 Jaichandar Subramanian
Jaichandar Subramanian Julie C. Savage
Julie C. Savage Marie-Ève Tremblay
Marie-Ève Tremblay- 1Department of Pharmacology & Toxicology, University of Kansas, Lawrence, KS, United States
- 2Axe Neurosciences, Centre de Recherche du CHU de Québec, Université Laval, Québec City, QC, Canada
- 3Neurology and Neurosurgery Department, McGill University, Montreal, QC, Canada
- 4Department of Molecular Medicine, Université Laval, Québec City, QC, Canada
- 5Division of Medical Sciences, University of Victoria, Victoria, BC, Canada
- 6Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, BC, Canada
Synapse loss is the strongest correlate for cognitive decline in Alzheimer's disease. The mechanisms underlying synapse loss have been extensively investigated using mouse models expressing genes with human familial Alzheimer's disease mutations. In this review, we summarize how multiphoton in vivo imaging has improved our understanding of synapse loss mechanisms associated with excessive amyloid in the living animal brain. We also discuss evidence obtained from these imaging studies for the role of cell-intrinsic calcium dyshomeostasis and cell-extrinsic activities of microglia, which are the immune cells of the brain, in mediating synapse loss.
Introduction
Immunohistochemistry of postmortem brain from Alzheimer's disease (AD) patients revealed that synapse loss is the strongest correlate for the cognitive deficit (Terry et al., 1991; DeKosky et al., 1996; Scheff and Price, 2006; Scheff et al., 2007; de Wilde et al., 2016). Mouse models that express familial AD-associated mutations in genes coding for amyloid precursor protein and presenilin that increase amyloid levels in the brain or that express mutated Tau leading to neurofibrillary tangles provide an entry point to study mechanisms of synapse loss associated with prominent AD related pathologies, such as amyloid plaques and neurofibrillary tangles (Jankowsky and Zheng, 2017). Morphological and electrophysiological studies done on ex vivo and postmortem preparations from these mice have confirmed that human familial AD associated mutations cause synaptic dysfunction, ranging from impaired plasticity to increased loss of synapses (Selkoe, 2002; Knobloch and Mansuy, 2008; Spires-Jones and Knafo, 2012; Yu and Lu, 2012; Pozueta et al., 2013; Forner et al., 2017). Postmortem preparations only offer a snapshot of pathology that does not capture dynamic events that precede or follow the observed deficits. Ex vivo preparations, on the other hand, allow for monitoring dynamic events but not in the context of the intact neural circuitry of a living brain. In addition, the functions of microglia, which are the immune cells of the brain, are severely affected by experimental procedures (Hellwig et al., 2013; Gosselin et al., 2017).
The advent of fluorescence labeling technologies and two-photon microscopy enabled direct visualization of synapse dynamics in vivo in the living mouse brain (Grutzendler et al., 2002; Trachtenberg et al., 2002). Typically, neurons are sparsely labeled with a fluorescent cell fill to visualize the morphology of subcellular structures, such as dendritic spines (postsynapse) or axonal boutons (presynapse). Fluorescence labeling is achieved by expressing fluorescent proteins through transgene integration, viral delivery, or in utero electroporation. Optical access to fluorescently labeled neurons is achieved by replacing part of the skull with a glass coverslip or thinning it. Though two-photon excitation provides higher depth resolution than traditional single-photon excitation, it is still limited to ~450 μm from the surface of the brain (Takasaki et al., 2020). Therefore, non-invasive two-photon imaging studies are restricted to superficial layers of the cortex, however synaptic imaging in the hippocampus has been achieved using more invasive approaches, such as removal of overlying cortical tissue or micro-endoscopy (Mizrahi et al., 2004; Gu et al., 2014; Attardo et al., 2015). Two-photon imaging in mouse models of AD has allowed for the interrogation of synaptic dysfunction associated with amyloid and tau pathology in the intact circuitry of living animals (Tables 1, 2). More importantly, in vivo imaging of synapses allows for chronic monitoring of synaptic changes in the same neurons over time, thereby enabling the visualization of synaptic dynamics. In this review, we focus on how in vivo imaging using two-photon microscopy has revealed the properties and mechanisms of synapse loss in mouse models of AD, emphasizing mouse models of amyloidosis. Due to the depth limitations of two-photon imaging, most of these studies tracked synaptic changes in the somatosensory cortex (unless otherwise mentioned).
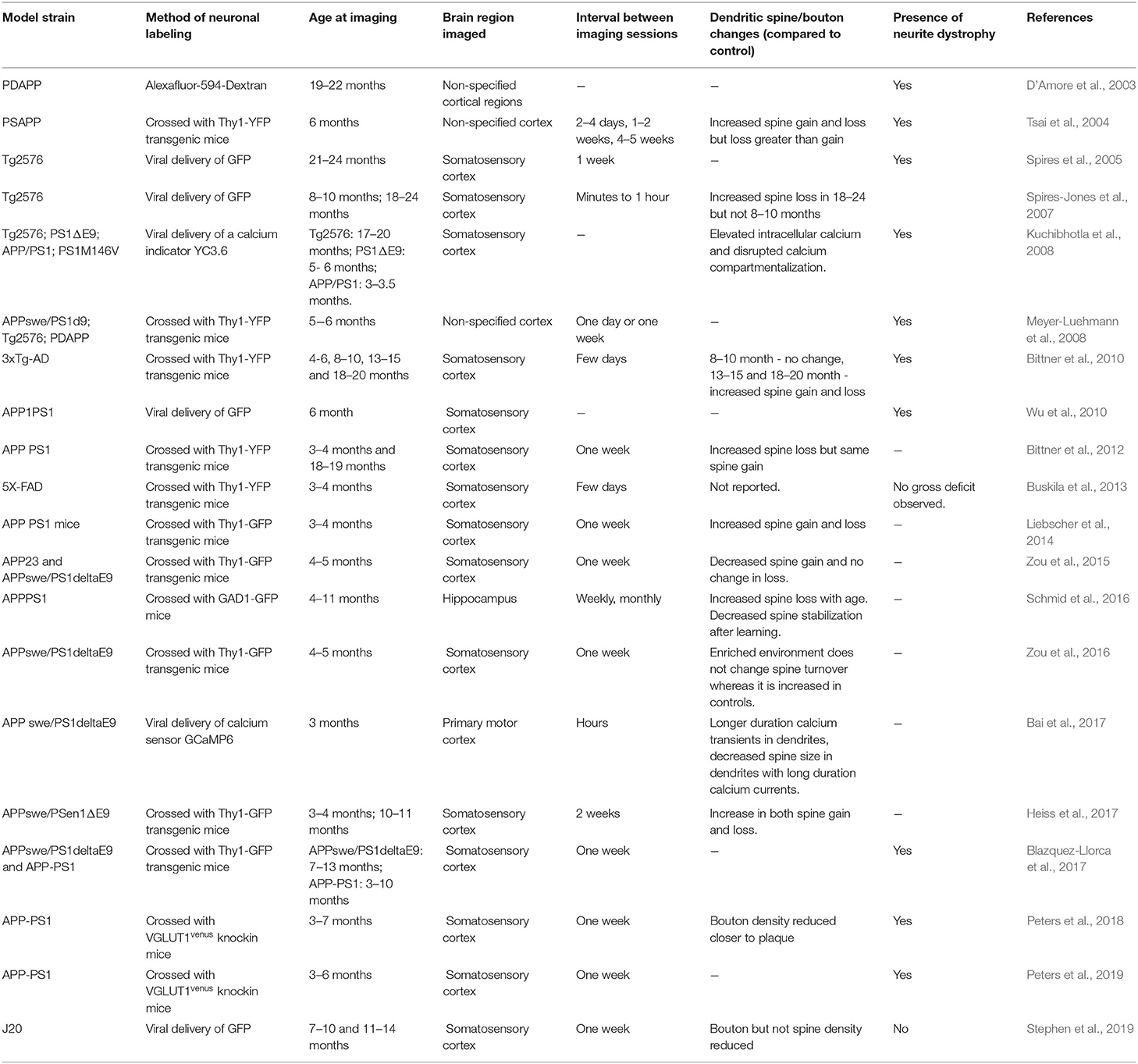
Table 1. Studies examining dendritic spine and neurite pathology in mouse models of amyloidosis using in vivo two-photon microscopy that are discussed in this review.
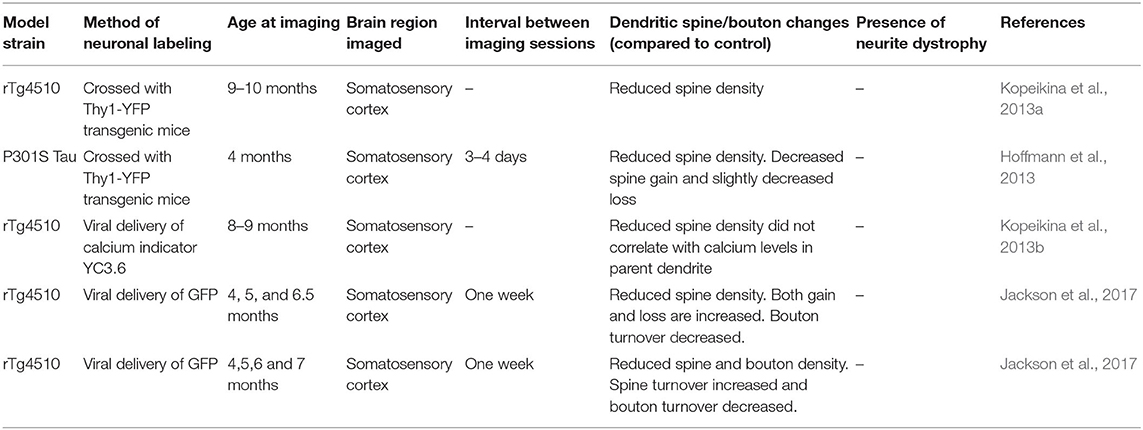
Table 2. Studies examining dendritic spine and neurite pathology in mouse models of Tauopathy using in vivo two-photon microscopy that are discussed in this review.
Amyloid Associated Neurite Pathology
After its development for in vivo imaging (Grutzendler et al., 2002; Trachtenberg et al., 2002), two-photon microscopy was quickly adopted to visualize neurite dystrophy associated with amyloidosis (D'Amore et al., 2003). The initial study confirmed findings from postmortem AD patient brains (Onorato et al., 1989) and amyloid mouse models (Richardson and Burns, 2002) that plaques alter the trajectory of neurites and are associated with their dystrophy (D'Amore et al., 2003). Consistently, many subsequent in vivo imaging studies confirmed the existence of dystrophic neurites (variably defined as swelling >2.5 μm or a volume 2-fold over normal neurites) near plaques (Tsai et al., 2004; Brendza et al., 2005; Spires et al., 2005; Spires-Jones et al., 2007, 2011; Kuchibhotla et al., 2008; Meyer-Luehmann et al., 2008; Bittner et al., 2010, 2012; Wu et al., 2010; Zou et al., 2015, 2016; Schmid et al., 2016; Blazquez-Llorca et al., 2017; Peters et al., 2018). The presence of dystrophic neurites near plaques could either mean that plaques cause neurite dystrophy or dystrophic neurites promote plaque formation. Addressing this question requires monitoring the dynamics of neurites and plaques over time, which is not possible with traditional imaging approaches on postmortem brain slices. Longitudinal imaging of dystrophic neurites and plaques shows that plaque formation precedes dystrophic neurites (Meyer-Luehmann et al., 2008; Blazquez-Llorca et al., 2017; Peters et al., 2018). In one of the mouse models, APPswe/PS1E9 mice, dystrophic dendrites were present in regions with no apparent plaques (Meyer-Luehmann et al., 2008). Longitudinal imaging of these regions did not identify any de novo plaque formation near the dystrophic dendrites. Interestingly, during this time, some of the neurites returned to normalcy or disappeared. Only 60% of the dystrophic neurites remained stable over 2 weeks (Meyer-Luehmann et al., 2008). If dystrophic neurites do not promote plaque formation, it is more likely that plaques increase the abundance of dystrophic neurites. This possibility is supported by the observations that the appearance of dystrophic neurites follows plaque appearance (Meyer-Luehmann et al., 2008; Blazquez-Llorca et al., 2017). The curvature of neurites across the plaque continues to increase following plaque formation, with damaged neurites appearing in the subsequent days (Tsai et al., 2004; Meyer-Luehmann et al., 2008). Only ~25% of neurites near plaque developed dystrophy, and more detailed analyses of their dynamics revealed that they are highly stable with a mean lifetime of 76 days (Blazquez-Llorca et al., 2017). The volume of dystrophy is highly variable, and interestingly, is also highly dynamic, with dystrophies undergoing both shrinkage and expansion (Blazquez-Llorca et al., 2017). Plaques smaller than 4 μm are not associated with dystrophic neurites, and as the plaque size increased, the extent of dystrophy also increased (Blazquez-Llorca et al., 2017; Peters et al., 2018).
Though dystrophic neurites do not appear to promote plaque formation, they may still contribute to the growth of preformed plaque. Plaque growth occurs over a long period, exhibiting sigmoidal growth kinetics (Christie et al., 2001; Yan et al., 2009; Burgold et al., 2011, 2014; Condello et al., 2011; Hefendehl et al., 2011; Bittner et al., 2012). One of the contributing factors for plaque growth is the local concentration of the enzyme BACE-1, whose cleavage of amyloid precursor protein results in amyloid peptides present in the plaque (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999; Lin et al., 2000). BACE-1 levels are increased in dystrophic neurites in cultured neurons and brain sections of AD patients, and 5XFAD amyloid mouse model (Zhang et al., 2009; Kandalepas et al., 2013; Sadleir et al., 2016), and therefore, dystrophic neurites can contribute to plaque growth. Consistent with this idea, inhibition of BACE-1 decreased the growth rate of plaques (Peters et al., 2018), which, in turn, reduced the formation of dystrophic neurites associated with plaque (Peters et al., 2018). The vicious cycle of amyloid plaque growth, the formation of neurite dystrophy, and accumulation of BACE-1 in dystrophic neurites also results in the formation of satellite plaques (Peters et al., 2018). Interestingly, the deletion of the gene coding for microtubule binding protein Tau reduces the accumulation of BACE-1 in dystrophic neurites and reduces the formation of satellite plaques (Peters et al., 2019). Thus, in vivo two-photon imaging approaches have allowed us to understand the kinetics of plaque formation and growth and its relevance to the appearance of dystrophic neurites in a manner not feasible with traditional postmortem analyses.
Amyloid Associated Dendritic Spine Pathology
Dystrophic neurites near plaques have a reduced density of dendritic spines (Spires et al., 2005). Since neurite dystrophy is increased after plaque appearance, is the reduction in synapses triggered only after plaque formation? One of the consensuses from the different in vivo imaging studies performed in mouse models of amyloidosis is that the density of dendritic spines is lower within 50 μm from the plaque compared to farther away from the plaque or non-transgenic controls (Spires-Jones et al., 2007; Kuchibhotla et al., 2008; Zou et al., 2015) but see Stephen et al. (2019). Though less pronounced, a reduction in spine density (Spires et al., 2005; Bittner et al., 2010) or a reduction in mature spine morphology (Zou et al., 2015) compared to non-transgenic controls was observed in dendrites > 50 μm away from plaque. A smaller reduction in spine density farther away from plaque could be due to reduced local concentration of soluble amyloid compared to the vicinity of a plaque. The reduction in spine density did not correlate with the size of the plaque (Spires et al., 2005), supporting the idea that soluble amyloid in the periphery of the plaque rather than the plaque itself is responsible for synapse loss (Mucke et al., 2000; Koffie et al., 2009). Consistently, spine loss is observed in ex vivo preparations following exposure to amyloid peptides (Hsieh et al., 2006; Shrestha et al., 2006; Shankar et al., 2007), and in vivo 1 day after the injection of soluble amyloid in non-transgenic mice (Arbel-Ornath et al., 2017).
Soluble amyloid is present even in the absence of plaques; therefore, if soluble amyloid alone were sufficient for spine loss, one would expect a significant reduction in spine loss prior to plaque formation. Longitudinal imaging of spines before and following de novo plaque formation reveals that spine density begins to reduce only 4.5 weeks after plaque formation (Bittner et al., 2012). Consistently, multiple imaging studies show normal spine density before plaque formation (Spires-Jones et al., 2007; Kuchibhotla et al., 2008; Bittner et al., 2010). Though these observations tend to support a role for amyloid plaque itself in spine loss, a more parsimonious explanation is that spine loss requires a high enough concentration of amyloid peptides that becomes available at an age when plaques are formed in the strains used for in vivo imaging (Maia et al., 2013). The less dramatic effect of soluble amyloid in transgenic mouse models of amyloidosis on overt spine loss in vivo compared to ex vivo preparations indicates possible resistance to spine loss when the buildup of amyloid is gradual compared to a sudden spike used in bath applications of ex vivo preparations. In addition, the lack or alteration of clearance mechanisms, such as microglial phagocytosis (Mandrekar et al., 2009), may exacerbate the synaptotoxic effect of amyloid in ex vivo preparations. These explanations, however, are at odds with the findings that brain slices from some mouse models of amyloidosis show synapse loss prior to plaque formation (Hsia et al., 1999; Moechars et al., 1999; Mucke et al., 2000; Lanz et al., 2003; Jacobsen et al., 2006). One plausible explanation for this discrepancy is that the brain regions examined for synapse loss using in vivo imaging (mostly, somatosensory cortex) and brain slices (mostly, hippocampus) are not the same. The vulnerability of synapses or the local concentration of amyloid could differ between brain regions and contribute to the plaque dependence for spine loss in brain regions imaged using two-photon microscopy. However, this may not be the sole reason because, in 3xTg-AD mouse strain, spine loss was observed in brain slices of the hippocampus and frontal cortex only after plaque formation, though these areas did not exhibit a characteristic plaque-distance dependence for spine loss (Bittner et al., 2010).
Pre- and postsynaptic terminals may exhibit differential plaque distance dependent vulnerability. The evidence for the vulnerability of presynaptic terminals in amyloid mouse models has been contradictory. Some histological studies found evidence for the loss of synaptophysin (Rutten et al., 2005; Dong et al., 2007; Tampellini et al., 2010) whereas others have not (King and Arendash, 2002; Rutten et al., 2003; Boncristiano et al., 2005; Hong et al., 2016), even in the same amyloid mouse model. In vivo imaging of presynaptic boutons in amyloid model mice show increased dynamics of boutons near the plaque compared to farther away from the plaque (Liebscher et al., 2014; Blazquez-Llorca et al., 2017; Stephen et al., 2019). The plaque distance-dependent synaptic loss and dystrophy holds up even when a presynaptic protein (Vglut1) is directly visualized in vivo (Peters et al., 2018). One of the reasons for the lack of consensus with respect to loss of presynaptic terminals in amyloid models could be due to variations in the sampling of different cell types between studies.
The plaque distance dependence of synapse loss observed in in vivo imaging studies using amyloid mouse models is in stark contrast to the observed loss of synapses affecting both pre- and postsynaptic elements across postmortem cortex (Terry et al., 1991; DeKosky et al., 1996; Sze et al., 1997; Scheff and Price, 2006; Scheff et al., 2007; de Wilde et al., 2016), despite plaques occupying only 5–10% of cortical volume in AD patients (Terry, 2000). Multiple differences between AD patients and mouse models of amyloidosis can account for the difference in plaque distance-dependent effect on synapses. One remarkable feature of mouse models of amyloidosis is that they exhibit limited or no neuronal death (Wirths and Bayer, 2010; Jankowsky and Zheng, 2017). The death of a neuron whose axons travel far will be accompanied by synapse loss farther from the soma. Thus, the lack of synapse loss farther away from plaque could be a consequence of limited or no plaque-associated cell death in these mouse models. Another possibility for preferential loss of synapses closer to plaques is that most mouse models of amyloidosis do not display the Tau pathology observed in human AD patients. When Tau mutation is combined with amyloidosis, as in the triple transgenic 3xTg-AD mice, spine loss was observed closer to and farther away from the plaque (Bittner et al., 2010).
Spine Loss and Cognitive Decline
If spine loss increases only at the age when plaques are present, then one would expect cognitive deficits to be apparent only after plaque formation. However, cognitive deficits are observed before plaque formation in many amyloid mouse models (Holcomb et al., 1998; Wisniewski and Sigurdsson, 2010). In one of the strains, APP23, where cognitive decline precedes plaque formation, it was found that amyloid precursor protein accumulates intracellularly, and the amount of intracellular accumulation correlates with spine loss (Zou et al., 2015). Another caveat in reconciling cognitive studies with in vivo spine imaging studies is that most in vivo imaging studies in amyloid mouse models are restricted to the somatosensory cortex. Typically used cognitive tests, such as spatial or contextual memory tasks, may not elicit synaptic remodeling in the somatosensory cortex. In contrast, changes to sensory experience have been shown to cause synaptic remodeling in the somatosensory cortex (Trachtenberg et al., 2002). In the one of the amyloid mouse models, synaptic remodeling elicited by exposure to an enriched environment is disrupted in the somatosensory cortex before plaque appearance when spine density is normal (Zou et al., 2016; Heiss et al., 2017).
One of the main reasons most in vivo imaging studies are restricted to the somatosensory cortex is that it is easily accessible for non-invasive imaging. Imaging hippocampus, located ~1 mm deep in the brain, is out of the range for conventional two-photon imaging. Two-photon imaging of dendritic spines in the hippocampus is achieved by microendoscopy or by removing the overlying cortex (Mizrahi et al., 2004; Gu et al., 2014; Attardo et al., 2015). To date, to our knowledge, spine imaging in vivo in the hippocampus has not been performed in excitatory neurons of amyloid mouse models. However, a group of inhibitory neurons in the hippocampus, positive for somatostatin, that possess dendritic spines, has been studied using in vivo two-photon imaging in an amyloid mouse model (Schmid et al., 2016). The density of spines on these interneurons is reduced near the plaque. This reduction is due to a decrease in input from cholinergic neurons, which are lost due to cell death. In these somatostatin positive interneurons, new spines are formed in response to fear conditioning; however, this process is disrupted in the amyloid model (Schmid et al., 2016).
Most in vivo imaging studies use dendritic spines as a proxy for excitatory synapses. The maturation status of the spine is inferred from their shape, stability, and age. Newly formed spines are usually thin and are transient. If they persist for more than 4 days, they are highly likely to carry synapses (Knott et al., 2006). Mature persistent spines resemble mushroom-like structures and are called mushroom spines (Bourne and Harris, 2007). Though age, shape, and stability of dendritic spines could be a good predictor for the maturation status of a synapse, they are not entirely reliable. Spines that persist for 4 days may not contain synaptic proteins associated with mature synapses (Subramanian et al., 2019). Even if the plaque-associated spine loss observed in the superficial cortical regions is globally true, structural alterations, such as changes to AMPA receptors concentration, not captured by imaging spine alone could underlie cognitive decline. Therefore, synaptic abnormalities associated with cognitive decline may not require overt spine loss and the mechanisms could differ depending on the brain region and the model strains.
Mechanisms of Amyloid Associated Spine Loss
In adult mice, spine formation and elimination are balanced to maintain spine density (Villa et al., 2016; Subramanian et al., 2019). A reduction in the density of spines under amyloid pathology could result from the decreased formation of new spines or increased elimination of preexisting spines or both. No consensus has emerged on whether amyloid pathology triggers spine reduction by regulating formation or elimination. In vivo imaging studies have found increased spine elimination with no change in formation or smaller increase in formation (Tsai et al., 2004; Spires-Jones et al., 2007; Bittner et al., 2012), decreased spine formation with no change or an increase in elimination (Zou et al., 2015), or an increase in both formation and elimination (Bittner et al., 2010; Liebscher et al., 2014; Heiss et al., 2017). How could an increase in both formation and elimination result in reduced spine density? Newly formed dendritic spines could mature into stable synapses or disappear without forming synapses (Subramanian et al., 2019). If amyloid pathology reduces synapse stabilization, neurons will continue to make futile attempts to form synapses, whereas under non-pathological conditions, due to synapse stabilization, there would be fewer futile attempts. Each futile attempt will be counted as a dynamic event, and therefore, APP mice would have higher spine dynamics and yet have lower spine density.
How could amyloid pathology induce neurite dystrophy and spine loss? Calcium signaling regulates synaptic plasticity and dendritic arbor development (Konur and Ghosh, 2005), and growing evidence suggests that amyloid pathology disrupts neuronal calcium homeostasis (Holscher, 1998; Kawahara, 2004; Small, 2009; Brawek and Garaschuk, 2014; Popugaeva et al., 2017). Consistent with ex vivo preparations (Mattson et al., 1992; Guo et al., 1999; Demuro et al., 2005; Smith et al., 2005; Mattson, 2007), in vivo imaging of calcium reveals that dendrites closer to plaque have increased intracellular neuronal calcium in the intact brain of amyloid mouse models (Kuchibhotla et al., 2008). Before plaque formation, dendritic calcium levels in an amyloid mouse model are not different from non-transgenic mice (Kuchibhotla et al., 2008). In non-transgenic mice, calcium concentration within the spine does not correlate with that of the parent dendrite, whereas under amyloid pathology, there is a linear relationship between the two, suggesting that the compartmentalization of calcium in spines is lost (Kuchibhotla et al., 2008). Amyloid peptides can increase intracellular calcium within an hour after exposure, but spine loss occurs only 24 h later (Arbel-Ornath et al., 2017). Interestingly, spines that shrink following the application of amyloid peptide in the motor cortex are the ones that were activated during the prolonged dendritic calcium current elicited by the amyloid peptide (Bai et al., 2017).
A Role for Tau in Synapse Loss
The in vivo imaging studies described thus far are mostly focused on how amyloidosis associated with familial AD cases influence synapses. The effect of accumulation of hyper-phosphorylated Tau, another prominent AD-related pathology, on synaptic structure integrity is relatively less studied using in vivo two-photon imaging. The effect of overexpressing mutant Tau on synapse density has yielded mixed results in the histological analysis (Shahani et al., 2006; Eckermann et al., 2007; Yoshiyama et al., 2007; Hoover et al., 2010; Rocher et al., 2010; Crimins et al., 2011; Jaworski et al., 2011; Kremer et al., 2011; Alldred et al., 2012). In contrast, there is consensus among in vivo two-photon imaging studies that dendritic spine density is reduced in Tau mouse models (Bittner et al., 2010; Hoffmann et al., 2013; Kopeikina et al., 2013a,b; Jackson et al., 2017, 2020). The first in vivo imaging study describing the effect of Tau on synapses was done in triple transgenic 3xTg-AD mice that also had mutations associated with amyloidosis (Bittner et al., 2010). However, later studies using Tau models also showed a decrease in spine density. In a Tau model with P301S mutation, spine gain is reduced and is not matched by an equivalent reduction in spine loss, whereas, in the control mice, formation and elimination of spines are balanced. As a consequence, spine density is reduced in the Tau mutant model. Interestingly, the spine deficits are present at an age when no neurofibrillary tangles are apparent, and Tau itself is not localized to dendritic spines (Hoffmann et al., 2013). In vivo imaging also revealed decreased spine density in 8–9 month-old rTg4510 mouse model with P301L mutation. Surprisingly, in the same study, array tomography revealed no difference in synaptic density compared to wild-type control mice (Kopeikina et al., 2013a). Later in vivo imaging studies using this model confirmed a progressive decline in spine density between 4 and 6.5 months of age (Jackson et al., 2017, 2020). The decline in spine density is not uniform in all dendrites, with some dendrites exhibiting complete loss of spines. Higher turnover of dendritic spines, in some instances, is followed by the loss of associated dendritic branches. Over 6 months, ~35% of the dendrites are lost. In contrast, the axonal loss is preceded by a decreased turnover of boutons. The loss of synapses in this model also occurred at an age when neurofibrillary tangles are not yet present (Jackson et al., 2017, 2020). Interestingly, unlike amyloid models, spine loss in the rTg4510 model is not associated with elevated intracellular neuronal calcium (Kopeikina et al., 2013b).
Multiple cellular pathologies, such as intracellular calcium homeostasis, mitochondrial dysfunction, energy metabolism, and reactive oxygen species, associated with amyloidosis, have been imaged in vivo using two-photon microscopy (McLellan et al., 2003; Xie et al., 2013; Arbel-Ornath et al., 2017; Bai et al., 2017; Gomez et al., 2018; Lerdkrai et al., 2018; Calvo-Rodriguez et al., 2020). However, the dynamics of synapse loss in relation to these different cellular deficits resulting from amyloidosis remains to be explored by in vivo two-photon imaging. Synapse loss may not occur solely due to these cell-intrinsic factors. Growing evidence indeed suggests that microglial phagocytosis may be a key player in synaptic loss in amyloid and/or Tau mouse models. Below, we discuss how two-photon in vivo imaging has also uncovered a novel role for microglia during amyloid and Tau pathology.
Microglial Implication in AD: Insights from Two-Photon in vivo Imaging Studies
Microglia, which are the resident innate immune cells of the brain, have been intimately associated with AD since the disease was first described. In his landmark studies, Dr. Alois Alzheimer described glial cells developing excess fibers and containing “adipose saccules” (Alzheimer et al., 1995). Over 80 years later, studies conducted on postmortem AD brain samples revealed that microglia surrounding plaques express elevated levels of histocompatibility antigens and the pro-inflammatory cytokine IL-1 (Rogers et al., 1988; Griffin et al., 1989; Overmyer et al., 1999). Ultrastructural studies further uncovered microglia's intimate relationship with plaques, and even posited that microglia were responsible for amyloid deposition (Wisniewski et al., 1989; Perlmutter et al., 1990). Though it was later discovered that neurons produce amyloid precursor protein and amyloid (Wisniewski et al., 1989; Alzheimer et al., 1995), the role of microglia in AD has remained largely elusive. Over the past 30 years, microglia in the AD brain have vacillated between valiant protectors, powerless observers, and indiscriminate destroyers. In this section of the review, we cover the insights into their roles in synaptic loss that were provided using two-photon in vivo imaging (Table 3) and complementary techniques.
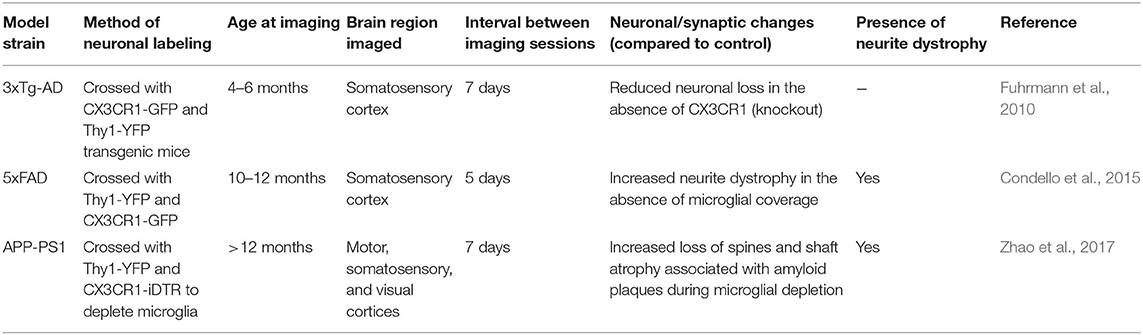
Table 3. Studies examining microglial interactions with neuronal or synaptic loss in mouse models of amyloid or Tau pathology using in vivo two-photon microscopy that are discussed in this review.
Loss of Microglial Phagocytic Abilities in AD
Two-photon in vivo studies in the APP/PS1Δe9 and Tg2576 models demonstrated that removing microglia during the chronic disease stage causes significant growth of existing plaques (Zhao et al., 2017). Combining these data with the fact that microglia (including the dark microglia, a subset identified by its markers of cellular stress including the condensation state of its cytoplasm and nucleoplasm resulting in a dark appearance under electron microscopy; Bisht et al., 2016) are occasionally seen containing amyloid deposits led to conclude that while microglia may be competent phagocytes, they are unable to control amyloid levels late during AD pathology. Of note, the dark microglia are rare in the healthy mature brain, but increase in number up to 10-fold with pathological conditions that include chronic stress, aging and amyloid deposition (in APP/PS1Δe9 mice; Bisht et al., 2016). This microglial subset discovered with electron microscopy displays hyper-ramified processes that extensively ensheath and engulf synaptic elements (pre- and postsynaptic), suggesting their role in the pathological remodeling of neuronal circuits in AD (Stratoulias et al., 2019; St-Pierre et al., 2020).
While neuroinflammation is a main hallmark of AD, alongside amyloid deposition and tangle formation, prolonged exposure to pro-inflammatory cytokines inhibits microglial phagocytic ability (Koenigsknecht and Landreth, 2004). Two-photon imaging in APP/PS1Δe9 mouse slices demonstrated that microglia lose their mobility and phagocytic capabilities as plaque deposition increases (Krabbe et al., 2013). Slice culture studies further determined that microglia from aged APP/PS1Δe9 and 5XFAD mice have reduced phagocytic capacity, possibly due to an impaired MerTK signaling (Savage et al., 2015). Work by others has determined that acute exposure to amyloid does not affect microglial phagocytic competence, whereas prolonged exposure in later disease stages results in reduced phagocytosis (Wendt et al., 2017). Together these data suggest that while microglial response to amyloid may be beneficial in the short-term, chronic exposure to amyloid may stunt microglia and prevent them from performing their normal phagocytic duties required to clear the brain from toxic or inflammatory debris.
Environmental enrichment has been shown using two-photon in vivo imaging to increase microglial amyloid clearing capacities thus preventing prion-like seeding of amyloid in a mouse model, while also reversing the deficits in neurogenesis and memory (Ziegler-Waldkirch et al., 2018). Environmental enrichment is well-known to promote beneficial neuroprotective microglial activities (Savage and Tremblay, 2019). However, increasing microglial phagocytosis with other strategies should be undertaken with extreme caution, as microglia could phagocytose the incorrect cargo. The same complement-mediated pathway which prunes excess synapses during normal brain development causes mistargeted phagocytosis of synapses early in mouse models of amyloid pathology, before plaques are deposited (Hong et al., 2016). Inhibiting C1q, C3, or microglial complement receptor CR3 in amyloid mouse models was shown to prevent synaptic loss, as well as cognitive impairment (Hammond et al., 2019). The microglial receptor TREM2, which displays various genetic variants in AD and is expressed by dark microglia, was also shown to mediate synaptic pruning during normal development, in cooperation with astrocytes (Filipello et al., 2018; Jay et al., 2019). Determining the outcome of TREM2 gene loss, haplodeficiency or variants on microglia-mediated synaptic loss in mouse models of amyloid and Tau pathology is an important topic of investigation (Yuan et al., 2016; Gratuze et al., 2020).
Similarly, fractalkine signaling between the neuronal chemokine fractalkine and its unique receptor CX3CR1, which is expressed by microglia, is a main mode of neuron-microglia communication in the brain. During normal physiological conditions, fractalkine signaling plays key roles in synaptic maturation, pruning, and plasticity, and mediates the adaptation of the brain and behavior to environmental challenges (Paolicelli et al., 2014; Tay et al., 2017). In AD pathology, evidence from complementary techniques revealed that fractalkine signaling is detrimental during amyloid pathology, yet beneficial in Tau pathology (Lee et al., 2010; Chen et al., 2016; Bemiller et al., 2018). In vivo, two-photon imaging indicates that 3xTg-AD mice deficient in fractalkine signaling are protected from neuronal loss, contrary to 3xTg-AD controls with an intact fractalkine signaling (Fuhrmann et al., 2010). In the 3xTg-AD mice, microglia were also shown to display an increased process velocity around the disappearing neurons, suggesting a possible involvement in their elimination, at early stages of pathology still devoid of plaques and Tau pathology (Fuhrmann et al., 2010). Similarly, using two-photon in vivo imaging in a zebrafish model of Tau pathology, in which neurons express hTauP301L, microglia were recently shown to transform their morphology (retraction and reduced number of processes, enlarged cell bodies), while increasing their migration and phagocytic activity toward dying neurons. Nevertheless, microglial phagocytosis failed in this context to remove all the dying neurons (Hassan-Abdi et al., 2019).
Emergence of Neuroprotective Phenotypes
While microglia are not sufficient to prevent AD pathology, synaptic loss, and cognitive impairment, they nonetheless form a barrier around plaques and slow disease progression, at least early on during disease pathogenesis. A major avenue of therapeutic research is focused on restoring proper microglial metabolism and physiological function, thus enabling them to clear amyloid, remove apoptotic neurons as well as provide trophic support for synaptic maintenance. Two-photon in vivo imaging of healthy mice revealed that microglia play an important role in the formation of dendritic spines, through their secretion of brain-derived neurotrophic factor, which is required for motor learning (Parkhurst et al., 2013). Microglia were also well-shown to dynamically contact pre- and postsynaptic elements by two-photon in vivo imaging during normal physiological conditions, and these interactions were frequently followed by the elimination of dendritic spines during experience-dependent plasticity (Tremblay et al., 2010). Whether microglia could exert similar beneficial roles at synapses in AD remains to be determined.
Recent studies identified a number of neuroprotective microglial phenotypes present on a subset of microglia within the AD brain, and these provide further potential for specific, microglia-targeted therapeutics. Two-photon in vivo imaging in 5XFAD mice revealed that microglia “wall off” plaques and likely promote neuronal and/or synaptic survival in regions affected by the amyloid pathology (Condello et al., 2015). In fact, dystrophic neurons were much more commonly seen in regions near plaques that were not covered by microglia, as these regions contained increased levels of toxic amyloid oligomeric and protofibrillar species. Microglial ability to seal off amyloid plaques from the surrounding neuropil is dependent on TREM2, as knockout mice had significantly reduced plaque area covered by microglia, while displaying increased plaque-associated neuronal dystrophy (Yuan et al., 2016).
Recent technical advances in the field now allow researchers to study populations of microglia from specific brain microenvironments, and thus characterize differences between microglia associated with plaques and those from nearby brain regions. These studies have begun to uncover the specifically and differentially-regulated transcriptome of microglial cells associated or not with plaques. When plaque-associated microglia were microdissected from amyloid mouse models, transcriptomic analysis identified increased genes associated with priming (i.e., immunological alert) and cellular metabolism as well as lysosomal activity (Kamphuis et al., 2016; Yin et al., 2017). While two-photon in vivo imaging of the primed microglia in particular and their dynamics at synapses is currently lacking, these studies indicate that plaque-associated microglia are not dystrophic or senescent, but rather highly active cells attempting to digest the amyloid, as supported by ultrastructural studies (El Hajj et al., 2019). These data support the idea that microglia surrounding plaques prevent plaque growth by removing oligomeric and protofibrillar amyloid from their immediate surroundings.
Additional work using single-cell RNAseq uncovered a disease-associated microglial (DAM) subtype in amyloid mouse models and human brain tissues (Keren-Shaul et al., 2017). A similar subtype was subsequently named the microglia-associated with neurodegeneration (MGnD) (Krasemann et al., 2017). These disease-associated subsets lose homeostatic microglial markers and upregulate genes associated with phagocytosis of apoptotic cells. The authors posit that MGnD microglia require contact with, if not phagocytosis of, dystrophic neurons like those associated with plaques, while depending on Trem2 and Apoe expression (Krasemann et al., 2017). These disease-associated phenotypes echo the ultrastructurally-defined dark microglia, but further in vivo research is required to determine the independent or concerted implication of these different microglial types in synaptic loss.
Conclusion and Perspectives
In vivo imaging approach using two-photon in vivo microscopy has found that dendritic spines are destabilized by the extracellular deposition or intracellular accumulation of amyloid. Cell intrinsic factors, such as elevated calcium, and extrinsic factors, like microglial phenotypic transformation elicited by the amyloid or Tau pathology, disrupt dendritic spine stability and might specifically trigger synaptic loss. Future two-photon imaging studies to simultaneously visualize the dynamics of axon terminals, dendritic spines, synaptic proteins, intracellular calcium, and microglia, together with amyloid and Tau pathology, in vivo in mouse models, will provide novel insights into the dynamic events preceding and causing loss of pre- and postsynaptic elements.
Author Contributions
JS and M-ÈT designed and wrote the manuscript. JCS contributed to the section on microglia. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the National Institutes of Health (Grant No. R01AG064067) to JS and a Canadian Institutes of Health Research–Foundation (Grant No. 353750) to M-ÈT. M-ÈT holds a Canada Research Chair (Tier 2) in Neurobiology of Aging and Cognition.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alldred, M. J., Duff, K. E., and Ginsberg, S. D. (2012). Microarray analysis of CA1 pyramidal neurons in a mouse model of tauopathy reveals progressive synaptic dysfunction. Neurobiol. Dis. 45, 751–762. doi: 10.1016/j.nbd.2011.10.022
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An english translation of alzheimer's 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Arbel-Ornath, M., Hudry, E., Boivin, J. R., Hashimoto, T., Takeda, S., Kuchibhotla, K. V., et al. (2017). Soluble oligomeric amyloid-beta induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener. 12:27. doi: 10.1186/s13024-017-0169-9
Attardo, A., Fitzgerald, J. E., and Schnitzer, M. J. (2015). Impermanence of dendritic spines in live adult CA1 hippocampus. Nature 523, 592–596. doi: 10.1038/nature14467
Bai, Y., Li, M., Zhou, Y., Ma, L., Qiao, Q., Hu, W., et al. (2017). Abnormal dendritic calcium activity and synaptic depotentiation occur early in a mouse model of alzheimer's disease. Mol. Neurodegener. 12:86. doi: 10.1186/s13024-017-0228-2
Bemiller, S. M., Maphis, N. M., Formica, S. V., Wilson, G. N., Miller, C. M., Xu, G., et al. (2018). Genetically enhancing the expression of chemokine domain of CX3CL1 fails to prevent tau pathology in mouse models of tauopathy. J. Neuroinflammation 15:278. doi: 10.1186/s12974-018-1310-6
Bisht, K., Sharma, K. P., Lecours, C., Sanchez, M. G., El Hajj, H., Milior, G., et al. (2016). Dark microglia: a new phenotype predominantly associated with pathological states. Glia 64, 826–839. doi: 10.1002/glia.22966
Bittner, T., Burgold, S., Dorostkar, M. M., Fuhrmann, M., Wegenast-Braun, B. M., Schmidt, B., et al. (2012). Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol. 124, 797–807. doi: 10.1007/s00401-012-1047-8
Bittner, T., Fuhrmann, M., Burgold, S., Ochs, S. M., Hoffmann, N., Mitteregger, G., et al. (2010). Multiple events lead to dendritic spine loss in triple transgenic alzheimer's disease mice. PLoS ONE 5:e15477. doi: 10.1371/journal.pone.0015477
Blazquez-Llorca, L., Valero-Freitag, S., Rodrigues, E. F., Merchan-Perez, A., Rodriguez, J. R., Dorostkar, M. M., et al. (2017). High plasticity of axonal pathology in alzheimer's disease mouse models. Acta Neuropathol. Commun. 5:14. doi: 10.1186/s40478-017-0415-y
Boncristiano, S., Calhoun, M. E., Howard, V., Bondolfi, L., Kaeser, S. A., Wiederhold, K. H., et al. (2005). Neocortical synaptic bouton number is maintained despite robust amyloid deposition in APP23 transgenic mice. Neurobiol. Aging 26, 607–613. doi: 10.1016/j.neurobiolaging.2004.06.010
Bourne, J., and Harris, K. M. (2007). Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 17, 381–386. doi: 10.1016/j.conb.2007.04.009
Brawek, B., and Garaschuk, O. (2014). Network-wide dysregulation of calcium homeostasis in alzheimer's disease. Cell Tissue Res. 357, 427–438. doi: 10.1007/s00441-014-1798-8
Brendza, R. P., Bacskai, B. J., Cirrito, J. R., Simmons, K. A., Skoch, J. M., Klunk, W. E., et al. (2005). Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J. Clin. Invest. 115, 428–433. doi: 10.1172/JCI23269
Burgold, S., Bittner, T., Dorostkar, M. M., Kieser, D., Fuhrmann, M., Mitteregger, G., et al. (2011). In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 121, 327–335. doi: 10.1007/s00401-010-0787-6
Burgold, S., Filser, S., Dorostkar, M. M., Schmidt, B., and Herms, J. (2014). In vivo imaging reveals sigmoidal growth kinetic of beta-amyloid plaques. Acta Neuropathol. Commun. 2:30. doi: 10.1186/2051-5960-2-30
Buskila, Y., Crowe, S. E., and Ellis-Davies, G. C. (2013). Synaptic deficits in layer 5 neurons precede overt structural decay in 5xFAD mice. Neuroscience 254, 152–159. doi: 10.1016/j.neuroscience.2013.09.016
Calvo-Rodriguez, M., Hou, S. S., Snyder, A. C., Kharitonova, E. K., Russ, A. N., Das, S., et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of alzheimer's disease. Nat. Commun. 11:2146. doi: 10.1038/s41467-020-16074-2
Chen, P., Zhao, W., Guo, Y., Xu, J., and Yin, M. (2016). CX3CL1/CX3CR1 in alzheimer's disease: a target for neuroprotection. Biomed. Res. Int. 2016:8090918. doi: 10.1155/2016/8090918
Christie, R. H., Bacskai, B. J., Zipfel, W. R., Williams, R. M., Kajdasz, S. T., Webb, W. W., et al. (2001). Growth arrest of individual senile plaques in a model of alzheimer's disease observed by in vivo multiphoton microscopy. J. Neurosci. 21, 858–864. doi: 10.1523/JNEUROSCI.21-03-00858.2001
Condello, C., Schain, A., and Grutzendler, J. (2011). Multicolor time-stamp reveals the dynamics and toxicity of amyloid deposition. Sci. Rep. 1:19. doi: 10.1038/srep00019
Condello, C., Yuan, P., Schain, A., and Grutzendler, J. (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat. Commun. 6:6176. doi: 10.1038/ncomms7176
Crimins, J. L., Rocher, A. B., Peters, A., Shultz, P., Lewis, J., and Luebke, J. I. (2011). Homeostatic responses by surviving cortical pyramidal cells in neurodegenerative tauopathy. Acta Neuropathol. 122, 551–564. doi: 10.1007/s00401-011-0877-0
D'Amore, J. D., Kajdasz, S. T., McLellan, M. E., Bacskai, B. J., Stern, E. A., and Hyman, B. T. (2003). In vivo multiphoton imaging of a transgenic mouse model of alzheimer disease reveals marked thioflavine-S-associated alterations in neurite trajectories. J. Neuropathol. Exp. Neurol. 62, 137–145. doi: 10.1093/jnen/62.2.137
de Wilde, M. C., Overk, C. R., Sijben, J. W., and Masliah, E. (2016). Meta-analysis of synaptic pathology in alzheimer's disease reveals selective molecular vesicular machinery vulnerability. Alzheimers. Dement. 12, 633–644. doi: 10.1016/j.jalz.2015.12.005
DeKosky, S. T., Scheff, S. W., and Styren, S. D. (1996). Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 5, 417–421. doi: 10.1006/neur.1996.0056
Demuro, A., Mina, E., Kayed, R., Milton, S. C., Parker, I., and Glabe, C. G. (2005). Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280, 17294–17300. doi: 10.1074/jbc.M500997200
Dong, H., Martin, M. V., Chambers, S., and Csernansky, J. G. (2007). Spatial relationship between synapse loss and beta-amyloid deposition in Tg2576 mice. J. Comp. Neurol. 500, 311–321. doi: 10.1002/cne.21176
Eckermann, K., Mocanu, M. M., Khlistunova, I., Biernat, J., Nissen, A., Hofmann, A., et al. (2007). The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J. Biol. Chem. 282, 31755–31765. doi: 10.1074/jbc.M705282200
El Hajj, H., Savage, J. C., Bisht, K., Parent, M., Vallieres, L., Rivest, S., et al. (2019). Ultrastructural evidence of microglial heterogeneity in alzheimer's disease amyloid pathology. J. Neuroinflammation 16:87. doi: 10.1186/s12974-019-1473-9
Filipello, F., Morini, R., Corradini, I., Zerbi, V., Canzi, A., Michalski, B., et al. (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48, 979–991.e978. doi: 10.1016/j.immuni.2018.04.016
Forner, S., Baglietto-Vargas, D., Martini, A. C., Trujillo-Estrada, L., and LaFerla, F. M. (2017). Synaptic impairment in alzheimer's disease: a dysregulated symphony. Trends Neurosci. 40, 347–357. doi: 10.1016/j.tins.2017.04.002
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of alzheimer's disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Gomez, C. A., Fu, B., Sakadzic, S., and Yaseen, M. A. (2018). Cerebral metabolism in a mouse model of alzheimer's disease characterized by two-photon fluorescence lifetime microscopy of intrinsic NADH. Neurophotonics 5:045008. doi: 10.1117/1.NPh.5.4.045008
Gosselin, D., Skola, D., Coufal, N. G., Holtman, I. R., Schlachetzki, J. C. M., Sajti, E., et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356:eaal3222. doi: 10.1126/science.aal3222
Gratuze, M., Leyns, C. E., Sauerbeck, A. D., St-Pierre, M. K., Xiong, M., Kim, N., et al. (2020). Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Invest. 30, 4654–4968. doi: 10.1172/JCI138179
Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., et al. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in down syndrome and alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 7611–7615. doi: 10.1073/pnas.86.19.7611
Grutzendler, J., Kasthuri, N., and Gan, W. B. (2002). Long-term dendritic spine stability in the adult cortex. Nature 420, 812–816. doi: 10.1038/nature01276
Gu, L., Kleiber, S., Schmid, L., Nebeling, F., Chamoun, M., Steffen, J., et al. (2014). Long-term in vivo imaging of dendritic spines in the hippocampus reveals structural plasticity. J. Neurosci. 34, 13948–13953. doi: 10.1523/JNEUROSCI.1464-14.2014
Guo, Q., Sebastian, L., Sopher, B. L., Miller, M. W., Ware, C. B., Martin, G. M., et al. (1999). Increased vulnerability of hippocampal neurons from presenilin-1 mutant knock-in mice to amyloid beta-peptide toxicity: central roles of superoxide production and caspase activation. J. Neurochem. 72, 1019–1029. doi: 10.1046/j.1471-4159.1999.0721019.x
Hammond, T. R., Marsh, S. E., and Stevens, B. (2019). Immune signaling in neurodegeneration. Immunity 50, 955–974. doi: 10.1016/j.immuni.2019.03.016
Hassan-Abdi, R., Brenet, A., Bennis, M., Yanicostas, C., and Soussi-Yanicostas, N. (2019). Neurons expressing pathological tau protein trigger dramatic changes in microglial morphology and dynamics. Front. Neurosci. 13:1199. doi: 10.3389/fnins.2019.01199
Hefendehl, J. K., Wegenast-Braun, B. M., Liebig, C., Eicke, D., Milford, D., Calhoun, M. E., et al. (2011). Long-term in vivo imaging of beta-amyloid plaque appearance and growth in a mouse model of cerebral beta-amyloidosis. J. Neurosci. 31, 624–629. doi: 10.1523/JNEUROSCI.5147-10.2011
Heiss, J. K., Barrett, J., Yu, Z., Haas, L. T., Kostylev, M. A., and Strittmatter, S. M. (2017). Early activation of experience-independent dendritic spine turnover in a mouse model of alzheimer's disease. Cereb. Cortex 27, 3660–3674. doi: 10.1093/cercor/bhw188
Hellwig, S., Heinrich, A., and Biber, K. (2013). The brain's best friend: microglial neurotoxicity revisited. Front. Cell. Neurosci. 7:71. doi: 10.3389/fncel.2013.00071
Hoffmann, N. A., Dorostkar, M. M., Blumenstock, S., Goedert, M., and Herms, J. (2013). Impaired plasticity of cortical dendritic spines in P301S tau transgenic mice. Acta Neuropathol. Commun. 1:82. doi: 10.1186/2051-5960-1-82
Holcomb, L., Gordon, M. N., McGowan, E., Yu, X., Benkovic, S., Jantzen, P., et al. (1998). Accelerated alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4, 97–100. doi: 10.1038/nm0198-097
Holscher, C. (1998). Possible causes of alzheimer's disease: amyloid fragments, free radicals, and calcium homeostasis. Neurobiol. Dis. 5, 129–141. doi: 10.1006/nbdi.1998.0193
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. doi: 10.1016/j.neuron.2010.11.030
Hsia, A. Y., Masliah, E., McConlogue, L., Yu, G. Q., Tatsuno, G., Hu, K., et al. (1999). Plaque-independent disruption of neural circuits in alzheimer's disease mouse models. Proc. Natl. Acad. Sci. U.S.A. 96, 3228–3233. doi: 10.1073/pnas.96.6.3228
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., et al. (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. doi: 10.1016/j.neuron.2006.10.035
Hussain, I., Powell, D., Howlett, D. R., Tew, D. G., Meek, T. D., Chapman, C., et al. (1999). Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 14, 419–427. doi: 10.1006/mcne.1999.0811
Jackson, J. S., Johnson, J. D., Meftah, S., Murray, T. K., Ahmed, Z., Fasiolo, M., et al. (2020). Differential aberrant structural synaptic plasticity in axons and dendrites ahead of their degeneration in tauopathy. bioRxiv [Preprint]. doi: 10.1101/2020.04.29.067629
Jackson, J. S., Witton, J., Johnson, J. D., Ahmed, Z., Ward, M., Randall, A. D., et al. (2017). Altered synapse stability in the early stages of tauopathy. Cell Rep. 18, 3063–3068. doi: 10.1016/j.celrep.2017.03.013
Jacobsen, J. S., Wu, C. C., Redwine, J. M., Comery, T. A., Arias, R., Bowlby, M., et al. (2006). Early-onset behavioral and synaptic deficits in a mouse model of alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5161–5166. doi: 10.1073/pnas.0600948103
Jankowsky, J. L., and Zheng, H. (2017). Practical considerations for choosing a mouse model of alzheimer's disease. Mol. Neurodegener. 12:89. doi: 10.1186/s13024-017-0231-7
Jaworski, T., Lechat, B., Demedts, D., Gielis, L., Devijver, H., Borghgraef, P., et al. (2011). Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am. J. Pathol. 179, 2001–2015. doi: 10.1016/j.ajpath.2011.06.025
Jay, T. R., von Saucken, V. E., Munoz, B., Codocedo, J. F., Atwood, B. K., Lamb, B. T., et al. (2019). TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 67, 1873–1892. doi: 10.1002/glia.23664
Kamphuis, W., Kooijman, L., Schetters, S., Orre, M., and Hol, E. M. (2016). Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for alzheimer's disease. Biochim. Biophys. Acta 1862, 1847–1860. doi: 10.1016/j.bbadis.2016.07.007
Kandalepas, P. C., Sadleir, K. R., Eimer, W. A., Zhao, J., Nicholson, D. A., and Vassar, R. (2013). The alzheimer's beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 126, 329–352. doi: 10.1007/s00401-013-1152-3
Kawahara, M. (2004). Disruption of calcium homeostasis in the pathogenesis of alzheimer's disease and other conformational diseases. Curr. Alzheimer Res. 1, 87–95. doi: 10.2174/1567205043332234
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of alzheimer's disease. Cell 169, 1276–1290.e1217. doi: 10.1016/j.cell.2017.05.018
King, D. L., and Arendash, G. W. (2002). Maintained synaptophysin immunoreactivity in Tg2576 transgenic mice during aging: correlations with cognitive impairment. Brain Res. 926, 58–68. doi: 10.1016/S0006-8993(01)03294-2
Knobloch, M., and Mansuy, I. M. (2008). Dendritic spine loss and synaptic alterations in alzheimer's disease. Mol. Neurobiol. 37, 73–82. doi: 10.1007/s12035-008-8018-z
Knott, G. W., Holtmaat, A., Wilbrecht, L., Welker, E., and Svoboda, K. (2006). Spine growth precedes synapse formation in the adult neocortex in vivo. Nat. Neurosci. 9, 1117–1124. doi: 10.1038/nn1747
Koenigsknecht, J., and Landreth, G. (2004). Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J. Neurosci. 24, 9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004
Koffie, R. M., Meyer-Luehmann, M., Hashimoto, T., Adams, K. W., Mielke, M. L., Garcia-Alloza, M., et al. (2009). Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U.S.A. 106, 4012–4017. doi: 10.1073/pnas.0811698106
Konur, S., and Ghosh, A. (2005). Calcium signaling and the control of dendritic development. Neuron 46, 401–405. doi: 10.1016/j.neuron.2005.04.022
Kopeikina, K. J., Polydoro, M., Tai, H. C., Yaeger, E., Carlson, G. A., Pitstick, R., et al. (2013a). Synaptic alterations in the rTg4510 mouse model of tauopathy. J. Comp. Neurol. 521, 1334–1353. doi: 10.1002/cne.23234
Kopeikina, K. J., Wegmann, S., Pitstick, R., Carlson, G. A., Bacskai, B. J., Betensky, R. A., et al. (2013b). Tau causes synapse loss without disrupting calcium homeostasis in the rTg4510 model of tauopathy. PLoS ONE 8:e80834. doi: 10.1371/journal.pone.0080834
Krabbe, G., Halle, A., Matyash, V., Rinnenthal, J. L., Eom, G. D., Bernhardt, U., et al. (2013). Functional impairment of microglia coincides with Beta-amyloid deposition in mice with alzheimer-like pathology. PLoS ONE 8:e60921. doi: 10.1371/journal.pone.0060921
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e569. doi: 10.1016/j.immuni.2017.08.008
Kremer, A., Maurin, H., Demedts, D., Devijver, H., Borghgraef, P., and Van Leuven, F. (2011). Early improved and late defective cognition is reflected by dendritic spines in Tau.P301L mice. J. Neurosci. 31, 18036–18047. doi: 10.1523/JNEUROSCI.4859-11.2011
Kuchibhotla, K. V., Goldman, S. T., Lattarulo, C. R., Wu, H. Y., Hyman, B. T., and Bacskai, B. J. (2008). Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59, 214–225. doi: 10.1016/j.neuron.2008.06.008
Lanz, T. A., Carter, D. B., and Merchant, K. M. (2003). Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis. 13, 246–253. doi: 10.1016/S0969-9961(03)00079-2
Lee, S., Varvel, N. H., Konerth, M. E., Xu, G., Cardona, A. E., Ransohoff, R. M., et al. (2010). CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two alzheimer's disease mouse models. Am. J. Pathol. 177, 2549–2562. doi: 10.2353/ajpath.2010.100265
Lerdkrai, C., Asavapanumas, N., Brawek, B., Kovalchuk, Y., Mojtahedi, N., Olmedillas Del Moral, M., et al. (2018). Intracellular Ca2+ stores control in vivo neuronal hyperactivity in a mouse model of alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 115, E1279–E1288. doi: 10.1073/pnas.1714409115
Liebscher, S., Page, R. M., Kafer, K., Winkler, E., Quinn, K., Goldbach, E., et al. (2014). Chronic gamma-secretase inhibition reduces amyloid plaque-associated instability of pre- and postsynaptic structures. Mol. Psychiatry 19, 937–946. doi: 10.1038/mp.2013.122
Lin, X., Koelsch, G., Wu, S., Downs, D., Dashti, A., and Tang, J. (2000). Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 97, 1456–1460. doi: 10.1073/pnas.97.4.1456
Maia, L. F., Kaeser, S. A., Reichwald, J., Hruscha, M., Martus, P., Staufenbiel, M., et al. (2013). Changes in amyloid-beta and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci. Transl. Med. 5:194re192. doi: 10.1126/scitranslmed.3006446
Mandrekar, S., Jiang, Q., Lee, C. Y., Koenigsknecht-Talboo, J., Holtzman, D. M., and Landreth, G. E. (2009). Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 29, 4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009
Mattson, M. P. (2007). Calcium and neurodegeneration. Aging Cell 6, 337–350. doi: 10.1111/j.1474-9726.2007.00275.x
Mattson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., and Rydel, R. E. (1992). β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 12, 376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992
McLellan, M. E., Kajdasz, S. T., Hyman, B. T., and Bacskai, B. J. (2003). In vivo imaging of reactive oxygen species specifically associated with thioflavine S-positive amyloid plaques by multiphoton microscopy. J. Neurosci. 23, 2212–2217. doi: 10.1523/JNEUROSCI.23-06-02212.2003
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., de Calignon, A., Rozkalne, A., et al. (2008). Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of alzheimer's disease. Nature 451, 720–724. doi: 10.1038/nature06616
Mizrahi, A., Crowley, J. C., Shtoyerman, E., and Katz, L. C. (2004). High-resolution in vivo imaging of hippocampal dendrites and spines. J. Neurosci. 24, 3147–3151. doi: 10.1523/JNEUROSCI.5218-03.2004
Moechars, D., Dewachter, I., Lorent, K., Reverse, D., Baekelandt, V., Naidu, A., et al. (1999). Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 274, 6483–6492. doi: 10.1074/jbc.274.10.6483
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000
Onorato, M., Mulvihill, P., Connolly, J., Galloway, P., Whitehouse, P., and Perry, G. (1989). Alteration of neuritic cytoarchitecture in alzheimer disease. Prog. Clin. Biol. Res. 317, 781–789.
Overmyer, M., Helisalmi, S., Soininen, H., Laakso, M., Riekkinen, P. Sr., and Alafuzoff, I. (1999). Reactive microglia in aging and dementia: an immunohistochemical study of postmortem human brain tissue. Acta Neuropathol. 97, 383–392. doi: 10.1007/s004010051002
Paolicelli, R. C., Bisht, K., and Tremblay, M. E. (2014). Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell. Neurosci. 8:129. doi: 10.3389/fncel.2014.00129
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III, Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Perlmutter, L. S., Barron, E., and Chui, H. C. (1990). Morphologic association between microglia and senile plaque amyloid in alzheimer's disease. Neurosci. Lett. 119, 32–36. doi: 10.1016/0304-3940(90)90748-X
Peters, F., Salihoglu, H., Pratsch, K., Herzog, E., Pigoni, M., Sgobio, C., et al. (2019). Tau deletion reduces plaque-associated BACE1 accumulation and decelerates plaque formation in a mouse model of alzheimer's disease. EMBO J. 38:e102345. doi: 10.15252/embj.2019102345
Peters, F., Salihoglu, H., Rodrigues, E., Herzog, E., Blume, T., Filser, S., et al. (2018). BACE1 inhibition more effectively suppresses initiation than progression of beta-amyloid pathology. Acta Neuropathol. 135, 695–710. doi: 10.1007/s00401-017-1804-9
Popugaeva, E., Pchitskaya, E., and Bezprozvanny, I. (2017). Dysregulation of neuronal calcium homeostasis in alzheimer's disease - a therapeutic opportunity? Biochem. Biophys. Res. Commun. 483, 998–1004. doi: 10.1016/j.bbrc.2016.09.053
Pozueta, J., Lefort, R., and Shelanski, M. L. (2013). Synaptic changes in alzheimer's disease and its models. Neuroscience 251, 51–65. doi: 10.1016/j.neuroscience.2012.05.050
Richardson, J. A., and Burns, D. K. (2002). Mouse models of alzheimer's disease: a quest for plaques and tangles. ILAR J. 43, 89–99. doi: 10.1093/ilar.43.2.89
Rocher, A. B., Crimins, J. L., Amatrudo, J. M., Kinson, M. S., Todd-Brown, M. A., Lewis, J., et al. (2010). Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp. Neurol. 223, 385–393. doi: 10.1016/j.expneurol.2009.07.029
Rogers, J., Luber-Narod, J., Styren, S. D., and Civin, W. H. (1988). Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of alzheimer's disease. Neurobiol. Aging 9, 339–349. doi: 10.1016/S0197-4580(88)80079-4
Rutten, B. P., Van der Kolk, N. M., Schafer, S., van Zandvoort, M. A., Bayer, T. A., Steinbusch, H. W., et al. (2005). Age-related loss of synaptophysin immunoreactive presynaptic boutons within the hippocampus of APP751SL, PS1M146L, and APP751SL/PS1M146L transgenic mice. Am. J. Pathol. 167, 161–173. doi: 10.1016/S0002-9440(10)62963-X
Rutten, B. P., Wirths, O., Van de Berg, W. D., Lichtenthaler, S. F., Vehoff, J., Steinbusch, H. W., et al. (2003). No alterations of hippocampal neuronal number and synaptic bouton number in a transgenic mouse model expressing the beta-cleaved C-terminal APP fragment. Neurobiol. Dis. 12, 110–120. doi: 10.1016/S0969-9961(02)00015-3
Sadleir, K. R., Kandalepas, P. C., Buggia-Prevot, V., Nicholson, D. A., Thinakaran, G., and Vassar, R. (2016). Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in alzheimer's disease. Acta Neuropathol. 132, 235–256. doi: 10.1007/s00401-016-1558-9
Savage, J. C., Jay, T., Goduni, E., Quigley, C., Mariani, M. M., Malm, T., et al. (2015). Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of alzheimer's disease. J. Neurosci. 35, 6532–6543. doi: 10.1523/JNEUROSCI.4586-14.2015
Savage, J. C., and Tremblay, M. E. (2019). Studying laboratory mice - into the wild. Trends Neurosci. 42, 566–568. doi: 10.1016/j.tins.2019.05.004
Scheff, S. W., and Price, D. A. (2006). Alzheimer's disease-related alterations in synaptic density: neocortex and hippocampus. J. Alzheimers. Dis. 9, 101–115. doi: 10.3233/JAD-2006-9S312
Scheff, S. W., Price, D. A., Schmitt, F. A., DeKosky, S. T., and Mufson, E. J. (2007). Synaptic alterations in CA1 in mild alzheimer disease and mild cognitive impairment. Neurology 68, 1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f
Schmid, L. C., Mittag, M., Poll, S., Steffen, J., Wagner, J., Geis, H. R., et al. (2016). Dysfunction of somatostatin-positive interneurons associated with memory deficits in an alzheimer's disease model. Neuron 92, 114–125. doi: 10.1016/j.neuron.2016.08.034
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Shahani, N., Subramaniam, S., Wolf, T., Tackenberg, C., and Brandt, R. (2006). Tau aggregation and progressive neuronal degeneration in the absence of changes in spine density and morphology after targeted expression of alzheimer's disease-relevant tau constructs in organotypic hippocampal slices. J. Neurosci. 26, 6103–6114. doi: 10.1523/JNEUROSCI.4245-05.2006
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007
Shrestha, B. R., Vitolo, O. V., Joshi, P., Lordkipanidze, T., Shelanski, M., and Dunaevsky, A. (2006). Amyloid beta peptide adversely affects spine number and motility in hippocampal neurons. Mol. Cell. Neurosci. 33, 274–282. doi: 10.1016/j.mcn.2006.07.011
Sinha, S., Anderson, J. P., Barbour, R., Basi, G. S., Caccavello, R., Davis, D., et al. (1999). Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537–540. doi: 10.1038/990114
Small, D. H. (2009). Dysregulation of calcium homeostasis in alzheimer's disease. Neurochem. Res. 34, 1824–1829. doi: 10.1007/s11064-009-9960-5
Smith, I. F., Green, K. N., and LaFerla, F. M. (2005). Calcium dysregulation in alzheimer's disease: recent advances gained from genetically modified animals. Cell Calcium 38, 427–437. doi: 10.1016/j.ceca.2005.06.021
Spires, T. L., Meyer-Luehmann, M., Stern, E. A., McLean, P. J., Skoch, J., Nguyen, P. T., et al. (2005). Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 25, 7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005
Spires-Jones, T., and Knafo, S. (2012). Spines, plasticity, and cognition in alzheimer's model mice. Neural Plast 2012:319836. doi: 10.1155/2012/319836
Spires-Jones, T. L., de Calignon, A., Meyer-Luehmann, M., Bacskai, B. J., and Hyman, B. T. (2011). Monitoring protein aggregation and toxicity in alzheimer's disease mouse models using in vivo imaging. Methods 53, 201–207. doi: 10.1016/j.ymeth.2010.12.009
Spires-Jones, T. L., Meyer-Luehmann, M., Osetek, J. D., Jones, P. B., Stern, E. A., Bacskai, B. J., et al. (2007). Impaired spine stability underlies plaque-related spine loss in an alzheimer's disease mouse model. Am. J. Pathol. 171, 1304–1311. doi: 10.2353/ajpath.2007.070055
Stephen, T. L., Tamagnini, F., Piegsa, J., Sung, K., Harvey, J., Oliver-Evans, A., et al. (2019). Imbalance in the response of pre- and post-synaptic components to amyloidopathy. Sci. Rep. 9:14837. doi: 10.1038/s41598-019-50781-1
St-Pierre, M. K., Simoncicova, E., Bogi, E., and Tremblay, M. E. (2020). Shedding light on the dark side of the microglia. ASN Neuro 12:1759091420925335. doi: 10.1177/1759091420925335
Stratoulias, V., Venero, J. L., Tremblay, M. E., and Joseph, B. (2019). Microglial subtypes: diversity within the microglial community. EMBO J. 38:e101997. doi: 10.15252/embj.2019101997
Subramanian, J., Michel, K., Benoit, M., and Nedivi, E. (2019). CPG15/neuritin mimics experience in selecting excitatory synapses for stabilization by facilitating PSD95 recruitment. Cell Rep. 28, 1584–1595.e1585. doi: 10.1016/j.celrep.2019.07.012
Sze, C. I., Troncoso, J. C., Kawas, C., Mouton, P., Price, D. L., and Martin, L. J. (1997). Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in alzheimer disease. J. Neuropathol. Exp. Neurol. 56, 933–944. doi: 10.1097/00005072-199708000-00011
Takasaki, K., Abbasi-Asl, R., and Waters, J. (2020). Superficial bound of the depth limit of two-photon imaging in mouse brain. eNeuro 7:ENEURO.0255-19.2019. doi: 10.1523/ENEURO.0255-19.2019
Tampellini, D., Capetillo-Zarate, E., Dumont, M., Huang, Z., Yu, F., Lin, M. T., et al. (2010). Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in alzheimer's disease transgenic mice. J. Neurosci. 30, 14299–14304. doi: 10.1523/JNEUROSCI.3383-10.2010
Tay, T. L., Savage, J. C., Hui, C. W., Bisht, K., and Tremblay, M. E. (2017). Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J. Physiol. 595, 1929–1945. doi: 10.1113/JP272134
Terry, R. D. (2000). Cell death or synaptic loss in alzheimer disease. J. Neuropathol. Exp. Neurol. 59, 1118–1119. doi: 10.1093/jnen/59.12.1118
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Trachtenberg, J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E., et al. (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794. doi: 10.1038/nature01273
Tremblay, M. E., Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Tsai, J., Grutzendler, J., Duff, K., and Gan, W. B. (2004). Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 7, 1181–1183. doi: 10.1038/nn1335
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). Beta-secretase cleavage of alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Villa, K. L., Berry, K. P., Subramanian, J., Cha, J. W., Oh, W. C., Kwon, H. B., et al. (2016). Inhibitory synapses are repeatedly assembled and removed at persistent sites in vivo. Neuron 89, 756–769. doi: 10.1016/j.neuron.2016.01.010
Wendt, S., Maricos, M., Vana, N., Meyer, N., Guneykaya, D., Semtner, M., et al. (2017). Changes in phagocytosis and potassium channel activity in microglia of 5xFAD mice indicate alterations in purinergic signaling in a mouse model of alzheimer's disease. Neurobiol. Aging 58, 41–53. doi: 10.1016/j.neurobiolaging.2017.05.027
Wirths, O., and Bayer, T. A. (2010). Neuron loss in transgenic mouse models of alzheimer's disease. Int. J. Alzheimers. Dis. 2010:723782. doi: 10.4061/2010/723782
Wisniewski, H. M., Wegiel, J., Wang, K. C., Kujawa, M., and Lach, B. (1989). Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can. J. Neurol. Sci. 16, 535–542. doi: 10.1017/S0317167100029887
Wisniewski, T., and Sigurdsson, E. M. (2010). Murine models of alzheimer's disease and their use in developing immunotherapies. Biochim. Biophys. Acta 1802, 847–859. doi: 10.1016/j.bbadis.2010.05.004
Wu, H. Y., Hudry, E., Hashimoto, T., Kuchibhotla, K., Rozkalne, A., Fan, Z., et al. (2010). Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 30, 2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010
Xie, H., Guan, J., Borrelli, L. A., Xu, J., Serrano-Pozo, A., and Bacskai, B. J. (2013). Mitochondrial alterations near amyloid plaques in an alzheimer's disease mouse model. J. Neurosci. 33, 17042–17051. doi: 10.1523/JNEUROSCI.1836-13.2013
Yan, P., Bero, A. W., Cirrito, J. R., Xiao, Q., Hu, X., Wang, Y., et al. (2009). Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J. Neurosci. 29, 10706–10714. doi: 10.1523/JNEUROSCI.2637-09.2009
Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., et al. (1999). Membrane-anchored aspartyl protease with alzheimer's disease beta-secretase activity. Nature 402, 533–537. doi: 10.1038/990107
Yin, Z., Raj, D., Saiepour, N., Van Dam, D., Brouwer, N., Holtman, I. R., et al. (2017). Immune hyperreactivity of Abeta plaque-associated microglia in alzheimer's disease. Neurobiol. Aging 55, 115–122. doi: 10.1016/j.neurobiolaging.2017.03.021
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010
Yu, W., and Lu, B. (2012). Synapses and dendritic spines as pathogenic targets in alzheimer's disease. Neural Plast 2012:247150. doi: 10.1155/2012/247150
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T. D., Paul, S. M., et al. (2016). TREM2 Haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. doi: 10.1016/j.neuron.2016.05.003
Zhang, X. M., Cai, Y., Xiong, K., Cai, H., Luo, X. G., Feng, J. C., et al. (2009). Beta-secretase-1 elevation in transgenic mouse models of alzheimer's disease is associated with synaptic/axonal pathology and amyloidogenesis: implications for neuritic plaque development. Eur. J. Neurosci. 30, 2271–2283. doi: 10.1111/j.1460-9568.2009.07017.x
Zhao, R., Hu, W., Tsai, J., Li, W., and Gan, W. B. (2017). Microglia limit the expansion of beta-amyloid plaques in a mouse model of alzheimer's disease. Mol. Neurodegener. 12:47. doi: 10.1186/s13024-017-0188-6
Ziegler-Waldkirch, S., d'Errico, P., Sauer, J. F., Erny, D., Savanthrapadian, S., Loreth, D., et al. (2018). Seed-induced Abeta deposition is modulated by microglia under environmental enrichment in a mouse model of alzheimer's disease. EMBO J. 37, 167–182. doi: 10.15252/embj.201797021
Zou, C., Montagna, E., Shi, Y., Peters, F., Blazquez-Llorca, L., Shi, S., et al. (2015). Intraneuronal APP and extracellular Abeta independently cause dendritic spine pathology in transgenic mouse models of alzheimer's disease. Acta Neuropathol. 129, 909–920. doi: 10.1007/s00401-015-1421-4
Keywords: dendritic spines, microglia, two-photon, in vivo imaging, amyloid mouse models
Citation: Subramanian J, Savage JC and Tremblay M-È (2020) Synaptic Loss in Alzheimer's Disease: Mechanistic Insights Provided by Two-Photon in vivo Imaging of Transgenic Mouse Models. Front. Cell. Neurosci. 14:592607. doi: 10.3389/fncel.2020.592607
Received: 07 August 2020; Accepted: 25 November 2020;
Published: 17 December 2020.
Edited by:
Carole Escartin, UMR9199 Laboratory of Neurodegenerative Diseases Mechanisms, Therapies, Imaging, FranceReviewed by:
Jochen Herms, German Center for Neurodegenerative Diseases (DZNE) and Zentrum fur Neuropathologie (ZNP) Ludwig-Maximilians-University Munich, GermanyEstibaliz Capetillo-Zarate, University of the Basque Country, Spain
Copyright © 2020 Subramanian, Savage and Tremblay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaichandar Subramanian, amFpY2hhbmRhckBrdS5lZHU=; Marie-Ève Tremblay, ZXZldHJlbWJsYXlAdXZpYy5jYQ==
†Present address: Julie C. Savage, Convelo Therapeutics, Cleveland, OH, United States