 Therese L. Dane
Therese L. Dane Anna L. Gill
Anna L. Gill Fernando G. Vieira
Fernando G. Vieira Kyle R. Denton
Kyle R. Denton- ALS Therapy Development Institute, Watertown, MA, United States
Introduction: Intronic repeat expansions in the C9orf72 gene are the most frequent known single genetic causes of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). These repeat expansions are believed to result in both loss-of-function and toxic gain-of-function. Gain-of-function results in the production of toxic arginine-rich dipeptide repeat proteins (DPRs), namely polyGR and polyPR. Small-molecule inhibition of Type I protein arginine methyltransferases (PRMTs) has been shown to protect against toxicity resulting from polyGR and polyPR challenge in NSC-34 cells and primary mouse-derived spinal neurons, but the effect in human motor neurons (MNs) has not yet been explored.
Methods: To study this, we generated a panel of C9orf72 homozygous and hemizygous knockout iPSCs to examine the contribution of C9orf72 loss-of-function toward disease pathogenesis. We differentiated these iPSCs into spinal motor neurons (sMNs).
Results: We found that reduced levels of C9orf72 exacerbate polyGR15 toxicity in a dose-dependent manner. Type I PRMT inhibition was able to partially rescue polyGR15 toxicity in both wild-type and C9orf72-expanded sMNs.
Discussion: This study explores the interplay of loss-of-function and gain-of-function toxicity in C9orf72 ALS. It also implicates type I PRMT inhibitors as a possible modulator of polyGR toxicity.
1. Introduction
The most frequent known causes of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are mutations in the C9orf72 gene, which account for 40% of familial and 5% of sporadic ALS cases, and act as a main genetic cause of FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Majounie et al., 2012; Suh et al., 2015; Van Mossevelde et al., 2017). These mutations manifest as expansions of a repeating hexanucleotide (GGGGCC) sequence in the first intron of the C9orf72 gene (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Translation of the C9orf72 hexanucleotide repeat expansion mutation (C9-HRE) is facilitated by an unconventional process termed repeat-associated non-AUG (RAN) translation (Ash et al., 2013; Mori et al., 2013). The role of the C9-HRE in ALS and FTD pathogenesis has been explored in the context of three possible disease mechanisms: loss-of-function, or decreased expression of endogenous C9orf72 protein, toxic gain-of-function of mutant sense and antisense repeat RNA, and toxic gain-of-function of RAN-translated dipeptide repeat proteins (DPRs): poly-Glycine-Arginine (polyGR), poly-Proline-Arginine (polyPR), poly-Glycine-Alanine (polyGA), poly-Glycine-Proline (polyGP), and poly-Proline-Alanine (polyPA) (Yang et al., 2020). A body of evidence surrounding each of these mechanisms introduces the possibility of a multiple-hit model, whereby loss-of-function and gain-of-function consequences might simultaneously contribute to C9-HRE-linked neurodegeneration (Braems et al., 2020).
Decreased expression of endogenous C9orf72 protein has been documented in both patient tissue samples and patient-derived induced pluripotent stem cell (iPSC) lines harboring the C9-HRE (Waite et al., 2014; Sivadasan et al., 2016; Frick et al., 2018). Resulting loss in the reported function of the C9orf72 protein has been associated with losses in axonal trafficking (Farg et al., 2014; Gendron and Petrucelli, 2018; Shi et al., 2018). In one study, heterozygous and homozygous deletion of C9orf72, as well as antisense oligonucleotide (ASO)-mediated knockdown, decreased survival of human iPSC-derived MNs, which was ameliorated with exogenous application of C9orf72 protein (Shi et al., 2018). Other studies using in vitro C9orf72 knockdown and knockout models have also demonstrated dysfunctional autophagic (Sellier et al., 2016; Webster et al., 2016; Yang et al., 2016; Aoki et al., 2017; Fomin et al., 2018; Leskelä et al., 2019), lysosomal (Amick et al., 2016; Aoki et al., 2017; Shi et al., 2018), and endosomal (Farg et al., 2014) behaviors. These autophagy deficits have also been observed in murine models deficient in C9orf72 (O’Rourke et al., 2016; Sullivan et al., 2016). Despite this, much evidence has been collected to assert that loss of endogenous C9orf72 protein alone is not sufficient to induce the neurodegeneration characteristic of ALS and FTD, instead, a combination of loss-of-function and gain-of-function is more likely (Braems et al., 2020; Pal et al., 2021).
Significant cytotoxic gain-of-function effects resulting from exposure to the arginine-rich DPRs (R-DPRs) polyGR and polyPR have also been extensively documented (Freibaum and Taylor, 2017). In in vitro systems, indications of detrimental cellular effects by R-DPRs have been detected in HEK293T cells, NSC-34 cells, iPSC-derived cortical and motor neurons, and others (Kwon et al., 2014; Wen et al., 2014; Tao et al., 2015; Lee et al., 2016; Kramer et al., 2018). We have previously reported that NSC-34 cells differentiated to exhibit more neuron-like characteristics are more sensitive to polyGR and polyPR challenge, potentially implicating neuronal cell physiology in R-DPR mechanisms of toxicity (Gill et al., 2019). Further, we have observed the abrogation of R-DPR cytotoxic effects in NSC-34 cells when exogenous R-DPR challenge is administered alongside a co-dose of small-molecule asymmetric (Type I) Protein Arginine Methyltransferase (PRMT) inhibitors (Premasiri et al., 2020; Gill et al., 2021). One study has approached an intersection between DPR toxicity and endogenous C9orf72 loss-of-function, where using an siRNA knockdown model of C9orf72 in GT1-7 and HEK293 cell models, the investigators reported that reduced C9orf72 expression substantially increased neuronal cell death induced by polyGR, polyGA, and polyGP accumulation, when the DPRs were expressed under their natural start codons (Boivin et al., 2020). Taken together, these studies implicate R-DPR toxicity in gain-of-function deficits, and even begin to associate changes in relationship to loss-of-function models. Our previous study links these DPR gain-of-function toxicity outcomes to PRMT activity, suggesting protein arginine methylation state of involved proteins may relate to how resulting toxicity manifests. However, none of the studies mentioned explore the intersection between all three: R-DPR gain-of-function, C9orf72 loss-of-function, and protein arginine methylation regulation.
Some findings point to a possible link between protein arginine methylation and C9orf72 protein activity in ALS models. One study has detailed the role of symmetric (Type II) arginine dimethylation in the association between C9orf72 and ALS-linked proteins p62, SMN, and FUS, which together initiate a cascade to recognize stress granules for autophagic degradation (Chitiprolu et al., 2018). It is also feasible that endogenous C9orf72 protein itself might be modified by PRMTs. The reported function of human C9orf72 is a guanine nucleotide exchange factor (GEF), and protein arginine methylation of another key human GEF, Vav1, has been reported to regulate its localization and function (Blanchet et al., 2005; Lawson et al., 2007; Peng and Wong, 2017). Other findings associate protein arginine methylation with R-DPR gain-of-function toxicity. One study has correlated the accumulation of symmetrically dimethylated polyGR in C9orf72 ALS patient post-mortem brain tissue with longer disease duration and age at death (Gittings et al., 2020). Further, protein arginine methylation activity has recently been implicated in ALS biomarker development. For example, higher levels of asymmetric dimethylarginine in cerebral spinal fluid (CSF) and serum have been shown to correlate faster progression of ALS symptoms (Ikenaka et al., 2019; Ikenaka et al., 2022). Consistent with these results, asymmetrically (Type I) dimethylated forms of polyGR detected in C9orf72 patient brain tissue did not correlate with later disease onset or longer disease duration, in contrast to symmetrically (Type II) dimethylated forms of polyGR, which did (Gittings et al., 2020). These studies associate Type I PRMT activity with worsening ALS disease outcomes, and even with C9orf72 R-DPR gain-of-function phenotypes. However, none of these studies have addressed how endogenous C9orf72 loss-of-function, R-DPR gain-of-function, and protein arginine methylation might all simultaneously contribute to negative outcomes in C9orf72-ALS models.
To explore this, we generated a panel of isogenic C9orf72 knockout iPSC lines using a CRISPR/Cas9 dual-guide approach, including one hemizygous patient-derived iPSC line with one copy of C9orf72 knocked out, and two homozygous iPSC lines with both copies of C9orf72 knocked out. Genotyping analysis, quantitative reverse transcription polymerase chain reactions (qRT-PCRs), and western blotting methods were used to confirm successful gene editing. These lines were differentiated into sMNs, and exogenous polyGR15 was titrated onto cells. Both hemizygous and homozygous C9orf72 knockout lines, as well as a patient line with the HRE, were the more sensitive to the polyGR15 challenge compared to wild-type. After confirming iPSC-derived sMNs with fewer copies of C9orf72 were more sensitive to polyGR15 challenge, we then introduced Type I PRMT inhibition to this assay framework to see how it might affect cytotoxic endpoints. We titrated the Type I PRMT inhibitor MS023 onto VACHT-tdTomato iPSC-derived sMNs concurrently with polyGR15 challenge, and measured resulting metabolic capacity at 24 and 48 h using a CellTiter-Blue assay. Results indicated that the Type I PRMT inhibitor MS023 significantly increased cell viability in response to polyGR15 challenge at doses from 0.3 to 3 μM as compared to cells treated with polyGR15 alone at both endpoints. This dose range required for cell rescue is consistent with previous results using the same dosing scheme in NSC-34 neuron-like mouse cells (Premasiri et al., 2020). These results begin to assess the intersections between PRMT activity, gain-of-function of arginine-rich C9-DPRs, and C9orf72 loss-of-function phenotypes in C9orf72 ALS models. Future study will explore the possible interplay between these three mechanisms, and their contributions to C9-linked neurodegeneration, further.
2. Materials and methods
2.1. iPSC cell culture
Cells were cultured in NutriStem hPSC XF Medium (ReproCell USA, Inc., Beltsville, MD, USA, 01–0005) on growth factor reduced Matrigel hESC-qualified matrix (Corning Incorporated – Life Sciences, Tewksbury, MA, USA, 354230). Cells were passaged every 4–7 days by rinsing once with Dulbecco’s phosphate-buffered Saline (DPBS) without calcium and magnesium (Thermo Fisher Scientific, Waltham, MA, USA, 14–190–250) then treated with pre-warmed Accutase (Stemcell Technologies, Cambridge, MA, USA, 07920) for 3 min in 37°C and 5% CO2 for dissociation. Dissociated cells were collected and centrifuged at 200 × g for 5 min. PBS was aspirated and the cell pellet was resuspended in NutriStem prior to replating.
2.2. Motor neuron differentiation and cell culture
Spinal motor neurons were generated from iPSCs following a previously reported protocol (Du et al., 2015). In short, iPSCs were plated on Matrigel-coated plates in NutriStem with 10 μM rho-associated protein kinase (ROCK) inhibitor (Tocris, Bio-Techne Corporation, Minneapolis, MN, USA, 1254) on day −1. The following day, the media was replaced with base neural differentiation media [1:1 Neurobasal:DMEM-F12 (Gibco USA, Jenks, OK, USA, 11330032), 1X NEAA (Gibco USA, Jenks, OK, USA, 11140050), 1X PenStrep (Gibco USA, Jenks, OK, USA, 15140163), 1X Glutamax (Gibco USA, Jenks, OK, USA, 35050079), 100 μM ascorbic acid (Sigma-Aldrich, Burlington, MA, USA, A4403-100MG), 0.5X N2 (Life Technologies, Carlsbad, CA, USA, A1370701), 0.5X B27 (Life Technologies, Carlsbad, CA, USA, 17504044) supplemented with 2 μM SB431542 (Selleck Chemicals, Houston, TX, USA, S1067), 2 μM DMH1 (Selleck Chemicals, Houston, TX, USA, S7146), 3 μM CHIR-99021 (Axon MedChem, Reston, VA, USA, 1386)]. Media was replaced every other day until day 6, when cells were passaged with Dispase-II (EMD Millipore, Burlington, MA, USA, SCM133) and plated at a 1–6 ratio. The media above was supplemented with 0.1 μM retinoic acid (Reprocell USA, Inc., Beltsville, MD, USA, 04–0021) and 0.5 μM purmorphamine (Reprocell USA, Inc., Beltsville, MD, USA, 04–0009). On day 12 of differentiation, cells were passaged again with Dispase-II and motor neuron progenitor spheres were formed on ultra-low attachment dishes (Corning Incorporated – Life Sciences, Tewksbury, MA, USA, 4615) in base NDM supplemented with 0.5 μM retinoic acid and 0.1 μM purmorphamine. On day 19, MNP spheres were dissociated with Accutase and plated on laminin/poly-ornithine coated plates in base NDM supplemented with 0.5 μM retinoic acid, 0.1 μM purmorphamine, 10 ng/mL CNTF (Peprotech US, Cranbury, NJ, USA, 450–13–100 μg), 10 ng/mL BDNF (R&D Systems, Inc., Minneapolis, MN, USA, 248-BDB-250/CF), 10 ng/mL GDNF (R&D Systems, Inc., Minneapolis, MN, USA, 212-GD-050), and 1 μM 5-Fluoro-2’-deoxyuridine (Sigma-Aldrich, Burlington, MA, USA, F0503). Media was replaced every 2–3 days using an EL406 automated plate washer (Agilent Technologies, Inc., Santa Clara, CA, USA) and neurons were allowed to mature until assay endpoint.
2.3. sgRNA guide design
Dual guides were designed using Benchling’s CRISPR Design tool in the second exon of the C9orf72 gene, and optimized for on-target efficiency and minimized for off-target score. Synthetic modified sgRNAs were ordered from Synthego (Redwood City, CA, USA) (sgRNA kit) and resuspended at 100 μM in TE Buffer and stored in the −80°C until use. N*N*N* indicate 2’-O-methyl analogs and 3’-phosphorothioate internucleotide linkages. The sequences are as follows:
Forward guide: U*U*A*ACACAUAUAAUCCGGAA
Reverse guide: C*A*C*ACACUCUAUGAAGUGGG.
2.4. Gene editing of iPSCs
Induced pluripotent stem cells were generated from an individual with 2 and 5 copies of the C9orf72 hexanucleotide repeat. To introduce double stranded DNA breaks in exon 2 of C9orf72, iPSCs were electroporated with ribonucleoprotein (RNP) complex of Alt-R SpCas9 Nuclease V3 (Integrated DNA Technologies, Ann Arbor, MI, USA, 1081059) and two different sgRNA guides (Synthego Corporation, Redwood City, CA, USA, sgRNA EZ kit) in a 1–3 molar ratio, respectively, to increase editing efficiency. In short, iPSCs were dissociated to single cells using Accutase for 3 min at in 37°C hypoxic incubator, collected with 10 mL of PBS, then spun at 200 × g for 5 min. The cell pellet was resuspended in PBS and counted using trypan blue on a Countess II (Thermo Fisher Scientific, Waltham, MA, USA). 1e^6 cells were resuspended per electroporation in Neon Buffer R. RNP complexes were formed by incubating Cas9 protein and both sgRNAs for 20 min at room temperature, then adding it to the dissociated cells in Neon Buffer R. A Neon electroporation system (Thermo Fisher Scientific, Waltham, MA, USA) was used to deliver the RNP complexes at two different settings- for TD4 pool: 1100 voltage/20 width/1 pulse, and for TD6 pool: 1,200 voltage/20width/2 pulses.
2.5. Clonal isolation
Each pool was sorted into clones by using a Sony SH800 Cell Sorter into 96-well plates pre-filled with 100 μL of NutriStem supplemented with 10 μM ROCK inhibitor at 1 cell/well. Single cells were allowed to grow for 2 weeks in culture. Surviving clones were passaged and reconsolidated into triplicate 96-well plates, one for continued cell culture, DNA isolation, and RNA isolation. Splitting cells was done using EDTA salt solution (Millipore Sigma, Burlington, MA, USA, 03690) into a new plate each time. Cells were banked at 1e6/vial or.5e6/vial in 10% dimethyl sulfoxide (DMSO) (sterile TC grade) and 90% filter-sterilized Knockout Serum Replacement (Thermo Fisher Scientific, Waltham, MA, USA, 10828028).
2.6. Genotyping of pools and clones
DNA isolation: 2 days after plating, individual clones were washed with DPBS, before Quick Extract Buffer (Lucigen Corporation Middleton, WI, USA, QE09050) was added. The solution was mixed ten times to lyse the cells prior to long-term storage at −80°C. PCR: 1 μL of DNA from Quick extract was loaded into a PCR using Hot start and the following 10 μM primers: Forward: GATGTCGACTCTTTGCCCAC Reverse: AGGCTCCCAAGAAGAATCCA that amplify a 727 base pair region around the cut site. PCR products were sent out for Sanger sequencing to Genewiz (South Plainfield, NJ, USA). Analysis was performed using Synthego Inference of CRISPR Edits (ICE) Analysis. 2019. v3.0. Synthego; (13 September, 2021) for editing efficiency and determination of knockout sequence.
2.7. qPCR for C9orf72 levels
For each cell line, 10,000 cells/well were plated onto Matrigel-coated 96-well plates. 24 h later, total RNA was collected using Cell-to-CT 1-Step TaqMan kit (Invitrogen, Waltham, MA, USA, A2560) following manufacturers specifications. Multiplexed TaqMan was run for C9orf72 (all variants) (HS00376619_m1, FAM) and GAPDH (Hs03929097_g1, VIC-MGB_PL) on a QuantStudio 7. Biological replicates were run in triplicate with four technical replicates each for all samples and plated using an OT2 (Opentrons Labworks, Inc., Brooklyn, NY, USA). Relative C9orf72 abundance was quantified using the delta delta CT method. Data was analyzed using GraphPad Prism.
2.8. Western blot
Cells were washed with DPBS, spun, and the cell pellet was stored in the −80 for long term storage. Cells pellets were lysed in RIPA (Thermo Fisher Scientific, Waltham, MA, USA, 89900) with Halt Protease inhibitor (Thermo Fisher Scientific, Waltham, MA, USA, 78441) and Dnase (Thermo Fisher Scientific, Waltham, MA, USA, 90083). Cell pellets were vortexed every 5 min on ice for a total of 30 min. Then pipetted up and down with a 32-gauge needle and spun at 12,000 RPM for 15 min at 4°C, and the supernatant was collected. Protein was quantified using a Pierce BCA Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA, 23225). A total of 20 μg of protein and 4X Loading Dye Solution (Thermo Fisher Scientific, Waltham, MA, USA, NP0007) were heated to 100°C for 5 min to break down tertiary bonds using 2-mercaptoethanol (Gibco USA, Jenks, OK, USA, 21985–023). Equal amounts of protein were loaded into a NuPAGE 4 to 12% Bis-Tris protein gel (Thermo Fisher Scientific, Waltham, MA, USA, NP0322BOX), and run at 100 V for 120 min in 1X MOPS Buffer (Thermo Fisher Scientific, Waltham, MA, USA, NP0001) on ice. The gel was transferred to PVDF using an iBlot2 at a standardized transfer called P0, which is 20 volts for 1 min, followed by 23 volts for 4 min, then 25 volts for 2 min. Primary antibody against Genetex C9orf72 (GeneTex, Irvine, CA, USA, GTX42591) was used at a 1:400 dilution, and GAPDH (Thermo Fisher Scientific, Waltham, MA, USA, PA1-987) at 1:10,000, separately, in blocking buffer comprised of 5% milk in 1xTBST (Thermo Fisher Scientific, Waltham, MA, USA, 28360) overnight at 4°C. The GeneTex C9orf72 antibody binds to the N-terminus in the longin domain of both the short and long C9orf72 isoforms (Xiao et al., 2016). The next day, three washes were performed using 1xTBST for 10 min each to remove the primary antibody. The membrane was incubated with anti-mouse secondary antibody (LI-COR Biosciences, Lincoln, NE, USA, 926–80010), or anti-rabbit secondary antibody (LI-COR Biosciences, Lincoln, NE, USA, 926–80011), for 1 h at room temperature at a 1:20,000 dilution in blocking buffer. The membrane was then washed three times for 10 min, using 1xTBST. Supersignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Waltham, MA, USA, 34095) was added onto the membrane (1:1). The membrane was incubated for 5 min, protected from light at room temperature, and imaged using the Licor C-Digit imager. Densitometric analysis was done using Image J.
2.9. Pluripotency staining for iPSCs
Cells were rinsed once with DPBS and fixed with 4% PFA for 10 min. PFA was removed with three DPBS rinses, prior to blocking with 3% donkey serum at 4°C overnight. The next day, primary antibodies for Oct3/4, Nanog, Tra-1–60, and Tra-1–81 were added and incubated overnight at 4°C. On the 3 day, primary antibody solutions were removed with three DPBS rinses, before AF488 or AF594 -conjugated secondary antibodies were added for 1 h at room temperature. After rinsing off the secondary antibody solutions, cells were stained with 1 μg/mL DAPI (Thermo Fisher Scientific, Waltham, MA, USA, 62248) for 10 min, before imaging on a Cytation3 automated microscope from BioTek with a 4X objective.
2.10. CellTracker Green CMFDA and Hoescht 333342 staining and imaging
Motor neurons progenitors were dissociated and plated on borate/poly-ornithine coated 384-well plates in laminin supplemented NDM. After 16 h, neurons were stained with 5 μM CellTracker Green and Hoechst 333342 (Thermo Fisher Scientific, Waltham, MA, USA, H3570) following the manufacturers protocol. In short, cells were washed once with NDM, then stained with 10 μM CellTracker Green CMFDA and 1.2 nM Hoechst 333342 for 30 min at 37°C. After incubation, cells were rinsed twice with NDM without phenol-red prior to imaging on a Cytation3 microscope using a 20X objective.
2.11. Storage and dilution of polyGR15
Synthesized polyGR15 (95.18% purity by HPLC, MW 3216.57 g/mol) (GenicBio Limited, Kowloon, Hong Kong SAR, China) was purchased as lyophilized powder and resuspended in DMSO at 10 mM. Aliquots were stored at −80°C until use.
2.12. Storage and dilution of PRMT inhibitor MS023 and inactive analog MS094
Protein arginine methyltransferase inhibitors MS023 (MedChem Express, HY-19615, Monmouth Junction, NJ, USA) and MS094 (Millipore Sigma, SML2548, Burlington, MA, USA) were both resuspended in DMSO at 10 mM and stored in the −80°C until use.
2.13. Preparation and dosing of polyGR15 and PRMT inhibitor
Separately, both polyGR15 and MS023, and MS094 were serially diluted into (for DMEM/F12) Gibco (Jenks, OK, USA) before combining in a 96-well source plate at a 2X concentration. 12 days post-plated MNs in 384-well plates were rinsed with complete NDM, and treated with the compound source plate at a 1:1 ratio. MNs were incubated for 24 or 48 h prior to assessing viability by CellTiter-Blue (Promega Corporation, Madison, WI, USA, G8081) fluorescence or live-cell imaging. Doses for exogenous application of polyGR15 were chosen based on previously established uptake profiles of synthetic DPR constructs in various cell models (Kwon et al., 2014; Kanekura et al., 2018; Gill et al., 2019; Lafarga et al., 2021).
2.14. CellTiter-Blue viability measurements
CellTiter-Blue (Promega Corporation, Madison, WI, USA, G8081) was thawed for 30 min in a water bath at 37°C. For cells in a 96-well format, 20 μL of undiluted CellTiter-Blue was added, and cells were incubated at 37°C for 2.5 h, followed by a top-well fluorescence read (Ex. 560, Em. 590) using a Cytation3. For 384-well plates, CellTiter-Blue was diluted 1:5 into DPBS, then plates were aspirated leaving 22 μL/well, followed by 10 μL diluted CTB addition and a 2.5 h incubation at 37°C. Background subtraction was performed across each plate using wells that did not contain cells.
2.15. Neurite outgrowth analysis
Neurite outgrowth was quantified using PerkinElmer Columbus software. Images were pre-processed with a sliding parabola filter (curvature 100) for the green channel. Nuclei were identified using Hoechst, and cytoplasm were segmented using the green channel. Neurites were identified using CSIRO neurite analysis 2.
2.16. C9orf72-HRE genotyping
Fibroblast genomic DNA was isolated using the Zymo Quick-DNA Miniprep Plus Kit (Zymo Research, Irvine, CA, USA, D4069) following manufacturers recommended protocol. Amplicon length analysis and RP-PCR were performed following (DeJesus-Hernandez et al., 2011), with slight modifications.
2.16.1. Amplicon length analysis
A total of 20 μL PCR reactions were composed of the following: 1 μL of 100 ng/μL genomic DNA, 1 μL 10X PCR Buffer (Qiagen, Germantown, MD, USA, 201203), 0.5 μL of 5 mM dATP, dCTP, dTTP mix (Invitrogen, Waltham, MA, USA, 10297117), 0.5 μL of 5 mM daeza-dGTP (New England Biolabs, Ipswich, MA, USA, N0445L), 1 μL of 10 μM forward and reverse primers (forward: Fam-CAAGGAGGGAAACAACCGCAGCC, reverse: GCAGGCACCGCAACCGCAG), 0.5 μL of 100% DMSO (Sigma-Aldrich, Burlington, MA, USA, D2650), 2 μL of 5M betaine (Sigma-Aldrich, Burlington, MA, USA, B0300-1VL), 0.1 μL Taq polymerase (Qiagen, Germantown, MD, USA, 201205), 9 μL of water. The following thermocycler protocol was performed: 98°C for 5 min, 11 cycles of the following: 97°C for 0:30 min, 55°C for 0:30 min (−1°C/cycle), 68°C for 1:30 min, 24 cycles of the following: 97°C for 0:30 min, 55°C for 0:30 min, 68°C for 1:30 min, a final extension at 68°C for 10 min, followed by a 4°C hold. The resulting PCR product was diluted 1 to 30 with water, and 2 μL was sent to the Georgia Genomics Facility for fragment analysis on an Applied Biosystems 3730xl DNA analyzer. ROX500 and formamide were added to the samples.
2.16.2. Repeat-primed PCR analysis (RP-PCR)
A total of 40 μL PCR reactions were composed of the following: 1 μL of 10 ng/μL genomic DNA, 2 μL 10X PCR Buffer (Qiagen, Germantown, MD, USA, 201203), 1 μL of 5 mM dATP, dCTP, dTTP mix (Invitrogen, Waltham, MA, USA, 10297117), 0.5 μL of 5 mM daeza-dGTP (New England Biolabs, Ipswich, MA, USA, N0445L), 2 μL of 10 μM primer mix (forward: FAM-TGTAAAACGACGGCCAGTCAAGGAGGGAAACAACCGCAG CC, anchor: CAGGAAACAGCTATGACC, reverse: CAGGAAA CAGCTATGACCGGGCCCGCCCCGACCACGCCCCGGCCCC GGCCCCGG), 1 μL of 100% DMSO (Sigma-Aldrich, Burlington, MA, USA, D2650), 4 μL of 5M betaine (Sigma-Aldrich, Burlington, MA, USA, B0300-1VL), 0.2 μL Taq polymerase (Qiagen, Germantown, MD, USA, 201205), 19 μL of water. The following thermocycler protocol was performed: 96.5°C for 12:30 min, 16 cycles of the following: 95°C for 1:00 min, 70°C for 1:00 min (−0.5°C/cycle), 72°C for 3:00 min, six cycles of the following: 95°C for 1:00 min, 61°C for 1:00 min (−1°C/cycle), 72°C for 3:00 min, 35 cycles of the following: 95°C for 1:00 min, 55°C for 1:00 min, 72°C for 3:00 min, a final extension at 72°C for 10 min, followed by a 4°C hold. The resulting PCR product was diluted 1 to 30 with water, and 2 μL was sent to the Georgia Genomics Facility for fragment analysis on an Applied Biosystems 3730xl DNA analyzer. ROX1000 and formamide were added to the samples. For both ALA and RP-PCR, trace files were analyzed using GeneMarker software (Softgenetics).
2.17. Longitudinal live-cell imaging
VACHT-tdTomato MNs were plated at a density of 700 cells per well in a 384-well. 13 days post-plating, an initial imaging timepoint of the entire plate was taken at 24 h prior to treatment with polyGR15 and MS023. Following 24 h post-treatment, VACHT-tdTomato cells were imaged every 6 h starting using a Cytation 3 microscope with a 4x objective. Four images were taken to capture the whole well, and the images were stitched into a composite image. Neuronal somas were quantified and normalized to the to the same well’s previous time point.
2.18. Statistical analysis
Statistical analyses were performed using GraphPad Prism v.9.4.1, Microsoft Excel, and TIBCO Spotfire v11.4.3. Statistical tests included one-way and two-way ANOVAs with Dunnett’s multiple comparisons test, and four parameter logistical regression models to calculate IC50s. Experiments were performed in technical quadruplicates, with biological replicate n values greater than three or four.
3. Results
A dual guide approach targeting exon 2 was used to successfully knockdown C9orf72 expression in pooled cells (Figure 1A). qPCR showed successful knockdown in C9orf72 mRNA levels with an 80–90% reduction in four pools (TD4, TD4 Duplicate, TD6, TD6 Duplicate) using an exon 2–3 C9orf72 probe set, which amplifies all three splice isoforms (Figure 1B). A different total C9orf72 probe set, which spans exons 3–4, showed a lower reduction (∼70%) in C9orf72 expression (Figure 1B). After isolation of isogenic clones, a qPCR revealed that the levels of detectable C9orf72 ranged from 0 to 50% compared to Tracr E1 edited control cells in many clones (Figure 1C). From this large screen of clones, a few candidate clones were selected (not all ∼150 clones shown). One hemizygous clone and two homozygous knockout clones were selected for further analysis. C9orf72 expression analyzed by qPCR showed that compared to the parental line (Pt. 337), total C9orf72 was reduced by 70% in the hemizygous clone (TD6 A2), and 100 and 85% in the two homozygous clones, TD6 F2 and TD6 G11, respectively (Figure 1C). Expression of C9orf72 splice variant 1 and 3 were dramatically lower than total C9orf72, suggesting that variant 2 is the most abundant splice isoform (Supplementary Figure 1). For hemizygous clone TD6 A2, ICE analysis following Sanger sequencing around the deletion showed edits on 1 allele, with 1 nucleotide insertion contributing 46%, and the wild-type allele contributing 42% (Figure 1D). The homozygous clone, TD6 G11 had a 41bp deletion contributing 57% and 4bp deletion at 40% and clone TD6 F2, had a 41bp deletion contributing 99% (Figure 1D). Western blot analysis showed no expression of the C9orf72 protein in the iPSC homozygous knockout clones (TD6 F2 and G11) and an expression lower than 50% in the iPSC hemizygous clone (TD6 A2) (Figures 1E, F). Western blot analysis using motor neuron progenitors at 44 days post-differentiation showed no C9orf72 protein expression in both homozygous knockout clones (TD6 F2 and G11) (Figures 1G, H). Full uncropped blots for Figures 1E, F are shown in Supplementary Figure 6. Following editing and clonal isolation, all cell lines retained expression of pluripotency markers Oct 3/4, Tra-1–60, Tra-1–81, and Nanog (Supplementary Figure 2).
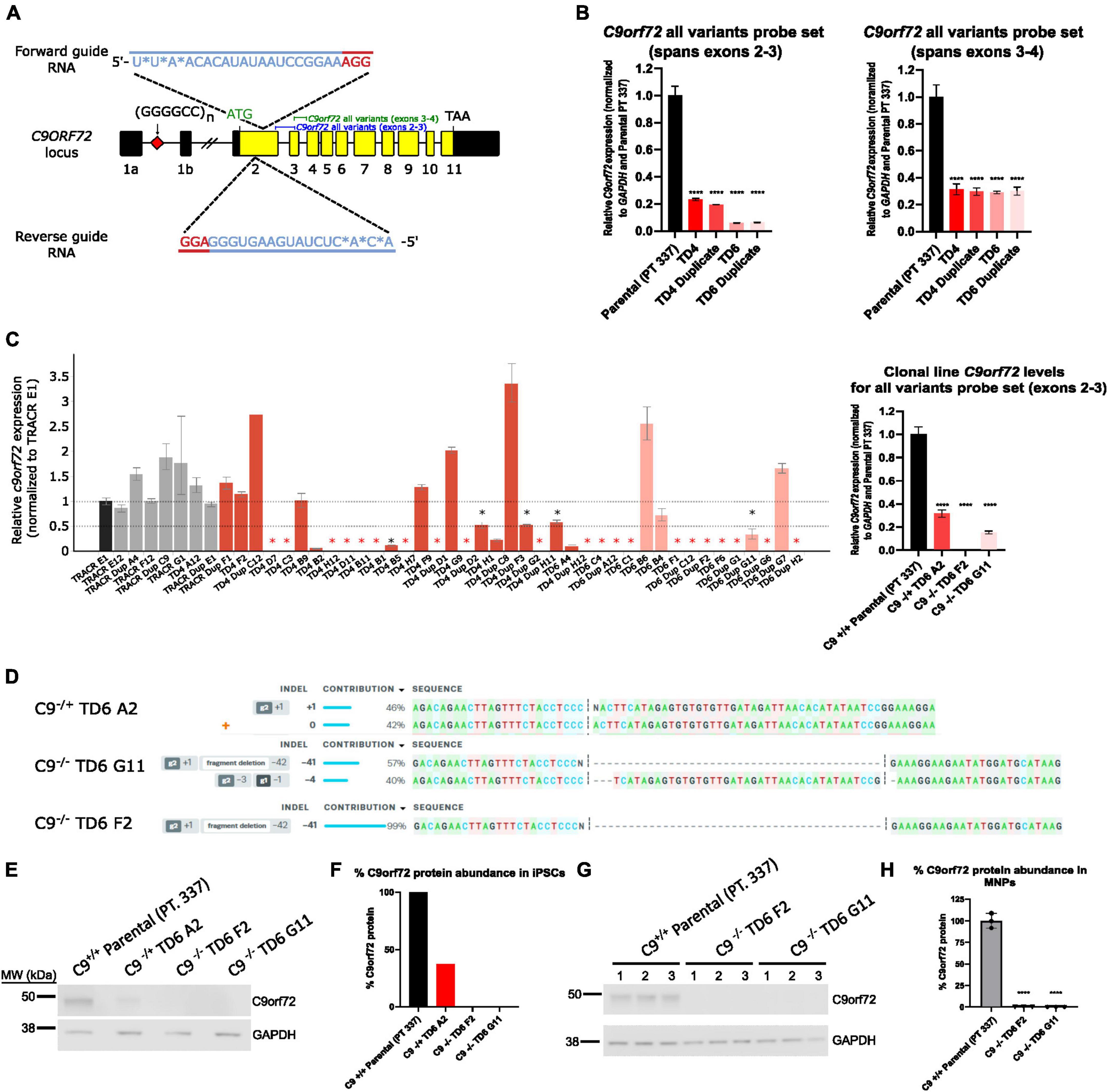
Figure 1. Generation of hemizygous and homozygous C9orf72 knockout induced pluripotent stem cells (iPSCs). (A) Schematic map of the C9orf72 genomic region and the relative binding positions of forward and reverse sgRNA guides used to introduce an INDEL mutation in exon 2, and TaqMan probe sets used to quantify expression. Diagram modified from Shi et al. (2018). (B) Expression of C9orf72 by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in pooled cell populations following Cas9:sgRNA RNP electroporation. Values were normalized to parental cells. ****Denotes p < 0.01 (comparing each pooled cell line to the parental pool by one-way ANOVA analysis of variance followed by Dunnett’s test). Data are presented as mean values ± standard deviations (error bars). (C) Expression of C9orf72 by qRT-PCR in a large screen of isolated clonal lines, as well as in a smaller subset of clonal lines that were used in this study. Black asterisk denotes potential heterozygous KO clones, and red asterisk denotes potential homozygous KO clones. (D) ICE analysis of Sanger sequencing of edited exon 2 region. (E) C9orf72 protein levels shown by Western blotting for iPSC clonal lines. (F) Relative quantification of the C9orf72 protein levels shown in (E). Data are presented as mean values ± standard deviations (error bars). (G) C9orf72 protein levels shown by Western blotting for motor neuron progenitor clonal lines run in triplicate. (H) Relative quantification of the C9orf72 protein levels shown in (G). C9–/– TD6 G11 lane three only contains 10 μg of the total protein (50%) due to insufficient volume.
In addition to the isogenic knockout clones, a wild-type NCRM1-TDT VACHT-ICRE or VACHT-tdTomato line was used (Garcia-Diaz et al., 2020). This line was selected due to its expression of the fluorescent tdTomato protein when the VACHT promotor is active, allowing longitudinal tracking of neuronal survival. An iPSC line from a patient with a C9orf72-expansion (Pt. 137) was also included. C9-HRE genotyping was performed using amplicon length analysis (ALA) and repeat-primed PCR (RP-PCR) (Supplementary Figure 3). All lines were then differentiated into spinal motor neurons using a chemically defined protocol (Du et al., 2015) that mimics normal embryonic development (Figure 2A). In C9+/+ motor neuron progenitors (MNPs), expression of C9orf72 increased by 5-fold compared to iPSCs (Supplementary Figure 1F). The levels of total C9orf72 transcript was reduced ∼100–80% in the homozygous KO lines compared to controls in MNPs. No significant difference in the ability to generate Hb9+ spinal motor neurons was observed between lines (Supplementary Figure 4). Neurons were stained with CellTracker Green 16 h after plating to assess early neuronal morphology (Figure 2B). The number of neurites per cell, number of roots per cell, maximum neurite length and total neurite length were calculated (Figures 2C–F). Compared to the parental wild-type cell line, homozygous knockout lines had significantly less neurite outgrowth. The hemizygous line had slightly less outgrowth compared to control lines, however, only differences in maximum neurite length were significant. The expanded patient line showed similar levels to that of our control Pt. 337 line for all of these neurite statistics across all quantifications. This may be due to differences in genetic background between the genome-edited neurons and the expanded patient line. After generating sMNs from each line, we applied a concentration range of polyGR15 and assessed cell viability after 24 and 48 h of exposure. Cell viability was measured using the resazurin-based fluorescent dye, CellTiter-Blue. We found that the parental Pt. 337 control neurons with intact C9orf72 were not as sensitive to polyGR15 compared to the hemizygous knockout line containing one copy of C9orf72, and compared to the homozygous knockout lines, which were the most sensitive to polyGR15 (Figures 3A, B). Statistical analysis is shown in Table 1. Interestingly, the expanded patient with one copy of C9orf72 intact and one copy of C9orf72 mutated performed the same in response to polyGR15 challenge as the homozygous knockout lines with no intact copies of C9orf72. The inverse correlation between C9orf72 expression and sensitivity to polyGR15 was present at both 24–48 h challenge endpoints.
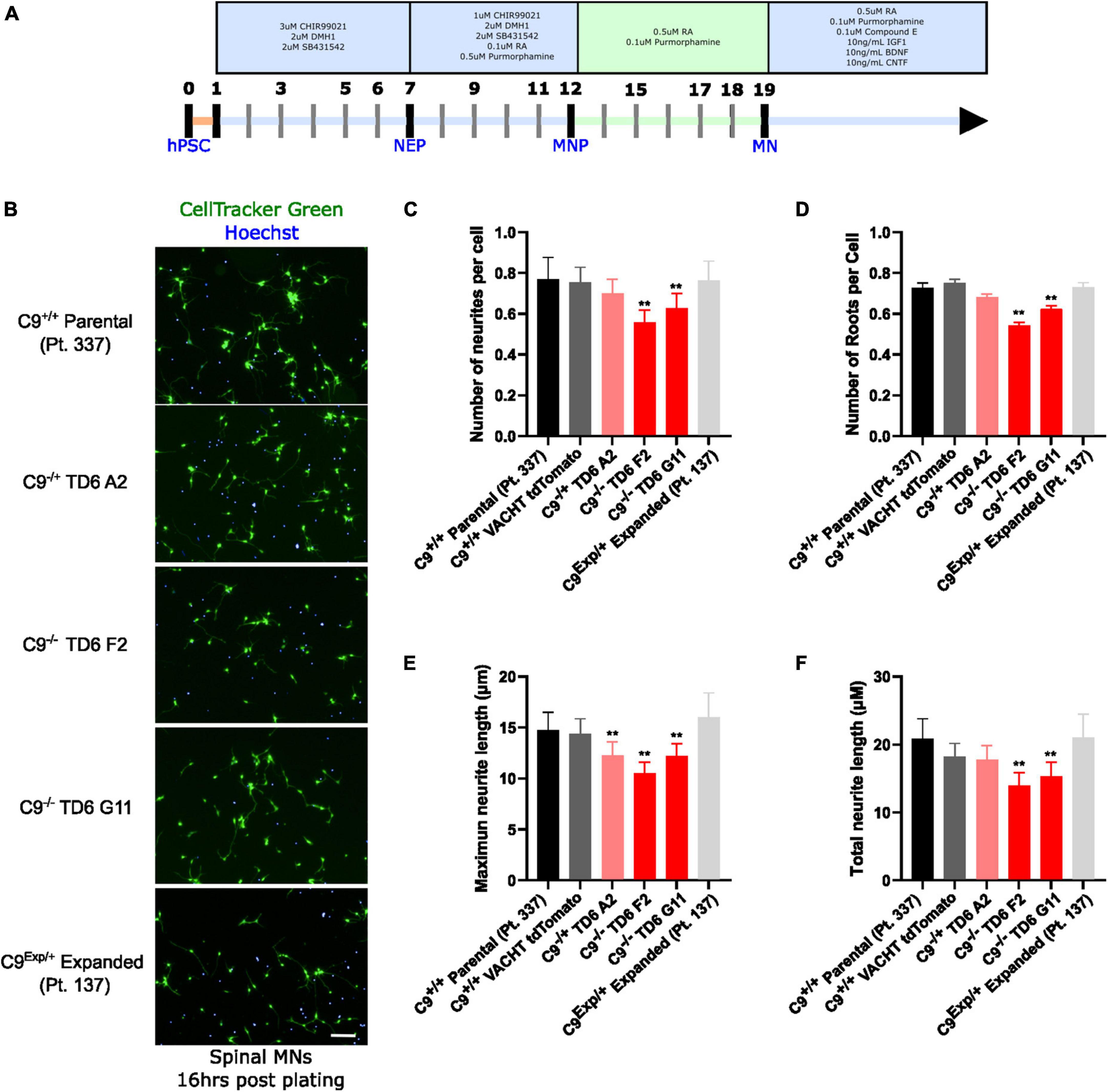
Figure 2. Reduced C9orf72 reduces neurite outgrowth. (A) Schematic timeline for spinal motor neuron differentiation. Adapted from Du et al. (2015) (B) live-cell imaging of spinal motor neurons stained with Cell Tracker Green and Hoechst 16 h after plating. Scale bar = 100 μm. (C) Quantification of the average number of neurite extremities per cell. (D) Quantification of the number of neurite roots per cell. (E) Quantification of the maximum neurite length per cell. (F) Quantification of total neurite length per cell. **Denotes p < 0.01 [comparing each cell line to C9(+/+) parental neurons by one-way ANOVA analysis of variance followed by Dunnett’s test]. Data are presented as mean values ± standard deviations (error bars).
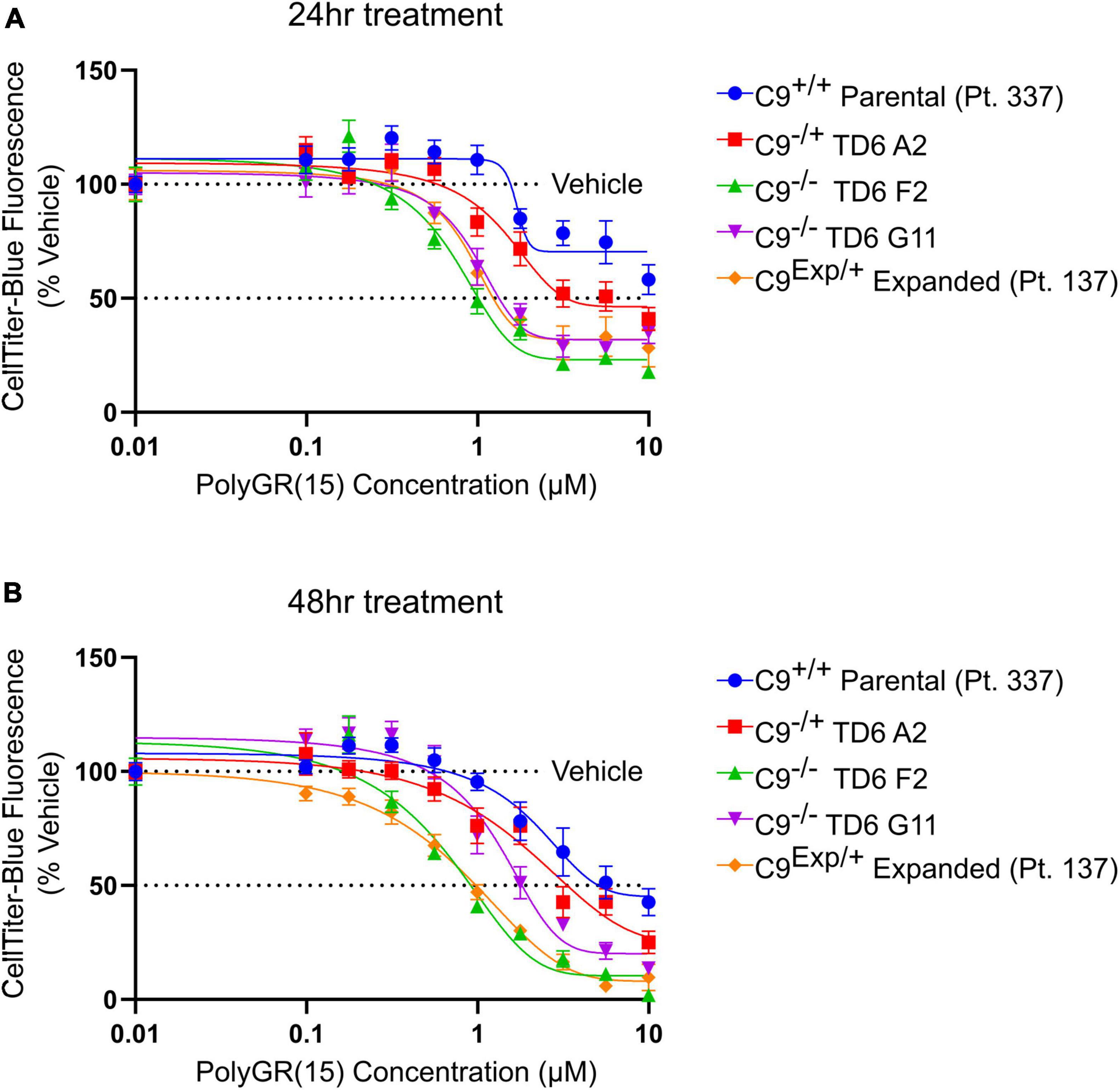
Figure 3. Reduced C9orf72 expression exacerbates polyGR15 toxicity in induced pluripotent stem cell (iPSC)-derived motor neurons. (A) Neuronal survival 24 h after exogenous treatment with polyGR15 ranging from 10 nM to 10 μM or 0.1% DMSO control. Values were normalized to vehicle treated wells for each cell line. (B) Neuronal survival 48 h after exogenous treatment with polyGR15 or 0.1% DMSO control. Values were normalized to vehicle treated wells for each cell line. Data are presented as mean values ± standard deviations (error bars).
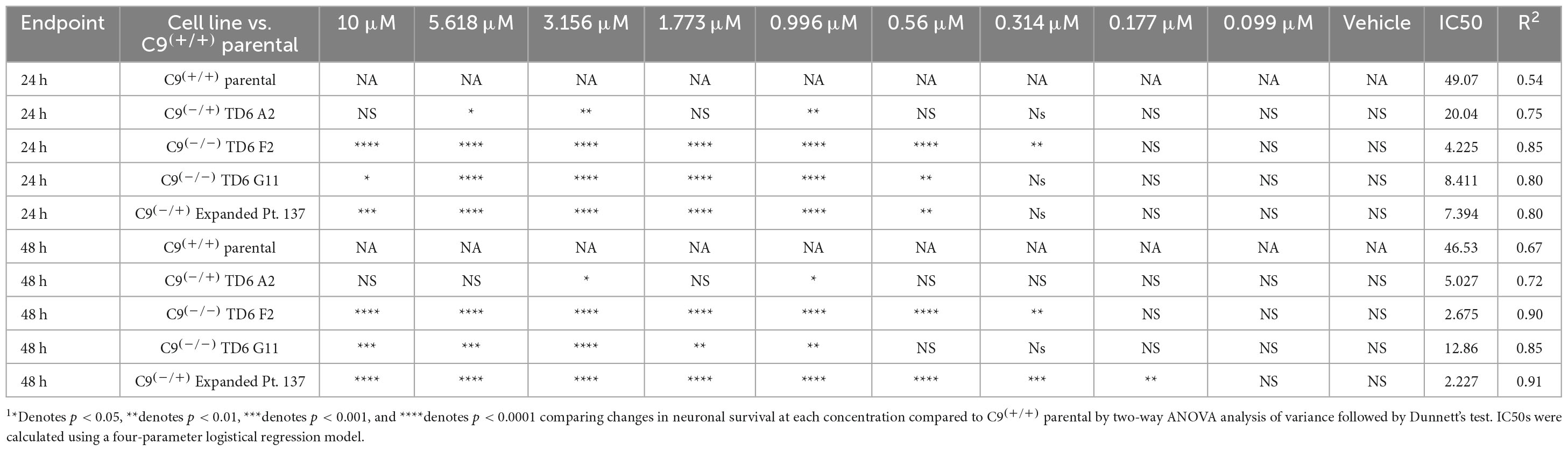
Table 1. Statistical testing of data in Figure 3 indicates significant differences C9(+/+) parental (Pt. 337) and edited lines following exposure to PolyGR151.
Previous work has shown that inhibition of Type I PRMTs reduces toxicity of arginine-rich DPRs in NSC-34 and primary mouse spinal motor neurons (Premasiri et al., 2020). To test if these results would replicate in iPSC-derived sMNs, the VACHT-tdTomato reporter line was used (Figure 4A). Following terminal differentiation, day 12 human sMNs were co-treated with polyGR15 and either Type I PRMT inhibitor MS023, or its inactive analog MS094 before measuring viability with CellTiter-Blue (Figure 4B). Addition of MS023 was able to rescue neuronal viability against 0.1–1.0 μM polyGR15 (Figure 4C). Surprisingly, the inactive analog MS094 was able to increase survival against 0.1–0.32 μM polyGR15 (Figure 4D). To assess cell viability using a different strategy, we employed longitudinal live-cell imaging to measure the number of tdTomato+ MNs every 6 h following application of 1–5.62 μM polyGR15 alone, or with co-treated with MS023 (Supplementary Figures 5A–D). Here, a concentration-dependent rescue of neuronal survival was seen with 0.1–1.0 μM MS023 against polyGR15 induced-toxicity. The preservation of intact neurites was seen 84 h post-polyGR15 when 1 μM MS023 was present (Supplementary Figure 5A). Lastly, we sought to determine if type I PRMT inhibition was beneficial in C9orf72-expanded MNs. Day 24 MNs were co-treated with 1 μM polyGR15 and 1 nM–10 μM MS023 for 24 h prior to cell survival assessment by CellTiter-Blue fluorescence (Figure 5). The increased sensitivity to 1 μM polyGR15 was again seen in the C9-expanded MNs. MS023 abrogated polyGR15 toxicity seen in the C9-expanded cells.
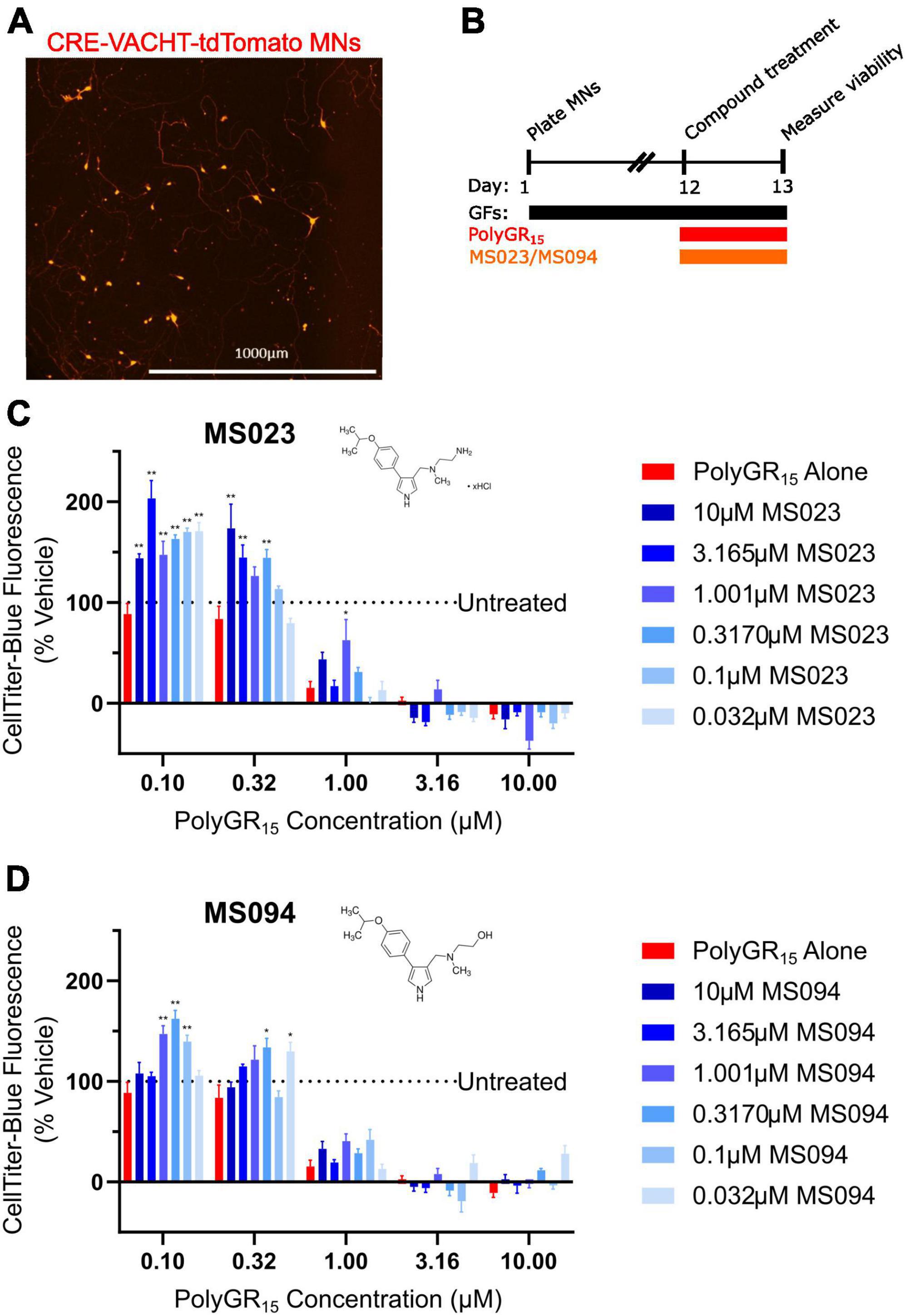
Figure 4. Protein arginine methyltransferase inhibition improves induced pluripotent stem cell (iPSC)-motor neuron (MN) survival in response to polyGR15 challenge. (A) Representative image of tdTomato+ CRE-VACHT-tdTomato control motor neurons employed in polyGR15–PRMT inhibitor cross-titration. (B) Timeline for cross-titration experiment. Neurons were differentiated to spinal motor neurons, and on day 1, dissociated and plated into 384-well plates. After 12 days, neurons were treated with polyGR15 concentrations ranging from 100 nM to 10 μM, and co-treated with MS023 or MS094 concentrations ranging from 32 nM to 10 μM. Cell survival was measured 24 or 48 h after treatment. (C) Neuronal survival following polyGR15 co-treated with MS023 normalized to vehicle treated wells (0.2% DMSO). (D) Neuronal survival following polyGR15 co-treated with MS023 normalized to vehicle treated wells (0.2% DMSO). *Denotes p < 0.05, and **denotes p < 0.01 (comparing MS023 or MS094 co-treated groups to polyGR15 alone by one-way ANOVA analysis of variance followed by Dunnett’s test). Data are presented as mean values ± standard deviations (error bars).
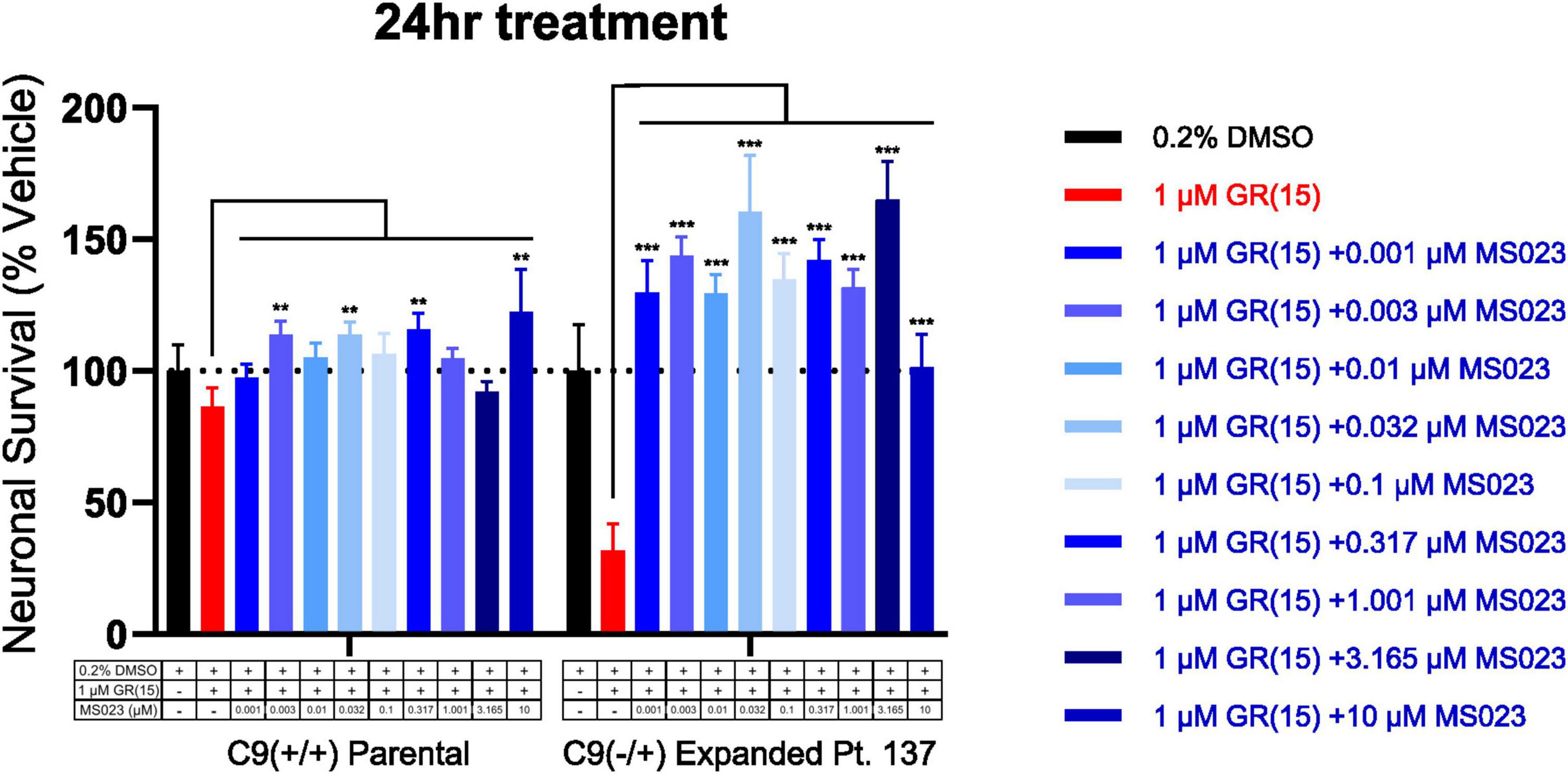
Figure 5. Protein arginine methyltransferase inhibition rescues polyGR15-induced toxicity in C9orf72-expanded motor neurons. Survival of spinal motor neurons 24 days post-plating following co-treatment with 1 μM polyGR15 and concentrations of MS023 ranging from 1 nM to 10 μM. **Denotes p < 0.01, and ***denotes p < 0.001 (comparing MS023 co-treated groups to polyGR15 alone by two-way ANOVA analysis of variance followed by Dunnett’s test). Data are presented as mean values ± standard deviations (error bars).
4. Discussion
A decade following the association of C9orf72 repeat expansion mutations in many cases of familial and sporadic ALS and FTD there have been tremendous strides in elucidating processes that may be implicated in these diseases (Balendra and Isaacs, 2018; Masrori and Van Damme, 2020). C9orf72 mutation-mediated neurodegeneration has proven to be complex, both biologically and clinically. Prominent hypotheses implicate both gain-of-function and loss-of-function mechanisms that lead to neuronal cell death. Thusly, treatment approaches should aim to address both, as few research studies have explored the additive toxic effects of both. Approaches to treat the C9orf72 HRE have mostly aimed at reducing toxic gain-of-function– with multiple antisense oligonucleotide therapeutics in clinical development now (Liu et al., 2021; Tran et al., 2022) or in recent months (Lagier-Tourenne et al., 2013). Biogen’s IONIS-C9Rx is an ASO targeting the C9orf72 open reading frame mRNA, which is aimed at reducing the DPRs translated from the expansion. Similarly, Wave Life Science’s WVE-3972–01 is a sterically pure ASO targeting downregulation of DPRs and pathogenic C9orf72-derived RNA transcripts and claims to have kept intact the healthy C9orf72 transcriptional V2 variants (Liu et al., 2022). These treatments aim to address downstream effects within the cell. However, it is important to consider a therapy that does not reduce levels of wild-type C9orf72. The association between increased levels of transcript variant 1 in the frontal cortex and cerebellum with longer survival in C9orf72 expansion carriers should caution against therapies that reduce C9orf72 levels (van Blitterswijk et al., 2015). Several small molecule strategies have also been explored for treatment of C9orf72-linked neurodegeneration. Metformin, an FDA-approved drug for the treatment of type 2 diabetes, is being repurposed as a small molecule approach to reduce production of toxic C9orf72 non-canonical translation products (Zu et al., 2020). A repositioned small molecule drug, TPN101, is being developed for C9orf72 ALS (NCT04993755) with an emphasis on its potential to reduce central nervous system inflammation (Zampatti et al., 2022). One challenge when developing these small molecule treatment approaches is careful consideration to avoid lowering levels of endogenous C9orf72 protein when targeting sequences adjacent to the hexanucleotide repeat, aiming to instead target repeat-containing RNAs (Zhu et al., 2020; Bush et al., 2022). One small molecule developed with dual function was able to bind the expansion and recruit an endonuclease to remove the expansion from the RNA without effecting the levels of C9orf72 protein (Bush et al., 2022).
We show that human iPSC-derived MNs lacking one or both of the C9orf72 alleles are more susceptible to polyGR15 repeat toxicity. Additionally, we demonstrate that iPSC-derived motor neurons from a person harboring one C9orf72-expanded allele are similarly sensitive to polyGR15 toxicity as cells completely lacking C9orf72 alleles. These findings may help elucidate the biological cascade of events that result in C9orf72 mutation-mediated neuronal cell death. One of the earliest observations about C9orf72 neurodegeneration was that people harboring repeat expansion mutations manifest reduced expression of C9orf72 transcript variants 1 and 2 (DeJesus-Hernandez et al., 2011; van Blitterswijk et al., 2015). This suggested the possibility of toxic loss-of-function playing a role in C9orf72-linked neurodegeneration. Our results are consistent with the idea that production of toxic DPRs in the context of reduced C9orf72 in people with repeat expansions would be more likely to result in neuronal cell death than the same exposure to toxic DPR in the context of normal C9orf72 expression levels. Interestingly, the fact that toxic DPR exposure was equivalently toxic to cells from a person with one expanded C9orf72 allele and cells with a C9orf72 double knockout, suggests that additional gain-of-function mechanisms might be playing a role as well. These might include toxic DPRs translated from the expanded repeat allele or toxic repeat expanded sense or antisense RNAs. In future studies, we will have to consider getting a baseline for the amount of endogenous polyGR present in each sample before addition of exogenous polyGR15 in order to determine background toxicity caused by endogenous polyGR.
Equally important in this study is the observation that inhibition of asymmetric arginine dimethylation by Type I PRMT inhibitor MS023 was protective against polyGR15 induced toxicity in iPSC-derived sMNs. We have previously observed that MS023 can completely abrogate toxicity resulting from polyGR15 and polyPR15 challenge in NSC-34 cells, a motor neuron-like cell line (Premasiri et al., 2020). Further, showing this phenomenon is present in iPSC-derived MNs from an expanded patient using polyGR15 implicates PRMTs in ALS disease pathology, and shows that it holds promise at elucidating further study into the mechanism of action of this rescue phenotype. This could shed light on how inhibiting PRMTs could lead to rescue of patient cells from toxicity from polyGR15. Our observation that MS023 rescues cell viability above vehicle-treated cells when measured using CellTiter-Blue is worth further investigation. Reduction of resazurin to resorufin present in CellTiter-Blue by the mitochondrial respiratory chain in live cells suggests metabolic activity is enhanced following MS023 treatment in the presence of polyGR15. It has been shown that PRMT inhibition by MS023 treatment results in global transcriptomic and proteomic alterations (Maron et al., 2021). The fact that MS023 also rescued neuron survival and preserved neurite integrity agrees with previous NSC-34, where PRMT inhibition rescued both metabolic activity and LDH release following DRP challenge (Premasiri et al., 2020). Further work to better understand the mechanism of rescue is critical to designing more selective PRMT inhibitors. Modulation of the levels of each Type I PRMT, through knockdown or knockout, will help shed light on which enzymes should be targeted.
We have previously described evidence supporting roles of protein arginine methylation in relation to cell stress responses such as splicing, chromatin remodeling, and stress granule dynamics in C9orf72 cell-based gain-of-function models (Gill et al., 2021). The current studies reveal a likely interaction between native C9orf72 protein and PRMTs in regulating these pathways in C9orf72-mutation-mediated neurodegeneration. Recent studies have highlighted the importance of stress granule formation and requisite liquid-liquid phase separation as modulators of C9orf72 mutation-mediated cytotoxicity in cell-based models (Liu et al., 2023; Millar et al., 2023). For example, C9orf72 has been demonstrated to be a regulator of the integrated stress response (ISR), facilitating stimulation of the unfolded protein response (UPR), inhibition of stress granule formation, and LLPS through interaction with eIF2a (Zheng et al., 2022). Accordingly, loss of C9orf72 results in enhanced stress granule formation and non-canonical translation of toxic DPRs in a feed-forward mechanism (Cheng et al., 2018). Additionally, protein arginine methylation of stress granule-associated proteins regulates phase separation, stress granule dynamics, and translation in stressed cells. Indeed, the type III PRMT, PRMT7, methylates eIF2a in a manner required for eIF2a-dependent stress granule formation (Haghandish et al., 2019). Additionally, arginine methylation of RGG/RG motifs within stress granule proteins can limit their interactions with RNA, poly (ADP-ribose), and other stress granule components (Tsang et al., 2019). Notably, methylation of key stress granule protein G3BP1 by PRMT1 prevents large stress granule formation (Tsai et al., 2016), and asymmetric dimethylation of several RNA binding proteins results in their recruitment to stress granules (Xie and Denman, 2011). Multiple described roles for PRMTs driving stress granule assembly and composition indicate that PRMT activity might influence the ISR in a manner protective to C9orf72-deficient cells.
Further, PRMT activity has been shown to modulate autophagy (Choi et al., 2019). In the case of simultaneous C9orf72 loss-of-function and DPR gain-of-function challenge modeled in the current experiments, pharmacological inhibition of Type I PRMT activity in C9orf72-deficient cells might increase autophagic clearance of DPRs by way of compensatory Type II PRMT substrate scavenging (Dhar et al., 2013; Stouth et al., 2017). Symmetric dimethylation of stress granule proteins by PRMT5 has been shown to drive the association of C9orf72 and p62 to the stress granule as a complex that works to eliminate stress granules by autophagy (Chitiprolu et al., 2018). In the present study, application of the Type I PRMT inhibitor MS023 concurrent with polyGR challenge most significantly rescued survival of iPSC-derived motor neurons harboring the C9orf72 repeat expansion mutation (Figure 5). Future studies should determine whether Type I PRMT inhibition enhances symmetric dimethylation of stress granule proteins, and subsequently drives more native C9orf72 protein to associate with p62 to promote protective autophagy of either accumulated DPRs or other deleterious inclusions. Such a mechanism could act to ameliorate reduced impaired autophagy observed in C9orf72 in vitro knockdown models (Sellier et al., 2016; Webster et al., 2016) and subsequent increases in DPR toxicity (Boivin et al., 2020; Zhu et al., 2020).
Other vital cellular mechanisms are co-regulated by C9orf72 protein and PRMT activity. One recent publication found that C9orf72 is involved in recruiting DNA repair enzymes to double-stranded breaks when localized to the nucleus, and that repair of double-stranded DNA breaks following polyGR overexpression is reduced in C9orf72-deficient cells (He et al., 2022). PRMTs have a vast set of roles in the DNA damage response that vary by PRMT enzyme and arginine methylation type (Lorton and Shechter, 2019; Urulangodi and Mohanty, 2020; Brobbey et al., 2022). Modulation of PRMT activity in the absence of C9orf72-mediated DNA damage repair may act to mitigate DNA damage response deficits in ALS loss-of-function and gain of function models. Recently, one study illustrated that loss of C9orf72 disrupts the Ran-GTPase gradient and nucleocytoplasmic transport in both in vitro and in vivo models (McGoldrick et al., 2023). Further, this study illustrated that abundance of importin β-1 granules is increased in states of C9orf72-deficiency. PRMT activity also modulates the Ran-GTPase gradient and nucleocytoplasmic transport. Namely, chromatin-bound RCC1 activates Ran-GTPase to initiate nucleocytoplasmic transport, and localization of RCC1 to chromatin requires methylation of RCC1 by PRMTs (Huang et al., 2021). Additionally, PRMT-mediated arginine methylation at nuclear transport domains (NTDs) and nuclear localization sequences (NLSs) regulates nucleocytoplasmic transport through interaction with importin proteins, including importin β (Smith et al., 2004). These significant overlaps in regulatory roles of PRMTs and C9orf72 warrant further investigation into whether modulation of PRMT behavior in C9orf72 mutation-mediated neurodegeneration might influence C9orf72 loss-of-function and gain-of-function consequences.
Data availability statement
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
TD and KD contributed to conception, design of the study, performed the statistical analysis and made figures. AG provided study design guidance and reagents based on previous data for formulations and dosages. KD, AG, FV, and TD wrote the first draft of the manuscript, introduction, and discussion. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was generously supported by the Augie’s Quest, Mindy Urlaub, and David Weinberg.
Acknowledgments
We thank people with ALS and their families and friends for their support- in particular, those living with C9orf72 repeat expansion-mediated ALS. We thank to Target ALS and the Columbia Stem Cell Core and its investigators, Barbara Corneo, Hynek Wichterle, and Alejandro Garcia Diaz provided us with the NCRM1-TDT VACHT-ICRE.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2023.1134090/full#supplementary-material
References
Amick, J., Roczniak-Ferguson, A., and Ferguson, S. M. (2016). C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol. Biol. Cell. 27, 3040–3051. doi: 10.1091/mbc.e16-01-0003
Aoki, Y., Manzano, R., Lee, Y., Dafinca, R., Aoki, M., Douglas, A. G. L., et al. (2017). C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140, 887–897. doi: 10.1093/brain/awx024
Ash, P. E. A., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., DeJesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Blanchet, F., Cardona, A., Letimier, F. A., Hershfield, M. S., and Acuto, O. (2005). CD28 costimulatory signal induces protein arginine methylation in T cells. J. Exp. Med. 202, 371–377. doi: 10.1084/jem.20050176
Boivin, M., Pfister, V., Gaucherot, A., Ruffenach, F., Negroni, L., Sellier, C., et al. (2020). Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 39:e100574. doi: 10.15252/embj.2018100574
Braems, E., Swinnen, B., and Van Den Bosch, L. (2020). C9orf72 loss-of-function: A trivial, stand-alone or additive mechanism in C9 ALS/FTD? Acta Neuropathol. 140, 625–643. doi: 10.1007/s00401-020-02214-x
Brobbey, C., Liu, L., Yin, S., and Gan, W. (2022). The role of protein arginine methyltransferases in DNA damage response. IJMS 23:9780. doi: 10.3390/ijms23179780
Bush, J. A., Meyer, S. M., Fuerst, R., Tong, Y., Li, Y., Benhamou, R. I., et al. (2022). A blood–brain penetrant RNA-targeted small molecule triggers elimination of r(G 4 C 2) exp in c9ALS/FTD via the nuclear RNA exosome. Proc. Natl. Acad. Sci. U. S. A. 119:e2210532119. doi: 10.1073/pnas.2210532119
Cheng, W., Wang, S., Mestre, A. A., Fu, C., Makarem, A., Xian, F., et al. (2018). C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2α phosphorylation. Nat. Commun. 9:51. doi: 10.1038/s41467-017-02495-z
Chitiprolu, M., Jagow, C., Tremblay, V., Bondy-Chorney, E., Paris, G., Savard, A., et al. (2018). A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat. Commun. 9:2794. doi: 10.1038/s41467-018-05273-7
Choi, S., Jeong, H. J., Kim, H., Choi, D., Cho, S. C., Seong, J. K., et al. (2019). Skeletal muscle-specific Prmt1 deletion causes muscle atrophy via deregulation of the PRMT6-FOXO3 axis. Autophagy 15, 1069–1081. doi: 10.1080/15548627.2019.1569931
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Dhar, S., Vemulapalli, V., Patananan, A. N., Huang, G. L., Di Lorenzo, A., Richard, S., et al. (2013). Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci. Rep. 3:1311. doi: 10.1038/srep01311
Du, Z. W., Chen, H., Liu, H., Lu, J., Qian, K., Huang, C. L., et al. (2015). Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 6:6626. doi: 10.1038/ncomms7626
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A. K., Levina, V., et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595. doi: 10.1093/hmg/ddu068
Fomin, V., Richard, P., Hoque, M., Li, C., Gu, Z., Fissore-O’Leary, M., et al. (2018). The C9ORF72 gene, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, encodes a protein that functions in control of endothelin and glutamate signaling. Mol. Cell. Biol. 38, e00155–18. doi: 10.1128/MCB.00155-18
Freibaum, B. D., and Taylor, J. P. (2017). The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front. Mol. Neurosci. 10:35. doi: 10.3389/fnmol.2017.00035
Frick, P., Sellier, C., Mackenzie, I. R. A., Cheng, C. Y., Tahraoui-Bories, J., Martinat, C., et al. (2018). Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol. Commun. 6:72. doi: 10.1186/s40478-018-0579-0
Garcia-Diaz, A., Efe, G., Kabra, K., Patel, A., Lowry, E. R., Shneider, N. A., et al. (2020). Standardized reporter systems for purification and imaging of human pluripotent stem cell-derived motor neurons and other cholinergic cells. Neuroscience 450, 48–56. doi: 10.1016/j.neuroscience.2020.06.028
Gendron, T. F., and Petrucelli, L. (2018). Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harb. Perspect. Med. 8:a024224. doi: 10.1101/cshperspect.a024224
Gill, A. L., Premasiri, A. S., and Vieira, F. G. (2021). Hypothesis and theory: Roles of arginine methylation in C9orf72-mediated ALS and FTD. Front. Cell. Neurosci. 15:633668. doi: 10.3389/fncel.2021.633668
Gill, A. L., Wang, M. Z., Levine, B., Premasiri, A., and Vieira, F. G. (2019). Primary neurons and differentiated NSC-34 cells are more susceptible to arginine-rich ALS dipeptide repeat protein-associated toxicity than non-differentiated NSC-34 and CHO cells. IJMS 20:6238. doi: 10.3390/ijms20246238
Gittings, L. M., Boeynaems, S., Lightwood, D., Clargo, A., Topia, S., Nakayama, L., et al. (2020). Symmetric dimethylation of poly-GR correlates with disease duration in C9orf72 FTLD and ALS and reduces poly-GR phase separation and toxicity. Acta Neuropathol. 139, 407–410. doi: 10.1007/s00401-019-02104-x
Haghandish, N., Baldwin, R. M., Morettin, A., Dawit, H. T., Adhikary, H., Masson, J.-Y., et al. (2019). PRMT7 methylates eukaryotic translation initiation factor 2α and regulates its role in stress granule formation. Mol. Biol. Cell. 30, 778–793. doi: 10.1091/mbc.E18-05-0330
He, L., Liang, J., Chen, C., Chen, J., Shen, Y., Sun, S., et al. (2022). C9orf72 functions in the nucleus to regulate DNA damage repair. Cell Death Differ. 30, 716–730. doi: 10.1038/s41418-022-01074-0
Huang, T., Yang, Y., Song, X., Wan, X., Wu, B., Sastry, N., et al. (2021). PRMT6 methylation of RCC1 regulates mitosis, tumorigenicity, and radiation response of glioblastoma stem cells. Mol. Cell 81, 1276–1291.e9. doi: 10.1016/j.molcel.2021.01.015
Ikenaka, K., Atsuta, N., Maeda, Y., Hotta, Y., Nakamura, R., Kawai, K., et al. (2019). Increase of arginine dimethylation correlates with the progression and prognosis of ALS. Neurology 92, e1868–e1877. doi: 10.1212/WNL.0000000000007311
Ikenaka, K., Maeda, Y., Hotta, Y., Nagano, S., Yamada, S., Ito, D., et al. (2022). Serum asymmetric dimethylarginine level correlates with the progression and prognosis of amyotrophic lateral sclerosis. Eur. J. Neurol. 29, 1410–1416. doi: 10.1111/ene.15254
Kanekura, K., Harada, Y., Fujimoto, M., Yagi, T., Hayamizu, Y., Nagaoka, K., et al. (2018). Characterization of membrane penetration and cytotoxicity of C9orf72-encoding arginine-rich dipeptides. Sci. Rep. 8:12740. doi: 10.1038/s41598-018-31096-z
Kramer, N. J., Haney, M. S., Morgens, D. W., Jovičić, A., Couthouis, J., Li, A., et al. (2018). CRISPR–Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 50, 603–612. doi: 10.1038/s41588-018-0070-7
Kwon, I., Xiang, S., Kato, M., Wu, L., Theodoropoulos, P., Wang, T., et al. (2014). Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345, 1139–1145. doi: 10.1126/science.1254917
Lafarga, V., Sirozh, O., Díaz-López, I., Galarreta, A., Hisaoka, M., Zarzuela, E., et al. (2021). Widespread displacement of DNA- and RNA-binding factors underlies toxicity of arginine-rich cell-penetrating peptides. EMBO J. 40:e103311. doi: 10.15252/embj.2019103311
Lagier-Tourenne, C., Baughn, M., Rigo, F., Sun, S., Liu, P., Li, H. R., et al. (2013). Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U. S. A. 110, E4530–E4539. doi: 10.1073/pnas.1318835110
Lawson, B. R., Manenkova, Y., Ahamed, J., Chen, X., Zou, J. P., Baccala, R., et al. (2007). Inhibition of transmethylation down-regulates CD4 T cell activation and curtails development of autoimmunity in a model system. J. Immunol. 178, 5366–5374. doi: 10.4049/jimmunol.178.8.5366
Lee, K. H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774–788.e17. doi: 10.1016/j.cell.2016.10.002
Leskelä, S., Huber, N., Rostalski, H., Natunen, T., Remes, A., Takalo, M., et al. (2019). C9orf72 proteins regulate autophagy and undergo autophagosomal or proteasomal degradation in a cell type-dependent manner. Cells 8:1233. doi: 10.3390/cells8101233
Liu, Y., Andreucci, A., Iwamoto, N., Yin, Y., Yang, H., Liu, F., et al. (2022). Preclinical evaluation of WVE-004, an investigational stereopure oligonucleotide for the treatment of C9orf72-associated ALS or FTD. Mol. Ther. Nucleic Acids 28, 558–570. doi: 10.1016/j.omtn.2022.04.007
Liu, Y., Dodart, J. C., Tran, H., Berkovitch, S., Braun, M., Byrne, M., et al. (2021). Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat. Commun. 12:847. doi: 10.1038/s41467-021-21112-8
Liu, Y., Huang, Z., Liu, H., Ji, Z., Arora, A., Cai, D., et al. (2023). DNA-initiated epigenetic cascades driven by C9orf72 hexanucleotide repeat. Neuron doi: 10.1016/j.neuron.2023.01.022 [Epub ahead of print].
Lorton, B. M., and Shechter, D. (2019). Cellular consequences of arginine methylation. Cell. Mol. Life Sci. 76, 2933–2956. doi: 10.1007/s00018-019-03140-2
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 11, 323–330. doi: 10.1016/S1474-4422(12)70043-1
Maron, M. I., Lehman, S. M., Gayatri, S., DeAngelo, J. D., Hegde, S., Lorton, B. M., et al. (2021). Independent transcriptomic and proteomic regulation by type I and II protein arginine methyltransferases. iScience 24:102971. doi: 10.1016/j.isci.2021.102971
Masrori, P., and Van Damme, P. (2020). Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 27, 1918–1929. doi: 10.1111/ene.14393
McGoldrick, P., Lau, A., You, Z., Durcan, T. M., and Robertson, J. (2023). Loss of C9orf72 perturbs the Ran-GTPase gradient and nucleocytoplasmic transport, generating compositionally diverse Importin β-1 granules. Cell Rep. 42:112134. doi: 10.1016/j.celrep.2023.112134
Millar, S. R., Huang, J. Q., Schreiber, K. J., Tsai, Y. C., Won, J., Zhang, J., et al. (2023). A new phase of networking: The molecular composition and regulatory dynamics of mammalian stress granules. Chem. Rev. doi: 10.1021/acs.chemrev.2c00608 [Epub ahead of print].
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
O’Rourke, J. G., Bogdanik, L., Yáñez, A., Lall, D., Wolf, A. J., Muhammad, A. K. M. G., et al. (2016). C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329. doi: 10.1126/science.aaf1064
Pal, A., Kretner, B., Abo-Rady, M., Glaβ, H., Dash, B. P., Naumann, M., et al. (2021). Concomitant gain and loss of function pathomechanisms in C9ORF72 amyotrophic lateral sclerosis. Life Sci. Alliance 4:e202000764. doi: 10.26508/lsa.202000764
Peng, C., and Wong, C. C. (2017). The story of protein arginine methylation: Characterization, regulation, and function. Expert Rev. Proteom. 14, 157–170. doi: 10.1080/14789450.2017.1275573
Premasiri, A. S., Gill, A. L., and Vieira, F. G. (2020). Type I PRMT inhibition protects against C9ORF72 arginine-rich dipeptide repeat toxicity. Front. Pharmacol. 11:569661. doi: 10.3389/fphar.2020.569661
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Sellier, C., Campanari, M. L., Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9orf72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W. H., Hung, S. T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Sivadasan, R., Hornburg, D., Drepper, C., Frank, N., Jablonka, S., Hansel, A., et al. (2016). C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat. Neurosci. 19, 1610–1618. doi: 10.1038/nn.4407
Smith, W. A., Schurter, B. T., Wong-Staal, F., and David, M. (2004). Arginine methylation of RNA helicase A determines Its subcellular localization. J. Biol. Chem. 279, 22795–22798. doi: 10.1074/jbc.C300512200
Stouth, D. W., vanLieshout, T. L., Shen, N. Y., and Ljubicic, V. (2017). Regulation of skeletal muscle plasticity by protein arginine methyltransferases and their potential roles in neuromuscular disorders. Front. Physiol. 8:870. doi: 10.3389/fphys.2017.00870
Suh, E., Lee, E. B., Neal, D., Wood, E. M., Toledo, J. B., Rennert, L., et al. (2015). Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration. Acta Neuropathol. 130, 363–372. doi: 10.1007/s00401-015-1445-9
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4:51. doi: 10.1186/s40478-016-0324-5
Tao, Z., Wang, H., Xia, Q., Li, K., Li, K., Jiang, X., et al. (2015). Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 24, 2426–2441. doi: 10.1093/hmg/ddv005
Tran, H., Moazami, M. P., Yang, H., McKenna-Yasek, D., Douthwright, C. L., Pinto, C., et al. (2022). Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 28, 117–124. doi: 10.1038/s41591-021-01557-6
Tsai, W. C., Gayatri, S., Reineke, L. C., Sbardella, G., Bedford, M. T., and Lloyd, R. E. (2016). Arginine demethylation of G3BP1 promotes stress granule assembly. J. Biol. Chem. 291, 22671–22685. doi: 10.1074/jbc.M116.739573
Tsang, B., Arsenault, J., Vernon, R. M., Lin, H., Sonenberg, N., Wang, L.-Y., et al. (2019). Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc. Natl. Acad. Sci. U. S. A. 116, 4218–4227. doi: 10.1073/pnas.1814385116
Urulangodi, M., and Mohanty, A. (2020). DNA damage response and repair pathway modulation by non-histone protein methylation: Implications in neurodegeneration. J. Cell Commun. Signal. 14, 31–45. doi: 10.1007/s12079-019-00538-2
van Blitterswijk, M., Gendron, T. F., Baker, M. C., DeJesus-Hernandez, M., Finch, N. A., Brown, P. H., et al. (2015). Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 130, 863–876. doi: 10.1007/s00401-015-1480-6
Van Mossevelde, S., van der Zee, J., Cruts, M., and Van Broeckhoven, C. (2017). Relationship between C9orf72 repeat size and clinical phenotype. Curr. Opin. Genet. Dev. 44, 117–124. doi: 10.1016/j.gde.2017.02.008
Waite, A. J., Bäumer, D., East, S., Neal, J., Morris, H. R., Ansorge, O., et al. (2014). Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 35, 1779.e5–1779.e13. doi: 10.1016/j.neurobiolaging.2014.01.016
Webster, C. P., Smith, E. F., Bauer, C. S., Moller, A., Hautbergue, G. M., Ferraiuolo, L., et al. (2016). The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 35, 1656–1676. doi: 10.15252/embj.201694401
Wen, X., Tan, W., Westergard, T., Krishnamurthy, K., Markandaiah, S. S., Shi, Y., et al. (2014). Antisense proline-arginine ran dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225. doi: 10.1016/j.neuron.2014.12.010
Xiao, S., MacNair, L., McLean, J., McGoldrick, P., McKeever, P., Soleimani, S., et al. (2016). C9orf72 isoforms in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain Res. 1647, 43–49. doi: 10.1016/j.brainres.2016.04.062
Xie, W., and Denman, R. B. (2011). Protein methylation and stress granules: Posttranslational remodeler or innocent bystander? Mol. Biol. Int. 2011:137459. doi: 10.4061/2011/137459
Yang, M., Liang, C., Swaminathan, K., Herrlinger, S., Lai, F., Shiekhattar, R., et al. (2016). A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2:e1601167. doi: 10.1126/sciadv.1601167
Yang, Q., Jiao, B., and Shen, L. (2020). The development of C9orf72-related amyotrophic lateral sclerosis and frontotemporal dementia disorders. Front. Genet. 11:562758. doi: 10.3389/fgene.2020.562758
Zampatti, S., Peconi, C., Campopiano, R., Gambardella, S., Caltagirone, C., and Giardina, E. (2022). C9orf72-related neurodegenerative diseases: From clinical diagnosis to therapeutic strategies. Front. Aging Neurosci. 14:907122. doi: 10.3389/fnagi.2022.907122
Zheng, W., Wang, K., Wu, Y., Yan, G., Zhang, C., Li, Z., et al. (2022). C9orf72 regulates the unfolded protein response and stress granule formation by interacting with eIF2α. Theranostics 12, 7289–7306. doi: 10.7150/thno.76138
Zhu, Q., Jiang, J., Gendron, T. F., McAlonis-Downes, M., Jiang, L., Taylor, A., et al. (2020). Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 23, 615–624. doi: 10.1038/s41593-020-0619-5
Keywords: amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), C9orf72, protein arginine methyltransferase (PRMT), asymmetric dimethylation, dipeptide repeat protein (DPR), poly-Glycine-Arginine (polyGR), hexanucleotide repeat expansion (HRE)
Citation: Dane TL, Gill AL, Vieira FG and Denton KR (2023) Reduced C9orf72 expression exacerbates polyGR toxicity in patient iPSC-derived motor neurons and a Type I protein arginine methyltransferase inhibitor reduces that toxicity. Front. Cell. Neurosci. 17:1134090. doi: 10.3389/fncel.2023.1134090
Received: 29 December 2022; Accepted: 27 March 2023;
Published: 17 April 2023.
Edited by:
Annakaisa Haapasalo, University of Eastern Finland, FinlandReviewed by:
Lydia Castelli, The University of Sheffield, United KingdomChristopher Webster, The University of Sheffield, United Kingdom
Copyright © 2023 Dane, Gill, Vieira and Denton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernando G. Vieira, ZnZpZWlyYUBhbHMubmV0