 Yasuhiro Kawasaki1†
Yasuhiro Kawasaki1† Mitsuru Saito1*†‡
Mitsuru Saito1*†‡ Jonghwa Won2
Jonghwa Won2 Jin Young Bae3
Jin Young Bae3 Hajime Sato1
Hajime Sato1 Hiroki Toyoda1
Hiroki Toyoda1 Eriko Kuramoto4‡Mikihiko Kogo1
Eriko Kuramoto4‡Mikihiko Kogo1 Takuma Tanaka5‡
Takuma Tanaka5‡ Takeshi Kaneko4
Takeshi Kaneko4 Seog Bae Oh2*Yong Chul Bae3
Seog Bae Oh2*Yong Chul Bae3 Youngnam Kang1*
Youngnam Kang1*- 1Department of Neuroscience and Oral Physiology, Graduate School of Dentistry, Osaka University, Osaka, Japan
- 2Department of Brain and Cognitive Sciences, College of Natural Sciences, Dental Research Institute and Department of Neurobiology and Physiology, School of Dentistry, Seoul National University, Seoul, South Korea
- 3Department of Oral Anatomy, School of Dentistry, Kyungpook National University, Daegu, South Korea
- 4Department of Morphological Brain Science, Graduate School of Medicine, Kyoto University, Kyoto, Japan
- 5Department of Computational Intelligence and Systems Science, Interdisciplinary Graduate School of Science and Engineering, Tokyo Institute of Technology, Yokohama, Japan
Glutamatergic dendritic EPSPs evoked in cortical pyramidal neurons are depressed by activation of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels expressed in dendritic spines. This depression has been attributed to shunting effects of HCN current (Ih) on input resistance or Ih deactivation. Primary sensory neurons in the rat mesencephalic trigeminal nucleus (MTN) have the somata covered by spine-like microvilli that express HCN channels. In rat MTN neurons, we demonstrated that Ih enhancement apparently diminished the glutamate receptor (GluR) current (IGluR) evoked by puff application of glutamate/AMPA and enhanced a transient outward current following IGluR (OT-IGluR). This suggests that some outward current opposes inward IGluR. The IGluR inhibition displayed a U-shaped voltage-dependence with a minimal inhibition around the resting membrane potential, suggesting that simple shunting effects or deactivation of Ih cannot explain the U-shaped voltage-dependence. Confocal imaging of Na+ revealed that GluR activation caused an accumulation of Na+ in the microvilli, which can cause a negative shift of the reversal potential for Ih (Eh). Taken together, it was suggested that IGluR evoked in MTN neurons is opposed by a transient decrease or increase in standing inward or outward Ih, respectively, both of which can be caused by negative shifts of Eh, as consistent with the U-shaped voltage-dependence of the IGluR inhibition and the OT-IGluR generation. An electron-microscopic immunohistochemical study revealed the colocalization of HCN channels and glutamatergic synapses in microvilli of MTN neurons, which would provide a morphological basis for the functional interaction between HCN and GluR channels. Mathematical modeling eliminated the possibilities of the involvements of Ih deactivation and/or shunting effect and supported the negative shift of Eh which causes the U-shaped voltage-dependent inhibition of IGluR.
Introduction
To date, many studies have reported that, not only in cortical pyramidal cells but also in various other neurons, the activation of hyperpolarization-activated cyclic nucleotide-gated (HCN) cation channels can decrease the amplitude and/or duration of EPSPs or depolarizations evoked by current pulses (Magee, 1998, 1999; Yamada et al., 2005; Carr et al., 2007; Ying et al., 2007; Harnett et al., 2015). Indeed, the blockade of HCN channels increased the amplitudes of EPSPs or depolarizations in these studies. Such modulation of EPSPs by HCN channels is crucially involved in a variety of brain functions, such as working memory (Wang et al., 2007), sleep/wakefulness (Postea and Biel, 2011), epilepsy (DiFrancesco et al., 2011), autism (Yi et al., 2016), and neuropathic pain (Harnett et al., 2015). Originally, the deactivation of HCN channels-mediated current (Ih) by EPSPs was considered to be responsible for the diminution of EPSPs and the generation of hyperpolarization following EPSPs, based on their sensitivity to Ih blockers (Magee, 1998, 1999). However, it is not clear whether the abolishment of the afterhyperpolarization by Ih blockers is a direct consequence of the abolishment of Ih deactivation or secondary to the Ih blocker-induced negative shift of the baseline membrane potential that consequently attenuates K+ channel-mediated afterhyperpolarization.
Since HCN channels were found to be expressed in the apical dendrites, especially on the shafts of dendritic spines, of cortical pyramidal cells (Lörincz et al., 2002; Notomi and Shigemoto, 2004), the inhibition of EPSPs by the activity of HCN channels has been considered to be due to the shunting effects of HCN channels on the input impedance of the spine that receives excitatory synaptic inputs (Carr et al., 2007; Tsay et al., 2007; Wang et al., 2007; Harnett et al., 2015). However, it is also not clear whether the peak level of EPSPs is lowered by a shunting effect of increases in the HCN channel conductance (Gh) although it certainly decreases the amplitudes of EPSPs, because Gh increases would depolarize the baseline membrane potential toward the reversal potential for Ih (Eh), near −40 mV. Indeed, the peak level of the EPSP observed following the blockade of Ih with ZD7288 was not higher than that of the control due to the hyperpolarization of the baseline potential, while this was not necessarily the case for that of summated EPSPs (Carr et al., 2007), indicating that the shunting effect is not always effective. Subsequently, it has been proposed in a mathematical simulation study that, in CA1 hippocampal pyramidal neurons, HCN-mediated depolarization can secondarily activate M-type K+ channels or some other K+ channels, which can produce a real shunting conductance with a more negative reversal potential (George et al., 2009; Migliore and Migliore, 2012). Thus, it remains unclear and controversial how Ih diminishes EPSPs despite its crucial involvement in various brain functions.
Among all the primary sensory neurons, those innervating muscle spindles in the jaw-closing muscles are uniquely and exceptionally located in the brain stem as the mesencephalic trigeminal nucleus (MTN), thereby receiving peptidergic, catecholaminergic, serotonergic, and nitrergic perineuronal arborizations in a basket-like manner in addition to glutamatergic input and expressing various receptors (Lazarov, 2002) including glutamate receptors (GluRs; Mineff et al., 1998; Turman et al., 2000) inducing DNQX/AP5-sensitive glutamatergic EPSPs (Verdier et al., 2004). MTN neurons have no dendrites but express numerous spine-like microvilli directly protruding from the somata (Liem et al., 1991), in which HCN channels are expressed (Kang et al., 2004). In the present study, we explored whether and how GluR-mediated currents (IGluR) are modified by concurrent activation of Ih, under voltage-clamp conditions in MTN neurons by taking advantages of their characteristic morphological structure of the round shaped soma covered by short spine-like microvilli, where space-clamp errors would not occur.
Materials and Methods
Ethical Approval
The experimental protocols were approved either by the Animal Ethics Committees of the Osaka University Graduate School of Dentistry for the Care and Use of Laboratory Animals or by Kyungpook National University Intramural Animal Care and Use Committee, and all experiments were performed in accordance with the relevant guidelines.
Slice Preparation
Wistar and Sprague-Dawley (SD) rats of both sexes at postnatal day (PND) 13–18 were used for the experiments shown in the results and the Supplemental Material, respectively. Ih has been reported to be matured at PND 10–12 in MTN neurons (Tanaka et al., 2003), and various synaptic inputs including glutamatergic one are developmentally mature by PND 11 (Paik et al., 2012). Therefore, rats at PND 13–18 can be used in place of adult preparations to investigate Ih and IGluR in MTN neurons. The rats were anesthetized with isoflurane, and the brains were quickly removed from the skull and immersed in ice-cold modified artificial cerebrospinal fluid (ACSF) containing the following (in mM): 210 sucrose, 1.8 KCl, 1.2 KH2PO4, 26 NaHCO3, 0.5 CaCl2, 2.5 MgCl2, and 50 D-glucose. With a microslicer (Super ZERO-1, Dosaka EM, Kyoto, Japan), coronal sections of 250 μm thickness including the MTN were cut and incubated at room temperature (20–24°C) for 30 min in 50% modified ACSF and 50% normal ACSF (N-ACSF, pH 7.3) containing the following (in mM): 124 NaCl, 1.8 KCl, 1.2 KH2PO4, 26 NaHCO3, 2.0 CaCl2, 1.0 MgCl2, and 10 D-glucose. The slices were then placed in N-ACSF at room temperature. N-ACSF was continuously gassed with a mixture of 95% O2-5% CO2.
Whole-Cell Patch-Clamp Recordings
Using an Axopatch 1D (MDS Analytical Technologies, Sunnyvale, CA), whole-cell voltage-clamp or current-clamp recordings were made from MTN neurons that were viewed under Nomarski optics (BX50WI-DIC, Olympus, Tokyo, Japan). The recording chamber, with a volume of 1.0 ml, was continuously perfused with the extracellular solution (N-ACSF) at a flow rate of 1.0–1.5 ml/min. The internal solution of the patch pipettes had the following ionic composition (in mM): 123 K-gluconate, 18 KCl, 10 NaCl, 2 MgCl2, 2 ATP-Na2, 0.3 GTP-Na3, 10 HEPES, and 0.2 EGTA; pH 7.3 adjusted with KOH (Tanaka et al., 2003; Kang et al., 2004). The membrane potential values given in the text were corrected for the junction potential (10 mV) between the internal solution for the whole-cell recording (negative) and the standard extracellular solution. The pipette resistances were 4–6 MΩ. The series resistance was <10 MΩ. All recordings were made at room temperature. Series resistance was compensated by ~70% when the I-V relationships were measured while it was not performed when current responses were recorded at a fixed holding potential. This is mainly because the activation time constants of puff-induced IGluR (>50 ms) or Ih (>100 ms) in MTN neurons (Tanaka et al., 2003) were much slower than the time constant of the capacitative current in MTN neurons (<5 ms). Records of currents and voltages were low-pass filtered at 5 kHz (3-pole Bessel filter), digitized at a sampling rate of 40 kHz (Digidata 1322A, MDS Analytical Technologies) and stored on a computer hard disk.
Drug Application
Using a pressure-pulsed microinjector (Picopump PV820, World Precision Instruments, Sarasota, FL), 50–200 μM glutamate or α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid hydrate (AMPA; Sigma-Aldrich, St. Louis, MO) was puff-applied for 50 or 20–500 ms, respectively, through a glass pipette, the tip of which was placed 10–20 μm apart from the soma. CsCl (an Ih blocker), ZD7288 (an Ih blocker), 8-bromoadenosine 3′,5′-cyclic monophosphate sodium salt (8-Br-cAMP; a membrane-permeable cAMP analog), 8-bromoguanosine 3′,5′-cyclic monophosphate sodium salt (8-Br-cGMP; a membrane-permeable cGMP analog), and ouabain octahydrate (a Na+/K+ pump inhibitor) were bath-applied at 5 mM, 10, 500, 200, and 50–100 μM, respectively. These chemicals were purchased from Sigma-Aldrich. ZD7288 is also known to block Nav1.4 (Wu et al., 2012) and T-type Ca2+ currents (Sánchez-Alonso et al., 2008). Because it has been reported that HCN channels localized in the presynaptic terminal are involved in the modulation of glutamate release (Huang et al., 2011; Huang and Trussell, 2014), we employed puff application of glutamate or AMPA to isolate the effects of postsynaptic HCN channels on the postsynaptic GluR, instead of examining the responses caused by activation of presynaptic input pathways. On the other hand, puff application of AMPA or glutamate may cause strong desensitization because the puff duration is much larger than the duration of synaptic transmission. Given the desensitization of AMPA currents depending on the concentration of AMPA or glutamate and the duration of puff application, we may have underestimated the effects of HCN channel activity on AMPA currents. However, such desensitization does not preclude our conclusion regarding whether HCN activity effectively inhibits GluR currents if it does despite the desensitization.
Fluorescence Imaging of Na+ Transient With Sodium Green Tetraacetate
Sodium Green tetraacetate and Pluronic F-127 were purchased from Thermo Fisher Scientific (Waltham, MA). The stock solution was prepared by dissolving 5 mM Sodium Green tetraacetate in DMSO and mixing it with an equal volume of 25% w/v Pluronic F-127 (Friedman and Haddad, 1994; Amorino and Fox, 1995). Slice preparations including the MTN neurons were incubated for 60 min in oxygenated ACSF containing 10 μM Sodium Green tetraacetate and then washed in the ACSF for 30 min before optical recording of the glutamate responses. Sodium Green-loaded slices were submerged in a chamber placed on the stage of a confocal microscope (LSM510; Carl Zeiss Microscopy GmbH, Jena, Germany). The sodium imaging was performed with an excitation of Sodium Green at 488 nm and its emission at >510 nm. We have not attempted the calibration of Sodium Green because it would largely underestimate the rapid and large changes in Na+ concentration in beneath the membrane in microvilli caused by activation of GluR due to the possible slow binding rate constant as a consequence of a large dissociation constant (6–21 mM) of Sodium Green.
Electron-Microscopic Immunohistochemistry
Three male SD rats weighing 300–320 g (8 weeks old) were used for this study. For tissue fixation, the rats were deeply anesthetized with sodium pentobarbital (80 mg/kg, i.p.) and perfused transcardially with 100 ml of heparinized normal saline followed by 500 ml of a freshly prepared mixture of 4% paraformaldehyde and 0.01% glutaraldehyde in 0.1 M phosphate buffer (PB; pH 7.4). The brainstem was removed and post-fixed in the same fixative for 2 h at 4°C. Sections were cut transversely on a vibratome at 60 μm and cryoprotected in 30% sucrose in PB overnight at 4°C. The sections were frozen on dry ice for 20 min and then thawed in 0.01 M phosphate-buffered saline (pH 7.2) to enhance penetration. The slices were pretreated with 1% sodium borohydride for 30 min to quench the glutaraldehyde and then blocked with 10% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) for 30 min to mask the secondary antibody binding sites. For single immunostaining for vesicular glutamate transporter 2 (VGLUT2), the sections of brainstem were incubated overnight in mouse anti-VGLUT2 (1:1,000; MAB5504, Merck Millipore, Billerica, MA) antibody. After rinsing in phosphate-buffered saline, the sections were incubated with 1 nm gold-conjugated donkey anti-rabbit (1:50; EMS, Hatfield, PA) antibody for 2–3 h. The sections were post-fixed with 1% glutaraldehyde in PB for 10 min, rinsed in PB several times, incubated for 4 min with HQ silver enhancement solution (Nanoprobes, Yaphank, NY) and rinsed in 0.1 M sodium acetate and PB. To control for the specificity of the antibody, the sections were processed as described above, except that the primary or secondary antibodies were omitted. Omission of the primary or secondary antibodies eliminated specific staining. Pre-adsorption with blocking peptides for VGLUT2 (15 mg/ml; #135-40P, Synaptic Systems) also completely abolished the respective staining. For immunostaining for HCN or glutamate was described in our previous studies (Cho et al., 2015; Park et al., 2016).
Sections were osmicated (in 0.5% osmium tetroxide in PB) for 30 min, dehydrated in graded alcohols, flat-embedded in Durcupan ACM (Fluka, Buchs, Switzerland) between strips of Aclar plastic film (EMS), and cured for 48 h at 60°C. Chips containing prominent staining for VGLUT2 in the brainstem containing MTN were cut out of the wafers and glued onto blank resin blocks with cyanoacrylate. Serially cut thin sections were collected on Formvar-coated single-slot nickel grids and stained with uranyl acetate and lead citrate. The grids were examined on a Hitachi H-7500 electron microscope (Hitachi, Tokyo, Japan) at 80 kV accelerating voltage. Images were captured with Digital Montage software driving a MultiScan cooled CCD camera (ES1000W; Gatan, Pleasanton, CA) attached to the microscope and saved as TIFF files.
Statistical Analysis
Normal distribution of data and homogeneity of variance were checked by Kolmogorov-Smirnov Lilliefors test and Levene's test, respectively (P > 0.05). Numerical data are expressed as the mean ± the SD (parametric) or the median with the interquartile range (IQR; non-parametric). Statistical significance of mean difference was assessed using paired Student's t-tests (*), while that of median difference was assessed using Wilcoxon signed-rank test (†). The Pearson correlation coefficient (#) was calculated to assess the strength of a linear association between the two variables. P < 0.05 was considered statistically significant.
Mathematical Modeling
IGluR can be expressed as follows:
where N0 is the maximum number of activated GluR channels, P(t) represents the time course of open probability change of the GluR channels (0 ≤ P(t) ≤ 1), τ1 and τ2 are the time constants for the rising and decay phases of open probability, respectively, and a single GluR current is expressed as the sum of iNa(t) and iK(t) because the GluR channel is equally permeable to Na+ and K+. iNa(t) and iK(t) should follow the Goldman-Hodgkin-Katz equation and can be expressed as follows:
where V(t), F, R, and T are the membrane potential, Faraday constant, gas constant and absolute temperature, respectively, and the coefficient k (= 1.22 × 10−17) was introduced to yield a single GluR current of 0.5 pA at −70 mV (Swanson et al., 1997). Provided that an MTN neuron is composed of the soma and microvilli compartments, the following first order differential equations can be formulated:
where VS and VV represent the membrane potential, RS and RV are the input resistance, CS and CV are the membrane capacitance, and Gh−S and Gh−V are the conductance of the HCN channels in the compartments of the soma and microvilli, respectively. ssGh and τh are the steady-state conductance and opening/closing time constant (250 ms) of the HCN channels. Ri is the resistance between the two compartments. Eh is the reversal potential for Ih, EK = −97 mV, and GhMax, Vhalf and Sf are the maximal conductance, half-activation potential (−100 mV) and slope factor (11 mV) for Ih, respectively. The mathematical model described by these formula can be represented by the equivalent circuit (Figure 8B). The Na+ concentration in the microvilli ([Na+]V) is expressed as follows:
where [Na+]S, L and τ are the Na+ concentration in the soma, the volume of the microvilli compartments, and the equalization time constant for the Na+ concentration between the soma and microvilli compartments, respectively. The value of [Na+]V under the resting condition at −70 mV is equal to [Na+]S (= [Na+]i). In addition to the Na+ microdomain model, we also simulated IGluR with the Ih deactivation model, in which [Na+]V remained constant (same as [Na+]S) and the Ih deactivation was caused by a large space-clamp error that was created by introducing a large resistance between the soma and microvillus compartments.
Results
Effects of 8-Br-cAMP On Spike Firings Induced by AMPA Puff Application or Current Pulse Injection
We previously demonstrated that HCN1/2 channels are expressed not only in cell membrane but also in microvilli together with Na+/K+ pump (Kang et al., 2004). To investigate the possible functional interactions between HCN and GluR channels during spike firing in MTN neurons, we first examined the effects of 8-Br-cAMP (an activator of cyclic nucleotide-gated channel) on the firing activities caused by a puff application (100 ms duration) of AMPA and current-pulse injections at a resting and a hyperpolarized membrane potentials (−70 and −90 mV, respectively) under the current-clamp condition. The mean resting membrane potential was −68.2 ± 3.4 mV (n = 11). The AMPA puff application induced high-frequency burst firings (Figures 1A1,D1) but caused no spike firings in the presence of 8-Br-cAMP, although 8-Br-cAMP slightly but significantly (*P < 0.001) depolarized the resting membrane potential (−65.4 ± 3.8 mV; n = 11) (Figures 1B1,E1). However, the burst firings were restored following the bath application of Cs+ (used as an Ih blocker) in addition to 8-Br-cAMP (Figure 1C1,F1), although Cs+ may also block K+ channels. In contrast, the bath application of 8-Br-cAMP did not affect the spike generation caused by injection of depolarizing current pulses despite the similar threshold for evoking the burst and the spike generation at the resting membrane potential (Figures 1A2–3,B2–3). On the other hand, when examined at −90 mV, which was brought about by increasing the negative DC level from −0.77 ± 0.30 to −0.97 ± 0.36 nA (n = 8), the threshold for inducing the burst by activation of GluR was lower than that for spikes evoked by the current pulse (Figures 1D2–3,E2–3). If the inhibition of spiking was due to the shunting effects of HCN channels, the spiking with the higher threshold would be more easily inhibited by the shunting effect, contrary to what was observed here. More importantly, 8-Br-cAMP never changed the current or voltage threshold for evoking spikes by injection of current pulses regardless of the baseline potentials of either −70 or −90 mV at which the current pulses were applied. This was also true for 8-bromoguanosine 3′,5′-cyclic monophosphate sodium salt (8-Br-cGMP; an activator of cyclic nucleotide-gated channel) that activates TASK1 leak K+ current as well as Ih (see Supplementary Figure 1). Given the activation of Ih by 8-Br-cAMP, these observations strongly suggest that, at least in MTN neurons, the shunting effects of Ih were not involved in the inhibition of the bursting by the activation of GluR, and the bursts appeared to be suppressed by a functional interaction between GluR and HCN channels. At the resting membrane potential (−70 mV), Cs+ application in the presence of 8-Br-cAMP restored the burst firing without changing the responses to current pulses (Figure 1C), whereas at −90 mV, Cs+ application caused stronger responses due to the blockade of Ih and IK, which had been more strongly activated at −90 mV than at −70 mV (Figure 1F). Cs+ application in addition to 8-Br-cAMP at −90 mV would have caused a further membrane hyperpolarization from −90 mV unless the negative DC level was decreased from −1.03 ± 0.38 nA to −0.56 ± 0.43 nA (n = 6).
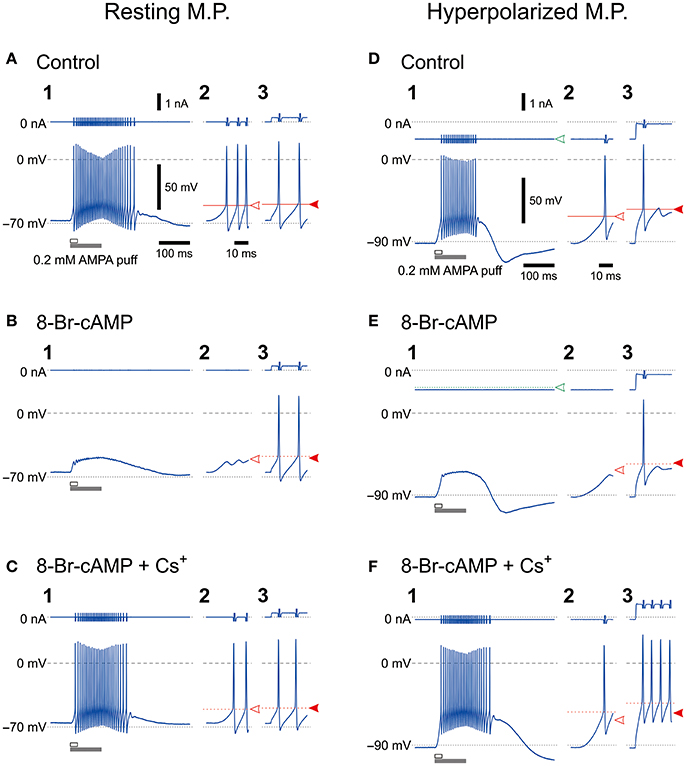
Figure 1. Effects of 8-Br-cAMP and Cs+ on spike firing induced by AMPA puff application or current pulse injection. Bottom, Membrane potential responses to a 100-ms puff of AMPA (1,2) or a 300-ms depolarizing current pulse (3) obtained before (A,D), during application of 8-Br-cAMP (B,E), and during the coapplication of 8-Br-cAMP and CsCl (C,F) under current-clamp conditions at baseline potentials of −70 mV (A–C) and −90 mV (D–F). Enlarged traces (2) seen during the respective time periods indicated with the open horizontal bars in 1. Top, Membrane currents. Panels labeled with 3 show the responses to the current pulses only for approximately 14 ms from the pulse onsets. Spikes that were evoked by current pulses were not affected by the possible shunting effects of Ih brought about by 8-Br-cAMP, which was consistent with the effects of 8-Br-cGMP that enhances leak K+ currents as well as Ih (Supplementary Figure 1). The calibrations in A1 also apply in all panels labeled with 1. The time calibration in A2 also applies in all panels labeled with 2 or 3.
Effect of Cs+ on Currents Induced by Glutamate Puffs
To exclude the possible shunting effects of HCN currents (Ih) or the effects of Ih deactivation, we further explored whether the HCN channels in MTN neurons can modify GluR-mediated currents under voltage-clamp conditions. We first tested the effect of blocking Ih with 5 mM extracellular Cs+, which is an effective blocker of Ih (Macri and Accili, 2004; Wu et al., 2012; Yang et al., 2015). We found that Cs+ enhanced the GluR currents (IGluR) in a highly voltage-dependent manner. In response to a short glutamate puff (0.2 mM, 50 ms duration), IGluR was evoked at various holding potentials in the absence and presence of 5 mM Cs+ under the voltage-clamp condition (Figure 2). In the absence of Cs+, the amplitude of the IGluR appeared to increase with membrane hyperpolarization up to −90 mV, whereas it was not increased but rather slightly decreased by further membrane hyperpolarization and was followed by an outward current that increased with membrane hyperpolarization (arrows, Figure 2A). In contrast, in the presence of Cs+, the IGluR monotonically increased in amplitude with membrane hyperpolarization and was not followed by any outward currents (Figure 2B). These features are well illustrated in the IGluR-V relationship obtained before and during the bath application of Cs+. As the holding potential was hyperpolarized from −50 to −90 mV, the IGluR measured at 95 ms after the puff application (Figure 2C) gradually increased (Figure 2A; blue open triangles, Figure 2D). However, at membrane potentials below −90 mV, the inward component of the IGluR (IN-IGluR) was decreased and the outward component measured at 395 ms after the puff application (Figure 2C) emerged (OT-IGluR; arrows, Figure 2A; blue open triangles and circles, respectively, Figure 2D). Thus, with a negative shift of the holding potential, IN-IGluR did not increase linearly despite the linear increase in the driving potential while OT-IGluR became more prominent. In view of the emergence of OT-IGluR and its increase with negative shifts of the holding potential, the IN-IGluR may have been curtailed by some outward current that increases as the holding potential is negatively shifted.
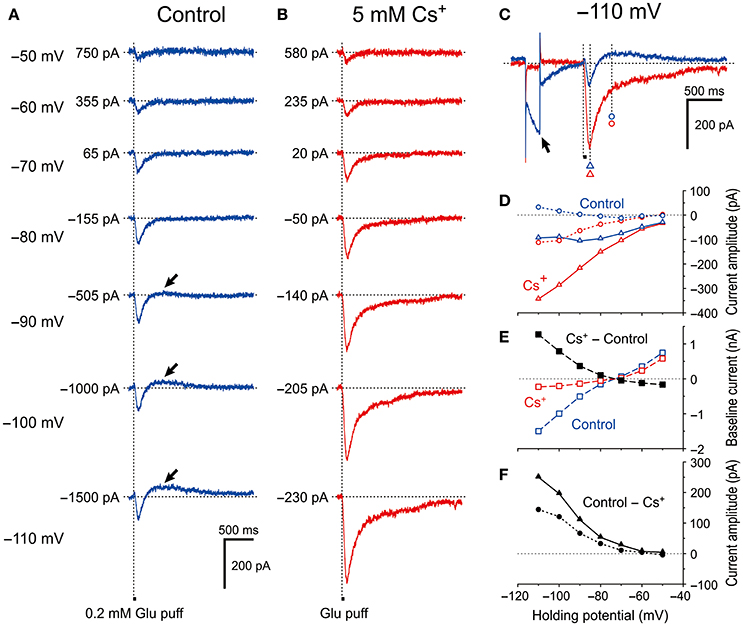
Figure 2. Voltage dependency of IGluR and effects of Cs+ on IGluR. (A) Current responses to 50-ms puff applications of 0.2 mM glutamate (IGluR) recorded under the voltage-clamp condition at holding potentials ranging between −110 anfd −50 mV in MTN neurons. (B) Bath application of 5 mM Cs+ outwardly shifted the baseline current level, enhanced the inward component of IGluR (IN-IGluR), and abolished the outward component of IGluR (OT-IGluR) seen at membrane potentials below −80 mV in the absence of Cs+. (C) Current responses at −110 mV obtained in the absence (blue trace) and presence (red trace) of Cs+. The baseline current levels were aligned. (D) I-V relationships of IGluR obtained in the absence (blue symbols) and the presence (red symbols) of Cs+. The triangles and circles represent the peak amplitudes of IGluR and the amplitudes of IGluR measured at 300 ms after the onset of the puff application of glutamate at which time IGluR exhibited a peak value of the outward component at −110 mV, respectively. (E) I-V relationships of the baseline current obtained in the absence (blue squares) and the presence (red squares) of Cs+. The black squares represent the amplitudes of outward shifts of the baseline current at respective holding potentials obtained by subtraction of the control response (blue squares) from that recorded during Cs+ application (red squares). (F) I-V relationships of the Cs+-sensitive components of IGluR that were obtained by subtraction of the response recorded during Cs+ application (red symbols) from the control (blue symbols) shown in D. The filled triangles and circles represent the amplitudes of Cs+-sensitive IGluR measured at its peak and at 300 ms after the onset of puff, respectively.
After bath application of Cs+ (Figure 2B), both the I-V relationships of IGluR measured at 95 and 395 ms after the puff application (Figure 2C) were almost linear (red open triangles and circles, respectively, Figure 2D). This linear I-V relationship of IGluR was invariably observed following Cs+ application in the 11 examined MTN neurons. Consequently, the amplitudes of the IN-IGluR at −70 mV were significantly increased by 26% ± 19% (*P < 0.002). Concomitantly, Cs+ abolished the Ih that was produced by a hyperpolarizing prepulse (arrow, Figure 2C), which is consistent with the outward shift of the baseline current that reflects the instantaneous or standing Ih at the respective membrane potentials (Figure 2E). The Cs+-sensitive outward component of IGluR (black filled triangles and circles, Figure 2F) that was obtained by subtraction of the response recorded after Cs+ application from the control revealed a voltage dependence similar to that of Ih. Giving the sensitivity of Ih to Cs+, this I-V relationship (black filled triangles, Figure 2F) suggests that Ih was involved in the apparent inhibition of IN-IGluR and in the generation of OT-IGluR.
Effect of Ih Activation With 8-Br-cAMP on IGluR
MTN neurons receive serotonergic synaptic inputs (Tanaka and Chandler, 2006) that activate 5-HT receptors to stimulate the production of cAMP, which in turn activates HCN channels through the binding with a cyclic nucleotide-binding domain (Wainger et al., 2001; Wang et al., 2007). To further investigate the involvement of HCN channels in the inhibition of IGluR, we next examined the effects of 0.5 mM 8-Br-cAMP on the IGluR evoked at −70 mV in response to a 500-ms puff applications of 0.2 mM AMPA (Figure 3). Bath application of 8-Br-cAMP shifted the baseline current inwardly from −149 ± 103 pA to −250 ± 148 pA (n = 6, *P < 0.007) and decreased the amplitude of the IN-IGluR while increasing the amplitude of the following OT-IGluR (blue and red traces, Figure 3A) concomitantly with an increase in Ih that was evoked by a negative pulse, as revealed by the superimposed traces aligned with their baseline levels (Figure 3B). In contrast, bath application of Cs+ right after the 8-Br-cAMP session shifted the baseline current outwardly to −76 ± 84 pA (n = 6, *P < 0.02) and increased the amplitude of the IN-IGluR but completely abolished the OT-IGluR concomitant with a marked inhibition of Ih (green traces, Figures 3A,B). These reciprocal changes between the IN-IGluR and Ih amplitudes that were observed during 8-Br-cAMP and Cs+ applications were represented by plotting the amplitudes of the IN-IGluR and Ih against time (blue and red circles, respectively, Figure 3C). Subsequently, plotting the amplitudes of the IN-IGluR (blue filled circles) and OT-IGluR (black open diamonds) against the amplitudes of the Ih revealed significantly negative (#P < 0.001, r = −0.96) and positive correlations (#P < 0.001, r = 0.95), respectively (Figure 3D). The inverse relationship of the normalized amplitudes between the Ih and the IN-IGluR and the proportional relationship between the Ih and the OT-IGluR were obtained in six MTN neurons following bath applications of 8-Br-cAMP and Cs+ (Figures 3E and F, respectively). The 8-Br-cAMP significantly decreased the IN-IGluR (*P < 0.001) but increased the OT-IGluR (*P < 0.006) concomitant with increases in Ih (*P < 0.002) (red symbols, n = 6; Figures 3E,F). In contrast, Cs+ significantly increased IN-IGluR (*P < 0.006) but decreased OT-IGluR (*P < 0.001) concomitant with decreases in Ih (*P < 0.001) (green symbols, n = 6; Figures 3E,F). Although Cs+ may block various K+ currents as well as the Ih, the inhibitory effect of Cs+ on K+ currents is very small at −70 mV. Indeed, consistent with these observations made with Cs+, the abolishment of the Ih by ZD7288 (Figure 3H) also significantly increased the amplitude of the IN-IGluR and concomitantly abolished the OT-IGluR completely (Figures 3G,I). These observations suggest that IN-IGluR was curtailed by an apparent outward current that presumably flowed through the HCN channels and emerged as OT-IGluR after the closure of the GluR channels at the offset of the AMPA puff.
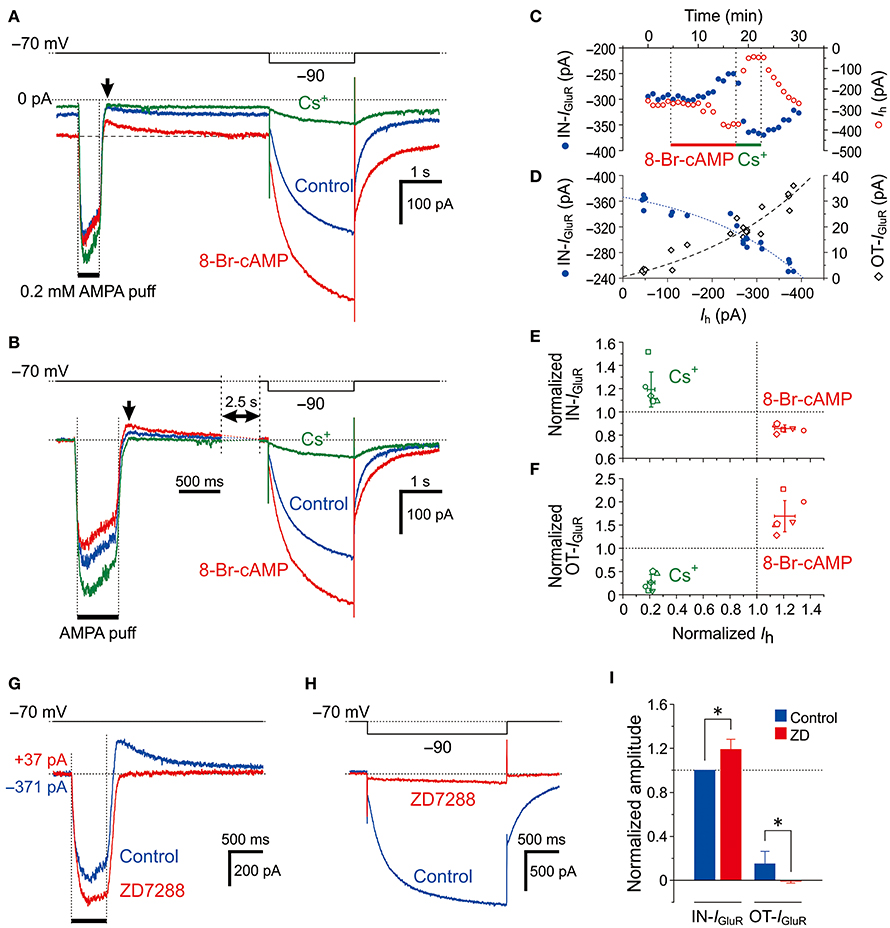
Figure 3. Effects of Ih activation by 8-Br-cAMP on IGluR. (A,B) Current responses to a 500-ms puff application of 0.2 mM AMPA and a hyperpolarizing pulse (−20 mV, 500 ms) recorded before (blue traces), during the application of 8-Br-cAMP (red traces), and during the application of Cs+ (green traces) under the voltage-clamp condition at −70 mV (A). The baseline current levels were aligned (B). The arrows indicate the outward components of IGluR (OT-IGluR). (C) Plot of the amplitudes of the inward component of IGluR (IN-IGluR; blue filled circles) and the Ih (red open circles) against time during applications of 8-Br-cAMP and Cs+. Note the reciprocal changes in the amplitudes of IN-IGluR and Ih. (D) Plot of the amplitudes of IN-IGluR (blue filled circles) and OT-IGluR (black open diamonds) against those of Ih obtained before and during 8-Br-cAMP and Cs+ applications. Note the negative correlation between the amplitudes of Ih and the IN-IGluR and the positive correlation between the amplitudes of Ih and the OT-IGluR. (E,F) Relationship between the normalized amplitudes (mean ± SD) of the IN-IGluR (E) and OT-IGluR (F) and that of Ih observed during 8-Br-cAMP (red symbols) and Cs+ (green symbols) applications in six MTN neurons (n = 6). A set of four (red and green, E,F) symbols with the same shape represent data obtained from a single neuron. 8-Br-cAMP: Ih, 1.21 ± 0.08 (*P < 0.002); IN-IGluR, 0.86 ± 0.03 (*P < 0.001); OT-IGluR, 1.69 ± 0.33 (*P < 0.006). Cs+: Ih, 0.21 ± 0.03 (*P < 0.001); IN-IGluR, 1.19 ± 0.15 (*P < 0.04); OT-IGluR, 0.26 ± 0.17 (*P < 0.001). (G,H) Current responses to a 500-ms puff application of 0.2 mM AMPA (G) and a negative voltage pulse (−20 mV, 2 s) (H) recorded before (blue traces) and during the application of ZD7288 (red traces). The baseline current levels were aligned. (I) Mean (± SD) normalized amplitudes of the IN-IGluR and OT-IGluR before (blue columns) and during the application of ZD7288 (red columns) (n = 6). *: P < 0.05.
Provided that Na+ ions flow intracellularly during IGluR and accumulate in a microdomain of spine-like microvilli, the reversal potential for Ih (Eh) should be transiently shifted in the negative direction resulting in a reduction of the driving potential of inward Ih, which in turn would shift the baseline inward Ih at −70 mV in the outward direction. Then, the IN-IGluR may be decreased due to the transient outward shift of the baseline Ih, and the OT-IGluR may become apparent following the cessation of the IN-IGluR at the puff offset because the baseline Ih is likely to recover slowly following the extrusion of Na+ ions from the microdomain. The outward shift of the baseline Ih during IGluR through the accumulation of Na+ ions may become larger as the conductance of HCN channels is increased by 8-Br-cAMP even if the reduction of the driving potential of Ih remains the same. Therefore, we hypothesized that 8-Br-cAMP decreases IN-IGluR and increases OT-IGluR by further outwardly shifting the baseline Ih at −70 mV during IGluR, which is consistent with the opposite effects of Cs+/ZD7288 on IGluR.
Differential Effects of Ouabain on Ih and IGluR
Because the Na+/K+ pump and HCN share a Na+ microdomain in the spine-like microvilli of MTN neurons as we previously reported (Kang et al., 2004), we next compared the effects of ouabain (Na+/K+ pump inhibitor) on Ih and IGluR evoked at −70 mV. Ouabain shifted the baseline current in the inward direction and increased the amplitudes of the inward Ih evoked by a negative voltage pulse to −90 mV as a result of the suppression of the outward Na+/K+ pump-mediated current that might have been induced in response to the activation of Ih (Figure 4A). This observation is consistent with our previous study (Kang et al., 2004). In contrast, ouabain did not increase but rather decreased the IN-IGluR whereas it increased the OT-IGluR (Figure 4Ba; also see Figure 4C) concomitant with an apparent enhancement of Ih (Figure 4A). Thus, the ouabain sensitive current which was acquired by subtraction of the response obtained after application of ouabain from the control response was composed of the ΔIN-IGluR and the slow inward tail-I as the ΔOT-IGluR (Figure 4Bb). In the 10 MTN neurons examined, the inhibition of Na+/K+ pump-mediated current with 50 μM ouabain significantly shifted the baseline current in the inward direction [from −25 (IQR 29) pA to −171 (IQR 44) pA, †P < 0.006, n = 10] and increased Ih [from −732 (IQR 94) pA to −791 (IQR 83) pA, †P < 0.006, n = 10], whereas it significantly decreased the amplitudes of the IN-IGluR at −70 mV from −676 (IQR 517) pA to −499 (IQR 456) pA (†P < 0.006, n = 10) and increased the amplitudes of the OT-IGluR at −70 mV from 30 (IQR 46) pA to 51 (IQR 27) pA (†P < 0.006, n = 10; Figure 4C).
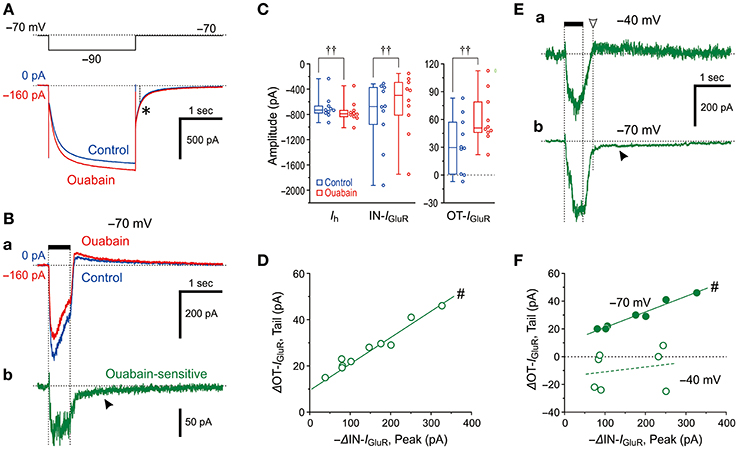
Figure 4. Effects of a Na+/K+ pump inhibitor on IGluR. (A,B) Current responses to a negative voltage pulse (−20 mV, 2 s) (A) and a 500-ms puff application of 0.2 mM AMPA (Ba) recorded before (blue traces) and during the application of 50 μM ouabain (red traces). The ouabain-sensitive IGluR, acquired by subtraction of the red trace from the blue trace shown in Ba (Bb). Note the presence of a slow tail component, reflecting an enhancement of OT-IGluR (arrowhead). The baseline current levels were aligned. The asterisk denotes the unchanged tail current before and during the application of ouabain. (C) Box-and-whisker plots represent the amplitudes of the steady-state Ih, IN-IGluR, and OT-IGluR obtained before (blue) and after 50 μM ouabain application (red). ††: P < 0.01 (Wilcoxon signed-rank test). (D) Plot of the increase in amplitude of OT-IGluR against the decreases in the amplitudes of IN-IGluR following ouabain application. #: P < 0.05 (Pearson correlation coefficient). (E) The ouabain-sensitive IGluR at −40 mV (upper trace) and at −70 mV (lower trace). Note the absence and presence (filled arrowhead) of slow tail component that reflects an enhancement of OT-IGluR. The amplitude of OT-IGluR was measured at the time indicated by downward open arrowhead. (F) Plot of changes in amplitude of OT-IGluR against decreases in amplitudes of IN-IGluR at −70 mV and −40 mV. Note the presence and absence of significant correlation between the two amplitudes, at −70 mV and −40 mV, respectively. #: P < 0.05 (Pearson correlation coefficient).
Such differential effects of ouabain on HCN and GluR would indicate that Na+ influx through HCN channels immediately and markedly activates Na+/K+ pumps, whereas Na+ influx through GluR would neither immediately nor markedly activate Na+/K+ pumps. Then, the apparent suppression of IGluR is either brought about directly by an accumulation of Na+ that causes the reduction of the driving potential of IGluR or caused by an enhancement of Ih as a result of the inhibition of Na+/K+ pump by ouabain, in a manner similar to the case with 8-Br-cAMP. Because the OT-IGluR was also enhanced by ouabain, the inhibition of IN-IGluR by ouabain was at least partly due to the generation of outward current mediated by a transient reduction of the enhanced (inwardly shifted) baseline Ih by ouabain during IGluR that might have led to the generation of OT-IGluR. Indeed, there was a significant positive correlation between the decrease in IN-IGluR and the increase in OT-IGluR (#P < 0.001, r = 0.98, n = 10; Figure 4D). Next, we aimed to examine if there is Ih-independent decrease in IGluR following ouabain application at −40 mV at which Ih is not active at all. As revealed by the ouabain sensitive component of IGluR at −40 mV, which was acquired by subtraction of IGluR obtained after ouabain application from that of the control (Figure 4Ea), ouabain decreased IGluR evoked at −40 mV but did not generate the slow inward tail component in contrast to the ouabain sensitive IGluR at −70 mV (Figure 4Eb). This also suggests that the functional interaction between GluR and Na+/K+ pump is very weak if any and the effect of Na+ accumulation on IGluR in microvilli overcame the interaction if any. As revealed by the presence or absence of the ouabain sensitive slow inward tail component of IGluR (Figures 4Ea,b), IGluR inhibition by ouabain at −70 mV was invariably accompanied by the enhancement of OT-IGluR, whereas IGluR inhibition by ouabain at −40 mV was not accompanied by enhancement of OT-IGluR. Indeed, there was a significant positive correlation between the decrease in IN-IGluR and the increase in OT-IGluR when examined at −70 mV (#P < 0.001, r = 0.98, n = 7) whereas no significant correlation between the decrease in IN-IGluR and the changes in OT-IGluR when examined at −40 mV in the same MTN neurons (#P > 0.6, r = 0.21, n = 7; Figure 4F). Thus, even in the absence of HCN activity, GluR activation did not apparently stimulate Na+/K+ pump, suggesting that the Euclidean distance between Na+/K+ pump and GluR is much larger than that between Na+/K+ pump and HCN channels.
These observations and notions suggest that Na+ homeostasis around active HCN channels is strictly regulated by Na+/K+ pump as long as GluR is not activated, whereas the homeostasis around active GluR is not regulated by Na+/K+ pump regardless of the activity of HCN channels. Indeed, ouabain increased the Ih amplitude but did not significantly (†P > 0.1, n = 7) increase its tail current (asterisk), as the amplitude measured 0.1 sec after the offset of the negative command pulse to −90 mV (interrupted line) was slightly changed from −187 ± 67 pA to −195 ± 67 pA following ouabain application (Figure 4A). This suggests the negative shift of the reversal potential of Ih due to the accumulation of Na+ through the breakdown of Na+ homeostasis around HCN channels by ouabain (see Discussion). This finding is in contrast to the case with 8-Br-cAMP (compare Figures 3A,B, 4A). Although the negative shift of Eh has not been reported, the activity-dependent shift of the reversal potential is not unusual for ligand gated channels such as GABAA (Fiumelli et al., 2005) or glycine (Kim and Trussell, 2009) receptor channels.
Effects of the Change in the Reversal Potential for Ih on IGluR
IGluR was evoked at the respective membrane potentials that ranged between −115 and −25 mV after the depolarizing (−25 mV; blue traces) or hyperpolarizing (−115 mV; red traces) prepulse that largely deactivated or activated Ih, respectively (Figure 5A). The amplitudes of the IN-IGluR obtained at the respective membrane potentials after the hyperpolarizing prepulse (red traces) were smaller than those obtained after the depolarizing prepulse (blue traces, Figure 5B). As revealed in the plot of the amplitudes of the IN-IGluR against the membrane potentials (Figure 5C), the IN-IGluR obtained below −55 mV after the hyperpolarizing prepulse was significantly (n = 5, *P < 0.05) smaller than those obtained after the depolarizing prepulse. In this experiment, an inhibition of IN-IGluR was observed following increases in the conductance of the HCN channels.
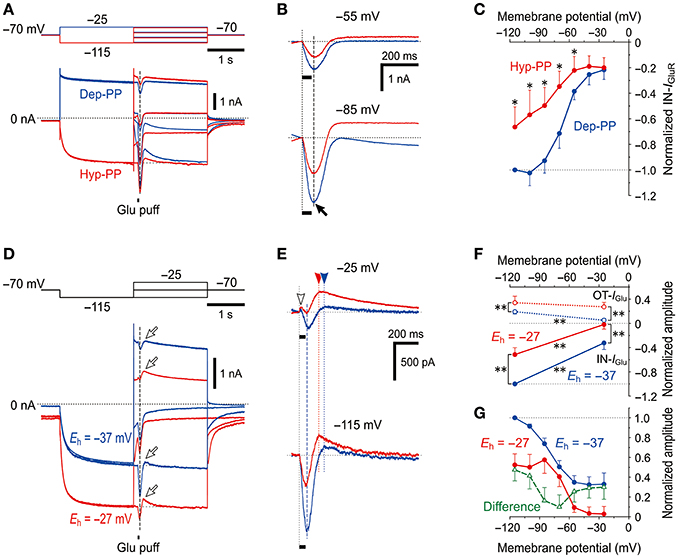
Figure 5. Voltage-dependent inhibitory effects of Ih on IGluR (A) IGluR responses evoked at various membrane potentials following either the activation of Ih by a hyperpolarizing prepulse (Hyp-PP; red traces) or the deactivation of Ih by a depolarizing prepulse (Dep-PP; blue traces). (B) Superimposed IGluR responses evoked at −55 (top) and −85 mV (bottom) showing the decrease in the amplitude of IGluR following the activation of Ih. (C) I-V relationships of IGluR obtained following hyperpolarizing (red) and depolarizing prepulses (blue circles). The amplitudes of IGluR were normalized to the amplitude of IGluR at −120 mV following a depolarizing prepulse. *P < 0.05. (D) Sample traces of IGluR responses evoked at various membrane potentials following activation of Ih by a hyperpolarizing prepulse obtained in the different extracellular solutions in which Eh is either −37 (blue) or −27 mV (red traces). (E) Superimposed IGluR responses evoked at −25 (top) and −115 mV (bottom) showing the decrease in the amplitude of IGluR following the activation of Ih with Eh of −27 mV compared with Eh of −37 mV. (F) I-V relationships of OT- and IN-IGluR obtained in the respective conditions. **P < 0.01. (G) The amplitudes of the IN-IGluR normalized to that at −115 mV under the condition of Eh = −37 mV. Note the U-shaped voltage-dependent inhibition of IN-IGluR following the increase in the driving potential for Ih (green open triangles).
In the next experiment, Ih was increased by increasing the driving potential without changing the conductance of HCN channels to directly clarify whether the inhibitory effects of Ih on glutamate responses were due to its shunting effect or the functional interaction between the two channels. [K+]o was increased from 3 to 21 mM by replacing 18 mM Na+ with equimolar K+ to shift the reversal potential for K+ currents (EK) from −97 to −47 mV and to shift the Eh from −37 to −27 mV while leaving the reversal potential for IGluR unchanged due to the equal permeability of GluR channels to K+ and Na+. Following an increase in [K+]o, the baseline current at −70 mV shifted inwardly, which suggests that the standing Ih reflected in the baseline current was increased by increasing the driving potential by 10 mV together with a generation of an inward leak K+ current. Concomitantly, the IN-IGluR was clearly decreased while the OT-IGluR (arrows) was clearly enhanced both at −25 and −115 mV (Figure 5D; also see Figure 5E). The amplitudes of IN-IGluR (filled circles) and OT-IGluR (open circles) obtained under the condition of Eh = −27 mV (red symbols) were significantly smaller (−25 mV, *P < 0.001 and −115 mV, *P < 0.001) and larger (−25 mV, *P < 0.001 and −115 mV, *P < 0.001), respectively, than those obtained under the control condition of Eh = −37 mV (n = 7; blue symbols, Figure 5F). The normalized decrease in the amplitude of IN-IGluR following the shift of Eh from −37 mV (blue filled circles) to −27 mV (red filled circles) was found to have a U-shaped voltage dependence with the minimal value at −70 to −60 mV (green open triangles, Figure 5G), although such an estimation for OT-IGluR was difficult due to its slower time-to-peak and the differential relaxation of Ih between the responses under the two different conditions of Eh.
Thus, without a conductance increase in HCN channels but with a positive shift of Eh by a [K+]o increase, IGluR was more strongly canceled. Simple voltage-dependent deactivation of Ih is not compatible with the U-shaped voltage dependence of inhibition of IN-IGluR because the deactivation of the possible outward baseline Ih generated at −25 mV would result in an increase in the IN-IGluR and a decrease in the OT-IGluR. These observations and notions clearly indicate that IGluR was suppressed neither by the shunting effects of Ih nor by the deactivation of Ih, but was rather canceled by a decrease in the inward baseline Ih or an increase in the outward baseline Ih that was induced during IGluR depending on the membrane potential at which IGluR was evoked (Figure 5G).
A possible negative shift of Eh due to accumulation of Na+ in the microvillus during IGluR can cause a decrease in the inward baseline Ih at −115 mV and an increase in the outward baseline Ih at −25 mV, both of which should result in the decrease in IN-IGluR and increase in OT-IGluR. This assumption is strongly supported by the U-shaped voltage dependence of the decrease in the amplitude of IN-IGluR (Figure 5G). Taken together, it is likely that IGluR can be decreased either by decreasing the driving potential for inward Ih or by increasing the driving potential for outward Ih depending on the holding potential, through the accumulation of Na+ ions in the microvillus which serves as a Na+ microdomain.
Na+ Accumulation in the Microvilli in Response to the Activation of GluR
Because a transient negative shift of the reversal potential for Ih is likely to be caused by a transient increase in Na+ concentration in microdomains following the activation of GluR, we next addressed whether Na+ concentration transiently increases in the microvilli in response to activation of GluR using a Na+ indicator, Sodium Green.
Using a confocal microscope, we performed fluorescence measurements of Na+ concentration changes in Sodium Green-loaded MTN neurons in response to the bath application of 1 mM glutamate (Figure 6). The Na+ concentration was gradually increased only just beneath the plasma membrane or presumably in the microvilli (Figure 6B), while the cytoplasm did not exhibit any marked increases in Na+ concentration in an MTN neuron (asterisk, Figure 6A). The first glutamate application for 1 min caused a Na+ transient that exhibited a more than 50% decay within 80 s from the offset of the glutamate application (Figures 6A,B,E). In contrast, the second application of glutamate for 3 min caused a larger increase in Na+ concentration not only in microvilli but also partly in the cytoplasm (Figures 6C,D), which exhibited a less than 25% decay after 80 sec from the offset of the glutamate application (Figure 6E), suggesting that the Na+/K+ pump activity was involved in the regulation of the decay time course in a manner dependent on its availability that was inversely proportional to the Na+ concentration. This notion further suggests that successive glutamatergic synaptic inputs may be more strongly depressed by HCN activity, as has been reported previously (Magee, 1999; Carr et al., 2007). In a total of seven MTN neurons, 1 min of glutamate application increased the ΔF/F0 by 31 ± 12% just beneath the cell membrane or microvilli. It should be noted that the rate constant for Na+ binding may be too slow to detect the rapid and large increase in Na+ in microvilli (Figure 6E; see section Materials and Methods).
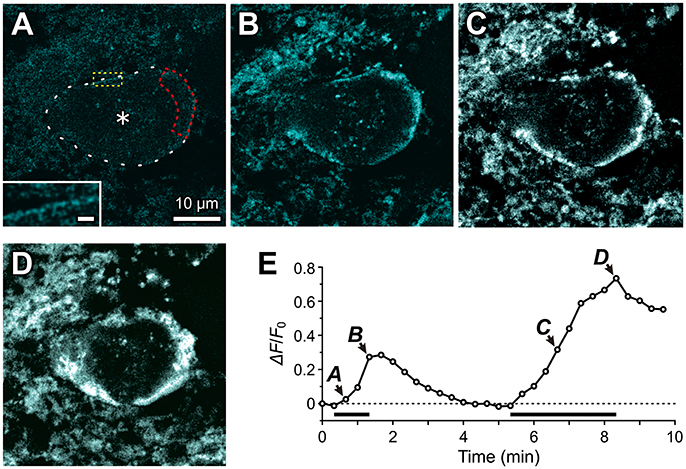
Figure 6. Fluorescence imaging of Na+ accumulation in the microvilli of MTN neurons using Sodium Green tetraacetate. Fluorescence images of Na+ concentration were captured every 20 sec in response to 1 mM glutamate application for 1 min and for 3 min in a Sodium Green-loaded MTN neuron. The respective fluorescence images (A–D) were obtained by subtraction of the frame 2 response from those of the respective frames (3rd, 5th, 21st, and 26th). (A) Twenty seconds after the onset of a 1 min glutamate application, apparent Na+ concentration increases can hardly be observed, but with a close look (inset) very thin or small dot-like fluorescence images can be observed along the contour of the MTN neuron (interrupted line). The region encircled with the red interrupted line represents the ROI. Scale bar in inset: 1 μm. (B) A Na+ concentration increase seen just beneath the plasma membrane or microvilli of a Sodium Green-loaded MTN neurons at the offset of a 1-min glutamate application. (C,D) A larger Na+ concentration increase captured at 80 sec (C) and 180 sec (D) after the onset of a 3-min glutamate application. (E) A time course of Na+ accumulation evoked twice in response to two successive application of 1 mM glutamate for 1 and 3 min separated by 4 min. The arrowheads with the letters A–D indicate the averaged fluorescence intensity in the ROI in the images A–D, respectively. The horizontal bars indicate the timing and duration of the glutamate application.
Glutamatergic Synapses on the Microvilli Expressing HCN2 Channels in MTN Neurons
Because we already demonstrated that in juvenile rats HCN1/2 are expressed in microvilli (Kang et al., 2004), we next confirmed that glutamatergic synapses are colocalized with HCN channels in microvilli of MTN neurons in adult rats. Electron-microscopic immunohistochemistry revealed that HCN2 immunoreactivity was observable as an electron-dense product that was localized in the spines of the MTN neuron (Figures 7A,B) and that a terminal bouton (asterisk) made a synaptic contact (arrowhead) with an HCN2-immunopositive spine of the MTN neuron (Figure 7C). Double immunostaining for HCN2 and vesicular glutamate transporter 2 (VGLUT2) revealed that a VGLUT2-immunopositive axon terminal (asterisk) made a synaptic contact on an HCN2-immunopositive spine (arrowhead) of the soma of the MTN neuron (Figure 7D). Furthermore, a terminal bouton (asterisk) of a glutamate-immunopositive axon formed asymmetrical synaptic contacts (arrowhead) with an HCN2-immunopositive spine (Figures 7E,F). These observations indicated that the glutamatergic axon arising from central neurons but not primary afferents (Pang et al., 2009) made synaptic contacts on the HCN-immunopositive spine that directly protrudes from the round shaped soma of the MTN neuron. Because it is known that in MTN neurons, HCN channels (Tanaka et al., 2003) and various synaptic inputs including glutamatergic one (Paik et al., 2012) are developmentally mature by PND 13 at the latest, these data obtained from adult rats can be extrapolated to juvenile rats at PND 13–18. Taking our previous study (Kang et al., 2004) into consideration together with the present morphological findings, the present electrophysiological findings obtained in juvenile rats can be extended to adult rats, eliminating the possibility that the functional interaction between HCN and GluR channels is a transient phenomenon accompanying the postnatal development of MTN neurons.
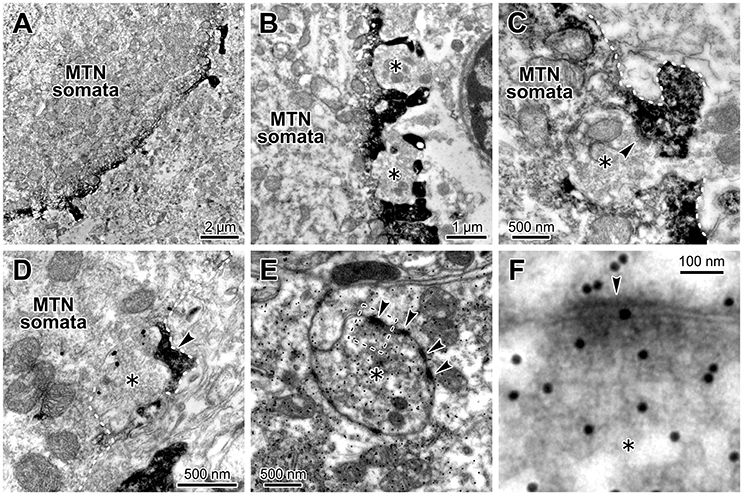
Figure 7. Glutamatergic axon terminals contact with HCN2-immunopositive small processes of the MTN neurons. (A,B) HCN2 immunoreactivity was localized in the small process and periphery of the MTN neuron. A terminal bouton (asterisk) contacted with an HCN2-immunopositive small process (arrowhead) of the MTN neuron (C). (C) The white dotted line demarcates the border of the small process. HCN2 immunoreactivity was observable as an electron-dense immunoreactive product. (D) A VGLUT2-immunopositive axon terminal (gold particles, asterisk) contacted with an HCN2-immunopositive small process (arrowhead) of the soma of the MTN neuron. The white dotted line demarcates the border of the small process. (E,F) Terminal boutons (asterisks) containing round vesicles that were immunopositive for an anti-glutamate antibody (E) contacted with the processes of the MTN neurons. Note that the glutamatergic terminal (silver grains) formed asymmetrical synaptic contacts (arrowheads) with a process. The area enclosed by a rectangle in E is enlarged in F.
A Mathematical Model of the IGluR Inhibition by the Activity of HCN Channels
As we previously reported the bidirectional interaction between HCN and Na+/K+ pump co-localized in the same microvillus in MTN neurons (Kang et al., 2004), Na+ influx/K+ efflux through HCN channels into the microvillus would not affect their own reversal potentials (Eh) due to the strict regulation of Na+/K+ homeostasis around the active HCN channels by the Na+/K+ pump (see Discussion section). This notion further suggests that HCN activity would also not affect GluR in the same microvillus. In contrast, GluR activity would affect HCN channels as well as the GluR channels themselves in the same microvillus because Na+/K+ homeostasis around the GluR was not strictly regulated by the Na+/K+ pump (Figure 4B). Then, as demonstrated using Sodium Green Na+ imaging (Figure 6), the Na+ influx during IGluR would transiently increase the Na+ concentration in the microvillus presumably because its volume is very small. However, K+ efflux through GluR channels during IGluR would not cause any marked reduction in the K+ concentration in the microvillus because of the following reason. The microvilli with diameters of 0.2–0.5 μm and lengths of only 1.0–1.5 μm directly protruded from the cell bodies of MTN neurons (Figure 7) where the Na+/K+ concentrations remain unchanged, and thereby the K+ efflux through the GluRs with far smaller pore sizes compared to the neck diameter of the microvillus would be instantaneously and easily compensated for by the equivalent K+ influx from the soma. These assumptions were made for the simplification of the Na+ microdomain model (Figure 8A). Numerical calculations were performed using a two-compartment model in which an MTN neuron is composed of the soma and the microvillus compartments (Figure 8B).
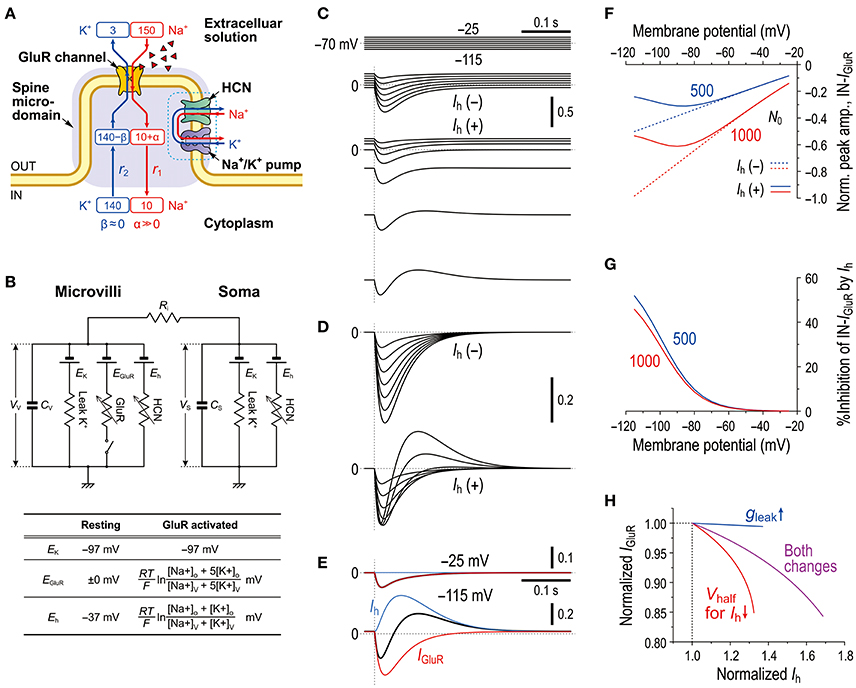
Figure 8. Mathematical modeling of IGluR inhibition with a negative shift of Eh through Na+ accumulation in the microvilli mimics the experimental results. (A) A schematic diagram showing a microvillus in which GluR and HCN were colocalized together with a Na+/K+ pump that has a functional coupling with the HCN channel. Only the Na+ concentration can be transiently increased via the activity of GluR, which consequently affects the reversal potentials for GluR and HCN channels. The HCN activity causes no changes in Na+ in a microvillus due to the coupling with Na+/K+ pump. The K+ concentration in the microvillus remains constant because K+ efflux through GluR is instantaneously and easily compensated from the soma. (B) Equivalent circuit of the mathematical model. (C) Simulated current responses to a glutamate puff obtained at the respective holding potentials (top panels, C) in the absence of HCN [Ih(–)] or in the presence of HCN [Ih(+)]. Maximum numbers of activated GluR channels N0 = 500. [Na+]o = 150 × 10−3 (M), [K+]o = 3 × 10−3 (M), [Na+]i = 14 × 10−3 (M), [K+]i = 141 × 10−3 (M), T = 293.15 (K), τ1 = 20 × 10−3 (s), τ2 = 40 × 10−3 (s), RS = 50 × 106 (Ω), RV = 400 × 106 (Ω), Ri = 5 × 105 (Ω), CS = 40 × 10−12 (F), CV = 5 × 10−12 (F), GhMax for Gh−S = 3.5 × 10−9 (S), GhMax for Gh−V = 35 × 10−9 (S), L = 210 × 10−18 (m3), and τ = 50 × 10−3 (s). (D) Superimposed traces of the simulated IGluR shown in C. (E) The current responses obtained at −25 and −115 mV shown in [D, Ih(+)] were separated into Ih and IGluR components (blue and red traces, respectively). (F) Non-linear (solid curves; under Ih) and linear (interrupted curves under no Ih) I-V relationships of the peak amplitudes of IN-IGluR normalized to that obtained at −115 mV under conditions of no Ih with N0 = 1,000 (red interrupted lines). The blue and red curves were obtained under the conditions of N0 = 500 and 1,000, respectively. (G) Voltage-dependent relative inhibitions of the peak IN-IGluR with the respective N0 calculated from (F). (H) The relationship between the normalized amplitudes of Ih and IGluR seen following the simultaneous increase in gLeak and the negative shift of Vhalf (magenta curve) and following the sole negative shift of Vhalf (red curve). The magenta and red curves simulated the effects of 8-Br-cAMP and 8-Br-cGMP on IGluR. In the case of the sole increase in gLeak (blue curve), leak K+ current was reflected in an instantaneous component of Ih. These results were experimentally confirmed (Supplementary Figure 2).
In the present mathematical simulation, the reversal potential for IGluR was variable because IGluR was expressed by a Goldman-Hodgkin-Katz equation, and Eh was also variable depending on the concentrations of intracellular Na+ and K+ (see the Materials and Methods section). First, our model correctly simulated the effects of voltage-dependent modulation of Ih on IGluR. Similar to the real experiments illustrated in Figure 2, the amplitude of the IGluR linearly increased with a negative shift of the holding potential when Ih was inactive [Ih(−), Figure 8C], whereas the amplitude of the IGluR increased with a negative shift of the holding potential to −80 or −90 and then turned to a decrease with further negative shifts of the holding potentials when Ih was active [Ih(+), Figure 8C]. The voltage-dependent inhibition of IGluR (Figures 8E–G) was also very similar to the real experiments (Figure 2E). IGluR promptly decayed, whereas the baseline Ih reduction lasted longer than IGluR, which led to the generation of a transient outward current as reflected in the differential time-to-peaks of the IGluR and Ih (Figure 8E). Under the voltage-clamp condition, the shunting effect no longer exists. Indeed, the gLeak increase did not cause any decrease in the IGluR (Figure 8H). However, the simultaneous increases in gLeak and Ih (magenta curve) were less effective in suppressing the apparent IGluR compared with the sole increase in Ih (red curve) (Figure 8H). This pattern was consistent with the comparison between the effect of 8-Br-cAMP (Figure 3D) and that of 8-Br-cGMP (see Supplementary Figure 2) which can activate TASK1 leak K+ current as well as Ih (Toyoda et al., 2010).
Similar to the real experiment illustrated in Figure 5, IGluR was smaller in amplitude and was followed by an outward current when IGluR was evoked right after the holding potential was positively stepped from −115 mV to varying potentials but before Ih was largely deactivated, which contrasts with the IGluR without Ih [compare Ih(+) and Ih(−) in Figures 9A,B,G]. This outward current was mediated either by a decrease in the baseline inward Ih or by an increase in the baseline outward Ih (Figure 9C), both of which are generated by negative shifts of Eh toward or away from the holding potentials (−115 and −25 mV; Figure 9J). Subsequently, a U-shaped voltage-dependent profile of the inhibition of IN-IGluR can be observed (blue curve, Figure 9H). The ratio of the peak amplitude of OT-IGluR to that of IN-IGluR also displayed a prominent U-shaped voltage dependence with a minimum ratio at −65 mV (blue curve, Figure 9I), which is clearly indicative of a U-shaped voltage-dependent mechanism for the generation of OT-IGluR. Furthermore, the effects of space-clamp error were also simulated by introducing a large Ri between the soma and microvillus compartments (Figures 9D–F) because space-clamp error, which allows IGluR to generate membrane depolarization, can consequently cause the deactivation of Ih. In the Ih deactivation model, a large diminution of IN-IGluR was accompanied only by a very small OT-IGluR (Figures 9D–G). Furthermore, the Ih deactivation failed to simulate the U-shaped voltage-dependent inhibition of IN-IGluR or the U-shaped voltage-dependent generation of OT-IGluR (Figures 9H,I). Instead, the inhibition of IN-IGluR decreased unidirectionally and nonlinearly with an increase in the membrane depolarization (red curve, Figures 9H), and the OT-IGluR evoked at hyperpolarized membrane potentials turned out to be a slow inward tail component of the preceding IN-IGluR at depolarized membrane potentials in the Ih deactivation model (red curve, Figure 9I). These observations clearly indicate that the present mechanism for the diminution of the IN-IGluR and the U-shaped voltage-dependent generation of OT-IGluR is distinct from the deactivation of Ih due to space-clamp error.
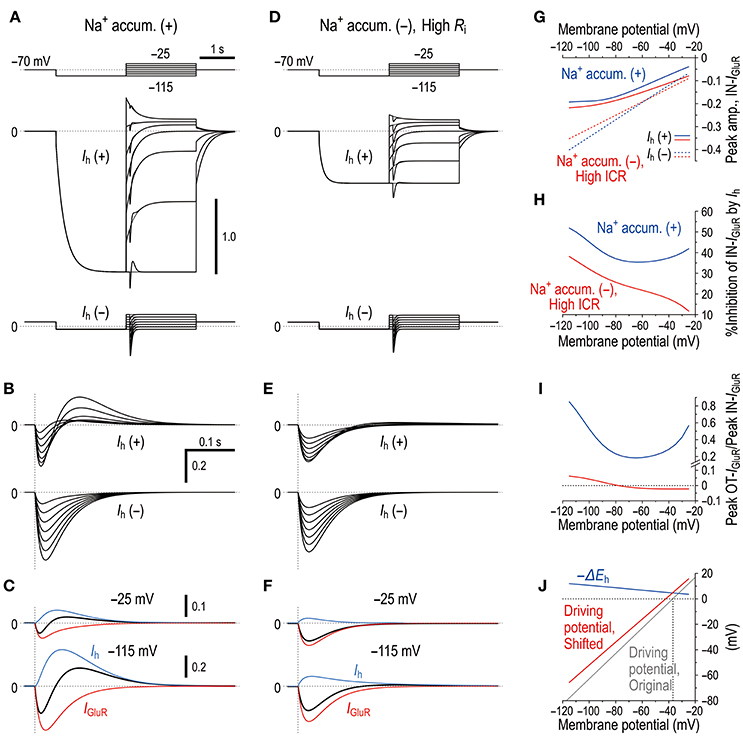
Figure 9. Microdomain model vs. Ih deactivation model. (A) In the Na+ microdomain model, IGluR was evoked immediately after the membrane potential was positively stepped from −115 mV to varying membrane potentials but before Ih was largely deactivated in the presence and absence of Ih (upper and lower panels, respectively). The uppermost panel shows the voltage command pulses. (B) Superimposed traces of the IGluR responses shown in A. (C) Separation of the pure IGluR and Ih components of the apparent biphasic IGluR composed of IN-IGluR and OT-IGluR obtained at −25 mV and −115 mV (upper and lower panels, respectively) as shown in B. (D) In the Ih deactivation model in which Ri was increased by 50 times from 5 × 105 to 250 × 105 (Ω) to create a space-clamp error. IGluR was immediately after the membrane potential was positively stepped from −115 mV to varying potentials but before Ih was largely deactivated in the presence and absence of Ih (upper and lower panels, respectively). The uppermost panel shows the voltage command pulses. (E) Superimposed traces of the IGluR responses shown in D. (F) Separation of the pure IGluR and Ih components of the IGluR composed of IN-IGluR and a much smaller OT-IGluR obtained at −25 mV and −115 mV (upper and lower panels, respectively) as shown in B. The biphasic profile of IGluR is much less clear compared with that obtained in the Na+ microdomain model. (G) Relationships between the holding potentials and the peak amplitudes of IN-IGluR obtained in the Na+ microdomain model and Ih deactivation model (blue and red curves, respectively) in the presence and absence of Ih (solid vs. interrupted curves, respectively). (H) A U-shaped profile of voltage-dependent inhibition of the peak IN-IGluR in the microdomain model (solid blue line) vs. a linear monophasic profile of voltage-dependent inhibition of the peak IN-IGluR in the Ih deactivation model (solid red line). (I) A U-shaped voltage-dependent change in the ratio of the peak amplitude of OT-IGluR to that of IN-IGluR with a minimal value of −65 mV in the Na+ microdomain model (blue curve) vs. a voltage-dependent non-linear monophasic change in the same ratio in the Ih deactivation model (red curve). Note the presence of negative ratios, which indicate that OT-IGluR decreased to zero and then turned into a slow inward tail component of the preceding IN-IGluR. (J) A voltage-dependent maximum negative shift of Eh (black curve) caused by Na+ accumulation in the microvilli during IGluR reduced the driving potential of Ih as revealed by a positive shift of the linear relationship between the membrane potential and the driving potential, which indicates that the generation of OT-IGluR is due to either the decrease in the inward Ih or the increase in the outward Ih. Compare the control (blue line) with that during IGluR (red line).
Discussion
In the present study, by taking advantages of the morphological structure of MTN neurons that have round shaped somata from which short spine-like microvilli of 1.0–1.5 μm length directly protruded (Figure 7; also see Kang et al., 2004), whole-cell voltage-clamp recordings of GluR responses and Ih were obtained from MTN neurons with little space-clamp errors (Figures 2–5) while we also showed current-clamp recordings (Figure 1). Therefore, deactivation of Ih due to unclamped depolarization would not occur in MTN neurons under voltage-clamped conditions. Furthermore, the shunting of the membrane resistance due to conductance increases in HCN channels is incompatible with voltage clamp. IGluR was markedly inhibited by the preceding activation of Ih in a U-shaped voltage-dependent manner with a minimal inhibition at approximately −60 mV (Figure 5), suggesting the existence of a mechanism distinct from the simple shunting effect or Ih deactivation mechanism. The U-shaped voltage-dependent generation of the outward current that curtails IN-IGluR appeared to be mediated by a decrease in the baseline inward Ih at hyperpolarized membrane potentials and by an increase in the baseline outward Ih at depolarized membrane potentials (Figure 9C), which can be generated by negative shifts of Eh either toward or away from the holding potentials, respectively (Figure 10). The negative shift of Eh that varies depending on the holding potentials should be brought about by a transient accumulation of Na+ in the microvilli of MTN neurons in response to activation of GluRs (Figure 6). Furthermore, the mathematical modeling validated the negative shift of Eh, and eliminated the possibilities of involvements of Ih deactivation or shunting in the inhibition of IGluR (Figures 8–10).
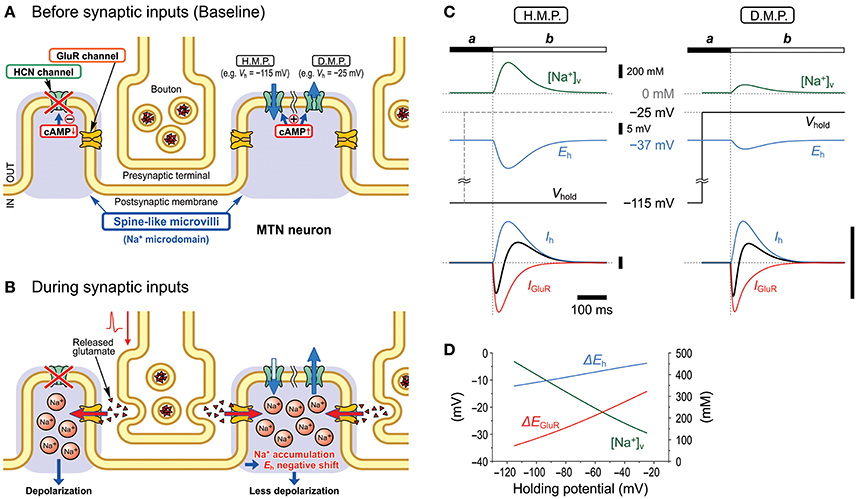
Figure 10. Schematic illustration showing the mechanisms underlying the inhibition of GluR responses by Ih. Under the condition that Ih is activated/upregulated by membrane hyperpolarization or cAMP production, Ih changes in the right microvilli before (A) and during (B) synaptic inputs are illustrated. Na+ accumulation in the microvilli produced by Na+ influx through GluR channels negatively shifts Eh and consequently causes a decrease or increase in the driving potential of the inward or outward baseline Ih, respectively, depending on the hyperpolarized (H.M.P.) or depolarized membrane potential (D.M.P.) levels, respectively (C). Such changes in the baseline Ih curtail IN-IGluR and generate the following OT-IGluR (C). The current calibration bars represent the same arbitrary unit. Peak [Na+]V changes in the microvillus in response to the GluR activation at varying holding potentials. Changes of peak shifts of Eh and EGluR following accumulation of Na+ in the microvillus at varying holding potentials (D).
Transient Accumulation of Na+ in the Microvilli during IGluR Causes a Negative Shift of Eh
In our previous study, we demonstrated that the Na+/K+ pump and HCN channels share a Na+ microdomain in spine-like microvilli and that there were bidirectional functional interactions between the Na+/K+ pump and HCN channels (Kang et al., 2004). The substitution of extracellular Na+ with Li+ increased Ih but almost abolished its tail current. This is because Li+ can flow through HCN channels into the microdomain but hardly or very slowly activates the Na+/K+ pump (Hermans et al., 1997; Féraille and Doucet, 2001), which consequently increases Ih but abolishes its tail-I due to the accumulation of Li+ that negatively shifts the Eh. In contrast, ouabain enhanced not only Ih but also tail-I when examined at −90 mV (Figure 5 in Kang et al., 2004). Enhancements of Ih and its tail-I by ouabain indicate that not only the Ih but also its tail-I was being opposed or contaminated by the outward current mediated by Na+/K+ pump. Then, the possible decrease in the tail-I by a negative shift of Eh as a consequence of Na+ accumulation in microvilli by ouabain would be masked by the blockade of Na+/K+ pump outward current by ouabain. This may be the reason why the amplitude of tail-I evoked at −70 mV remained almost unchanged in spite of increase in Ih after application of ouabain (Figure 4A). Thus, depending on the balance between the degree of negative shift of Eh due to the accumulation of Na+ in the microvilli and the degree of inhibition of Na+/K+ pump current, the amplitude of tail-I would be changed following application of ouabain. Therefore, it is strongly suggested that Na+/K+ homeostasis around HCN channels is strictly regulated by the Na+/K+ pump, and the activity of HCN channels did not affect their reversal potential as long as the Na+/K+ pump was active in the same microdomain. However, this appears not to be the case with GluR because IGluR was not enhanced but depressed by ouabain (Figure 4B). The present study demonstrated that GluR were co-localized with HCN channels in spine-like microvilli (Figure 7) and that GluR activation produced a transient accumulation of Na+ ions in the microvilli (Figure 6). Thus, when the Na+ influx through GluR is generated in addition to HCN activity, the Na+/K+ pump may not be able to afford to maintain Na+ concentration constant in the microvillus probably due to either the limited availability of the Na+/K+ pump or the differences in the Euclidean distance among the three channels that are colocalized in the same microvillus. Then, the Eh would be transiently shifted in the negative direction in response to GluR activation, which would lead to a reduction of the driving potential of the standing inward Ih at the resting or holding potential (< −70 mV). Because the standing inward Ih is reflected in the baseline current, the baseline current would shift outwardly during IGluR, and thereby cancel IGluR.
The diameter of spine neck in layer 2/3 pyramidal cells in the visual cortex ranged between 100 and 500 nm with a mean value of 200 nm (Arellano et al., 2007). Also in our study, spine-like microvilli neck diameters in electron microscopic observations in Figures 7C,D are about 200–250 and about 100 nm, respectively, although that appear larger in Figure 7E. The microvilli with lengths of 1.0–1.5 μm directly protruded from the cell bodies of MTN neurons. Regardless of the presence or absence of diffusion barrier, Na+ accumulation actually occurred in microvilli as demonstrated by Na+ imaging in the present study (Figure 6), although the diffusion barrier can modulate the time course of Na+ accumulation in microvilli. Furthermore, it is also known that stubby or mushroom type spines with large heads and thick necks can display larger responses to uncaged glutamate compared to thin spines which hardly display glutamate responses detectable in soma in spite of similar electrotonic distances (Matsuzaki et al., 2001). Given a transient accumulation of Na+ in dendritic spines, a similar modulation of IGluR by HCN channels may occur in dendritic spines of cortical pyramidal neurons.
Non-synaptic GluRs may also exist in MTN neurons as glutamate responses have been found in the soma of spinal dorsal root ganglion neurons (Huettner, 1990) or trigeminal ganglion neurons (Sahara et al., 1997). If non-synaptic GluRs are expressed in extra-microvilli in MTN neurons, glutamate puff would also activate these GluRs. Then, this may preclude us from drawing the present conclusion. However, an electron microscopic study demonstrated the embracement of MTN neurons by astrocytic processes that only allow synaptic contacts on the neuronal surface and protect MTN neurons from non-synaptic input (Copray et al., 1990a). This is in contrast to the somatic synapses on most brainstem motoneurons and interneurons lacking any astrocytic wrapping (Copray et al., 1990a), which would allow ambient GABA or glutamate to activate extra-synaptic receptors, causing tonic currents. This would not happen in MTN neurons due to the embracement by astrocytic processes, and therefore extra-synaptic receptors would not exist in MTN neurons. Nevertheless, a further study to selectively activate synaptic GluRs may be necessary to draw a definitive conclusion.
Na+ Accumulation in Response to GluR Activation by Puff Application vs. Physiological Activity of Presynaptic Terminals
Puff application of glutamate or AMPA may activate most of GluRs expressed in a single MTN neuron, the number of which may be much larger than that of GluRs activated in response to glutamate release from presynaptic terminals of a certain specific input. Subsequently, the total Na+ influx from GluRs in response to puff application may be much larger than that caused by physiological activity of glutamatergic inputs. Given a negative shift of Eh due to a large accumulation of Na+ by puff application, a question arises whether physiological activity of glutamatergic inputs can cause a similar negative shift of Eh because the accumulation of Na+ might be much smaller than that caused by puff application of glutamate or AMPA. Especially, a large accumulation of Na+ as a result of many GluRs activation by puff application may lead to a saturation of Na+/K+ pump activity that facilitates Na+ accumulation, whereas such saturation would not occur under physiological activity.
However, Na+ accumulation in microvilli by GluR activation is not a result of saturation of Na+/K+ pump activity, as revealed by the differential effects of ouabain on IGluR and Ih (Figure 4), and by Na+ imaging (Figure 6). The soma of MTN neurons is covered by numerous microvilli (Liem et al., 1991; Lazarov, 2002), and synaptic GluRs are sparsely distributed in respective microvilli (Figure 7; Paik et al., 2012). Activation of these GluRs by bath application of glutamate at 1 mM caused increases in Na+ concentration only beneath the cytoplasmic membrane in the microvilli distributed over the soma, leaving the Na+ concentration in the cytoplasm almost unchanged (Figure 6). Saturation of Na+/K+ pump activity occurred after 3 min bath application of glutamate as revealed by the increase in Na+ concentration not only in the microvilli but also partly in the cytoplasm close to cytoplasmic membrane, but hardly occurred in response to 30-s bath application (Figure 6E). Therefore, saturation of Na+/K+ pump activity would not occur in response to 50 or 200 ms puff application of glutamate or AMPA at 5–20 times smaller concentration (50–200 μM) than that of bath application.
Furthermore, ouabain differentially affected Ih and IGluR (Figure 4): Enhancement of Ih by ouabain indicates that Na+/K+ pump was activated immediately by the influx of Na+ through HCN channels. In contrast, inhibition of IGluR by ouabain indicates that Na+ influx through GluRs did not apparently stimulate Na+/K+ pump. IGluR is either decreased due to an increase in the basal level of Na+ concentration caused by inhibition of Na+/K+ pump by ouabain or opposed by an enhancement of Ih as a result of the inhibition of Na+/K+ pump by ouabain, in a manner similar to the case with 8-Br-cAMP. Because the OT-IGluR was also enhanced by ouabain, the inhibition of IN-IGluR by ouabain was at least partly due to the generation of outward current mediated by a transient reduction of the enhanced (inwardly shifted) baseline Ih by ouabain during IGluR that might have led to the generation of OT-IGluR. This strongly suggests that the activity of Na+/K+ pump is not saturated yet even during or at the offset of IGluR. Therefore, it is likely that the accumulation of Na+ during IGluR is not caused by the saturation of Na+/K+ pump activity.
The observed inhibition of IGluR by Ih is a whole-cell current generated as a result of summation of the small current changes independently occurring in the respective microvilli. Even if most of GluRs in an MTN neuron are activated by puff application of glutamate or AMPA, Na+ concentrations in the respective microvilli would not increase proportionally with the total number of activated GluRs in an MTN neuron, because respective GluRs in respective microvilli do not contribute to the accumulation of Na+ in the cytoplasm, but separately and independently causing Na+ increase in the respective microvilli. Thus, the total number of activated GluRs is not reflected in the concentration of Na+ in the cytoplasm or respective microvilli. Repetitive stimulation which can be mimicked by 50- or 200-ms puff application is more effective in inducing IGluR inhibition by HCN channels compared to single stimulation of GluR (Carr et al., 2007), suggesting that HCN2, rather than or in addition to HCN1, may be involved in this inhibition.
Deactivation of Ih Is Not Compatible With the U-Shaped Voltage-Dependent Inhibition of EPSCs
The outward current that follows the inward glutamatergic current is very similar to the hyperpolarization that follows EPSPs in many cell types, and the hyperpolarization was considered to be caused by deactivation of Ih due to EPSPs (Magee, 1998, 1999; Santello and Nevian, 2015). However, this idea is not necessarily correct but yet to be addressed. It is certain that the peak amplitudes of EPSPs would be decreased due to the deactivation of HCN channels during the rising phase of EPSPs if EPSPs are evoked from a potential where HCN channels are active. However, EPSPs would not be followed by HCN-mediated afterhyperpolarizations. This is because during the decay phase of EPSPs, the deactivation of Ih would be replaced with the voltage-dependent activation of Ih. The apparent sensitivity of the hyperpolarization following EPSPs to the HCN blocker ZD7288 may be simply due to the ZD7288-induced hyperpolarization of the baseline membrane potential, which consequently decreases the membrane hyperpolarization even if K+ channels are responsible for the hyperpolarization. Thus, the underlying mechanism is not clear in cortical pyramidal cells.
There may be a possibility that the transient outward current following the glutamate puffs at negative voltages is artificially caused by a space-clamp error that allowed a transient deactivation of Ih due to a possible unclamped membrane depolarization evoked by IGluR. However, this possibility is very small because the space-clamp error can be considered to be negligible due to a very short electrotonic length of the microvilli of 1.0–1.5 μm in length and 0.2–0.5 μm in diameter that protrude directly from the soma (Figures 7B,C,E). More importantly, the transient outward current following the glutamate puffs displayed a U-shaped voltage dependence (Figures 5E,F). Because the baseline Ih is likely to be very close to zero or an outward current at such depolarized membrane potentials when the reversal potential for Ih is −27 or −37 mV, its deactivation due to the possible membrane depolarization by IGluR would have resulted in the generation of either no outward current or an apparent inward current, contrary to what was observed in the present study (Figures 5E,F). Indeed, the mathematical modeling of the Ih deactivation as was the case with pyramidal cells revealed that despite a large diminution of IN-IGluR only a negligibly small outward current could have been caused by the deactivation of Ih brought about by creating a large space-clamp error (Figures 9E,F), and neither the diminution of IN-IGluR nor the generation of OT-IGluR displayed a U-shaped voltage-dependent nature (Figures 9H,I).
Possible Shunting Effects on IGluR Under Voltage-Clamp Conditions
Under the current-clamp condition, the amplitudes of the EPSPs would be decreased by decreasing the input resistance. However, under the voltage-clamp conditions, EPSCs would remain constant despite changes in input resistance unless there was charge redistribution that can be seen in cortical pyramidal cells during the activation of synaptic inputs onto the spines of the apical dendrites due to the space-clamp problem. Because in MTN neurons spine-like microvilli directly protrude from the soma, space clamp in spine-like microvilli is much more stringent than that in dendritic spines that protrude from the apical dendrites of cortical pyramidal cells. Nevertheless, IGluR was reduced by the activation of HCN channels in MTN neurons. In the present study, there were three lines of evidence against the possible involvement of the shunting effects of Ih. First, shunting effects would not cause outward currents following IGluR (Figures 2–5). Second, 8-Br-cAMP application suppressed the GluR-activated bursting without affecting the current threshold for evoking spikes by current pulse injections (Figure 1). Third, the mathematical modeling of the simultaneous activation of Ih and leak K+ current, which certainly has shunting effects, revealed decreases in the inhibitory effect of Ih on IGluR (Figure 8H), which was consistent with the results of the experiment (Supplementary Figure 2). Contrary to the present findings, it has been reported that activation of NO-cGMP signaling pathway enhanced NMDA current through gating HCN channels in CA1 hippocampal pyramidal neurons (Neitz et al., 2014).
Functional Significance of IGluR Inhibition by Ih in MTN Neurons
In MTN neurons, Ih activation hampered glutamatergic synaptic impacts and thereby suppressed glutamate-induced burst firing (Figure 1). Activation of serotonin receptors in MTN neurons has been reported to cause cAMP production and inhibit persistent INa (INaP) that mediates bursting in MTN neurons (Tanaka and Chandler, 2006). Therefore, such synaptic action would also enhance Ih to inhibit IGluR and prevent MTN neurons from INaP-mediated bursting. Subsequently, MTN neurons would be kept in primary sensory neuron mode, which faithfully conveys proprioceptive information to the central nervous system. This notion is consistent with the previously proposed mechanism (Saito et al., 2006; Kang et al., 2007) for voltage-dependent switching of the functional modes of MTN neurons between the primary sensory neuron single spiking and the premotor neuron bursting modes through the voltage-dependent activities of 4-aminopyridine-sensitive A-type K+ currents in the soma and riluzole-sensitive low-threshold INaP in the stem axon.
Recently, we have reported that protein kinase C activation by metabotropic glutamate receptors enhanced burst firing through the enhancement of resonance by upregulating INaP in MTN neurons (Chung et al., 2015). Taken together, it is suggested that the activities of Ih, 4-aminopyridine-sensitive A-type K+ currents, and INaP cooperatively contribute to switching between the two modes. It is of interest to investigate whether there are any synaptic inputs to inhibit HCN in MTN neurons. It is already known that terminals arising from the nucleus locus coeruleus exert noradrenergic synaptic action on MTN neurons (Copray et al., 1990b; Takahashi et al., 2010). These noradrenergic inputs may inhibit HCN activity by downregulating cAMP production through the activation of α2A adrenergic receptors (Wang et al., 2007) and consequently facilitate burst firing in response to glutamatergic inputs in MTN neurons, which is implicated in the attack behavior by biting enemies (Copray et al., 1990b; Takahashi et al., 2010). These functions are crucially mediated by Na+ microdomain in which the Na+/K+ pump, HCN and GluR functionally interact one another. This novel mechanism highlights a possible involvement of an impaired functional coupling between HCN channels and the Na+/K+ pump in a variety of neurological disorders also in other brain regions.
Author Contributions
YoK: Conceived and designed the research; MS, YaK, JW, HS, and HT: Conducted the electrophysiological experiments; JB, EK, TK, and YB: Conducted the immunohistochemical experiments; TT, MS, and YoK: Performed the numerical simulation study; All authors analyzed the data; MS, YaK, MK, SO, YB, and YoK: Wrote the manuscript. All authors have given approval to the final version of the manuscript.
Funding
This work was supported by Japan Society for the Promotion of Science grant KAKENHI JP26290006, JP17K07055 (YoK), and JP16K11482 (MS), National Research Foundation of Korea grants MSIP 2008-0062282 (YB) and MSIP 2016M3A9B6021209 and NRF-2017M3C7A1025602 (SBO). YoK was partly supported by Brain Pool Program through the Korean Federation of Science and Technology Societies (KOFST) funded by the Korea Ministry of Science, ICT and Future Planning. The funding body has no role in the design of the study and collection, analysis and interpretation of data and in writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2018.00113/full#supplementary-material
Abbreviations
8-Br-cAMP, 8-bromoadenosine 3′,5′-cyclic monophosphate sodium salt; 8-Br-cGMP, 8-bromoguanosine 3′,5′-cyclic monophosphate sodium salt; Eh, reversal potential for Ih; EK, reversal potential for K+ currents; Gh, conductance of HCN channels; GluR, glutamate receptor; HCN, hyperpolarization-activated cyclic nucleotide-gated; IGluR, GluR-mediated currents; Ih, current mediated by HCN channels; INaP, persistent Na+ current; IN-IGluR, inward component of the IGluR; IQR, interquartile range; MTN, mesencephalic trigeminal nucleus; N-ACSF, normal ACSF; OT-IGluR, outward component of the IGluR; PB, phosphate buffer; PND, postnatal day; VGLUT, vesicular glutamate transporter.
References
Amorino, G. P., and Fox, M. H. (1995). Intracellular Na+ measurements using sodium green tetraacetate with flow cytometry. Cytometry 21, 248–256. doi: 10.1002/cyto.990210305
Arellano, J. I., Benavides-Piccione, R., Defelipe, J., and Yuste, R. (2007). Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front. Neurosci. 1, 131–143. doi: 10.3389/neuro.01.1.1.010.2007
Carr, D. B., Andrews, G. D., Glen, W. B., and Lavin, A. (2007). α2-Noradrenergic receptors activation enhances excitability and synaptic integration in rat prefrontal cortex pyramidal neurons via inhibition of HCN currents. J. Physiol. 584, 437–450. doi: 10.1113/jphysiol.2007.141671
Cho, Y. S., Kim, Y. S., Moozhayil, S. J., Yang, E. S., and Bae, Y. C. (2015). The expression of hyperpolarization-activated cyclic nucleotide-gated channel 1 (HCN1) and HCN2 in the rat trigeminal ganglion, sensory root, and dental pulp. Neuroscience 291, 15–25. doi: 10.1016/j.neuroscience.2015.01.066
Chung, G., Saito, M., Kawasaki, Y., Kawano, T., Yin, D., Lee, S., et al. (2015). Generation of resonance-dependent oscillation by mGluR-I activation switches single spiking to bursting in mesencephalic trigeminal sensory neurons. Eur. J. Neurosci. 41, 998–1012. doi: 10.1111/ejn.12858
Copray, J. C., Liem, R. S., and Van Willigen, J. D. (1990a). Morphological arrangement between astrocytes and trigeminal mesencephalic primary afferent neurons in the rat. Exp. Brain Res. 83, 215–218. doi: 10.1007/BF00232211
Copray, J. C., Ter Horst, G. J., Liem, R. S., and Van Willigen, J. D. (1990b). Neurotransmitters and neuropeptides within the mesencephalic trigeminal nucleus of the rat: an immunohistochemical analysis. Neuroscience 37, 399–411. doi: 10.1016/0306-4522(90)90410-6
DiFrancesco, J. C., Barbuti, A., Milanesi, R., Coco, S., Bucchi, A., Bottelli, G., et al. (2011). Recessive loss-of-function mutation in the pacemaker HCN2 channel causing increased neuronal excitability in a patient with idiopathic generalized epilepsy. J. Neurosci. 31, 17327–17337. doi: 10.1523/JNEUROSCI.3727-11.2011
Féraille, E., and Doucet, A. (2001). Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: hormonal control. Physiol. Rev. 81, 345–418. doi: 10.1152/physrev.2001.81.1.345
Fiumelli, H., Cancedda, L., and Poo, M. M. (2005). Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron 48, 773–786. doi: 10.1016/j.neuron.2005.10.025
Friedman, J. E., and Haddad, G. G. (1994). Anoxia induces an increase in intracellular sodium in rat central neurons in vitro. Brain Res. 663, 329–334. doi: 10.1016/0006-8993(94)91281-5
George, M. S., Abbott, L. F., and Siegelbaum, S. A. (2009). HCN hyperpolarization-activated cation channels inhibit EPSPs by interactions with M-type K+ channels. Nat. Neurosci. 12, 577–584. doi: 10.1038/nn.2307
Harnett, M. T., Magee, J. C., and Williams, S. R. (2015). Distribution and function of HCN channels in the apical dendritic tuft of neocortical pyramidal neurons. J. Neurosci. 35, 1024–1037. doi: 10.1523/JNEUROSCI.2813-14.2015
Hermans, A. N., Glitsch, H. G., and Verdonck, F. (1997). Activation of the Na+/K+ pump current by intra- and extracellular Li ions in single guinea-pig cardiac cells. Biochim. Biophys. Acta 1330, 83–93. doi: 10.1016/S0005-2736(97)00143-0
Huang, H., and Trussell, L. O. (2014). Presynaptic HCN channels regulate vesicular glutamate transport. Neuron 84, 340–346. doi: 10.1016/j.neuron.2014.08.046
Huang, Z., Lujan, R., Kadurin, I., Uebele, V. N., Renger, J. J., Dolphin, A. C., et al. (2011). Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat. Neurosci. 14, 478–486. doi: 10.1038/nn.2757
Huettner, J. E. (1990). Glutamate receptor channels in rat DRG neurons: activation by kainate and quisqualate and blockade of desensitization by Con A. Neuron 5, 255–266. doi: 10.1016/0896-6273(90)90163-A
Kang, Y., Notomi, T., Saito, M., Zhang, W., and Shigemoto, R. (2004). Bidirectional interactions between h-channels and Na+-K+ pumps in mesencephalic trigeminal neurons. J. Neurosci. 24, 3694–3702. doi: 10.1523/JNEUROSCI.5641-03.2004
Kang, Y., Saito, M., Sato, H., Toyoda, H., Maeda, Y., Hirai, T., et al. (2007). Involvement of persistent Na+ current in spike initiation in primary sensory neurons of the rat mesencephalic trigeminal nucleus. J. Neurophysiol. 97, 2385–2393. doi: 10.1152/jn.01191.2006
Kim, Y., and Trussell, L. O. (2009). Negative shift in the glycine reversal potential mediated by a Ca2+- and pH-dependent mechanism in interneurons. J. Neurosci. 29, 11495–11510. doi: 10.1523/JNEUROSCI.1086-09.2009
Lazarov, N. E. (2002). Comparative analysis of the chemical neuroanatomy of the mammalian trigeminal ganglion and mesencephalic trigeminal nucleus. Prog. Neurobiol. 66, 19–59. doi: 10.1016/S0301-0082(01)00021-1
Liem, R. S., Copray, J. C., and Van Willigen, J. D. (1991). Ultrastructure of the rat mesencephalic trigeminal nucleus. Acta Anat. 140, 112–119. doi: 10.1159/000147045
Lörincz, A., Notomi, T., Tamás, G., Shigemoto, R., and Nusser, Z. (2002). Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat. Neurosci. 5, 1185–1193. doi: 10.1038/nn962
Macri, V., and Accili, E. A. (2004). Structural elements of instantaneous and slow gating in hyperpolarization-activated cyclic nucleotide-gated channels. J. Biol. Chem. 279, 16832–16846. doi: 10.1074/jbc.M400518200
Magee, J. C. (1998). Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J. Neurosci. 18, 7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998
Magee, J. C. (1999). Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat. Neurosci. 2:848. doi: 10.1038/12229
Matsuzaki, M., Ellis-Davies, G. C., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H. (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092. doi: 10.1038/nn736
Migliore, M., and Migliore, R. (2012). Know your current Ih: interaction with a shunting current explains the puzzling effects of its pharmacological or pathological modulations. PLoS ONE 7:e36867. doi: 10.1371/journal.pone.0036867
Mineff, E. M., Popratiloff, A., Usunoff, K. G., and Marani, E. (1998). Immunocytochemical localization of the AMPA receptor subunits in the mesencephalic trigeminal nucleus of the rat. Arch. Physiol. Biochem. 106, 203–209. doi: 10.1076/apab.106.3.203.4383
Neitz, A., Mergia, E., Imbrosci, B., Petrasch-Parwez, E., Eysel, U. T., Koesling, D., et al. (2014). Postsynaptic NO/cGMP increases NMDA receptor currents via hyperpolarization-activated cyclic nucleotide-gated channels in the hippocampus. Cereb. Cortex 24, 1923–1936. doi: 10.1093/cercor/bht048
Notomi, T., and Shigemoto, R. (2004). Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J. Comp. Neurol. 471, 241–276. doi: 10.1002/cne.11039
Paik, S. K., Kwak, M. K., Bae, J. Y., Yi, H. W., Yoshida, A., Ahn, D. K., et al. (2012). γ-Aminobutyric acid-, glycine-, and glutamate-immunopositive boutons on mesencephalic trigeminal neurons that innervate jaw-closing muscle spindles in the rat: ultrastructure and development. J. Comp. Neurol. 520, 3414–3427. doi: 10.1002/cne.23110
Pang, Y. W., Ge, S. N., Nakamura, K. C., Li, J. L., Xiong, K. H., Kaneko, T., et al. (2009). Axon terminals expressing vesicular glutamate transporter VGLUT1 or VGLUT2 within the trigeminal motor nucleus of the rat: origins and distribution patterns. J. Comp. Neurol. 512, 595–612. doi: 10.1002/cne.21894
Park, S. K., Lee, D. S., Bae, J. Y., and Bae, Y. C. (2016). Central connectivity of the chorda tympani afferent terminals in the rat rostral nucleus of the solitary tract. Brain Struct. Funct. 221, 1125–1137. doi: 10.1007/s00429-014-0959-6
Postea, O., and Biel, M. (2011). Exploring HCN channels as novel drug targets. Nat. Rev. Drug Discov. 10, 903–914. doi: 10.1038/nrd3576
Sahara, Y., Noro, N., Iida, Y., Soma, K., and Nakamura, Y. (1997). Glutamate receptor subunits GluR5 and KA-2 are coexpressed in rat trigeminal ganglion neurons. J. Neurosci. 17, 6611–6620. doi: 10.1523/JNEUROSCI.17-17-06611.1997
Saito, M., Murai, Y., Sato, H., Bae, Y. C., Akaike, T., Takada, M., et al. (2006). Two opposing roles of 4-AP-sensitive K+ current in initiation and invasion of spikes in rat mesencephalic trigeminal neurons. J. Neurophysiol. 96, 1887–1901. doi: 10.1152/jn.00176.2006
Sánchez-Alonso, J. L., Halliwell, J. V., and Colino, A. (2008). ZD 7288 inhibits T-type calcium current in rat hippocampal pyramidal cells. Neurosci. Lett. 439, 275–280. doi: 10.1016/j.neulet.2008.05.016
Santello, M., and Nevian, T. (2015). Dysfunction of cortical dendritic integration in neuropathic pain reversed by serotoninergic neuromodulation. Neuron 86, 233–246. doi: 10.1016/j.neuron.2015.03.003
Swanson, G. T., Kamboj, S. K., and Cull-Candy, S. G. (1997). Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J. Neurosci. 17, 58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997
Takahashi, T., Shirasu, M., Shirasu, M., Kubo, K. Y., Onozuka, M., Sato, S., et al. (2010). The locus coeruleus projects to the mesencephalic trigeminal nucleus in rats. Neurosci. Res. 68, 103–106. doi: 10.1016/j.neures.2010.06.012
Tanaka, S., and Chandler, S. H. (2006). Serotonergic modulation of persistent sodium currents and membrane excitability via cyclic AMP-protein kinase A cascade in mesencephalic V neurons. J. Neurosci. Res. 83, 1362–1372. doi: 10.1002/jnr.20822
Tanaka, S., Wu, N., Hsaio, C. F., Turman, J. Jr., and Chandler, S. H. (2003). Development of inward rectification and control of membrane excitability in mesencephalic v neurons. J. Neurophysiol. 89, 1288–1298. doi: 10.1152/jn.00850.2002
Toyoda, H., Saito, M., Okazawa, M., Hirao, K., Sato, H., Abe, H., et al. (2010). Protein kinase G dynamically modulates TASK1-mediated leak K+ currents in cholinergic neurons of the basal forebrain. J. Neurosci. 30, 5677–5689. doi: 10.1523/JNEUROSCI.5407-09.2010
Tsay, D., Dudman, J. T., and Siegelbaum, S. A. (2007). HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron 56, 1076–1089. doi: 10.1016/j.neuron.2007.11.015
Turman, J. E. Jr., Macdonald, A. S., Pawl, K. E., Bringas, P., and Chandler, S. H. (2000). AMPA receptor subunit expression in trigeminal neurons during postnatal development. J. Comp. Neurol. 427, 109–123. doi: 10.1002/1096-9861(20001106)427:1<109::AID-CNE7>3.0.CO;2-T
Verdier, D., Lund, J. P., and Kolta, A. (2004). Synaptic inputs to trigeminal primary afferent neurons cause firing and modulate intrinsic oscillatory activity. J. Neurophysiol. 92, 2444–2455. doi: 10.1152/jn.00279.2004
Wainger, B. J., Degennaro, M., Santoro, B., Siegelbaum, S. A., and Tibbs, G. R. (2001). Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411, 805–810. doi: 10.1038/35081088
Wang, M., Ramos, B. P., Paspalas, C. D., Shu, Y., Simen, A., Duque, A., et al. (2007). α2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell 129, 397–410. doi: 10.1016/j.cell.2007.03.015
Wu, S., Gao, W., Xie, C., Xu, X., Vorvis, C., Marni, F., et al. (2012). Inner activation gate in S6 contributes to the state-dependent binding of cAMP in full-length HCN2 channel. J. Gen. Physiol. 140, 29–39. doi: 10.1085/jgp.201110749
Yamada, R., Kuba, H., Ishii, T. M., and Ohmori, H. (2005). Hyperpolarization-activated cyclic nucleotide-gated cation channels regulate auditory coincidence detection in nucleus laminaris of the chick. J. Neurosci. 25, 8867–8877. doi: 10.1523/JNEUROSCI.2541-05.2005
Yang, Q., Kuzyk, P., Antonov, I., Bostwick, C. J., Kohn, A. B., Moroz, L. L., et al. (2015). Hyperpolarization-activated, cyclic nucleotide-gated cation channels in Aplysia: contribution to classical conditioning. Proc. Natl. Acad. Sci. U.S.A. 112, 16030–16035. doi: 10.1073/pnas.1501731113
Yi, F., Danko, T., Botelho, S. C., Patzke, C., Pak, C., Wernig, M., et al. (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352:aaf2669. doi: 10.1126/science.aaf2669
Keywords: glutamate receptor, hyperpolarization-activated cyclic nucleotide-gated cation channel, Na+/K+ pump, primary sensory neuron, mesencephalic trigeminal nucleus, microvilli
Citation: Kawasaki Y, Saito M, Won J, Bae JY, Sato H, Toyoda H, Kuramoto E, Kogo M, Tanaka T, Kaneko T, Oh SB, Bae YC and Kang Y (2018) Inhibition of GluR Current in Microvilli of Sensory Neurons via Na+-Microdomain Coupling Among GluR, HCN Channel, and Na+/K+ Pump. Front. Cell. Neurosci. 12:113. doi: 10.3389/fncel.2018.00113
Received: 25 October 2017; Accepted: 06 April 2018;
Published: 24 April 2018.
Edited by:
Tommaso Pizzorusso, Consiglio Nazionale Delle Ricerche (CNR), ItalyReviewed by:
Thomas Budde, Universität Münster, GermanyGia Michele Ratto, Consiglio Nazionale Delle Ricerche (CNR), Italy
Peter A. Goldstein, Cornell University, United States
Copyright © 2018 Kawasaki, Saito, Won, Bae, Sato, Toyoda, Kuramoto, Kogo, Tanaka, Kaneko, Oh, Bae and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitsuru Saito, bXRyc2FpdG9AZGVudC5rYWdvc2hpbWEtdS5hYy5qcA==
Seog Bae Oh, b2RvbGJhZUBzbnUuYWMua3I=
Youngnam Kang, a2FuZ0BkZW50Lm9zYWthLXUuYWMuanA=
†These authors have contributed equally to this work.
‡Present Address: Mitsuru Saito, Department of Oral Physiology, Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima, Japan
Eriko Kuramoto, Department of Anatomy for Oral Sciences, Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima, Japan
Takuma Tanaka, The Center for Data Science Education and Research, Shiga University, Hikone, Shiga, Japan