalvi et al., 2011; Ribeiro et al., 2011). However, the secretome analysis still faces some problem mainly related to cell preparation, protein extraction, enrichment, and conjugation. To facilitate ongoing research involving secreted proteins, we revisited cell culture protocols and whole secreted protein enrichment. We identified the optimal fibroblast cells culture protocol, secreted protein collection, as well as precipitation method for the subsequent secretomic screening experiments. The maximum recovery yield was obtained when collecting the conditioned supernatants 48 h after induction when fibroblasts were at 95% of confluence with the TCA-DOC precipitation method. Washing the cells three times with PBS before serum deprivation ruled out possible contaminants present in the bovine serum (Rogers et al., 2014). It cannot be excluded however that some intracellular proteins, released in the medium by cellular lysis, might be present in the collected supernatants. Nonetheless, since the serum-deprived protocol used to extract proteins was the same for all tested conditions, it is unlikely that the presence of lysis-derived contaminants would interfere with the analysis and give false-positive results. Moreover, it has been previously shown that the removal of bovine protein, especially when using PBS, seems not to compromise the number of living cells and to minimize the effects on cell lysis and viability (Rogers et al., 2014). A comparative approach between the conditioned-medium and cytosolic extract should however be performed in order to normalize the results for the presence of minimized lysis-derived contaminants. The concept of DIGE was originally developed by Unlü and colleagues (Unlü et al., 1997). This technique allows higher reproducibility and provides information about more subtle changes in protein expression than conventional 2D electrophoresis as three different sample types can be run on the same gel and be directly compared to one another, one being the internal standard (i.e., a pool of all the samples of the experiment). This technique, relying on pre-electrophoretic labeling of samples with one of three spectrally resolvable fluorescent cyanine dyes (Cy2, Cy3, and Cy5), also allows multiplexing of samples into the same gel, and has become the method of choice over the last years for comparative proteomic profiling in a cost-effective manner. With the different 2-D gel image analysis software available commercially, spot detection as well as statistical analysis, proteome maps and spot picking, for mass spectrometry (MS) analysis, can easily be done. Table 4 presents a list of available software. Therefore, we have identified the optimal technical parameters that shall be used in the subsequent 2D fibroblast secretome profiling experiments. According to our results, we therefore propose that protein precipitation should be performed by TCA-DOC precipitation method after 48 h at 95% of confluence in a serum-deprived culture medium.
Table 4. Available 2D gel analysis software and spot quantification.
Comparative proteomic profiling is a relevant approach to biomarker discovery and is becoming extensively utilized in secretome analysis. Mainly, two prominent sources of secreted proteins have been utilized in secretomic studies: cell line supernatants derived from patients and proximal biological fluids (blood, sera, cerebral spinal fluid, etc.). The availability of skin fibroblasts is particularly advantageous for research involving neurological diseases, since it is difficult to obtain affected tissues from living patients. The main function of fibroblasts is to maintain the structural integrity of connective tissues by continuously secreting precursors of the extracellular matrix. Fibroblasts are readily cultured in research laboratories and incorporation of fibroblasts into tissue-engineered skin substitutes has produced encouraging results in the treatment of severely burned patients, symptomatic pain relief, more rapid healing of acute, chronic wounds, and even to model different diseases such as ALS and psoriasis (Auger et al., 1998, 2009; Moulin et al., 2000; Auxenfans et al., 2009; Jean et al., 2009; Paré et al., 2015). As a matter of fact, this novel tissue-engineered skin model derived from both sporadic and familial ALS patients was reported to be the first to present cytoplasmic TDP-43 accumulation in cells outside of the nervous system. Furthermore, generation of human induced pluripotent stem cells (iPSCs) from fibroblast cells provides a new method to elucidate the molecular basis of human diseases and a novel approach to disease modeling and drug discovery (Takahashi et al., 2007; Yu et al., 2007). Actually, an increasing number of studies have employed disease-specific human iPSCs to model neurological diseases, including Parkinson's disease, Autism spectrum disorders, Friedreich's Ataxia, spinal muscular atrophy, ALS, Angelman syndrome and Prader-Willi syndrome, and only few of them have demonstrated disease-specific phenotypes (Dimos et al., 2008; Park et al., 2008; Ebert et al., 2009; Soldner et al., 2009; Chamberlain et al., 2010; Ku et al., 2010; Marchetto et al., 2010; Nguyen et al., 2011; Ryan et al., 2013; Velasco et al., 2014). The potential of iPSCs technology is enormous, but major obstacles to iPSC studies for late-onset neurodegenerative diseases remains. Their production is still very expensive and time consuming. iPSC also show genetic and epigenetic abnormalities after time in culture, sometimes even showing cancer-related mutations (Gore et al., 2011; Bhartiya et al., 2013). This present study can overcome these mutations-related problems since the skin fibroblasts are used without any genetic modifications. They are also easier to work with than iPSC-derived neurons since the culture medium needed is less expensive and doesn't need to be supplemented of various chemical and biological compounds such as nerve growth factors. The identical germ layer origin of both the skin and neural tissue leads us to think that skin can also be used to study neurodegenerative diseases. For instance, cytoplasmic accumulation of TDP-43, a pathological signature of ALS and ALS/FTD, was observed in the skin of ALS patients and in ALS-derived tissue engineered skins (Paré et al., 2015). Thus, fibroblast cells, able to secrete extracellular matrix molecules, as the matrix metalloproteinases (MMPs), which were shown to be associated with neural morphogenesis as well as being important in the synaptic remodeling, on both the structural and functional levels (McFarlane et al., 2003; Rivera et al., 2010; Huntley, 2012), could be of major importance in understanding pathophysiological mechanisms of neurodegenerative diseases.
Conclusion
Comparative proteomic profiling of patient-derived fibroblasts to elucidate the pathogenesis of rare diseases, especially neurological disorders, presents an attractive alternative to neural tissue biopsies from living patients that are of limited access. These new techniques will hopefully help researchers and clinicians to better understand these yet untreatable diseases. Conceivably, the proposed approach based on the study of patient-derived skin fibroblasts to facilitate the identification of biomarkers for early diagnosis or to monitor disease progression becomes highly attractive and of high importance in clinical neurology. A comprehensive survey of patient-derived whole secreted proteins in fibroblast-conditioned media will certainly lead to more translational research to facilitate the identification of specific disease biomarkers and new molecular targets for therapy or molecular signatures to improve early diagnosis, to find a cure or to slow down the course of this yet untreatable diseases.
Author Contributions
Experimental and technical inputs: BP, LD. Study design: BP, RP, and FGL. Recruitment and clinical assessement of patients: ND. Writing of the article: BP, RP, ND, and FGL. Operational grant money: FGL.
Funding
This work was supported by ALS Canada, the Canadian Institutes for Health Research and the W. Garfield Weston Foundation through the Weston Brain Institute. Part of this work was also supported by the Fondation des Hôpitaux Enfant-Jésus—St-Sacrement.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
FGL is the recipient of a tier 2 Canada research Chair. BP is the recipient of a Laval University—Faculty of Medicine scholarship, CHU de Québec Foundation scholarship and Thé Cell research network scholarship (Cell and Tissue Therapy Network—FRQS). We thank Lily-Ann Franche, Sébastien Larochelle, Marie-Pascale Rousseau, Vincent Roy and François-Dominique Scott for experimental input.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fncel.2016.00070
References
Auger, F. A., Lacroix, D., and Germain, L. (2009). Skin substitutes and wound healing. Ski Pharmacol. Physiol. 22, 94–102. doi: 10.1159/000178868
Auger, F. A., Rouabhia, M., Goulet, F., Berthod, F., Moulin, V., and Germain, L. (1998). Tissue-engineered human skin substitutes developed from collagen-populated hydrated gels: clinical and fundamental applications. Med. Biol. Eng. Comput. 36, 801–812. doi: 10.1007/BF02518887
Auxenfans, C., Fradette, J., Lequeux, C., Germain, L., Kinikoglu, B., Bechetoille, N., et al. (2009). Evolution of three dimensional skin equivalent models reconstructed in vitro by tissue engineering. Eur. J. Dermatol. 19, 107–113. doi: 10.1684/ejd.2008.0573
Beckett, P. (2012). The basics of 2D DIGE. Methods Mol. Biol. 854, 9–19. doi: 10.1007/978-1-61779-573-2_2
Bensadoun, A., and Weinstein, D. (1976). Assay of proteins in the presence of interfering materials. Anal. Biochem. 70, 241–250. doi: 10.1016/S0003-2697(76)80064-4
Brown, K. J., Seol, H., Pillai, D. K., Sankoorikal, B. J., Formolo, C. A., Mac, J., et al. (2013). The human secretome atlas initiative: implications in health and disease conditions. Biochim. Biophys. Acta 1834, 2454–2461. doi: 10.1016/j.bbapap.2013.04.007
Bhartiya, D., Nagvenkar, P., Sriraman, K., and Shaikh, A. (2013). “An overview of pluripotent stem cells”, Pluripotent Stem Cells, ed D. Bhartiya (InTech). doi: 10.5772/55130. Available online at: http://www.intechopen.com/books/pluripotent-stem-cells/an-overview-of-pluripotent-stem-cells
Chamberlain, S. J., Chen, P., Ng, K. Y., Bourgois-rocha, F., Lemtiri-chlieh, F., Levine, E. S., et al. (2010). Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader – Willi syndromes. Proc. Natl. Acad. Sci. U.S.A. 107, 17668–17673. doi: 10.1073/pnas.1004487107
Chevallet, M., Diemer, H., Van Dorssealer, A., Villiers, C., and Rabilloud, T. (2007). Toward a better analysis of secreted proteins: the example of the myeloid cells secretome. Proteomics 7, 1757–1770. doi: 10.1002/pmic.200601024
Del Galdo, F., Shaw, M. A., and Jimenez, S. A. (2010). Proteomic analysis identification of a pattern of shared alterations in the secretome of dermal fibroblasts from systemic sclerosis and nephrogenic systemic fibrosis. Am. J. Pathol. 177, 1638–1646. doi: 10.2353/ajpath.2010.091095
De Schepper, S., Boucneau, J., Lambert, J., Messiaen, L., and Naeyaert, J.-M. (2005). Pigment cell-related manifestations in neurofibromatosis type 1: an overview. Pigment Cell Res. 18, 13–24. doi: 10.1111/j.1600-0749.2004.00206.x
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218–1221. doi: 10.1126/science.1158799
Ebert, A. D., Yu, J., Rose, F. F., Mattis, V. B., Lorson, C. L., Thomson, J. A., et al. (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277–280. doi: 10.1038/nature07677
Feist, P., and Hummon, A. (2015). Proteomic challenges: sample preparation techniques for microgram-quantity protein analysis from biological samples. Int. J. Mol. Sci. 16, 3537–3563. doi: 10.3390/ijms16023537
García-Romero, M. T., Parkin, P., and Lara-Corrales, I. (2016). Mosaic neurofibromatosis type 1: a systematic review. Pediatr. Dermatol. 33, 9–17. doi: 10.1111/pde.12673
Gelman, J. S., Dasgupta, S., Berezniuk, I., and Fricker, L. D. (2013). Analysis of peptides secreted from cultured mouse brain tissue. Biochim. Biophys. Acta 1834, 2408–2417. doi: 10.1016/j.bbapap.2013.01.043
Gore, A., Li, Z., Fung, H.-L., Young, J. E., Agarwal, S., Antosiewicz-Bourget, J., et al. (2011). Somatic coding mutations in human induced pluripotent stem cells. Nature 471, 63–67. doi: 10.1038/nature09805
Hartwig, S., Raschke, S., Knebel, B., Scheler, M., Irmler, M., Passlack, W., et al. (2013). Secretome profiling of primary human skeletal muscle cells. Biochim. Biophys. Acta 1844, 1011–1017. doi: 10.1016/j.bbapap.2013.08.004
Huntley, G. W. (2012). Synaptic circuit remodelling by matrix disease. Nat. Rev. Neurosci. 13, 743–757. doi: 10.1038/nrn3320
Jean, J., Lapointe, M., Soucy, J., and Pouliot, R. (2009). Development of an in vitro psoriatic skin model by tissue engineering. J. Dermatol. Sci. 53, 19–25. doi: 10.1016/j.jdermsci.2008.07.009
Jung, M. E., Kim, W., Avliyakulov, N. K., Oztug, M., and Haykinson, M. J. (2012). “Synthesis and validation of cyanine-based dyes for DIGE,” in Difference Gel Electrophoresis (DIGE): Methods and Protocols, Vol. 854, 2012th Edn., eds R. Cramer and R. Westermeier (Humana Press), 67–85.
Katz-Jaffe, M. G., McReynolds, S., Gardner, D. K., and Schoolcraft, W. B. (2009). The role of proteomics in defining the human embryonic secretome. Mol. Hum. Reprod. 15, 271–277. doi: 10.1093/molehr/gap012
Klose, J. (1975). Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik 26, 231–243. doi: 10.1007/BF00281458
Ku, S., Soragni, E., Campau, E., Thomas, E. A., Altun, G., Laurent, L. C., et al. (2010). Friedreich's ataxia induced pluripotent stem cells model intergenerational GAA•TTC triplet repeat instability. Cell Stem Cell 7, 631–637. doi: 10.1016/j.stem.2010.09.014
Leroy, B., Toubeau, G., Falmagne, P., and Wattiez, R. (2006). Identification and characterization of new protein chemoattractants in the frog skin secretome. Mol. Cell. Proteomics 5, 2114–2123. doi: 10.1074/mcp.M600205-MCP200
Luo, J., Yu, L., Guo, Y., and Li, M. (2012). Functional classification of secreted proteins by position specific scoring matrix and auto covariance. Chemom. Intell. Lab. Syst. 110, 163–167. doi: 10.1016/j.chemolab.2011.11.008
Maertens, O., De Schepper, S., Vandesompele, J., Brems, H., Heyns, I., Janssens, S., et al. (2007). Molecular dissection of isolated disease features in mosaic neurofibromatosis type 1. Am. J. Hum. Genet. 81, 243–251. doi: 10.1086/519562
Makridakis, M., Roubelakis, M. G., and Vlahou, A. (2013). Stem cells: insights into the secretome. Biochim. Biophys. Acta 1834, 2380–2384. doi: 10.1016/j.bbapap.2013.01.032
Marchetto, M. C. N., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., et al. (2010). A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. doi: 10.1016/j.cell.2010.10.016
Meissner, F., Scheltema, R. A., Mollenkopf, H.-J., and Mann, M. (2013). Direct proteomic quantification of the secretome of activated immune cells. Science 340, 475–478. doi: 10.1126/science.1232578
McFarlane, S., Tn, C., and Adam, M. (2003). Metalloproteases : carving out a role in axon guidance. Neuron 37, 559–562. doi: 10.1016/S0896-6273(03)00089-8
Minden, J. (2007). Comparative proteomics and difference gel electrophoresis. Biotechniques 43, 739–745. doi: 10.2144/000112653
Moulin, V., Auger, F. A., Garrel, D., and Germain, L. (2000). Role of wound healing myofibroblasts on re-epithelialization of human skin. Burns 26, 3–12. doi: 10.1016/S0305-4179(99)00091-1
Mukherjee, P., and Mani, S. (2013). Methodologies to decipher the cell secretome. Biochim. Biophys. Acta 1834, 2226–2232. doi: 10.1016/j.bbapap.2013.01.022
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P., et al. (2011). LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Paré, B., Touzel-Deschênes, L., Lamontagne, R., Lamarre, M.-S., Scott, F.-D., Khuong, H. T., et al. (2015). Early detection of structural abnormalities and cytoplasmic accumulation of TDP-43 in tissue-engineered skins derived from ALS patients. Acta Neuropathol. Commun. 3:5. doi: 10.1186/s40478-014-0181-z
O'Farrell, P. H. (1975). High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 250, 4007–4021. doi: 10.1016/j.bbi.2008.05.010
Park, I., Arora, N., Huo, H., Maherali, N., Shimamura, A., Lensch, M. W., et al. (2008). Disease-specific induced pluripotent stem (iPS) cells. Cell 134, 877–886. doi: 10.1016/j.cell.2008.07.041
Pavlou, M. P., and Diamandis, E. P. (2010). The cancer cell secretome: a good source for discovering biomarkers? J. Proteomics 73, 1896–1906. doi: 10.1016/j.jprot.2010.04.003
Peterson, G. L. (1977). A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 83, 346–356. doi: 10.1016/0003-2697(77)90043-4
Pocsfalvi, G., Votta, G., De Vincenzo, A., Fiume, I., Raj, D. A. A., Marra, G., et al. (2011). Analysis of secretome changes uncovers an autocrine/paracrine component in the modulation of cell proliferation and motility by c-Myc. J. Proteome Res. 10, 5326–5337. doi: 10.1021/pr200584y
Ribeiro, C. A., Salgado, A. J., Fraga, J. S., Silva, N. A., Reis, R. L., and Sousa, N. (2011). The secretome of bone marrow mesenchymal stem cells-conditioned media varies with time and drives a distinct effect on mature neurons and glial cells (primary cultures). J. Tissue Eng. Regen. Med. 5, 668–672. doi: 10.1002/term.365
Rivera, S., Khrestchatisky, M., Kaczmarek, L., Rosenberg, G. A., and Jaworski, D. M. (2010). Metzincin proteases and their inhibitors, foes or friends in nervous system physiology? J. Neurosci. 30, 15337–15357. doi: 10.1523/JNEUROSCI.3467-10.2010
Rogers, L. J., Suchowerska, N., Khan, A., Polaskova, V., and McKenzie, D. R. (2014). Profiling of the secretome of human cancer cells: preparation of supernatant for proteomic analysis. Electrophoresis 35, 2626–2633. doi: 10.1002/elps.201400100
Ryan, S. D., Dolatabadi, N., Chan, S. F., Zhang, X., Waseem, M., Parker, J., et al. (2013). Isogenic human iPSC Parkinson's model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155, 1351–1364. doi: 10.1016/j.cell.2013.11.009
Soldner, F., Hockemeyer, D., Beard, C., Gao, Q., Bell, G. W., Cook, E. G., et al. (2009). Parkinson's diseasepatient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977. doi: 10.1016/j.cell.2009.02.013
Stastna, M., and Van Eyk, J. E. (2012). Secreted proteins as a fundamental source for biomarker discovery. Proteomics 12, 722–735. doi: 10.1002/pmic.201100346
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Unlü, M., Morgan, M. E., and Minden, J. S. (1997). Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis 18, 2071–2077. doi: 10.1002/elps.1150181133
Velasco, I., Salazar, P., Giorgetti, A., Ramos-Mejía, V., Castaño, J., Romero-Moya, D., et al. (2014). Generation of neurons from somatic cells of healthy individuals and neurological patients through induced pluripotency or direct conversion. Stem Cells 32, 2811–2817. doi: 10.1002/stem.1782
Villarreal, L., Méndez, O., Salvans, C., Gregori, J., Baselga, J., and Villanueva, J. (2013). Unconventional secretion is a major contributor of cancer cell line secretomes. Mol. Cell. Proteomics 12, 1046–1060. doi: 10.1074/mcp.M112.021618
Keywords: secretomic, 2D-DIGE, secreted proteins, secretome, fibrolast-conditioned media
Citation: Paré B, Deschênes LT, Pouliot R, Dupré N and Gros-Louis F (2016) An Optimized Approach to Recover Secreted Proteins from Fibroblast Conditioned-Media for Secretomic Analysis. Front. Cell. Neurosci. 10:70. doi: 10.3389/fncel.2016.00070
Received: 24 November 2015; Accepted: 04 March 2016;
Published: 31 March 2016.
Edited by:
Pier Giorgio Mastroberardino, Erasmus MC University Medical Center Rotterdam, NetherlandsReviewed by:
Grzegorz Kreiner, Polish Academy of Sciences, PolandMichelle A. Clark, Nova Southeastern University, USA
Copyright © 2016 Paré, Deschênes, Pouliot, Dupré and Gros-Louis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francois Gros-Louis, ZnJhbmNvaXMuZ3Jvcy1sb3Vpc0BmbWVkLnVsYXZhbC5jYQ==