 Christoph Ufer1*
Christoph Ufer1* Chi Chiu Wang2,3,4*
Chi Chiu Wang2,3,4*- 1 Institute of Biochemistry, Charité – University Medicine Berlin, Berlin, Germany
- 2 Department of Obstetrics and Gynaecology, The Chinese University of Hong Kong, Shatin, Hong Kong
- 3 Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Shatin, Hong Kong
- 4 Neuro-Degeneration and -Development and Repair, Institute of Biomedical Sciences, The Chinese University of Hong Kong, Shatin, Hong Kong
Embryo development relies on the complex interplay of the basic cellular processes including proliferation, differentiation, and apoptotic cell death. Precise regulation of these events is the basis for the establishment of embryonic structures and the organ development. Beginning with fertilization of the oocyte until delivery the developing embryo encounters changing environmental conditions such as varying levels of oxygen, which can give rise to reactive oxygen species (ROS). These challenges are met by the embryo with metabolic adaptations and by an array of anti-oxidative mechanisms. ROS can be deleterious by modifying biological molecules including lipids, proteins, and nucleic acids and may induce abnormal development or even embryonic lethality. On the other hand ROS are vital players of various signaling cascades that affect the balance between cell growth, differentiation, and death. An imbalance or dysregulation of these biological processes may generate cells with abnormal growth and is therefore potentially teratogenic and tumorigenic. Thus, a precise balance between processes generating ROS and those decomposing ROS is critical for normal embryo development. One tier of the cellular protective system against ROS constitutes the family of selenium-dependent glutathione peroxidases (GPx). These enzymes reduce hydroperoxides to the corresponding alcohols at the expense of reduced glutathione. Of special interest within this protein family is the moonlighting enzyme glutathione peroxidase 4 (Gpx4). This enzyme is a scavenger of lipophilic hydroperoxides on one hand, but on the other hand can be transformed into an enzymatically inactive cellular structural component. GPx4 deficiency – in contrast to all other GPx family members – leads to abnormal embryo development and finally produces a lethal phenotype in mice. This review is aimed at summarizing the current knowledge on GPx isoforms during embryo development and tumor development with an emphasis on GPx4.
Introduction
The availability of molecular dioxygen is the basis of eukaryotic cell function by serving as electron acceptor of the mitochondrial electron transport chain. This biological process that reduces oxygen to water is not perfect but produces a significant amount of reactive oxygen species (ROS) such as the superoxide anion, hydrogen peroxide, and the hydroxyl radical (Balaban et al., 2005). In addition, ROS can be generated by an array of other enzymatic and non-enzymatic reactions (Ufer et al., 2010). On one hand ROS pose a hazard for cell survival by being able to modify virtually all biological molecules. Oxidative modifications may alter the properties of affected molecules and impair normal cellular processes. Most importantly, oxidative modifications of nuclear DNA can alter the genetic information of cells, which may subsequently lead to the development of cancer (Poulsen, 2005). On the other hand ROS can be vital for maintaining normal cell function including growth, differentiation and cell death (Van Leyen et al., 1998; Giorgio et al., 2007). To facilitate precise ROS action cellular mechanisms have evolved to detect cellular ROS levels and to counteract ROS production. This intricate system of ROS homeostasis employs redox-sensitive signaling molecules and transcription factors on one hand and a robust firewall of protective anti-oxidative enzymes on the other hand.
The aim of this review is firstly to outline the current knowledge on the biological significance of ROS during mammalian embryo development. Secondly, the various contributions of selenium-dependent GPxs to embryonic ROS homeostasis will be discussed. Most of the available knowledge has been gained from experiments in mice. Thus, this review we will mostly focus on mice, but will make excursions to the human organism when appropriate.
Embryo Development
The developmental sequence begins with a fertilized egg, proceeds to adulthood, and then repeats itself through reproduction is known as the life cycle. Developmental biologists divide the animal life cycle into three major periods: embryo development, postembryonic development, and adulthood. The term embryo is generally used to describe the developing individual from fertilization through the formation of differentiation tissues. Embryogenesis is a period of embryo development, which can be subdivided into the stages of fertilization, cleavage, gastrulation, organogenesis, and histogenesis (Gilbert, 2000).
Fertilization is the union of oocyte and sperm. The oocyte is an extremely large cell, loaded with nutrients to support early embryo development. The sperm is a cell that is highly specialized for its function of finding and fertilizing an oocyte. After fertilization by a sperm, the oocyte is called a zygote. During embryonic cleavage, the zygote undergoes a series of mitotic divisions that result in the formation of the blastomere. The early embryo develops in the oviduct before migrating to the uterus for implantation. Compaction and blastulation occur in the transition from oviduct to uterus before post-hatching development (Boyd and Hamilton, 1952). During gastrulation, embryos undergo a complex series of morphogenetic movements, in which embryonic cells rearrange and migrate. At the end of gastrulation the embryo consists of three germ layers which will undergo further morphogenetic movements and interactions to form the rudiments of primitive organs. During organogenesis, the developing embryo shows the basic body plan and finally acquires functional specialization. These developmental processes involve a series of proliferation and differentiation processes that are well balanced by spatial and temporal controls of molecular and cellular programming.
Oxygen and Development
Oxygen is essential during embryo development (Tuuli et al., 2011). Following fertilization in the oviduct, the early embryo encounters a decreasing oxygen gradient when moving down the reproductive tract. The oviductal oxygen concentration is less than 40% of the atmospheric concentration (Leese, 1995). In the uterus, oxygen concentration is just about 3–5%. Thus, preimplantation and early postimplantation embryogenesis takes place under hypoxic and even almost anaerobic conditions (Fischer and Bavister, 1993; Figure 1).
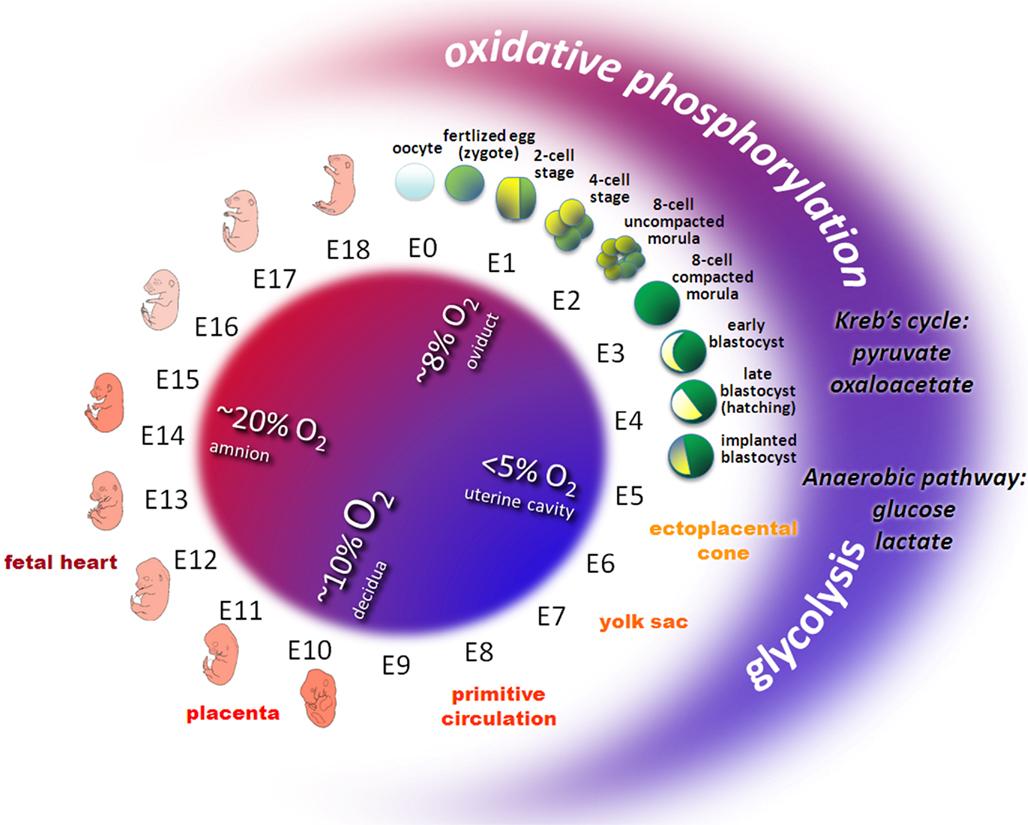
Figure 1. Oxygen and metabolic status during embryo development in mouse. Owing to the variable environmental conditions including oxygen and glucose concentrations in different regions of the female reproductive tract, the developing early embryo has to adapt its energy metabolism. Before the circulation system in the ectoplacental cone, the yolk sac, the placenta and the embryonic heart develop, early embryos transit from relative aerobic (red area, ~8% O2) and low glucose to anaerobic (blue area, < 5% O2) and high glucose conditions. Redox metabolism transits from oxidative phosphorylation (low glucose, high oxygen) to glycolysis (high glucose, low oxygen). Pyruvate or oxaloacetate are essential substrates during early cleavage whereas glucose and lactate prevail from the eight-cell stage embryos to peri-implanted blastocyst.
The yolk sac plays an important role for early embryo development since it delivers oxygen and nutrients to the embryo during the postimplantation period. During early implantation, hypoxic and even anoxic conditions confront the invasion of the trophectoderm into the endometrium of the uterus (Leese, 1995). Later on, the yolk sac regresses and the allantoic placenta takes over its function to support further embryonic development. These sudden alterations in uteroplacental circulation expose the embryos to high oxygen concentrations (New and Coppola, 1970). Once the uteroplacental and embryonic circulation systems are established, the embryo becomes less sensitive for the maternal oxidative status.
Energy Metabolism
Energy is required for most control mechanisms of development and is based on the production of high-energy adenosine triphosphate (ATP) by either glycolysis or oxidative phosphorylation. In glycolysis glucose is used as substrate and converted to pyruvate. In oxidative phosphorylation pyruvate and oxaloacetate are used as substrates. Reduced nicotinamide adenine dinucleotide (NADH) as an electron donor and oxygen as an electron acceptor in the electron transport chain convert adenosine diphosphate (ADP) to ATP. Following ovulation, mammalian oocytes rely on internal energy resources and on the few metabolites supplied by secretions of the Fallopian tube to survive. However, the oviductal fluid generally has relatively low oxygen and glucose concentrations (< 1 mmol/L) which favor glycolytic activity.
The ability of the early embryo to maintain an appropriate energy metabolism is necessary for successful development. Changes in energy metabolism pathways are involved in the control of early preimplantation embryo development (Wales, 1975; Bavister, 1995). In the eight-cell stage glucose is an effective substrate, while in the blastocyst stage pyruvate and/or oxaloacetate are essential for early cleavage. An increasing role for energy derived from glycolysis is required during compaction and blastulation. Such a shift in metabolic pathways is essential as the embryo faces an increasingly hypoxic environment in utero (Harvey et al., 2002). Later on, the transition from anaerobic glycolysis to oxidative metabolism coincides with the time at which the chorioallantoic circulation is established and the heart starts to function (Clough, 1985).
Although oxygen is required for embryo development, the use of oxygen as an energy substrate also poses a potential hazard via the formation of ROS and reactive nitrogen species (RNS) as natural by-products of oxygen metabolism, particularly the superoxide anion 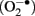 and the hydroxyl radical (OH• ). ROS and RNS are highly active electron acceptors, able to strip electrons from other molecules to become free radicals. Thus, they are able to modify all biological macromolecules including lipids, proteins, RNA and DNA, and impair their function. Hydrogen peroxide (H2O2) is not a radical per se, but is a product of and metal ion catalysis. However, both H2O2 and can form the extremely reactive OH• The cells must protect themselves from the damage caused by ROS.
and the hydroxyl radical (OH• ). ROS and RNS are highly active electron acceptors, able to strip electrons from other molecules to become free radicals. Thus, they are able to modify all biological macromolecules including lipids, proteins, RNA and DNA, and impair their function. Hydrogen peroxide (H2O2) is not a radical per se, but is a product of and metal ion catalysis. However, both H2O2 and can form the extremely reactive OH• The cells must protect themselves from the damage caused by ROS.
Redox Responses
Oxidative stress mediated by ROS results in an imbalance of the intracellular oxidative potential toward an oxidized potential (Balaban et al., 2005). Oxidative stress is associated with impaired early development and fragmented embryos (Johnson and Nasr-Esfahani, 1994; Yang et al., 1998). It can induce apoptosis of the oocyte and the early embryo (Liu and Keefe, 2000; Liu et al., 2000) and is associated with postovulatory aging of the oocyte (Tarin, 1996; Fissore et al., 2002). Oxidative stress also is a natural process needed to maintain normal cell function. ROS actively mediate cellular responses to a variety of stimuli directly or indirectly regulated by oxidative stress. ROS and RNS metabolites at lower concentrations function as signal transducers acting on various regulatory levels and thus affect the cellular phenotype (D’Autreaux and Toledano, 2007). ROS have a specific function during hatching of the mouse blastocyst from the zona pellucida (Thomas et al., 1997). Embryos undergo a specific burst of ROS production at hatching. The production of ROS may also be an important regulatory system for programmed cell death in the blastocyst (Pierce et al., 1991).
The balance of reductive and oxidative (redox) homeostasis is the key to the orchestration of regulations during embryo development (Covarrubias et al., 2008). Precise control of cellular redox homeostasis is essential for regular cell function, and this is of particular importance for the complex developmental processes during embryogenesis (Castagne et al., 1999). Cellular redox homeostasis affects a variety of basic cellular functions such as energy supply, proliferation, differentiation, maturation, and apoptosis (Salas-Vidal et al., 1998). It also plays an important role in the regulation of the gene expression. Embryogenesis involves a series of differentiation and maturation processes that are well balanced by spatial and temporal controls of cellular gene expression involving mechanisms on epigenetic, transcriptional, and post-transcriptional levels according to an inbuilt biological program (Ufer et al., 2010).
Oxidative Damage
Reactive oxygen species/RNS at higher concentration pose a potential hazard in modifying macromolecules and altering their biological functions. The modifications often lead to cellular dysfunction and may have deleterious consequences. Dysregulations in the redox equilibrium induce developmental retardations, organ malformations, teratogenesis, and even embryo lethality (Chen et al., 1999; Hansen, 2006). The processes controlling embryonic redox homeostasis are important determinants of teratological risk. Individual risk of ROS mediated teratogenesis depends on both genetic as well as environmental factors (Wells et al., 2005). During certain time windows of embryogenesis, the embryo is more susceptible to oxidative stress and excessive ROS formation (Hansen, 2006).
Under normal circumstances, cells are able to defend themselves against redox-mediated damage. At certain developmental periods, including early postimplantation stage, and the completion of embryonic circulation, the embryo is particularly susceptible to redox alterations and at these stages a functional anti-oxidative defense system is of particular importance. As for the adult organism, both small molecule antioxidants and anti-oxidative enzymes play an important role in protecting the developing embryos from oxidative damage. Unfortunately, the enzymatic activity of many anti-oxidative enzymes in embryos is much lower than in adults. Thus, embryos are particularly sensitive to oxidative damage (Parman et al., 1999; Winn and Wells, 1999).
Antioxidants
Small molecule antioxidants include ascorbic acid, tocopherol, uric acid, and glutathione. Glutathione (γ-glutamylcysteinylglycine) is the major cellular non-protein sulfhydryl compound. This thiol plays a prominent role in detoxification of exogenous and endogenous compounds, anti-oxidative defense as well as maintaining the intracellular redox status (Munday and Winterbourn, 1989). Glutathione exists in its oxidized (GSSG) and its reduced (GSH) form and plays a major role in regulating ROS concentrations within the cytoplasm, both directly as a free-radical scavenger and indirectly as a substrate with NADPH for detoxifying ROS (Guerin et al., 2001).
GSH is involved in the protection against oxidative damage in both the male and female gametes (Luberda, 2005). GSH concentrations in the maturing spermatozoa gradually decline during spermatogenesis. GSH has been also implicated in maintaining the meiotic spindle morphology of the oocyte. It plays an important role in the preparation of a mature oocyte to receive a sperm and in oocyte maturation (Luberda, 2005). During preimplantation development embryonic GSH levels continuously decrease (Gardiner and Reed, 1995). After fertilization, GSH plays an active role in the formation of the male pronucleus, and is crucial for early embryogenesis up to the blastocyst stage. Following preimplantation stage embryos have little capacity for GSH synthesis, GSH concentrations decrease as early cleavage proceeds and as the late blastocyst prepares for implantation (Gardiner and Reed, 1995).
Another important group of antioxidants are selenoproteins such as glutathione peroxidases (GPxs) and thioredoxin reductases (TrxRs; Brigelius-Flohe et al., 2003). TrxRs are crucial to maintain the small antioxidant protein thioredoxin in its reduced state (Arner, 2009). Overexpression of thioredoxin renders embryos more resistant to oxidative stress (Kobayashi-Miura et al., 2002), whereas thioredoxin null mutants exhibit dramatically reduced proliferation of the inner cell mass resulting in lethality during the early postimplantation period (reviewed in Ufer et al., 2010). Accordingly, when thioredoxin function is compromised by blocking expression of TrxR enzymes TrxR knockout embryos fail to survive beyond midgestation (reviewed in Conrad, 2009; Ufer et al., 2010). This indicates an important role for these selenoproteins in early embryo development.
Antioxidant enzymes remove various ROS produced by free-radical reactions. Superoxide dismutases (SODs) scavenge , converting it into H2O2 and oxygen (Maier and Chan, 2002). Mice lacking Sod1 have increased age-related muscle mass loss, early development of cataracts, macular degeneration, thymic involution, hepatocellular carcinoma, and a shortened lifespan (Muller et al., 2007). Mice lacking Sod2 die shortly after birth, indicating that uncontrolled levels of H2O2 are incompatible with mammalian life (Li et al., 1995). Heterozygous Sod2 knockout mice have a normal lifespan and minimal phenotypic defects but do suffer increased DNA damage and increased incidence of cancer (Van Remmen et al., 2003). Data relating to the biological role of Sod3 in embryo development are still missing. Catalase and peroxidases, on the other hand, convert H2O2 into water (O’Brien, 2000; Kirkman and Gaetani, 2007). Mammals have many peroxidases but only one catalase (Jin et al., 2003). Mice genetically engineered to lack catalase are phenotypically normal, indicating that this enzyme is dispensable during development (Ho et al., 2004).
Selenium-Dependent Glutathione Peroxidases and Development
Besides catalases and SODs the family of glutathione-dependent peroxidases (GPx) constitutes the major player to keep oxidative stress at bay. GPx1–4 in mice and humans and GPx6 in humans out of eight known GPx isoforms in mammals contain selenocysteine at their active sites and thus are categorized as selenoproteins. This review is focused on the murine selenium containing GPx isoenzymes GPx1–4 and the interested reader is referred to other publications for non-selenium GPx isoenzymes GPx5–8 (Kryukov et al., 2003; Chabory et al., 2009; Nguyen et al., 2011). The selenocysteine is coded for by an opal codon and incorporated by an unusual mechanism that recodes this stop codon and facilitates co-translational insertion of the biogenic amino acid selenocysteine into the nascent polypeptide chain (Hatfield and Gladyshev, 2002). GPxs catalyze the reduction of hydroperoxides to the corresponding alcohols at the expense of the reducing equivalent glutathione. Despite a common catalytic mechanism each GPx isoform bears unique features with respect to their subcellular localization, tissue distribution, substrate specificity and their apparent biological function. An interesting characteristic of mammalian selenoproteins is the so-called selenium hierarchy, which determines whether a selenoprotein is expressed or not under selenium deficiency (Behne and Kyriakopoulos, 2001). Within this hierarchy GPx4 for instance ranks very high permitting stable GPx4 expression when selenium is rare which indicates a strong dependence of the organism on this selenoprotein. The underlying molecular mechanisms of the selenium hierarchy however remain somewhat unclear.
To assess the impact of selenoproteins on the survival of mammalian organisms different transgenic mouse models were employed. Selenoprotein biosynthesis crucially depends on the availability of the selenocysteine-specific tRNA[Ser]Sec (Trsp). Targeted deletion of the gene coding for Trsp results in early embryonic lethality at the egg cylinder stage (gestational day E6.0) which highlights the importance of selenoproteins for embryonic survival (Bosl et al., 1997). In mammals selenium can be stored and transported by the plasma protein selenoprotein P (SePP; Motsenbocker and Tappel, 1982), but SePP-/- mice are viable (Schomburg et al., 2003). In addition to SePP-dependent storage, selenium can also be made available through nutrition and it was suggested that selenium uptake via nutrition prevents the development of a more severe knockout phenotype (Schweizer et al., 2004b). Nevertheless, SePP-/- develop ataxia and a wide and clumsy gait, which points to the importance of selenium for the functionality of the brain. This is in line with a phenotype associated to a neuron-specific knockout of the Trsp gene, which induces abnormal neuronal development and progressive neurodegeneration (Wirth et al., 2010). Glutathione peroxidases not only depend on selenium but also on the availability of reduced glutathione (GSH) for their enzymatic activity. GSH is ubiquitously present in the organism at varying redox states and constitutes a vital component of the cellular antioxidant system (Mari et al., 2009). Targeted genetic abrogation of GSH biosynthesis causes early embryonic death in mice and impaired biosynthesis of GSH in humans has been correlated with neurological disorders such as schizophrenia (Shi et al., 2000; Gysin et al., 2007). To exert its antioxidant function GSH needs to be constantly converted from its oxidized GSSH to its reduced GSH by a process that requires NADPH. A major source of NADPH is the oxidative pentose phosphate shunt. When this metabolic pathway is interrupted and the capacity of the organism to maintain sufficient levels of reduced GSH is impaired embryos do not survive past midgestation (Longo et al., 2002). Embryonic death induced by abrogation of the oxidative pentose phosphate shunt is considerably delayed (gestational day E8.5 versus E11.5) to what is observed in embryos lacking GSH de novo biosynthesis. Thus, other sources for NADPH such as malic enzyme or isocitrate dehydrogenase may contribute cellular NADPH levels (Dumollard et al., 2007).
Glutathione Peroxidase 1
Glutathione peroxidase 1 (GPx1) is ubiquitously expressed and dominantly found in the cytosol, where it scavenges hydrophilic peroxide species such as H2O2 (Brigelius-Flohe, 1999). In addition, GPx1 expression has been detected in mitochondria where it is thought to protect mitochondrial DNA from oxidative damage (Legault et al., 2000; Kienhofer et al., 2009). GPx1 is considered the major antioxidant enzyme within the GPx family. However, GPx1-/- mice develop normally and are able to compensate mild oxidative stress (Ho et al., 1997). In that respect GPx1 shares the fate of many other anti-oxidative genes such as catalases and superoxide dismutases that also fail to induce significant prenatal phenotypes (Ufer et al., 2010). These findings emphasize the fact that the cellular anti-oxidative defense system contains many compensatory mechanisms to allow survival. By controlling cellular levels of H2O2 GPx1 is also implicated in the regulation of the cell cycle and artificial GPx1 overexpression is paralleled by faster growth of tumor cells (Lu et al., 1997; Li et al., 2000). In contrast, GPx1 deficiency renders cells more sensitive to certain stressors, which results in elevated induction of apoptotic cell death (Fu et al., 2001; Flentjar et al., 2002).
The promoter of the GPx1 gene has not yet been systematically characterized but scattered data on GPx1 expression regulation are available. The GPx1 promoter lacks a canonical TATA-box and contains a proximal CpG island, which are typical characteristics for the promoter a housekeeping genes. In silico analysis (Cartharius et al., 2005) indicates the presence of a number of potential stimulatory/specificity protein 1 (Sp1) binding sites. In addition, GPx1 was found to be under the control of the tumor suppressor protein p53 (Gladyshev et al., 1998; Tan et al., 1999). The transcription factor p53 is a vital regulator that translates the occurrence of genotoxic stress into alterations of gene expression (Tishler et al., 1993). Thus, by inducing GPx1 expression p53 increases the cellular anti-oxidative capacity when DNA damage is detected. A frequently observed prerequisite for the onset of cancer is genomic instability which is affected by aberrant methylation patterns of the genome (Negrini et al., 2010; Sharma et al., 2010). Interestingly, the promoters of GPx1 as well as GPx3 were found to be hypermethylated in gastric carcinoma cell lines (Jee et al., 2009). Promoter hypermethylation is commonly associated with a silencing of gene expression. DNA methylation of CpG islands is an epigenetic modification in higher eukaryotes that affects transcriptional regulation vital for embryonic and neural development (Bogdanovic and Veenstra, 2009). Dysregulation of CpG methylation has been related to genetic instability, cancerogenesis, and neuronal degeneration (Nagarajan and Costello, 2009). On the other hand, epigenetic modifications themselves are sensitive to changes of the cellular redox tone implying a regulatory loop that modulates epigenetic mechanisms of gene expression regulation (Zawia et al., 2009). Thus, expression silencing of GPx1 and GPx3 reduces the capacity of the affected cells to detoxify genotoxic substances and potentially enhances the development of cancerous abnormalities.
On the post-translational level, GPx1 activity is positively affected by phosphorylation of tyrosine residue (Tyr-96) by the tyrosine kinases c-abl and arg (Cao et al., 2003). C-abl and arg are activated by an array of extracellular stimuli including growth factors and cytoplasmic c-abl has been shown to be required for cell proliferation by promoting G1 progression (Sirvent et al., 2008). De-regulation of c-abl signaling is observed in various forms of human cancer. This is in line with the mitogenic effect of GPx1 in GPx1 overexpression in tumor cells (Lu et al., 1997; Li et al., 2000).
Glutathione Peroxidase 2
In the gastrointestinal system of mammals and in humans also in the liver the second GPx family member, namely glutathione peroxidase 2 (GPx2), is expressed (Chu et al., 1993; Dreher et al., 1997). Its expression pattern suggests a protective role of this isoform against ROS derived from the gut. GPx2 ranks very high within the selenium hierarchy pointing to a strong dependence of the organism on GPx2 expression (Brigelius-Flohe, 1999). However, GPx2 knockout mice do not develop an aberrant phenotype before birth (Esworthy et al., 2000), but exhibit an elevated incidence of squamous cell carcinomas induced by ultraviolet irradiation (Walshe et al., 2007). Moreover, a GPx1/GPx2 double knockout is paralleled by a high incidence of intestinal inflammation as well as increased tumor development (Esworthy et al., 2001; Chu et al., 2004). These data indicate that ubiquitously expressed GPx1 can at least partially substitute for a lack of intestinal GPx2 expression. The underlying mechanisms of accelerated tumor development appear to involve an accumulation of mutations of the genetic and epigenetic code (Lee et al., 2006; Hahn et al., 2008). Thus, whereas GPx2 may be dispensable during embryo development it is a vital factor for maintaining a functional intestinal epithelium in adults and protects the organism from genotoxic substances that are produced in the gut.
The strong tissue specific expression of GPx2 requires the presence of tissue specific regulatory expression mechanisms. Similar to the human GPx1 gene no canonical TATA-box is found in the proximal GPx2 promoter region. In silico analysis predicts the presence of a number of nuclear factor-Y (NF-Y) and Sp1-binding sites, but their functional significance remains to be shown (Cartharius et al., 2005). Despite the lack of knowledge on the basic regulation of GPx2 expression a number of regulatory mechanisms were identified that affect GPx2 expression. For instance, the GPx2 promoter contains so-called antioxidant response elements that confer sensitivity to oxidative stress via the Nrf2/Kelch-like ECH-associated protein 1 (Keap1) system (Banning et al., 2005). The transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) is sequestered in the cytoplasm by Keap1 and released into the nucleus following oxidative modification of Keap1 (Lau et al., 2008). Thereby, the Nrf2/Keap1 system poses a key sensor of oxidative stress in the cell and translates oxidative stress into gene expression regulation (Kaspar et al., 2009). In addition, GPx2 expression appears to be under the control of a number of transcription factors that are involved in the regulation of cell proliferation and differentiation, such as p63 isoforms, β-catenin, Nkx3.1, and retinoic acid (Chu et al., 1999; Ouyang et al., 2005; Yan and Chen, 2006; Kipp et al., 2007). The impact of these regulatory proteins on GPx2 expression during embryo development has not yet been determined. However, the GPx2 expression was asserted to be under negative control of disabled-2 in murine embryonic stem cells (Huang et al., 2010). Disabled-2 is a regulatory adaptor protein involved in various process during development including mesenchymal differentiation and mesoderm formation (Rula et al., 2007; Martin et al., 2010). The underlying mechanisms of GPx2 repression and the functional significance of this regulatory circuit however remain to be investigated.
Glutathione Peroxidase 3
Glutathione peroxidase 3 (GPx3) is a secreted plasma protein that is mainly produced in the proximal tubuli of the kidney (Avissar et al., 1994b) but found in most extracellular fluids (reviewed in Brigelius-Flohe, 1999). During embryo development GPx3 expression was detected in many embryonic tissues but also in the placenta (Avissar et al., 1994a; Kingsley et al., 1998). Unfortunately, very little is known about the biological role of this GPx isoform. Interestingly, GPx3 uses thioredoxin or glutaredoxin as reducing equivalents instead of glutathione because of the very low glutathione levels found in plasma (Bjornstedt et al., 1994). Recently published knockout data indicate that GPx3-/- mice show no abnormal phenotype throughout their lifetime (Olson et al., 2010). The fast reduction of GPx3 expression following selenium depletion places GPx3 in the lower ranks of the selenium hierarchy (Brigelius-Flohe, 1999). Very little is known about the mechanisms driving GPx3 expression. The proximal promoter region lacks a TATA-box but contains several NF-Y and Sp1-binding sites as well as a CpG island. Indeed, hypermethylation of the GPx3 promoter which is associated with reduced GPx3 expression was observed in prostate carcinoma and tumors associated with Barrett’s esophagus (Lee et al., 2005; Yu et al., 2007). However, the functional significance of low GPx3 expression in the progression of cancer has not yet been sufficiently addressed.
Glutathione Peroxidase 4
Glutathione peroxidase 4 (GPx4) was originally identified as an inhibitor of lipid peroxidation in pig livers (Ursini et al., 1982, 1985). Within the protein family GPx4 takes up a special position for a number of reasons. Whereas the other GPx enzymes exist in the tetrameric form GPx4 was found to be a monomeric enzyme containing a single selenocysteine moiety (Ursini et al., 1985). Moreover, in contrast to the other GPx family members GPx4 exhibits a broad affinity toward its substrate hydroperoxides as well as its reducing equivalents. GPx4 shares the structural preconditions for its enzymatic activity with the other GPx isoforms (Scheerer et al., 2007). In contrast to other GPx isoforms however, the active site of GPx4 is not framed by an exposed surface loop that potentially limits the accessibility of the active site and its largely hydrophobic surface allow GPx4 to closely associate with membranes and lipoproteins. Thus, GPx4 not only reduces small hydrophilic peroxides such as H2O2 but also more complex substrates such as phospholipid or cholesterol hydroperoxides even if these are incorporated into membranes or lipoproteins (Ursini et al., 1985; Thomas et al., 1990; Sattler et al., 1994). When glutathione concentrations are sufficient GPx4 uses glutathione as reducing equivalent. However, when glutathione becomes limiting such as in developing sperm cells GPx4 accepts thiol groups in proteins as reducing equivalent (Godeas et al., 1997). This thiol-oxidase activity toward protein thiols subsequently induces protein cross-links. In fact, the GPx4 protein itself possesses a number of cysteine residues on its protein surface, which have been implicated in its ability to form extensive enzymatically inactive protein polymers (Scheerer et al., 2007). This very particular feature of GPx4 is crucial for sperm maturation (Godeas et al., 1997). In addition, the presence of cysteine residues within the GPx4 protein has been suggested to be critical for its catalytic cycle (Mannes et al., 2011).
In mammalian cells the GPx4 protein is found in most subcellular compartments including the cytoplasm, the nucleus, mitochondria, and the endoplasmic reticulum (Arai et al., 1999). Accordingly, various biological functions have been attributed to GPx4 including cellular anti-oxidative defense, anti-apoptotic activities, eicosanoid signaling, and chromatin condensation (Kuhn and Borchert, 2002; Imai and Nakagawa, 2003; Conrad et al., 2005). In mammals the GPx4 gene gives rise to three different isoenzymes that exhibit similar enzymatic properties but that can be distinguished by their specific N-terminal sequences (Kelner and Montoya, 1998; Borchert et al., 1999; Pfeifer et al., 2001; Figure 2). These N-terminal sequences are attained by alternative usage of three translational initiation sites (AUGs) that reside in two alternative exons 1 (exon 1a and 1b). Exon 1a contains two in-frame translational start sites (5′AUG and 3′AUG) and codes for an N-terminal mitochondrial leader sequence, which confers post-translational import into mitochondria (Arai et al., 1999). Translation initiation at the 5′AUG results in the generation of a GPx4 isoenzyme, that is targeted for mitochondrial import (mitochondrial GPx4, m-GPx4). In contrast, translation from the 3′AUG yields a GPx4 isoenzyme, that lacks this leader signal (cytosolic GPx4, c-GPx4). Because the mitochondrial leader peptide is cleaved off after import into mitochondria, c-GPx4 and m-GPx4 cannot be distinguished on the protein level anymore. Another translational initiation site is found in exon 1b. This exon codes for an N-terminal peptide that causes nuclear localization (Pfeifer et al., 2001) of the third GPx4 isoform (nuclear GPx4, n-GPx4).
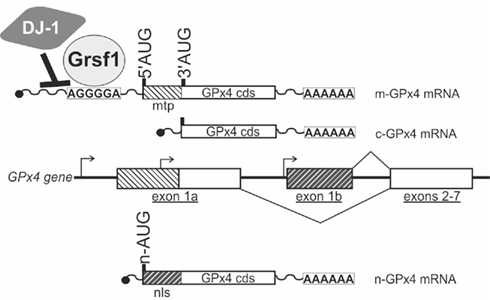
Figure 2. Coding multiplicity of the GPx4 gene. The GPx4 gene gives rise to three isoenzymes designated m-GPx4, c-GPx4, and n-GPx4. They can be distinguished by their N-terminal sequences that determine their subcellular localization [mtp, mitochondrial targeting peptide (light hatching); nls, nuclear localization sequence (dark hatching)]. The mammalian GPx4 gene consists of seven exons and contains three windows of transcriptional (arrows) and translational (5′AUG, 3′AUG, n-AUG) initiation, that are specific to the isoenzymes. Two protein factors (DJ-1, Grsf1) have been identified that affect post-transcriptional regulation of the GPx4 gene. cds, coding sequence.
In addition to the coding multiplicity of the GPx4 gene a transcribed c-GPx4 pseudogene was identified in mice (Boschan et al., 2002). However, the biological significance of this pseudogene has not yet been established.
GPx4 expression regulation
Three different mRNA species have been shown to be transcribed from the GPx4 gene (Figure 2; Esworthy et al., 1994; Pushpa-Rekha et al., 1995; Knopp et al., 1999; Moreno et al., 2003). Transcripts starting from the most upstream transcription initiation site of the GPx4 gene contain translational start sites for both, the mitochondrial as well as the cytosolic GPx4. This start site is dominantly used in spermatogenic cells, but the mechanisms that induce transcription from this site remain largely unknown. A second window of transcription initiation is located between the 5′AUG and the 3′AUG in exon 1a. This mRNA species is ubiquitously expressed in most mammalian cell types and only contains the translational start site for the c-GPx4 isoform. The corresponding regulatory region upstream exon 1a represents the promoter of a housekeeping gene, that is devoid of a classical TATA-box and instead under the control of general transcription factors Sp1 and NF-Y (Ufer et al., 2003). The third window of transcription initiation was found just upstream exon 1b and generates mRNA species coding for the n-GPx4 (Moreno et al., 2003). Transcription from this site is dominantly initiated in developing spermatids and appears to be under the control of the cAMP-response element modulator tau (CREM-τ; Borchert et al., 2003; Tramer et al., 2004).
GPx4 isoforms in embryos
Messenger RNAs for all three GPx4 isoforms were detected in mouse embryos early on during development from gestational day E6.5 (Borchert et al., 2006; Schneider et al., 2006). In the developing embryo GPx4 mRNA is expressed in the early neural tube at E8.0 and later extends into the developing neuroepithelium of the fore-, mid-, and hind-brain (Borchert et al., 2006; Figure 3). At later stages GPx4 expression is detected throughout most developing organs in particular in the developing limbs (Schneider et al., 2006). Interestingly, whereas the mRNA levels for c-GPx4 remain largely constant throughout embryo development, expression profiles of the m-GPx4 and n-GPx4 isoforms appear to be under stage-dependent control and both enzymes obey to similar expression kinetics (Borchert et al., 2006).
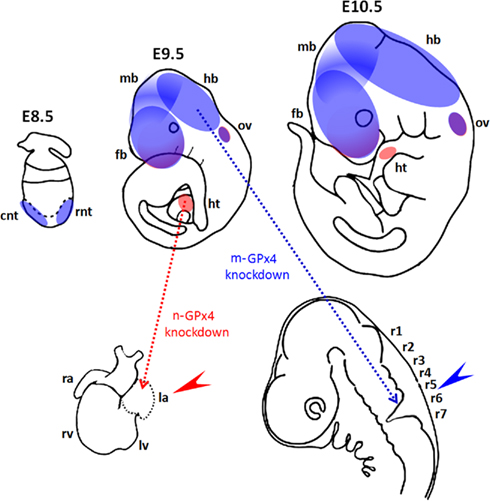
Figure 3. Functional roles of GPx4 in mouse development. (Upper panel) Diagrams show spatial expression of mitochondrial GPx (m-GPx4, blue) and nuclear GPx (n-GPx4, red) in developing mouse embryos at gestational day E8.5–E10.5. Expression of m-GPx4 and n-GPx4 are overlapping in frontal forebrain (fb) and otic vesicles (ov). m-GPx4 is mainly expressed in both rostral and caudal neural tube (rnt and cnt) in early stage and at forebrain (fb), midbrain (mb), and hindbrain (hb) in later stages. n-GPx4 is expressed in the developing heart (ht). (Lower panel) Diagrams show the abnormal embryo development following isoform specific GPx4 knockdown by RNA interference in whole mouse embryo culture. m-GPx4 siRNA constructs induce minor microencephaly and abnormal hindbrain development (blue arrow) in the fifth and sixth rhombomeres (r5 and r6). n-GPx4 siRNA constructs induce growth restriction and abnormal heart formation (red arrow) of the left atrium (la).
To explore the role of GPx4 during embryo development targeted constitutive GPx4 knockout mice were created (Imai et al., 2003; Yant et al., 2003). In contrast to the other GPx isoenzymes GPx1-3 (see above) all GPx4 knockout strategies failed to produce viable homozygous offspring. Homozygous GPx4 knockout embryos died by gestational day E8.5 and underwent intrauterine resorption (Imai et al., 2003; Yant et al., 2003). Extraembryonic structures remained largely unaffected by GPx4 ablation. Interestingly, a similar phenotype was observed when de novo biosynthesis of GSH was interrupted by targeted inactivation of γ-glutamylcysteine synthase (Shi et al., 2000). This implies that GPx4 enzymatic activity is the vital glutathione-dependent process at this embryonic stage.
The key features of GPx4 function are its anti-oxidative and anti-apoptotic activities. In developing embryos GPx4 expression correlates with areas of reduced apoptosis in developing limbs (Schnabel et al., 2006). Indeed, impaired GPx4 expression in developing embryos resulted in increased DNA fragmentation as an indicator of increased apoptotic cell death (Imai et al., 2003; Borchert et al., 2006). Similar observations were made in transgenic embryos that are incapable of de novo GSH biosynthesis (Shi et al., 2000).
The developing brain appears to be the dominant site for GPx4 expression in embryonic mice and rats (Schweizer et al., 2004a). GPx4 mRNA and protein expression were detected in various areas of the brain such as pyramidal neurons of the frontal and entorhinal cortex, dentate granule cells and CA pyramidal neurons in the hippocampus of embryonic rat brains but not in glial cells (Savaskan et al., 2007). In mice neural GPx4 expression was most dominant in the cerebral cortex, tectum, and in the olfactory epithelium (Schneider et al., 2006). Thus, when GPx4 expression was suppressed by siRNA-mediated knockdown between gestational day E7.5 and E10.5 abnormal development of mid and hindbrain structures was observed (Figure 3; Borchert et al., 2006).
To further dissect the roles of the three GPx4 isoforms for embryo development different transgenic mouse models were developed, which aimed at selective inactivation of the GPx4 isoforms. Its localization in the mitochondrial intermembrane space has implicated the m-GPx4 isoform as the GPx4 isoform modulating apoptotic cell death (Imai and Nakagawa, 2003) and this has been suggested as the major cause for embryonic death in GPx4 deficient mice. Hence, one would expect that targeted inactivation of m-GPx4 expression induces apoptosis leading to embryonic death similar to the GPx4-/- mouse model. The m-GPx4 specific knockout was achieved by the introduction of an ochre codon within the mitochondrial targeting peptide of m-GPx4, which prevented m-GPx4 protein translation whilst permitting c-GPx4 expression. Surprisingly, m-GPx4-/- mice were found to be fully viable suggesting that m-GPx4 is not viable for embryo development (Schneider et al., 2009). However, m-GPx4-/- males were infertile confirming the vital role of m-GPx4 expression for sperm development. This raises the question of whether the c-GPx4 isoform is able to compensate for its so-called mitochondrial sibling to facilitate normal embryo development. This question was answered by Liang et al. (2009a,b) by means of a rescue experiment using GPx4-/- mice. For this purpose modified GPx4 gene constructs were re-introduced into a GPx4-/- background. These transgene constructs permitted selective expression of the m-GPx4 or the c-GPx4 isoform by alternative mutation of the translational start sites for the individual isoforms. Indeed, the expression of a c-GPx4 transgene in GPx4-/- mice rescued the homozygous knockout phenotype by restoring embryonic viability whereas transgenic m-GPx4 expression failed to restore embryonic viability. Moreover, this group showed that c-GPx4 is targeted into the mitochondrial intermembrane space although it lacks a mitochondrial leader sequence (Liang et al., 2009b). The mechanisms causing mitochondrial localization of the so-called cytosolic GPx4 isoform however remain unsolved.
Taken together, these data suggest that the 5′AUG is only employed in post-pubertal sperm cells whereas in developing embryos only the 3′AUG giving rise to the c-GPx4 isoform is used (Figure 2). However, in the brains of developing embryos about half of all GPx4 mRNA species carry translational start sites for both the m-GPx4 and the c-GPx4 isoforms as well as an 5′-untranslated region (5′UTR) which has been shown to serve as binding platform for translational regulators (Borchert et al., 2006; Ufer et al., 2008). The strong expression of mRNA containing the 5′AUG is rather surprising considering the apparent redundancy of the m-GPx4 isoform for embryo development. However, upstream starting codons and extended 5′UTRs are frequently employed as regulatory elements that affect translation or messenger stability (Wilkie et al., 2003). Thus, these extended GPx4 mRNA sequences are likely to serve regulatory purposes rather than as template for translation of an N-terminal mitochondrial targeting peptide. The discovery of the RNA-binding proteins Guanine-rich sequence binding factor 1 (Grsf1) and Parkinson’s disease-related protein DJ-1, which both bind the extended 5′UTR of the GPx4 mRNA supports this idea (Ufer et al., 2008; van der Brug et al., 2008). Targeted knockdown of Grsf1 expression in developing embryos induces a phenotype that mimics a GPx4 knockdown phenotype paralleled by increased apoptotic cell death and lipid oxidation (Ufer et al., 2008). The translational silencer DJ-1 loses its affinity to RNA when cysteine residues of the DJ-1 protein become oxidized (Blackinton et al., 2009). Thus, oxidative stress liberates GPx4 mRNA from DJ-1 mediated translational inhibition subsequently permitting GPx4 protein translation. However, regulation through DJ-1 seems to be more complex. DJ-1 deficiency as it is frequently observed in Parkinson’s disease or as obtained by targeted knockout of the DJ-1 gene is paralleled by increased GPx4 protein translation but renders DJ-1 deficient embryonic cortical neurons more sensitive to oxidative stress (Kim et al., 2005). This data indicates that other DJ-1 targets are involved in this phenotype. Hence, phenotypic alterations observed when the function of the GPx4 5′UTR is compromised either by siRNA targeted at these sequences (Borchert et al., 2006) or by manipulating the expression of RNA-binding proteins such as Grsf1(Ufer et al., 2008) that bind these sequences reflect the importance of this regulatory mechanism for GPx4 expression. In fact, this may at least partly explain the diverging phenotypes observed in the different experimental set-ups of targeted knockout versus knockdown of m-GPx4 expression.
Similar to the m-GPx4 isoform the n-GPx4 isoform was also found to be dispensable for embryo development (Conrad et al., 2005). However, the c-GPx4 isoform can also be found in the nucleus (Arai et al., 1999). This raises the question whether c-GPx4 is also able to compensate for lacking n-GPx4 expression in embryos. At least this is not the case when sudden repression of n-GPx4 expression is induced by n-GPx4 specific siRNA in developing embryos at midgestation. In fact, n-GPx4 knockdown embryos exhibit abnormal development of the heart lacking the left atrium (Figure 3; Borchert et al., 2006).
GPx4 in cell signaling
The precise molecular mechanisms underlying early embryonic death and neuronal cell death following ablation of GPx4 expression have so far remained unclear. The anti-apoptotic impact of GPx4 activity by suppressing cytochrome c release from the mitochondria is the most appealing mechanism (Imai and Nakagawa, 2003). However, GPx4 expression has been shown to affect various cellular signaling events that may at least partly contribute to the developmental abnormalities observed in embryos with impaired GPx4 expression. A major hallmark of GPx4 deficiency is extensive membrane oxidation (Seiler et al., 2008; Ufer et al., 2008) and oxidative modification of the mitochondrial lipid cardiolipin facilitates the release of cytochrome c into the cytosol to unleash apoptotic signaling cascades activating caspases 3 (Imai and Nakagawa, 2003). Interestingly, neuronal degeneration in GPx4 deficient cells does not involve caspase-3 activation, instead the apoptosis-inducing factor (AIF)-dependent pathway of apoptosis is pursued (Seiler et al., 2008). AIF is attached to the inner mitochondrial membrane by its N-terminal anchoring peptide. Oxidative modification of AIF exposes an internal calpain cleavage site near its membrane anchor. Following proteolytic cleavage by calpain AIF is released from its membrane anchor, transported into the cytosol and subsequently translocated into the nucleus, where AIF contributes to DNA fragmentation and chromatin condensation (Norberg et al., 2010). By counteracting mitochondrial generation of ROS GPx4 appears to protect neuronal cells from oxidative modification of AIF, which prevents AIF cleavage and release into the cytosol (Seiler et al., 2008).
Cytoplasmic GPx4 activity is considered important for the regulation of the cellular redox tone by reducing lipid hydroperoxides (Imai et al., 1996; Kuhn and Borchert, 2002). Thereby GPx4 activity affects enzymes such as lipoxygenases and cyclooxygenase, which require a certain hydroperoxide tonus for their enzymatic activity (Schilstra et al., 1992). Indeed, GPx4 has been shown to suppress leukotriene and prostaglandin biosynthesis by inhibiting lipoxygenase and cyclooxygenase activities (Weitzel and Wendel, 1993; Imai et al., 1998; Chen et al., 2003). Lipoxygenase activity has been implicated in a broad array of normal physiological and patho-physiological processes (Kuhn and O’Donnell, 2006). For instance, the impact of lipoxygenase activity on angiogenesis has been a controversially discussed matter (Kuhn and O’Donnell, 2006; Mochizuki and Kwon, 2008). Recent experiments indicated a role for GPx4 in tumor angiogenesis by suppressing lipid peroxidation derived from 12/15-lipoxygenase activity or cycloxygenase-2 possibly via modulation of ROS sensitive tyrosine kinase signaling pathways (Heirman et al., 2006; Conrad et al., 2010; Schneider et al., 2010). This raises the question, whether GPx4 plays a role in the formation of the embryonic vasculature. Strategies of a targeted conditional knockout of GPx4 expression restricted to the cardio-vascular system may help to answer that question in the future. Conditional abrogation of selenoprotein synthesis using an endothelial-specific knockout of the Trsp gene induced multiple developmental abnormalities (Shrimali et al., 2007) and correlates with a higher incidence of colon (Irons et al., 2006) and prostate cancer (Diwadkar-Navsariwala et al., 2006). From gestational day E14.5 on these embryos suffered massive necrosis in the parenchyma of the brain and the spinal cord as well as greatly retarded development of the vasculature and erythrocytes. However this experimental approach does not allow the identification of a specific selenoprotein which is responsible for this severe phenotype.
Cellular signaling cascades ultimately affect gene expression regulation. This involves modulation of transcriptional, post-transcriptional as well as post-translational events, which all depend on regulatory protein factors. Many of these regulatory proteins contain structures such as thiol groups that render them sensitive to the cellular redox state (Brigelius-Flohe, 2006; Ufer et al., 2010). Thereby the redox state of a cell can be monitored and modulated by gene expression regulation. Indeed, GPx4 has previously been shown to modulate the activity of transcription factors including nuclear factor κB (NF-κB) and Nrf2 (Brigelius-Flohe et al., 1997; Banning et al., 2004; Wenk et al., 2004; Banning and Brigelius-Flohe, 2005). By dampening NF-κB signaling GPx4 reduces expression of matrix metalloproteinase-1 (MMP-1), a protein frequently upregulated during invasion and metastasis of various tumors (Wenk et al., 2004). Both, Nrf2 and NF-κB are expressed during murine brain development from around midgestation (Chan et al., 1996; Schmidt-Ullrich et al., 1996). Whereas Nrf2 appears to be dispensable for prenatal development, dysfunctional NF-κB expression causes embryonic lethality around gestational day E15 paralleled by increased apoptosis of the liver parenchyma and impaired embryonic hematopoiesis (Beg et al., 1995; Ma et al., 2006). The impact of GPx4 expression on NF-κB activity in embryo development has not yet been tested. However, expression of the manganese SOD, a known NF-κB target gene (Mattson and Camandola, 2001), is not changed in mice that artificially overexpress GPx4 (Ran et al., 2004). Hence, the impact of GPx4 on NF-κB mediated signaling most likely depends on further preconditions.
Perspectives
Reliable determinations of ROS concentration in embryos are still lacking. Evidences to support an actual developmental role of ROS are very limited. It is technically very challenging to measure ROS in situ in the developing embryos. Although the activity of the respiratory chain and the levels of antioxidants during development are not indicative for specific ROS functions, they could serve as reporters for regions and/or times at which ROS potentially have a function. Unfortunately, the instability of most ROS species in vivo precludes a confident determination of their concentration in developing cells. Taking advantage of the damage caused by oxidative stress, an alternative indirect way to determine ROS levels is by measuring the oxidation level of macromolecules. It is important to consider the half-life of the damaged molecule and repair mechanisms, as these will determine whether the measurements indicate a short-term increase in ROS or an accumulative damage due to continuous ROS production. The detection of the expression patterns of anti-oxidative enzymes will become important because they could either define the areas of high and low ROS levels or reveal the areas exposed to elevated levels of oxidative stress, as several of those genes respond to this condition. However, the complexity of the events that take place in such close timely succession makes it difficult to draw satisfactory conclusions. Moreover, as shown for the GPx isoenzymes proteins or enzymes might behave differently than the general consensus predicts, such as differing subcellular localizations or enzymatic activities. And such moonlighting activities may be identified for other gene products. Moreover, the lethal phenotype of a homozygous GPx4 knockout makes it difficult to study the physiological role of GPx4 during embryo development. The creation of inducible or tissue specific knockouts or knock-ins may provide valuable tools to tackle this issue and a plethora of such mouse models is available.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arai, M., Imai, H., Koumura, T., Yoshida, M., Emoto, K., Umeda, M., Chiba, N., and Nakagawa, Y. (1999). Mitochondrial phospholipid hydroperoxide glutathione peroxidase plays a major role in preventing oxidative injury to cells. J. Biol. Chem. 274, 4924–4933.
Arner, E. S. (2009). Focus on mammalian thioredoxin reductases – important selenoproteins with versatile functions. Biochim. Biophys. Acta 1790, 495–526.
Avissar, N., Eisenmann, C., Breen, J. G., Horowitz, S., Miller, R. K., and Cohen, H. J. (1994a). Human placenta makes extracellular glutathione peroxidase and secretes it into maternal circulation. Am. J. Physiol. 267, E68–E76.
Avissar, N., Ornt, D. B., Yagil, Y., Horowitz, S., Watkins, R. H., Kerl, E. A., Takahashi, K., Palmer, I. S., and Cohen, H. J. (1994b). Human kidney proximal tubules are the main source of plasma glutathione peroxidase. Am. J. Physiol. 266, C367–C375.
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495.
Banning, A., and Brigelius-Flohe, R. (2005). NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid. Redox Signal. 7, 889–899.
Banning, A., Deubel, S., Kluth, D., Zhou, Z., and Brigelius-Flohe, R. (2005). The GI-GPx gene is a target for Nrf2. Mol. Cell. Biol. 25, 4914–4923.
Banning, A., Schnurr, K., Bol, G. F., Kupper, D., Muller-Schmehl, K., Viita, H., Yla-Herttuala, S., and Brigelius-Flohe, R. (2004). Inhibition of basal and interleukin-1-induced VCAM-1 expression by phospholipid hydroperoxide glutathione peroxidase and 15-lipoxygenase in rabbit aortic smooth muscle cells. Free Radic. Biol. Med. 36, 135–144.
Bavister, B. D. (1995). Culture of preimplantation embryos: facts and artifacts. Hum. Reprod. Update 1, 91–148.
Beg, A. A., Sha, W. C., Bronson, R. T., Ghosh, S., and Baltimore, D. (1995). Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 376, 167–170.
Behne, D., and Kyriakopoulos, A. (2001). Mammalian selenium-containing proteins. Annu. Rev. Nutr. 21, 453–473.
Bjornstedt, M., Xue, J., Huang, W., Akesson, B., and Holmgren, A. (1994). The thioredoxin and glutaredoxin systems are efficient electron donors to human plasma glutathione peroxidase. J. Biol. Chem. 269, 29382–29384.
Blackinton, J., Kumaran, R., van der Brug, M. P., Ahmad, R., Olson, L., Galter, D., Lees, A., Bandopadhyay, R., and Cookson, M. R. (2009). Post-transcriptional regulation of mRNA associated with DJ-1 in sporadic Parkinson disease. Neurosci. Lett. 452, 8–11.
Bogdanovic, O., and Veenstra, G. J. (2009). DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma 118, 549–565.
Borchert, A., Savaskan, N. E., and Kuhn, H. (2003). Regulation of expression of the phospholipid hydroperoxide/sperm nucleus glutathione peroxidase gene. Tissue-specific expression pattern and identification of functional cis- and trans-regulatory elements. J. Biol. Chem. 278, 2571–2580.
Borchert, A., Schnurr, K., Thiele, B. J., and Kuhn, H. (1999). Cloning of the mouse phospholipid hydroperoxide glutathione peroxidase gene. FEBS Lett. 446, 223–227.
Borchert, A., Wang, C. C., Ufer, C., Schiebel, H., Savaskan, N. E., and Kuhn, H. (2006). The role of phospholipid hydroperoxide glutathione peroxidase isoforms in murine embryogenesis. J. Biol. Chem. 281, 19655–19664.
Boschan, C., Borchert, A., Ufer, C., Thiele, B. J., and Kuhn, H. (2002). Discovery of a functional retrotransposon of the murine phospholipid hydroperoxide glutathione peroxidase: chromosomal localization and tissue-specific expression pattern. Genomics 79, 387–394.
Bosl, M. R., Takaku, K., Oshima, M., Nishimura, S., and Taketo, M. M. (1997). Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp). Proc. Natl. Acad. Sci. U.S.A. 94, 5531–5534.
Boyd, J. D., and Hamilton, W. J. (eds). (1952). Cleavage, Early Development and Implantation of the Egg. London: Longmans, Green and Co.
Brigelius-Flohe, R. (1999). Tissue-specific functions of individual glutathione peroxidases. Free Radic. Biol. Med. 27, 951–965.
Brigelius-Flohe, R. (2006). Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem. 387, 1329–1335.
Brigelius-Flohe, R., Banning, A., and Schnurr, K. (2003). Selenium-dependent enzymes in endothelial cell function. Antioxid. Redox Signal. 5, 205–215.
Brigelius-Flohe, R., Friedrichs, B., Maurer, S., Schultz, M., and Streicher, R. (1997). Interleukin-1-induced nuclear factor kappa B activation is inhibited by overexpression of phospholipid hydroperoxide glutathione peroxidase in a human endothelial cell line. Biochem. J. 328(Pt 1), 199–203.
Cao, C., Leng, Y., Huang, W., Liu, X., and Kufe, D. (2003). Glutathione peroxidase 1 is regulated by the c-Abl and Arg tyrosine kinases. J. Biol. Chem. 278, 39609–39614.
Cartharius, K., Frech, K., Grote, K., Klocke, B., Haltmeier, M., Klingenhoff, A., Frisch, M., Bayerlein, M., and Werner, T. (2005). MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21, 2933–2942.
Castagne, V., Lefevre, K., Natero, R., Clarke, P. G., and Bedker, D. A. (1999). An optimal redox status for the survival of axotomized ganglion cells in the developing retina. Neuroscience 93, 313–320.
Chabory, E., Damon, C., Lenoir, A., Kauselmann, G., Kern, H., Zevnik, B., Garrel, C., Saez, F., Cadet, R., Henry-Berger, J., Schoor, M., Gottwald, U., Habenicht, U., Drevet, J. R., and Vernet, P. (2009). Epididymis seleno-independent glutathione peroxidase 5 maintains sperm DNA integrity in mice. J. Clin. Invest. 119, 2074–2085.
Chan, K., Lu, R., Chang, J. C., and Kan, Y. W. (1996). NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. U.S.A. 93, 13943–13948.
Chen, C. J., Huang, H. S., and Chang, W. C. (2003). Depletion of phospholipid hydroperoxide glutathione peroxidase up-regulates arachidonate metabolism by 12S-lipoxygenase and cyclooxygenase 1 in human epidermoid carcinoma A431 cells. FASEB J. 17, 1694–1696.
Chen, E. Y., Fujinaga, M., and Giaccia, A. J. (1999). Hypoxic microenvironment within an embryo induces apoptosis and is essential for proper morphological development. Teratology 60, 215–225.
Chu, F. F., Doroshow, J. H., and Esworthy, R. S. (1993). Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI. J. Biol. Chem. 268, 2571–2576.
Chu, F. F., Esworthy, R. S., Chu, P. G., Longmate, J. A., Huycke, M. M., Wilczynski, S., and Doroshow, J. H. (2004). Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Res. 64, 962–968.
Chu, F. F., Esworthy, R. S., Lee, L., and Wilczynski, S. (1999). Retinoic acid induces Gpx2 gene expression in MCF-7 human breast cancer cells. J. Nutr. 129, 1846–1854.
Clough, J. R. (1985). Energy metabolism during mammalian embryogenesis. Biochem. Soc. Trans. 13, 77–79.
Conrad, M. (2009). Transgenic mouse models for the vital selenoenzymes cytosolic thioredoxin reductase, mitochondrial thioredoxin reductase and glutathione peroxidase 4. Biochim. Biophys. Acta 1790, 1575–1585.
Conrad, M., Moreno, S. G., Sinowatz, F., Ursini, F., Kolle, S., Roveri, A., Brielmeier, M., Wurst, W., Maiorino, M., and Bornkamm, G. W. (2005). The nuclear form of phospholipid hydroperoxide glutathione peroxidase is a protein thiol peroxidase contributing to sperm chromatin stability. Mol. Cell. Biol. 25, 7637–7644.
Conrad, M., Sandin, A., Forster, H., Seiler, A., Frijhoff, J., Dagnell, M., Bornkamm, G. W., Radmark, O., Hooft Van Huijsduijnen, R., Aspenstrom, P., Bohmer, F., and Ostman, A. (2010). 12/15-lipoxygenase-derived lipid peroxides control receptor tyrosine kinase signaling through oxidation of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 107, 15774–15779.
Covarrubias, L., Hernandez-Garcia, D., Schnabel, D., Salas-Vidal, E., and Castro-Obregon, S. (2008). Function of reactive oxygen species during animal development: passive or active? Dev. Biol. 320, 1–11.
D’Autreaux, B., and Toledano, M. B. (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 8, 813–824.
Diwadkar-Navsariwala, V., Prins, G. S., Swanson, S. M., Birch, L. A., Ray, V. H., Hedayat, S., Lantvit, D. L., and Diamond, A. M. (2006). Selenoprotein deficiency accelerates prostate carcinogenesis in a transgenic model. Proc. Natl. Acad. Sci. U.S.A. 103, 8179–8184.
Dreher, I., Schmutzler, C., Jakob, F., and Kohrle, J. (1997). Expression of selenoproteins in various rat and human tissues and cell lines. J. Trace Elem. Med. Biol. 11, 83–91.
Dumollard, R., Ward, Z., Carroll, J., and Duchen, M. R. (2007). Regulation of redox metabolism in the mouse oocyte and embryo. Development 134, 455–465.
Esworthy, R. S., Aranda, R., Martin, M. G., Doroshow, J. H., Binder, S. W., and Chu, F. F. (2001). Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G848–G855.
Esworthy, R. S., Doan, K., Doroshow, J. H., and Chu, F. F. (1994). Cloning and sequencing of the cDNA encoding a human testis phospholipid hydroperoxide glutathione peroxidase. Gene 144, 317–318.
Esworthy, R. S., Mann, J. R., Sam, M., and Chu, F. F. (2000). Low glutathione peroxidase activity in Gpx1 knockout mice protects jejunum crypts from gamma-irradiation damage. Am. J. Physiol. Gastrointest. Liver Physiol. 279, G426–G436.
Fischer, B., and Bavister, B. D. (1993). Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J. Reprod. Fertil. 99, 673–679.
Fissore, R. A., Kurokawa, M., Knott, J., Zhang, M., and Smyth, J. (2002). Mechanisms underlying oocyte activation and postovulatory ageing. Reproduction 124, 745–754.
Flentjar, N. J., Crack, P. J., Boyd, R., Malin, M., De Haan, J. B., Hertzog, P., Kola, I., and Iannello, R. (2002). Mice lacking glutathione peroxidase-1 activity show increased TUNEL staining and an accelerated inflammatory response in brain following a cold-induced injury. Exp. Neurol. 177, 9–20.
Fu, Y., Sies, H., and Lei, X. G. (2001). Opposite roles of selenium-dependent glutathione peroxidase-1 in superoxide generator diquat- and peroxynitrite-induced apoptosis and signaling. J. Biol. Chem. 276, 43004–43009.
Gardiner, C. S., and Reed, D. J. (1995). Synthesis of glutathione in the preimplantation mouse embryo. Arch. Biochem. Biophys. 318, 30–36.
Giorgio, M., Trinei, M., Migliaccio, E., and Pelicci, P. G. (2007). Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 8, 722–728.
Gladyshev, V. N., Factor, V. M., Housseau, F., and Hatfield, D. L. (1998). Contrasting patterns of regulation of the antioxidant selenoproteins, thioredoxin reductase, and glutathione peroxidase, in cancer cells. Biochem. Biophys. Res. Commun. 251, 488–493.
Godeas, C., Tramer, F., Micali, F., Soranzo, M., Sandri, G., and Panfili, E. (1997). Distribution and possible novel role of phospholipid hydroperoxide glutathione peroxidase in rat epididymal spermatozoa. Biol. Reprod. 57, 1502–1508.
Guerin, P., El Mouatassim, S., and Menezo, Y. (2001). Oxidative stress and protection against reactive oxygen species in the pre-implantation embryo and its surroundings. Hum. Reprod. Update 7, 175–189.
Gysin, R., Kraftsik, R., Sandell, J., Bovet, P., Chappuis, C., Conus, P., Deppen, P., Preisig, M., Ruiz, V., Steullet, P., Tosic, M., Werge, T., Cuenod, M., and Do, K. Q. (2007). Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc. Natl. Acad. Sci. U.S.A. 104, 16621–16626.
Hahn, M. A., Hahn, T., Lee, D. H., Esworthy, R. S., Kim, B. W., Riggs, A. D., Chu, F. F., and Pfeifer, G. P. (2008). Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res. 68, 10280–10289.
Hansen, J. M. (2006). Oxidative stress as a mechanism of teratogenesis. Birth Defects Res. C Embryo Today 78, 293–307.
Harvey, A. J., Kind, K. L., and Thompson, J. G. (2002). REDOX regulation of early embryo development. Reproduction 123, 479–486.
Hatfield, D. L., and Gladyshev, V. N. (2002). How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 22, 3565–3576.
Heirman, I., Ginneberge, D., Brigelius-Flohe, R., Hendrickx, N., Agostinis, P., Brouckaert, P., Rottiers, P., and Grooten, J. (2006). Blocking tumor cell eicosanoid synthesis by GP x 4 impedes tumor growth and malignancy. Free Radic. Biol. Med. 40, 285–294.
Ho, Y. S., Magnenat, J. L., Bronson, R. T., Cao, J., Gargano, M., Sugawara, M., and Funk, C. D. (1997). Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 272, 16644–16651.
Ho, Y. S., Xiong, Y., Ma, W., Spector, A., and Ho, D. S. (2004). Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 279, 32804–32812.
Huang, C. L., Cheng, J. C., Kitajima, K., Nakano, T., Yeh, C. F., Chong, K. Y., and Tseng, C. P. (2010). Disabled-2 is required for mesoderm differentiation of murine embryonic stem cells. J. Cell. Physiol. 225, 92–105.
Imai, H., Hirao, F., Sakamoto, T., Sekine, K., Mizukura, Y., Saito, M., Kitamoto, T., Hayasaka, M., Hanaoka, K., and Nakagawa, Y. (2003). Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 305, 278–286.
Imai, H., and Nakagawa, Y. (2003). Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 34, 145–169.
Imai, H., Narashima, K., Arai, M., Sakamoto, H., Chiba, N., and Nakagawa, Y. (1998). Suppression of leukotriene formation in RBL-2H3 cells that overexpressed phospholipid hydroperoxide glutathione peroxidase. J. Biol. Chem. 273, 1990–1997.
Imai, H., Sumi, D., Sakamoto, H., Hanamoto, A., Arai, M., Chiba, N., and Nakagawa, Y. (1996). Overexpression of phospholipid hydroperoxide glutathione peroxidase suppressed cell death due to oxidative damage in rat basophile leukemia cells (RBL-2H3). Biochem. Biophys. Res. Commun. 222, 432–438.
Irons, R., Carlson, B. A., Hatfield, D. L., and Davis, C. D. (2006). Both selenoproteins and low molecular weight selenocompounds reduce colon cancer risk in mice with genetically impaired selenoprotein expression. J. Nutr. 136, 1311–1317.
Jee, C. D., Kim, M. A., Jung, E. J., Kim, J., and Kim, W. H. (2009). Identification of genes epigenetically silenced by CpG methylation in human gastric carcinoma. Eur. J. Cancer 45, 1282–1293.
Jin, L. H., Kim, D. W., Eum, W. S., Yoon, C. S., Jang, S. H., Choi, H. S., Choi, S. H., Kim, Y. H., Kim, S. Y., Jung, M. R., Kang, T. C., Won, M. H., Lee, H. Y., Kang, J. H., Kwon, O. S., Cho, S. W., Lee, K. S., Park, J., and Choi, S. Y. (2003). Human liver catalase: cloning, expression and characterization of monoclonal antibodies. Mol. Cells 15, 381–386.
Johnson, M. H., and Nasr-Esfahani, M. H. (1994). Radical solutions and cultural problems: could free oxygen radicals be responsible for the impaired development of preimplantation mammalian embryos in vitro? Bioessays 16, 31–38.
Kaspar, J. W., Niture, S. K., and Jaiswal, A. K. (2009). Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 47, 1304–1309.
Kelner, M. J., and Montoya, M. A. (1998). Structural organization of the human selenium-dependent phospholipid hydroperoxide glutathione peroxidase gene (GPX4): chromosomal localization to 19p13.3. Biochem. Biophys. Res. Commun. 249, 53–55.
Kienhofer, J., Haussler, D. J., Ruckelshausen, F., Muessig, E., Weber, K., Pimentel, D., Ullrich, V., Burkle, A., and Bachschmid, M. M. (2009). Association of mitochondrial antioxidant enzymes with mitochondrial DNA as integral nucleoid constituents. FASEB J. 23, 2034–2044.
Kim, R. H., Smith, P. D., Aleyasin, H., Hayley, S., Mount, M. P., Pownall, S., Wakeham, A., You-Ten, A. J., Kalia, S. K., Horne, P., Westaway, D., Lozano, A. M., Anisman, H., Park, D. S., and Mak, T. W. (2005). Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 102, 5215–5220.
Kingsley, P. D., Whitin, J. C., Cohen, H. J., and Palis, J. (1998). Developmental expression of extracellular glutathione peroxidase suggests antioxidant roles in deciduum, visceral yolk sac, and skin. Mol. Reprod. Dev. 49, 343–355.
Kipp, A., Banning, A., and Brigelius-Flohe, R. (2007). Activation of the glutathione peroxidase 2 (GPx2) promoter by beta-catenin. Biol. Chem. 388, 1027–1033.
Kirkman, H. N., and Gaetani, G. F. (2007). Mammalian catalase: a venerable enzyme with new mysteries. Trends Biochem. Sci. 32, 44–50.
Knopp, E. A., Arndt, T. L., Eng, K. L., Caldwell, M., Leboeuf, R. C., Deeb, S. S., and O’Brien, K. D. (1999). Murine phospholipid hydroperoxide glutathione peroxidase: cDNA sequence, tissue expression, and mapping. Mamm. Genome 10, 601–605.
Kobayashi-Miura, M., Nakamura, H., Yodoi, J., and Shiota, K. (2002). Thioredoxin, an anti-oxidant protein, protects mouse embryos from oxidative stress-induced developmental anomalies. Free Radic. Res. 36, 949–956.
Kryukov, G. V., Castellano, S., Novoselov, S. V., Lobanov, A. V., Zehtab, O., Guigo, R., and Gladyshev, V. N. (2003). Characterization of mammalian selenoproteomes. Science 300, 1439–1443.
Kuhn, H., and Borchert, A. (2002). Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes. Free Radic. Biol. Med. 33, 154–172.
Kuhn, H., and O’Donnell, V. B. (2006). Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 45, 334–356.
Lau, A., Villeneuve, N. F., Sun, Z., Wong, P. K., and Zhang, D. D. (2008). Dual roles of Nrf2 in cancer. Pharmacol. Res. 58, 262–270.
Lee, D. H., Esworthy, R. S., Chu, C., Pfeifer, G. P., and Chu, F. F. (2006). Mutation accumulation in the intestine and colon of mice deficient in two intracellular glutathione peroxidases. Cancer Res. 66, 9845–9851.
Lee, O. J., Schneider-Stock, R., Mcchesney, P. A., Kuester, D., Roessner, A., Vieth, M., Moskaluk, C. A., and El-Rifai, W. (2005). Hypermethylation and loss of expression of glutathione peroxidase-3 in Barrett’s tumorigenesis. Neoplasia 7, 854–861.
Leese, H. J. (1995). Metabolic control during preimplantation mammalian development. Hum. Reprod. Update 1, 63–72.
Legault, J., Carrier, C., Petrov, P., Renard, P., Remacle, J., and Mirault, M. E. (2000). Mitochondrial GPx1 decreases induced but not basal oxidative damage to mtDNA in T47D cells. Biochem. Biophys. Res. Commun. 272, 416–422.
Li, S., Yan, T., Yang, J. Q., Oberley, T. D., and Oberley, L. W. (2000). The role of cellular glutathione peroxidase redox regulation in the suppression of tumor cell growth by manganese superoxide dismutase. Cancer Res. 60, 3927–3939.
Li, Y., Huang, T. T., Carlson, E. J., Melov, S., Ursell, P. C., Olson, J. L., Noble, L. J., Yoshimura, M. P., Berger, C., Chan, P. H., Wallace, D. C., and Epstein, C. J. (1995). Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 11, 376–381.
Liang, H., Ran, Q., Jang, Y. C., Holstein, D., Lechleiter, J., Mcdonald-Marsh, T., Musatov, A., Song, W., Van Remmen, H., and Richardson, A. (2009a). Glutathione peroxidase 4 differentially regulates the release of apoptogenic proteins from mitochondria. Free Radic. Biol. Med. 47, 312–320.
Liang, H., Yoo, S. E., Na, R., Walter, C. A., Richardson, A., and Ran, Q. (2009b). Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J. Biol. Chem. 284, 30836–30844.
Liu, L., and Keefe, D. L. (2000). Cytoplasm mediates both development and oxidation-induced apoptotic cell death in mouse zygotes. Biol. Reprod. 62, 1828–1834.
Liu, L., Trimarchi, J. R., and Keefe, D. L. (2000). Involvement of mitochondria in oxidative stress-induced cell death in mouse zygotes. Biol. Reprod. 62, 1745–1753.
Longo, L., Vanegas, O. C., Patel, M., Rosti, V., Li, H., Waka, J., Merghoub, T., Pandolfi, P. P., Notaro, R., Manova, K., and Luzzatto, L. (2002). Maternally transmitted severe glucose 6-phosphate dehydrogenase deficiency is an embryonic lethal. EMBO J. 21, 4229–4239.
Lu, Y. P., Lou, Y. R., Yen, P., Newmark, H. L., Mirochnitchenko, O. I., Inouye, M., and Huang, M. T. (1997). Enhanced skin carcinogenesis in transgenic mice with high expression of glutathione peroxidase or both glutathione peroxidase and superoxide dismutase. Cancer Res. 57, 1468–1474.
Ma, Q., Battelli, L., and Hubbs, A. F. (2006). Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 168, 1960–1974.
Maier, C. M., and Chan, P. H. (2002). Role of superoxide dismutases in oxidative damage and neurodegenerative disorders. Neuroscientist 8, 323–334.
Mannes, A. M., Seiler, A., Bosello, V., Maiorino, M., and Conrad, M. (2011). Cysteine mutant of mammalian GPx4 rescues cell death induced by disruption of the wild-type selenoenzyme. FASEB J. 25, 2135–2144.
Mari, M., Morales, A., Colell, A., Garcia-Ruiz, C., and Fernandez-Checa, J. C. (2009). Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 11, 2685–2700.
Martin, J. C., Herbert, B. S., and Hocevar, B. A. (2010). Disabled-2 downregulation promotes epithelial-to-mesenchymal transition. Br. J. Cancer 103, 1716–1723.
Mattson, M. P., and Camandola, S. (2001). NF-kappaB in neuronal plasticity and neurodegenerative disorders. J. Clin. Invest. 107, 247–254.
Mochizuki, N., and Kwon, Y. G. (2008). 15-lipoxygenase-1 in the vasculature: expanding roles in angiogenesis. Circ. Res. 102, 143–145.
Moreno, S. G., Laux, G., Brielmeier, M., Bornkamm, G. W., and Conrad, M. (2003). Testis-specific expression of the nuclear form of phospholipid hydroperoxide glutathione peroxidase (PHGPx). Biol. Chem. 384, 635–643.
Motsenbocker, M. A., and Tappel, A. L. (1982). A selenocysteine-containing selenium-transport protein in rat plasma. Biochim. Biophys. Acta 719, 147–153.
Muller, F. L., Lustgarten, M. S., Jang, Y., Richardson, A., and Van Remmen, H. (2007). Trends in oxidative aging theories. Free Radic. Biol. Med. 43, 477–503.
Munday, R., and Winterbourn, C. C. (1989). Reduced glutathione in combination with superoxide dismutase as an important biological antioxidant defence mechanism. Biochem. Pharmacol. 38, 4349–4352.
Nagarajan, R. P., and Costello, J. F. (2009). Molecular epigenetics and genetics in neuro-oncology. Neurotherapeutics 6, 436–446.
Negrini, S., Gorgoulis, V. G., and Halazonetis, T. D. (2010). Genomic instability – an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228.
New, D. A., and Coppola, P. T. (1970). Effects of different oxygen concentrations on the development of rat embryos in culture. J. Reprod. Fertil. 21, 109–118.
Nguyen, V. D., Saaranen, M. J., Karala, A. R., Lappi, A. K., Wang, L., Raykhel, I. B., Alanen, H. I., Salo, K. E., Wang, C. C., and Ruddock, L. W. (2011). Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 406, 503–515.
Norberg, E., Orrenius, S., and Zhivotovsky, B. (2010). Mitochondrial regulation of cell death: processing of apoptosis-inducing factor (AIF). Biochem. Biophys. Res. Commun. 396, 95–100.
Olson, G. E., Whitin, J. C., Hill, K. E., Winfrey, V. P., Motley, A. K., Austin, L. M., Deal, J., Cohen, H. J., and Burk, R. F. (2010). Extracellular glutathione peroxidase (Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am. J. Physiol. Renal Physiol. 298, F1244–F1253.
Ouyang, X., Deweese, T. L., Nelson, W. G., and Abate-Shen, C. (2005). Loss-of-function of Nkx3.1 promotes increased oxidative damage in prostate carcinogenesis. Cancer Res. 65, 6773–6779.
Parman, T., Wiley, M. J., and Wells, P. G. (1999). Free radical-mediated oxidative DNA damage in the mechanism of thalidomide teratogenicity. Nat. Med. 5, 582–585.
Pfeifer, H., Conrad, M., Roethlein, D., Kyriakopoulos, A., Brielmeier, M., Bornkamm, G. W., and Behne, D. (2001). Identification of a specific sperm nuclei selenoenzyme necessary for protamine thiol cross-linking during sperm maturation. FASEB J. 15, 1236–1238.
Pierce, G. B., Parchment, R. E., and Lewellyn, A. L. (1991). Hydrogen peroxide as a mediator of programmed cell death in the blastocyst. Differentiation 46, 181–186.
Pushpa-Rekha, T. R., Burdsall, A. L., Oleksa, L. M., Chisolm, G. M., and Driscoll, D. M. (1995). Rat phospholipid-hydroperoxide glutathione peroxidase. cDNA cloning and identification of multiple transcription and translation start sites. J. Biol. Chem. 270, 26993–26999.
Ran, Q., Liang, H., Gu, M., Qi, W., Walter, C. A., Roberts, L. J. Il, Herman, B., Richardson, A., and Van Remmen, H. (2004). Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J. Biol. Chem. 279, 55137–55146.
Rula, M. E., Cai, K. Q., Moore, R., Yang, D. H., Staub, C. M., Capo-Chichi, C. D., Jablonski, S. A., Howe, P. H., Smith, E. R., and Xu, X. X. (2007). Cell autonomous sorting and surface positioning in the formation of primitive endoderm in embryoid bodies. Genesis 45, 327–338.
Salas-Vidal, E., Lomeli, H., Castro-Obregon, S., Cuervo, R., Escalante-Alcalde, D., and Covarrubias, L. (1998). Reactive oxygen species participate in the control of mouse embryonic cell death. Exp. Cell Res. 238, 136–147.
Sattler, W., Maiorino, M., and Stocker, R. (1994). Reduction of HDL- and LDL-associated cholesterylester and phospholipid hydroperoxides by phospholipid hydroperoxide glutathione peroxidase and Ebselen (PZ 51). Arch. Biochem. Biophys. 309, 214–221.
Savaskan, N. E., Borchert, A., Brauer, A. U., and Kuhn, H. (2007). Role for glutathione peroxidase-4 in brain development and neuronal apoptosis: specific induction of enzyme expression in reactive astrocytes following brain injury. Free Radic. Biol. Med. 43, 191–201.
Scheerer, P., Borchert, A., Krauss, N., Wessner, H., Gerth, C., Hohne, W., and Kuhn, H. (2007). Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4). Biochemistry 46, 9041–9049.
Schilstra, M. J., Veldink, G. A., Verhagen, J., and Vliegenthart, J. F. (1992). Effect of lipid hydroperoxide on lipoxygenase kinetics. Biochemistry 31, 7692–7699.
Schmidt-Ullrich, R., Memet, S., Lilienbaum, A., Feuillard, J., Raphael, M., and Israel, A. (1996). NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development 122, 2117–2128.
Schnabel, D., Salas-Vidal, E., Narvaez, V., Sanchez-Carbente Mdel, R., Hernandez-Garcia, D., Cuervo, R., and Covarrubias, L. (2006). Expression and regulation of antioxidant enzymes in the developing limb support a function of ROS in interdigital cell death. Dev. Biol. 291, 291–299.
Schneider, M., Forster, H., Boersma, A., Seiler, A., Wehnes, H., Sinowatz, F., Neumuller, C., Deutsch, M. J., Walch, A., Hrabe De Angelis, M., Wurst, W., Ursini, F., Roveri, A., Maleszewski, M., Maiorino, M., and Conrad, M. (2009). Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 23, 3233–3242.
Schneider, M., Vogt Weisenhorn, D. M., Seiler, A., Bornkamm, G. W., Brielmeier, M., and Conrad, M. (2006). Embryonic expression profile of phospholipid hydroperoxide glutathione peroxidase. Gene Expr. Patterns 6, 489–494.
Schneider, M., Wortmann, M., Mandal, P. K., Arpornchayanon, W., Jannasch, K., Alves, F., Strieth, S., Conrad, M., and Beck, H. (2010). Absence of glutathione peroxidase 4 affects tumor angiogenesis through increased 12/15-lipoxygenase activity. Neoplasia 12, 254–263.
Schomburg, L., Schweizer, U., Holtmann, B., Flohe, L., Sendtner, M., and Kohrle, J. (2003). Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 370, 397–402.
Schweizer, U., Brauer, A. U., Kohrle, J., Nitsch, R., and Savaskan, N. E. (2004a). Selenium and brain function: a poorly recognized liaison. Brain Res. Brain Res. Rev. 45, 164–178.
Schweizer, U., Michaelis, M., Kohrle, J., and Schomburg, L. (2004b). Efficient selenium transfer from mother to offspring in selenoprotein-P-deficient mice enables dose-dependent rescue of phenotypes associated with selenium deficiency. Biochem. J. 378, 21–26.
Seiler, A., Schneider, M., Forster, H., Roth, S., Wirth, E. K., Culmsee, C., Plesnila, N., Kremmer, E., Radmark, O., Wurst, W., Bornkamm, G. W., Schweizer, U., and Conrad, M. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248.
Shi, Z. Z., Osei-Frimpong, J., Kala, G., Kala, S. V., Barrios, R. J., Habib, G. M., Lukin, D. J., Danney, C. M., Matzuk, M. M., and Lieberman, M. W. (2000). Glutathione synthesis is essential for mouse development but not for cell growth in culture. Proc. Natl. Acad. Sci. U.S.A. 97, 5101–5106.
Shrimali, R. K., Weaver, J. A., Miller, G. F., Starost, M. F., Carlson, B. A., Novoselov, S. V., Kumaraswamy, E., Gladyshev, V. N., and Hatfield, D. L. (2007). Selenoprotein expression is essential in endothelial cell development and cardiac muscle function. Neuromuscul. Disord. 17, 135–142.
Sirvent, A., Benistant, C., and Roche, S. (2008). Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol. Cell 100, 617–631.
Tan, M., Li, S., Swaroop, M., Guan, K., Oberley, L. W., and Sun, Y. (1999). Transcriptional activation of the human glutathione peroxidase promoter by p53. J. Biol. Chem. 274, 12061–12066.
Tarin, J. J. (1996). Potential effects of age-associated oxidative stress on mammalian oocytes/embryos. Mol. Hum. Reprod. 2, 717–724.
Thomas, J. P., Geiger, P. G., Maiorino, M., Ursini, F., and Girotti, A. W. (1990). Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochim. Biophys. Acta 1045, 252–260.
Thomas, M., Jain, S., Kumar, G. P., and Laloraya, M. (1997). A programmed oxyradical burst causes hatching of mouse blastocysts. J. Cell Sci. 110(Pt 14), 1597–1602.
Tishler, R. B., Calderwood, S. K., Coleman, C. N., and Price, B. D. (1993). Increases in sequence specific DNA binding by p53 following treatment with chemotherapeutic and DNA damaging agents. Cancer Res. 53, 2212–2216.
Tramer, F., Vetere, A., Martinelli, M., Paroni, F., Marsich, E., Boitani, C., Sandri, G., and Panfili, E. (2004). cAMP-response element modulator-tau activates a distinct promoter element for the expression of the phospholipid hydroperoxide/sperm nucleus glutathione peroxidase gene. Biochem. J. 383, 179–185.
Tuuli, M. G., Longtine, M. S., and Nelson, D. M. (2011). Review: oxygen and trophoblast biology – a source of controversy. Placenta 32(Suppl. 2), S109–S118.
Ufer, C., Borchert, A., and Kuhn, H. (2003). Functional characterization of cis- and trans-regulatory elements involved in expression of phospholipid hydroperoxide glutathione peroxidase. Nucleic Acids Res. 31, 4293–4303.
Ufer, C., Wang, C. C., Borchert, A., Heydeck, D., and Kuhn, H. (2010). Redox control in mammalian embryo development. Antioxid. Redox Signal. 13, 833–875.
Ufer, C., Wang, C. C., Fahling, M., Schiebel, H., Thiele, B. J., Billett, E. E., Kuhn, H., and Borchert, A. (2008). Translational regulation of glutathione peroxidase 4 expression through guanine-rich sequence-binding factor 1 is essential for embryonic brain development. Genes Dev. 22, 1838–1850.
Ursini, F., Maiorino, M., and Gregolin, C. (1985). The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim. Biophys. Acta 839, 62–70.
Ursini, F., Maiorino, M., Valente, M., Ferri, L., and Gregolin, C. (1982). Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 710, 197–211.
van der Brug, M. P., Blackinton, J., Chandran, J., Hao, L. Y., Lal, A., Mazan-Mamczarz, K., Martindale, J., Xie, C., Ahmad, R., Thomas, K. J., Beilina, A., Gibbs, J. R., Ding, J., Myers, A. J., Zhan, M., Cai, H., Bonini, N. M., Gorospe, M., and Cookson, M. R. (2008). RNA binding activity of the recessive parkinsonism protein DJ-1 supports involvement in multiple cellular pathways. Proc. Natl. Acad. Sci. U.S.A. 105, 10244–10249.
Van Leyen, K., Duvoisin, R. M., Engelhardt, H., and Wiedmann, M. (1998). A function for lipoxygenase in programmed organelle degradation. Nature 395, 392–395.
Van Remmen, H., Ikeno, Y., Hamilton, M., Pahlavani, M., Wolf, N., Thorpe, S. R., Alderson, N. L., Baynes, J. W., Epstein, C. J., Huang, T. T., Nelson, J., Strong, R., and Richardson, A. (2003). Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics 16, 29–37.
Wales, R. G. (1975). Maturation of the mammalian embryo: biochemical aspects. Biol. Reprod. 12, 66–81.
Walshe, J., Serewko-Auret, M. M., Teakle, N., Cameron, S., Minto, K., Smith, L., Burcham, P. C., Russell, T., Strutton, G., Griffin, A., Chu, F. F., Esworthy, S., Reeve, V., and Saunders, N. A. (2007). Inactivation of glutathione peroxidase activity contributes to UV-induced squamous cell carcinoma formation. Cancer Res. 67, 4751–4758.
Weitzel, F., and Wendel, A. (1993). Selenoenzymes regulate the activity of leukocyte 5-lipoxygenase via the peroxide tone. J. Biol. Chem. 268, 6288–6292.
Wells, P. G., Bhuller, Y., Chen, C. S., Jeng, W., Kasapinovic, S., Kennedy, J. C., Kim, P. M., Laposa, R. R., Mccallum, G. P., Nicol, C. J., Parman, T., Wiley, M. J., and Wong, A. W. (2005). Molecular and biochemical mechanisms in teratogenesis involving reactive oxygen species. Toxicol. Appl. Pharmacol. 207, 354–366.
Wenk, J., Schuller, J., Hinrichs, C., Syrovets, T., Azoitei, N., Podda, M., Wlaschek, M., Brenneisen, P., Schneider, L. A., Sabiwalsky, A., Peters, T., Sulyok, S., Dissemond, J., Schauen, M., Krieg, T., Wirth, T., Simmet, T., and Scharffetter-Kochanek, K. (2004). Overexpression of phospholipid-hydroperoxide glutathione peroxidase in human dermal fibroblasts abrogates UVA irradiation-induced expression of interstitial collagenase/matrix metalloproteinase-1 by suppression of phosphatidylcholine hydroperoxide-mediated NFkappaB activation and interleukin-6 release. J. Biol. Chem. 279, 45634–45642.
Wilkie, G. S., Dickson, K. S., and Gray, N. K. (2003). Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem. Sci. 28, 182–188.
Winn, L. M., and Wells, P. G. (1999). Maternal administration of superoxide dismutase and catalase in phenytoin teratogenicity. Free Radic. Biol. Med. 26, 266–274.
Wirth, E. K., Conrad, M., Winterer, J., Wozny, C., Carlson, B. A., Roth, S., Schmitz, D., Bornkamm, G. W., Coppola, V., Tessarollo, L., Schomburg, L., Kohrle, J., Hatfield, D. L., and Schweizer, U. (2010). Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J. 24, 844–852.
Yan, W., and Chen, X. (2006). GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J. Biol. Chem. 281, 7856–7862.
Yang, H. W., Hwang, K. J., Kwon, H. C., Kim, H. S., Choi, K. W., and Oh, K. S. (1998). Detection of reactive oxygen species (ROS) and apoptosis in human fragmented embryos. Hum. Reprod. 13, 998–1002.
Yant, L. J., Ran, Q., Rao, L., Van Remmen, H., Shibatani, T., Belter, J. G., Motta, L., Richardson, A., and Prolla, T. A. (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 34, 496–502.
Yu, Y. P., Yu, G., Tseng, G., Cieply, K., Nelson, J., Defrances, M., Zarnegar, R., Michalopoulos, G., and Luo, J. H. (2007). Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 67, 8043–8050.
Keywords: reactive oxygen species, teratogenesis, anti-oxidative defense, selenium
Citation: Ufer C and Wang CC (2011) The roles of glutathione peroxidases during embryo development. Front. Mol. Neurosci. 4:12. doi: 10.3389/fnmol.2011.00012
Received: 04 May 2011;
Accepted: 13 July 2011;
Published online: 28 July 2011.
Edited by:
Michael Fähling, Charité – Universitätsmedizin Berlin, GermanyReviewed by:
Michael Fähling, Charité – Universitätsmedizin Berlin, GermanyMarcus Conrad, German Center for Neurodegenerative Disease, Germany
Copyright: © 2011 Ufer and Wang. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Christoph Ufer, Institut für Biochemie, Universitätsmedizin Berlin – Charité, Oudenarder Str. 16, 13347 Berlin, Germany. e-mail:Y2hyaXN0b3BoLnVmZXJAY2hhcml0ZS5kZQ==; Chi Chiu Wang, Department of Obstetrics and Gynaecology, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong. e-mail:Y2N3YW5nQGN1aGsuZWR1Lmhr