
- Division of Oral Biology and Medicine, School of Dentistry, University of California, Los Angeles, CA, USA
Chronic alcohol exposure causes marked changes in reinforcement mechanisms and motivational state that are thought to contribute to the development of cravings and relapse during protracted withdrawal. The nucleus accumbens (NAcc) is a key structure of the mesolimbic dopaminergic reward system. Although the NAcc plays an important role in mediating alcohol-seeking behaviors, little is known about the molecular mechanisms underlying alcohol-induced neuroadaptive changes in NAcc function. The aim of this study was to investigate the effects of chronic intermittent ethanol (CIE) treatment, a rat model of alcohol withdrawal and dependence, on intrinsic electrical membrane properties and glutamatergic synaptic transmission of medium spiny neurons (MSNs) in the NAcc core during protracted withdrawal. We show that CIE treatment followed by prolonged withdrawal increased the inward rectification of MSNs observed at hyperpolarized potentials. In addition, MSNs from CIE-treated animals displayed a lower input resistance, faster action potentials (APs), and larger fast afterhyperpolarizations (fAHPs) than MSNs from vehicle-treated animals, all suggestive of increases in K+-channel conductances. Significant increases in the Cs+-sensitive inwardly rectifying K+-current accounted for the increased input resistance, while increases in the A-type K+-current accounted for the faster APs and increased fAHPs in MSNs from CIE rats. We also show that the amplitude and the conductance of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-mediated mEPSCs were enhanced in CIE-treated animals due to an increase in a small fraction of functional postsynaptic GluA2-lacking AMPARs. These long-lasting modifications of excitability and excitatory synaptic receptor function of MSNs in the NAcc core could play a critical role in the neuroadaptive changes underlying alcohol withdrawal and dependence.
Introduction
Alcohol (ethanol, EtOH) is lawfully consumed in our society for recreational purposes. Yet, excessive alcohol-drinking can lead to alcohol dependence and loss of control over alcohol consumption, with serious detrimental health consequences. Alcohol consumption is a major risk factor for burden of disease (Rehm et al., 2009). An important factor contributing to alcohol-related disease is the addictive property of alcohol. Alcohol addiction is a chronic relapsing disorder characterized by craving for alcohol and compulsive alcohol-seeking behaviors. Chronic alcohol exposure causes profound long-lasting neuroadaptations in brain reward systems that produce a negative affect state contributing to an increased susceptibility to relapse during protracted withdrawal (Koob et al., 2004; Koob, 2008; Koob and Le Moal, 2008). Relapse is a critical problem in treating alcoholism and the three federally approved medications for the treatment of alcohol use disorders – acamprosate, disulfiram, and naltrexone (Ross and Peselow, 2009) have shown limited efficacy in terms of craving, relapse, and abstinence rates reduction (Hughes and Cook, 1997; Mann et al., 2004; Snyder and Bowers, 2008), suggesting that these treatments may not be targeting the neuroadaptive changes that underlie the negative emotional alcohol-dependent state.
Nucleus accumbens (NAcc) is a key structure of the mesolimbic dopaminergic rewarding system involved in alcohol-induced neuroadaptations underlying alcohol-seeking behaviors. Indeed, pharmacological inactivation of the NAcc attenuates alcohol reinstatement and cue-induced relapse (Rassnick et al., 1992; Chaudhri et al., 2010). The NAcc is mainly composed of GABAergic medium spiny neurons (MSNs). MSNs receive major glutamatergic inputs from the ventral subiculum of the hippocampus, prefrontal cortex, basolateral amygdala, and thalamus (Sesack and Grace, 2010). Activation of these inputs can modulate the release of dopamine from the ventral tegmental area (VTA) projections to the NAcc (Floresco et al., 2001; Howland et al., 2002). Extensive studies have shown that dysregulation of glutamatergic transmission in the NAcc during protracted withdrawal from drugs plays a critical role in the relapse to drug-seeking behavior (Thomas et al., 2001; LaLumiere and Kalivas, 2008; Kasanetz et al., 2010). For instance, potentiation of AMPA receptor (AMPAR) transmission has been observed in NAcc core after prolonged withdrawal from cocaine self-administration (Conrad et al., 2008). Increased glutamatergic synaptic strength is associated with increased conductance of AMPARs due to an insertion of postsynaptic calcium-permeable GluA2-lacking AMPARs (Conrad et al., 2008; Mameli et al., 2009). Blockade of GluA2-lacking AMPARs in the NAcc reduces cue-induced cocaine seeking (Conrad et al., 2008). By contrast, little is known about the molecular mechanisms underlying alcohol-mediated modulation of synaptic excitability in the NAcc during protracted withdrawal.
The regulation of electrical membrane properties of MSNs is critical in the integration and processing of drug-related information converging on the NAcc (Mu et al., 2010; Kim et al., 2011). Recently, decreased Ca2+-dependent SK channel activity in NAcc MSNs was implicated in increased alcohol-seeking behavior during protracted withdrawal (Hopf et al., 2010).
The goal of the present study was to investigate the neuroadaptations induced by alcohol in the NAcc. To this end, we examined the intrinsic electrical membrane properties and glutamatergic synaptic transmission of MSNs in the NAcc core of rats during protracted withdrawal from chronic intermittent ethanol (CIE) treatment.
Materials and Methods
Animals
All experiments were performed in accordance with the guidance of the National Institutes of Health on animal care and use and the formal approval of the UCLA Animal Research Committee. Male Sprague Dawley rats (200–250 g) were housed in the vivarium under a 12 h light/dark cycle and had ad libitum access to food and water. A total of 70 rats were used in the present study. Ethanol (EtOH; Pharmo Products, Brookfield, CT, USA) was administrated by oral intubation. A long-term and short-term CIE treatment was used. In the long-term CIE treatment, rats received 5 g/kg of body weight of EtOH as a 25% (w/v) EtOH solution in drinking water once every other day for the first five doses, and they received 6 g/kg of body weight of EtOH as a 30% (w/v) EtOH once every day for the following 55 doses. In the short-term CIE treatment, rats received 2.5 g/kg of body weight of EtOH as a 12.5% (w/v) EtOH solution in drinking water once every other day for the first five doses, and they received 3 g/kg of body weight of EtOH as a 15% (w/v) EtOH once every day for the following seven doses. With these EtOH regimens, rats experience multiple cycles of intoxication and withdrawal phases. The chronic intermittent vehicle (CIV) groups, corresponding to each CIE treatment, received drinking water (20 ml/kg of body weight) instead of EtOH. In other studies we showed that both short- and long-term CIE treatments resulted in similar long-lasting (>40 days) tolerance to the sedative/anesthetic effects of diazepam (Abriam et al., 2009). After the treatment and 40–60 days of withdrawal, rats were anesthetized with isoflurane (Phoenix, St. Joseph, MO, USA) and decapitated to obtain tissues for experiments. Both long-term and short-term CIE treatments were used in current-clamp recordings, whereas only the short-term CIE treatment was used in the voltage-clamp recordings and biochemical studies.
Slice Preparation
Coronal brain slices (400 μm) containing the NAcc were obtained with a vibrating slicer (Leica VT 1200S, Nussloch, Germany) from a block of tissue. Slices were allowed to recover for at least 1 h at room temperature in artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, and 10 glucose, saturated with 95% O2 and 5% CO2.
Electrophysiology
Whole-cell patch clamp recordings were obtained from MSNs in the NAcc core, which could be identified by anatomical landmarks, at 34 ± 0.5°C during perfusion with ACSF. MSNs were identified based on well-defined electrophysiological characteristics (Uchimura et al., 1989; O’Donnell and Grace, 1993). Patch clamp recording pipettes (TW150F-3, WPI, Sarasota, FL, USA) with 5–7 MΩ resistance were filled with a solution containing (in mM): 135 KMeSO4 or K-gluconate, 5 NaCl, 0.3 CaCl2, 1.1 EGTA, 2 MgATP, 0.2 NaGTP, and 10 HEPES, pH adjusted to 7.3 with KOH. To investigate Ca2+-activated K+ currents, the concentrations of CaCl2 and EGTA were reduced to 0.08 and 0.3 mM, respectively. Once in whole-cell configuration, the cell was allowed to recover for at least 10 min before applying any experimental protocol to allow equilibration and stabilization of ionic conductances. Access resistance (<30 MΩ) was continuously monitored and recordings were accepted for analysis if changes were <20%. AMPAR-mediated miniature EPSCs (mEPSCs) were recorded at a holding membrane potential of −70 mV in the presence of 0.5 μM tetrodotoxin (TTX), 50 μM picrotoxin (Ascent, Bristol, UK), and 1 μM CGP 54626 (5,7,8,9-tetrahydro-5-hydroxy-6H-benzocyclohepten-6-ylideneacetic acid) hydrochloride (Tocris, Park Ellisville, MO, USA) in ACSF. The antagonist of GluA2-lacking AMPARs, IEM-1460 (N,N,H,-trimethyl-5-[(tricyclo[3.3.1.13,7]dec-1-ylmethyl) amino]-1-pentanaminiumbromide hydrobromide; Tocris, Park Ellisville, MO, USA), was bath applied for at least 7 min at a concentration of 50 μM.
Electrophysiological signals were amplified using the Multiclamp 700B amplifier (Molecular Devices, Union City, CA, USA), low-pass filtered at 1 kHz, and digitized at 10 kHz with the Digidata 1440A (Molecular Devices). Data were acquired using pClamp 10 software (Molecular Devices), and analyzed using Clampfit software (Molecular Devices) and the Mini Analysis program (Synaptosoft, Decatur, GA, USA). To obtain current-voltage (I-V) responses and to test the excitability of MSNs, a series of hyperpolarizing and depolarizing current steps of 1 s were applied. Input resistance (Rin) was calculated in the linear range of the I-V curve around the resting membrane potential (RMP). Action potential (AP) waveform parameters including AP half-width were analyzed at the current step where the first AP was fired. Fast afterhyperpolarization (fAHP) and slow afterhyperpolarization (sAHP) magnitude were estimated relative to the AP threshold (4 and 15 ms after the AP threshold, respectively). Interspike interval values were obtained from the first depolarizing response to currents that evoked at least two APs. To measure the inwardly rectifying potassium (KIR) current, MSNs were voltage-clamped at a holding potential of −70 mV ad a 50-ms step to −150 mV was applied; then, MSNs were depolarized to −45 mV with a ramp pulse of 0.6 mV/ms (Shen et al., 2007; Gertler et al., 2008). To investigate SK tail currents, MSNs were held at −70 mV, and depolarizing current steps (800 ms, from −60 to −10 mV in 10 mV steps) were applied, with 5 s between successive steps.
AMPA receptor-mediated mEPSCs were detected (Mini Analysis program) using a threshold equivalent to 2.5 standard deviations from baseline noise. The mean amplitude of mEPSCs was determined by the average of 100–1000 events per cell. The mean frequency of mEPSCs was determined for each cell as the number of events per second during a 100–300 s recording period. Kolmogorov–Smirnov tests were used for comparing two cumulative distributions. Cumulative histograms of mEPSC amplitude were plotted with 1 pA bins.
A peak-scaled non-stationary fluctuation analysis was performed from an ensemble of mEPSCs at a holding potential of −70 mV to estimate the mean single-channel conductance of AMPARs using the Mini Analysis software (Benke et al., 2001; Hartveit and Veruki, 2007). Unitary mEPSCs were carefully selected for analysis by visual inspection based on the following criteria: fast rise time to allowing for precise alignment, stable baseline, and exponential decay. The mEPSCs were aligned by their point of maximal rise and averaged. The average response waveform was scaled to the peak of individual responses and the variance of the fluctuation of the decay around the mean was calculated. This variance was binned (100 bins of equal current decrement) and plotted versus the mean current amplitude of the decay. The shape of the variance-amplitude relationship was fitted with the following equation: σ2 = iI – I2/N + b where i was the mean single-channel AMPAR current, I the mean current, N the number of channels activated at the peak, and b the baseline variance. i was estimated as the slope of the linear fit of the first portion of the parabola, since the equation becomes linear when AMPAR open probability gets close to zero. Unitary current was converted to conductance as Γ = i/V (assumed reversal potential of 0 mV). The number of open channels at the peak was calculated by dividing the average mEPSC amplitude by the unitary current i. Any correlation between conductance and average mEPSC amplitude, mean rise time, mean decay time, access resistance, or background noise variance was observed (p > 0.05; data not shown). This indicates that the range of values for Γ was mainly due to variations of AMPAR conductance between synapses (Benke et al., 1998; Hartveit and Veruki, 2007).
Synaptoneurosome Preparation
The preparation of synaptoneurosomes was based on previous studies (Johnson et al., 1997; Williams et al., 2003; Figure 7A). NAcc was microdissected from coronal brain slices obtained from experimental rats in cold ACSF, saturated with 95% O2/5% CO2. Isolated NAcc was homogenized on ice in 150 μl of iced cold homogenate buffer containing (in mM): 124 NaCl, 3.2 KCl, 1.06 NaH2PO4, 2.5 CaCl2, 1.3 MgCl2, 26 NaHCO3, and 10 glucose, bulled with 95% O2/5% CO2 to adjust pH at 7.4. Protease inhibitors (1:20; P8340, Sigma-Aldrich, St. Louis, MO, USA) were included in the buffer to minimize proteolysis. The homogenate was passed through three layers of a pre-wetted 100 μm pore nylon net filter (NY1H09000, Millipore, Billerica, MA, USA) contained in a 37 mm diameter syringe filter holder (SX0002500, Millipore, Billerica, MA, USA). The filtrate was collected and passed through a pre-wetted 5 μm pore nitrocellulose membrane filter (LSWP02500, Millipore, Billerica, MA, USA). The final filtrate was spun at 1,000 × g for 15 min at 4°C. The pellet obtained corresponded to the synaptoneurosome fraction. Isolated synaptoneurosomes were stored at −20°C prior to SDS-PAGE Western blot analysis.
Western Blot Analysis
Synaptoneurosome pellets were re-suspended in 50 μl of homogenate buffer containing protease inhibitors. Protein concentrations were determined with BCA Protein Assay Kit (Pierce, Rockford, IL, USA) according to the manufacturer’s instructions. Equal quantities of proteins (20 μg/lane) were electrophoresed onto a 10% SDS-PAGE (Bio-Rad, Hercules, CA, USA). Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad). After saturation in Tris-buffered saline-Tween (0.05%) containing 5% non-fat dry milk (Bio-Rad), the PVDF membranes were probed with a rabbit polyclonal anti-GluA1 (AB1504, Millipore, Billerica, MA, USA), or with a rabbit polyclonal anti-GluA2 (AB1768, Millipore, Billerica, MA, USA), or with a rabbit polyclonal anti-GluN1, alternative CT (06-314, Upstate, Lake Placid, NY, USA) at 1:1000, or with a mouse monoclonal anti-GluN2B (clone N59/20, UC Davis/NIH NeuroMab Facility), or with a mouse monoclonal anti-PSD-95 (clone K28/43, NeuroMab) at 1:500, or with rabbit polyclonal β-actin (ab8227, Abcam, Cambridge, MA, USA) at 1:2000, and were incubated overnight at 4°C. After washing in Tris-buffered saline-Tween (0.05%), PVDF membranes were incubated with the corresponding peroxidase-conjugated secondary antibody for 1 h at room temperature. Revelation was obtained by chemiluminescence reaction (ECL, Pierce) and pictures were acquired with a camera system of luminescent image analyzer (LAS-3000, Fujifilm, Tokyo, Japan). Optical density analysis of the signals was performed using ImageJ64 software (ImageJ 1.43u, NIH). The integral of the entire optical density profile was calculated as the final value.
Statistics
The investigator performing the experiments and analysis was blind to the treatment (CIV or CIE) that the rats received. Data were expressed as mean ± standard error (SEM). Statistical analyses were performed with two-way ANOVA followed by Bonferroni posttest, or paired or unpaired Student’s t-test, or non-parametric Mann–Whitney test as appropriate. A value of p < 0.05 was considered statistically significant.
Results
CIE Treatment Alters Intrinsic Electrical Membrane Properties of MSNs
To investigate whether CIE treatment affects the intrinsic electrical membrane of MSNs in the NAcc core, we obtained whole-cell patch recordings in current-clamp mode.
All recorded neurons showed the general electrophysiological characteristics of MSNs, including a hyperpolarized RMP, a slow ramp depolarization preceding the first AP and the presence of inward rectification of the current-voltage relationship at hyperpolarized voltages (Figure 1A) (Belleau and Warren, 2000).
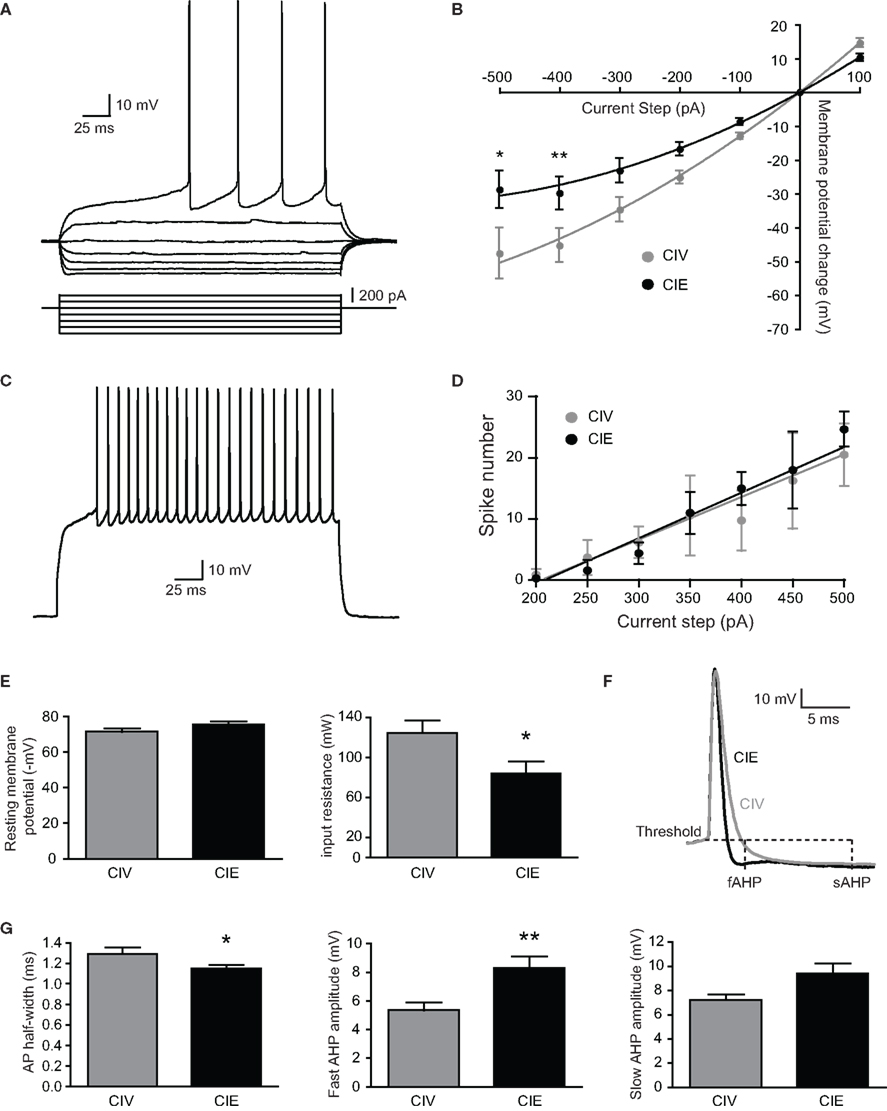
Figure 1. Electrical membrane properties of medium spiny neurons (MSNs) from CIV- and CIE-treated rats. (A) typical MSN responses to depolarizing and hyperpolarizing currents. Note the small voltage sag in responses to hyperpolarizing currents, the inward rectification of subthreshold voltage responses, and the ramp depolarization with a delayed action potential. (B) summary of current-voltage (I-V) curves shows that the inward rectification detectable at hyperpolarized membrane potentials was significantly increased in CIE-treated rats. (C) Typical spike discharge pattern in response to a 500 pA depolarizing current step (D) summary of current-firing curves indicate that the number of evoked action potentials (APs) in response to depolarizing current steps was not significantly different between CIV- and CIE-treated rats. (E) resting membrane potential was more hyperpolarized in MSNs from CIE-treated rats. In addition, input resistance was significantly decreased in CIE-treated rats. (F) representative APs from CIV- (gray) and CIE-treated (black) rats. Fast afterhyperpolarization (fAHP) and slow afterhyperpolarization (sAHP) were measured at 4 and 15 ms, respectively after the AP threshold. (G) AP half-width and fAHP recorded in CIE-treated rats were significantly different compared to CIV-treated rats. However, the sAHP was not modified. (**p < 0.01, *p < 0.05).
The inward rectification, shown as the shift from linearity in the I-V curve relationship, was significantly different in both long-term and short-term CIE treatments compared to their respective CIV controls (long-term CIV vs. CIE, *p < 0.05; short-term CIV vs. CIE, *p < 0.05; two-way ANOVA with Bonferroni posttest). Since the results of the patch clamp recording experiments between the long- and short-duration CIE treatments were not different, all current-clamp data were pooled together. The inward rectification was significantly increased in MSNs from CIE-treated animals compared to CIV-treated animals (−500 pA-step: CIV: −47.68 ± 7.57 mV, n = 6 from five rats; CIE: −28.81 ± 5.48 mV, n = 13 from six rats; *p < 0.05; −400 pA-step: CIV: −45.32 ± 5 mV, n = 16 from 10 rats; CIE: −29.87 ± 4.86 mV, n = 17 from 11 rats; **p < 0.01; two-way ANOVA with Bonferroni posttest; Figure 1B). The number of APs evoked by somatic positive current steps was similar in both groups (Figures 1C,D). The increase in inward rectification in CIE rats was associated with a decrease in input resistance (CIV: 124.7 ± 12.73 MΩ, n = 16 from 10 rats; CIE: 84.05 ± 12.18 MΩ, n = 17 from 11 rats; *p = 0.027, unpaired t-test; Figure 1E). The RMP tended to be more hyperpolarized in MSNs from CIE-treated rats but the difference was not statistically significant (CIV: −71.63 ± 1.99 mV, n = 16 from 10 rats; CIE: −75.8 ± 1.69 mV, n = 17 from 11 rats; p = 0.12, unpaired t-test; Figure 1E). Membrane capacitance of MSNs was not different between groups (data not shown).
Analysis of AP parameters revealed a significant decrease in AP half-width (CIV: 1.29 ± 0.06 ms, n = 14 from 10 rats; CIE: 1.15 ± 0.04 mV, n = 15 from 12 rats; *p = 0.04, unpaired t-test) associated with an increase in the fAHP amplitude in CIE-treated rats (CIV: 5.36 ± 0.5 mV, n = 14 from 10 rats; CIE: 8.28 ± 0.84 mV, n = 15 from 12 rats; **p = 0.008, unpaired t-test; Figures 1F,G). However, neither the sAHP (Figure 1G; CIV: 7.24 ± 0.44 mV, n = 14 from 10 rats; CIE: 9.39 ± 0.84 mV, n = 15 from 12 rats; p = 0.06, Mann–Whitney test) nor the AP amplitude (CIV: 48.8 ± 3.8 mV, n = 14 from 10 rats; CIE: 54.4 ± 4.3 mV, n = 15 from 12 rats; p = 0.33, unpaired t-test) was modified. The latency to the first-evoked spike (CIV: 0.33 ± 0.03 s, n = 14 from 10 rats; CIE: 0.36 ± 0.05 s, n = 15 from 12 rats, p = 0.68, unpaired t-test) and the interspike interval between the first- and the second-evoked spikes (CIV: 0.1 ± 0.01 s, n = 14 from 10 rats; CIE: 0.11 ± 0.02 s, n = 15 from 12 rats, p = 0.94, Mann–Whitney test) were not different between treatments.
CIE Treatment Increases the KIR Current of MSNs
The inward rectification of the I-V relationship in MSNs is attributed to the activation of KIR channels during hyperpolarizing potentials (Uchimura et al., 1989; Nisenbaum and Wilson, 1995). In addition, KIR channels are the major determinants of the input resistance and the hyperpolarized RMP of MSNs during the down-state (Nisenbaum and Wilson, 1995). Cs+ has been shown to potently block inward rectification in MSNs suggesting that KIR channels are highly sensitive to Cs+ (Nisenbaum and Wilson, 1995; Mermelstein et al., 1998; Belleau and Warren, 2000). Indeed, as shown in Figure 2A, the inwardly rectifying currents induced by voltage steps in presence of TTX were reduced by CsCl (3 mM). To investigate whether CIE treatment modifies inwardly rectifying KIR current, we used voltage ramps from −150 to −45 mV (Figure 2B). The total current elicited by the voltage ramps was Cs+-sensitive (Figure 2B) and was modeled as the sum of the inwardly rectifying KIR current and the linear leaky K+ current (Kleak) (Shen et al., 2007; Gertler et al., 2008). The I-V relationship showed a similar activation profile and reversal potential of MSN KIR + Kleak current in both CIV and CIE groups (Figure 2D). However, the KIR + Kleak peak current amplitude was significantly increased in CIE-treated rats (Figure 2C; CIV: 1.11 ± 0.16 nA, n = 13 from two rats; CIE: 2.18 ± 0.23 nA, n = 10 from two rats, ***p = 0.0009, unpaired t-test). When KIR + Kleak current amplitude was normalized with membrane capacitance, the differences between the groups remained (in nA/pF; CIV: 24.74 ± 3.6, n = 13; CIE: 37.79 ± 4.1, n = 10, *p = 0.025, unpaired t-test). This result suggests that differences in density of K+ channels, rather than cell membrane surface, accounted for the enhanced whole-cell KIR + Kleak current observed in CIE-treated rats. All together, these results suggest that CIE treatment may increase the surface expression of inwardly rectifying KIR channels in MSNs, which could be responsible for the observed decreases in the input resistance of MSNs.
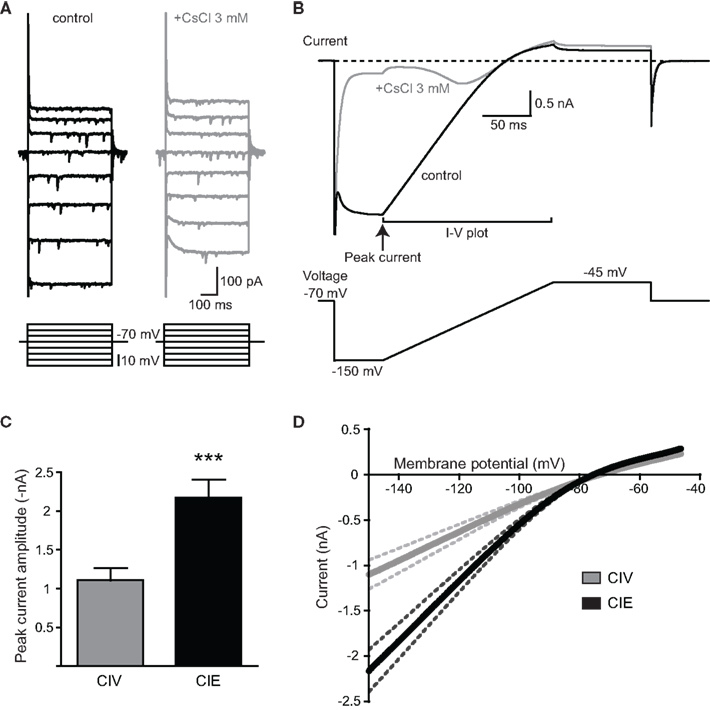
Figure 2. Enhanced activation of KIR current in MSNs of CIE-treated rats. (A) Representative current responses to depolarizing and hyperpolarizing voltages in control condition (black) and during application of CsCl (3 mM; gray). Note that the inwardly rectifying current was reduced by CsCl. (B) Representative averaged current response of five sweeps from the same MSN showed in (A) to a voltage step from −70 to −150 mV followed by a voltage ramp from −150 to −45 mV in control condition (black). The current induced by the voltage protocol was sensitive to the application of CsCl (3 mM; gray). The inwardly rectifying potassium (KIR) current was measured during the voltage ramp from −150 to −45 mV. Time points used to estimate the peak current and I-V relationship are indicated. (C) The peak current was significantly increased in CIE- compared to CIV-treated rats (***p < 0.001). (D) Mean I-V relationship characteristic of KIR current in MSNs measured during the voltage ramp. Note the increased peak current in MSNs recorded from CIE (black) compared to CIV-treated rats (gray), and the similar reversal profile in both groups. Dashed lines represent SEM.
It has been recently demonstrated that repeated EtOH self-administration followed by withdrawal reduces the amplitude of sAHP due to a decrease in small conductance voltage- and Ca2+-activated potassium (SK) channel function in MSNs from the NAcc core (Hopf et al., 2010). However, as described above we did not find any modification of the sAHP amplitude in MSNs from CIE-treated rats compared to CIV-treated rats. These differences could be explained by our use of higher concentration of EGTA (1.1 mM), a Ca2+ chelator, than that used by Hopf et al. (2010) (0.3 mM). Therefore, additional experiments were performed on naïve rats with an internal solution containing 0.3 mM EGTA. Under these conditions, neither the fAHP nor the sAHP were significantly affected by the application of two different Ca2+-activated K+ channel blockers, apamin (100 nM; change in amplitude: fAHP: −1.3 ± 0.1 mV, n = 2; sAHP: −2.4 ± 0.8 mV, n = 2 from one rat) or (in separate recordings) cadmium (0.2 mM) (change in amplitude: fAHP: −1.1 ± 0.3 mV, n = 3 MSNs from two naïve rats; p > 0.05, paired t-test; sAHP: −1.8 ± 0.7 mV, n = 3 from two naïve rats; p > 0.05, paired t-test; Figure 3A). These results suggest that Ca2+-activated K+ channels do not contribute significantly to AHP amplitude under these conditions. To directly examine the current mediated by SK channels, we used voltage steps to depolarize the membrane potential of the neuron from −70 to −20 mV prior to bring it back to −70 mV to generate a tail current previously shown to be mediated by apamin-sensitive SK channels in the NAcc core (Hopf et al., 2010). However, we did not observe any tail current upon returning to −70 mV in MSNs from the NAcc core (n = 3 MSNs in two naïve rats; Figure 3B). To confirm that our experimental conditions were sufficient to generate an apamin-sensitive Ca2+-activated K+ current, we used the same experimental protocol, with the same internal solution, in recordings from hippocampal CA1 pyramidal neurons that express apamin-sensitive SK channels (Mulholland et al., 2011; Wang et al., 2011). A large tail current was observed in CA1 neurons that was reduced by application of apamin and abolished by cadmium (n = 2 in one rat; Figure 3C). This indicates that our experimental conditions were sufficient to generate and record currents mediated by SK channels. All together, these results suggest that (a) apamin/cadmium-sensitive SK channels do not significantly contribute to the fAHP and sAHP in MSNs from the NAcc core, and (b) that the CIE treatment-induced decreases in AP duration and increases in fAHP are not be due to modifications in Ca2+-activated K+ channels.
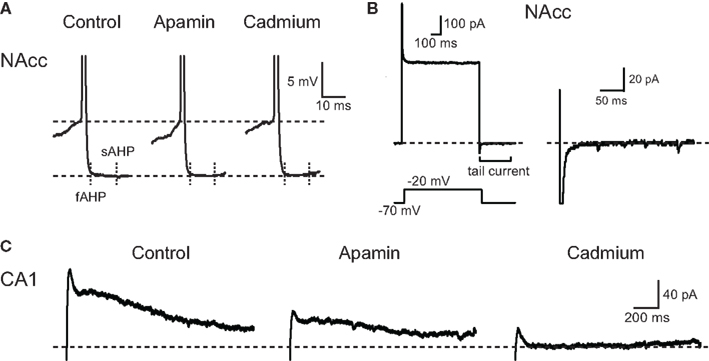
Figure 3. AHPs of NAcc core MSNs are insensitive to Ca2+-activated K+ current modulators. (A) Representative AHPs from a NAcc core MSN recorded before (control), and during application of apamin (100 nM), followed by cadmium (0.2 mM). Note the absence of effect of apamin and cadmium on the amplitude of fAHP and sAHP. (B) Representative current response to a voltage depolarization to −20 mV from a −70 mV holding potential from another NAcc core MSN (left). Note the absence of a tail current after returning to −70 mV following depolarization (right). (C) Representative tail current elicited in a CA1 pyramidal neuron after returning from −20 to −70 mV. Note that both apamin (100 nM) and cadmium (0.2 mM) reduced the amplitude of the tail current.
CIE Treatment Increases the A-type K+ Current of MSNs
To investigate possible alterations in other voltage-gated K+ currents we compared the activation kinetics of the A-type and delayed rectifier K+ currents, both of which are implicated in AP repolarization, in MSNs from rats treated exclusively with the short-term CIV and CIE treatments. To examine the activation properties of these currents, we used voltage steps to briefly hyperpolarize neurons to −120 mV to remove inactivation of voltage-gated channels followed by depolarizing steps from −70 to 60 mV in the presence of TTX (Figure 4A) (Deng et al., 2011). The amplitude of the transient K+ current measured at 10 ms after the depolarizing step onset, corresponding to the sum of A-type and delayed rectifier K+ currents, was significantly increased in CIE-treated animals (+60 mV-step: CIV: 2.84 ± 0.89 nA, n = 5 from two rats; CIE: 4.66 ± 0.64 nA, n = 8 from two rats; *p < 0.05, two-way ANOVA with Bonferroni posttest) (Figures 4A,B). The amplitude of the sustained K+ current measured at the end of the depolarizing steps, corresponding to the delayed rectifier K+ current, was not significantly modified by CIE treatment (+60 mV-step: CIV: 2.60 ± 0.78 nA, n = 5 from two rats; CIE: 3.99 ± 0.57 nA, n = 8 from two rats; p > 0.05, two-way ANOVA with Bonferroni posttest; Figures 4A,C). The inactivation properties of A-type and delayed rectifier K+ currents were not altered by CIE treatment (data not shown). These results suggest that CIE treatment increases the amplitude of the A-type K+ current generated at depolarizing membrane potentials, thereby contributing to faster AP repolarization and increased amplitude of fAHP observed in current-clamp recordings of MSNs from CIE rats.
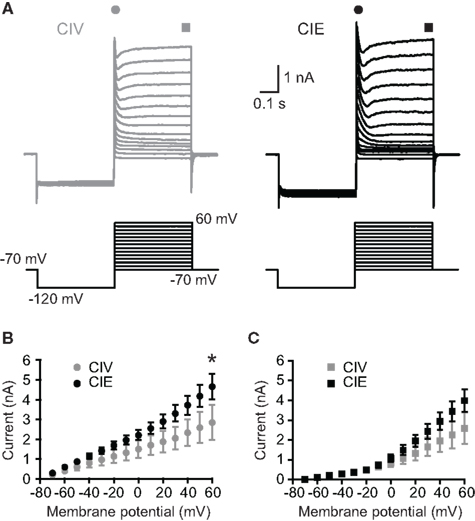
Figure 4. Enhanced activation of A-type K+ current in MSNs of CIE-treated rats. (A) representative current responses voltage-gated A-type and delayed rectifier K+ currents to voltage steps from −70 to 60 mV preceded by a prepulse to −120 mV in MSNs from CIV- and CIE-treated rats. (B) Mean I-V relationship of the transient K+ current measured within the first milliseconds of the depolarizing steps. The amplitude of the transient K+ current was significantly increased in CIE-treated rats (*p < 0.05). (C) Mean I-V relationship of the sustained K+ current measured at the end of the depolarizing steps. The amplitude of the sustained K+ current was not significantly different between groups.
CIE Treatment Increases the Amplitude of AMPAR MEPSCs
To investigate whether CIE treatment alters glutamatergic synaptic transmission of MSNs, we obtained whole-cell patch recordings and examined mEPSCs in voltage-clamp mode from MSNs of rats treated exclusively with the short-term CIE treatment. At −70 mV, mEPSCs in MSNs are completely blocked by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), indicating that the current is exclusively mediated by the activation of AMPA/kainate receptors (data not shown). We observed that glutamatergic transmission was enhanced in MSNs from CIE-treated rats (Figures 5A,B). The mean amplitude of mEPSCs was increased (CIV: −8.77 ± 0.46 pA, n = 20 from nine rats; CIE: −10.79 ± 0.85 pA, n = 11 from seven rats; *p = 0.028, unpaired t-test) and the cumulative probability distribution of mEPSC amplitudes was significantly shifted to the right toward higher values in CIE-treated rats [Kolmogorov-Smirnov (KS) test, ***p = 0.001; Figures 5B–D]. However, no change in mEPSC frequency was observed between CIV- and CIE-treated rats (Figure 5D; CIV: 4.96 ± 0.83 Hz, n = 20 from nine rats; CIE: 4.98 ± 1.03 Hz, n = 11 from seven rats; p = 0.99, unpaired t-test) suggesting that CIE-induced increase in glutamate transmission is mediated by a postsynaptic mechanism. Furthermore, we observed that the kinetics of AMPAR mEPSCs from CIE-treated rats were slightly faster compared to CIV-treated rats (Figure 5E). However the differences in mEPSC rise time (CIV: 2.13 ± 0.15 ms, n = 20 from nine rats; CIE: 1.84 ± 0.18 ms, n = 11 from seven rats; p = 0.25, unpaired t-test) and mEPSC decay time (CIV: 5.47 ± 0.31 ms, n = 20 from nine rats; CIE: 4.92 ± 0.31 ms, n = 11 from seven rats; p = 0.26, unpaired t-test) were not statistically significant (Figures 5F,G).
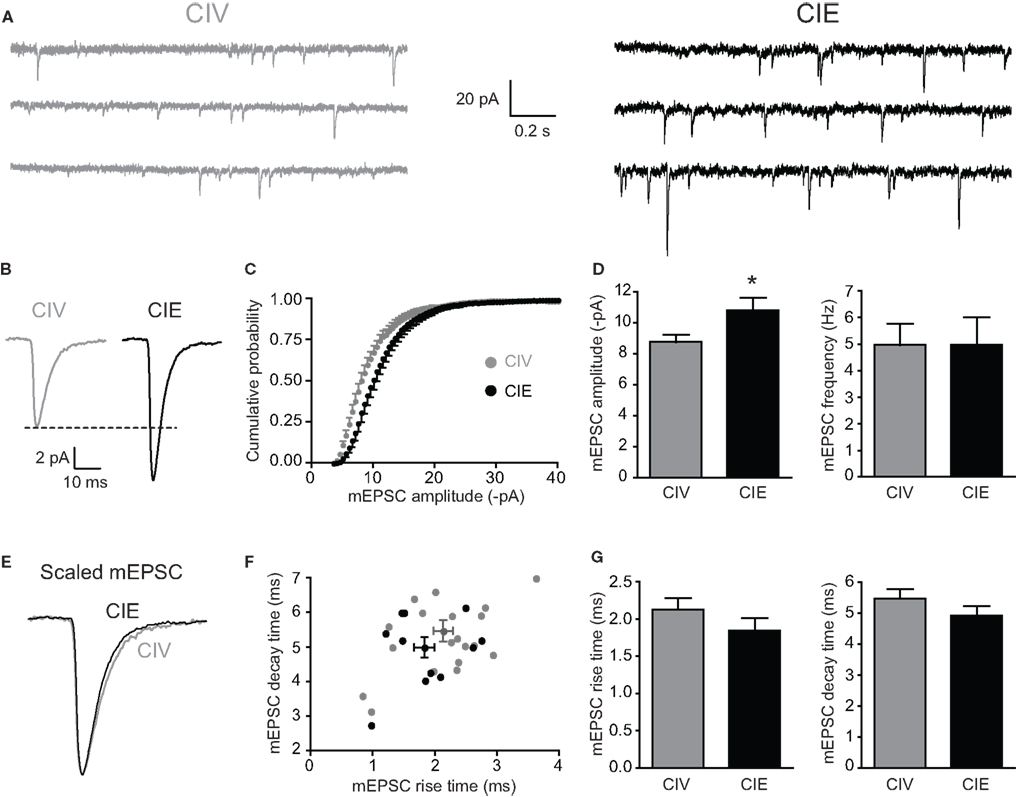
Figure 5. Excitatory glutamatergic transmission is enhanced in CIE-treated rats. (A) Representative current traces of miniature EPSCs recorded from MSNs in CIV- (gray) and CIE-treated (black) rats. (B) Average mEPSC traces from two representative MSNs in CIV- (gray) and CIE-treated (black) rats. (C) Cumulative probability plot of amplitude distribution of mEPSCs showing a significant shift to the right in CIE-treated compared to CIV-treated rats (***p < 0.001, Kolmogorov–Smirnov test). (D) Summary graphs showing the significant increase in mEPSC amplitude in CIE-treated rats (*p < 0.05) while the frequency of mEPSCs was unchanged. (E) Normalized average mEPSC traces from CIV- and CIE-treated rats. (F) Plot of mEPSC rise time and decay time for MSNs from CIV- (gray) and CIE-treated (black) rats. Population average is shown as mean ± SEM. (G) summary graphs showing mEPSC rise time and decay time in CIV- and CIV-treated rats.
CIE Treatment Increases the Unitary Conductance of AMPARs
To investigate the postsynaptic mechanisms underlying the increased glutamatergic transmission in CIE rats, we examined the functional properties and the expression of AMPARs in MSNs. Several studies have shown that synaptic expression of GluA1 and GluA2 plays a crucial role in mediating drug-induced synaptic plasticity underlying addictive behaviors associated with withdrawal (Boudreau and Wolf, 2005; Boudreau et al., 2007; Conrad et al., 2008; Ghasemzadeh et al., 2009; Mameli et al., 2009). Potentiation of NAcc AMPAR transmission that occurs after cocaine withdrawal has been shown to depend on the insertion of synaptic GluA2-lacking AMPARs resulting in an increase in the unitary conductance of AMPARs (Γ) (Benke et al., 2001; Isaac et al., 2007; Conrad et al., 2008; Mameli et al., 2009). To determine whether unitary conductance of synaptic AMPARs was changed by CIE treatment we applied non-stationary fluctuation analysis to mEPSCs. Unitary conductance Γ was significantly higher in MSNs from CIE-treated rats compared to MSNs from CIV-treated rats (CIV: 8.86 ± 0.98 pS, n = 12 from six rats; CIE: 14.43 ± 1.28 pS, n = 9 from seven rats; **p = 0.004, Mann–Whitney test; Figures 6A,B). The calculated number of open channels at the peak of mIPSCs was not significantly different between groups (CIV: 14.43 ± 1.2, n = 12 from six rats; CIE: 11.36 ± 0.4, n = 9 from seven rats; p = 0.07, Mann–Whitney test; Figure 6C). Therefore, we next performed a series of experiments to investigate whether the CIE-induced increase in Γ was associated with a modification in the subunit composition of synaptic AMPARs.
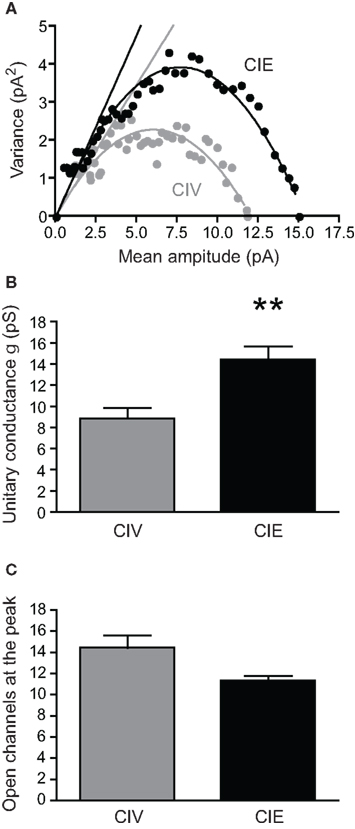
Figure 6. CIE treatment increases AMPAR unitary conductance (Γ). (A) Representative current-variance relationship from CIV- (gray) and CIE- (black) treated rats using the non-stationary fluctuation analysis. The conductance Γ was estimated from the initial slope (straight lines) of the parabolic fits. Note the greater slope from the CIE compared to CIV recordings. (B) Summary graph showing a significant increase in Γ in CIE compared to CIV-treated rats (**p < 0.01). (C) Summary graph showing the calculated number of open channels at the peak in CIV- and CIE-treated rats.
Lack of Significant Changes in the Expression of Synaptic AMPAR or NMDAR Subunit Proteins after CIE Treatment
First, we used Western blot analysis on synaptoneurosomes isolated from microdissected NAcc to investigate the expression of AMPAR and NMDAR subunits in CIV- and CIE-treated rats (Figure 7A). The synaptoneurosome is a biochemical preparation enriched with synaptic proteins (Johnson et al., 1997; Williams et al., 2003). As expected, synaptoneurosome preparations isolated from NAcc were enriched with PSD-95, a postsynaptic scaffolding protein associated with glutamate receptors, compared to whole extract of NAcc (Figure 7B). The expression of PSD-95 was similar in each group (PSD-95/β-actin: CIV: 100 ± 3.2%, n = 10 rats; CIE: 104 ± 1% of CIV control, n = 12 rats; p = 0.58, Mann–Whitney test; Figure 7C). This suggests that the synaptoneurosome purification process was consistent and reliable to allow an accurate analysis of synaptic AMPAR subunit expression in each group. Immunoblotting analysis revealed no significant changes in the expression of GluA1 or GluA2 in synaptoneurosomes isolated from CIE-treated rats compared to CIV-treated rats (GluA1/β-actin: CIV: 103 ± 3%, n = 10 rats; CIE: 116 ± 7% of CIV control, n = 11 rats; p = 0.14, unpaired t-test; GluA2/β-actin: CIV: 100 ± 3%, n = 10 rats; CIE: 110 ± 5% of CIV control, n = 12 rats; p = 0.11, unpaired t-test; Figures 7C,D). In addition, there was no modification in the expression of NMDA receptor (NMDAR) subunits GluN1 (GluN1/β-actin: CIV: 100 ± 7%, n = 6 rats; CIE: 114 ± 8% of CIV control, n = 8 rats; p = 0.27, unpaired t-test) and GluN2B (GluN2B/β-actin: CIV: 100 ± 11%, n = 6 rats; CIE: 104 ± 8% of CIV control, n = 8 rats; p = 0.73, unpaired t-test; Figure 7D). These data suggest that CIE treatment does not affect the expression of synaptic AMPARs and NMDARs in the NAcc. However, this does not necessarily rule out the possibility of a functionally significant increase in GluA2-lacking AMPARs occurred which was not properly detected by our biochemical techniques (Guire et al., 2008; Ferrario et al., 2011). Therefore, in the next series of experiments, we used selective pharmacological tools to further investigate the presence of GluA2-lacking AMPARs on MSNs from CIE-treated animals.
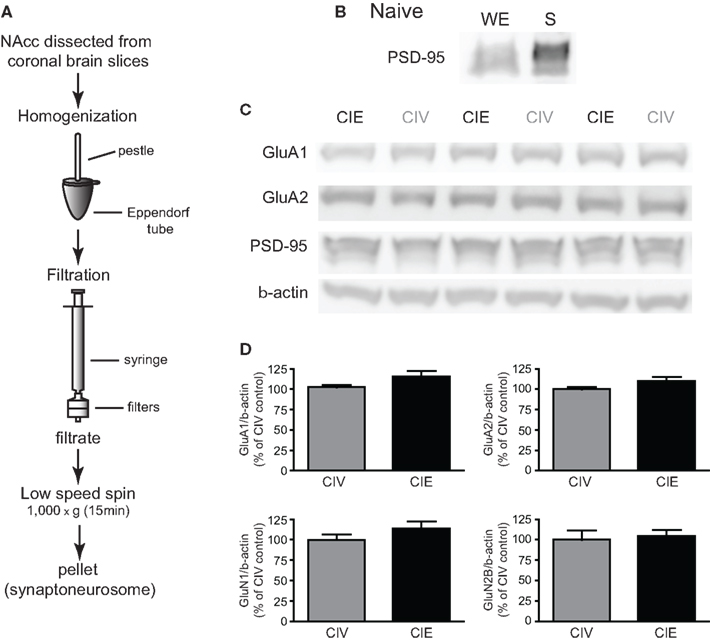
Figure 7. Effects of CIE treatment on the synaptic expression of AMPAR and NMDAR subunits. (A) Illustration of the technique used to isolate synaptoneurosomes from microdissected NAcc (adapted from Johnson et al., 1997). (B) Representative Western blot from NAcc of a naïve rat showing the enrichment in PSD-95 in synaptoneurosomes (S) compared to the whole extract (WE). (C) Representative Western blots of AMPAR subunits GluA1 and GluA2 from synaptoneurosomes isolated from NAcc of CIV- and CIE-treated rats. PSD-95 and β-actin were used as controls. (D) Summary graphs showing the lack of significant effect of CIE treatment on the synaptic expression of AMPAR subunits GluA1 and GluA2, and NMDA subunits GluN1 and GluN2 compared with CIV treatment. Results are presented as a ratio with β-actin.
Pharmacological Evidence for Accumulation of Synaptic GluA2-lacking AMPARs after CIE Treatment
To test for the presence of GluA2-lacking AMPARs, we studied the sensitivity of AMPAR mEPSCs to a selective antagonist of GluA2-lacking AMPARs, IEM-1460, which does not affect GluA2-containing AMPARs (Magazanik et al., 1997; Buldakova et al., 2007). Bath application of IEM-1460 (50 μM) did not significantly modify mEPSCs amplitude of MSNs from CIV-treated rats (96.92 ± 2.83% of baseline, n = 8 from three rats; p = 0.35 baseline vs. IEM-1460, paired t-test; Figures 8A,B) indicating that functional GluA2-lacking AMPARs are not significantly expressed at MSN synapses of CIV-treated rats. In contrast, IEM-1460 significantly decreased mEPSC amplitude of MSNs from CIE-treated rats (84.74 ± 3.05% of baseline, n = 8 from two rats; **p = 0.002 baseline vs. IEM-1460, paired t-test; #p = 0.011 CIV vs. CIE, unpaired t-test; Figures 8A,B). This effect of IEM-1460 on mEPSC amplitude was reversible (IEM-1460 washout: 92.01 ± 9.4% of baseline, n = 3 from three rats, p = 0.75 baseline vs. IEM-1460 washout, Wilcoxon signed rank test, data not shown). No significant effects of IEM-1460 were observed on the frequency of mEPSCs (CIV: 88.6 ± 6.4% of baseline, n = 8 from three rats, p > 0.05 baseline vs. IEM-1460, paired t-test; CIE: 108.9 ± 6.4% of baseline, n = 8 from two rats, p > 0.05 baseline vs. IEM-1460, paired t-test; Figure 8C). In the presence of IEM-1460, the mean AMPA mEPSC amplitude of MSNs from CIE rats was similar to the mean AMPA mEPSC amplitude from CIV rats (Figure 8D). These results suggest that CIE treatment-induced a significant increase in the proportion of functional synaptic GluA2-lacking AMPARs in MSNs of the NAcc core.
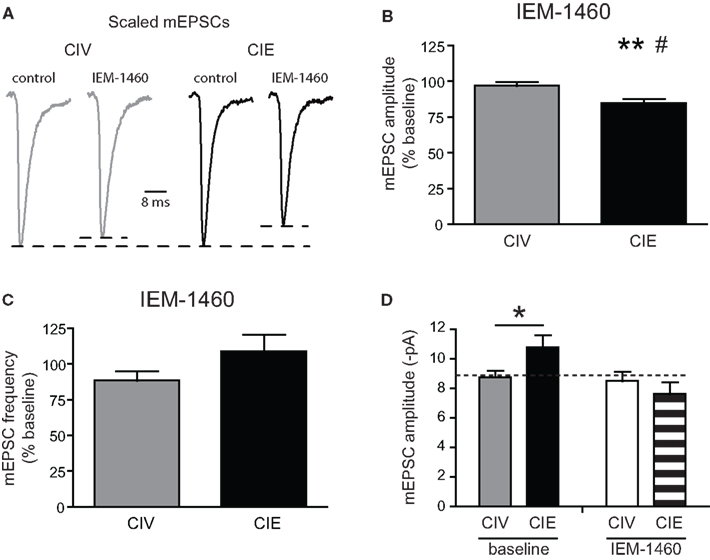
Figure 8. CIE treatment increases sensitivity to a selective antagonist of synaptic GluA2-lacking AMPARs. (A) Examples of normalized average mEPSC traces from CIV- and CIE-treated rats before and after bath application of a selective GluA2-lacking AMPAR antagonist, IEM-1460 (50 μM). (B) Summary graph of IEM-1460-induced decreases in mEPSC amplitudes of CIE- but not CIV-treated rats (**p < 0.01, baseline vs. IEM-1460; #p < 0.05 CIV vs. CIE). (C) Summary graph of the absence of effects of IEM-1460 on mEPSC frequency of CIV- and CIE-treated rats. (D) Summary graph of AMPA mEPSC amplitude of NAcc core MSNs from CIV- and CIE-treated rats (*p < 0.05) before (baseline) and during application of IEM-1460.
Discussion
Alcohol-seeking behaviors observed during protracted withdrawal are most likely dictated by long-term neuroadaptive changes in neuronal excitability and synaptic plasticity. CIE rats treated by oral intubation have previously been shown to exhibit signs of alcohol withdrawal and dependence including anxiety, hyperactivity, and decreased seizure thresholds (Kokka et al., 1993; Cagetti et al., 2003). It has also been demonstrated that CIE treatment induces profound and persistent changes in pharmacological properties and subunit composition of inhibitory postsynaptic GABAA receptors in hippocampal neurons (Liang et al., 2006, 2007). These changes persist for at least 120 days of withdrawal (Liang et al., 2007). Here we demonstrate that CIE treatment followed by protracted withdrawal alters the intrinsic membrane properties of MSNs in the NAcc core, specifically via increases in the KIR and A-type K+ currents. We also show that glutamatergic synaptic transmission is potentiated in CIE-treated rats. Specifically, we demonstrate that the enhanced amplitude and conductance of AMPAR mEPSCs in MSNs of CIE-treated animals is mediated by increases in a small fraction of functional postsynaptic GluA2-lacking AMPARs. These results may represent novel mechanisms underlying the neuroadaptive changes that persist long after CIE exposure and withdrawal.
Alteration of Intrinsic Electrical Membrane Properties of MSNs
The inward rectification in the I-V relationship of MSNs has been attributed to the activation of inwardly rectifying K+ (KIR) channels (Uchimura et al., 1989; Nisenbaum and Wilson, 1995; Belleau and Warren, 2000). KIR channels are the major determinants of the input resistance and the hyperpolarized RMP of MSNs during the down-state (Nisenbaum and Wilson, 1995). Interestingly, acute EtOH drinking modulates the expression of mRNAs coding for the KIR channel subunits in the NAcc of alcohol-preferring mice (Mulligan et al., 2011). In vitro, acute application of EtOH induces a hyperpolarization associated with a decrease in input resistance of MSNs in the striatum (Blomeley et al., 2011). This EtOH-induced hyperpolarization is abolished in the presence of Ba2+ suggesting that the effect of EtOH may be due to an increase in K+ conductance through Ba2+-sensitive KIR channels (Blomeley et al., 2011). Here, we show that CIE treatment produced an increase in inward rectification of the current-voltage relationship associated with a decrease in input resistance of MSNs in CIE-treated rats. In addition, we found that the RMP of MSNs tended to be more hyperpolarized in CIE-treated rats. These results suggest that CIE treatment may induce a long-lasting increase in KIR channel activity of MSNs in the NAcc core. Voltage-clamp experiments confirmed a significant increase in KIR current in MSNs from CIE rats. Increased KIR channel conductance has been shown to reduce AP firing rate of MSNs at depolarized potentials (Sears et al., 2010). However, we did not observe any modification of the AP firing rate in the MSNs of CIE-treated rats compared to CIV-treated rats. These results suggest that CIE treatment may only affect the activation properties of KIR channels without modifying the inactivation properties involved in the depolarization of MSNs.
Presently we found that MSNs from CIE-treated rats display faster AP half-width and increased fAHP amplitude. Lack of alterations in AP amplitude or threshold suggested that voltage-gated Na+ were not modified. Large conductance voltage- and Ca2+-activated K+ (BK) channels are potent regulators of spike repolarization and fAHP (Shao et al., 1999; Ishikawa et al., 2009). Inhibition of BK channels has been shown to significantly reduce fAHP in MSNs of the NAcc shell (Ishikawa et al., 2009). Further, another study demonstrated that MSNs in the NAcc core expressed SK channels whose function was reduced after repeated EtOH self-administration followed by withdrawal (Hopf et al., 2010). This decrease in SK channels function was responsible for the decrease in the apamin-sensitive sAHP amplitude resulting in an increase in membrane excitability of MSNs (Hopf et al., 2010). However, despite adopting experimental conditions similar to Hopf et al. (2010) and sufficient to induce an apamin- or Cd2+-sensitive SK current in CA1 pyramidal neurons, we found that both the fAHP and sAHP in NAcc core MSNs are insensitive to apamin or Cd2+ application. Interestingly, it has been shown that the sAHP of MSNs in the NAcc shell was also insensitive to apamin (Ishikawa et al., 2009). This suggests that the CIE-induced increases in spike repolarization and fAHP amplitude observed in our study could be due to changes in other voltage-dependent K+ channels involved in AP kinetics, such as the fast-inactivating A-type K+ channels (Nisenbaum et al., 1994). Voltage-clamp experiments revealed an increased A-type K+ current at depolarizing membrane potentials suggesting that A-type K+ channels may be responsible for the faster AP repolarization and increased fAHP in MSNs of CIE-treated rats. Since we studied K+ current kinetics in the presence of TTX, we cannot exclude the possibility that Na+-dependent K+ currents (Budelli et al., 2009) were also altered by CIE treatment and contributed to the faster AP repolarization and larger fAHP of CIE rats.
All together, these results suggest that chronic alcohol exposure induces long-lasting alterations of voltage-gated K+ channels which regulate the electrical membrane properties of MSNs. These persistent neuroadaptations could lead to the alteration of processing and synaptic integration of information converging on the NAcc (Nisenbaum et al., 1994; Steephen and Manchanda, 2009), which may have important implications for alcohol-related behaviors.
CIE-Induced Plasticity of AMPAR Synaptic Transmission
The short-term effects of chronic alcohol exposure on glutamatergic receptor function in several brain regions have largely been studied during early withdrawal (Stuber et al., 2010). In the NAcc of rats, it has been shown recently that CIE exposure induces the activation of AKT kinase and its intracellular target named mammalian target of rapamycin complex 1 (mTORC1) kinase (Neasta et al., 2010). Activation of the mTORC1-related pathway is thought to be responsible for the increased expression level of AMPAR subunit GluA1 and Homer protein, involved in the regulation of glutamate signaling in the postsynaptic density, in the NAcc observed at 24 h withdrawal after 3 months of CIE exposure (Neasta et al., 2010). Interestingly, inhibition of AKT or mTORC1 reduces alcohol-seeking behavior (Neasta et al., 2010, 2011). Further, it has been shown that Homer2 protein expression in the NAcc plays an important role in EtOH-induced neuroplasticity (Szumlinski et al., 2005, 2008). These studies suggest that altered expression of proteins involved in the regulation of glutamatergic transmission in the NAcc is critical in the development of behavioral and neurochemical plasticity after repeated ethanol administration.
Only one study to date has shown a potentiation of glutamatergic synaptic transmission mediated by an increase in AMPA mEPSC frequency, but not amplitude, in the putamen of chronic alcohol-drinking monkeys after 28 days of EtOH abstinence (Cuzon Carlson et al., 2011). In contrast with Cuzon Carlson et al. (2011), we found that CIE treatment-induced an increase in AMPAR mEPSC amplitude without modification of the frequency, which strongly suggests a postsynaptic effect of CIE treatment on glutamatergic transmission. The differences between these two studies can be explained by the difference in animal models and brain regions examined. Potentiation of AMPAR-mediated synaptic transmission has been observed after 24 h of EtOH withdrawal in the basolateral amygdala, likely due to an alteration of postsynaptic AMPAR function (Lack et al., 2007). In the NAcc shell, the NMDA-dependent long-term depression (LTD) of AMPAR EPSCs of MSNs induced by pairing stimulation is abolished after 24 h withdrawal from chronic alcohol exposure. Instead, a long-term potentiation (LTP) of EPSC amplitude was observed following pairing stimulation. After 72 h withdrawal, although the NMDA-induced LTD was still strongly reduced, the EPSC potentiation was no longer observed (Jeanes et al., 2011). This time-dependent effect of alcohol during withdrawal may be due to distinct regulation of glutamatergic neurotransmission during short- and long-term withdrawal periods. The neuroadaptive changes observed during early withdrawal may lead to distinct long-term neuroadaptations involving different neuronal mechanisms that are more likely to underlie alcohol-seeking behaviors that promote craving and relapse. Metabotropic glutamate receptor 1 (mGlu1), NMDAR subunit GluN2B, and synaptic protein Homer2B protein expression increases in the NAcc for 2 weeks of withdrawal following 3 months of alcohol-drinking. However, after 2 months of withdrawal, only the Homer2B expression increases remain (Szumlinski et al., 2008). In agreement, we found that CIE treatment did not affect the synaptic expression of NMDAR subunits GluN1 and GluN2B during protracted withdrawal suggesting that NMDAR may not be affected. However, this result does not exclude the possibility of a functional modification of NMDARs independently of subunit expression.
Increasing evidence suggests that there are commonalities between alterations induced by different drugs of abuse in several brain regions. For example, prolonged withdrawal from cocaine induces a long-lasting potentiation of AMPAR EPSC amplitude in the NAcc core associated with an increase in the expression of postsynaptic Ca2+-permeable GluA2-lacking AMPARs (Boudreau and Wolf, 2005; Boudreau et al., 2007; Conrad et al., 2008; Mameli et al., 2009; Ferrario et al., 2011). The expression of synaptic GluA2 in isolated MSN synaptoneurosomes was not modified by the CIE treatment, ruling out the possibility of a down-regulation of GluA2-containing AMPARs in favor of GluA2-lacking AMPARs (Mameli et al., 2007). Although the expression of GluA1 subunit was increased by ∼16% in CIE-treated rats, the increases were not statistically significant when compared to CIV controls. However, increases in a small fraction (<5%) of functional synaptic GluA1-containing AMPARs, which is below the limit of detection of the usual biochemical techniques, has been shown to be sufficient to produce a potentiation of AMPAR synaptic transmission (Guire et al., 2008). GluA2-lacking AMPARs are characterized by a higher single-channel conductance (Isaac et al., 2007). Our findings show an increase in AMPAR unitary conductance after prolonged withdrawal from EtOH. In addition, we found significant inhibition of mEPSCs by the selective antagonist of GluA2-lacking AMPARs only in MSNs from CIE-treated rats. These results suggest that CIE treatment and prolonged withdrawal induces a redistribution of AMPAR subunit composition via increases in a small fraction of functional postsynaptic GluA2-lacking AMPARs.
Potentiation of excitatory glutamatergic transmission of MSNs could alter the integration and processing of pertinent information converging on the NAcc. Depolarized potentials (up-state) of MSNs in the NAcc are driven by excitatory inputs (O’Donnell and Grace, 1995; Goto and O’Donnell, 2001; Gruber and O’Donnell, 2009). These glutamatergic inputs to MSNs do not systematically trigger an AP but a wide range of subthreshold postsynaptic depolarizations (Wilson and Kawaguchi, 1996; Stern et al., 1997; Gruber and O’Donnell, 2009). Thus, enhanced excitatory synaptic transmission efficacy may facilitate the occurrence of suprathreshold postsynaptic depolarizations thereby increasing the firing of MSNs. The modulation of the NAcc neuronal network plasticity by alcohol could lead to important alterations in the integration and transmission of pertinent information to output structures, such as the VTA, which plays a major role in the behavioral effects of alcohol (Nestler, 2001; Hyman et al., 2006).
Homeostatic synaptic scaling is a form of synaptic plasticity that adjusts the strength of all synapses on neurons in response to prolonged or chronic blockade of neuronal activity to maintain stable neuronal function (Turrigiano, 2008). Several studies have demonstrated that prolonged blockade of glutamatergic transmission with a glutamate receptor antagonist triggers the mechanisms of synaptic scaling that induce relatively selective increases in GluA2-lacking AMPARs (Ju et al., 2004; Thiagarajan et al., 2005; Sutton et al., 2006). Interestingly, acute EtOH reduces presynaptic release of glutamate within the NAcc and inhibits postsynaptic NMDAR function (Nie et al., 1994; Zhang et al., 2005). The recurring cycles of EtOH intoxication and withdrawal produced by CIE treatment could induce the mechanisms of synaptic scaling, thus promoting the accumulation of GluA2-lacking AMPARs on MSNs during withdrawal.
In summary, we demonstrated for the first time that CIE treatment induces a long-lasting alteration in membrane properties consistent with increases in KIR and A-type K+ currents of MSNs. We also demonstrated for the first time that prolonged withdrawal from CIE treatment potentiated the glutamatergic transmission of MSNs via a postsynaptic mechanism that involves synaptic incorporation of GluA2-lacking AMPARs characterized by an increased unitary conductance. This enhanced glutamatergic transmission observed in CIE-treated animals may produce an increase in the reactivity of the NAcc neurons to EtOH-related cues leading to an increased alcohol consumption following reinstatement (Rimondini et al., 2003). Clinical studies have shown that pharmacological agents acting on glutamatergic signaling, either to antagonize glutamate receptors or to reduce the release of glutamate, tend to reduce the reinforcing effects of drugs of abuse, and can attenuate the reinstatement of drug-seeking behavior (Olive et al., 2012). Better understanding of the long-lasting cellular and molecular mechanisms involved in alcohol-induced neuroadaptations, such as demonstrated in the present study, will be helpful in the development of innovative effective treatments for alcoholism and other drugs of abuse.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Claire Tu, Katie Jeong, Bill Kwon, Steve Perales, and Dr. Jing Liang for their helpful assistance in the gavage treatment performed on the animals. This work was supported by the National Institute of Health grant AA016100.
Abbreviations
ACSF, artificial cerebrospinal fluid; AHP, afterhyperpolarization; AMPAR, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor; AP, action potential; BK channels, large conductance voltage- and calcium-activated potassium channels; CIE, chronic intermittent ethanol; CIV, chronic intermittent vehicle; EtOH, ethanol; Γ, conductance of AMPARs; GABA, Γ-amino butyric acid; KIR channels, inward rectifying potassium channels; LTD, long-term depression; LTP, long-term potentiation; mEPSC, miniature excitatory postsynaptic current; MSN, medium spiny neuron; NAcc, nucleus accumbens; NMDAR, N-methyl-d-aspartate receptor; Rin, input resistance; RMP, resting membrane potential; SK channels, small conductance voltage- and calcium-activated potassium channels; STDP, spike-time-dependent plasticity; STREX, stress axis-regulated exon; TTX, tetrodotoxin; VTA, ventral tegmental area.
References
Abriam, A., Finzen, E., Bito-Onon, J., Liang, J., Olsen, R. W., and Spigelman, I. (2009). Dose- and frequency dependence of cross-tolerance to diazepam after chronic intermittent ethanol treatment of rats. Abstr. Soc. Neurosci. 34, 64.66.
Belleau, M. L., and Warren, R. A. (2000). Postnatal development of electrophysiological properties of nucleus accumbens neurons. J. Neurophysiol. 84, 2204–2216.
Benke, T. A., Luthi, A., Isaac, J. T., and Collingridge, G. L. (1998). Modulation of AMPA receptor unitary conductance by synaptic activity. Nature 393, 793–797.
Benke, T. A., Luthi, A., Palmer, M. J., Wikstrom, M. A., Anderson, W. W., Isaac, J. T., and Collingridge, G. L. (2001). Mathematical modelling of non-stationary fluctuation analysis for studying channel properties of synaptic AMPA receptors. J. Physiol. (Lond.) 537, 407–420.
Blomeley, C. P., Cains, S., Smith, R., and Bracci, E. (2011). Ethanol affects striatal interneurons directly and projection neurons through a reduction in cholinergic tone. Neuropsychopharmacology 36, 1033–1046.
Boudreau, A. C., Reimers, J. M., Milovanovic, M., and Wolf, M. E. (2007). Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J. Neurosci. 27, 10621–10635.
Boudreau, A. C., and Wolf, M. E. (2005). Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J. Neurosci. 25, 9144–9151.
Budelli, G., Hage, T. A., Wei, A., Rojas, P., Jong, Y. J., O’Malley, K., and Salkoff, L. (2009). Na+-activated K+ channels express a large delayed outward current in neurons during normal physiology. Nat. Neurosci. 12, 745–750.
Buldakova, S. L., Kim, K. K., Tikhonov, D. B., and Magazanik, L. G. (2007). Selective blockade of Ca2+ permeable AMPA receptors in CA1 area of rat hippocampus. Neuroscience 144, 88–99.
Cagetti, E., Liang, J., Spigelman, I., and Olsen, R. W. (2003). Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol. Pharmacol. 63, 53–64.
Chaudhri, N., Sahuque, L. L., Schairer, W. W., and Janak, P. H. (2010). Separable roles of the nucleus accumbens core and shell in context- and cue-induced alcohol-seeking. Neuropsychopharmacology 35, 783–791.
Conrad, K. L., Tseng, K. Y., Uejima, J. L., Reimers, J. M., Heng, L. J., Shaham, Y., Marinelli, M., and Wolf, M. E. (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121.
Cuzon Carlson, V. C., Seabold, G. K., Helms, C. M., Garg, N., Odagiri, M., Rau, A. R., Daunais, J., Alvarez, V. A., Lovinger, D. M., and Grant, K. A. (2011). Synaptic and morphological neuroadaptations in the putamen associated with long-term, relapsing alcohol drinking in primates. Neuropsychopharmacology 36, 2513–2528.
Deng, P., Pang, Z. P., Lei, Z., Shikano, S., Xiong, Q., Harvey, B. K., London, B., Wang, Y., Li, M., and Xu, Z. C. (2011). Up-regulation of A-type potassium currents protects neurons against cerebral ischemia. J. Cereb. Blood Flow Metab. 31, 1823–1835.
Ferrario, C. R., Loweth, J. A., Milovanovic, M., Ford, K. A., Galinanes, G. L., Heng, L. J., Tseng, K. Y., and Wolf, M. E. (2011). Alterations in AMPA receptor subunits and TARPs in the rat nucleus accumbens related to the formation of Ca(2+)-permeable AMPA receptors during the incubation of cocaine craving. Neuropharmacology 61, 1141–1151.
Floresco, S. B., Todd, C. L., and Grace, A. A. (2001). Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J. Neurosci. 21, 4915–4922.
Gertler, T. S., Chan, C. S., and Surmeier, D. J. (2008). Dichotomous anatomical properties of adult striatal medium spiny neurons. J. Neurosci. 28, 10814–10824.
Ghasemzadeh, M. B., Mueller, C., and Vasudevan, P. (2009). Behavioral sensitization to cocaine is associated with increased glutamate receptor trafficking to the postsynaptic density after extended withdrawal period. Neuroscience 159, 414–426.
Goto, Y., and O’Donnell, P. (2001). Synchronous activity in the hippocampus and nucleus accumbens in vivo. J. Neurosci. 21, RC131.
Gruber, A. J., and O’Donnell, P. (2009). Bursting activation of prefrontal cortex drives sustained up states in nucleus accumbens spiny neurons in vivo. Synapse 63, 173–180.
Guire, E. S., Oh, M. C., Soderling, T. R., and Derkach, V. A. (2008). Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J. Neurosci. 28, 6000–6009.
Hartveit, E., and Veruki, M. L. (2007). Studying properties of neurotransmitter receptors by non-stationary noise analysis of spontaneous postsynaptic currents and agonist-evoked responses in outside-out patches. Nat. Protoc. 2, 434–448.
Hopf, F. W., Bowers, M. S., Chang, S. J., Chen, B. T., Martin, M., Seif, T., Cho, S. L., Tye, K., and Bonci, A. (2010). Reduced nucleus accumbens SK channel activity enhances alcohol seeking during abstinence. Neuron 65, 682–694.
Howland, J. G., Taepavarapruk, P., and Phillips, A. G. (2002). Glutamate receptor-dependent modulation of dopamine efflux in the nucleus accumbens by basolateral, but not central, nucleus of the amygdala in rats. J. Neurosci. 22, 1137–1145.
Hughes, J. C., and Cook, C. C. (1997). The efficacy of disulfiram: a review of outcome studies. Addiction 92, 381–395.
Hyman, S. E., Malenka, R. C., and Nestler, E. J. (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci. 29, 565–598.
Isaac, J. T., Ashby, M. C., and Mcbain, C. J. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871.
Ishikawa, M., Mu, P., Moyer, J. T., Wolf, J. A., Quock, R. M., Davies, N. M., Hu, X. T., Schluter, O. M., and Dong, Y. (2009). Homeostatic synapse-driven membrane plasticity in nucleus accumbens neurons. J. Neurosci. 29, 5820–5831.
Jeanes, Z. M., Buske, T. R., and Morrisett, R. A. (2011). In vivo chronic intermittent ethanol exposure reverses the polarity of synaptic plasticity in the nucleus accumbens shell. J. Pharmacol. Exp. Ther. 336, 155–164.
Johnson, M. W., Chotiner, J. K., and Watson, J. B. (1997). Isolation and characterization of synaptoneurosomes from single rat hippocampal slices. J. Neurosci. Methods 77, 151–156.
Ju, W., Morishita, W., Tsui, J., Gaietta, G., Deerinck, T. J., Adams, S. R., Garner, C. C., Tsien, R. Y., Ellisman, M. H., and Malenka, R. C. (2004). Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat. Neurosci. 7, 244–253.
Kasanetz, F., Deroche-Gamonet, V., Berson, N., Balado, E., Lafourcade, M., Manzoni, O., and Piazza, P. V. (2010). Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science 328, 1709–1712.
Kim, J., Park, B. H., Lee, J. H., Park, S. K., and Kim, J. H. (2011). Cell type-specific alterations in the nucleus accumbens by repeated exposures to cocaine. Biol. Psychiatry 69, 1026–1034.
Kokka, N., Sapp, D. W., Taylor, A. M., and Olsen, R. W. (1993). The kindling model of alcohol dependence: similar persistent reduction in seizure threshold to pentylenetetrazol in animals receiving chronic ethanol or chronic pentylenetetrazol. Alcohol. Clin. Exp. Res. 17, 525–531.
Koob, G. F. (2008). Hedonic homeostatic dysregulation as a driver of drug-seeking behavior. Drug Discov. Today Dis. Models 5, 207–215.
Koob, G. F., Ahmed, S. H., Boutrel, B., Chen, S. A., Kenny, P. J., Markou, A., O’Dell, L. E., Parsons, L. H., and Sanna, P. P. (2004). Neurobiological mechanisms in the transition from drug use to drug dependence. Neurosci. Biobehav. Rev. 27, 739–749.
Koob, G. F., and Le Moal, M. (2008). Review. Neurobiological mechanisms for opponent motivational processes in addiction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 3113–3123.
Lack, A. K., Diaz, M. R., Chappell, A., Dubois, D. W., and Mccool, B. A. (2007). Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J. Neurophysiol. 98, 3185–3196.
LaLumiere, R. T., and Kalivas, P. W. (2008). Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J. Neurosci. 28, 3170–3177.
Liang, J., Suryanarayanan, A., Abriam, A., Snyder, B., Olsen, R. W., and Spigelman, I. (2007). Mechanisms of reversible GABAA receptor plasticity after ethanol intoxication. J. Neurosci. 27, 12367–12377.
Liang, J., Zhang, N., Cagetti, E., Houser, C. R., Olsen, R. W., and Spigelman, I. (2006). Chronic intermittent ethanol-induced switch of ethanol actions from extrasynaptic to synaptic hippocampal GABAA receptors. J. Neurosci. 26, 1749–1758.
Magazanik, L. G., Buldakova, S. L., Samoilova, M. V., Gmiro, V. E., Mellor, I. R., and Usherwood, P. N. (1997). Block of open channels of recombinant AMPA receptors and native AMPA/kainate receptors by adamantane derivatives. J. Physiol. (Lond.) 505(Pt 3), 655–663.
Mameli, M., Balland, B., Lujan, R., and Luscher, C. (2007). Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317, 530–533.
Mameli, M., Halbout, B., Creton, C., Engblom, D., Parkitna, J. R., Spanagel, R., and Luscher, C. (2009). Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat. Neurosci. 12, 1036–1041.
Mann, K., Lehert, P., and Morgan, M. Y. (2004). The efficacy of acamprosate in the maintenance of abstinence in alcohol-dependent individuals: results of a meta-analysis. Alcohol. Clin. Exp. Res. 28, 51–63.
Mermelstein, P. G., Song, W. J., Tkatch, T., Yan, Z., and Surmeier, D. J. (1998). Inwardly rectifying potassium (IRK) currents are correlated with IRK subunit expression in rat nucleus accumbens medium spiny neurons. J. Neurosci. 18, 6650–6661.
Mu, P., Moyer, J. T., Ishikawa, M., Zhang, Y., Panksepp, J., Sorg, B. A., Schluter, O. M., and Dong, Y. (2010). Exposure to cocaine dynamically regulates the intrinsic membrane excitability of nucleus accumbens neurons. J. Neurosci. 30, 3689–3699.
Mulholland, P. J., Becker, H. C., Woodward, J. J., and Chandler, L. J. (2011). Small conductance calcium-activated potassium type 2 channels regulate alcohol-associated plasticity of glutamatergic synapses. Biol. Psychiatry 69, 625–632.
Mulligan, M. K., Rhodes, J. S., Crabbe, J. C., Mayfield, R. D., Adron Harris, R., and Ponomarev, I. (2011). Molecular profiles of drinking alcohol to intoxication in C57BL/6J mice. Alcohol. Clin. Exp. Res. 35, 659–670.
Neasta, J., Ben Hamida, S., Yowell, Q., Carnicella, S., and Ron, D. (2010). Role for mammalian target of rapamycin complex 1 signaling in neuroadaptations underlying alcohol-related disorders. Proc. Natl. Acad. Sci. U.S.A. 107, 20093–20098.
Neasta, J., Ben Hamida, S., Yowell, Q. V., Carnicella, S., and Ron, D. (2011). AKT signaling pathway in the nucleus accumbens mediates excessive alcohol drinking behaviors. Biol. Psychiatry 70, 575–582.
Nie, Z., Madamba, S. G., and Siggins, G. R. (1994). Ethanol inhibits glutamatergic neurotransmission in nucleus accumbens neurons by multiple mechanisms. J. Pharmacol. Exp. Ther. 271, 1566–1573.
Nisenbaum, E. S., and Wilson, C. J. (1995). Potassium currents responsible for inward and outward rectification in rat neostriatal spiny projection neurons. J. Neurosci. 15, 4449–4463.
Nisenbaum, E. S., Xu, Z. C., and Wilson, C. J. (1994). Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projection neurons. J. Neurophysiol. 71, 1174–1189.
O’Donnell, P., and Grace, A. A. (1993). Physiological and morphological properties of accumbens core and shell neurons recorded in vitro. Synapse 13, 135–160.
O’Donnell, P., and Grace, A. A. (1995). Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J. Neurosci. 15, 3622–3639.
Olive, M. F., Cleva, R. M., Kalivas, P. W., and Malcolm, R. J. (2012). Glutamatergic medications for the treatment of drug and behavioral addictions. Pharmacol. Biochem. Behav. 100, 801–810.
Rassnick, S., Pulvirenti, L., and Koob, G. F. (1992). Oral ethanol self-administration in rats is reduced by the administration of dopamine and glutamate receptor antagonists into the nucleus accumbens. Psychopharmacology (Berl.) 109, 92–98.
Rehm, J., Mathers, C., Popova, S., Thavorncharoensap, M., Teerawattananon, Y., and Patra, J. (2009). Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 373, 2223–2233.
Rimondini, R., Sommer, W., and Heilig, M. (2003). A temporal threshold for induction of persistent alcohol preference: behavioral evidence in a rat model of intermittent intoxication. J. Stud. Alcohol 64, 445–449.
Ross, S., and Peselow, E. (2009). Pharmacotherapy of addictive disorders. Clin. Neuropharmacol. 32, 277–289.
Sears, R. M., Liu, R. J., Narayanan, N. S., Sharf, R., Yeckel, M. F., Laubach, M., Aghajanian, G. K., and Dileone, R. J. (2010). Regulation of nucleus accumbens activity by the hypothalamic neuropeptide melanin-concentrating hormone. J. Neurosci. 30, 8263–8273.
Sesack, S. R., and Grace, A. A. (2010). Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology 35, 27–47.
Shao, L. R., Halvorsrud, R., Borg-Graham, L., and Storm, J. F. (1999). The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J. Physiol. (Lond.) 521(Pt 1), 135–146.
Shen, W., Tian, X., Day, M., Ulrich, S., Tkatch, T., Nathanson, N. M., and Surmeier, D. J. (2007). Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat. Neurosci. 10, 1458–1466.
Snyder, J. L., and Bowers, T. G. (2008). The efficacy of acamprosate and naltrexone in the treatment of alcohol dependence: a relative benefits analysis of randomized controlled trials. Am. J. Drug Alcohol Abuse 34, 449–461.
Steephen, J. E., and Manchanda, R. (2009). Differences in biophysical properties of nucleus accumbens medium spiny neurons emerging from inactivation of inward rectifying potassium currents. J. Comput. Neurosci. 27, 453–470.
Stern, E. A., Kincaid, A. E., and Wilson, C. J. (1997). Spontaneous subthreshold membrane potential fluctuations and action potential variability of rat corticostriatal and striatal neurons in vivo. J. Neurophysiol. 77, 1697–1715.
Stuber, G. D., Hopf, F. W., Tye, K. M., Chen, B. T., and Bonci, A. (2010). Neuroplastic alterations in the limbic system following cocaine or alcohol exposure. Curr. Top. Behav. Neurosci. 3, 3–27.
Sutton, M. A., Ito, H. T., Cressy, P., Kempf, C., Woo, J. C., and Schuman, E. M. (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125, 785–799.
Szumlinski, K. K., Ary, A. W., Lominac, K. D., Klugmann, M., and Kippin, T. E. (2008). Accumbens Homer2 overexpression facilitates alcohol-induced neuroplasticity in C57BL/6J mice. Neuropsychopharmacology 33, 1365–1378.
Szumlinski, K. K., Lominac, K. D., Oleson, E. B., Walker, J. K., Mason, A., Dehoff, M. H., Klugmann, M., Cagle, S., Welt, K., During, M., Worley, P. F., Middaugh, L. D., and Kalivas, P. W. (2005). Homer2 is necessary for EtOH-induced neuroplasticity. J. Neurosci. 25, 7054–7061.
Thiagarajan, T. C., Lindskog, M., and Tsien, R. W. (2005). Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47, 725–737.
Thomas, M. J., Beurrier, C., Bonci, A., and Malenka, R. C. (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat. Neurosci. 4, 1217–1223.
Turrigiano, G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435.
Uchimura, N., Cherubini, E., and North, R. A. (1989). Inward rectification in rat nucleus accumbens neurons. J. Neurophysiol. 62, 1280–1286.
Wang, W., Zhang, K., Yan, S., Li, A., Hu, X., Zhang, L., and Liu, C. (2011). Enhancement of apamin-sensitive medium afterhyperpolarization current by anandamide and its role in excitability control in cultured hippocampal neurons. Neuropharmacology 60, 901–909.
Williams, J. M., Guevremont, D., Kennard, J. T., Mason-Parker, S. E., Tate, W. P., and Abraham, W. C. (2003). Long-term regulation of N-methyl-D-aspartate receptor subunits and associated synaptic proteins following hippocampal synaptic plasticity. Neuroscience 118, 1003–1013.
Wilson, C. J., and Kawaguchi, Y. (1996). The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J. Neurosci. 16, 2397–2410.
Keywords: alcoholism, withdrawal, reward, GluA2-lacking AMPARs, chronic intermittent ethanol treatment, synaptic transmission
Citation: Marty VN and Spigelman I (2012) Long-lasting alterations in membrane properties, K+ currents, and glutamatergic synaptic currents of nucleus accumbens medium spiny neurons in a rat model of alcohol dependence. Front. Neurosci. 6:86. doi: 10.3389/fnins.2012.00086
Received: 16 March 2012; Accepted: 22 May 2012;
Published online: 08 June 2012.
Edited by:
A. Leslie Morrow, University of North Carolina School of Medicine, USAReviewed by:
Kuei Y. Tseng, Rosalind Franklin University of Medicine and Science, USAJohn Woodward, Medical University of South Carolina, USA
Copyright: © 2012 Marty and Spigelman. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Igor Spigelman, Division of Oral Biology and Medicine, UCLA School of Dentistry, 10833 Le Conte Avenue, 63-078 CHS, Los Angeles, CA 90095-1668, USA. e-mail: igor@ucla.edu