
 Somaira Nowsheen1†
Somaira Nowsheen1†
- 1 Department of Radiation Oncology, Comprehensive Cancer Center, University of Alabama-Birmingham School of Medicine, Birmingham, AL, USA
- 2 Department of Cell Biology, University of Alabama-Birmingham, Birmingham, AL, USA
- 3 Department of Pharmacology and Toxicology, University of Alabama-Birmingham, Birmingham, AL, USA
The constitutively active protein glycogen synthase kinase 3 (GSK3), a serine/threonine kinase, acts paradoxically as a tumor suppressor in some cancers while potentiates growth in others. Deciphering what governs its actions is vital for understanding many pathological conditions, including brain cancer. What are seemingly disparate roles of GSK3 stems from the complex regulation of many cellular functions by GSK3. This review focuses on the regulation of GSK3, its role in survival, apoptosis and DNA damage, and finally its potential therapeutic impact in brain cancer. A thorough understanding of this versatile protein is critical for improving the outcome of various diseases, especially cancer.
Introduction
Glycogen synthase kinase 3 (GSK3) has been shown over the past three decades to regulate a myriad of cellular functions including cell polarity, cell fate, metabolic homeostasis, development, apoptosis, microtubule function, and neuronal growth and differentiation. Elegant cellular checkpoints and highly regulated equilibrium of protein levels are responsible for the precise control of GSK3 activity. GSK3, while involved in many critical functions, still remains a nearly untapped source of modulation therapeutically. The emergence of GSK3 as an attractive therapeutic agent could stem from its involvement in several of the critical signaling pathways. Multiple genes involved in these pathways are often susceptible to activating mutations which lead to multiple tumor types and/or tumor resistance (Brugge et al., 2007). By targeting GSK3, one could potentially attenuate these dysregulated pathways to improve tumor response in patients.
More importantly, GSK3 inhibition may also impact quality of life. The standard curative treatment regimen for primary and metastatic brain tumors typically involves cranial irradiation (IR), which often results in long-term neurological side effects, especially in the pediatric population (Roman and Sperduto, 1995; Anderson et al., 2000; Boehme et al., 2008). There have been reports of intellectual impairment, reduction in performance IQ, memory loss, and dementia after exposure of the brain to radiation. This cognitive decline observed may be due to IR-induced damage to the hippocampus, a critical area of the brain responsible for learning and memory (Yazlovitskaya et al., 2006). Consistent with these findings, radiation to the hippocampus is associated with more pronounced cognitive deficits compared with radiation to other areas of the brain (Abayomi, 2002). As GSK3 inhibition has been shown to protect neurocognitive function following cranial irradiation, the therapeutic value of GSK3 in cancer may also be related, in part, to its effects on DNA repair and its emerging role in neuroprotection of normal neuronal tissues from radiation induced apoptosis (Thotala et al., 2008; Yang et al., 2009, 2011). These combined effects of GSK3 inhibition, namely inducing tumor cell death while protecting normal cells, again make GSK3 an attractive therapeutic target for brain tumors.
In this review, we will discuss the rationale for therapeutic targeting of GSK3 in oncology, due to the involvement of GSK3 in apoptosis, DNA damage/repair, and autophagy. Obstacles to targeting GSK3 include its varied and highly regulated role in cellular homeostasis in addition to the existence of highly homologous GSK3 isoforms, which will be discussed. Despite these hurdles, GSK3 nevertheless has been tagged as a potential target in human cancers, and more importantly, as a protector of normal tissues at high risk for treatment-related side effects. These characteristics may ultimately improve the therapeutic ratio and enhance patient quality of life.
GSK3 Modulation of Tumorigenic Signals
Glycogen synthase kinase 3 is essentially expressed ubiquitously in all mammalian tissues and is almost always in an active state in cells, even when not stimulated by mitogenic or hormonal signals. There are two mammalian GSK3 isoforms encoded by distinct genes: GSK3α and GSK3β. GSK3α and GSK3β, although structurally similar, are functionally diverse. GSK3α and GSK3β are highly conserved and widely expressed kinases that share 98% sequence homology within their catalytic domains. Interestingly, loss of GSK3β isoform is embryonically lethal due to liver degeneration caused by extensive hepatocyte apoptosis (Hoeflich et al., 2000). Moreover, GSK3α is unable to rescue this phenotype. It is also well established that GSK3 activity is regulated primarily at the posttranslational level, chiefly by protein–protein interactions or posttranslational modifications (Cohen and Frame, 2001). Phosphorylation of GSK3 N-terminally at S9 lowers its activity toward its substrates (Cohen and Frame, 2001). We refer the readers to some excellent reviews in this special issue for further details on GSK3, including its isoforms, regulation of these isoforms, and their distinct functions.
Dysregulation in GSK3 activity has been linked to multiple cancers. However, the direction by which GSK3 is dysregulated, i.e., suppressed vs. activated, is heterogeneous among tumor types as discussed below. In general, GSK3 primarily functions by inactivating its substrates via phosphorylation, thus altering their conformation, localization, and/or degradation (Figures 1 and 2). This, in turn, can affect the subsequent ability of these substrates to interact and trigger downstream signaling events. Usually, the substrates of GSK3 need to be “primed” by a separate kinase to allow GSK3 to bind and subsequently phosphorylate the target molecule. Here we look at the direct and indirect roles of GSK3 in cancer.
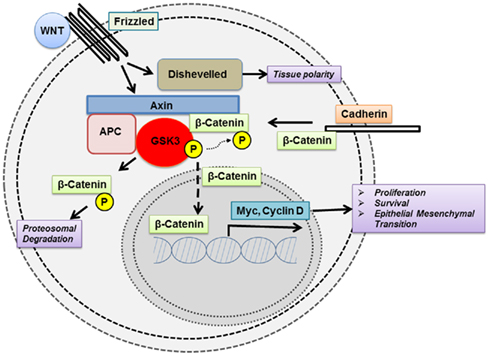
Figure 1. Glycogen synthase kinase 3 modulates the function of key signaling proteins in the wnt pathway.
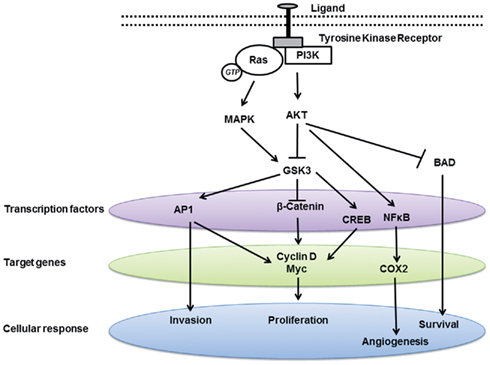
Figure 2. Glycogen synthase kinase 3 modulates multiple signaling pathways involved in carcinogenesis.
Direct Roles of GSK3 in Cancer
Though various roles of GSK3 in cancer have been proposed, the direct vs. indirect roles of GSK3 in this disease are difficult to tease out due to the embryonic lethality of GSK3 loss. In addition, the significant cross talk between different signaling pathways and varied role of GSK3 in these pathways makes it all the more difficult to pinpoint one player. However, we will detail in the next section the direct roles of GSK3 in cancer as reported in the literature.
Expression of GSK3β is drastically diminished in multiple cancers as listed in Table 1. Ma et al. (2007) have demonstrated that normal patient skin tissue specimens express higher GSK3β and pGSK3β expression when compared to cancer. Moreover, utilizing different constructs, they show that modulation of GSK3β activity negatively regulates epidermal cell transformation. In the complex web underlying skin tumorigenesis and involving interactions among multiple signaling cascades and various transcription factors, GSK3β appears to be an important component in the signaling cascade since modulation of GSK3β expression/activity is sufficient to alter the transformation potential of epidermal cells. Thus, GSK3β is a target for skin cancer prevention and treatment strategies.
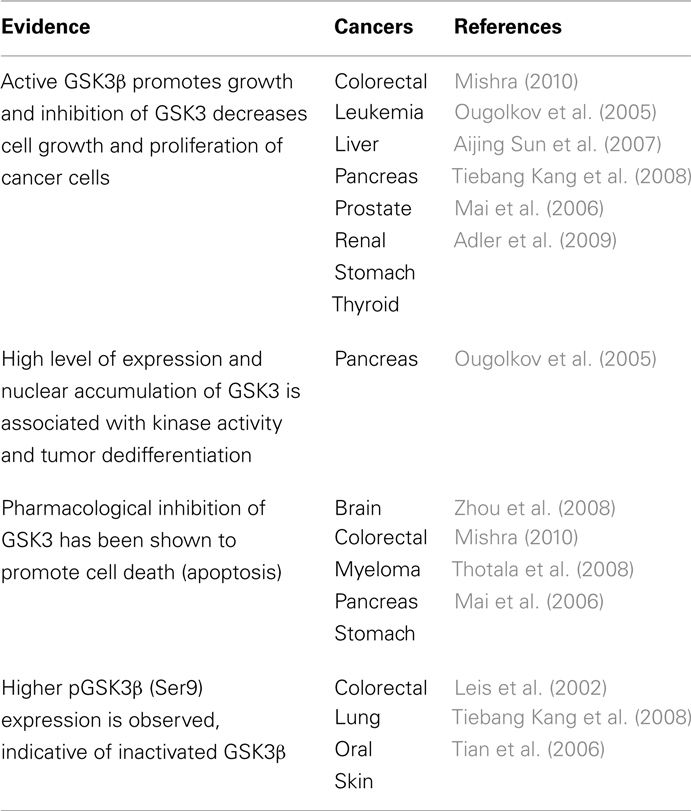
Table 1. Evidence for GSK3 involvement in distinct cancers.
Interestingly, in pancreatic cancer, pharmacological inhibition of GSK3 decreased proliferation and survival of these cancer cells (Ougolkov et al., 2005). The mechanism was shown to be related to blockade of GSK3β-mediated upregulation of NF-κB-mediated gene transcription. Similarly, brain, bladder, colorectal, myeloma, leukemia, and stomach cancers were also responsive to inhibition of GSK3 (Mishra, 2010; Naito et al., 2010). For example, pharmacological inhibition of GSK3 downregulated BCL-2 and XIAP protein expression levels leading to decreased survival of bladder cancer cells. Furthermore, aberrant nuclear accumulation of GSK3β in urothelial and bladder cancer was observed. Nuclear expression of GSK3β has been associated with high grade tumors, metastasis and worse survival in bladder cancer patients and thus may be a prognostic marker for this cancer (Naito et al., 2010).
In addition, GSK3 inhibition has been shown to promote apoptosis in leukemic cells via modulation of chromatin structure through histone modifications that abrogate the transcriptional activity of NFκB (Ougolkov et al., 2007). GSK3 can also promote mixed lineage leukemia (MLL) cell proliferation and transformation via destabilization of the cyclin-dependent kinase inhibitor p27Kip1 (Wang et al., 2008). Similarly, inhibition of GSK3β has been shown to promote apoptosis in neuroblastoma by promoting G2/M cell cycle arrest, accumulation of β-catenin and subsequent inhibition of NFκB activity (Dickey et al., 2011). Furthermore, recent studies in prostate, pancreatic, and colorectal cancer cell lines indicate that GSK3 inhibitors significantly decrease cell growth and proliferation (Martinez et al., 2006). Thus, taken together, these results suggest GSK3 as a potential new therapeutic target in human cancer.
In the brain specifically, cancers result from abnormal cell growth and may arise from different cell lineages: neuronal or glial progenitor cells. Medulloblastomas originate from neuronal progenitors which ultimately gives rise to neurons. On the other hand, glial progenitors are far more plastic and may give rise to astrocytoma or glioblastoma (from astrocyte progenitor) or oligodendroglioma (from oligodendrocyte progenitor). We refer the readers to an excellent review on brain cancer by Huse and Holland (2010).
Medulloblastoma, one of the more common malignant brain tumors in children which originates in the area between the brainstem and the cerebellum, remains responsible for a significant level of morbidity and mortality among cancer patients. A proposed pathway involved in the development of medulloblastoma is the sonic hedgehog (SHH) pathway (Weiner et al., 2002). Approximately 15% of sporadic medulloblastomas present with genomic alterations in components of the SHH signaling pathway (Taylor et al., 2002). Normally, the SHH mitogen enables the cerebellar granule neuron precursors (CGNPs) to exhibit rapid cellular growth that eventually progresses to terminal differentiation into glutamatergic neurons. Dysregulation of SHH activation is implicated in medulloblastoma and its proliferative effects are enabled by its transcription factor target, N-myc (Knoepfler and Kenney, 2006). N-myc has been shown to be critical for normal brain growth as well as being a key player in medulloblastoma. Importantly, GSK3 is reported to be a regulator of N-myc phosphorylation (Knoepfler and Kenney, 2006). Phosphorylation of N-myc leads to its destabilization and degradation. Thus, treatment of CGNPs with LiCl or GSK3 chemical inhibitor leads to a rapid decrease in N-myc phosphorylation. Furthermore, inhibition of PI3K leads to an increase in GSK3 activity resulting in a rapid turnover of N-myc (Kenney et al., 2004). Thus, dysregulation of GSK3 is a key player in medulloblastoma and targeting this protein may be a novel therapeutic approach.
Inhibition of GSK3β activity also induces tumor cell differentiation and enhances apoptosis in glioblastoma. Targeted inhibition of GSK3β results in a decrease in progenitor markers while inducing expression of GFAP, β-tubulin III, CNPase, all being cellular differentiation markers. In addition, GSK3 inhibition has a negative effect on the subpopulation of cancer stem cell-like cells, depleting them and pushing into differentiation. These observations strengthen the claim that GSK3 inhibition in glioma cells results in decreased proficiency in anchorage-independent growth compared to wild-type. Impairment in the formation of neurospheres and decrease in stem cell markers are also observed following GSK3β inhibition (Kotliarova et al., 2008; Korur et al., 2009).
Indirect Roles of GSK3 in Cancer
In addition to the aforementioned direct roles of GSK3 in cancer, it may also indirectly be involved in cancer due to its participation in dysregulated signaling pathways. For example, one of the major upstream protein kinases known to phosphorylate and inactivate GSK3 is protein kinase B (PKB)/AKT, which is often dysregulated in tumors (Robinson et al., 2011). AKT is able to enhance growth-factor mediated survival by multiple mechanisms. One method is the phosphorylation of the pro-apoptotic protein, BCL2-associated agonist of cell death (BAD), leading to inhibition of complex formation between BAD and BCL-2. This dissociation causes a shift from apoptosis to cellular survival (Downward, 2004). Another possibility is the ability of AKT to activate NF-κB by the non-canonical pathway involving activation of IKKα which increases p53 production. This increase in NF-κB activity is implicated in transformation of murine fibroblasts as well as involvement in breast and skin cancers (Gustin et al., 2006). Additionally, AKT promotes nuclear translocation of mouse double minute 2 (MDM2), an E3 ubiquitin ligase, thereby down-regulating p53-mediated apoptosis (Mayo and Donner, 2001). The tumor suppressor phosphatase and tensin homolog (PTEN) antagonizes this PI3K/Akt pathway. For example, in certain brain cancers, PTEN dysfunction may contribute to the formation of gliomas in multiple ways including having effects on stem cell self-renewal, migration, invasion as well as by enhanced degradation of p53 via increased expression of MDM2 (Endersby and Baker, 2008).
Glycogen synthase kinase 3 can also phosphorylate the pro-oncogenic molecules β-catenin, c-myc, and c-Jun, targeting them for degradation or inactivation. This results in inhibition of cell proliferation and self-renewal. Dysregulation in these pathways and gain-of-function mutations in these three proteins interfere with the function of GSK3 and have been linked to cancers of the skin, colon, prostate, and liver (Polakis, 2007).
Dysregulation in the GSK3 pathway has also been implicated in oral cancer. Tumor recurrence and metastasis pose a challenge to controlling this malignancy. Emerging research suggests a critical role of GSK3β as a tumor suppressor in oral cancer. Furthermore, GSK3β may be vital in regulating key players that control transcription, accelerated cell cycle progression, activation of invasion/metastasis, and anti-apoptosis (Goto et al., 2002; Chien et al., 2010; Iwai et al., 2010). P-cadherin triggers GSK3β mediated inactivation and subsequent cytoplasmic translocation of Snail in oral squamous cell carcinoma, thus conferring mesenchymal cells with epithelial features (Bauer et al., 2009). These results contradict the idea of GSK3 as a therapeutic target. However, as discussed below, in a subset of cancers, GSK3 inhibition can induce death or halt tumor progression.
Specifically, in brain tumors, GSK3β may have an indirect role in glioblastomas which is one of the most lethal brain tumors. Glioblastomas are notoriously difficult to treat and have a median survival time of ~14 months. As mentioned above cancers are driven by cells with stem cell properties. Expression of GSK3β, which is consistently expressed in primary glioblastoma, is functionally linked to Bmi1, a protein involved in neural stem cell self-renewal and anti-oxidant defense in neurons (Kotliarova et al., 2008; Korur et al., 2009). Research has also shown that inhibition of Bmi1, a member of the polycomb family of proteins, results in reduced GSK3β as well as increased differentiation of glioma (Korur et al., 2009). While fully differentiated normal brain tissue has no Bmi1 expression, it is marked in 84% of primary glioblastomas and 71% of primary oligodendroglioma. Downregulation of Bmi1 in glioma cells results in an increase in cell differentiation toward an astrocytic fate and a decrease in expression of Nestin, Sox2, and GSK3β.
Furthermore, inactivation of GSK3α and GSK3β has been reported to block differentiation in brain progenitor cells, and instead lead to substantial hyperproliferation of the neural progenitor cells, thus, expanding the population (Kim et al., 2009). These observations were attributed to dysregulation of β-catenin, SHH, Notch, and fibroblast growth factor signaling. This critical GSK3-mediated homeostatic control was only removed by a major reduction of GSK3 signaling. Since brain development requires a balance of progenitor proliferation and differentiation this might, in a more sporadic background, lead to amplification of stem cells. This may, at least theoretically, lead to subsequent tumor formation. Thus, GSK3 signaling is integral to homeostatic controls and further dissection of these intricate processes will elucidate the therapeutic role of GSK3 in cancer.
Another potential mechanism of GSK3 activation in cancer may be via the epidermal growth factor receptor (EGFR) pathways. The EGFR family of proteins (EGFR/HER/ErbB2) plays an essential role in modulating proliferation, differentiation, and survival. EGFR has become a heavily targeted pathway as a novel cancer therapeutic strategy since its overexpression confers resistance to therapy. It has also been recently reported that lapatinib, a small molecule tyrosine kinase inhibitor targeting EGFR and HER2, is associated with changes in Akt and GSK3 (Li et al., 2011). Accordingly, EGFR and phosphorylated GSK3β have been validated as good prognostic markers for EGFR overexpressing lung carcinoma (Zheng et al., 2007). Thus, it is intriguing to hypothesize that there may be significant cross-talk between GSK3 and EGFR signaling pathways. Interestingly, dysregulation in the EGFR family of proteins is observed in glioblastoma (Furnari et al., 2007; Chin, 2008; Parsons et al., 2008; Bredel et al., 2011). The EGFR signaling pathway may be activated alternatively via NF-κB and can be repressed by the nuclear factor of κ-light polypeptide gene enhancer in B-cells inhibitor-α (NFκBIA). It has been suggested that NFκBIA may function as a tumor suppressor in multiple cancers (Osborne et al., 2005; Sjöblom et al., 2006; Bu et al., 2007). Not surprisingly, NFκBIA is often deleted in glioblastomas and confers resistance to chemotherapy (Bredel et al., 2011). Since NF-κB is also known to be an important player in the survival of glioma cells, it is not surprising that attenuation of GSK3 inhibits NF-κB leading to decreased glioma cell growth (Kasuga et al., 2004; Robe et al., 2004). TRAIL, DR4/5, and c-myc are induced upon GSK3 inhibition in a dose-dependent manner. Furthermore, GSK3 inhibition has been reported to have synergistic effects in combination with the chemotherapeutic drug, carboplatin, on glioma cytotoxicity.
Further evidence of this link lies in the signal transducer and activator of transcription (STAT) family of transcription factors, which is downstream of EGFR and plays a central role in cancer. STAT3 is constitutively activated in many human cancers where it functions as a critical mediator of oncogenic signaling through transcriptional activation of genes encoding apoptosis inhibitors [e.g., BCL-x(L) and survivin], cell-cycle regulators (e.g., cyclin D1 and c-myc) and inducers of angiogenesis (e.g., vascular endothelial growth factor). The STAT3 gene has been reported to regulate cancer stem cells in brain cancer. Importantly, GSK3 inhibitors have been demonstrated to block STAT3 DNA binding activity and reduce the expression of STAT3-induced Glial fibrillary acidic protein (GFAP) and B-cell CLL/lymphoma 3 (BCL-3; Beurel and Jope, 2008).
The above findings suggest that GSK3 inhibition indeed may be beneficial in brain tumors. However, one must be cautioned and consider the actions of GSK3 in normal neurons. Dysregulation of GSK3 in a variety of brain abnormalities supports its function as the switch between basic mechanisms of neuronal functions. It plays a pivotal role in brain bioenergetics, establishment of neuronal circuits, modulation of neuronal polarity, migration, neuronal proliferation, and survival (Frame and Cohen, 2001). In particular, the role of GSK3 in phosphorylation of cytoskeletal proteins impacts neuronal plasticity. Since cytoskeletal constituents are involved in the development and maintenance of neurites, alterations in the rate of stabilization/destabilization of microtubules influences major cellular compartments of neurons, such as dendrites, spines, axons, and synapses. This modulates synaptic plasticity in the adult brain (Peineau et al., 2008). Due to the effects of GSK3 on synaptic plasticity, the use of GSK3 inhibitors for cancer therapy must be approached with caution. However, a subset of cancer patients may benefit from GSK3 inhibition, due to the paradoxical effect of GSK3 activity on tumor growth. Further investigation to determine the full extent of GSK3 dysregulation in cancer is thus warranted.
GSK3 Inhibition and Protection of Irradiated Neurons
Nevertheless, the role of GSK3 inhibition in brain tumor therapy may extend beyond tumoricidal effects and provide neuroprotection following irradiation, which increases the therapeutic index and improves patient quality of life (Figure 3). Cranial irradiation therapy is a standard method for treatment of brain cancer. Since radiation cannot be selectively delivered to cancer cells, it leads to many destructive cellular processes including apoptosis, genomic instability, and autophagy. Cranial irradiation is known to cause long-term cognitive defects especially in the developing brains of children (Meadows et al., 1981). The subgranular zone of the hippocampus, housing the actively proliferating neuronal progenitor cells, is thought to be the area most affected by cranial irradiation leading to neurocognitive deficits (Nagai et al., 2000).
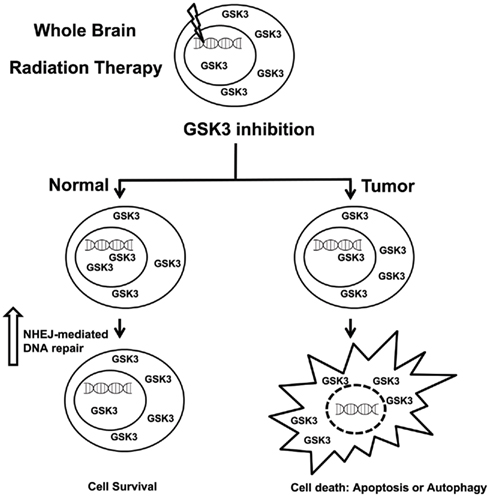
Figure 3. Inhibitors of GSK3 can selectively protect normal tissues from radiation induced toxicities.
Interestingly, it has been known for some time that lithium treatment has neuroprotective effects against various insults to neuronal tissues (Nonaka et al., 1998; Cimarosti et al., 2001; Hongisto et al., 2003). As previously mentioned, lithium is known to modulate GSK3 activity by attenuating GSK3 activity at therapeutic concentrations via the PI3K/AKT pathway (~1 mM; De Sarno et al., 2002; Jope, 2003). These neuroprotective effects of GSK3 inhibition have led to the hypothesis that GSK3 can potentially be targeted to attenuate the side effects of cranial irradiation (Inouye et al., 1995; Yazlovitskaya et al., 2006; Thotala et al., 2008). Treatment with LiCl increases phosphorylation of GSK3β at S9 and concomitantly decreases the activating phosphorylation of GSK3β, GSK3β-T216. The major kinase responsible for the inhibitory phosphorylation of GSK3 at S9 is Akt which is activated upon LiCl treatment as well as treatment with ionizing radiation. A two-fold increase in protein levels of the downstream targets of GSK3, β-catenin, and cyclin D1, has been reported following treatment with LiCl. Together these show that LiCl treatment has a direct effect on GSK3 activity, which may be explained by an increase in Akt activity. Importantly, both lithium prophylaxis and prophylaxes with small molecule GSK3β inhibitors prior to cranial irradiation maintained neurocognitive function in irradiated mice (Thotala et al., 2008). Thus lithium can potentially improve neurocognitive function following irradiation and the mechanism of action may be attributed to reduction of radiation-induced apoptosis of hippocampal neurons.
While apoptosis is one consequence of this type of treatment, radiation induced DNA damage is certainly another potential trigger leading to neuronal death and subsequent neurocognitive decline. There are multiple types of DNA damage that can occur in a given cell, the most lethal and critical being the double-strand break (DSB; Rich et al., 2000). There are two major DSB repair pathways, homologous recombination (HR) and non-homologous end-joining (NHEJ; van Gent et al., 2001). Cells with radiation-induced DSBs utilize the NHEJ pathway predominantly. During NHEJ-mediated repair, DNA-dependent protein kinase catalytic subunit (DNA-PKcs) undergoes autophosphorylation upon exposure to ionizing radiation. Thus, another mechanism of neuroprotection by GSK3 inhibition may involve augmenting DNA repair via one of these major repair pathways to attenuate DNA damage.
Supporting this notion, GSK3 inhibition using LiCl selectively augments DSB repair following IR in normal tissues but not cancer (Yang et al., 2009). Moreover, the accelerated repair of DSB in normal tissues but not tumor involves the NHEJ-mediated repair pathway. These findings support the rationale for the use of lithium as a neuroprotector for normal cells exposed to ionizing radiation.
Since LiCl has multiple targets, neuroprotection via specific inhibition of GSK3β was subsequently investigated. Thotala et al. (2008) used specific GSK3β inhibitors or a kinase-inactive GSK3β to show that GSK3β is indeed required for apoptosis and neurocognitive decline in hippocampal neurons as a result of radiation treatment. Additionally, it has been shown that GSK3 inhibition using these selective inhibitors augment DSB repair following IR in normal tissues but not cancer (Yang et al., 2011). Moreover, the accelerated repair of DSB in normal tissues but not tumor involves the NHEJ-mediated repair pathway. These results were validated using genetic models as well and support the rationale for the use of GSK3 inhibitors as neuroprotectors for normal cells exposed to ionizing radiation.
The role of GSK3 in genomic instability is only recently gaining attention. It follows that since LiCl and other inhibitors of GSK3 exhibits neuroprotective effects manifesting as a decrease in apoptosis resulting from radiation treatment, and DNA damage is another known characteristic of radiation exposure, that GSK3 could potentially modulate DNA damage or repair. It should be noted that lithium has its drawbacks as a neuroprotector. Besides being non-specific, lithium requires a long 7-day prophylaxis, has a narrow therapeutic window, and lacks specificity. Thus, it is not feasible for the majority of brain tumors which require immediate treatment. This necessitates efforts to discover novel GSK3 inhibitors that have greater therapeutic potential. Nevertheless, several clinical trials have been initiated to test the efficacy of lithium in neuroprotection during the treatment of brain tumors (Table 2). It should be noted that the trials are still in their early stage. It remains to be seen if, indeed, lithium can improve the lives of cancer patients.

Table 2. Lithium in clinical trials as a potential neuroprotector.
It is perplexing that inhibition of GSK3 selectively upregulates DNA repair in normal cells. One explanation may be that GSK3 is already maximally inhibited in the majority of cancer. It is also intriguing to postulate that, perhaps, the status of p53 determines cellular response to GSK3 inhibition. Perhaps, there are different roles of GSK3 in p53 wild-type vs. mutated tumors and the p53-independent component also mediates repair that is only apparent when p53 is mutated as is the case in the majority of cancer. Further research is warranted in this avenue.
The mechanisms by which GSK3 is involved in DNA repair are still forthcoming. Since GSK3 is implicated in several of the pathways that contribute to successful DNA repair, it is likely GSK3 could act as a direct regulator. One method could involve GSK3 interaction with key repair proteins. Our lab is currently exploring this and other avenues to determine the full extent by which GSK3 is involved DNA damage/repair and how this relates to GSK3-induced neuroprotection. This has the potential to improve the quality of life of cancer patients, especially in the pediatric population.
Summary and Conclusion
Glycogen synthase kinase 3 is proving to be an even more complex regulator of cellular function than was previously thought. Its role in programmed cellular death pathways including apoptosis and autophagy as well as its effects on DNA damage/repair and subsequent neuroprotection provide rationale for novel and specific methods of targeting GSK3, especially for brain tumors. This would be the ideal therapeutic strategy which would maximize the therapeutic index by promoting tumor cell kill while protecting normal tissues. Nevertheless, the utility of GSK3 as a therapeutic target must be approached with caution. Firstly, one must verify the direction of GSK3 dysregulation as the impetus for tumor development to ensure that benefit can be realized with GSK3 inhibition. Second, due to the effects of GSK3 on synaptic plasticity in the adult brain, the use of GSK3 inhibitors for cancer therapy must be approached with caution. Third, effects on both peripheral tissue and tumor from using GSK3 inhibitor as neuro-protector need to be teased out. Expression profile and subcellular localization of GSK3 in different cancers may provide a lead in this aspect. Furthermore, since GSK3 has multiple critical roles in cellular metabolism, inhibition of its activity may have unwanted side effects. Further thorough evaluation is necessitated in this field before GSK3 can be embraced as a target for cancer therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the IMPACT Award from the Department of Radiation Oncology, University of Alabama-Birmingham Comprehensive Cancer Center, the Fighting Children’s Cancer Foundation, and the Gabrielle’s Angel Foundation (to Eddy S. Yang). We recognize that we were unable to cover all aspects of GSK3 in cancer in this review. We apologize to those whom we have been unable to cite owing to space constraints.
References
Abayomi, O. K. (2002). Pathogenesis of cognitive decline following therapeutic irradiation for head and neck tumors. Acta Oncol. 41, 346–351.
Adler, J. T., Cook, M., Luo, Y., Pitt, S. C., Ju, J., Li, W., Shen, B., Kunnimalaiyaan, M., and Chen, H. (2009). Tautomycetin and tautomycin suppress the growth of medullary thyroid cancer cells via inhibition of glycogen synthase kinase-3beta. Mol. Cancer Ther. 8, 914–920.
Anderson, V. A., Godber, T., Smibert, E., Weiskop, S., and Ekert, H. (2000). Cognitive and academic outcome following cranial irradiation and chemotherapy in children: a longitudinal study. Br. J. Cancer 82, 255–262.
Bauer, K., Dowejko, A., Bosserhoff, A. K., Reichert, T. E., and Bauer, R. J. (2009). P-cadherin induces an epithelial-like phenotype in oral squamous cell carcinoma by GSK-3beta-mediated Snail phosphorylation. Carcinogenesis 30, 1781–1788.
Beurel, E., and Jope, R. S. (2008). Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 283, 21934–21944.
Boehme, K. A., Kulikov, R., and Blattner, C. (2008). p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc. Natl. Acad. Sci. U.S.A. 105, 7785–7790.
Bredel, M., Scholtens, D. M., Yadav, A.‘K., Alvarez, A. A., Renfrow, J. J., Chandler, J. P., Yu, I. L. Y., Carro, M. S., Dai, F., Tagge, M. J., Ferrarese, R., Bredel, C., Phillips, H. S., Lukac, P. J., Robe, P. A., Weyerbrock, A., Vogel, H., Dubner, S., Mobley, B., He, X., Scheck, A. C., Sikic, B. I., Aldape, K. D. Chakravarti, A., and Harsh, G. R. (2011). NFKBIA deletion in glioblastomas. N. Engl. J. Med. 364, 627–637.
Brugge, J., Hung, M.-C., and Mills, G. B. (2007). A new mutational AKTivation in the PI3K pathway. Cancer Cell 12, 104–107.
Bu, H., Rosdahl, I., Sun, X.-F., and Zhang, H. (2007). Importance of polymorphisms in NF-kappaB1 and NF-kappaBIalpha genes for melanoma risk, clinicopathological features and tumor progression in Swedish melanoma patients. J. Cancer Res. Clin. Oncol. 133, 859–866.
Chien, C.-M., Lin, K.-L., Su, J.-C., Chuang, P.-W., Tseng, C.-H., Chen, Y.-L., Chang, L.-S., and Lin, S.-R. (2010). Naphtho[1,2-b]furan-4,5-dione induces apoptosis of oral squamous cell carcinoma: involvement of EGF receptor/PI3K/Akt signaling pathway. Eur. J. Pharmacol. 636, 52–58.
Chin, L. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068.
Cimarosti, H., Rodnight, R., Tavares, A., Paiva, R., Valentim, L., Rocha, E., and Salbego, C. (2001). An investigation of the neuroprotective effect of lithium inorganotypic slice cultures of rat hippocampus exposed to oxygen and glucose deprivation. Neurosci. Lett. 315, 33–36.
De Sarno, P., Li, X., and Jope, R. S. (2002) Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuropharmacology 43, 1158–1164.
Dickey, A., Schleicher, S., Leahy, K., Hu, R., Hallahan, D., and Thotala, D. K. (2011). GSK-3beta inhibition promotes cell death, apoptosis, and in vivo tumor growth delay in neuroblastoma Neuro-2A cell line. J. Neurooncol. 104, 145–153.
Endersby, R., and Baker, S. J. (2008). PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene 27, 5416–5430.
Frame, S., and Cohen, P. (2001). GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359(Pt 1), 1–16.
Furnari, F. B., Fenton, T., Bachoo, R. M., Mukasa, A., Stommel, J. M., Stegh, A., Hahn, W. C., Ligon, K. L., Louis, D. N., Brennan, C., Chin, L., DePinho, R. A., and Cavenee, W. K. (2007). Malignant astrocyticglioma: genetics, biology, and paths to treatment. Genes Dev. 21, 2683–2710.
Goto, H., Kawano, K., Kobayashi, I., Sakai, H., and Yanagisawa, S. (2002). Expression of cyclin D1 and GSK-3beta and their predictive value of prognosis in squamous cell carcinomas of the tongue. Oral Oncol. 38, 549–556.
Gustin, J. A., Korgaonkar, C. K., Pincheira, R., Li, Q., and Donner, D. B. (2006). Akt regulates basal and induced processing of NF-kappaB2 (p100) to p52. J. Biol. Chem. 281, 16473–16481.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90.
Hongisto, V., Smeds, N., Brecht, S., Herdegen, T., Courtney, M. J., and Coffey, E. T. (2003). Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell. Biol. 23, 6027.
Huse, J. T., and Holland, E. C. (2010). Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 10, 319–331.
Inouye, M., Yamamura, H., and Nakano, A. (1995). Lithium delays the radiation-induced apoptotic process in external granule cells of mouse cerebellum. J. Radiat. Res. 36, 203–208.
Iwai, S., Yonekawa, A., Harada, C., Hamada, M., Katagiri, W., Nakazawa, M., and Yura, Y. (2010). Involvement of the Wnt-β-catenin pathway in invasion and migration of oral squamous carcinoma cells. Int. J. Oncol. 37, 1095–1103.
Jope, R. S. (2003). Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol. Sci. 24, 441–443.
Kang, T., Wei, Y., Honaker, Y., Yamaguchi, H., Appella, E., Hung, M.-C., and Piwnica-Worms, H. (2008). GSK-3 beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer cell 13, 36–47.
Kasuga, C., Ebata, T., Kayagaki, N., Yagita, H., Hishii, M., Arai, H., Sato, K., and Okumura, K. (2004). Sensitization of human glioblastomas to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) by NF-kappaB inhibitors. Cancer Sci. 95 840–844.
Kenney, A. M., Widlund, H. R., and Rowitch, D. H. (2004). Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 131, 217–228.
Kim, W.-Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., and Snider, W. D. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12 1390–1397.
Knoepfler, P. S., and Kenney, A. M. (2006). Neural precursor cycling at sonic speed: N-Myc pedals, GSK-3 brakes. Cell Cycle 5, 47–52.
Korur, S., Huber, R. M., Sivasankaran, B., Petrich, M., Morin, P., Hemmings, B. A., Merlo, A., and Lino, M. M. (2009). GSK3beta regulates differentiation and growth arrest in glioblastoma. PLoS ONE 4, e7443. doi:10.1371/journal.pone.0007443
Kotliarova, S., Pastorino, S., Kovell, L. C., Kotliarov, Y., Song, H., Zhang, W., Bailey, R., Maric, D., Zenklusen, J. C., Lee, J., and Fine, H. A. (2008). Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-kappaB, and glucose regulation. Cancer Res. 68, 6643–6651.
Leis, H., Segrelles, C., Ruiz, S., Santos, M., and Paramio, J. M. (2002). Expression, localization, and activity of glycogen synthase kinase 3beta during mouse skin tumorigenesis. Mol. Carcinog. 35, 180–185.
Li, J., Cho, Y.-Y., Langfald, A., Carper, A., Lubet, R. A., Grubbs, C. J., Ericson, M. E., and Bode, A. M. (2011). Lapatinib, a preventive/therapeutic agent against mammary cancer, suppresses RTK-mediated signaling through multiple signaling pathways. Cancer Prev. Res. (Phila.) 4, 1190–1197.
Ma, C., Wang, J., Gao, Y., Gao, T.-W., Chen, G., Bower, K. A., Odetallah, M., Ding, Ke, Z., and Luo, J. (2007). The role of glycogen synthase kinase 3beta in the transformation of epidermal cells. Cancer Res. 67, 7756–7764.
Mai, W., Miyashita, K., Shakoori, A., Zhang, B., Yu, Z. W., Takahashi, Y., Motoo, Y., Kawakami, K., and Minamoto, T. (2006) Detection of active fraction of glycogen synthase kinase 3beta in cancer cells by nonradioisotopic in vitro kinase assay. Oncology 71, 297–305.
Martinez, A., Castro, A., and Medina, M. (2006). Glycogen synthase kinase 3 (GSK-3) and its inhibitors: drug discovery and development, 1st Edn, eds A. Martinez, A. Castro, and M. Medina (Wiley). Available at: http://www.wiley.com/WileyCDA/WileyTitle/productCd-0470052155.html
Mayo, L. D., and Donner, D. B. (2001). A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. U.S.A. 98, 11598–115603.
Meadows, A. T., Gordon, J., Massari, D. J., Littman, P., Fergusson, J., and Moss, K. (1981). Declines in IQ scores and cognitive dysfunctions in children with acute lymphocytic leukaemia treated with cranial irradiation. Lancet 2, 1015–1018.
Mishra, R. (2010). Glycogen synthase kinase 3 beta: can it be a target for oral cancer. Mol. Cancer 9, 144.
Nagai, R., Tsunoda, S., Hori, Y., and Asada, H. (2000). Selective vulnerability to radiation in the hippocampal dentate granule cells. Surg. Neurol. 53, 503–506; discussion 506–507.
Naito, S., Bilim, V., Yuuki, K., Ugolkov, A., Motoyama, T., Nagaoka, A., Kato, T., and Tomita, Y. (2010). Glycogen synthase kinase-3beta: a prognostic marker and a potential therapeutic target in human bladder cancer. Clin. Cancer Res. 16, 5124–5132.
Nonaka, S., Hough, C. J. J., and Chuang, D. M. M. (1998). Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc. Natl. Acad. Sci. U.S.A. 95. 2642–2647.
Osborne, J., Lake, A., Alexander, F. E., Taylor, G. M., and Jarrett, R. F. (2005). Germline mutations and polymorphisms in the NFKBIA gene in Hodgkin lymphoma. Int. J. Cancer 116, 646–651.
Ougolkov, A. V., Bone, N. D., Fernandez-Zapico, M. E., Kay, N. E., and Billadeau, D. D. (2007). Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor kappaB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood 110, 735–742.
Ougolkov, A. V., Fernandez-Zapico, M. E., Savoy, D. N., Urrutia, R. A., and Billadeau, D. D. (2005). Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 65, 2076–2081.
Parsons, D. W., Jones, S., Zhang, X., Lin, J. C., Leary, R. J., Angenendt, P., Mankoo, P., Carter, H., Siu, I. M., Gallia, G. L., Olivi, A., McLendon, R., Rasheed, B. A., Keir, S., Nikolskaya, T., Nikolsky, Y., Busam, D. A., Tekleab, H., Diaz, L. A. Jr., Hartigan, J., Smith, D. R., Strausberg, R. L., Marie, S. K., Shinjo, S. M., Yan, H., Riggins, G. J., Bigner, D. D., Karchin, R., Papadopoulos, N., Parmigiani, G., Vogelstein, B., Velculescu, V. E., and Kinzler, K. W. (2008). An integrated genomic analysis of human glioblastomamultiforme. Science 321, 1807–1812.
Peineau, S., Bradley, C., Taghibiglou, C., Doherty, A., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2008). The role of GSK-3 in synaptic plasticity. Br. J. Pharmacol. 153(Suppl.), S428–S437.
Rich, T., Allen, R. L., and Wyllie, A. H. (2000). Defying death after DNA damage. Nature 407, 777–783.
Robe, P. A., Bentires-Alj, M., Bonif, M., Rogister, B., Deprez, M., Haddada, H., Khac, M. T., Jolois, O., Erkmen, K., Merville, M. P., Black, P. M., and Bours, V. (2004). In vitro and in vivo activity of the nuclear factor-kappaB inhibitor sulfasalazine in human glioblastomas. Clin. Cancer Res. 10, 5595–5603.
Robinson, J. P., Vanbrocklin, M. W., McKinney, A. J., Gach, H. M., and Holmen, S. L. (2011) Akt signaling is required for glioblastoma maintenance in vivo. Am. J. Cancer Res. 1, 155–167.
Roman, D., and Sperduto, P. (1995). Neuropsychological effects of cranial radiation: current knowledge and future directions. Int. J. Radiat. Oncol. Biol. Phys. 31, 983–998.
Sjöblom, T., Jones, S., Wood, L. D., Parsons, D. W., Lin, J., Barber, T., Mandelker, D., Leary, R. J., Ptak, J., Silliman, N., Szabo, S., Buckhaults, P., Farrell, C., Meeh, P., Markowitz, S. D., Willis, J., Dawson, D., Willson, J. K. V., Gazdar, A. F., Hartigan, J., Wu, L., Liu, C., Parmigiani, G., Park, B. H., Bachman, K. E., Papadopoulos, N., Vogelstein, B., Kinzler, K. W., and Velculescu, V. E. (2006). The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274.
Sun, A., Shanmugam, I., Song, J., Terranova, P. F., Thrasher, J. B., and Li, B. (2007). Lithium suppresses cell proliferation by interrupting E2F-DNA interaction and subsequently reducing S-phase gene expression in prostate cancer. The Prostate 67, 976–988.
Taylor, M. D., Liu, L., Raffel, C., Hui, C.-C., Mainprize, T. G., Zhang, X., Agatep, R., Chiappa, S., Gao, L., Lowrance, A., Hao, A., Goldstein, A. M., Stavrou, T., Scherer, S. W., Dura, W. T., Wainwright, B., and Squire, J. A. (2002) Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 31, 306–310.
Thotala, D. K., Hallahan, D. E., and Yazlovitskaya, E. M. (2008). Inhibition of glycogen synthase kinase 3 beta attenuates neurocognitive dysfunction resulting from cranial irradiation. Cancer Res. 68, 5859–5868.
Tian, D., Zhu, M., Chen, W.-S., Li, J.-S., Wu, R.-L., and Wang, X. (2006). Role of glycogen synthase kinase 3 in squamous differentiation induced by cigarette smoke in porcine tracheobronchial epithelial cells. Food Chem. Toxicol. 44, 1590–1596.
van Gent, D. C., Hoeijmakers, J. H., and Kanaar, R. (2001) Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2, 196–206.
Wang, Z., Smith, K. S., Murphy, M., Piloto, O., Somervaille, T. C. P., and Cleary, M. L. (2008). Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 455, 1205–1209.
Weiner, H. L., Bakst, R., Hurlbert, M. S., Ruggiero, J., Ahn, E., Lee, W. S., Stephen, D., Zagzag, D., Joyner, A. L., and Turnbull, D. H. (2002). Induction of medulloblastomas in mice by sonic hedgehog, independent of Gli1. Cancer Res. 62, 6385–6389.
Yang, E. S., Nowsheen, S., Wang, T., Thotala, D. K., and Xia, F. (2011). Glycogen synthase kinase 3{beta} inhibition enhances repair of DNA double-strand breaks in irradiated hippocampal neurons. Neuro Oncol. 5671.
Yang, E. S., Wang, H., Jiang, G., Nowsheen, S., Fu, A., Hallahan, D. E., and Xia, F. (2009). Lithium-mediated protection of hippocampal cells involves enhancement of DNA-PK–dependent repair in mice. J. Clin. Invest. 119, 1124–1135.
Yazlovitskaya, E. M., Edwards, E., Thotala, D., Fu, A., Osusky, K. L., Whetsell, W. O., Boone, B., Shinohara, E. T., and Hallahan, D. E. (2006). Lithium treatment prevents neurocognitive deficit resulting from cranial irradiation. Cancer Res. 66, 11179–11186.
Zheng, H., Saito, H., Masuda, S., Yang, Z., and Takano, Y. (2007). Phosphorylated GSK3beta-ser9 and EGFR are good prognostic factors for lung carcinomas. Anticancer Res. 27, 3561–3569.
Keywords: glycogen synthase kinase 3, neuroprotection, lithium, brain cancer, DNA damage, epidermal growth factor receptor, NFκB, apoptosis and autophagy
Citation: Mills CN, Nowsheen S, Bonner JA and Yang ES (2011) Emerging roles of glycogen synthase kinase 3 in the treatment of brain tumors. Front. Mol. Neurosci. 4:47. doi: 10.3389/fnmol.2011.00047
Received: 10 August 2011;
Accepted: 06 November 2011;
Published online: 25 November 2011.
Edited by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaReviewed by:
Karl P. Giese, University of London, UKJim Robert Woodgett, Mount Sinai Hospital, Canada
Copyright: © 2011 Mills, Nowsheen, Bonner and Yang. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Eddy S. Yang, Department of Radiation Oncology, University of Alabama at Birmingham, 1700 6th Avenue South, 176F, HSROC Suite 2232, Birmingham, AL 35249-6832, USA. e-mail:ZXlhbmdAdWFiLmVkdQ==
†Caroline N. Mills and Somaira Nowsheen have contributed equally to this work.