 Yu-Ju Liu
Yu-Ju Liu Po-Yi Tsai
Po-Yi Tsai Yijuang Chern
Yijuang Chern- Division of Neuroscience, Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron disease that is clinically characterized by progressive muscle weakness and impaired voluntary movement due to the loss of motor neurons in the brain, brain stem and spinal cord. To date, no effective treatment is available. Ample evidence suggests that impaired RNA homeostasis and abnormal energy status are two major pathogenesis pathways in ALS. In the present review article, we focus on recent studies that report molecular insights of both pathways, and discuss the possibility that energy dysfunction might negatively regulate RNA homeostasis via the impairment of cytoplasmic-nuclear shuttling in motor neurons and subsequently contribute to the development of ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a rare motor neuron disease that occurs in adults with a prevalence of ~5 per 100,000. Patients with ALS develop muscle weakness and impaired voluntary movement, largely because of degeneration of the upper and lower motor neurons (Bento-Abreu et al., 2010; Kiernan et al., 2011; Al-Chalabi et al., 2016). Although substantial efforts have been made to investigate ALS pathogenesis in the past decade, no effective treatment is currently available. Most patients with ALS (~90%) have the sporadic form of the disease. Familial ALS (approximately 10% of cases) is caused by various mutations in more than 20 genes (Sreedharan and Brown, 2013; Steinberg et al., 2015; Taylor et al., 2016), including Cu-Zn superoxide dismutase (SOD1, Bruijn et al., 1998), TAR DNA-binding protein of 43 kDa (TDP-43, Neumann et al., 2006), fused in sarcoma/translocated in liposarcoma (FUS/TLS Lagier-Tourenne et al., 2010) and the expansion of (G4C2) repeat in C9ORF72 (DeJesus-Hernandez et al., 2011). The list is growing. It is intriguing that a significant portion of these ALS-associated genes are functionally important for the homeostasis of RNA- and DNA-binding proteins (Taylor et al., 2016). Another interesting observation is that abnormal energy metabolism (including weight loss, hyperlipidemia and mitochondrial dysfunction) has been observed in patients with ALS and in ALS mouse models, thus suggesting that abnormal energy homeostasis is an important hallmark of ALS (Ahmed et al., 2016). Moreover, abnormal activation of an energy sensor (AMP kinase, AMPK) is negatively associated with the survival of motor neurons and the impaired nuclear-cytoplasmic shuttling of ALS-associated genes/proteins (Lim et al., 2012; Liu et al., 2015a,b). This review is not intended to comprehensively describe RNA toxicity in ALS. Instead, it focuses on the emerging possibility that energy dysfunction in ALS might contribute to RNA toxicity and proposes a potential working hypothesis for the etiology of sporadic ALS.
Abnormal Energy Homeostasis in AlS
Body Weight, Hyperlipidemia and Energy Homeostasis in ALS
The balance between energy consumption and storage is critical in both healthy individuals, and individuals with diseases. There is substantial evidence demonstrating that energy homeostasis in patients with ALS is disrupted, because these patients usually exhibit a low body-mass index and abnormal energy metabolism (including hypermetabolism and hyperlipidemia), probably as a result of higher energy consumption and lower food intake (Desport et al., 1999; Vaisman et al., 2009; Dupuis et al., 2011). It is important to note that weight loss is negatively associated with survival and is therefore an important prognostic factor for patients with ALS (Stambler et al., 1998; Chio et al., 2009b; Jawaid et al., 2010). In addition, weight loss in patients with ALS is tightly linked to disease progression (Kasarskis et al., 1996; Desport et al., 1999, 2000). Weight loss in patients with ALS may be caused by dysphagia resulting from bulbar muscle weakness (Kühnlein et al., 2008). Another factor that may contribute to weight loss in patients with ALS is hypermetabolism. Given that patients with ALS have muscle weakness, and muscle tissue is a major site of energy consumption, hypermetabolism in patients with ALS is somewhat unexpected (Desport et al., 2001, 2005). Interestingly, patients with ALS have increased amounts of low-density lipoprotein (LDL), cholesterol and apolipoprotein E (APOE) in the blood (Lacomblez et al., 2002; Dupuis et al., 2008). Although hyperlipidemia in ALS is protective regarding respiratory function (Dorst et al., 2011), in a specific group of patients in Italy it has not been found to correlate with survival (Chio et al., 2009a). Lower body weight and increased metabolism, which occur before the onset of motor impairment, have also been observed in a widely used ALS mouse model that expresses disease-causing Cu/Zn-SOD1 mutations (i.e., G93A and G86R; Dupuis et al., 2004b). The causes of hypermetabolism and hyperlipidemia in ALS are currently unknown. It appears that the peripheral clearance of lipoproteins in ALS mice is enhanced (Fergani et al., 2007), suggesting that peripheral usage of lipids in ALS might be protective. Interestingly, feeding ALS mice (SOD1-G93A and SOD1-G86R) with a high-fat diet reduces the degeneration of motor neurons, delays disease onset and increases lifespan (Dupuis et al., 2004b; Mattson et al., 2007). Thus, therapeutic interventions for abnormal energy homeostasis in ALS may provide a strategy for the treatment of ALS.
Because some ALS patients tend to have higher levels of blood cholesterol and triglycerides (Dorst et al., 2011), the long-term effect of chronic administration of lipid-lowering drugs (such as statins) in patients with ALS has been a heated topic of debate in the past decade. Several studies reported a plausible association between chronic intake of statins and ALS (Edwards et al., 2007; Golomb et al., 2009). It was proposed that the detrimental effects of statins in patients with ALS might result from the suppression of cholesterol synthesis and the reduction of LDL availability to skeletal muscles. However, this claim regarding the association between ALS and statins has been contested (Colman et al., 2008; Sørensen and Lash, 2009) and requires further investigation.
To date, the mechanism underlying the association between abnormal metabolism and neurodegeneration remains to be determined. As Ahmed et al. (2016) have noted, it is important to clarify whether the atrophy of certain brain areas directly or indirectly affects metabolic homeostasis or if impaired metabolic metabolism causes degeneration of motor neurons.
Aberrant Mitochondrial Function and AMP-Activated Protein Kinase (AMPK) Activation in ALS
Ample evidence has demonstrated that dysregulation of mitochondrial function is a central event in ALS pathogenesis and is closely associated with disease progression (Echaniz-Laguna et al., 2002; Crugnola et al., 2010). Mitochondrial dysfunction usually occurs via dysregulation of the electron transport chain machinery and/or decreased mitochondria cytochrome oxidase activity (Borthwick et al., 1999; Ricquier and Bouillaud, 2000), which significantly contributes to neuronal cell death (Gorman et al., 2000). Expression levels of some regulators of mitochondrial biogenesis (e.g., PGC-1α) are also decreased in the spinal cords and muscles of mice and patients with ALS (Thau et al., 2012).
ALS Genes and Mitochondria
Many ALS-associated genes have been implicated in the regulation of mitochondrial function. For example, overexpression of mutant SOD1 (mSOD1) impairs mitochondrial function by disrupting the respiratory transport chain machinery (Beretta et al., 2003; Coussee et al., 2011), inhibiting the transportation of proteins into mitochondria by interacting with the mitochondrial channel protein (VDAC1, Israelson et al., 2010) and impairing mitochondria fission and fusion through alteration of the expression of DRP1 or OPA1 (Ferri et al., 2010; Liu et al., 2013). Furthermore, Wang et al. (2013) reported that overexpression of wildtype TDP43 or ALS-associated TDP-43 mutants causes the impairment of mitochondrial dynamics through increased localization of TDP-43 on mitochondria in motor neurons. FUS/TLS is also an important disease protein in ALS. Expression of FUS/TLS was found to trigger mitochondrial damage by interacting with the mitochondria chaperone protein, Hsp60 (Deng et al., 2015). Valosin-containing protein (VCP) is also involved in ALS pathology. Mutations in VCP cause mitochondrial dysfunctions, including impaired clearance of damaged mitochondria and decreased ATP levels (Chang et al., 2011; Kim et al., 2013). Recently, the C9ORF72 repeat expansion has been shown to increase oxygen consumption and mitochondrial hyperpolarization in patients with ALS (Onesto et al., 2016). iPSC-derived motor neurons from patients with a C9ORF72 repeat expansion also have a lower mitochondrial membrane potential than those from non-ALS controls, thus supporting the involvement of C9ORF72-induced mitochondrial dysfunction in ALS pathogenesis (Dafinca et al., 2016). Biochemical evidence suggests that the C9ORF72 repeat expansion causes mitochondrial damage by interacting with mitochondrial proteins, such as ATP synthase subunits alpha and beta (Rossi et al., 2015). Collectively, although via distinct pathways, mutations in multiple ALS genes result in mitochondrial dysfunctions that contribute to ALS pathogenesis. Improved mitochondrial functions as well as energy homeostasis in motor neurons and muscles have thus become the key therapeutic targets for ALS in recent years.
Beyond the degeneration of motor neurons, another major hallmark of ALS is muscle atrophy. Muscle atrophy is of great interest because the energy metabolism of skeletal muscle is an essential component of overall energy homeostasis. In an ALS mouse model, exogenous expression of human SOD1-G93A in the muscle induces severe muscle atrophy, reduced muscle strength and abnormal muscle structure (Dobrowolny et al., 2008). Similarly, overexpression of another disease-causing gene (TAR DNA-binding protein-43, TDP-43) also disturbs energy metabolism in mice by disrupting mitochondrial function and morphology in neurons (Shan et al., 2010; Xu et al., 2010). Genetic removal of TDP-43 also leads to defective energy homeostasis by decreasing adipose tissue (Chiang et al., 2010), suggesting that TDP-43 is involved in tightly tuned energy homeostasis, which is important in ALS. Collectively, these findings suggest that impairment of energy metabolism is a critical event in the pathogenesis of ALS. Further investigation is required to understand and block the pathogenic mechanisms underlying this energy dysfunction caused by mitochondrial defects.
Mitochondria and AMP-Activated Protein Kinase (AMPK)
Outside of the mitochondria, the major component that regulates energy homeostasis at the cellular level is AMPK (Long and Zierath, 2006; Hardie, 2008), which has recently been implicated in ALS and other degenerative diseases. Specifically, abnormal AMPK activation was observed in Huntington’s disease (HD; Chou et al., 2005; Ju et al., 2011, 2012, 2014), Alzheimer’s disease (AD; Thornton et al., 2011; Mairet-Coello et al., 2013) and ALS (Lim et al., 2012; Liu et al., 2015a). The impact of AMPK activation during the progression of several neurodegenerative diseases (e.g., HD and AD) is seemingly complex and appears to depend on the disease stage and the specific context of the experimental model used in the study (Ju et al., 2011; Ma et al., 2014; Domise et al., 2016; Ng et al., 2016; Tulino et al., 2016; Vázquez-Manrique et al., 2016). In ALS, higher AMPK activity was found in the spinal cord of an ALS mouse model (SOD1-G93A; Lim et al., 2012). Suppression of AMPK activity either pharmacologically or genetically decreases the mSOD1-evoked death of motor neurons. Consistent with the hypothesis that abnormal activation of AMPK is detrimental, overexpression of a disease-causing SOD1 mutation (mSOD1) in C. elegans leads to locomotor dysfunction in an AMPK-dependent manner, whereas deletion of the ortholog of AMPK (i.e., aak-2) has been found to improve motor function in mSOD1-expressing or TDP-43 mutant-expressing worms (Lim et al., 2012). These findings were the first to suggest that abnormal AMPK activation, which is expected to cause energy dysregulation, may be relevant to ALS. Moreover, abnormal AMPK activation has been detected in motor neurons of patients with ALS and TDP-43-ALS mice and subsequently cause cytoplasmic mislocalization of TDP-43 and motor dysfunction (Liu et al., 2015a). Collectively, these results suggest that cellular energy status is critical for the function of motor neurons. Activity of the energy sensor AMPK appears to interfere with the proper cellular localization of an RNA-binding protein (TDP-43), which is considered an early event in ALS pathogenesis (Giordana et al., 2010). The loss of TDP-43 in nuclei due to its redistribution under stress and inclusion formation in the cytoplasmic region might, at least partly, contribute to the observed TDP-43 proteinopathy (Lagier-Tourenne et al., 2010).
Consistent with the abovementioned hypothesis that energy status is critical for ALS, previous studies have suggested that traumas affecting energy homeostasis can contribute to ALS pathogenesis. For example, hypoxia is a proposed risk factor for ALS (Vanacore et al., 2010). This possibility is interesting because hypoxia is known to induce AMPK activation via the production of reactive oxygen species (ROS) and downstream pathways (Mungai et al., 2011). Sufficient blood supply and oxygen availability to the spinal cord are critical. Lower neural vascular perfusion caused by the suppression of vascular endothelial growth factor (VEGF) induced degeneration of motor neurons in the spinal cord (Oosthuyse et al., 2001), while administration of VEGF increased the lifespan of an ALS mouse model (mSOD1, Azzouz et al., 2004). Beyond the importance of VEGF in the protection of motor neurons, these findings also support the hypothesis that proper maintenance of energy homeostasis is critical for ALS.
Dysfunctional RNA Metabolism and Energy Homeostasis in ALS
Abnormal RNA metabolism may lead to RNA toxicity, which is usually caused by accumulation of toxic RNAs and dysfunction of RNA-binding proteins (Sicot and Gomes-Pereira, 2013). The latter has been well documented in various pathogenic events involving RNA metabolism, including RNA splicing, RNA localization, RNA transcription and miRNA biogenesis. Importantly, RNA toxicity has been implicated in various genetic diseases including fragile-X syndrome (Oberlé et al., 1991), spinobulbar muscular atrophy (La Spada et al., 1991), myotonic dystrophy type 1 (Brook et al., 1992) and HD (Bañez-Coronel et al., 2012). More recently, overexpansion of G4C2 hexanucleotide repeats in the noncoding region of C9ORF72 has been found in patients with ALS and frontotemporal lobar degeneration (FTLD; DeJesus-Hernandez et al., 2011; Renton et al., 2011; Gijselinck et al., 2012) and has been shown to play a critical role in ALS pathogenesis (Rohrer et al., 2015; Jiang et al., 2016; Lee et al., 2016; Lin et al., 2016; Taylor et al., 2016). In the following sections, we will focus on the literature suggesting potential aspects of RNA homeostasis that might be sensitive to impaired energy homeostasis in ALS.
ALS Genes and Stress Granules (SGs)
Many ALS genes (e.g., TDP-43, FUS/TLS, hnRNPA1, MATR3) encode RNA-binding proteins that have been found to affect multiple levels of RNA processing, including mRNA transcription, RNA transport, mRNA stabilization and miRNA biogenesis (Ling et al., 2013; Taylor et al., 2016). Recent studies have suggested that disturbed RNA homeostasis is a central pathogenic pathway in ALS (Weishaupt et al., 2016).
Importantly, several ALS-related RNA-binding proteins are found in stress granules (SGs; Colombrita et al., 2009; Dewey et al., 2011; Bentmann et al., 2012; Li et al., 2013). Eukaryotic cells commonly respond to stress by forming SGs, which are composed of untranslating messenger ribonucleoproteins (mRNPs) and a variety of other proteins. The key function of SGs is to temporally inhibit translation and store mRNA during stress. It has been well documented that SGs recruit RNA-binding proteins and various RNAs, thus controlling the post-transcriptional regulation of RNA, RNA stability and translation. Translation initiation of mRNA-protein complexes existing in SGs is usually limited (Buchan and Parker, 2009; Jain et al., 2016). It has been proposed that the formation of SGs is closely associated with neurodegenerative diseases, because expression of disease-causing proteins (e.g., TDP-43, FUS/TLS) facilitates and/or sustains the formation of SGs (Andersson et al., 2008; Colombrita et al., 2009; Dewey et al., 2011; Bentmann et al., 2012; Li et al., 2013; Ramaswami et al., 2013; Aulas and Vande Velde, 2015). The dynamic state of SGs can be regulated by various stressors via energy-dependent pathways involving ATP-dependent remodeling complexes (Protter and Parker, 2016).
An earlier study has suggested that TDP-43 modulates the formation of SGs by controlling the amount of SG proteins (i.e., G3BP1 and TIA-1; McDonald et al., 2011). Exogenous overexpression of ALS-associated TDP-43 mutants enhanced the number and size of SGs when compared with wild-type TDP-43 (Liu-Yesucevitz et al., 2010). Similarly, FUS/TLS is also localized or recruited to SGs during stress exposure (Sama et al., 2013; Lenzi et al., 2015). A few ALS-associated FUS/TLS mutants were shown to affect the assembly and dynamics of SGs and might contribute to the pathogenesis of ALS (Baron et al., 2013). The detailed roles of TDP-43 and FUS/TLS in regulating SGs appear different and will be discussed in more detail in the following sections.
An interesting question emerging from recent studies is whether the TDP-43 inclusions observed in ALS motor neurons are derived from SGs (Li et al., 2013; Aulas and Vande Velde, 2015). This is an important question because multiple laboratories have reported that, after exposure to various stresses (including ER stress, mitochondrial stress and proteasome inhibition), TDP-43 is recruited to SGs (Colombrita et al., 2009; Volkening et al., 2009; Dewey et al., 2011). Once accumulated in SGs, TDP-43 and FUS/TLS may gradually evolve into stable protein aggregates as observed in diseased motor neurons (Parker et al., 2012). Moreover, mislocalization of FUS/TLS and TDP-43 in the cytoplasm is likely to recruit and sequester their interacting proteins into either SGs or inclusions and subsequently lead to disease pathogenesis induced by loss-of-function in motor neurons (Kamelgarn et al., 2016). Mislocalization of RNA-binding proteins (such as TDP-43 and FUS/TLS) and the formation/assembly of SGs have therefore attracted a great deal of attention in ALS research in recent years. Of note, because not all stress models show SG formation (Takahashi et al., 2013) and SG formation may be protective for cells (Anderson and Kedersha, 2008), whether the formation of TDP-43 inclusion is detrimental remains unknown (Baloh, 2011; Cragnaz et al., 2014), and the detailed function and regulation of SGs in ALS requires continued investigation.
In the following section, we will focus on the roles of TDP-43, FUS/TLS and the C9ORF72 hexanucleotide repeat expansion in the regulation of RNA metabolism in ALS. Although the compositions and properties of SGs have not been well characterized, the discovery of AMPK holoenzymes in SGs is intriguing (Mahboubi et al., 2015a,b). The abnormalities of energy metabolism and its downstream signals in modulating RNA homeostasis will also be discussed.
TDP-43
In ALS, TDP-43 is a key pathological protein that forms cytosolic aggregates. TDP-43 consists of 414 amino acids and contains two RNA recognition motifs, RRM1 and RRM2. It is normally localized to nuclei and affects several RNA-related processes, including RNA splicing, RNA transcription, RNA transport and miRNA production. Previous findings have indicated that TDP-43 binds to more than 6000 RNA targets in the brain (Tollervey et al., 2011; Colombrita et al., 2012; Polymenidou et al., 2012). Earlier studies have suggested that TDP-43 is a transcriptional regulator that regulates the expression of many proteins (including CHMP2B, OPTN, VAPB and VCP) that are involved in protein homeostasis (Polymenidou et al., 2011) and in autophagy (e.g., the autophagy-related 7, Bose et al., 2011). TDP-43 also affects RNA stability by binding to the 3′-untranslated regions (3′UTRs) of specific mRNAs and regulating mRNA degradation (Polymenidou et al., 2011). Neuronal activation induces the translocation of TDP-43 to dendrites, where TDP-43 regulates RNA targets critical for synaptic functions (Wang et al., 2008).
In addition, TDP-43 is involved in miRNA biogenesis (Kawahara and Mieda-Sato, 2012). For example, down-regulation of TDP-43 increases the expression of miR-633 but decreases that of let-7b miRNA (Buratti et al., 2010). TDP-43 also controls the splicing pattern of many important genes, including amyloid beta precursor protein (APP), presenilin, huntingtin, multiple ataxins, α-synuclein, progranulin, FUS/TLS and TDP-43 itself (Polymenidou et al., 2011; Sephton et al., 2011). In addition, TDP-43 binds to long (>200 base) non-coding RNAs (ncRNAs), including the two highly expressed ncRNAs, nuclear-enriched autosomal transcript 1 (NEAT1) and metastasis-associated lung adenocarcinoma transcript 1 (MALAT1; Tollervey et al., 2011). Although the functional roles of this interaction remain unclear, both of these ncRNAs appear to be important because their levels are increased in patients with FTLD and ALS (Tollervey et al., 2011; Nishimoto et al., 2013). In post-mortem cortical tissues of FTLD patients, the amount of NEAT1 that binds to TDP-43 is also enhanced (Tollervey et al., 2011). NEAT1 is a component of a subnuclear structure (i.e., paraspeckles, huge RNP particles) and functions as a scaffold for RNA-binding proteins and RNAs. The functions of paraspeckles have not been clearly defined yet. Previous studies suggest that paraspeckles may be involved in the nucleocytoplasmic shuttling of specific mRNAs, transcription and pre-miRNA splicing under pathophysiological conditions (Naganuma and Hirose, 2013). Analyses of the spinal cords of ALS patients have shown that paraspeckles with high levels of NEAT1 and TDP-43 are found primarily in motor neurons of patients with early-stage ALS (Nishimoto et al., 2013). Significant recruitment of TDP-43 to paraspeckles might interfere with TDP-43-mediated RNA processing and disrupt RNA homeostasis in ALS motor neurons.
TDP-43 has been reported to co-localize with SG proteins, such as poly-A binding protein-1 (PABP-1; McGurk et al., 2014). Furthermore, several reports have demonstrated that TDP-43 localizes within SGs after stress treatment, including oxidative stress, heat shock and osmotic stress (Colombrita et al., 2009; Freibaum et al., 2010; Liu-Yesucevitz et al., 2010; Dewey et al., 2011). The recruitment of TDP-43 into SGs may be an initiating event, which could trigger pathological inclusion formation (Dewey et al., 2012; Li et al., 2013). In addition, TDP-43 inclusions are known to recruit important proteins into the inclusions and consequently affect their normal functions. For example, the RNA-binding motif 45 (RBM45) co-localizes with cytosolic TDP-43 inclusions in patients with ALS (Collins et al., 2012). Previous studies have suggested that RBM45 normally exists in nuclei, binds poly(C) RNAs and mediates the antioxidant response (Bakkar et al., 2015). Therefore, TDP-43 may regulate RNA homeostasis by controlling the amount of other RNA-binding proteins.
Mislocalization of TDP-43 has been reported in motor neurons of ALS patients (Arai et al., 2006; Neumann et al., 2006; Liu et al., 2015a). Giordana et al. (2010) have proposed the intriguing hypothesis that TDP-43 redistribution might be an early event in sporadic ALS). Indeed, mislocalization of TDP-43 in the cytoplasm during stress (e.g., oxidative stress, inflammation, energy dysfunction), as reported by several laboratories (Ayala et al., 2011; Correia et al., 2015; Liu et al., 2015a,b; Xia et al., 2016), might subsequently promote the formation of detrimental TDP-43 oligomers, decrease the normal amount of nuclear TDP-43, and cause detrimental effects. Of note, whether mislocalization of TDP-43 was observed in motor neurons of ALS-mSOD1 mouse models remains controversial. While no TDP-43 mislocalization was detected in motor neurons of three mSOD1 ALS mouse models (i.e., G93A, G37R and G85R; Robertson et al., 2007). Shan et al. (2009) observed that mislocalization of TDP-43 occurs in motor neurons of mSOD1-G93A mice having advanced disease). Importantly, redistribution of TDP-43 to the cytoplasmic region in motor neurons was observed in familial ALS patients carrying mutations in SOD1 (Robertson et al., 2007). These seemingly contradictory findings suggest that mSOD1-ALS mouse models may not fully recapitulate human ALS.
Lim et al. (2012) first reported the importance of AMPK in ALS, by using several genetic experimental models, and suggested that suppression of AMPK activity is protective in ALS. Liu et al. (2015a) have further reported higher activity of AMPK in motor neurons of ALS patients and animals. Pharmacological and genetic approaches have demonstrated that AMPK activation positively contributes to TDP-43 mislocalization. Given that AMPK is a major energy sensor and is usually activated in cells when the cellular ATP level is low or the cells are stressed (Long and Zierath, 2006; Hardie, 2008), these studies link energy dysfunction with TDP-43 mislocalization, which inevitably impairs RNA homeostasis, owing to the altered amount or/and cellular localization of TDP-43 in motor neurons as described above. Given that mitochondrial defects have been implicated in ALS and AMPK activation in studies during the past two decades (Dupuis et al., 2004a; Duffy et al., 2011; Watanabe et al., 2016), further investigations into the role of AMPK and its downstream pathways involved in the mislocalization of TDP-43 and RNA homeostasis would pave the way for a better understanding of the initiation of sporadic ALS.
Collectively, depletion of nuclear TDP-43, either by mislocalization to the cytoplasmic region or/and chelation by paraspeckles/Neat 1, TDP-43 inclusion or SGs would induce the dysregulation of TDP43-related RNA targets and contribute to ALS pathology.
FUS/TLS
In addition to TDP-43, FUS/TLS is another important disease-causing gene in ALS. FUS/TLS-containing inclusions are also found in ALS motor neurons (Bäumer et al., 2010; Huang et al., 2010; Yamashita et al., 2012). FUS/TLS consists of 526 amino acids and is mainly located in nuclei. Several mutant forms of FUS/TLS, which are associated with ALS, are located in the cytoplasmic region (Kwiatkowski et al., 2009; Vance et al., 2009). Similarly to TDP-43, FUS/TLS is an RNA-binding protein that mediates multiple cellular pathways, including transcriptional regulation, mRNA splicing and miRNA production. For example, FUS/TLS interacts with several nuclear hormone receptors and modulates their functions (Powers et al., 1998). FUS/TLS also acts as a co-modulator for certain transcription factors (including NF-κB, SPI1 and RUNX (Runbox) transcription factor; Hallier et al., 1998; Uranishi et al., 2001; Li et al., 2010). Genome-wide approaches have revealed more than 5000 human RNA targets and more than 8000 mouse RNA targets for FUS/TLS (Lagier-Tourenne et al., 2012; Rogelj et al., 2012). Because FUS/TLS is a component of the hnRNP complex, it plays an important role in the splicing mechanism (Iko et al., 2004) and may alter the splicing patterns of more than 900 mRNAs (Lagier-Tourenne et al., 2012). Biochemical evidence has suggested that FUS/TLS regulates several mRNA targets that are important for spine and dendritic morphology (Fujii et al., 2005). Down-regulation of FUS/TLS causes abnormal synaptic function. Moreover, FUS/TLS is a component of the microprocessor complex and mediates the biogenesis of miRNA (Gregory et al., 2004). Similarly to loss of TDP-43, loss of FUS/TLS nuclear function is accompanied by the cytosolic inclusion of FUS/TLS in ALS motor neurons (Kwiatkowski et al., 2009; Vance et al., 2009).
During stress, FUS/TLS is recruited into SGs and co-localizes with an SG marker, PABP-1 (Andersson et al., 2008; Gal et al., 2011). Importantly, FUS/TLS with an ALS-linked mutation in its nuclear localization signal (NLS) causes the FUS/TLS variant to localize to the SGs more often (Bosco et al., 2010; Dormann et al., 2010; Ito et al., 2011). Like TDP-43, FUS/TLS also binds to several mRNAs of ALS-related genes (including VCP, VAPB, ubiquilin-2 and OPTN, Hoell et al., 2011; Colombrita et al., 2012; Lagier-Tourenne et al., 2012) and further modulates their expression. In addition, FUS/TLS is a key factor that regulates NEAT 1 stability and maintains nuclear body structure (Shelkovnikova et al., 2014). Interaction between FUS/TLS and NEAT 1 may contribute to the development of neuronal dysfunction in ALS.
Interestingly, activation of AMPK with AICAR (an AMPK activator, Corton et al., 1995) has been found to induce mislocalization of FUS/TLS in a motor neuron cell line (Lim et al., 2012; Liu et al., 2015a, b). Thus, similarly to TDP-43 (Liu et al., 2015a), abnormal activation of AMPK caused by impaired energy homeostasis in motor neurons might substantially alter RNA homeostasis by changing the cellular distribution of FUS/TLS.
C9ORF72
The hexanucleotide repeat expansion of C9ORF72 is a frequent mutation observed in patients with ALS (5%–20% of sporadic ALS and 20%–50% of familial ALS; Boeve et al., 2012; Chio et al., 2012; Cooper-Knock et al., 2012). Patients with the C9ORF72 repeat expansion usually have earlier disease onset and accelerated disease progression, as compared with patients without the C9 repeat expansion (Byrne et al., 2012; Chio et al., 2012; Millecamps et al., 2012). The (G4C2) expansion transcripts are produced from the non-coding exons 1a and 1b of the C9ORF72 gene (C9, Gunnarsson et al., 1991) and the repeat number can be expanded to 700–1600 copies (DeJesus-Hernandez et al., 2011; Gijselinck et al., 2012). Although the function of the C9ORF72 gene remains unclear, expansion of the G4C2 repeats induces degeneration of motor neurons. Bioinformatic analysis has suggested that the C9ORF72 protein may be involved in autophagy and membrane trafficking (Zhang et al., 2012; Levine et al., 2013). Ciura et al. (2013) have shown that a decrease in C9ORF72 in zebrafish causes a locomotion deficit and reduces axon length). In addition, the G4C2 transcript frequently co-localizes with TDP-43 inclusions (McGurk et al., 2014).
RNA foci of the C9ORF72 repeat expansion were initially observed in the spinal cord of ALS/FTD patients (DeJesus-Hernandez et al., 2011; Renton et al., 2011), although the RNA level of C9ORF72 is decreased in patients with ALS and the C9ORF72 repeat expansion (Renton et al., 2011). The formation of RNA foci facilitates the recruitment of RNA-binding proteins and thus might interfere with their normal functions (Miller et al., 2000; Simón-Sánchez et al., 2012). For example, G4C2 RNA repeats interact with hnRNP-H, which mediates TARBP2 RNA splicing. Exogenous expression of G4C2 repeats decreases the levels of TARBP2 transcripts. This finding indicates that the C9ORF72 repeat expansion induces RNA toxicity by disrupting the function of hnRNP-H (Lee et al., 2013). Formation of the C9ORF72 repeat expansion-containing RNA foci also sequesters hnRNP-A3 and represses its RNA processing function (Mori et al., 2013a). Down-regulation of C9ORF72 RNA by antisense oligonucleotides decreases the number of RNA foci and RNA toxicity in human motor neurons derived from induced pluripotent stem cells (iPSCs) of ALS patients (Donnelly et al., 2013; Lagier-Tourenne et al., 2013). Collectively, these studies suggest that the C9ORF72 repeat expansion disturbs cellular RNA homeostasis by formation of RNA foci that recruit RNA-binding proteins. In addition, similarly to TDP-43, the C9ORF72 repeat expansion has been found in PABP-1-containing SG in patients with ALS (McGurk et al., 2014).
Importantly, C9ORF72 repeat expansion had been implicated in the impairment of nuclear import by interacting with a Ran GTPase-activating protein (RanGAP) in a Drosophila model and in motor neurons derived from iPSCs of C9-ALS patients. This impairment of nuclear transport can be rescued by antisense oligonucleotides against the (G4C2) G-quadruplexes, thus suggesting the importance of the hexanucleotide repeat expansion (Zhang et al., 2015). Another study also suggested that G4C2 repeat expansion causes the abnormalities of the nuclear envelope and results in dysfunction of nucleocytoplasmic transport (Freibaum et al., 2015). Consistent with the importance of nucleocytoplasmic transport in ALS, screening for modifiers of C9ORF72 toxicity using either Drosophila or yeast also identified a group of genes (such as importins, proteins of nuclear pore complex and Ran-GTP cycle regulators) involved in nucleocytoplasmic transport (Jovičić et al., 2015; Boeynaems et al., 2016a; Figure 1). Moreover, the C9ORF72 repeat expansion also induces protein toxicity by the repeat-associated non-ATG translation (RAN translation), which allows C9ORF72 repeat transcripts to be translated into dipeptide repeat (DPR) proteins consisting of di-amino acids. These DPRs contain poly-(glycine-alanine, GA), poly-(glycine-proline, GP), poly-(glycine-arginine, GR) and poly-(proline-alanine, PA) and form pathological inclusions in ALS (Mori et al., 2013b). Notably, impairment of nuclear transport by one specific DPR aggregate (i.e., poly-GA) has recently been shown to cause TDP-43 mislocalization in primary neurons (Khosravi et al., 2017), which further worsen RNA toxicity as described above. These findings further highlight the important role of nucleo-cytoplasmic transport in C9-ALS.
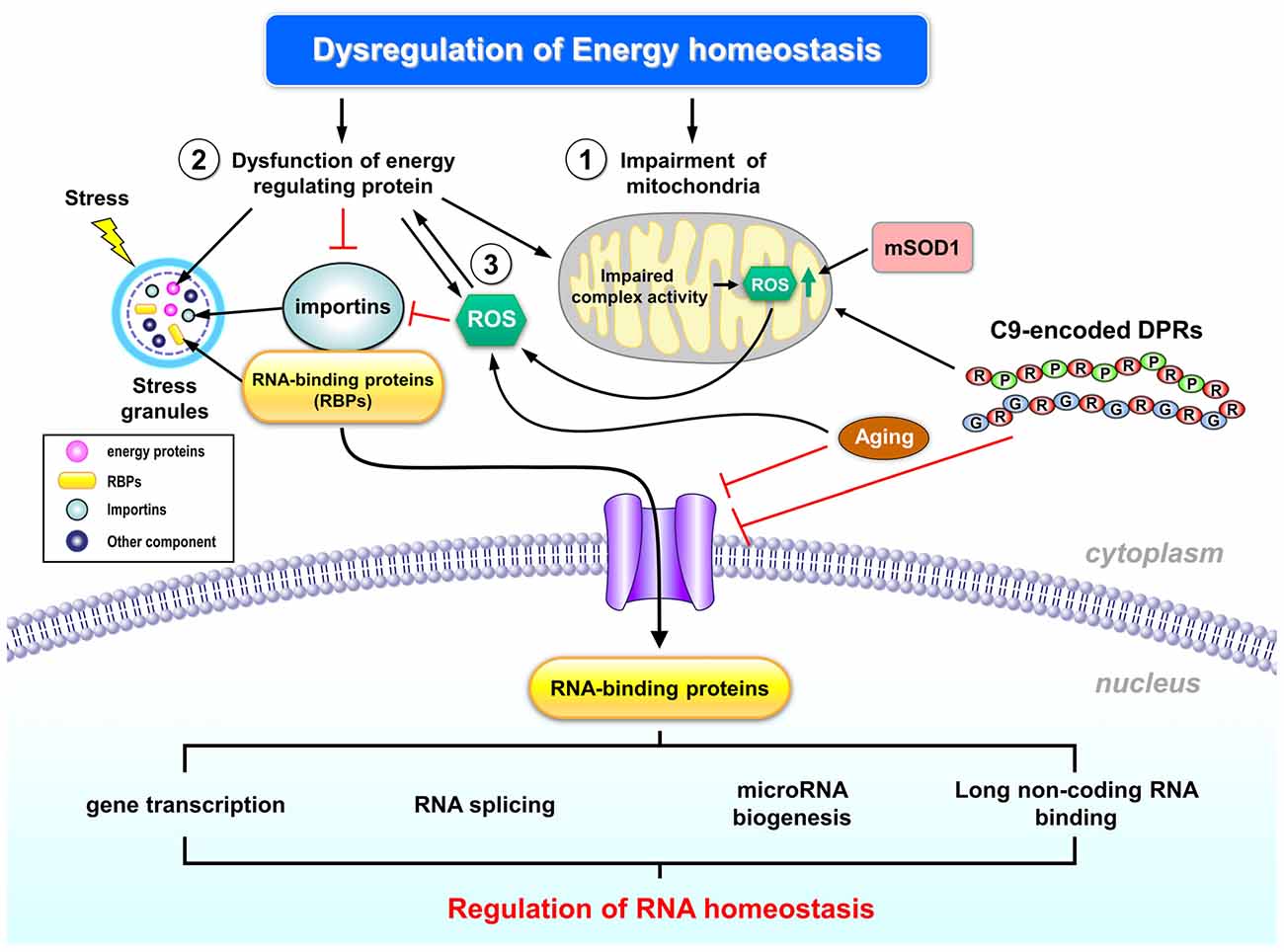
Figure 1. Schematic representation of the potential regulation of RNA homeostasis by abnormal energy metabolism in amyotrophic lateral sclerosis (ALS). Dysregulated energy homeostasis might result from dysregulation of mitochondrial function, which has been well documented in ALS pathogenesis (1). Many ALS- associated genes (including TDP-43, FUS/TLS and C9ORF72) have been implicated in the regulation of mitochondrial functions and RNA metabolism, thus suggesting that altered mitochondrial functions and RNA toxicity might contribute to ALS pathogenesis. In addition, dysfunction of energy-regulating proteins may also contribute to dysregulated energy homeostasis in ALS (2). Specifically, recent studies suggest that abnormal cellular energy status in ALS enhances activity of an energy sensor (AMP kinase, AMPK) that interferes with the transport of several RNA-binding proteins (RBPs) into nuclei of motor neurons, which might be one of the early events in ALS pathogenesis. The impaired transport of nuclear RBPs might disrupt cellular RNA homeostasis. Moreover, reactive oxygen species (ROS) production might also cause the abnormal activation of energy regulating proteins (3), and subsequently led to the mislocalization of RBPs. Further investigation is required to delineate the potential regulation of RNA homeostasis by energy dysfunction, to explore the key players involved in such regulation, and to consolidate the role of such abnormality in ALS pathogenesis. See text for additional details.
Expression of the C9ORF72 repeat expansion and the resultant DPRs is associated with abnormal mitophagy and impaired mitochondrial function (Lopez-Gonzalez et al., 2016; Onesto et al., 2016), thus supporting the potential involvement of energy dysregulation in C9-associated ALS. Further investigations are required to evaluate whether and how the RNA toxicity and energy dysfunction caused by C9ORF72 repeat expansion cross-regulate each other.
Importins and AMPK in Nucleocytoplasmic Transport
As noted by several recent studies and reviews, the loss of nuclear functions of many RNA-binding proteins and nuclear transport components and the gain of function and/or aggregate formation of these proteins in the cytoplasm may contribute to ALS pathogenesis (Zhang et al., 2015, 2016; Boeynaems et al., 2016a,b; Jovičić et al., 2016; Taylor et al., 2016). Mutations in several major ALS genes (e.g., TDP-43, FUS/TLS) and the aging process appear to trigger these progressive impairments (Jovičić et al., 2016). Recent studies have demonstrated that aggregates located in the cytoplasm are likely to interfere with nucleocytoplasmic transport functions (Khosravi et al., 2017; Woerner et al., 2016). Nucleocytoplasmic transport has become one of the most heavily researched topics in ALS pathogenesis, because the disturbance of this process might be the crucial and initial step that triggers ALS pathogenesis in motor neurons (Dormann and Haass, 2011). Among all of the proteins involved in nucleocytoplasmic transport, we are most interested in importins because they are regulated by AMPK (Wang et al., 2004).
Importins are key molecules in the control of nucleocytoplasmic transport because they are needed for transporting macromolecules larger than 40 kDa (such as proteins; Görlich and Kutay, 1999; Depping et al., 2015). Most proteins transported by importin α/β contain specific NLSs. A total of six different NLS classes have been reported, and each of them binds to a different binding site on the importin α isoform (Kosugi et al., 2009). Interestingly, there are six importin α subtypes in mice, and seven importin α subtypes in human, but only one importin β is present in both organisms (Miyamoto et al., 2016). These importin α subtypes not only function in the control of nucleocytoplasmic transport but also have been implicated in mRNA biogenesis (Wen and Shatkin, 2000), and protein degradation (Kim et al., 2014).
Multiple lines of evidence suggest the involvement of importins in ALS. First, we have previously demonstrated that AMPK activity is higher in motor neurons of the ALS spinal cord when compared with that of non-ALS subjects (Liu et al., 2015a). Second, cellular localization of importin-α1 in the motor neurons of patients with ALS is altered. Most importantly, phosphorylation of importin-α1 by AMPK caused the mislocalization of importin and human antigen R (HuR, a major RNA stability factor, Nabors et al., 2001; Doller et al., 2008) in the cytoplasm (Liu et al., 2015b). Notably, HuR has been implicated in ALS (Lu L. et al., 2009; Lu et al., 2014). It is also intriguing that importins are located in SGs and have been implicated in ALS pathology by bioinformatic analysis (Boeynaems et al., 2016b). Mislocalization of HuR in motor neurons is closely associated with a lower level of VEGF in patients with ALS and is known to regulate the expression of TDP-43 and FUS/TLS (Lu et al., 2014). This is also an interesting finding because VEGF is known to protect against neurotoxicity and delay disease progression in a mouse model of ALS (Lladó et al., 2013; Wang et al., 2016). It is very likely that the AMPK-mediated phosphorylation of importin-α1 may lead to mislocalization of HuR, thus negatively affecting the normal function of HuR in RNA processing and eventually causing ALS pathology. For example, it has been demonstrated that mislocalization of HuR by abnormal activation of AMPK in a motor neuron cell line (NSC34) reduces the half-life of its target mRNA (i.e., VEGF; Liu et al., 2015b). This finding is consistent with an earlier study that mislocalization of HuR in motor neurons of ALS patients is closely associated with a lower level of VEGF (Lu et al., 2014). Together, these observations suggest that energy dysfunction in ALS might alter the cellular distribution of RBPs (such as HuR) and subsequently alter homeostasis of their target mRNAs (e.g., VEGF).
This alteration in the function of importin-α1 induced by AMPK phosphorylation is likely to be a common pathogenic pathway that may contribute, at least partially, to the RNA toxicity observed in ALS. In accordance with the importance of importins in ALS, up-regulation of importin α has been found to be beneficial in a fly model of ALS that exogenously expresses the C9ORF72 repeat expansion (Zhang et al., 2015). In contrast, suppression of importin β1 enhances the toxicity of DRPs (Boeynaems et al., 2016a). These data collectively suggest that importins and abnormal activation of AMPK are important therapeutic targets for ALS.
RNA Transport
Proper RNA transport is critical for cellular RNA homeostasis. In highly polarized cells, such as motor neurons, mRNA can be transported to neurites. Motor neurons are known to have local protein synthesis in their synapses after exposure to stimuli (Yoo et al., 2010). Although TDP-43 and FUS/TLS are primarily considered to be nuclear proteins, several studies have suggested that these proteins have cytoplasmic functions, including local translation with synaptic plasticity, mRNA transport and RNA stabilization. Several studies demonstrated that cytosolic TDP-43 and FUS/TLS can transport mRNA to dendritic spines after stimulus exposure (Kanai et al., 2004; Belly et al., 2005; Fujii and Takumi, 2005; Wang et al., 2008). In addition, FUS/TLS may recruit mRNA to RNA granules and transport them to dendritic spines after activation of the metabotropic glutamate receptor 5 (mGluR5; Fujii et al., 2005). After post-synaptic activation of mGluR5, the FUS/TLS-containing complex may facilitate the activity of the actin-based motor by interacting with myosin (Yoshimura et al., 2006; Takarada et al., 2009). Similarly, cytoplasmic TDP-43 has been demonstrated to trigger dendrite formation (Feiguin et al., 2009; Lu Y. et al., 2009). The TDP-43-containing RNA complex is also known to interact with PSD-95 and to regulate neuronal activity (Wang et al., 2008). TDP-43 and FUS/TLS also regulate transport ribonucleoprotein particles (tRNPs; Sephton and Yu, 2015). Additional functions of TDP-43 include the stabilization of low molecular weight neurofilament mRNA by binding to the UG motif (Strong et al., 2007; Volkening et al., 2009) and the regulation of the transport and local translation of important transcripts (e.g., fragile mental retardation protein and Staufen) in neurons (Wang et al., 2008). Given that RNA transport to dendrites requires energy (Davis et al., 1987), dysregulation of energy homeostasis in motor neurons is expected to contribute to the RNA toxicity observed in ALS because the correct localization of RNAs is critical for neuronal activity. RNAs localize to dendrites and translation of these mRNAs are important for synaptic remodeling and plasticity (Martin and Zukin, 2006). The abnormal localization of RNAs caused by impaired energy homeostasis may disrupt RNA homeostasis and further cause RNA toxicity.
Concluding Remarks
As described above, many ALS genes encode RNA-binding proteins (RBPs, Figure 1) that control RNA homeostasis and have been implicated in the regulation of mitochondrial function. During various cellular stresses (including elevated oxidative stress, impaired mitochondrial function, ER stress and aging), mislocalization of some RBPs (e.g., TDP-43, FUS/TLS and HuR) might occur in motor neurons. This aberrant cellular distribution of RBPs is likely to disrupt RNA homeostasis in motor neurons, and was proposed to be one of the early events in the development of ALS. Future investigations should further delineate the pathway(s) and explore the crucial molecular players underlying the potential regulation of RNA homeostasis by energy dysfunction, and consolidate whether such abnormality plays an important role in ALS pathogenesis.
Author Contributions
Y-JL wrote the first draft from section Abnormal Energy Homeostasis in ALS to section C9ORF72, P-YT wrote the first draft of section Importins and AMPK in Nucleocytoplasmic Transport and RNA Transport, YC organized the manuscript and wrote the final draft.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from Ministry of Science and Technology (Taiwan; 102-2321-B-001-068-MY3) and Academia Sinica (Taiwan; AS-106-TP-B13). We are grateful to the Art Rom of Institute of Biomedical Sciences at Academia Sinica for its assistance in preparing the illustration.
References
Ahmed, R. M., Irish, M., Piguet, O., Halliday, G. M., Ittner, L. M., Farooqi, S., et al. (2016). Amyotrophic lateral sclerosis and frontotemporal dementia: distinct and overlapping changes in eating behaviour and metabolism. Lancet Neurol. 15, 332–342. doi: 10.1016/S1474-4422(15)00380-4
Al-Chalabi, A., Hardiman, O., Kiernan, M. C., Chiò, A., Rix-Brooks, B., and van den Berg, L. H. (2016). Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 15, 1182–1194. doi: 10.1016/S1474-4422(16)30199-5
Anderson, P., and Kedersha, N. (2008). Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33, 141–150. doi: 10.1016/j.tibs.2007.12.003
Andersson, M. K., Ståhlberg, A., Arvidsson, Y., Olofsson, A., Semb, H., Stenman, G., et al. (2008). The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 9:37. doi: 10.1186/1471-2121-9-37
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Aulas, A., and Vande Velde, C. (2015). Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Front. Cell. Neurosci. 9:423. doi: 10.3389/fncel.2015.00423
Ayala, V., Granado-Serrano, A. B., Cacabelos, D., Naudi, A., Ilieva, E. V., Boada, J., et al. (2011). Cell stress induces TDP-43 pathological changes associated with ERK1/2 dysfunction: implications in ALS. Acta Neuropathol. 122, 259–270. doi: 10.1007/s00401-011-0850-y
Azzouz, M., Ralph, G. S., Storkebaum, E., Walmsley, L. E., Mitrophanous, K. A., Kingsman, S. M., et al. (2004). VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 429, 413–417. doi: 10.1038/nature02544
Bakkar, N., Kousari, A., Kovalik, T., Li, Y., and Bowser, R. (2015). RBM45 modulates the antioxidant response in amyotrophic lateral sclerosis through interactions with KEAP1. Mol. Cell. Biol. 35, 2385–2399. doi: 10.1128/MCB.00087-15
Baloh, R. H. (2011). TDP-43: the relationship between protein aggregation and neurodegeneration in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. FEBS J. 278, 3539–3549. doi: 10.1111/j.1742-4658.2011.08256.x
Bañez-Coronel, M., Porta, S., Kagerbauer, B., Mateu-Huertas, E., Pantano, L., Ferrer, I., et al. (2012). A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 8:e1002481. doi: 10.1371/journal.pgen.1002481
Baron, D. M., Kaushansky, L. J., Ward, C. L., Sama, R. R. K., Chian, R.-J., Boggio, K. J., et al. (2013). Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 8:30. doi: 10.1186/1750-1326-8-30
Bäumer, D., Hilton, D., Paine, S. M. L., Turner, M. R., Lowe, J., Talbot, K., et al. (2010). Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology 75, 611–618. doi: 10.1212/WNL.0b013e3181ed9cde
Belly, A., Moreau-Gachelin, F., Sadoul, R., and Goldberg, Y. (2005). Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci. Lett. 379, 152–157. doi: 10.1016/j.neulet.2004.12.071
Bentmann, E., Neumann, M., Tahirovic, S., Rodde, R., Dormann, D., and Haass, C. (2012). Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094. doi: 10.1074/jbc.M111.328757
Bento-Abreu, A., Van Damme, P., Van Den Bosch, L., and Robberecht, W. (2010). The neurobiology of amyotrophic lateral sclerosis. Eur. J. Neurosci. 31, 2247–2265. doi: 10.1111/j.1460-9568.2010.07260.x
Beretta, S., Sala, G., Mattavelli, L., Ceresa, C., Casciati, A., Ferri, A., et al. (2003). Mitochondrial dysfunction due to mutant copper/zinc superoxide dismutase associated with amyotrophic lateral sclerosis is reversed by N-acetylcysteine. Neurobiol. Dis. 13, 213–221. doi: 10.1016/s0969-9961(03)00043-3
Boeve, B. F., Boylan, K. B., Graff-Radford, N. R., DeJesus-Hernandez, M., Knopman, D. S., Pedraza, O., et al. (2012). Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Acta Neuropathol. Commun. 135, 765–783. doi: 10.1093/brain/aws004
Boeynaems, S., Bogaert, E., Michiels, E., Gijselinck, I., Sieben, A., Jovičić, A., et al. (2016a). Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 6:20877. doi: 10.1038/srep20877
Boeynaems, S., Bogaert, E., Van Damme, P., and Van Den Bosch, L. (2016b). Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 132, 159–173. doi: 10.1007/s00401-016-1586-5
Borthwick, G. M., Johnson, M. A., Ince, P. G., Shaw, P. J., and Turnbull, D. M. (1999). Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 46, 787–790. doi: 10.1002/1531-8249(199911)46:5<787::AID-ANA17>3.0.CO;2-8
Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J. Jr., et al. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19, 4160–4175. doi: 10.1093/hmg/ddq335
Bose, J. K., Huang, C. C., and Shen, C. K. (2011). Regulation of autophagy by neuropathological protein TDP-43. J. Biol. Chem. 286, 44441–44448. doi: 10.1074/jbc.M111.237115
Brook, J. D., McCurrach, M. E., Harley, H. G., Buckler, A. J., Church, D., Aburatani, H., et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 69:385. doi: 10.1016/0092-8674(92)90154-5
Bruijn, L. I., Houseweart, M. K., Kato, S., Anderson, K. L., Anderson, S. D., Ohama, E., et al. (1998). Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854. doi: 10.1126/science.281.5384.1851
Buchan, J. R., and Parker, R. (2009). Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36, 932–941. doi: 10.1016/j.molcel.2009.11.020
Buratti, E., De Conti, L., Stuani, C., Romano, M., Baralle, M., and Baralle, F. (2010). Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J. 277, 2268–2281. doi: 10.1111/j.1742-4658.2010.07643.x
Byrne, S., Elamin, M., Bede, P., Shatunov, A., Walsh, C., Corr, B., et al. (2012). Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 11, 232–240. doi: 10.1016/S1474-4422(12)70014-5
Chang, Y.-C., Hung, W.-T., Chang, H.-C., Wu, C.-L., Chiang, A.-S., Jackson, G. R., et al. (2011). Pathogenic VCP/TER94 alleles are dominant actives and contribute to neurodegeneration by altering cellular ATP level in a Drosophila IBMPFD model. PLoS Genet. 7:e1001288. doi: 10.1371/journal.pgen.1001288
Chiang, P. M., Ling, J., Jeong, Y. H., Price, D. L., Aja, S. M., and Wong, P. C. (2010). Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc. Natl. Acad. Sci. U S A 107, 16320–16324. doi: 10.1073/pnas.1002176107
Chio, A., Calvo, A., Ilardi, A., Cavallo, E., Moglia, C., Mutani, R., et al. (2009a). Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology 73, 1681–1685. doi: 10.1212/WNL.0b013e3181c1df1e
Chio, A., Logroscino, G., Hardiman, O., Swingler, R., Mitchell, D., Beghi, E., et al. (2009b). Prognostic factors in ALS: a critical review. Amyotroph. Lateral Scler. 10, 310–323. doi: 10.3109/17482960802566824
Chio, A., Borghero, G., Restagno, G., Mora, G., Drepper, C., Traynor, B. J., et al. (2012). Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 135, 784–793. doi: 10.1093/brain/awr366
Chou, S. Y., Lee, Y. C., Chen, H. M., Chiang, M. C., Lai, H. L., Chang, H. H., et al. (2005). CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J. Neurochem. 93, 310–320. doi: 10.1111/j.1471-4159.2005.03029.x
Ciura, S., Lattante, S., Le Ber, I., Latouche, M., Tostivint, H., Brice, A., et al. (2013). Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 74, 180–187. doi: 10.1002/ana.23946
Collins, M., Riascos, D., Kovalik, T., An, J., Krupa, K., Hood, B. L., et al. (2012). The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 124, 717–732. doi: 10.1007/s00401-012-1045-x
Colman, E., Szarfman, A., Wyeth, J., Mosholder, A., Jillapalli, D., Levine, J., et al. (2008). An evaluation of a data mining signal for amyotrophic lateral sclerosis and statins detected in FDA’s spontaneous adverse event reporting system. Pharmacoepidemiol. Drug Saf. 17, 1068–1076. doi: 10.1002/pds.1643
Colombrita, C., Onesto, E., Megiorni, F., Pizzuti, A., Baralle, F. E., Buratti, E., et al. (2012). TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 287, 15635–15647. doi: 10.1074/jbc.M111.333450
Colombrita, C., Zennaro, E., Fallini, C., Weber, M., Sommacal, A., Buratti, E., et al. (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x
Cooper-Knock, J., Hewitt, C., Highley, J. R., Brockington, A., Milano, A., Man, S., et al. (2012). Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 135, 751–764. doi: 10.5772/30897
Correia, A. S., Patel, P., Dutta, K., and Julien, J. P. (2015). Inflammation induces TDP-43 mislocalization and aggregation. PLoS One 10:e0140248. doi: 10.1371/journal.pone.0140248
Corton, J. M., Gillespie, J. G., Hawley, S. A., and Hardie, D. G. (1995). 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229, 558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x
Coussee, E., De Smet, P., Bogaert, E., Elens, I., Van Damme, P., Willems, P., et al. (2011). G37R SOD1 mutant alters mitochondrial complex I activity, Ca2+ uptake and ATP production. Cell Calcium 49, 217–225. doi: 10.1016/j.ceca.2011.02.004
Cragnaz, L., Klima, R., Skoko, N., Budini, M., Feiguin, F., and Baralle, F. E. (2014). Aggregate formation prevents dTDP-43 neurotoxicity in the Drosophila melanogaster eye. Neurobiol. Dis. 71, 74–80. doi: 10.1016/j.nbd.2014.07.009
Crugnola, V., Lamperti, C., Lucchini, V., Ronchi, D., Peverelli, L., Prelle, A., et al. (2010). Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch. Neurol. 67, 849–854. doi: 10.1001/archneurol.2010.128
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
Davis, L., Banker, G. A., and Steward, O. (1987). Selective dendritic transport of RNA in hippocampal neurons in culture. Nature 330, 477–479. doi: 10.1038/330477a0
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deng, J., Yang, M., Chen, Y., Chen, X., Liu, J., Sun, S., et al. (2015). FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 11:e1005357. doi: 10.1371/journal.pgen.1005357
Depping, R., Jelkmann, W., and Kosyna, F. K. (2015). Nuclear-cytoplasmatic shuttling of proteins in control of cellular oxygen sensing. J. Mol. Med. 93, 599–608. doi: 10.1007/s00109-015-1276-0
Desport, J. C., Preux, P. M., Magy, L., Boirie, Y., Vallat, J. M., Beaufrere, B., et al. (2001). Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 74, 328–334.
Desport, J. C., Preux, P. M., Truong, C. T., Courat, L., Vallat, J. M., and Couratier, P. (2000). Nutritional assessment and survival in ALS patients. Amyotroph. Lateral Scler Other Motor Neuron Disord. 1, 91–96. doi: 10.1080/14660820050515386
Desport, J. C., Preux, P. M., Truong, T. C., Vallat, J. M., Sautereau, D., and Couratier, P. (1999). Nutritional status is a prognostic factor for survival in ALS patients. Neurology 53, 1059–1063. doi: 10.1212/WNL.53.5.1059
Desport, J. C., Torny, F., Lacoste, M., Preux, P. M., and Couratier, P. (2005). Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener. Dis. 2, 202–207. doi: 10.1159/000089626
Dewey, C. M., Cenik, B., Sephton, C. F., Dries, D. R., Mayer, P. III., Good, S. K., et al. (2011). TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 31, 1098–1108. doi: 10.1128/MCB.01279-10
Dewey, C. M., Cenik, B., Sephton, C. F., Johnson, B. A., Herz, J., and Yu, G. (2012). TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain Res. 1462, 16–25. doi: 10.1016/j.brainres.2012.02.032
Dobrowolny, G., Aucello, M., Rizzuto, E., Beccafico, S., Mammucari, C., Boncompagni, S., et al. (2008). Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 8, 425–436. doi: 10.1016/j.cmet.2008.09.002
Doller, A., Pfeilschifter, J., and Eberhardt, W. (2008). Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell. Signal. 20, 2165–2173. doi: 10.1016/j.cellsig.2008.05.007
Domise, M., Didier, S., Marinangeli, C., Zhao, H., Chandakkar, P., Buée, L., et al. (2016). AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci. Rep. 6:26758. doi: 10.1038/srep26758
Donnelly, C. J., Zhang, P. W., Pham, J. T., Haeusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.015
Dormann, D., and Haass, C. (2011). TDP-43 and FUS: a nuclear affair. Trends Neurosci. 34, 339–348. doi: 10.1016/j.tins.2011.05.002
Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 29, 2841–2857. doi: 10.1038/emboj.2010.143
Dorst, J., Kuhnlein, P., Hendrich, C., Kassubek, J., Sperfeld, A. D., and Ludolph, A. C. (2011). Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J. Neurol. 258, 613–617. doi: 10.1007/s00415-010-5805-z
Duffy, L. M., Chapman, A. L., Shaw, P. J., and Grierson, A. J. (2011). Review: the role of mitochondria in the pathogenesis of amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 37, 336–352. doi: 10.1111/j.1365-2990.2011.01166.x
Dupuis, L., Corcia, P., Fergani, A., Gonzalez De Aguilar, J. L., Bonnefont-Rousselot, D., Bittar, R., et al. (2008). Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70, 1004–1009. doi: 10.1212/01.wnl.0000285080.70324.27
Dupuis, L., Gonzalez de Aguilar, J. L., Oudart, H., de Tapia, M., Barbeito, L., and Loeffler, J. P. (2004a). Mitochondria in amyotrophic lateral sclerosis: a trigger and a target. Neurodegener. Dis. 1, 245–254. doi: 10.1159/000085063
Dupuis, L., Oudart, H., René, F., Gonzalez de Aguilar, J. L., and Loeffler, J. P. (2004b). Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. U S A 101, 11159–11164. doi: 10.1073/pnas.0402026101
Dupuis, L., Pradat, P. F., Ludolph, A. C., and Loeffler, J. P. (2011). Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 10, 75–82. doi: 10.1016/S1474-4422(10)70224-6
Echaniz-Laguna, A., Zoll, J., Ribera, F., Tranchant, C., Warter, J. M., Lonsdorfer, J., et al. (2002). Mitochondrial respiratory chain function in skeletal muscle of ALS patients. Ann. Neurol. 52, 623–627. doi: 10.1002/ana.10357
Edwards, I. R., Star, K., and Kiuru, A. (2007). Statins, neuromuscular degenerative disease and an amyotrophic lateral sclerosis-like syndrome: an analysis of individual case safety reports from vigibase. Drug Saf. 30, 515–525. doi: 10.2165/00002018-200730060-00005
Feiguin, F., Godena, V. K., Romano, G., D’Ambrogio, A., Klima, R., and Baralle, F. E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592. doi: 10.1016/j.febslet.2009.04.019
Fergani, A., Oudart, H., Gonzalez De Aguilar, J. L., Fricker, B., René, F., Hocquette, J. F., et al. (2007). Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J. Lipid Res. 48, 1571–1580. doi: 10.1194/jlr.m700017-jlr200
Ferri, A., Fiorenzo, P., Nencini, M., Cozzolino, M., Pesaresi, M. G., Valle, C., et al. (2010). Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 19, 4529–4542. doi: 10.1093/hmg/ddq383
Freibaum, B. D., Chitta, R. K., High, A. A., and Taylor, J. P. (2010). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9, 1104–1120. doi: 10.1021/pr901076y
Freibaum, B. D., Lu, Y., Lopez-Gonzalez, R., Kim, N. C., Almeida, S., Lee, K. H., et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. doi: 10.1038/nature14974
Fujii, R., Okabe, S., Urushido, T., Inoue, K., Yoshimura, A., Tachibana, T., et al. (2005). The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol. 15, 587–593. doi: 10.3410/f.1009219.325127
Fujii, R., and Takumi, T. (2005). TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci. 118, 5755–5765. doi: 10.1242/jcs.02692
Gal, J., Zhang, J., Kwinter, D. M., Zhai, J., Jia, H., Jia, J., et al. (2011). Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging 32, 2323.e27–2323.e40. doi: 10.1016/j.neurobiolaging.2010.06.010
Gijselinck, I., Van Langenhove, T., van der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., et al. (2012). A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65. doi: 10.1016/S1474-4422(11)70261-7
Giordana, M. T., Piccinini, M., Grifoni, S., De Marco, G., Vercellino, M., Magistrello, M., et al. (2010). TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 20, 351–360. doi: 10.1111/j.1750-3639.2009.00284.x
Golomb, B. A., Kwon, E. K., Koperski, S., and Evans, M. A. (2009). Amyotrophic lateral sclerosis-like conditions in possible association with cholesterol-lowering drugs: an analysis of patient reports to the university of California, San Diego (UCSD) statin effects study. Drug Saf. 32, 649–661. doi: 10.2165/00002018-200932080-00004
Görlich, D., and Kutay, U. (1999). Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15, 607–660. doi: 10.1146/annurev.cellbio.15.1.607
Gorman, A. M., Ceccatelli, S., and Orrenius, S. (2000). Role of mitochondria in neuronal apoptosis. Dev. Neurosci. 22, 348–358. doi: 10.1159/000017460
Gregory, R. I., Yan, K. P., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., et al. (2004). The microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240. doi: 10.1038/nature03120
Gunnarsson, L. G., Dahlbom, K., and Strandman, E. (1991). Motor neuron disease and dementia reported among 13 members of a single family. Acta Neurol. Scand. 84, 429–433. doi: 10.1111/j.1600-0404.1991.tb04983.x
Hallier, M., Lerga, A., Barnache, S., Tavitian, A., and Moreau-Gachelin, F. (1998). The transcription factor Spi-1/PU.1 interacts with the potential splicing factor TLS. J. Biol. Chem. 273, 4838–4842. doi: 10.1074/jbc.273.9.4838
Hardie, D. G. (2008). AMPK: a key regulator of energy balance in the single cell and the whole organism. Int. J. Obes. (Lond) 32, S7–S12. doi: 10.1038/ijo.2008.116
Hoell, J. I., Larsson, E., Runge, S., Nusbaum, J. D., Duggimpudi, S., Farazi, T. A., et al. (2011). RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 18, 1428–1431. doi: 10.1038/nsmb.2163
Huang, E. J., Zhang, J., Geser, F., Trojanowski, J. Q., Strober, J. B., Dickson, D. W., et al. (2010). Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol. 20, 1069–1076. doi: 10.1111/j.1750-3639.2010.00413.x
Iko, Y., Kodama, T. S., Kasai, N., Oyama, T., Morita, E. H., Muto, T., et al. (2004). Domain architectures and characterization of an RNA-binding protein, TLS. J. Biol. Chem. 279, 44834–44840. doi: 10.1074/jbc.M408552200
Israelson, A., Arbel, N., Da Cruz, S., Ilieva, H., Yamanaka, K., Shoshan-Barmatz, V., et al. (2010). Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67, 575–587. doi: 10.1016/j.neuron.2010.07.019
Ito, D., Seki, M., Tsunoda, Y., Uchiyama, H., and Suzuki, N. (2011). Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol. 69, 152–162. doi: 10.1002/ana.22246
Jain, S., Wheeler, J. R., Walters, R. W., Agrawal, A., Barsic, A., and Parker, R. (2016). ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164, 487–498. doi: 10.1016/j.cell.2015.12.038
Jawaid, A., Murthy, S. B., Wilson, A. M., Qureshi, S. U., Amro, M. J., Wheaton, M., et al. (2010). A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph. Lateral Scler. 11, 542–548. doi: 10.3109/17482968.2010.482592
Jiang, J., Zhu, Q., Gendron, T. F., Saberi, S., McAlonis-Downes, M., Seelman, A., et al. (2016). Gain of Toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550. doi: 10.1016/j.neuron.2016.04.006
Jovičić, A., Mertens, J., Boeynaems, S., Bogaert, E., Chai, N., Yamada, S. B., et al. (2015). Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 18, 1226–1229. doi: 10.1038/nn.4085
Jovičić, A., Paul, J. W. III., and Gitler, A. D. (2016). Nuclear transport dysfunction: a common theme in amyotrophic lateral sclerosis and frontotemporal dementia. J. Neurochem. 138, 134–144. doi: 10.1111/jnc.13642
Ju, T. C., Chen, H. M., Chen, Y. C., Chang, C. P., Chang, C., and Chern, Y. (2014). AMPK-α1 functions downstream of oxidative stress to mediate neuronal atrophy in Huntington’s disease. Biochim. Biophys. Acta 1842, 1668–1680. doi: 10.1016/j.bbadis.2014.06.012
Ju, T. C., Chen, H. M., Lin, J. T., Chang, C. P., Chang, W. C., Kang, J. J., et al. (2011). Nuclear translocation of AMPK-α1 potentiates striatal neurodegeneration in Huntington’s disease. J. Cell Biol. 194, 209–227. doi: 10.1083/jcb.201105010
Ju, T. C., Lin, Y. S., and Chern, Y. (2012). Energy dysfunction in Huntington’s disease: insights from PGC-1α, AMPK and CKB. Cell. Mol. Life Sci. 69, 4107–4120. doi: 10.1007/s00018-012-1025-2
Kamelgarn, M., Chen, J., Kuang, L., Arenas, A., Zhai, J., Zhu, H., et al. (2016). Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta 1862, 2004–2014. doi: 10.1016/j.bbadis.2016.07.015
Kanai, Y., Dohmae, N., and Hirokawa, N. (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525. doi: 10.1016/j.neuron.2004.07.022
Kasarskis, E. J., Berryman, S., Vanderleest, J. G., Schneider, A. R., and McClain, C. J. (1996). Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death. Am. J. Clin. Nutr. 63, 130–137.
Kawahara, Y., and Mieda-Sato, A. (2012). TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. U S A 109, 3347–3352. doi: 10.1073/pnas.1112427109
Khosravi, B., Hartmann, H., May, S., Möhl, C., Ederle, H., Michaelsen, M., et al. (2017). Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 26, 790–800. doi: 10.1093/hmg/ddw432
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kim, J., Kwak, H. J., Cha, J. Y., Jeong, Y. S., Rhee, S. D., Kim, K. R., et al. (2014). Metformin suppresses lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages via activating transcription factor-3 (ATF-3) induction. J. Biol. Chem. 289, 23246–23255. doi: 10.1074/jbc.m114.577908
Kim, N. C., Tresse, E., Kolaitis, R. M., Molliex, A., Thomas, R. E., Alami, N. H., et al. (2013). VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78, 65–80. doi: 10.1016/j.neuron.2013.02.029
Kosugi, S., Hasebe, M., Matsumura, N., Takashima, H., Miyamoto-Sato, E., Tomita, M., et al. (2009). Six classes of nuclear localization signals specific to different binding grooves of importin α. J. Biol. Chem. 284, 478–485. doi: 10.1074/jbc.M807017200
Kühnlein, P., Gdynia, H.-J., Sperfeld, A.-D., Lindner-Pfleghar, B., Ludolph, A. C., Prosiegel, M., et al. (2008). Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat. Clin. Pract. Neurol. 4, 366–374. doi: 10.1038/ncpneuro0853
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Lacomblez, L., Doppler, V., Beucler, I., Costes, G., Salachas, F., Raisonnier, A., et al. (2002). APOE: a potential marker of disease progression in ALS. Neurology 58, 1112–1114. doi: 10.1212/wnl.58.7.1112
Lagier-Tourenne, C., Baughn, M., Rigo, F., Sun, S., Liu, P., Li, H. R., et al. (2013). Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U S A 110, E4530–E4539. doi: 10.1073/pnas.1318835110
Lagier-Tourenne, C., Polymenidou, M., and Cleveland, D. W. (2010). TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64. doi: 10.1093/hmg/ddq137
Lagier-Tourenne, C., Polymenidou, M., Hutt, K. R., Vu, A. Q., Baughn, M., Huelga, S. C., et al. (2012). Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15, 1488–1497. doi: 10.1038/nn.3230
La Spada, A. R., Wilson, E. M., Lubahn, D. B., Harding, A. E., and Fischbeck, K. H. (1991). Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79. doi: 10.1038/352077a0
Lee, Y. B., Chen, H. J., Peres, J. N., Gomez-Deza, J., Attig, J., Stalekar, M., et al. (2013). Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 5, 1178–1186. doi: 10.1016/j.celrep.2013.10.049
Lee, K.-H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774.e17–788.e17. doi: 10.1016/j.cell.2016.10.002
Lenzi, J., De Santis, R., de Turris, V., Morlando, M., Laneve, P., Calvo, A., et al. (2015). ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis. Model. Mech. 8, 755–766. doi: 10.1242/dmm.020099
Levine, T. P., Daniels, R. D., Gatta, A. T., Wong, L. H., and Hayes, M. J. (2013). The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503. doi: 10.1093/bioinformatics/bts725
Li, X., Decker, M., and Westendorf, J. J. (2010). TEThered to Runx: novel binding partners for runx factors. Blood Cells Mol. Dis. 45, 82–85. doi: 10.1016/j.bcmd.2010.03.002
Li, Y. R., King, O. D., Shorter, J., and Gitler, A. D. (2013). Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 201, 361–372. doi: 10.1083/jcb.201302044
Lim, M. A., Selak, M. A., Xiang, Z., Krainc, D., Neve, R. L., Kraemer, B. C., et al. (2012). Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J. Neurosci. 32, 1123–1141. doi: 10.1523/JNEUROSCI.6554-10.2012
Lin, Y., Mori, E., Kato, M., Xiang, S., Wu, L., Kwon, I., et al. (2016). Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167, 789.e12–802.e12. doi: 10.1016/j.cell.2016.10.003
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu, Y. J., Ju, T. C., Chen, H. M., Jang, Y. S., Lee, L. M., Lai, H. L., et al. (2015a). Activation of AMP-activated protein kinase α1 mediates mislocalization of TDP-43 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 24, 787–801. doi: 10.1093/hmg/ddu497
Liu, Y. J., Lee, L. M., Lai, H. L., and Chern, Y. (2015b). Aberrant activation of AMP-activated protein kinase contributes to the abnormal distribution of HuR in amyotrophic lateral sclerosis. FEBS Lett. 589, 432–439. doi: 10.1016/j.febslet.2014.12.029
Liu, W., Yamashita, T., Tian, F., Morimoto, N., Ikeda, Y., Deguchi, K., et al. (2013). Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 10, 222–230. doi: 10.2174/15672026113109990060
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y. J., Vanderweyde, T., Citro, A., Mehta, T., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250. doi: 10.1371/journal.pone.0013250
Lladó, J., Tolosa, L., and Olmos, G. (2013). Cellular and molecular mechanisms involved in the neuroprotective effects of VEGF on motoneurons. Front. Cell. Neurosci. 7:181. doi: 10.3389/fncel.2013.00181
Long, Y. C., and Zierath, J. R. (2006). AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Invest. 116, 1776–1783. doi: 10.1172/JCI29044
Lopez-Gonzalez, R., Lu, Y., Gendron, T. F., Karydas, A., Tran, H., Yang, D., et al. (2016). Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92, 383–391. doi: 10.1016/j.neuron.2016.09.015
Lu, Y., Ferris, J., and Gao, F. B. (2009). Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol. Brain 2:30. doi: 10.1186/1756-6606-2-30
Lu, L., Wang, S., Zheng, L., Li, X., Suswam, E. A., Zhang, X., et al. (2009). Amyotrophic lateral sclerosis-linked mutant SOD1 sequesters Hu antigen R (HuR) and TIA-1-related protein (TIAR): implications for impaired post-transcriptional regulation of vascular endothelial growth factor. J. Biol. Chem. 284, 33989–33998. doi: 10.1074/jbc.M109.067918
Lu, L., Zheng, L., Si, Y., Luo, W., Dujardin, G., Kwan, T., et al. (2014). Hu antigen R (HuR) is a positive regulator of the RNA-binding proteins TDP-43 and FUS/TLS: implications for amyotrophic lateral sclerosis. J. Biol. Chem. 289, 31792–31804. doi: 10.1074/jbc.M114.573246
Ma, T., Chen, Y., Vingtdeux, V., Zhao, H., Viollet, B., Marambaud, P., et al. (2014). Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid β. J. Neurosci. 34, 12230–12238. doi: 10.1523/JNEUROSCI.1694-14.2014
Mahboubi, H., Barisé, R., and Stochaj, U. (2015a). 5′-AMP-activated protein kinase α regulates stress granule biogenesis. Biochim. Biophys. Acta 1853, 1725–1737. doi: 10.1016/j.bbamcr.2015.03.015
Mahboubi, H., Barisé, R., and Stochaj, U. (2015b). Data in support of 5′AMP-activated protein kinase α regulates stress granule biogenesis. Data Brief 4, 54–59. doi: 10.1016/j.dib.2015.04.010
Mairet-Coello, G., Courchet, J., Pieraut, S., Courchet, V., Maximov, A., and Polleux, F. (2013). The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron 78, 94–108. doi: 10.1016/j.neuron.2013.02.003
Martin, K. C., and Zukin, R. S. (2006). RNA trafficking and local protein synthesis in dendrites: an overview. J. Neurosci. 26, 7131–7134. doi: 10.1523/jneurosci.1801-06.2006
Mattson, M. P., Cutler, R. G., and Camandola, S. (2007). Energy intake and amyotrophic lateral sclerosis. Neuromolecular Med. 9, 17–20. doi: 10.1385/NMM:9:1:17
McDonald, K. K., Aulas, A., Destroismaisons, L., Pickles, S., Beleac, E., Camu, W., et al. (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 20, 1400–1410. doi: 10.1093/hmg/ddr021
McGurk, L., Lee, V. M., Trojanowksi, J. Q., Van Deerlin, V. M., Lee, E. B., and Bonini, N. M. (2014). Poly-A binding protein-1 localization to a subset of TDP-43 inclusions in amyotrophic lateral sclerosis occurs more frequently in patients harboring an expansion in C9orf72. J. Neuropathol. Exp. Neurol. 73, 837–845. doi: 10.1097/NEN.0000000000000102
Millecamps, S., Boillée, S., Le Ber, I., Seilhean, D., Teyssou, E., Giraudeau, M., et al. (2012). Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J. Med. Genet. 49, 258–263. doi: 10.1136/jmedgenet-2011-100699
Miller, J. W., Urbinati, C. R., Teng-Umnuay, P., Stenberg, M. G., Byrne, B. J., Thornton, C. A., et al. (2000). Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 19, 4439–4448. doi: 10.1093/emboj/19.17.4439
Miyamoto, Y., Yamada, K., and Yoneda, Y. (2016). Importin α: a key molecule in nuclear transport and non-transport functions. J. Biochem. 160, 69–75. doi: 10.1093/jb/mvw036
Mori, K., Lammich, S., Mackenzie, I. R., Forné, I., Zilow, S., Kretzschmar, H., et al. (2013a). hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 125, 413–423. doi: 10.1007/s00401-013-1088-7
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013b). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Mungai, P. T., Waypa, G. B., Jairaman, A., Prakriya, M., Dokic, D., Ball, M. K., et al. (2011). Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol. 31, 3531–3545. doi: 10.1128/MCB.05124-11
Nabors, L. B., Gillespie, G. Y., Harkins, L., and King, P. H. (2001). HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 61, 2154–2161.
Naganuma, T., and Hirose, T. (2013). Paraspeckle formation during the biogenesis of long non-coding RNAs. RNA Biol. 10, 456–461. doi: 10.4161/rna.23547
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Ng, R. C., Cheng, O. Y., Jian, M., Kwan, J. S., Ho, P. W., Cheng, K. K., et al. (2016). Chronic adiponectin deficiency leads to Alzheimer’s disease-like cognitive impairments and pathologies through AMPK inactivation and cerebral insulin resistance in aged mice. Mol. Neurodegener. 11:71. doi: 10.1186/s13024-016-0136-x
Nishimoto, Y., Nakagawa, S., Hirose, T., Okano, H. J., Takao, M., Shibata, S., et al. (2013). The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 6:31. doi: 10.1186/1756-6606-6-31
Oberlé, I., Rousseau, F., Heitz, D., Kretz, C., Devys, D., Hanauer, A., et al. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102. doi: 10.1126/science.252.5009.1097
Onesto, E., Colombrita, C., Gumina, V., Borghi, M. O., Dusi, S., Doretti, A., et al. (2016). Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 4:47. doi: 10.1186/s40478-016-0316-5
Oosthuyse, B., Moons, L., Storkebaum, E., Beck, H., Nuyens, D., Brusselmans, K., et al. (2001). Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 28, 131–138. doi: 10.1038/88842
Parker, S. J., Meyerowitz, J., James, J. L., Liddell, J. R., Crouch, P. J., Kanninen, K. M., et al. (2012). Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem. Int. 60, 415–424. doi: 10.1016/j.neuint.2012.01.019
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Bennett, C. F., Cleveland, D. W., and Yeo, G. W. (2012). Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 1462, 3–15. doi: 10.1016/j.brainres.2012.02.059
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
Powers, C. A., Mathur, M., Raaka, B. M., Ron, D., and Samuels, H. H. (1998). TLS (translocated-in-liposarcoma) is a high-affinity interactor for steroid, thyroid hormone and retinoid receptors. Mol. Endocrinol. 12, 4–18. doi: 10.1210/mend.12.1.0043
Protter, D. S., and Parker, R. (2016). Principles and properties of stress granules. Trends Cell Biol. 26, 668–679. doi: 10.1016/j.tcb.2016.05.004
Ramaswami, M., Taylor, J. P., and Parker, R. (2013). Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 154, 727–736. doi: 10.1016/j.cell.2013.07.038
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Ricquier, D., and Bouillaud, F. (2000). Mitochondrial uncoupling proteins: from mitochondria to the regulation of energy balance. J. Physiol. 529, 3–10. doi: 10.1111/j.1469-7793.2000.00003.x
Robertson, J., Sanelli, T., Xiao, S., Yang, W., Horne, P., Hammond, R., et al. (2007). Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci. Lett. 420, 128–132. doi: 10.1016/j.neulet.2007.03.066
Rogelj, B., Easton, L. E., Bogu, G. K., Stanton, L. W., Rot, G., Curk, T., et al. (2012). Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2:603. doi: 10.1038/srep00603
Rohrer, J. D., Isaacs, A. M., Mizielinska, S., Mead, S., Lashley, T., Wray, S., et al. (2015). C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol. 14, 291–301. doi: 10.1016/S1474-4422(14)70233-9
Rossi, S., Serrano, A., Gerbino, V., Giorgi, A., Di Francesco, L., Nencini, M., et al. (2015). Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J. Cell Sci. 128, 1787–1799. doi: 10.1242/jcs.165332
Sama, R. R., Ward, C. L., Kaushansky, L. J., Lemay, N., Ishigaki, S., Urano, F., et al. (2013). FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Physiol. 228, 2222–2231. doi: 10.1002/jcp.24395
Sephton, C. F., Cenik, C., Kucukural, A., Dammer, E. B., Cenik, B., Han, Y., et al. (2011). Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 286, 1204–1215. doi: 10.1074/jbc.M110.190884
Sephton, C. F., and Yu, G. (2015). The function of RNA-binding proteins at the synapse: implications for neurodegeneration. Cell. Mol. Life Sci. 72, 3621–3635. doi: 10.1007/s00018-015-1943-x
Shan, X., Chiang, P. M., Price, D. L., and Wong, P. C. (2010). Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. U S A 107, 16325–16330. doi: 10.1073/pnas.1003459107
Shan, X., Vocadlo, D., and Krieger, C. (2009). Mislocalization of TDP-43 in the G93A mutant SOD1 transgenic mouse model of ALS. Neurosci. Lett. 458, 70–74. doi: 10.1016/j.neulet.2009.04.031
Shelkovnikova, T. A., Robinson, H. K., Troakes, C., Ninkina, N., and Buchman, V. L. (2014). Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 23, 2298–2312. doi: 10.1093/hmg/ddt622
Sicot, G., and Gomes-Pereira, M. (2013). RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochim. Biophys. Acta 1832, 1390–1409. doi: 10.1016/j.bbadis.2013.03.002
Simón-Sánchez, J., Dopper, E. G., Cohn-Hokke, P. E., Hukema, R. K., Nicolaou, N., Seelaar, H., et al. (2012). The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 135, 723–735. doi: 10.1093/brain/awr353
Sørensen, H. T., and Lash, T. L. (2009). Statins and amyotrophic lateral sclerosis–the level of evidence for an association. J. Intern. Med. 266, 520–526. doi: 10.1111/j.1365-2796.2009.02173.x
Sreedharan, J., and Brown, R. H. Jr. (2013). Amyotrophic lateral sclerosis: problems and prospects. Ann. Neurol. 74, 309–316. doi: 10.1002/ana.24012
Stambler, N., Charatan, M., and Cedarbaum, J. M. (1998). Prognostic indicators of survival in ALS. ALS CNTF Treatment Study Group. Neurology 50, 66–72. doi: 10.1212/wnl.50.1.66
Steinberg, K. M., Yu, B., Koboldt, D. C., Mardis, E. R., and Pamphlett, R. (2015). Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci. Rep. 5:9124. doi: 10.1038/srep09124
Strong, M. J., Volkening, K., Hammond, R., Yang, W., Strong, W., Leystra-Lantz, C., et al. (2007). TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci. 35, 320–327. doi: 10.1016/j.mcn.2007.03.007
Takahashi, M., Higuchi, M., Matsuki, H., Yoshita, M., Ohsawa, T., Oie, M., et al. (2013). Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol. Cell. Biol. 33, 815–829. doi: 10.1128/MCB.00763-12
Takarada, T., Tamaki, K., Takumi, T., Ogura, M., Ito, Y., Nakamichi, N., et al. (2009). A protein-protein interaction of stress-responsive myosin VI endowed to inhibit neural progenitor self-replication with RNA binding protein, TLS, in murine hippocampus. J. Neurochem. 110, 1457–1468. doi: 10.1111/j.1471-4159.2009.06225.x
Taylor, J. P., Brown, R. H. Jr., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Thau, N., Knippenberg, S., Körner, S., Rath, K. J., Dengler, R., and Petri, S. (2012). Decreased mRNA expression of PGC-1α and PGC-1α-regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 71, 1064–1074. doi: 10.1097/NEN.0b013e318275df4b
Thornton, C., Bright, N. J., Sastre, M., Muckett, P. J., and Carling, D. (2011). AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid β-peptide exposure. Biochem. J. 434, 503–512. doi: 10.1042/BJ20101485
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458. doi: 10.1038/nn.2778
Tulino, R., Benjamin, A. C., Jolinon, N., Smith, D. L., Chini, E. N., Carnemolla, A., et al. (2016). SIRT1 activity is linked to its brain region-specific phosphorylation and is impaired in Huntington’s disease mice. PLoS One 11:e0145425. doi: 10.1371/journal.pone.0150682
Uranishi, H., Tetsuka, T., Yamashita, M., Asamitsu, K., Shimizu, M., Itoh, M., et al. (2001). Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in nuclear factor-κ B p65-mediated transcription as a coactivator. J. Biol. Chem. 276, 13395–13401. doi: 10.1074/jbc.M011176200
Vaisman, N., Lusaus, M., Nefussy, B., Niv, E., Comaneshter, D., Hallack, R., et al. (2009). Do patients with amyotrophic lateral sclerosis (ALS) have increased energy needs? J. Neurol. Sci. 279, 26–29. doi: 10.1016/j.jns.2008.12.027
Vanacore, N., Cocco, P., Fadda, D., and Dosemeci, M. (2010). Job strain, hypoxia and risk of amyotrophic lateral sclerosis: results from a death certificate study. Amyotroph. Lateral Scler. 11, 430–434. doi: 10.3109/17482961003605796
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vázquez-Manrique, R. P., Farina, F., Cambon, K., Sequedo Dolores, M., Parker, A. J., Millan, J. M., et al. (2016). AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 25, 1043–1058. doi: 10.1093/hmg/ddv513
Volkening, K., Leystra-Lantz, C., Yang, W., Jaffee, H., and Strong, M. J. (2009). Tar DNA binding protein of 43 kDa (TDP-43), 14–3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 1305, 168–182. doi: 10.1016/j.brainres.2009.09.105
Wang, Y., Duan, W., Wang, W., Di, W., Liu, Y., Liu, Y., et al. (2016). scAAV9-VEGF prolongs the survival of transgenic ALS mice by promoting activation of M2 microglia and the PI3K/Akt pathway. Brain Res. 1648, 1–10. doi: 10.1016/j.brainres.2016.06.043
Wang, W., Li, L., Lin, W. L., Dickson, D. W., Petrucelli, L., Zhang, T., et al. (2013). The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 22, 4706–4719. doi: 10.1093/hmg/ddt319
Wang, I. F., Wu, L. S., Chang, H. Y., and Shen, C. K. (2008). TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem. 105, 797–806. doi: 10.1111/j.1471-4159.2007.05190.x
Wang, W., Yang, X., Kawai, T., López de Silanes, I., Mazan-Mamczarz, K., Chen, P., et al. (2004). AMP-activated protein kinase-regulated phosphorylation and acetylation of importin α1: involvement in the nuclear import of RNA-binding protein HuR. J. Biol. Chem. 279, 48376–48388. doi: 10.1074/jbc.m409014200
Watanabe, S., Ilieva, H., Tamada, H., Nomura, H., Komine, O., Endo, F., et al. (2016). Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 8, 1421–1437. doi: 10.15252/emmm.201606403
Weishaupt, J. H., Hyman, T., and Dikic, I. (2016). Common molecular pathways in amyotrophic lateral sclerosis and Frontotemporal dementia. Trends Mol. Med. 22, 769–783. doi: 10.1016/j.molmed.2016.07.005
Wen, Y., and Shatkin, A. J. (2000). Cap methyltransferase selective binding and methylation of GpppG-RNA are stimulated by importin-α. Genes Dev. 14, 2944–2949. doi: 10.1101/gad.848200
Woerner, A. C., Frottin, F., Hornburg, D., Feng, L. R., Meissner, F., Patra, M., et al. (2016). Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351, 173–176. doi: 10.1126/science.aad2033
Xia, Q., Wang, G., Wang, H., Hu, Q., and Ying, Z. (2016). Folliculin, a tumor suppressor associated with Birt-Hogg-Dube (BHD) syndrome, is a novel modifier of TDP-43 cytoplasmic translocation and aggregation. Hum. Mol. Genet. 25, 83–96. doi: 10.1093/hmg/ddv450
Xu, Y. F., Gendron, T. F., Zhang, Y. J., Lin, W. L., D’Alton, S., Sheng, H., et al. (2010). Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits and early mortality in transgenic mice. J. Neurosci. 30, 10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010
Yamashita, S., Mori, A., Sakaguchi, H., Suga, T., Ishihara, D., Ueda, A., et al. (2012). Sporadic juvenile amyotrophic lateral sclerosis caused by mutant FUS/TLS: possible association of mental retardation with this mutation. J. Neurol. 259, 1039–1044. doi: 10.1007/s00415-011-6292-6
Yoo, S., van Niekerk, E. A., Merianda, T. T., and Twiss, J. L. (2010). Dynamics of axonal mRNA transport and implications for peripheral nerve regeneration. Exp. Neurol. 223, 19–27. doi: 10.1016/j.expneurol.2009.08.011
Yoshimura, A., Fujii, R., Watanabe, Y., Okabe, S., Fukui, K., and Takumi, T. (2006). Myosin-Va facilitates the accumulation of mRNA/protein complex in dendritic spines. Curr. Biol. 16, 2345–2351. doi: 10.1016/j.cub.2006.10.024
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973
Zhang, Y. J., Gendron, T. F., Grima, J. C., Sasaguri, H., Jansen-West, K., Xu, Y. F., et al. (2016). C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 19, 668–677. doi: 10.1038/nn.4272
Keywords: amyotrophic lateral sclerosis, AMPK, TDP-43, importin, RNA binding proteins
Citation: Liu Y-J, Tsai P-Y and Chern Y (2017) Energy Homeostasis and Abnormal RNA Metabolism in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 11:126. doi: 10.3389/fncel.2017.00126
Received: 09 January 2017; Accepted: 18 April 2017;
Published: 04 May 2017.
Edited by:
Ho Yin Edwin Chan, The Chinese University of Hong Kong, Hong KongReviewed by:
Shyuan Ngo, The University of Queensland, AustraliaMargarida Gama-Carvalho, Universidade de Lisboa, Portugal
Copyright © 2017 Liu, Tsai and Chern. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yijuang Chern, Ym15Y2hlcm5AaWJtcy5zaW5pY2EuZWR1LnR3