 Nikolas Layer1
Nikolas Layer1 Lukas Sonnenberg2
Lukas Sonnenberg2 Emilio Pardo González1
Emilio Pardo González1 Jan Benda2,3
Jan Benda2,3 Ulrike B. S. Hedrich1
Ulrike B. S. Hedrich1 Holger Lerche1
Holger Lerche1 Henner Koch1,4
Henner Koch1,4 Thomas V. Wuttke1,5*
Thomas V. Wuttke1,5*- 1Department of Neurology and Epileptology, Hertie Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany
- 2Institute for Neurobiology, Eberhard Karls University Tübingen, Tübingen, Germany
- 3Bernstein Center for Computational Neuroscience, Eberhard Karls Universitat, Tübingen, Germany
- 4Department of Epileptology, Neurology, Rheinisch-Westfälische Technische Hochschule Aachen, Aachen, Germany
- 5Department of Neurosurgery, University of Tübingen, Tübingen, Germany
Dravet syndrome (DS) is a developmental epileptic encephalopathy mainly caused by functional NaV1.1 haploinsufficiency in inhibitory interneurons. Recently, a new conditional mouse model expressing the recurrent human p.(Ala1783Val) missense variant has become available. In this study, we provided an electrophysiological characterization of this variant in tsA201 cells, revealing both altered voltage-dependence of activation and slow inactivation without reduced sodium peak current density. Based on these data, simulated interneuron (IN) firing properties in a conductance-based single-compartment model suggested surprisingly similar firing deficits for NaV1.1A1783V and full haploinsufficiency as caused by heterozygous truncation variants. Impaired NaV1.1A1783V channel activation was predicted to have a significantly larger impact on channel function than altered slow inactivation and is therefore proposed as the main mechanism underlying IN dysfunction. The computational model was validated in cortical organotypic slice cultures derived from conditional Scn1aA1783V mice. Pan-neuronal activation of the p.Ala1783V in vitro confirmed a predicted IN firing deficit and revealed an accompanying reduction of interneuronal input resistance while demonstrating normal excitability of pyramidal neurons. Altered input resistance was fed back into the model for further refinement. Taken together these data demonstrate that primary loss of function (LOF) gating properties accompanied by altered membrane characteristics may match effects of full haploinsufficiency on the neuronal level despite maintaining physiological peak current density, thereby causing DS.
Introduction
Dravet syndrome (DS) is a developmental epileptic encephalopathy (DEE) with early childhood febrile seizures followed by a high-frequency seizure period with a large diversity of seizure types (Dravet, 1978). The epilepsy phenotype is accompanied by a multitude of comorbidities, including intellectual disability, sleep disorder, motor dysfunction, and an increased incidence of sudden unexpected death in epilepsy. More than 80% of diagnosed Dravet cases are caused by de-novo loss of function (LOF) variants causing functional haploinsufficiency of the SCN1A gene coding for the alpha-subunit of the voltage-gated sodium channel NaV1.1 (Marini et al., 2011). SCN1A variants are associated with multiple neurological diseases including generalized epilepsy with febrile seizures plus, sporadic/familial hemiplegic migraine, and DS resembling a clinical phenotype at the severe end of the spectrum (Gambardella and Marini, 2009). The high diversity of SCN1A associated disorders can be explained by a variety of gain and loss of function variants (Catterall et al., 2010), which impair the channel on different levels including transcriptional changes (Lange et al., 2019), reduced protein expression, altered membrane trafficking (Thompson et al., 2012), impaired ß-subunit interaction (Spampanato et al., 2004), and channel gating dysfunction (Kluckova et al., 2020).
In the past years, different cell cultures and animal models have been established based on patient variants to explore the different pathomechanisms of NaV1.1. Most frequently, heterozygous NaV1.1 knockout mice, mimicking protein-truncating non-sense variants found in patients with DS, have been used to investigate pathophysiological disease mechanisms (Yu et al., 2006; Ogiwara et al., 2007; Mistry et al., 2014; Favero et al., 2018). Such studies revealed a predominant role of LOF effects in certain inhibitory neuron classes including fast-spiking interneurons (FS-INs) (Bechi et al., 2012; Tai et al., 2014; Rubinstein et al., 2015; Favero et al., 2018; Tiraboschi et al., 2020; Tran et al., 2020). However, non-sense mutations only account for a subset of pathogenic variants leading to epileptic phenotypes. Especially milder epilepsy syndromes are often caused by missense variants within the protein-coding sequence and were studied in IPSC-derived neurons (Xie et al., 2020) and mouse models (Hedrich et al., 2014; Das et al., 2021). However, DS may also be caused by missense variants (Marini et al., 2011). Although protein truncation (non-sense variants) or diminished surface expression (certain missense variants) result in a pronounced reduction of sodium peak current, the mechanisms of remaining missense variants can be more complex, as physiological gating properties of channels may be affected on multiple levels. Although full haploinsufficiency is modeled well by heterozygous Scn1a knockout mice or knockin of a truncating variant (Yu et al., 2006; Ogiwara et al., 2007), they may not generally reflect the disease status of patients with DS for carriers of missense variants. To approach this uncertainty, conditional mice expressing the recurrent human Dravet missense variant p. Ala1783Val (A1783V) (Depienne et al., 2009; Klassen et al., 2014) have recently been developed by the Dravet Syndrome European Federation (Ricobaraza et al., 2019). A limited number of studies provide the first insight into the effects of mutant Scn1aA1783V channels on neuron excitability after breeding conditional Scn1aA1783V mice with Cre-driver lines. Depending on the exact genetic background of offspring animals, the subtype of studied neurons and the brain areas in which neurons were recorded, and the differential effects on neuron firing have been described (Kuo et al., 2019; Almog et al., 2021). Although LOF of NaV1.1A1783V has been assumed, the biophysical effects of the variant on NaV1.1 channel function have not yet been delineated to date. In this study, we provided a detailed comparative characterization of variant p.(Ala1783Val) and wild type (WT) gating properties in tsA201 cells. Although full haploinsufficiency with reduced Na+ peak currents is a common feature in DS, our recordings revealed clear LOF mechanisms, albeit with preserved overall sodium peak current density. Building on these data, a single-compartment computational neuron model was developed to predict firing deficits in inhibitory and excitatory neurons for p.Ala1783Val in comparison to full haploinsufficiency by heterozygous knockout. The model was electrophysiologically validated in cortical organotypic brain slice cultures derived from Scn1aA1783V mice. Interestingly, despite overall normal sodium peak current density, p.(Ala1783Val) was predicted to cause a fairly strong Interneuron (IN) firing deficit comparable to the heterozygous knockout condition. As for numerous other channelopathies, correlations of the nature of SCN1A variants (such as location in the channel, genetic mechanism, and associated impact on encoded protein function) with disease severity have been described (Zuberi et al., 2011). Our data demonstrate that primarily altered channel gating and accompanying changes in membrane properties can outweigh maintained physiological sodium peak current density and translate into pronounced functional impairment on neuronal level as frequently associated with DS.
Methods
Mutagenesis
The human SCN1A sequence cloned in the pCDM8 vector as described earlier (Hedrich et al., 2014) was corrected for two-point mutations, namely, Glu650Val and Ser1969Ala (Peters et al., 2016) by site-directed mutagenesis (Agilent Technologies, Santa Clara, CA, USA). The WT open reading frame included the canonical SCN1A adult isoform 2 (total length of 5997bp) equivalent to transcript NM_006920.5. The missense variant p.(Ala1783Val) was engineered into the human NaV1.1 channel by site-directed mutagenesis using PCR with Pfu polymerase (Promega; mutagenesis primers 5′ to 3′; F: ATG TAC ATC GTG GTC ATC CTG GAG AAC TTC AGT, R: AGG ATG ACC ACG ATG TAC ATG TTC ACC ACA). The introduced variant was verified, and further variants were excluded by sequencing the whole SCN1A cDNA. Plasmid purification was performed using Escherichia coli One Shot TOP10/P3 (Thermo Scientific, Waltham, MA, USA).
tsA201 Cell Culture and Transfection
tsA201 cells were cultured in the Dulbecco's modified Eagle nutrient medium (Thermo Scientific, Waltham, MA, USA) supplemented with pyruvate, 10% v/v fetal bovine serum (Pan Biotech, Aidenbach, Germany), and 2 mM L-glutamine (Merck Millipore, Burlington, MA, USA) at 37°C in a 5% CO2 humidified atmosphere. For transfection, cells with a passage from P15 to P22 were used. Before transfection, 800,000–1,000,000 cells were split in 35-mm Petri dishes. Six hours later, cells were transfected by the following standard protocols: 4 μg of WT or mutant SCN1A cDNA, encoding the NaV1.1 channel-α-subunit, and 0.4 μg each of the human β1- and β2-subunits of voltage-gated NaV channels, which had been previously modified to express either green fluorescent protein (pCLH-hb1-EGFP) or a CD8 marker (pCLH-hb2-CD8) to label cells expressing both subunits (Liao et al., 2010), were added to 250 μl of Opti-MEM (Gibco) supplemented with 7.5 μl of Mirus TransIT reagent (Mirus Bio LLC, Madison, WI, USA). After 20 min, the transfection mixture was added to the cells.
Cortical Brain Slice Cultures
Coronal brain slices from conditional B6(Cg)-Scn1atm1.1Dsf/J mice (JAX stock #026133) of either sex were obtained at P4-5. Mice were quickly decapitated, and cranium, brain stem, and hindbrain were removed. The forebrain was placed in ice-cold artificial cerebrospinal fluid (aCSF) bubbled with carbogen (95% O2/5% CO2). The aCSF contained the following [in mM]: 118 NaCl, 3 KCl, 1.5 CaCl2, 1 MgCl2, 25 NaHCO3, 1 NaH2PO4, 30 glucose, and pH 7.4 with an osmolarity of 310–320 mOsm/kg. Forebrains were glued with the cerebellar end facing downward on an agar block, and 350 μm thick slices were cut using a vibratome (Microm, H650 V) in ice-cold aCSF. Slices were placed in 36°C warm aCSF (bubbled with carbogen) for 10 min. Coronal slices containing the somatosensory cortex were transferred to Millicell cell culture inserts (Merck Millipore, Burlington, MA, USA) floating on the slice culture medium (Minimum Essential Medium Eagle with 20% horse serum, 1 mM L-glutamine, 0.00125% ascorbic acid, 0.001 mg/ml insulin, 1 mM CaCl2, 2 mM MgSO4, 1% penicillin/streptomycin, 13 mM glucose, pH 7.28, and osmolarity 320 mOsm/kg).
Electrophysiological Recordings in tsA Cells and Slice Cultures
Whole-cell voltage-clamp recordings of tsA201 cells were performed 48 h after transfection from cells expressing all three sodium channel subunits indicated by an inward sodium current (α-subunit), anti-CD8 antibody-coated microbeads (Dynabeads M450, Dynal) (β1-subunit), and green fluorescence (β2-subunit). Cells were split in 35-mm Petri dishes 1 h prior to recordings, and each dish was used for up to 1 h after transfer to the patch-clamp setup. The extracellular bath solution contained the following [in mM]: 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 5 HEPES, and 4 glucose. pH was adjusted with HCl to 7.4, and osmolarity was 300–305 mOsm/kg. Patch pipettes were pulled from borosilicate glass (Science Products GmbH) using a P97 Puller (Sutter Instruments), with resistances of 1.5–3.0 MΩ. Intracellular solutions for patch pipettes contained the following [in mM]: 5 NaCl, 2 MgCl2, 5 EGTA, 10 HEPES, 130 CsF, pH 7.4 (adjusted with CsOH), and osmolarity 290–295 mOsm/kg.
Whole-cell current-clamp recordings of excitatory pyramidal cells (PCs) and FS-INs in the somatosensory cortex of organotypic cortical slice cultures of conditional or WT animals were performed 7–14 days after adeno-associated viral transduction with 1 μl/slice of AAV8-hSyn-Cre-GFP (1 × 1013 VG/ml; SignaGen Laboratories, Frederick, MD, USA). Slices were positioned in a submerged-type recording chamber (Luigs & Neumann, Ratingen, Germany), continuously superfused with oxygenated recording aCSF, and maintained at a temperature of 33 ± 1°C. Transduced neurons were visualized using an Axioskop 2FS microscope (Carl Zeiss, Oberkochen, Germany). Patch pipettes had resistances of 2.5–5.5 MΩ. Intracellular solutions contained the following [in mM]: 140 K-gluconate, 1 CaCl2, 10 EGTA, 2 MgCl2, 4 Na2-ATP, 10 HEPES, 0.45% biocytin (Sigma-Aldrich, B4261), pH 7.2, and osmolarity of 300–310 mOsm/kg. PCs and FS-INs were used for current-clamp recordings. Cell identity was determined via morphological and electrophysiological properties. To block postsynaptic AMPA receptor-driven depolarization waves, 20 μM CNQX (Sigma-Aldrich, St. Louis, MO, USA) was added to the bath solution before recording.
Immunohistological Stainings of Cortical Slice Cultures and Imaging
Following recordings, slices were fixed for 24 h in 4% paraformaldehyde (Morphisto GmbH, Offenbach am Main, Germany), washed in DPBS (Thermo Fisher Scientific, Waltham, MA, USA, Cat. 12559069), and blocked with 1% normal goat serum with 0.2% Triton X-100 for 2 h. AAV-positive cells were stained with rabbit anti-GFP IgG primary antibody (dilution 1:1,000, Invitrogen, Cat. A-11122) overnight at 4°C. Subsequently, slices were washed in DPBS and counterstained with goat anti-rabbit IgG secondary antibody Alexa Fluor-488 (dilution 1:200, Invitrogen, Cat. A-11008) and Streptavidin-Cy3 (dilution 1:100, Sigma, Cat. S6402) for 3 h at room temperature. After washing, slices were mounted with DAPI Fluoromount-G (Southern Biotech, Birmingham, AL, USA, Cat. 011-20). Immunohistological images of stained slices were acquired on a Zeiss Axiovert 200M microscope (widefield images) and a Leica SP8 microscope (confocal images).
In silico Modeling of Cortical Neurons/Computational Modeling
Simulations were run using custom Python 3.7 software. A single-compartment conductance-based model with a cylindrical shape with length L and diameter d was used. The model is based on Pospischil et al. (2008) and consists of two sodium currents (INa,wt and INa,mut), a delayed rectifier potassium current IK, a M-type potassium current IM, and a leak current Ileak:
with membrane capacitance , input current Iinput, maximal conductance gi, reversal potential Ei, gating variables m, h, n, and p with dynamics
and the slow inactivating gating variable s with
where ẋi is the derivative of the gating parameter with respect to time, αi(V) is the opening rate, and βi(V) is the closing rate of the respective gate xi. Steady-state curves and time constants are given by x∞, i(V) = αi(V)/[αi(V) + βi(V)] and τi(V) = 1/[αi(V) + βi(V)]. The dynamics of the slow inactivating gating variable s followed
As suggested in the study by Pospischil et al. (2008), parameters for gating variables m, h, and n were taken from Traub and Miles (1991) and for gating variable p from Yamada (1989). We constructed the slow inactivation gate s to be similar to observed kinetics as suggested by Vilin and Ruben (2001). To simulate different neuron types, the conductivities of the ionic currents were adapted from the study by Pospischil et al. (2008). All parameters that differ from the study by Pospischil et al. (2008) are summarized in Table 1. Effects of variants were simulated with changes in the shifting parameter shifti for the activation and slow inactivation sodium gates with shifti = 0 mv unless stated otherwise.

Table 1. Simulation parameters for the cortical neuron models.
To simulate the effects of heterozygous variants in SCN1A on the firing behavior of PCs and interneurons, different ratios of NaV1.1 and NaV1.6 expression were modeled in the respective neuron types with the assumption of equal gating properties in both WT sodium channels (gNa,wt = gNaV1.1/2 + gNaV1.6) and potentially altered NaV1.1 channels (gNa,mut = gNaV1.1/2). The ratio of assumed sodium conductance was gNaV1.6 = 5gNaV1.1 for pyramidal neurons and gNaV1.6 = 0.8gNaV1.1 for FS-INs based on expression data from cortical mouse neurons (Yao et al., 2020). To investigate how each observed change in gating parameters contributes to the observed change in firing behavior, we constructed several variant models, one combined variant model with all parameter changes, and for each adapted parameter, one model with only one respective change.
The excitability of these models was computed with the f−I curve. We simulated square pulse stimuli between 0 and 500 pA with increments of 5 pA. Action potential (AP) frequency of each step was computed by dividing the number of measured spikes, reduced by 1, with the time difference between the first and the last spike:
Statistical Analysis
All data are displayed as mean ± SEM. Statistical testing was performed via unpaired, two-tailed t-test for normally distributed data and Mann–Whitney U test for non-normally distributed data and n indicates the number of cells. Data normality was tested by using the D'Agostino and Pearson normality test. Mean-, n- and p-values are shown in Supplementary Tables 1–3.
All data were analyzed using pClamp 10.703 (Molecular Devices, LLC, San Jose, CA, USA), Microsoft Excel (Microsoft Corporation, Redmond, WA, USA). Statistics were performed using GraphPad 7 (GraphPad Prism, San Diego, CA, USA).
Results
Biophysical Characterization of SCN1AA1783V Channel Function in tsA201 Cells Reveals LOF With Maintained Peak Sodium Currents
Wild-type or mutant NaV1.1 channels were transfected into tsA201 cells, and whole-cell patch recordings were performed. Representative raw current traces are shown in Figure 1A. While peak current density was not changed for mutant channels in comparison to WT (Figure 1B), activation and inactivation properties were markedly altered. We found a significant right shift of the half-maximal conductance indicating that mutant channels open and reach their maximal activation at more depolarized potentials (Figure 1C). Fast inactivation was unaffected as revealed by comparable voltage-dependence of steady-state fast inactivation (Figure 1C), time constant τ (Figure 1E), and recovery from fast inactivation (Figure 1D) for WT and NaV1.1A1783V channels. In contrast, entry into slow inactivation was accelerated (Figure 1F), and voltage-dependence was shifted to more hyperpolarized potentials (Figure 1G). Overall, NaV1.1A1783V channel LOF became very obvious on rapid successive depolarizing stimulations at 40 Hz revealing a markedly increased use-dependence with consecutively pronounced “rundown” of the sodium current (Figure 1H).
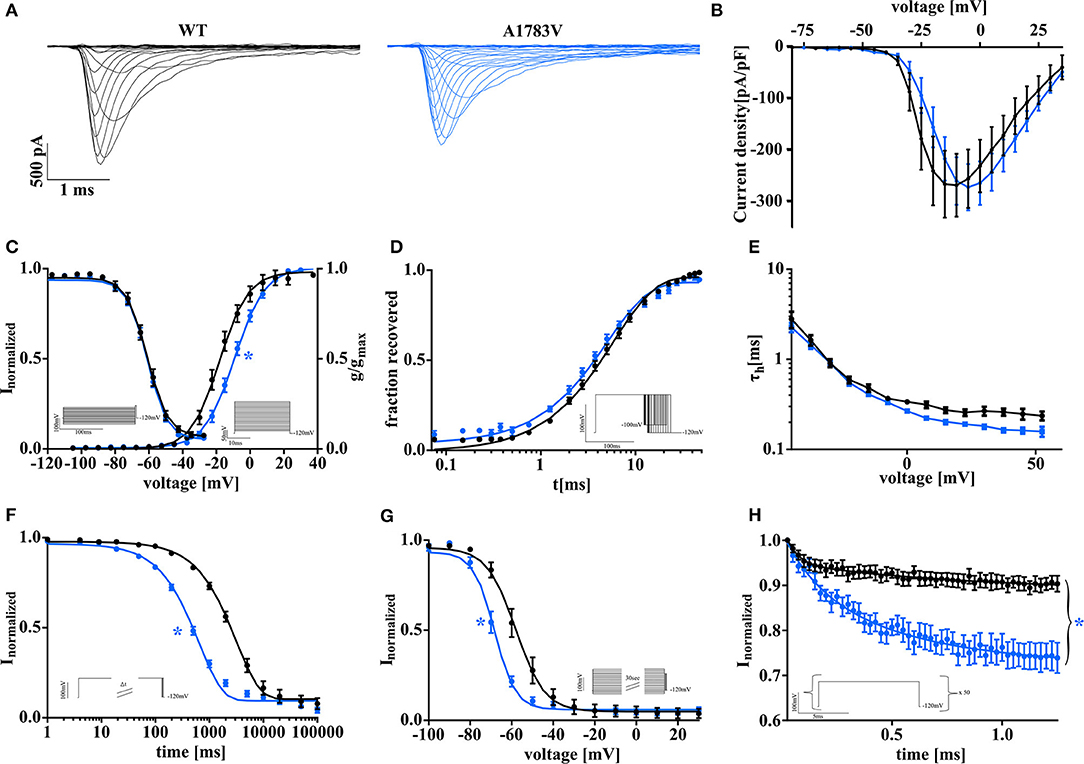
Figure 1. Biophysical properties of SCN1A wild type and mutant NaV1.1A1783V channels recorded in tsA201 cells display a clear loss of function. (A) Representative current traces were recorded from tsA201 cells transfected with NaV1.1WT (black traces) or NaV1.1A1783V (blue traces). (B) Peak Na+ currents normalized by cell capacitance were plotted vs. command voltage and revealed no changes between wild-type (WT) and mutant NaV1.1A1783V channels. (C, right) Voltage-dependence of steady-state activation curves represented as normalized conductance fit by a Boltzmann function to the data points (lines). (C, left curve) Voltage-dependent state of fast inactivation fit by a Boltzmann function to the data points (lines). (D) Time course of recovery from fast inactivation at −100 mV. (E) Voltage-dependence of the time constant of fast inactivation τh analyzed by a one exponential fit to inactivating phase of the Na+ currents. (G) Entry into slow inactivation is represented as a first-order exponential fit to the data points. (H) Voltage-dependent steady-state of slow inactivation fit by a Boltzmann function as in (C). (F) Na-current use dependence evoked by 50 pulses at a frequency of 40 Hz as normalized peak currents plotted against the time and fit by a second-order exponential function. The number of recorded cells, exact values of statistically analyzed parameters, and p-values are listed in Supplementary Table 1.
In silico Modeling Reveals Pronounced Interneuron Firing Impairment by NaV1.1A1783V
Full haploinsufficiency is considered as the genetic mechanism underlying the majority of SCN1A Dravet variants. About half of the described variants lead to protein truncation with reduced protein expression in affected neurons and likely markedly reduced conductance (Marini et al., 2011). Our recordings revealed clear biophysical LOF changes of NaV1.1A1783V compared with WT, however no reduction of peak current density. Then, we asked how these combined functional alterations translate to the neuronal level and how they compare to the heterozygous knockout condition. To address these questions, we built a single-compartment conductance-based model with the combined biophysical alterations reflecting intrinsic and firing properties of either cortical FS-INs or excitatory PCs neurons. Differential NaV1.1–NaV1.6 expression ratios of FS-IN and PCs and either the WT (NaV1.1+/+), the mutant (NaV1.1+/A1783V), or the full haploinsufficiency (NaV1.1+/−) condition were implemented. The parameters for the different model types are summarized in Table 1. Additionally, we simulated altered voltage-dependence of activation as well as altered voltage-dependence and kinetics of slow inactivation separately to dissect their joint or exclusive impact on neuron action potential firing in comparison to the WT and the full haploinsufficiency condition (Figure 2A).
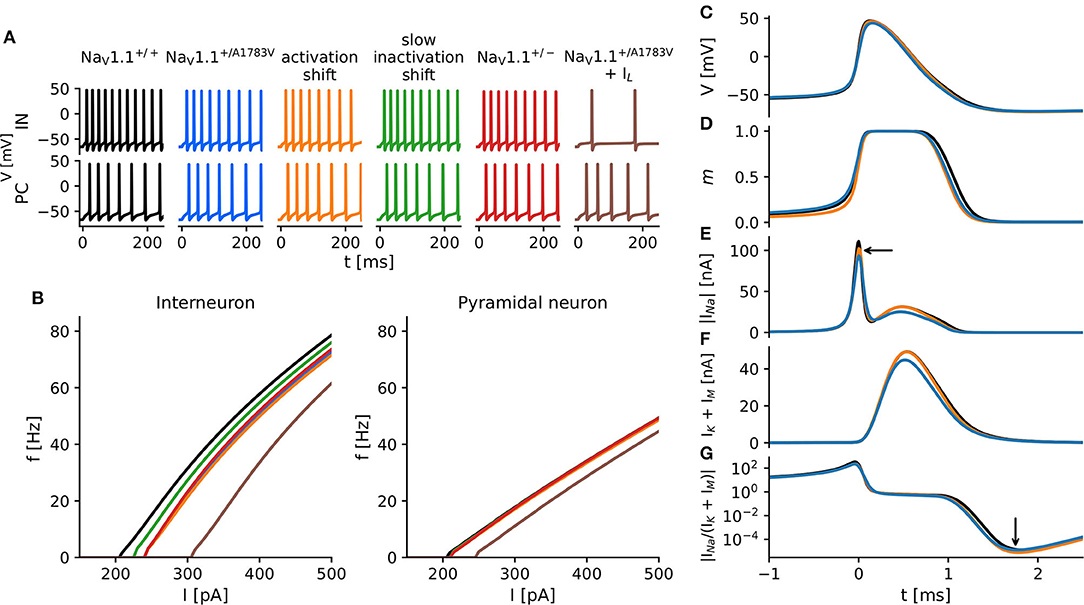
Figure 2. In silico modeling of neuron excitability. (A) Action potentials of simulated cortical fast-spiking interneurons (upper traces) and cortical pyramidal cells (PCs) (lower traces) in response to square pulse current injections of 320 pA with various modifications of NaV1.1 current kinetics based on findings in tsA recordings. Colors indicate different model conditions: WT (black), the heterozygous shift of the voltage-dependence of steady-state activation (orange), the heterozygous shift of the voltage-dependence of the steady-state slow inactivation (green), the combination of both changes (blue), a heterozygous protein knockout (red), and a combination of respective changes in sodium current and a decreased input resistance of the cell (brown). The exact parameters for all simulated conditions are summarized in Table 1. (B) f−I curves of interneurons (left) and PCs (right) constructed from simulation studies displayed in (A). (C–G) Time courses of model variables during the first action potential after a 300 pA current injection, t = 0 indicates the peak of the action potential. (C) Membrane voltage; (D) activation gate m—ratio of activated sodium channels; (E) absolute INa; (F) IK; and (G) the absolute ratio of INa to IK.
In the interneuron model, solely shifting the activation curve to more depolarized membrane potentials caused the most severe reductions of the firing rates in comparison to the WT, closely followed by the NaV1.1+/A1783V and NaV1.1+/− conditions. Shifting the slow inactivation curve to more hyperpolarized potentials reduced firing rates to a lesser extent (Figure 2B, left). Although the faster slow inactivation model was the only simulation yielding an accelerated rundown of sodium current amplitudes as had been observed in tsA201 cells expressing NaV1.1A1783V (Supplementary Figure 1C), it showed almost no impact on firing rates in comparison to WT (Supplementary Figures 1A,B). The firing properties of all pyramidal models remained largely unaffected (Figure 2B, right; Supplementary Figure 1B).
To better understand why shifting the activation curve for half of the NaV1.1. channels (to mimic the heterozygous patient situation) had a stronger effect on neuronal firing than the simulation of the heterozygous protein knockout, we analyzed the current dynamics of the first action potential in more detail (Figures 2C–G). Although on a first glance, the action potential waveforms seemed to be rather similar (Figure 2C), the sodium activation gate (Figure 2D) of the model with a shifted activation curve (orange) opened later and closed earlier compared with the WT (black) and the NaV1.1+/− models (blue). The resulting sodium current was reduced during AP initiation and rising phase of the action potential compared to the WT simulation but was similar during the falling phase of the action potential (AP). Only at the very end of the AP, the sodium current appeared again slightly reduced. In contrast, the NaV1.1+/− model depicted a generally reduced sodium current (Figure 2E), which was followed by a pronounced reduction of the delayed rectifier potassium current. This effect on the potassium current was almost absent when modeling a right-shifted activation curve of NaV1.1. (Figure 2F). In combination with the observed effects on the sodium current, the right-shifted activation curve model showed a greater reduction of the sodium-to-potassium current ratio in the final phase of the afterhyperpolarization than the NaV1.1+/− model (Figure 2G). This increased after-hyperpolarization lead then to a delay of the next action potential.
NaV1.1A1783V LOF Is Restricted to Inhibitory Neurons in Cortical Mouse Brain Slice Cultures
To confirm the simulated effects of NaV1.1A1783V on neuronal firing, we performed whole-cell patch-clamp recordings in cortical brain slice cultures derived from P4-5 heterozygous B6(Cg)-Scn1atm1.1Dsf/J mice and WT littermates. Cultures were virally transduced at day 1 in vitro with AAV8 Cre-recombinase-GFP under the human synapsin promoter to induce pan-neuronal recombination and subsequent expression of the NaV1.1+/A1783V variant. Since Scn1a is upregulated in the postnatal period only from P11 onward (Cheah et al., 2013), this early expression of Cre-recombinase allowed for recombination of transduced neurons carrying the floxed Scn1aA1783V allele before endogenous upregulation of the Scn1a gene. This approach ensured activation of the variant following the endogenous expression time course of the NaV1.1 channel. Notably, 7–14 days after transduction, GFP+ neurons were patched (Supplementary Figure 2). While the firing of PCs was not altered (Figures 3A,B), we found a LOF in FS-INs (Figures 3C,D). NaV1.1+/A1783V expressing FS-INs displayed a significantly increased rheobase, reduced maximum firing frequency, and reduced input resistance compared to WT controls (Supplementary Table 2). Similarly, we could detect a trend for a reduction of the input resistance of PCs (Supplementary Table 3). However, the firing properties of PCs were not significantly altered. AP threshold, AP amplitude, AP rise time, AP half-width, and afterhyperpolarization amplitude within evoked AP trains were indistinguishable between neurons (FS-INs and PCs) recorded from slices of WT and mutant animals. We applied the observed changes in input resistance to the in silico model (Figure 2, brown). These simulations combining changed gating and membrane properties predicted an even stronger LOF in FS-INs while only tentatively affecting the firing of simulated PCs.
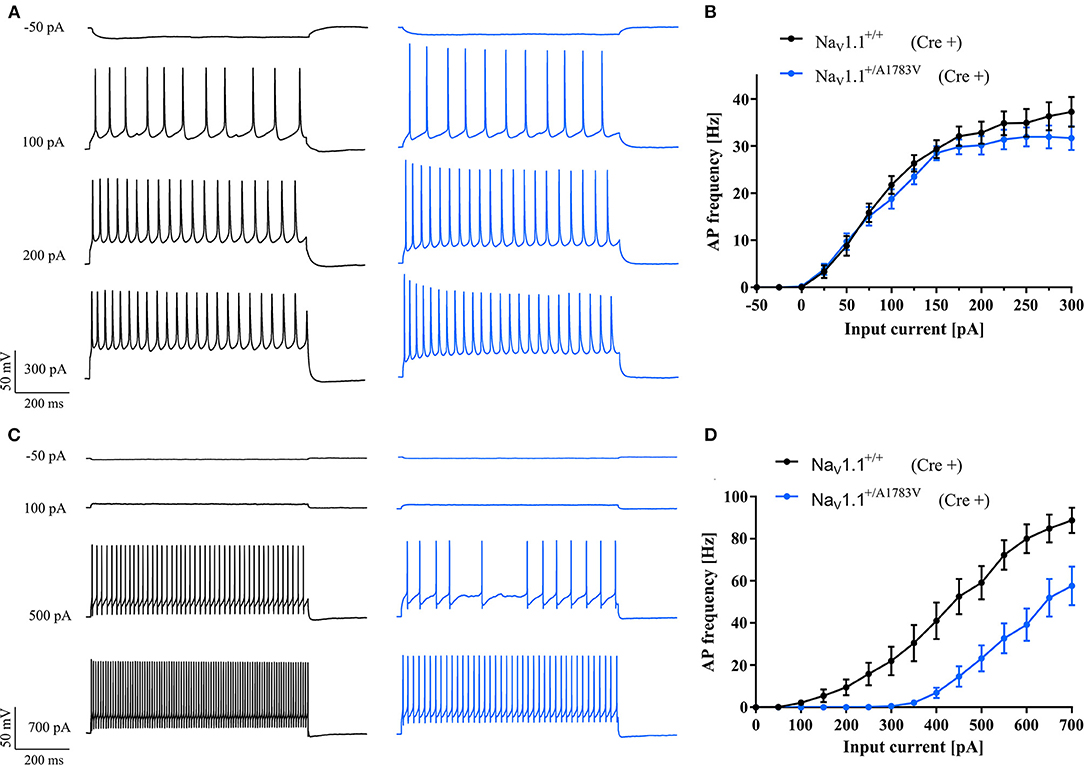
Figure 3. In vitro studies of neuronal excitability. (A) Representative AP trains in response to 800 ms step current injections from cortical PCs and (C) fast-spiking interneurons recorded in brain slice cultures of WT (black traces) and heterozygous NaV1.1+/A1783V animals (blue traces). (B) f−I curves of PCs from floxed heterozygous mice showed the same frequency of action potential firing while (D) fast-spiking inhibitory neurons showed higher and maintained lower AP frequencies compared to cells of WT animals. A number of recorded cells, exact values of statistically analyzed parameters, and p-values are listed in Supplementary Tables 2, 3.
Discussion
Full haploinsufficiency is considered the leading genetic cause of DS, resulting in impaired excitability predominantly of interneurons (Catterall et al., 2008). In this study, we functionally characterized the human recurrent SCN1AA1783V DS variant. Although reduced-sodium peak current density is a common feature associated with DS, our analysis revealed altered activation and slow inactivation properties, albeit with preserved sodium peak current density. However, an enhanced use-dependent rundown of Na+ current was observed on repetitive stimulations. Although an interference of the variant (localized at the end of segment 6 in domain 4) with local determinants of fast inactivation would have been conceivable (Ulbricht, 2005), fast inactivation was found to be unchanged. Since fast inactivation was unaffected, use-dependence can only be attributed to slow inactivation characteristics of the variant. In fact, this could be confirmed by in silico modeling of Na+ current with physiological or accelerated slow inactivation kinetics (Supplementary Figure 1C). Interestingly, modeling of neuronal excitability predicted a fairly strong overall interneuron AP firing deficit for NaV1.1+/A1783V at comparable levels to the simulated full haploinsufficiency condition (NaV1.1+/−). Since our tsA201 cell recordings had revealed a ~25% sodium current “rundown” on 40 Hz stimulation, we speculated that altered slow inactivation and associated enhanced use-dependence might largely account for the predicted interneuron firing deficit. Importantly, the model correctly predicted enhanced use-dependence of NaV1.1A1783V currents and confirmed accelerated slow inactivation as the only underlying mechanism (Supplementary Figure 1C). However, when simulating f−I curves for FS-INs with different time constants of slow inactivation, an acceleration of τ only had a marginal effect on neuronal firing (Supplementary Figure 1A) and shifted voltage-dependence of fast activation was predicted as the main driver of the impaired interneuron firing. Another observation from our recordings in cortical brain slice cultures was a significant reduction of input resistance in neurons expressing p.A1783V. These changes in passive membrane properties might also contribute to the observed IN LOF and are consistent with recordings of hippocampal INs in brain slices of mice harboring p.A1783V (Almog et al., 2021). However, the underlying mechanisms of altered input resistance remain still to be addressed in a follow-up study. At this point, it is hard to project how the pA1783V variant would directly account for this change, and secondary cellular changes affecting membrane leak properties may have to be considered.
In our simulations, the comparison of NaV1.1+/A1783V with the simulated NaV1.1+/− condition revealed a similar action potential deficit. Although surprising at first, these findings could at least partially be explained when putting the activation properties and relative levels of conducted currents of NaV1.1+/A1783V and NaV1.1+/− in context with the different phases of the action potential. Conductance-based modeling suggested a reduced number of sodium channels available to participate in action potential initiation due to the shifted activation curve and subsequently delayed opening of the channels. This effect was qualitatively similar to NaV1.1+/− (with only a limited number of available channels due to the simulated heterozygous knockout condition) in this early phase of an action potential and was predicted to account for most of the overall LOF. However, in the falling phase of the action potential, NaV1.1+/A1783V yielded larger sodium currents than NaV1.1+/− and accordingly recruited more potassium channels. Our simulations predicted that the induced potassium current outweighed the conducted sodium current in the final hyperpolarizing phase in neurons of the heterozygous NaV1.1+/A1783V condition. This relative surplus of K+ current led to a slightly pronounced afterhyperpolarization (0.5–1.0 mV) and a prolonged time window until the next spike could occur, altogether increasing the LOF effect. This predicted change on afterhyperpolarization was reflected as a nonsignificant tendency in our brain slice culture recordings (Supplementary Table 2). Consistent with our data, pronounced shifts of voltage-dependence of activation have also been described for other LOF mutations associated with epilepsy, such as SCN1AI1656M (Lossin et al., 2003), SCN1AD249E (Kluckova et al., 2020), and SCN1AR859C (Barela et al., 2006). Similar to SCN1AA1783V, these variants also feature unaltered current density in comparison to the WT.
NaV1.1 is predominantly expressed in interneurons (Yao et al., 2020), while PCs mainly rely on NaV1.6 for action potential initiation. Therefore, a leading dysfunction of interneurons was expected and was indeed reflected in silico. These data were confirmed in cortical brain slice cultures derived from conditional Scn1a+/A1783V mice. Our findings are consistent with data on hippocampal interneurons expressing NaV1.1 +/A1783V, which also displayed a shift in rheobase and a firing deficit at higher current injections (Almog et al., 2021).
Conclusion
SCN1AA1783V results in altered voltage-dependence of activation and slow inactivation while maintaining sodium peak current density. Interneuron excitability was simulated in silico by using a Hodgkin-Huxley one-compartment model suggesting largely similar firing impairment for NaV1.1+/A1783V in comparison to NaV1.1+/− resembling full haploinsufficiency. Impaired NaV1.1A1783V voltage-dependent activation was suggested as the main primary mechanism underlying interneuron dysfunction.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by local Animal Care and Use Committee (Regierungspraesidium Tuebingen, Tuebingen, Germany).
Author Contributions
NL did the molecular biology, cultured brain slices, and performed current-clamp recordings in brain slice cultures. NL and EP performed voltage-clamp recordings in tsA201 cells. LS and JB developed in silico model. NL and LS did the data analysis. NL, LS, JB, HK, and TW wrote the manuscript. NL, LS, EP, JB, UBSH, HL, HK, and TW did the proof reading and study design. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the German Research Foundation (DFG/FNR INTER research unit FOR2715 grants Ko4877/3-1, Le1030/15-1, He8155/1-1). TW was supported by an intramural Clinician Scientist Fellowship granted by the Faculty of Medicine, University of Tübingen (419-0-0). We acknowledge support by Open Access Publishing Fund of University of Tübingen.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2021.754530/full#supplementary-material
References
Almog, Y., Fadila, S., Brusel, M., Mavashov, A., Anderson, K., and Rubinstein, M. (2021). Developmental alterations in firing properties of hippocampal CA1 inhibitory and excitatory neurons in a mouse model of Dravet syndrome. Neurobiol. Dis. 148:105209. doi: 10.1016/j.nbd.2020.105209
Barela, A. J., Waddy, S. P., Lickfett, J. G., Hunter, J., Anido, A., Helmers, S. L., et al. (2006). An epilepsy mutation in the sodium channel SCN1A that decreases channel excitability. J. Neurosci. 26, 2714–2723. doi: 10.1523/JNEUROSCI.2977-05.2006
Bechi, G., Scalmani, P., Schiavon, E., Rusconi, R., Franceschetti, S., and Mantegazza, M. (2012). Pure haploinsufficiency for Dravet syndrome Na V1.1 (SCN1A) sodium channel truncating mutations. Epilepsia 53, 87–100. doi: 10.1111/j.1528-1167.2011.03346.x
Catterall, W. A., Dib-Hajj, S., Meisler, M. H., and Pietrobon, D. (2008). Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J. Neurosci. 28, 11768–11777. doi: 10.1523/JNEUROSCI.3901-08.2008
Catterall, W. A., Kalume, F., and Oakley, J. C. (2010). NaV1.1 channels and epilepsy. J. Physiol. 588, 1849–1859. doi: 10.1113/jphysiol.2010.187484
Cheah, C. S., Westenbroek, R. E., Roden, W. H., Kalume, F., Oakley, J. C., Jansen, L. A., et al. (2013). Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels 7. doi: 10.4161/chan.26023
Das, A., Zhu, B., Xie, Y., Zeng, L., Pham, A. T., Neumann, J. C., et al. (2021). Interneuron dysfunction in a new mouse model of scn1a gefs+. eNeuro 8, 1–16. doi: 10.1523/ENEURO.0394-20.2021
Depienne, C., Trouillard, O., Saint-Martin, C., Gourfinkel-An, I., Bouteiller, D., Carpentier, W., et al. (2009). Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J. Med. Genet. 46, 183–191. doi: 10.1136/jmg.2008.062323
Favero, M., Sotuyo, N. P., Lopez, E., Kearney, J. A., and Goldberg, E. M. (2018). A transient developmental window of fast-spiking interneuron dysfunction in a mouse model of Dravet syndrome. J. Neurosci. 38, 7912–7927. doi: 10.1523/JNEUROSCI.0193-18.2018
Gambardella, A., and Marini, C. (2009). Clinical spectrum of SCN1A mutations. Epilepsia 50, 20–23. doi: 10.1111/j.1528-1167.2009.02115.x
Hedrich, U. B., Liautard, C., Kirschenbaum, D., Pofahl, M., Lavigne, J., Liu, Y., et al. (2014). Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human Na(V)1.1 mutation. J. Neurosci. 34, 14874–14889. doi: 10.1523/JNEUROSCI.0721-14.2014
Klassen, T. L., Bomben, V. C., Patel, A., Drabek, J., Chen, T. T., Gu, W., et al. (2014). High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia 55, 6–12. doi: 10.1111/epi.12489
Kluckova, D., Kolnikova, M., Lacinova, L., Jurkovicova-tarabova, B., Foltan, T., Demko, V., et al. (2020). OPEN a study among the genotype, functional alternations, and phenotype of 9 SCN1A mutations in epilepsy patients. Sci. Rep. 501:217. doi: 10.1038/s41598-020-67215-y
Kuo, F.-S., Cleary, C. M., LoTurco, J. J., Chen, X., and Mulkey, D. K. (2019). Disordered breathing in a mouse model of Dravet syndrome. doi: 10.7554/elife.43387
Lange, I. M., Weuring, W., ‘t Slot, R., Gunning, B., Sonsma, A. C. M., McCormack, M., et al. (2019). Influence of common SCN1A promoter variants on the severity of SCN1A-related phenotypes. Mol. Genet. Genomic Med. 7. doi: 10.1002/mgg3.727
Liao, Y., Anttonen, A. K., Liukkonen, E., Gaily, E., Maljevic, S., Schubert, S., et al. (2010). SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 75, 1454–1458. doi: 10.1212/WNL.0b013e3181f8812e
Lossin, C., Rhodes, T. H., Desai, R. R., Vanoye, C. G., Wang, D., Carniciu, S., et al. (2003). Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A. J. Neurosci. 23, 11289–11295. doi: 10.1523/JNEUROSCI.23-36-11289.2003
Marini, C., Scheffer, I. E., Nabbout, R., Suls, A., De Jonghe, P., Zara, F., et al. (2011). The genetics of Dravet syndrome. Epilepsia 52, 24–29. doi: 10.1111/j.1528-1167.2011.02997.x
Mistry, A. M., Thompson, C. H., Miller, A. R., Vanoye, C. G., George, A. L., and Kearney, J. A. (2014). Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol. Dis. 65, 1–11. doi: 10.1016/j.nbd.2014.01.006
Ogiwara, I., Miyamoto, H., Morita, N., Atapour, N., Mazaki, E., Inoue, I., et al. (2007). Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007
Peters, C., Rosch, R. E., Hughes, E., and Ruben, P. C. (2016). Temperature-dependent changes in neuronal dynamics in a patient with an SCN1A mutation and hyperthermia induced seizures. Sci. Rep. 6, 1–12. doi: 10.1038/srep31879
Pospischil, M., Toledo-Rodriguez, M., Monier, C., Piwkowska, Z., Bal, T., Frégnac, Y., et al. (2008). Minimal Hodgkin-Huxley type models for different classes of cortical and thalamic neurons. Biol. Cybern. 99, 427–441. doi: 10.1007/s00422-008-0263-8
Ricobaraza, A., Mora-jimenez, L., Puerta, E., Sanchez-carpintero, R., Mingorance, A., Artieda, J., et al. (2019). Epilepsy and neuropsychiatric comorbidities in mice carrying a recurrent Dravet syndrome SCN1A missense mutation. Sci. Rep. 9:14172. doi: 10.1038/s41598-019-50627-w
Rubinstein, M., Westenbroek, R. E., Yu, F. H., Jones, C. J., Scheuer, T., and Catterall, W. A. (2015). Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome. Neurobiol. Dis. 73, 106–117. doi: 10.1016/j.nbd.2014.09.017
Spampanato, J., Kearney, J. A., De Haan, G., McEwen, D. P., Escayg, A., Aradi, I., et al. (2004). A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for β subunit interaction. J. Neurosci. 24, 10022–10034. doi: 10.1523/JNEUROSCI.2034-04.2004
Tai, C., Abe, Y., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, 3139–3148. doi: 10.1073/pnas.1411131111
Thompson, C. H., Porter, J. C., Kahlig, K. M., Daniels, M. A., and George, A. L. (2012). Nontruncating SCN1A mutations associated with severe myoclonic epilepsy of infancy impair cell surface expression. J. Biol. Chem. 287, 42001–42008. doi: 10.1074/jbc.M112.421883
Tiraboschi, E., Martina, S., van der Ent, W., Grzyb, K., Gawel, K., Cordero-Maldonado, M. L., et al. (2020). New insights into the early mechanisms of epileptogenesis in a zebrafish model of Dravet syndrome. Epilepsia 61, 549–560. doi: 10.1111/epi.16456
Tran, C. H., Vaiana, M., Nakuci, J., Somarowthu, A., Goff, K. M., Goldstein, N., et al. (2020). Interneuron desynchronization precedes seizures in a mouse model of Dravet syndrome. J. Neurosci. 40, 2764–2775. doi: 10.1523/JNEUROSCI.2370-19.2020
Traub, R. D., and Miles, R. (1991). Neuronal Networks of the Hippocampus. Cambridge: Cambridge University Press.
Ulbricht, W. (2005). Sodium channel inactivation: molecular determinants and modulation. Physiol. Rev. 85, 1271–1301. doi: 10.1152/physrev.00024.2004
Vilin, Y. Y., and Ruben, P. C. (2001). Slow inactivation in voltage-gated sodium channels: Molecular substrates and contributions to channelopathies. Cell Biochem. Biophys. 35, 171–190. doi: 10.1385/CBB:35:2:171
Xie, Y., Ng, N. N., Safrina, O. S., Ramos, C. M., Ess, K. C., Schwartz, P. H., et al. (2020). Comparisons of dual isogenic human iPSC pairs identify functional alterations directly caused by an epilepsy associated SCN1A mutation. Neurobiol. Dis. 134:104627. doi: 10.1016/j.nbd.2019.104627
Yao, Z., Nguyen, T. N., van Velthoven, C. T. J., Goldy, J., Sedeno-Cortes, A. E., Baftizadeh, F., et al. (2020). A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation Zizhen. bioRxiv 10:3. doi: 10.1101/2020.03.30.015214
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Keywords: Dravet syndrome (SMEI), developmental epileptic encephalopathy, SCN1A, Nav1.1, in silico modeling, electrophysiology
Citation: Layer N, Sonnenberg L, Pardo González E, Benda J, Hedrich UBS, Lerche H, Koch H and Wuttke TV (2021) Dravet Variant SCN1AA1783V Impairs Interneuron Firing Predominantly by Altered Channel Activation. Front. Cell. Neurosci. 15:754530. doi: 10.3389/fncel.2021.754530
Received: 06 August 2021; Accepted: 28 September 2021;
Published: 28 October 2021.
Edited by:
Dominique Debanne, INSERM U1072 Neurobiologie des canaux Ioniques et de la Synapse, FranceReviewed by:
Ilya A. Fleidervish, Ben-Gurion University of the Negev, IsraelPierre Apostolides, University of Michigan, United States
Copyright © 2021 Layer, Sonnenberg, Pardo González, Benda, Hedrich, Lerche, Koch and Wuttke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas V. Wuttke, dGhvbWFzLnd1dHRrZUBtZWQudW5pLXR1ZWJpbmdlbi5kZQ== orcid.org/0000-0001-5655-8490