 Elisa Ren1†
Elisa Ren1† Vincenzo Roncacé2†
Vincenzo Roncacé2† Stefania Trazzi1†
Stefania Trazzi1† Claudia Fuchs1
Claudia Fuchs1 Giorgio Medici1
Giorgio Medici1 Laura Gennaccaro1Manuela Loi1Giuseppe Galvani1Keqiang Ye3Roberto Rimondini4
Laura Gennaccaro1Manuela Loi1Giuseppe Galvani1Keqiang Ye3Roberto Rimondini4 Giorgio Aicardi2,5*
Giorgio Aicardi2,5* Elisabetta Ciani1*
Elisabetta Ciani1*- 1Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy
- 2Department for Life Quality Studies, University of Bologna, Bologna, Italy
- 3School of Medicine, Emory University, Atlanta, GA, United States
- 4Department of Biomedical and Clinical Sciences, University of Bologna, Bologna, Italy
- 5Interdepartmental Center “Luigi Galvani” for Integrated Studies of Bioinformatics, Biophysics and Biocomplexity, University of Bologna, Bologna, Italy
Cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD) is a severe X-linked neurodevelopmental encephalopathy caused by mutations in the CDKL5 gene and characterized by early-onset epilepsy and intellectual and motor impairments. No cure is currently available for CDD patients, as limited knowledge of the pathology has hindered the development of therapeutics. Cdkl5 knockout (KO) mouse models, recently created to investigate the role of CDKL5 in the etiology of CDD, recapitulate various features of the disorder. Previous studies have shown alterations in synaptic plasticity and dendritic pattern in the cerebral cortex and in the hippocampus, but the knowledge of the molecular substrates underlying these alterations is still limited. Here, we have examined for the first time synaptic function and plasticity, dendritic morphology, and signal transduction pathways in the perirhinal cortex (PRC) of this mouse model. Being interconnected with a wide range of cortical and subcortical structures and involved in various cognitive processes, PRC provides a very interesting framework for examining how CDKL5 mutation leads to deficits at the synapse, circuit, and behavioral level. We found that long-term potentiation (LTP) was impaired, and that the TrkB/PLCγ1 pathway could be mechanistically involved in this alteration. PRC neurons in mutant mice showed a reduction in dendritic length, dendritic branches, PSD-95-positive puncta, GluA2-AMPA receptor levels, and spine density and maturation. These functional and structural deficits were associated with impairment in visual recognition memory. Interestingly, an in vivo treatment with a TrkB agonist (the 7,8-DHF prodrug R13) to trigger the TrkB/PLCγ1 pathway rescued defective LTP, dendritic pattern, PSD-95 and GluA2-AMPA receptor levels, and restored visual recognition memory in Cdkl5 KO mice. Present findings demonstrate a critical role of TrkB signaling in the synaptic development alterations due to CDKL5 mutation, and suggest the possibility of TrkB-targeted pharmacological interventions.
Introduction
Cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD) is a rare encephalopathy characterized by early-onset intractable epileptic seizures, severe intellectual disability, gross motor impairment, stereotypies, visual impairments and autistic-like features (Kalscheuer et al., 2003; Weaving et al., 2004; Bahi-Buisson et al., 2008; Bahi-Buisson and Bienvenu, 2012; Moseley et al., 2012; Paine et al., 2012; Stalpers et al., 2012; Fehr et al., 2013). CDD is caused by mutations in the X-linked gene, CDKL5, a member of a highly conserved family of serine-threonine kinases (Tao et al., 2004). To date, several different mutations have been described in the CDKL5 gene, mainly located within the CDKL5 catalytic domain (Kilstrup-Nielsen et al., 2012; Das et al., 2013; Fehr et al., 2013), suggesting that impaired CDKL5 kinase activity plays an important role in the pathogenesis of CDD (Tao et al., 2004; Bahi-Buisson et al., 2012).
No cure is currently available for CDD patients, as limited knowledge of the pathology has hindered the development of therapeutics. Cdkl5 knockout (KO) mice (Wang et al., 2012; Amendola et al., 2014; Okuda et al., 2017) have recently been created to investigate the role of CDKL5 in the etiology of CDD. Cdkl5 KO mice recapitulate different features of CDD, exhibiting severe impairment in learning and memory, visual and respiratory deficits, and motor stereotypies (Wang et al., 2012; Amendola et al., 2014; Fuchs et al., 2014, 2015; Mazziotti et al., 2017). The neuropathology of CDD points to arrested neuronal development rather than neurodegeneration or severe malformation of nervous tissue. Reduced neuronal branching and spine density have been observed in the visual and somatosensory cortex (Della Sala et al., 2016; Pizzo et al., 2016), and in the hippocampal region (Amendola et al., 2014; Fuchs et al., 2014) of Cdkl5 KO mice. In addition, immunocytochemical studies have demonstrated alterations in synaptic connectivity, which might lead to an excitation-inhibition imbalance (Pizzo et al., 2016; Sivilia et al., 2016). Together, these observations have led to the suggestion that there is an overall reduction in the number of synaptic inputs to neurons in the CDKL5-deficient brain. It has been shown that CDKL5 is localized at excitatory synapses (Ricciardi et al., 2012) where it binds to the scaffolding postsynaptic density protein 95 (PSD-95) and to the synaptic cell adhesion molecule NGL-1 (Ricciardi et al., 2012; Zhu et al., 2013). Cdkl5 deficiency in primary hippocampal neurons leads to deranged expression of the GluA2 subunit of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (GluA2-AMPAR), and it is probable that this prompts an alteration of synaptic functions (Tramarin et al., 2018).
To date, only a few studies have investigated the functional consequences of these changes at the synaptic level (Della Sala et al., 2016; Okuda et al., 2017; Tang et al., 2017). Long-term potentiation (LTP) is the most widely studied form of synaptic plasticity in the mammalian nervous system. It provides a neuronal substrate for learning and memory and is impaired in several models of psychiatric and neurologic disorders. LTP was found to be altered in opposing ways in two brain regions of Cdkl5 KO mice: it was strongly reduced in the somatosensory cortex (Della Sala et al., 2016) and slightly increased in the hippocampal CA1 region (Okuda et al., 2017). The latter effect appears to be mediated by upregulation of GluN2B-containing NMDA receptors (Okuda et al., 2017). Interestingly, the ablation of Cdkl5 expression specifically from forebrain glutamatergic neurons leads to impairment in hippocampal neuronal maturation, synaptic function, and impairment in hippocampal-dependent learning and memory (Tang et al., 2017). Despite this well-documented evidence, knowledge of the molecular substrates underlying CDKL5-related alterations in synaptic plasticity is still limited.
The perirhinal cortex (PRC) is located at the boundary between the medial temporal lobe and the ventral visual pathway. It has several interconnections with a wide range of cortical and subcortical structures and is involved in various cognitive processes. In particular, it plays an essential role in visual recognition memory, that is critical to the ability to record events and to guide prospective behavior (Kealy and Commins, 2011; Suzuki and Naya, 2014; Brown and Banks, 2015). A previous study has shown that male Cdkl5 KO mice tested in a sociability protocol are more interested than littermate controls in a novel object (Wang et al., 2012). This might suggest that visual recognition memory is not affected by CDKL5 mutation, but a novel object recognition (NOR) test is required to exclude this possibility.
Perirhinal cortex has never been investigated in Cdkl5 KO mice. Here we provide evidence for LTP impairment, associated with reduced dendritic length, dendritic branches, PSD-95-positive puncta, GluA2-AMPA receptor levels, and spine density and maturation. Data obtained in a four-object NOR indicate that also visual recognition memory is impaired. Notably, most of these alterations, including LTP and visual recognition memory impairments, were rescued by triggering the TrKB/PLCγ1 pathway using the 7,8-DHF prodrug R13.
Materials and Methods
Colony
The mice were produced by crossing Cdkl5 KO +/- females with Cdkl5 KO Y/- males (Amendola et al., 2014). Littermate controls were used for all experiments. Animals were karyotyped using PCR on genomic DNA as previously described (Amendola et al., 2014). The day of birth was designated as postnatal day (P) zero and animals with 24 h of age were considered as 1-day-old animals (P1). Mice were housed three to five per cage on a 12-h light/dark cycle in a temperature-controlled environment with food and water provided ad libitum. Experiments were performed in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) for the use of experimental animals, and were approved by the Italian Ministry of Public Health (approval n 114/2018-PR). All efforts were made to minimize animal suffering and to keep the number of animals used to a minimum.
Experimental Protocol
Experiments were carried out on a total of 85 Cdkl5 -/Y and 70 Cdkl5 +/Y mice. Treated Cdkl5 -/Y and Cdkl5 +/Y mice received a daily intraperitoneal injection (at 9–10 am) of R13 (7,8-DHF prodrug (Chen et al., 2018), 5.0 mg/kg in vehicle: PBS with 1% DMSO) or vehicle from P35 to P50. The dosage of 5.0 mg/kg R13 (7,8-DHF) was chosen on the basis of previous in vivo studies (Jang et al., 2010; Liu et al., 2010; Andero et al., 2011, 2012; Devi and Ohno, 2012), which demonstrated central TrkB activation enhanced neurogenesis and related behavioral changes in rodents treated with systemic 7,8-DHF administration. Intraperitoneal administration was chosen since prolonged oral gavage administration was stressful and harmful for Cdkl5 KO mice. At the end of the treatment, (P50) mice were sacrificed for electrophysiological or histological analyses or behavioral testing.
Electrophysiology
Preparation of horizontal brain slices (400 μm-thick, including the PRC, the entorhinal cortex and the hippocampus), electrophysiological recording of evoked field excitatory postsynaptic potentials (fEPSP), measurements of fEPSP amplitude and calculation of paired-pulse ratio (PPR) were performed as previously reported (Ziakopoulos et al., 1999; Aicardi et al., 2004; Roncacé et al., 2017) and are described in detail in the online Supplementary Data. Theta burst stimulation (TBS; four trains every 15 s, each train comprising 10 bursts of 5 pulses at 100 Hz, inter-burst interval 150 ms) (Ziakopoulos et al., 1999) was used to induce LTP. Synaptic plasticity was further investigated by delivering four consecutive TBS stimulations at 15-min intervals (Weng et al., 2011).
Immunohistochemistry
Some animals were deeply anesthetized and transcardially perfused with ice cold phosphate-buffered saline (PBS, 100 mM, pH 7.4), followed by a 4% solution of paraformaldehyde in PBS. The right hemisphere was cut with a freezing microtome into 30 μm-thick coronal sections.
Synaptic Terminals
One out of four sections (3–6 per mouse) of the PRC was used for immunohistochemistry. Free-floating sections were stained overnight at 4°C with the primary antibodies post-synaptic density protein 95 (1:1000, anti-PSD-95 rabbit polyclonal Ab, Abcam, Cambridge, United Kingdom) or glutamate vesicular transporter 1 (1:500, anti-VGlut1 rabbit polyclonal Ab, Thermo Scientific), and then stained in fluorescent secondary antibody (Cy3-conjugated anti-rabbit secondary antibody 1:200; Jackson ImmunoResearch Laboratories Inc., West Grove, PA, United States) for 2 h at room temperature. For quantification of synaptic puncta, images immunoprocessed for PSD-95 and VGlut1 were acquired with a Leica TCS SL confocal microscope. In each section four images from the PRC were captured and the density of individual puncta exhibiting VGlut1 or PSD-95 immunoreactivity was evaluated as previously described (Guidi et al., 2013).
P-PLCγ1 Intensity
Horizontal brain slices were cut from both hemispheres, and LTP was induced in the PRC by TBS as described above and in online Supplementary Data (Electrophysiology). Ten minutes after TBS, the slices were fixed in a 4% solution of paraformaldehyde in PBS, then cryoprotected in 15–20% sucrose. Brain slices were cut using a freezing microtome into 30 μm-thick transversal sections. Free-floating sections were stained overnight at 4°C with the primary antibodies P-PLCγ1 (1:500, anti-P-PLCγ (Y783) rabbit polyclonal Ab, Cell Signaling Technology, Danvers, MA, United States), and then stained in fluorescent secondary antibody (Cy3-conjugated anti-rabbit secondary antibody 1:200; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, United States) for 2 h at room temperature. Nuclei were counterstained with Hoechst-33342 (Sigma-Aldrich) and fluorescence images were acquired at the same intensity. To assess P-PLCγ1 cytoplasmic intensity a cytoplasmic area was traced and the Cy3-staining corresponding to the P-PLCγ1 signal was quantified by determining the mean intensity of positive (bright) pixels inside this area. The intensity of the cytoplasmic signal was normalized to the background outside the same cell by calculating the ratio between the intensity of cytoplasmic versus extra cellular signal.
Golgi Staining
Golgi staining, measurement of the dendritic tree, and dendritic spine analysis and calculation were performed as previously reported (Guidi et al., 2013; Risher et al., 2014), and are described in detail in the online Supplementary Data.
Western Blotting
In order to obtain samples of the PRC in isolation, it was micro-dissected from slices (400 μm-thick) taken at the same levels as those used for electrophysiological recording. In homogenates of the PRC of P50 mice, total proteins were obtained as previously described (Trazzi et al., 2011) and the antibodies used are listed in Supplementary Table. For TrkB, PLCγ1, and Erk phosphorylation levels, membranes were probed with the antibody for the phosphorylated form of the analyzed protein, stripped with the RestoreTM Stripping Buffer (Thermo Fisher Scientific) following manufacturer’s instructions, and then re-probed with the antibody for the un-phosphorylated form of the same protein. Densitometric analysis of digitized images was carried out with ChemiDoc XRS Imaging Systems and Image LabTM Software (Bio-Rad Laboratories, Hercules, CA, United States).
Behavioral Testing
The animal behavioral test was performed by operators blind to genotype and treatment. Mice were allowed to habituate to the testing room for at least 1 h before the test, and testing was always performed at the same time of day. The test was performed in an open field-arena (50 × 50 cm) and the behavior of the mice was monitored using a video camera placed above the center of the arena. The experiments were scored using EthoVision XT ver. 14 software (Noldus, Netherlands). Test chambers were cleaned with 70% ethanol between test subjects.
Pretraining Habituation
The animals were habituated in the open field arena without stimuli for 2 days before the commencement of the behavioral testing. Each animal was placed in the center of the arena and allowed to freely explore the open field for 20 min.
Novel Object Preference Task
The procedure involved a familiarization phase, followed by a preference test phase (Supplementary Figure 1A). In the familiarization phase (10 min duration), each animal was placed in the same arena (of the pre-training habituation), in which four copies of the same object (a plastic tube, too heavy for the animal to displace; objects 1–4) were located near the four corners of the arena (15 cm from each adjacent wall). After 1 h delay, during which one of the four objects (object 1) was replaced by a novel object (a wooden cube; objects 2–4 remained in the same positions), the animal was returned to the arena for the preference test phase (10 min duration; Supplementary Figure 1A).
Behavioral Measures
Exploration behavior was defined as the animal directing its nose toward the object at a distance ≤ 2 cm or touching it with the nose, while turning around or sitting on the object was not considered as an exploration. Discrimination among the objects was calculated using the Exploratory Preference Index, i.e., the percentage of time spent exploring any of the four objects over the total time spent exploring the four objects (Wang et al., 2007). Therefore, a preference index of above 25% indicates the preference for an object. Data obtained from the 10 min of the test period are presented.
Statistical Analysis
Results are presented as the mean ± standard error of the mean (SE). Statistical testing was performed using the two-tailed Student’s t-test or two-way ANOVA with genotype (Cdkl5 -/Y, Cdkl5 +/Y) and treatment (R13, vehicle) as factors, followed by Fisher’s LSD or the Tukey post hoc test. For categorical data, that is, percentages of spines, we used a chi-squared test. A probability level of p < 0.05 was considered to be statistically significant.
Results
LTP Is Impaired in the Perirhinal Cortex of Cdkl5 KO Mice
To evaluate possible differences in PRC functional connectivity between Cdkl5 -/Y and wild-type (+/Y) mice, we compared fEPSPs evoked in layers II–III of the PRC by stimuli applied in the same layers (Figure 1A). Stimulus-response curves (input-output relationships) obtained from Cdkl5 -/Y slices were not significantly different from those of wild-type mice over a wide range of stimulus intensities (Figure 1B). Thus, the basic properties of synaptic function in response to single stimuli appear to be unaltered by Cdkl5 deletion. This conclusion is strengthened by the observation that the slices from Cdkl5 -/Y and wild-type (+/Y) mice exhibited a similar relationship between the magnitude of the afferent volley (“non-synaptic” component of the response) and the magnitude of the synaptic response (Figure 1C). Also the responses to paired stimuli obtained in slices from Cdkl5 -/Y mice were not significantly different from those of wild-type mice (Figure 1D), suggesting that Cdkl5 deletion does not affect short-term synaptic plasticity.
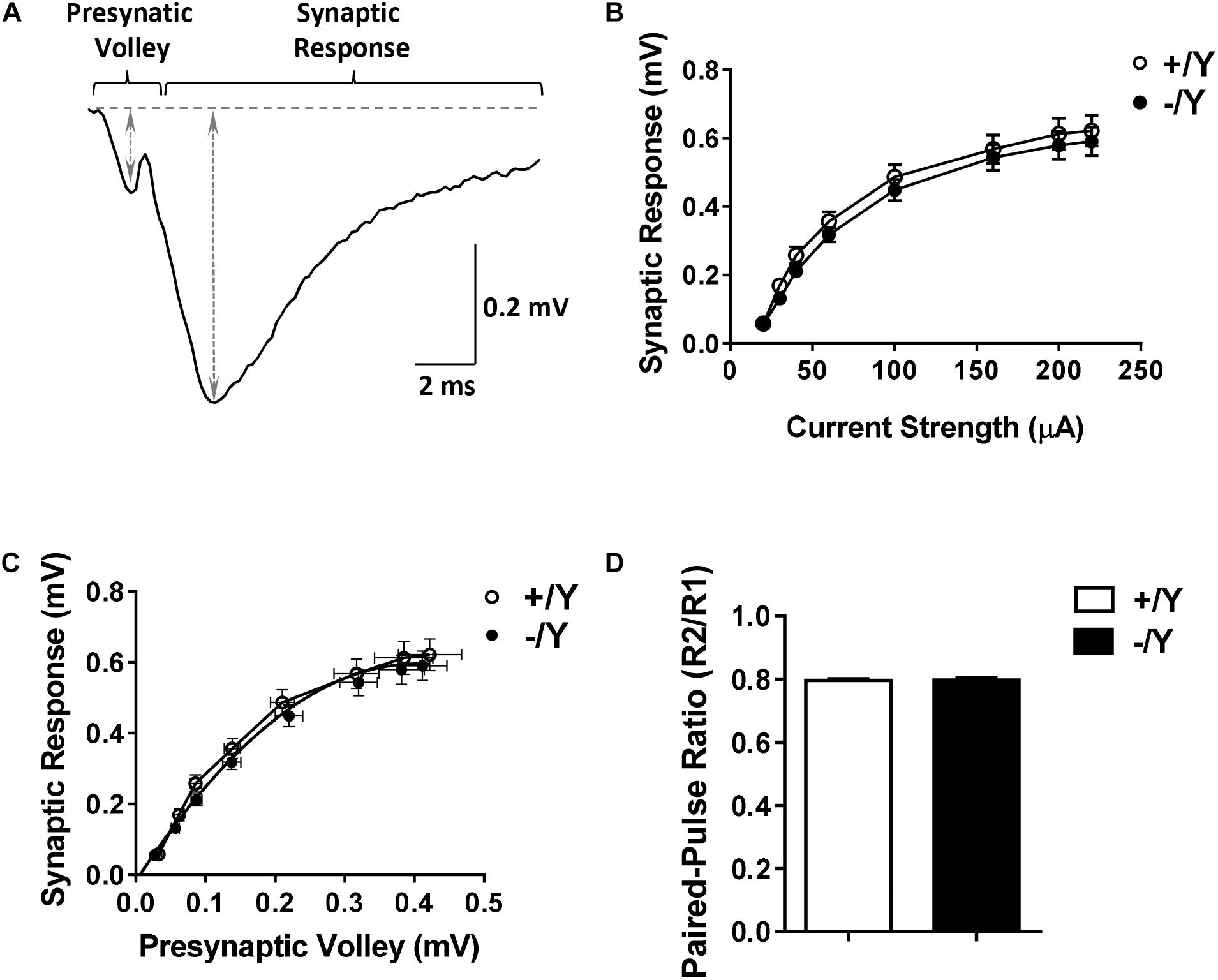
Figure 1. Input-output relations and responses to paired-pulse stimulation in the perirhinal cortex (PRC) of Cdkl5 +/Y and Cdkl5 -/Y mice. (A) Example of the response evoked in layers II/III of the PRC by stimulation of layers II/III. The double-headed arrows indicate how the magnitude of the presynaptic volley and synaptic response were measured. (B) Magnitude of the synaptic response as a function of the stimulus strength in P50 Cdkl5 +/Y (n = 16; 10 animals) and Cdkl5 -/Y (n = 21; 10 animals) mice. (C) Magnitude of the synaptic response as a function of the magnitude of the presynaptic volley. (D) Ratio of the synaptic responses (R2/R1) evoked by a pair of stimuli with an interpulse interval of 200 ms in P50 Cdkl5 +/Y (n = 11; 9 animals) and Cdkl5 -/Y (n = 9; 9 animals) mice. Values represent mean ± SE.
To evaluate the possibility that Cdkl5 deletion may affect long-term synaptic plasticity, we induced LTP in layers II–III of the PRC using TBS, i.e., short trains of 100 Hz pulses delivered at theta (5 Hz) frequency. We found that the magnitude of TBS-induced LTP was significantly smaller in slices from Cdkl5 -/Y mice (Figures 2A,B). Figure 2B shows that 55–60 min after TBS the LTP magnitude was 165 ± 3.1% in slices from wild-type (+/Y) mice, and only 139.0 ± 1.6% in slices from Cdkl5 -/Y mice. Further investigation of synaptic plasticity was conducted by delivering four consecutive TBS stimulations at 15-min intervals, in order to reveal possible saturation effects. Figures 2C,D show that the four TBS stimulations induced a significantly smaller progressive increase of LTP magnitude in slices from Cdkl5 -/Y mice compared to slices from wild-type (+/Y) mice, revealing a saturation effect after the second stimulation (+/Y: 151.3 ± 2.1%, 180.8 ± 2.5%, 203.7 ± 3.3% and 221.4 ± 4.2%; -/Y: 131.7 ± 1.2%, 150.7 ± 1.8%, 153.6 ± 2.3% and 161.5 ± 3.3% of baseline response; Figure 2D).
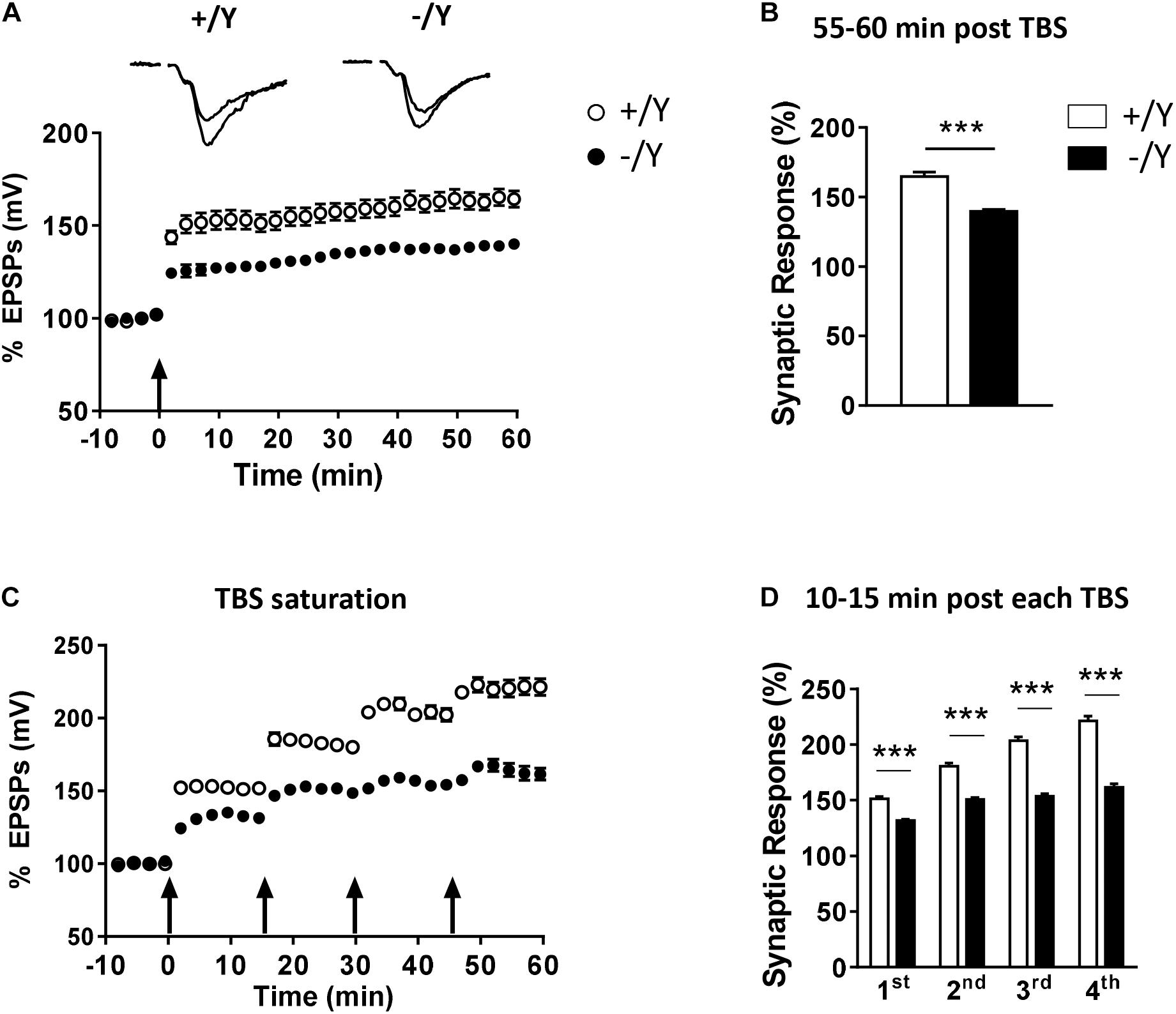
Figure 2. LTP in the perirhinal cortex of Cdkl5 +/Y and Cdkl5 -/Y mice. (A) Amplitude of the synaptic responses evoked before and after theta burst stimulation (TBS; four trains every 15 s, each train comprising 10 bursts of 5 pulses at 100 Hz, inter-burst interval 150 ms). Here and in the following panels (B–D), the amplitude of the responses is expressed as a percentage of the average amplitude of responses recorded 10 min before LTP induction. Here, and in panel C, the arrows indicate the time of delivery of the TBS. The traces at the top are examples of responses recorded before, and 55–60 min after, LTP induction. (B) The histograms indicate the averaged amplitude of the responses recorded 55–60 min after TBS. Same data as in (A). Recordings were carried out in slices from P50 Cdkl5 +/Y (n = 9; 5 animals) and Cdkl5 -/Y (n = 11; 9 animals) mice. (C) Amplitude of the synaptic response evoked before and after four consecutive TBS stimulations delivered at 15-min intervals. (D) The histograms indicate the averaged amplitude of the responses recorded 10–15 min after each TBS. Same data as in (C). Recordings were carried out in slices from P50 Cdkl5 +/Y (n = 7; 7 animals) and Cdkl5 -/Y (n = 7; 7 animals) mice. ∗∗∗p < 0.001 (Student’s two-tailed t-test).
TrkB/PLCγ1 Signaling Is Impaired in the Perirhinal Cortex of Cdkl5 KO Mice
A previous study in the PRC has shown that TBS elicits a large increase in brain-derived neurotrophic factor (BDNF) secretion, which is necessary for LTP induction (Aicardi et al., 2004). As shown in Figure 3A, BDNF acts on TrkB receptors leading to dimerization and autophosphorylation of tyrosine residues at position Tyr515 and Tyr816. Phosphorylation and recruitment of adaptors at position 515 activate Erk and Akt pathways, which promote survival and growth of neurons and other cells through Ras or GRB-associated binder G1 (GAB1). Phosphorylation at position 816 recruits and activates phospholipase Cγ1 (PLCγ1), which results in the generation of inositol-1,4,5-triphosphate and diacylglycerol, leading to synaptic plasticity and dendritic maturation (Minichiello et al., 2002; Gartner et al., 2006; Minichiello, 2009).
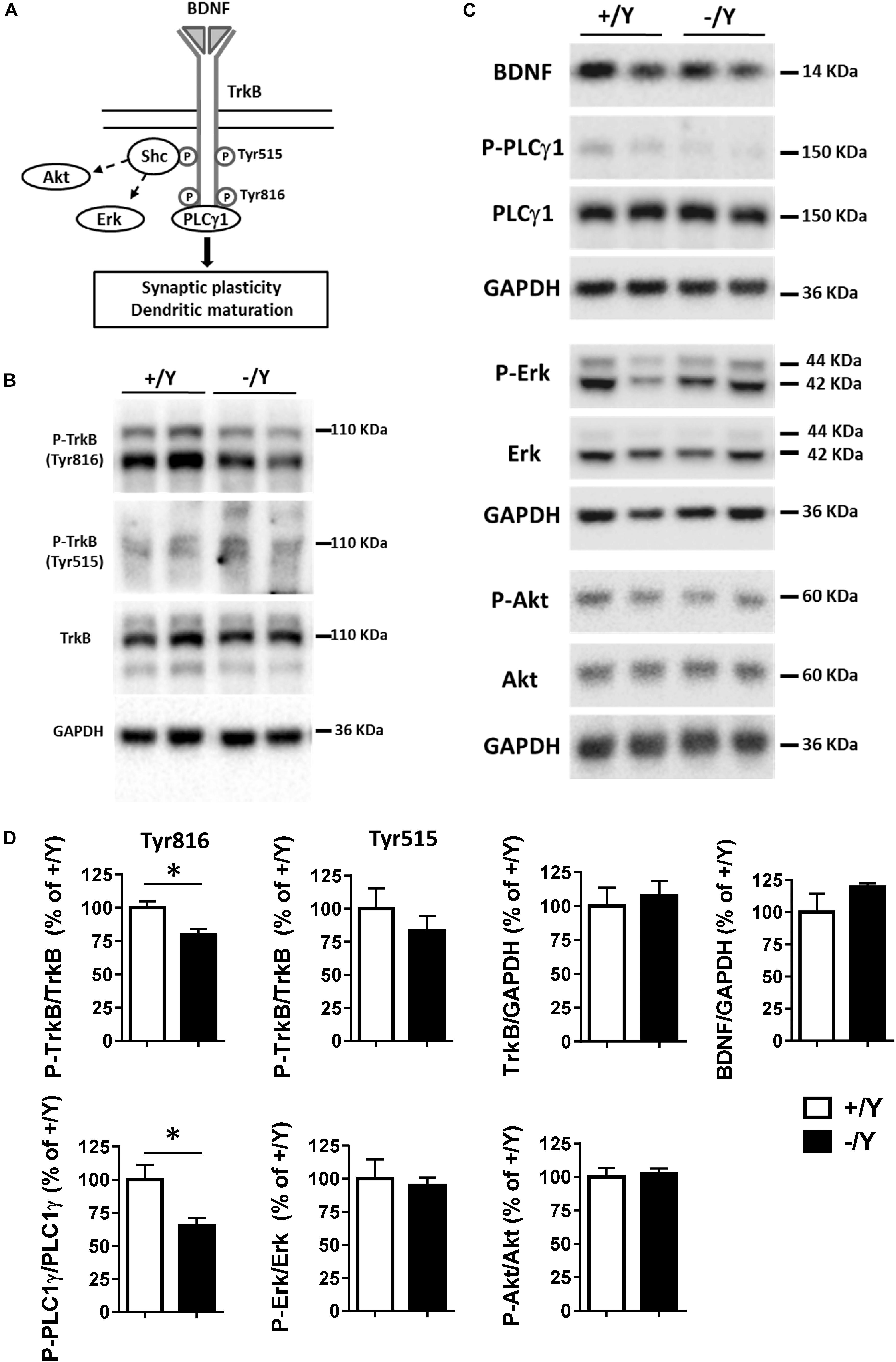
Figure 3. TrkB signaling pathways in the perirhinal cortex of Cdkl5 +/Y and Cdkl5 -/Y mice. (A) Diagram of brain-derived neurotrophic factor (BDNF) and TrkB signaling pathways. BDNF binds to the extra cellular domain of TrkB forming homodimers to activate downstream intracellular signaling cascades, including Shc/Erk, Shc/Akt, and phospholipase C (PLC) γ1 pathways. (B) Western blots examples of P-TrkB (Tyr816 and Tyr515) and TrkB in PRC homogenates of P50 Cdkl5 +/Y and Cdkl5 -/Y mice. (C) Western blot examples of BDNF, P-PLCγ1 (Tyr783), PLCγ1, P-Erk, Erk, P-Akt (Ser437), Akt and GAPDH levels in PRC homogenates of P50 Cdkl5 +/Y and Cdkl5 -/Y mice. (D) The histograms show western blot analysis of samples as in B,C. Phosphoprotein levels were normalized to corresponding total protein levels, and total protein levels were normalized to GAPDH (Cdkl5 +/Y n = 5; Cdkl5 +/Y n = 5). Data are expressed as % of those obtained in Cdkl5 +/Y mice. Values represent mean ± SE. ∗p < 0.05 (Student’s two-tailed t-test).
To investigate the molecular mechanisms underlying TBS-induced LTP impairment, we first analyzed the levels of TrkB phosphorylation using Western blot in tissue homogenates isolated from the PRC. Cdkl5 -/Y mice showed no differences in the P-TrkB(Tyr515) or total TrkB protein levels (Figures 3B,D). On the contrary, we found a significantly lower level of P-TrkB(Tyr816) protein in Cdkl5 -/Y mice compared to wild-type (+/Y) mice (Figures 3B,D). No significant differences in BDNF protein level were observed (Figures 3C,D), suggesting a specific effect of Cdkl5 deletion on the autophosphorylation process at position Tyr816 of TrkB, rather than on BDNF availability. We then examined the main downstream effectors of the TrkB pathway. Predictably, we found a significantly lower PLCγ1 phosphorylation in the PRC of Cdkl5 KO mice in comparison with wild-type mice, whereas no significant differences in Erk and Akt phosphorylation were observed (Figures 3C,D).
Impaired PLCγ1 Phosphorylation and LTP in the Perirhinal Cortex of Cdkl5 KO Mice Are Rescued by Treatment With the TrkB Agonist R13
In order to investigate whether TrkB/PLCγ1 signaling alteration underlies LTP impairment in Cdkl5 KO mice, we quantified P-PLCγ1 cellular intensity in PRC slices from Cdkl5 KO and wild-type mice before and after TBS. Figures 4A–C show that 10 min after TBS, PLCγ1 phosphorylation was increased in PRC slices from both Cdkl5 and wild-type mice. Interestingly, PLCγ1 phosphorylation after TBS was still significantly lower in Cdkl5 KO slices than in wild-type slices.
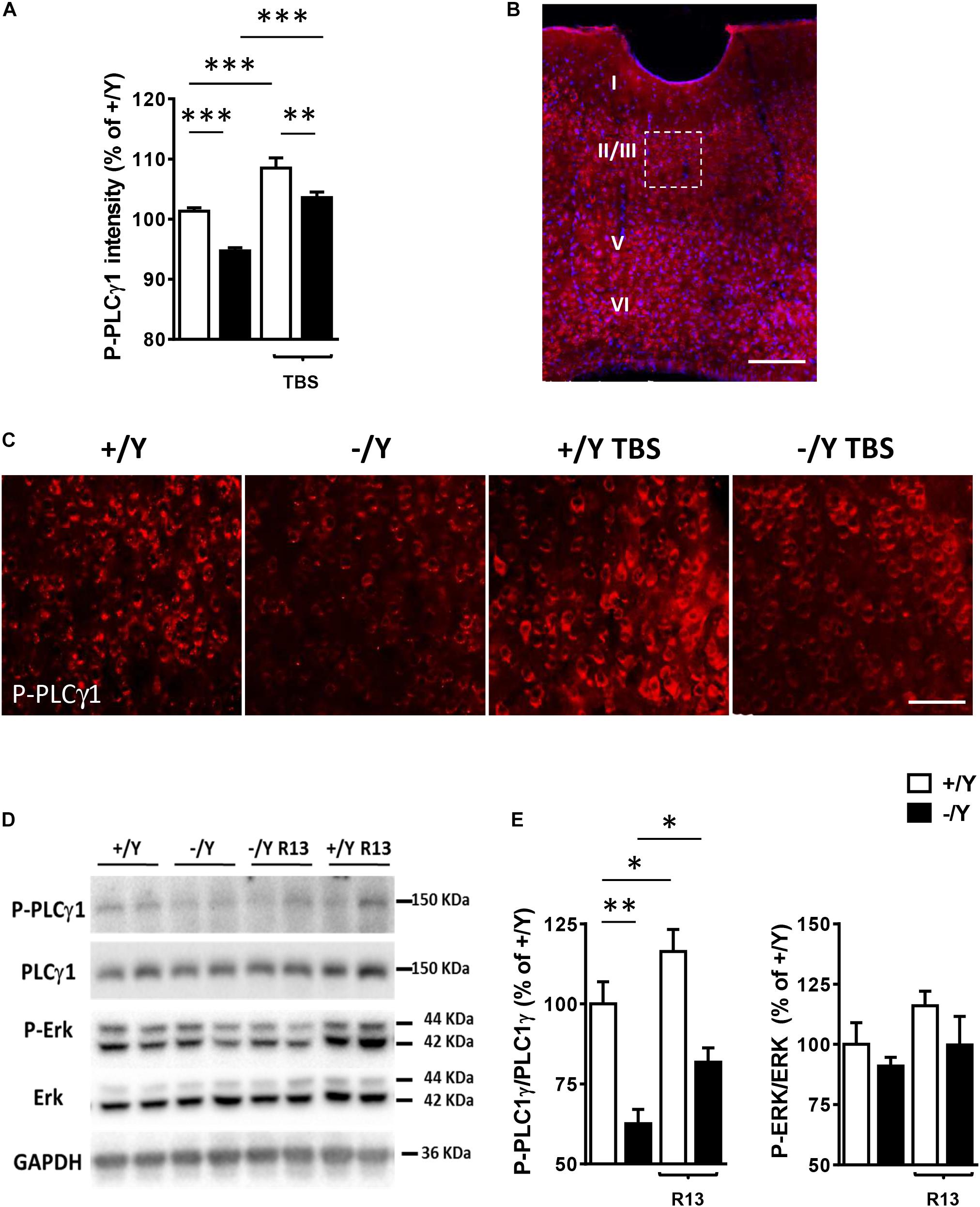
Figure 4. Effect of TBS or treatment with R13 on TrkB/ PLCγ1 signaling in the perirhinal cortex of Cdkl5 -/Y mice. (A) Quantification of P-PLCγ1 signal intensity in PRC slices before and after theta burst stimulation (TBS; four trains every 15 s, each train comprising 10 bursts of 5 pulses at 100 Hz, inter-burst interval 150 ms) of Cdkl5 +/Y (n = 4) and Cdkl5 -/Y (n = 4) mice. (B) A representative image of PRC processed for fluorescent P-PLCγ1 immunostaining (red) of a wild-type (+/Y) mouse. Nuclei are stained with Hoechst (blue). The dotted box indicates the region shown at a higher magnification in (C). Scale bar = 50 μm. Roman numerals indicate PRC cytoarchitectonic layers. (C) Representative images of layer II-III PRC neurons of Cdkl5 +/Y and Cdkl5 -/Y PRC slices as in (A). (D,E) Quantification of PLCγ1 and Erk phosphorylation levels before and after treatment with R13. Cdkl5 -/Y and Cdkl5 +/Y mice were treated for 15 days (5 mg/Kg IP) from postnatal day 35 (P35) to 50 (P50), the day of sacrifice. Western blot examples (D) and analyses (E) of P-PLCγ1 (Tyr783), PLCγ1, P-Erk and Erk levels in PRC homogenates of vehicle-treated Cdkl5 +/Y (n = 5) and Cdkl5 -/Y (n = 5) mice, and R13-treated Cdkl5 +/Y (n = 6) and Cdkl5 -/Y (n = 6) mice. Values represent mean ± SE. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 (Fisher’s LSD test after two-way ANOVA).
In order to investigate the effect of chronic activation of TrkB/PLCγ1 signaling in Cdkl5 KO mice, we treated Cdkl5 -/Y mice for 15 days with R13, a prodrug of 7,8-dihydroxyflavone (7,8-DHF) (Chen et al., 2018). 7,8-DHF is a potent molecular TrkB agonist that specifically binds to the TrkB receptor extra cellular domain. A recent study in striatal neurons has shown that it acts through selective phosphorylation of the 816 residue of TrkB, leading to activation of the PLCγ1 pathway (Garcia-Diaz Barriga et al., 2017). Accordingly, we found that R13 treatment increased PLCγ1 phosphorylation levels but not P-Erk levels in both Cdkl5 and wild-type mice (Figures 4D,E), suggesting a selective action of 7,8-DHF on the TrkB(Y816)-PLCγ1 pathway also in the PRC.
Interestingly, R13 treatment restored LTP in Cdkl5 -/Y mice (Figures 5A–D), suggesting a critical role of TrkB/PLCγ1 signaling in the defective synaptic plasticity. R13 treatment affected neither LTP in Cdkl5 +/Y mice (Figures 5A–D) nor basal synaptic transmission (input-output responses; responses to paired stimuli) in Cdkl5 -/Y and Cdkl5 +/Y mice (not shown). Vehicle treatment did not affect any functional parameter considered in Cdkl5 -/Y and Cdkl5 +/Y mice (Figures 5A–D).
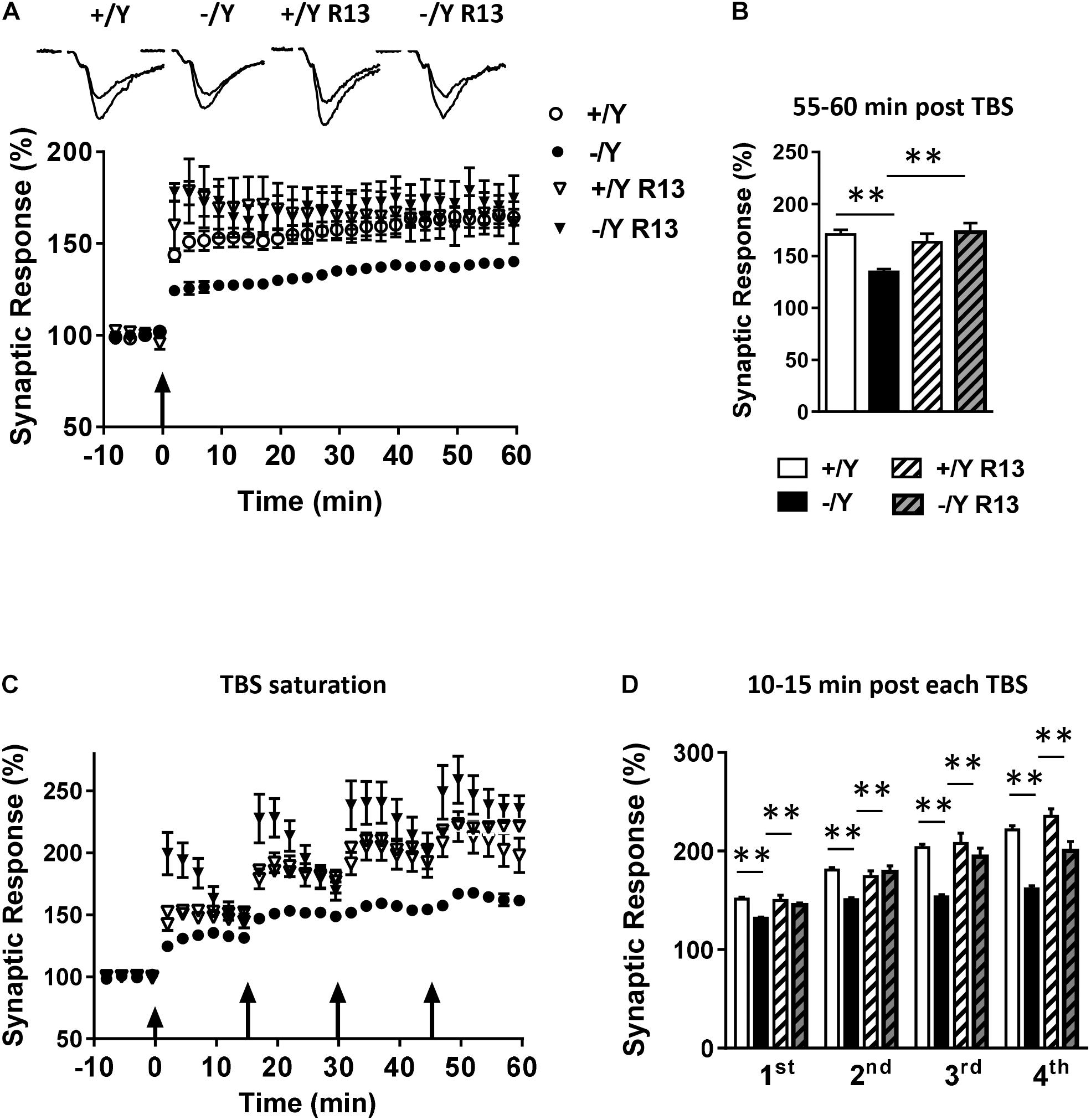
Figure 5. Effect of treatment with R13 on LTP in the perirhinal cortex of Cdkl5 -/Y mice. (A) Amplitude of the synaptic responses evoked before and after TBS. Here, and in the following panels (B–D), the amplitude of the responses is expressed as a percentage of the average amplitude of responses recorded 10 min before TBS. The traces at the top are examples of responses recorded before, and 55–60 min after, TBS. Here, and in panel C, the arrows indicate the time of TBS delivery. Recordings were carried out in slices from P50 vehicle-treated Cdkl5 +/Y (n = 13; 7 animals) and Cdkl5 -/Y (n = 13; 6 animals), and R13-treated Cdkl5 +/Y (n = 6; 4 animals) and Cdkl5 -/Y (n = 7; 7 animals) mice. (B) The histograms indicate the averaged amplitude of the responses recorded 55–60 min after TBS. Same data as in (A). (C) Amplitude of the synaptic responses evoked before and after four consecutive TBS stimulations delivered at 15-min intervals. Recordings were carried out in slices from P50 vehicle-treated Cdkl5 +/Y (n = 6; 6 animals) and Cdkl5 -/Y (n = 7; 7 animals), and R13-treated Cdkl5 +/Y (n = 7; 4 animals) and Cdkl5 -/Y (n = 6; 6 animals) mice. (D) The histograms indicate the averaged amplitude of the responses recorded 10–15 min after each TBS. Same data as in (C). Values represent mean ± SE. ∗p < 0.05; ∗∗p < 0.01 (Tukey test after two-way ANOVA).
Alteration of Dendritic Pattern in the Perirhinal Cortex of Cdkl5 KO Mice Is Rescued by Treatment With the TrkB Agonist R13
BDNF/TrkB signaling participates in the regulation of dendritic differentiation, and in the formation and maturation of dendritic spines during postnatal development (Chapleau et al., 2009). Abnormalities in dendritic and synaptic differentiation are thought to underlie altered synaptic function and network connectivity, thus contributing to the impaired neuronal function. Previous studies in Cdkl5 -/Y mice have shown a reduction in dendritic pattern in granule cells and CA1 pyramidal neurons of the hippocampal region (Fuchs et al., 2015; Trazzi et al., 2016).
In order to establish whether these alterations also occur in PRC neurons, we examined apical and basal dendritic branches of layer II-III PRC neurons in Golgi-stained brain sections (Figure 6A). We found that both basal and apical dendrites were shorter (Figure 6B), and there was a reduced number of branches (Figure 6C) in PRC neurons of Cdkl5 -/Y compared to Cdkl5 +/Y mice. Interestingly, R13 treatment restored dendritic length and number of branches in Cdkl5 -/Y mice, but had no effect in control mice (+/Y) (Figures 6B,C). Figure 6D shows that the difference in number of branches was significant in the fourth and fifth order in apical dendrites, and in the second, third, and fourth order in basal dendrites; moreover, it shows a lack of branches of the seventh order in basal dendrites. All these defects were corrected by R13 treatment. These results indicate that activation of the TrkB/PLCγ1 pathway restores the dendritic pattern of the PRC neurons in Cdkl5 -/Y mice. Vehicle treatment did not affect any morphological parameter considered in Cdkl5 -/Y and Cdkl5 +/Y mice (Figures 6B–D).
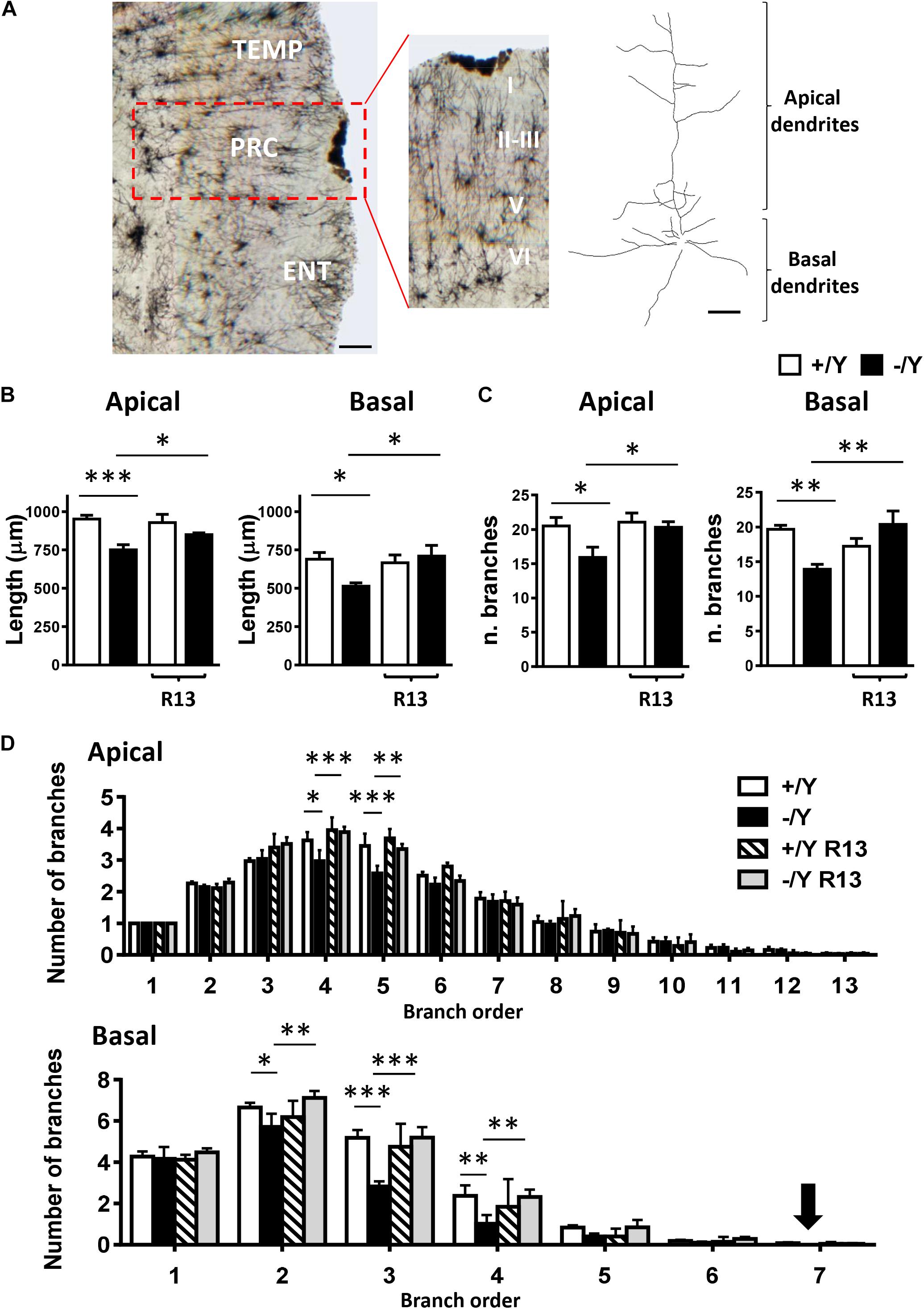
Figure 6. Effect of treatment with R13 on dendritic morphology in the perirhinal cortex of Cdkl5 -/Y mice. (A) Example of Golgi-stained section at the level of the PRC of a P50 Cdkl5 -/Y mouse (panel on the left; scale bar = 100 μm). The central panel shows a higher magnification of the area enclosed in the red square. Roman numerals indicate PRC cytoarchitectonic layers. The panel on the right shows an example of the apical and basal dendritic tree of a Golgi-stained PRC neuron from layers II-III (scale bar = 30 μm). (B) Apical and basal mean total dendritic length of layer II-III PRC neurons of vehicle-treated P50 Cdkl5 +/Y (n = 4) and Cdkl5 -/Y (n = 4) mice, and R13-treated P50 Cdkl5 +/Y (n = 4) and Cdkl5 -/Y (n = 5) mice. (C) Mean number of dendritic segments of the apical and basal dendrites of layer II-III PRC neurons in the same mice as in (B). (D) Mean number of apical and basal branches of the different orders of vehicle-treated Cdkl5 +/Y (n = 4) and Cdkl5 -/Y (n = 4) mice, and R13-treated Cdkl5 -/Y (n = 5) mice. The numbers on the X axis indicate the branch order. The arrow indicates the absence of branches in Cdkl5 -/Y mice. Values in (B–D) represent mean ± SE. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 (Fisher’s LSD test after two-way ANOVA).
Alteration of Dendritic Spine Density in the Perirhinal Cortex of Cdkl5 KO Mice Is Rescued by Treatment With the TrkB Agonist R13
In Golgi-stained brain sections we examined spine density in the apical and basal dendritic branches of layer II-III neurons of the PRC. PRC neurons had a reduced spine density in both basal and apical dendrites in untreated Cdkl5 -/Y mice in comparison with wild-type (+/Y) mice (Figures 7A,B). Cdkl5 -/Y mice treated with R13 underwent a restoration of spine density (Figures 7A,B). Dendritic spines are heterogeneous in size and shape, and can be classified as immature spines (filopodia, thin-shaped, and stubby-shaped) and mature spines (mushroom and cup shapes) (Risher et al., 2014). Separate counts of different classes of dendritic spines revealed that PRC neurons of Cdkl5 -/Y mice had a higher percentage of immature spines and a reduced percentage of mature spines compared to Cdkl5 +/Y mice (Figure 7C). Treatment with R13 improved the balance between immature and mature spines (Figure 7C), suggesting that activation of the TrkB/PLCγ1 pathway improves PRC dendritic spine maturation. Vehicle treatment did not affect spine density in Cdkl5 -/Y and Cdkl5 +/Y mice (Figures 7A,B).
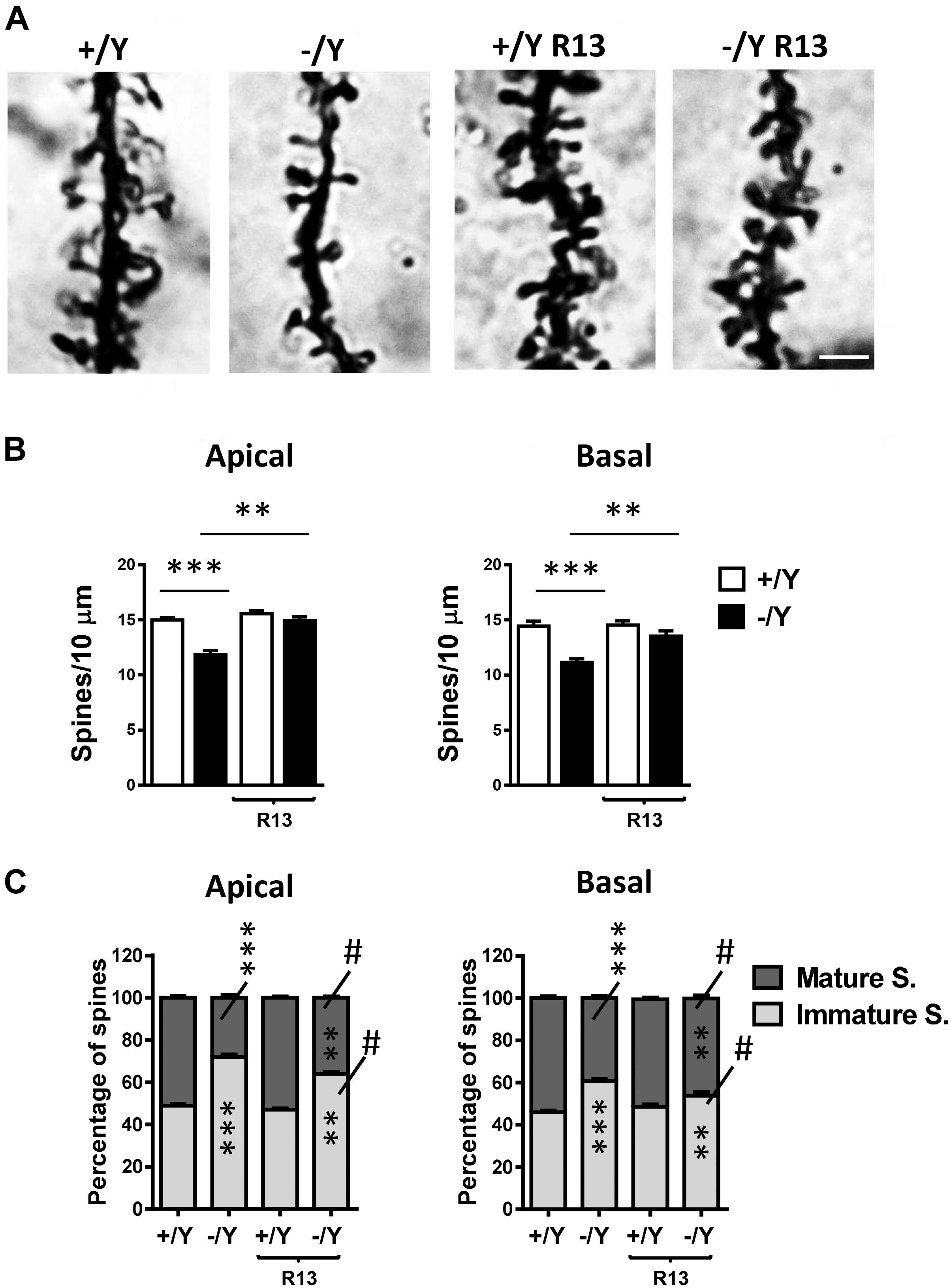
Figure 7. Effect of treatment with R13 on dendritic spines in the perirhinal cortex of Cdkl5 -/Y mice. (A) Images of Golgi-stained dendritic branches of layer II-III perirhinal neurons of a P50 Cdkl5 +/Y and a Cdkl5 -/Y mouse, and of a Cdkl5 -/Y mouse treated with R13. Scale bar = 1.5 μm. (B) Dendritic spine density (number of spines per 10 μm) in apical and basal dendrites from vehicle-treated Cdkl5 +/Y (n = 4) and Cdkl5 -/Y (n = 4) mice, and Cdkl5 +/Y (n = 4) and Cdkl5 -/Y (n = 4) mice treated with R13. (C) Percentage of immature and mature spines in relation to the total number of protrusions of layer II-III perirhinal neurons in mice as in (B). Values represent mean ± SE. ∗∗P < 0.01; ∗∗∗P < 0.001; #p < 0.01 compared to untreated Cdkl5 -/Y mice in (C) [data in (B) Fisher’s LSD test after two-way ANOVA; data in (C) Chi-squared test].
Altered Expression of PSD-95 Positive Synaptic Puncta and GluA2-AMPAR in the Perirhinal Cortex of Cdkl5 KO Mice Is Rescued by Treatment With the TrkB Agonist R13
Most excitatory synapses in the mature mammalian brain occur on spines in which postsynaptic density protein 95 (PSD-95) clusters are localized. Evaluation of PSD-95 immunoreactivity showed a strong reduction in the number of PSD-95-positive puncta in the PRC of Cdkl5 -/Y mice (Figures 8A,B), which is consistent with the reduced number of mature spines (Figure 7C) and suggests loss of excitatory synaptic contacts. To clarify this issue, we examined the immunoreactivity for the vesicular glutamate transporter-1 (VGlut1), a marker of glutamatergic (excitatory) terminals. We found no difference in the number of VGlut1-positive immunoreactive puncta between Cdkl5 -/Y and wild-type mice (Figures 8A,C), indicating a similar number of glutamatergic terminals in the PRC. Treatment with R13 restored the number of PSD-95-positive immunoreactive puncta in Cdkl5 -/Y mice, but did not affect immunoreactivity for PSD-95 in Cdkl5 +/Y mice (Figures 8A,B) or for VGlut1 in Cdkl5 +/Y and Cdkl5 -/Y mice (Figures 8A,C). Vehicle treatment did not affect immunoreactivity for PSD-95 or VGlut1 in Cdkl5 +/Y and Cdkl5 -/Y mice (Figures 8A,C).
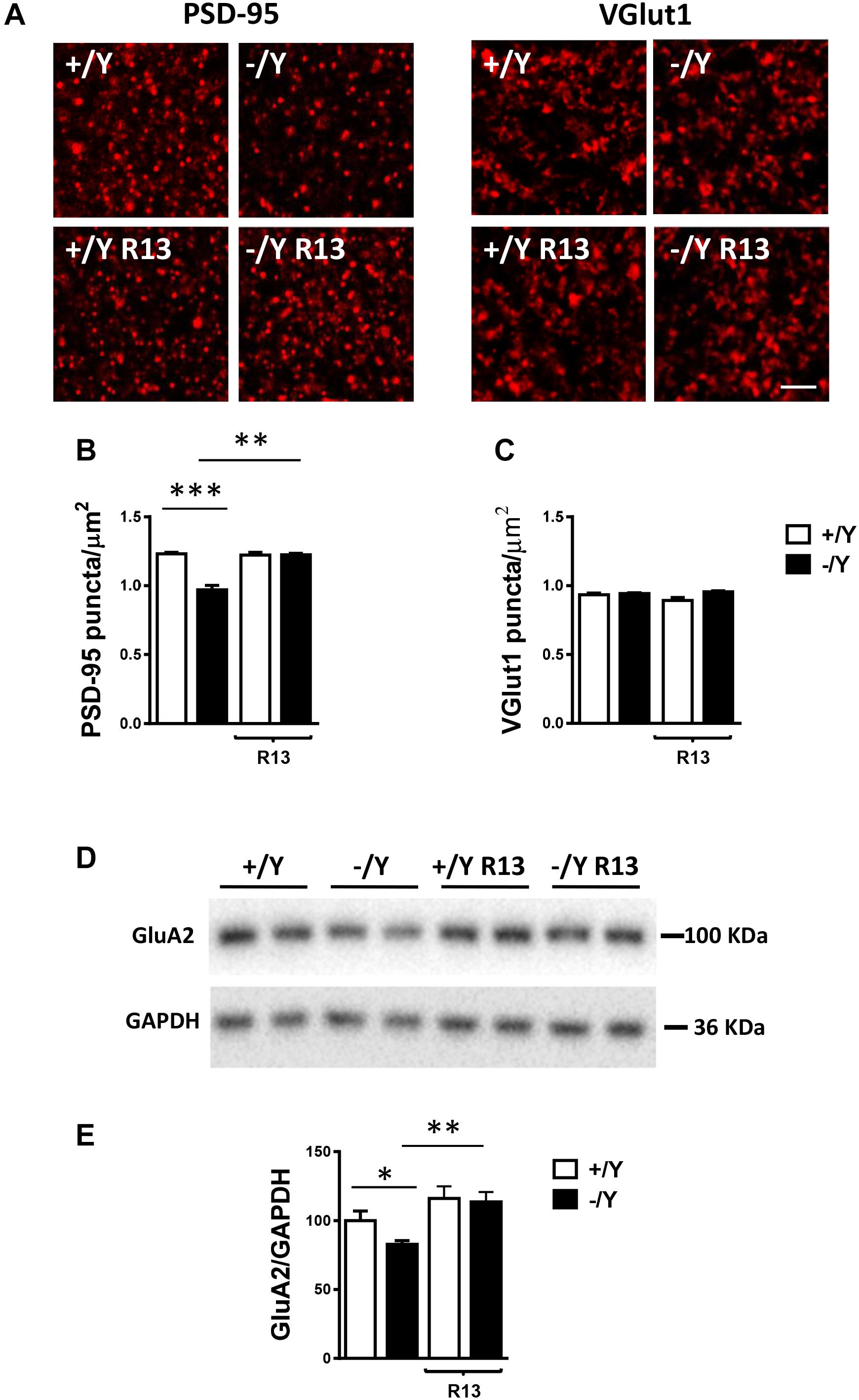
Figure 8. Effect of treatment with R13 on neuronal connectivity in the perirhinal cortex of Cdkl5 -/Y mice. (A) Representative fluorescence image of a PRC section processed for PSD-95 or VGlut1 immunoreactivity, showing examples of PSD-95 and VGlut1 immunoreactive puncta. Scale bar: 3 μm. (B,C) Number of fluorescent puncta per μm2 exhibiting PSD-95 (B) or VGlut1 (C) immunoreactivity in layers II-III of the PRC of vehicle-treated P50 Cdkl5 +/Y (n = 4) and Cdkl5 -/Y (n = 4) mice, and P50 Cdkl5 +/Y (n = 4) and Cdkl5 -/Y (n = 4) mice treated with R13. (D,E) Western blot analysis of GluA2-AMPA levels in PRC homogenates of vehicle-treated P50 Cdkl5 +/Y (n = 6) and Cdkl5 -/Y (n = 6) mice and R13-treated P50 Cdkl5 -/Y (n = 5) mice. Values represent mean ± SE. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 (data in (B,C) Fisher’s LSD test after two-way ANOVA; data in (E) Student’s two-tailed t-test).
The TrkB/PLCγ1 pathway is involved in diverse postsynaptic events, such as the modulation of AMPAR expression and trafficking, which in turn contributes to synaptic plasticity (Jang et al., 2013). As previously observed in hippocampal neurons (Tramarin et al., 2018), we found a lower expression of GluA2-AMPAR in the PRC of Cdkl5 -/Y mice in comparison with wild-type mice (Figures 8D,E). Notably, GluA2-AMPAR levels in Cdkl5 -/Y mice were rescued by treatment with R13 (Figures 8D,E).
Altered Visual Recognition Memory in Cdkl5 KO Mice Is Rescued by Treatment With the TrkB Agonist R13
In the rodent brain, PRC plays an essential role in visual object recognition memory, that can be evaluated using the NOR test. A 4-object NOR test was performed in an open-field arena, preceded by a 2-day habituation phase (Supplementary Figure 1A). During the familiarization phase, animals normally show equal preference for the objects that are placed in an arena, while they exhibit a higher preference for the new object during the subsequent test phase. Discrimination among the objects (Preference Index) was calculated by taking into account the percentage of time spent exploring any of the four objects over the total time spent exploring the objects. As expected, the preference index in wild-type (+/Y) mice was significantly larger for the novel object during the test phase (Figures 9A,B). However, we did not observe any increase in preference index in Cdkl5 -/Y mice (Figure 9B), indicating a deficit in remembering the identity of an object in an open field. Interestingly, treatment with R13 rescued visual recognition memory in Cdkl5 -/Y mice (Figure 9B). R13 treatment did not affect locomotor activity in Cdkl5 -/Y or Cdkl5 +/Y mice (Supplementary Figure 1B); nor was it able to further increase the preference index in Cdkl5+/Y mice (Figure 9B).
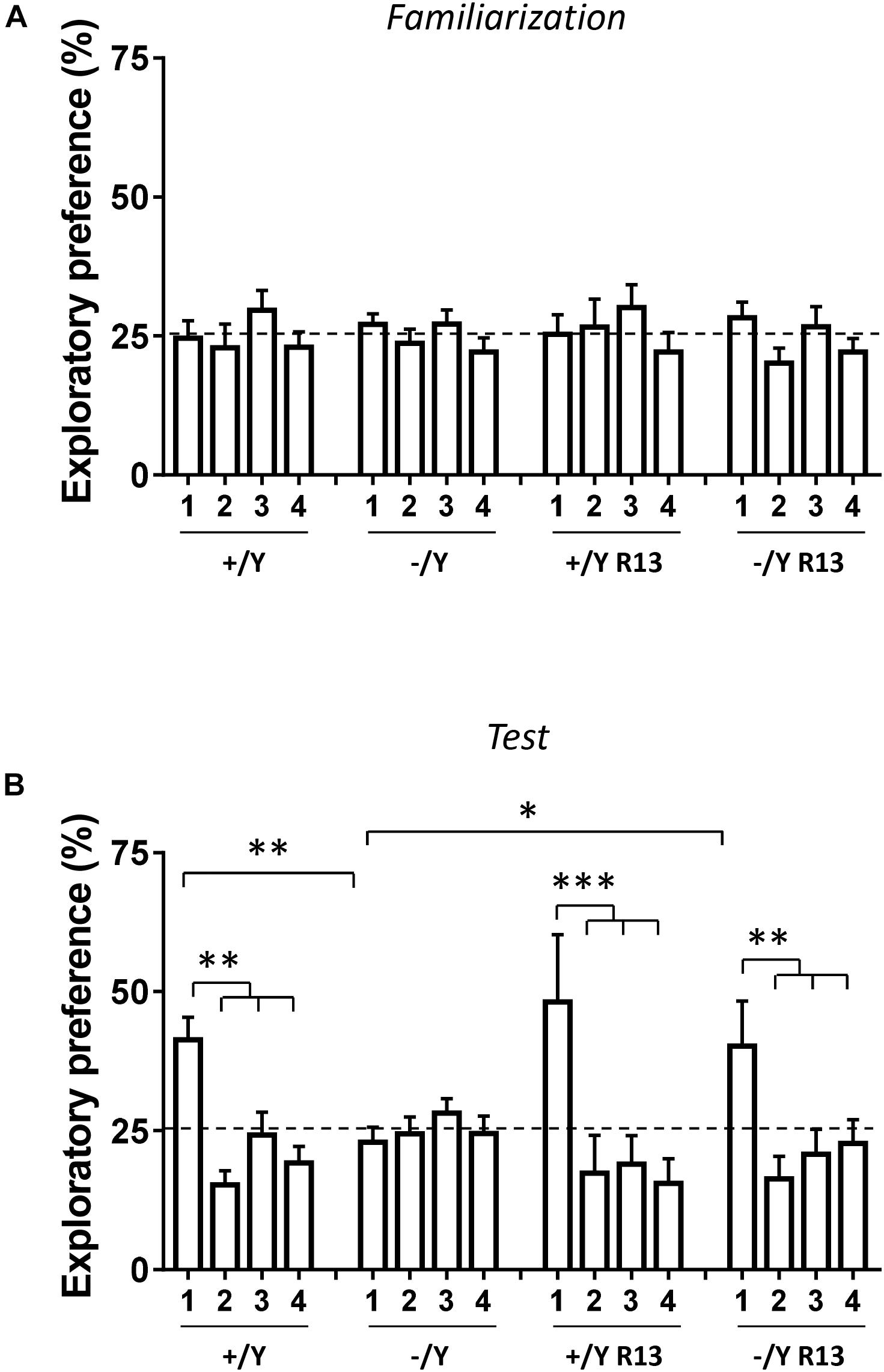
Figure 9. Effect of treatment with R13 on novel object recognition memory in Cdkl5 -/Y mice. (A) Familiarization phase. Percentage of time spent exploring the four identical objects during the familiarization phase (10 min duration) of vehicle-treated Cdkl5 -/Y (n = 13) and Cdkl5 +/Y (n = 13) mice, and R13-treated Cdkl5 -/Y (n = 9) and Cdkl5 +/Y (n = 9) mice. (B) Exploratory Preference Test phase (one hour after the familiarization phase, 10 min duration). Percentage of time spent exploring new object 1 (that replaced the previous object 1) and the previous identical objects 2–4. Same mice as in (A). Values represent mean ± SE. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 (Fisher’s LSD test after two-way ANOVA).
Discussion
Functional and Morphological Impairments in the Perirhinal Cortex of Cdkl5 KO Mice
Our results indicate that the magnitude of LTP is reduced in the PRC of Cdkl5 KO mice, consistently with the strong reduction previously observed in the somatosensory cortex (Della Sala et al., 2016). To investigate the molecular mechanisms underlying LTP reduction, we analyzed some elements of the signaling cascade activated by BDNF. Previous studies in the hippocampus have shown that the PLCγ1 pathway mediated by TrkB plays a predominant role in LTP (Minichiello et al., 2002), whereas the Erk/Akt pathway is of lesser, if of any, importance (Korte et al., 2000). Accordingly, we found a lower PLCγ1 phosphorylation in the PRC of Cdkl5 KO mice in comparison with wild-type mice, and no significant differences in Erk and Akt phosphorylation. This evidence suggests that the TrkB/PLCγ1 pathway plays a critical role in the observed alteration of LTP in the PRC.
Brain-derived neurotrophic factor levels were not reduced in Cdkl5 KO mice, suggesting a specific effect of Cdkl5 deletion on the autophosphorylation process at position Tyr816, rather than on BDNF availability.
At variance with the results obtained in the PRC (present data) and somatosensory cortex (Della Sala et al., 2016), LTP was previously found to be slightly increased in the hippocampal CA1 region of Cdkl5 KO mice (Okuda et al., 2017), suggesting that CDKL5 action might be region-specific. These conflicting results might also be explained, at least in part, by different sensitivity to LTP inducing protocols, TBS [present data, and (Della Sala et al., 2016)] versus HFS (Okuda et al., 2017).
Previous studies in Cdkl5 KO mice have shown reduced neuronal branching accompanied by reduced spine density and maturation in the hippocampus, in the visual cortex, and in the somatosensory cortex (Amendola et al., 2014; Fuchs et al., 2014; Della Sala et al., 2016; Pizzo et al., 2016; Trazzi et al., 2016). Accordingly, we found that PRC neurons of Cdkl5 KO mice are characterized by a shorter dendritic length, a reduced number of branches, a lower spine density and a higher percentage of immature spines. Moreover, we observed a reduced number of PSD-95 immunoreactive puncta in dendritic spines. Since PSD-95 localization in the spine correlates with activity-driven synapse stabilization (Ehrlich et al., 2007), the decreased number of PSD-95 puncta in Cdkl5 KO mice correlates with the observed increased number of immature spines. Evaluation of the number of presynaptic excitatory terminals (VGlut1 immunopositive puncta) showed no difference between Cdkl5 KO and wild-type mice, in accordance with the observation that basic properties of synaptic function are unaltered in the PRC (present data) and in the hippocampus (Okuda et al., 2017). As observed in hippocampal neurons (Tramarin et al., 2018), we found a lower expression of GluA2-AMPAR in the PRC of Cdkl5 -/Y mice. Most neuronal AMPARs contain this critical subunit, but in certain restricted neuronal populations and under certain physiological or pathological conditions, AMPARs that lack this subunit are expressed. This subunit determines many of the major biophysical properties of AMPARs (including Ca2+ permeability, single-channel conductance, and receptor kinetics), strongly influences receptor assembly and trafficking, and plays pivotal roles in various forms of synaptic plasticity (Isaac et al., 2007). In particular, GluA2-AMPAR is required for spine changes during synaptic plasticity (Asrar and Jia, 2013). Thus, it is reasonable to suppose that the lower expression of GluA2-AMPAR in the PRC might play a role in the impaired functional and morphological synaptic plasticity observed in the PRC of Cdkl5 -/Y mice.
Visual recognition memory is the ability to judge the prior occurrence of stimuli and is fundamental to our ability to record events and to guide prospective behavior. Studies in humans and animals indicate that PRC plays an essential role in recognition memory and familiarity discrimination for individual items (Kealy and Commins, 2011; Suzuki and Naya, 2014; Brown and Banks, 2015). Our finding that short-term object recognition memory is impaired in Cdkl5 KO mice is in line with the observed neuroanatomical defects and LTP impairment in the PRC. This behavioral evidence suggests that specific PRC defects may contribute, along with hippocampal impairments, to poor memory performance in individuals with CDD.
Treatment With the TrkB Agonist R13 Rescues Functional and Morphological Impairments in the Perirhinal Cortex of Cdkl5 KO Mice
The natural flavonoid 7,8-DHF, a potent small molecular TrkB agonist, displays beneficial effects on the brain in health and disease (Spencer, 2008; Williams and Spencer, 2012; Roncacé et al., 2017), but has only modest oral bioavailability and a moderate pharmacokinetic profile. To efficiently mimic the actions of BDNF, we used the recently synthesized 7,8-DHF prodrug R13, which is hydrolyzed into 7,8-DHF in liver microsomes and is characterized by a longer half-life and a higher plasma concentration, and higher brain exposure (Chen et al., 2018). We found that treatment with R13 activated TrkB/PLCγ1 signaling, rescued the impaired TBS-induced LTP, restored dendritic pattern as well as PSD-95 and GluA2-AMPAR levels, and improved the balance between immature and mature spines in PRC neurons of Cdkl5 KO mice. Importantly, restoration of morphological and synaptic impairments leaded to recovery of PRC-dependent visual recognition memory in Cdkl5 KO mice.
In wild type +/Y mice, treatment with R13 caused an activation of TrkB/PLCγ1 signaling in the PRC (Figure 4D), which, however, did not induce effects on PRC neuroanatomy or synaptic function and, consequently, on behavior. The finding that treatment with R13 has relatively scarce or no effects in normal animals is in line with a previous evidence (Garcia-Diaz Barriga et al., 2017) and suggests that TrkB/PLCγ1 signaling activation may help brain development under abnormal, but not normal, brain conditions.
Indeed, an increasing body of evidence suggests that PLCγ1 plays a pivotal role in the regulation of synaptic plasticity and maturation; in particular, PLCγ1 signaling is required for structural and functional changes in spine actin, PSD scaffolding, and AMPAR trafficking (Horne and Dell’Acqua, 2007). Consistent with its critical role, abnormal expression and activation of PLCγ1 has been observed in various brain disorders (Jang et al., 2013).
Conclusion
In conclusion, present results provide the first evidence for morphological and functional impairments in the PRC of Cdkl5 KO mice, associated with a deficit in visual recognition memory. The TrkB agonist R13 rescued most of these alterations, including LTP and visual recognition memory impairments, by triggering the TrkB/PLCγ1 pathway. If R13 induced a widespread positive effect in other brain areas, it might represent a promising candidate for a targeted therapeutic strategy aimed at restoring synaptic development and plasticity in CDD patients.
Ethics Statement
Experiments were performed in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) for the use of experimental animals, and were approved by the Italian Ministry of Public Health (approval in 114/2018-PR). All efforts were made to minimize animal suffering and to keep the number of animals used to a minimum.
Author Contributions
EC, GA, ST, and VR designed the study. ER, VR, ST, CF, GM, LG, ML, and GG performed the experiments. ER, VR, ST, and RR analyzed the data. KY executed the R13 synthesis. EC and GA wrote the manuscript.
Funding
This work was supported by the Telethon Foundation (Grant No. GGP15098 to EC) and by the Italian parent association “CDKL5 insieme verso la cura.”
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2019.00169/full#supplementary-material
References
Aicardi, G., Argilli, E., Cappello, S., Santi, S., Riccio, M., Thoenen, H., et al. (2004). Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. U.S.A. 101, 15788–15792. doi: 10.1073/pnas.0406960101
Amendola, E., Zhan, Y., Mattucci, C., Castroflorio, E., Calcagno, E., Fuchs, C., et al. (2014). Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS One 9:e91613. doi: 10.1371/journal.pone.0091613
Andero, R., Daviu, N., Escorihuela, R. M., Nadal, R., and Armario, A. (2012). 7,8-dihydroxyflavone, a TrkB receptor agonist, blocks long-term spatial memory impairment caused by immobilization stress in rats. Hippocampus 22, 399–408. doi: 10.1002/hipo.20906
Andero, R., Heldt, S. A., Ye, K., Liu, X., Armario, A., and Ressler, K. J. (2011). Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am. J. Psychiatry 168, 163–172. doi: 10.1176/appi.ajp.2010.10030326
Asrar, S., and Jia, Z. (2013). Molecular mechanisms coordinating functional and morphological plasticity at the synapse: role of GluA2/N-cadherin interaction-mediated actin signaling in mGluR-dependent LTD. Cell. Signal. 25, 397–402. doi: 10.1016/j.cellsig.2012.11.007
Bahi-Buisson, N., and Bienvenu, T. (2012). CDKL5-related disorders: from clinical description to molecular genetics. Mol. Syndromol. 2, 137–152.
Bahi-Buisson, N., Nectoux, J., Rosas-Vargas, H., Milh, M., Boddaert, N., Girard, B., et al. (2008). Key clinical features to identify girls with CDKL5 mutations. Brain 131(Pt 10), 2647–2661. doi: 10.1093/brain/awn197
Bahi-Buisson, N., Villeneuve, N., Caietta, E., Jacquette, A., Maurey, H., Matthijs, G., et al. (2012). Recurrent mutations in the CDKL5 gene: genotype-phenotype relationships. Am. J. Med. Genet. A 158A, 1612–1619. doi: 10.1002/ajmg.a.35401
Brown, M. W., and Banks, P. J. (2015). In search of a recognition memory engram. Neurosci. Biobehav. Rev. 50, 12–28. doi: 10.1016/j.neubiorev.2014.09.016
Chapleau, C. A., Larimore, J. L., Theibert, A., and Pozzo-Miller, L. (2009). Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking: fundamental roles in neurodevelopmental disorders associated with mental retardation and autism. J. Neurodev. Disord. 1, 185–196. doi: 10.1007/s11689-009-9027-6
Chen, C., Wang, Z., Zhang, Z., Liu, X., Kang, S. S., Zhang, Y., et al. (2018). The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 115, 578–583. doi: 10.1073/pnas.1718683115
Das, D. K., Mehta, B., Menon, S. R., Raha, S., and Udani, V. (2013). Novel mutations in cyclin-dependent kinase-like 5 (CDKL5) gene in Indian cases of Rett syndrome. Neuromolecular Med. 15, 218–225. doi: 10.1007/s12017-012-8212-z
Della Sala, G., Putignano, E., Chelini, G., Melani, R., Calcagno, E., Michele Ratto, G., et al. (2016). Dendritic spine instability in a mouse model of CDKL5 disorder is rescued by insulin-like growth factor 1. Biol. Psychiatry 80, 302–311. doi: 10.1016/j.biopsych.2015.08.028
Devi, L., and Ohno, M. (2012). 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 37, 434–444. doi: 10.1038/npp.2011.191
Ehrlich, I., Klein, M., Rumpel, S., and Malinow, R. (2007). PSD-95 is required for activity-driven synapse stabilization. Proc. Natl. Acad. Sci. U.S.A. 104, 4176–4181. doi: 10.1073/pnas.0609307104
Fehr, S., Wilson, M., Downs, J., Williams, S., Murgia, A., Sartori, S., et al. (2013). The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 21, 266–273. doi: 10.1038/ejhg.2012.156
Fuchs, C., Rimondini, R., Viggiano, R., Trazzi, S., De Franceschi, M., Bartesaghi, R., et al. (2015). Inhibition of GSK3beta rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol. Dis. 82, 298–310. doi: 10.1016/j.nbd.2015.06.018
Fuchs, C., Trazzi, S., Roberta, T., Viggiano, R., De Franceschi, M. E., et al. (2014). Loss of Cdkl5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3beta signaling. Neurobiol. Dis. 70, 53–68. doi: 10.1016/j.nbd.2014.06.006
Garcia-Diaz Barriga, G., Giralt, A., Anglada-Huguet, M., Gaja-Capdevila, N., Orlandi, J. G., Soriano, J., et al. (2017). 7,8-dihydroxyflavone ameliorates cognitive and motor deficits in a Huntington’s disease mouse model through specific activation of the PLCgamma1 pathway. Hum. Mol. Genet. 26, 3144–3160. doi: 10.1093/hmg/ddx198
Gartner, A., Polnau, D. G., Staiger, V., Sciarretta, C., Minichiello, L., Thoenen, H., et al. (2006). Hippocampal long-term potentiation is supported by presynaptic and postsynaptic tyrosine receptor kinase B-mediated phospholipase Cgamma signaling. J. Neurosci. 26, 3496–3504. doi: 10.1523/JNEUROSCI.3792-05.2006
Guidi, S., Stagni, F., Bianchi, P., Ciani, E., Ragazzi, E., Trazzi, S., et al. (2013). Early pharmacotherapy with fluoxetine rescues dendritic pathology in the Ts65Dn mouse model of down syndrome. Brain Pathol. 23, 129–143. doi: 10.1111/j.1750-3639.2012.00624.x
Horne, E. A., and Dell’Acqua, M. L. (2007). Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J. Neurosci. 27, 3523–3534. doi: 10.1523/JNEUROSCI.4340-06.2007
Isaac, J. T., Ashby, M. C., and McBain, C. J. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871. doi: 10.1016/j.neuron.2007.06.001
Jang, H. J., Yang, Y. R., Kim, J. K., Choi, J. H., Seo, Y. K., Lee, Y. H., et al. (2013). Phospholipase C-gamma1 involved in brain disorders. Adv. Biol. Regul. 53, 51–62. doi: 10.1016/j.jbior.2012.09.008
Jang, S. W., Liu, X., Yepes, M., Shepherd, K. R., Miller, G. W., Liu, Y., et al. (2010). A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. U.S.A. 107, 2687–2692. doi: 10.1073/pnas.0913572107
Kalscheuer, V. M., Tao, J., Donnelly, A., Hollway, G., Schwinger, E., Kubart, S., et al. (2003). Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am. J. Hum. Genet. 72, 1401–1411. doi: 10.1086/375538
Kealy, J., and Commins, S. (2011). The rat perirhinal cortex: a review of anatomy, physiology, plasticity, and function. Prog. Neurobiol. 93, 522–548. doi: 10.1016/j.pneurobio.2011.03.002
Kilstrup-Nielsen, C., Rusconi, L., La Montanara, P., Ciceri, D., Bergo, A., Bedogni, F., et al. (2012). What we know and would like to know about CDKL5 and its involvement in epileptic encephalopathy. Neural Plast. 2012:728267. doi: 10.1155/2012/728267
Korte, M., Minichiello, L., Klein, R., and Bonhoeffer, T. (2000). Shc-binding site in the TrkB receptor is not required for hippocampal long-term potentiation. Neuropharmacology 39, 717–724. doi: 10.1016/s0028-3908(99)00273-7
Liu, X., Chan, C. B., Jang, S. W., Pradoldej, S., Huang, J., He, K., et al. (2010). A synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J. Med. Chem. 53, 8274–8286. doi: 10.1021/jm101206p
Mazziotti, R., Lupori, L., Sagona, G., Gennaro, M., Della Sala, G., Putignano, E., et al. (2017). Searching for biomarkers of CDKL5 disorder: early-onset visual impairment in CDKL5 mutant mice. Hum. Mol. Genet. 26, 2290–2298. doi: 10.1093/hmg/ddx119
Minichiello, L. (2009). TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 10, 850–860. doi: 10.1038/nrn2738
Minichiello, L., Calella, A. M., Medina, D. L., Bonhoeffer, T., Klein, R., and Korte, M. (2002). Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36, 121–137. doi: 10.1016/s0896-6273(02)00942-x
Moseley, B. D., Dhamija, R., Wirrell, E. C., and Nickels, K. C. (2012). Historic, clinical, and prognostic features of epileptic encephalopathies caused by CDKL5 mutations. Pediatr. Neurol. 46, 101–105. doi: 10.1016/j.pediatrneurol.2011.11.007
Okuda, K., Kobayashi, S., Fukaya, M., Watanabe, A., Murakami, T., Hagiwara, M., et al. (2017). CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis. 106, 158–170. doi: 10.1016/j.nbd.2017.07.002
Paine, S. M., Munot, P., Carmichael, J., Das, K., Weber, M. A., Prabhakar, P., et al. (2012). The neuropathological consequences of CDKL5 mutation. Neuropathol. Appl. Neurobiol. 38, 744–747. doi: 10.1111/j.1365-2990.2012.01292.x
Pizzo, R., Gurgone, A., Castroflorio, E., Amendola, E., Gross, C., Sassoe-Pognetto, M., et al. (2016). Lack of Cdkl5 disrupts the organization of excitatory and inhibitory synapses and parvalbumin interneurons in the primary visual cortex. Front. Cell Neurosci. 10:261. doi: 10.3389/fncel.2016.00261
Ricciardi, S., Ungaro, F., Hambrock, M., Rademacher, N., Stefanelli, G., Brambilla, D., et al. (2012). CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 14, 911–923. doi: 10.1038/ncb2566
Risher, W. C., Ustunkaya, T., Singh Alvarado, J., and Eroglu, C. (2014). Rapid golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS One 9:e107591. doi: 10.1371/journal.pone.0107591
Roncacé, V., Burattini, C., Stagni, F., Guidi, S., Giacomini, A., Emili, M., et al. (2017). Neuroanatomical alterations and synaptic plasticity impairment in the perirhinal cortex of the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 106, 89–100. doi: 10.1016/j.nbd.2017.06.017
Sivilia, S., Mangano, C., Beggiato, S., Giuliani, A., Torricella, R., Baldassarro, V. A., et al. (2016). CDKL5 knockout leads to altered inhibitory transmission in the cerebellum of adult mice. Genes Brain Behav. 15, 491–502. doi: 10.1111/gbb.12292
Spencer, J. P. (2008). Flavonoids: modulators of brain function? Br. J. Nutr. 99(E Suppl. 1), ES60–ES77. doi: 10.1017/S0007114508965776
Stalpers, X. L., Spruijt, L., Yntema, H. G., and Verrips, A. (2012). Clinical phenotype of 5 females with a CDKL5 mutation. J. Child Neurol. 27, 90–93. doi: 10.1177/0883073811413832
Suzuki, W. A., and Naya, Y. (2014). The perirhinal cortex. Annu. Rev. Neurosci. 37, 39–53. doi: 10.1146/annurev-neuro-071013-014207
Tang, S., Wang, I. J., Yue, C., Takano, H., Terzic, B., Pance, K., et al. (2017). Loss of CDKL5 in glutamatergic neurons disrupts hippocampal microcircuitry and leads to memory impairment in mice. J. Neurosci. 37, 7420–7437. doi: 10.1523/JNEUROSCI.0539-17.2017
Tao, J., Van Esch, H., Hagedorn-Greiwe, M., Hoffmann, K., Moser, B., Raynaud, M., et al. (2004). Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 75, 1149–1154. doi: 10.1086/426460
Tramarin, M., Rusconi, L., Pizzamiglio, L., Barbiero, I., Peroni, D., Scaramuzza, L., et al. (2018). The antidepressant tianeptine reverts synaptic AMPA receptor defects caused by deficiency of CDKL5. Hum. Mol. Genet. 27, 2052–2063. doi: 10.1093/hmg/ddy108
Trazzi, S., Fuchs, C., Viggiano, R., De Franceschi, M., Valli, E., Jedynak, P., et al. (2016). HDAC4: a key factor underlying brain developmental alterations in CDKL5 disorder. Hum. Mol. Genet. 25, 3887–3907. doi: 10.1093/hmg/ddw231
Trazzi, S., Mitrugno, V. M., Valli, E., Fuchs, C., Rizzi, S., Guidi, S., et al. (2011). APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum. Mol. Genet. 20, 1560–1573. doi: 10.1093/hmg/ddr033
Wang, D., Noda, Y., Zhou, Y., Mouri, A., Mizoguchi, H., Nitta, A., et al. (2007). The allosteric potentiation of nicotinic acetylcholine receptors by galantamine ameliorates the cognitive dysfunction in beta amyloid25-35 i.c.v.-injected mice: involvement of dopaminergic systems. Neuropsychopharmacology 32, 1261–1271. doi: 10.1038/sj.npp.1301256
Wang, I. T., Allen, M., Goffin, D., Zhu, X., Fairless, A. H., Brodkin, E. S., et al. (2012). Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. U.S.A. 109, 21516–21521. doi: 10.1073/pnas.1216988110
Weaving, L. S., Christodoulou, J., Williamson, S. L., Friend, K. L., McKenzie, O. L., Archer, H., et al. (2004). Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 75, 1079–1093. doi: 10.1086/426462
Weng, S. M., McLeod, F., Bailey, M. E., and Cobb, S. R. (2011). Synaptic plasticity deficits in an experimental model of rett syndrome: long-term potentiation saturation and its pharmacological reversal. Neuroscience 180, 314–321. doi: 10.1016/j.neuroscience.2011.01.061
Williams, R. J., and Spencer, J. P. (2012). Flavonoids, cognition, and dementia: actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic. Biol. Med. 52, 35–45. doi: 10.1016/j.freeradbiomed.2011.09.010
Zhu, Y. C., Li, D., Wang, L., Lu, B., Zheng, J., Zhao, S. L., et al. (2013). Palmitoylation-dependent CDKL5-PSD-95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc. Natl. Acad. Sci. U.S.A. 110, 9118–9123. doi: 10.1073/pnas.1300003110
Keywords: CDKL5, synaptic plasticity, TrkB, PLCγ1, dendritic pattern, GluA2, perirhinal cortex, rett syndrome
Citation: Ren E, Roncacé V, Trazzi S, Fuchs C, Medici G, Gennaccaro L, Loi M, Galvani G, Ye K, Rimondini R, Aicardi G and Ciani E (2019) Functional and Structural Impairments in the Perirhinal Cortex of a Mouse Model of CDKL5 Deficiency Disorder Are Rescued by a TrkB Agonist. Front. Cell. Neurosci. 13:169. doi: 10.3389/fncel.2019.00169
Received: 29 October 2018; Accepted: 09 April 2019;
Published: 30 April 2019.
Edited by:
João Peça, University of Coimbra, PortugalReviewed by:
Nicoletta Landsberger, University of Milan, ItalyNicola Berretta, Fondazione Santa Lucia (IRCCS), Italy
Copyright © 2019 Ren, Roncacé, Trazzi, Fuchs, Medici, Gennaccaro, Loi, Galvani, Ye, Rimondini, Aicardi and Ciani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgio Aicardi, Z2lvcmdpby5haWNhcmRpQHVuaWJvLml0 Elisabetta Ciani, ZWxpc2FiZXR0YS5jaWFuaUB1bmliby5pdA==
†These authors have contributed equally to this work