 Chang-Mei Liu1
Chang-Mei Liu1
- 1 Department of Orthopaedic Surgery, The Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 2 The Solomon H. Snyder Department of Neuroscience, The Johns Hopkins University School of Medicine, Baltimore, MD, USA
Axon growth requires the coordinated regulation of gene expression in the neuronal soma, local protein translation in the axon, anterograde transport of synthesized raw materials along the axon, and assembly of cytoskeleton and membranes in the nerve growth cone. Glycogen synthase kinase 3 (GSK3) signaling has recently been shown to play key roles in the regulation of axonal transport and cytoskeletal assembly during axon growth. GSK3 signaling is also known to regulate gene expression via controlling the functions of many transcription factors, suggesting that GSK3 may be an important regulator of gene transcription supporting axon growth. We review signaling pathways that control local axon assembly at the growth cone and gene expression in the soma during developmental or regenerative axon growth and discuss the potential involvement of GSK3 signaling in these processes, with a particular focus on how GSK3 signaling modulates the function of axon growth-associated transcription factors.
Introduction
During development of the nervous system, an important step in forming the neural circuitry is directed axon extension from post-mitotic neurons to reach their synaptic targets. To induce axon extension, the cellular machinery at the nerve growth cone controls the assembly of cytoskeletal proteins and membrane components into new axons. For sustained axon growth over long distance, gene expression at the neuronal soma is also required to provide signaling molecules that control the intrinsic axon growth ability and raw materials that support axon assembly (Zhou and Snider, 2006). Active axon extension is also important in adult animals after nerve injuries, when axotomized axons need to regenerate back to their original synaptic targets for functional recovery. It is well-known that neurons in the mature mammalian central nervous system (CNS) do not regenerate their axons after the injury, largely because of their reduced intrinsic axon growth ability and the hostile environment that blocks local axon assembly at the growth cone (Yiu and He, 2006; Liu et al., 2011). Therefore, understanding the two fundamental aspects of axon growth, i.e., local axon assembly at the growth cone and gene expression in the soma, is very important in the study of how axon growth is regulated in development and why axons in the mature CNS fail to regenerate after injuries.
During the past decades, extensive studies have shown how extracellular factors (neurotrophic or guidance), through their receptors and downstream signaling mediators, control axon assembly or pathfinding locally at the growth cone (Lowery and Van Vactor, 2009). In contrast, we know relatively less about the signaling events occurring at the neuronal soma that controls gene expression during axon growth. For instance, little is known about the transcription factors and the targeted genes they control that specifically regulate axon growth downstream of neurotrophic factors. We know even less about how axon growth-associated gene expression at the soma is coordinated with local axon assembly at the growth cone. In this review, we summarize signaling pathways that regulate local axon assembly or gene expression during axon growth and discuss the potential role of glycogen synthase kinase 3 (GSK3) signaling in these two processes, with a particular focus on regulation of gene expression.
Model Systems for Studying Axon Growth
To study axon growth during development, many types of primary cultured neurons have been used, such as sensory and sympathetic neurons in the peripheral nervous system (PNS), as well as retinal ganglion cells (RGCs), cortical neurons, hippocampal neurons, and cerebellar granule cells in the CNS. In vitro, these embryonic neurons can be cultured easily, and they extend axons in response to defined neurotrophic factors. In vivo, genetic deletion of neurotrophic factors or their receptors has been used to study the regulation of axon growth during development (Patel et al., 2000, 2003; Kuruvilla et al., 2004).
For studying axon growth in adult neurons, the models are limited to a few PNS neuronal types that can regenerate after injuries. Among them, dorsal root ganglion (DRG) neurons regenerate robustly after peripheral nerve injuries. Each DRG neuron possesses one axon stemming from the cell body that branches into two axons: a peripheral descending axon branch innervating peripheral targets and an ascending central branch that projects into the dorsal column of the spinal cord. The peripheral branches of the DRG neurons regenerate readily after peripheral nerve injury, and therefore a crush model of this nerve is widely to study the mechanisms that regulate axon regeneration in vivo. In contrast, the central branches of DRG neurons do not regenerate after the spinal cord injury. However, if peripheral axotomy occurs before the dorsal column injury (a process called conditioning lesion), the central branches can grow beyond the injury site in the spinal cord (Neumann and Woolf, 1999). Importantly, the central axons of DRG neurons share the same hostile CNS environment with descending corticospinal axons in the spinal cord, making the study relevant for CNS axon regeneration. As a result, injury of the DRG central axons in the spinal cord is a well-accepted model for investigating approaches that promote axon regeneration. After a conditioning lesion, dissociated adult DRG neurons can extend long axons in culture, which is increasingly used as an in vitro model system for studying regenerative axon growth (Zhou et al., 2006).
Signaling Pathways Mediating Developmental and Regenerative Axon Growth: Coordination between Gene Expression and Local Axon Assembly
Extracellular Factors Supporting Axon Growth during Development
Axon extension does not occur autonomously from the newly differentiated neurons, and extracellular signals are absolutely necessary to induce and support long distance axon growth (Goldberg et al., 2002; Goldberg, 2003). These extracellular factors induce axon growth by activating gene expression in the soma and local axon assembly at the growth cone. Among all the characterized neurotrophic factors, the most extensively studied are growth factors, especially the neurotrophin family proteins, such as the nerve growth factor (NGF), neurotrophin-3 (NT3), and brain-derived neurotrophic factor (BDNF). In cultured sympathetic ganglion neurons or DRG neurons, NGF and NT3 induce robust axon growth by activation of their corresponding receptors, TrkA and TrkC, respectively (Lentz et al., 1999; Markus et al., 2002). However, in vivo study of developing DRG neurons lacking TrkA has shown that NGF signaling is required only for terminal axon arborization, not for initial axon extension (Patel et al., 2000). Similar results were also reported in developing trigeminal ganglion neurons and sympathetic neurons (O’Connor and Tessier-Lavigne, 1999; Kuruvilla et al., 2004). To date, the identities of neurotrophic factors that mediate initial axon growth from these PNS neurons in vivo remain unknown. BDNF is able to induce axon growth of hippocampal neurons via its receptor TrkB in vitro (Labelle and Leclerc, 2000; Cheng et al., 2011). However, whether it regulates axon growth of these CNS neurons in vivo is not clear. Insulin-like growth factor 1 (IGF-1) has been shown to control axon growth of corticospinal motor neurons both in vitro and in vivo (Ozdinler and Macklis, 2006).
By using a compartmentalized culture system (i.e., the Campenot chamber) that can separate neuronal soma from their axons, studies of sympathetic neurons have revealed that application of NGF only at the soma compartment is not sufficient to promote axon growth into the axonal compartment, indicating that local NGF signaling at the growth cone is required for inducing axon growth (Campenot, 1977, 1982a,b). In addition, gene transcription, which occurs in the neuronal soma, is necessary to support sustained axon growth (Bodmer et al., 2011). Together, these studies have established that activation of neurotrophic signaling both in the neuronal soma and locally at the growth cone is necessary for efficient axon extension.
Intracellular Signaling Regulating Axon Growth during Development
Both the Trk receptors and the IGF-1 receptor belong to the family of receptor tyrosine kinases (RTKs), which transduce their signals through two major downstream mediators, MAPK and PI3K. A compartmentalized culture study has shown that both MAPK and PI3K are required locally at the growth cone to regulate axon assembly (Atwal et al., 2000). PI3K pathway is well recognized as a major regulator of local axon assembly. In support, PI3K activity is necessary for the local application of NGF-induced axonal branching or axon pathfinding (Gallo and Letourneau, 1998; Ming et al., 1999). PI3K controls local axon assembly largely through its ability to regulate the neuronal cytoskeleton. For instance, PI3K acts upstream of the small GTPases Rac and Cdc42, both of which are key regulators of actin filaments (Cantley, 2002). In line with its role in the regulation of actin dynamics, activated PI3K is localized at the leading edge of the growth cone, where actin polymerization occurs (Zhou et al., 2004). Moreover, PI3K can also regulate microtubule dynamics in the growth cone via GSK3 and its downstream microtubule-binding proteins (Zhou et al., 2004). How MAPK pathway regulates local axon assembly at the growth cone is less clear because a direct link between the MAPK pathway and the growth cone cytoskeleton is still lacking. However, MAPK pathway has been shown to control local protein synthesis in axons in response to guidance cues (Campbell and Holt, 2003), which may indirectly contribute to regulation of local axon assembly. At the neuronal soma, MAPK signaling has been shown to control neurotrophin-induced axon growth by regulation of gene expression via the transcription factor cyclic AMP response element-binding protein (CREB; Lonze et al., 2002), and/or the serum response factor (SRF; Wickramasinghe et al., 2008). In contrast, there is no evidence that PI3K regulates gene transcription during neurotrophin-induced axon growth. Taken together, the evidence to date suggests that in neurotrophin-induced developmental axon growth MAPK and PI3K share a division of labor with MAPK mainly controlling gene expression in the soma and PI3K mainly controlling local axon assembly at the growth cone.
Intracellular Signaling Regulating Axon Regeneration
In contrast to axon growth during embryonic development, only limited neuronal types in the mammalian PNS can regenerate after injuries. As a result, very little is known about the signaling pathways that control naturally occurring axon regeneration. An attractive hypothesis is that mature neurons may control axon regeneration by recapitulating signaling pathways that control developmental axon growth. Indeed, many axon growth-associated proteins are re-expressed in adult neurons to support axon regeneration after the injury (i.e., GAP43). However, the environment in which axon growth occurs in development is drastically different from that during axon regeneration. In addition, the regenerating axons need to travel much longer distances than developing axons because of their larger body size. These differences suggest that regenerating neurons may use distinct signaling pathways to control axon growth. In support, two studies have compared axon growth from the same DRG neurons during development and regeneration (Liu and Snider, 2001; Zhou et al., 2006). They showed that in adult DRG neurons neither MAPK nor PI3K pathway was necessary for conditioning lesion-induced local axon assembly, which is in strong contrast to that of neurotrophin-induced developmental axon assembly. Instead, both in vitro and in vivo studies (Werner et al., 2000; Zhou et al., 2006) have suggested that the integrin pathway play an important role in mediating PNS axon regeneration by controlling local axon assembly. Moreover, the transcription factor c-Jun has been shown to specifically regulate axon growth during regeneration but not development (Raivich et al., 2004; Saijilafu et al., 2011). Together, these studies indicate that mature neurons use different signaling pathways to control gene expression and local axon assembly during axon regeneration, although these pathways may control developmental and regenerative axon growth via similar axon growth-associated proteins (i.e., GAP43 and cytoskeletal proteins).
Comparing to developmental axon growth, we know much less about pathways that specifically regulate axon regeneration. In a mouse genetic study (Eickholt et al., 2007), in which the catalytic subunit of PI3K p110δ was mutated to an inactivated form, axon regeneration of adult DRG neurons was significantly reduced. Because PI3K is not required for local axon assembly of regenerating adult DRG neurons (Liu and Snider, 2001; Zhou et al., 2006), this result suggests that PI3K signaling is specifically involved in regulation of gene expression in the neuronal soma during axon regeneration. Indeed, in the p110δ mutant mice the peripheral nerve axotomy-induced up-regulation of the regeneration-associated gene (RAG) SPRR1A was substantially reduced compared with wild-type mice. Additional studies, in which PI3K activity is specifically manipulated in the neuronal soma or axons, are needed to test this hypothesis.
Extracellular Factors Promoting Axon Regeneration
Several recent studies have shown that after optical nerve injury, astrocyte-derived ciliary neurotrophic factor (CNTF) or macrophage-derived oncomodulin can promote RGN axon regeneration (Yin et al., 2006, 2009; Muller et al., 2007; Leibinger et al., 2009). CNTF promotes axon regeneration via the JAK–STAT signaling pathway (Muller et al., 2007), which is negatively regulated by the suppressor of cytokine signaling 3 (SOCS3; Smith et al., 2009). Pharmacological inhibition of JAK2 has been shown to block regenerative axon growth of adult DRG neurons induced by conditioning lesion (Liu and Snider, 2001). Because such in vitro axon regeneration is transcription independent (Zhou et al., 2006), this result suggests that JAK2 plays a local role in regulation of axon assembly at the growth cone. Moreover, genetic deletion of STAT3 in DRG neurons impairs peripheral axon regeneration (Qiu et al., 2005). Conversely, overexpression of STAT3 promotes the regeneration of DRG central axons in the spinal cord (Bareyre et al., 2011). These studies indicate that the JAK–STAT pathway controls axon regeneration by coordinating gene expression (via STAT3) and local axon assembly (via JAK2).
GSK3 Signaling in Axon Growth and Regeneration
Glycogen synthase kinase 3 is a unique kinase because it is usually activated in resting cells, which acts to suppress cell signaling. In response to extracellular stimuli, it is inactivated to allow propagation of the signal transduction cascades (Hur and Zhou, 2010). PI3K is one of the important upstream regulators of GSK3. Activation of the PI3K pathway is believed to inactivate GSK3 by phosphorylating its amino-terminal serine residue (Ser21 in GSK3α and Ser9 in GSK3β) via Akt. However, when such serine residue is replaced with alanine, the resulted mutant mice (GSK3α-Ser21Ala/GSK3β-Ser9Ala double knockin mice) develop normally without obvious phenotype in the nervous system (McManus et al., 2005), suggesting that GSK3 inactivation downstream of PI3K might be mediated by alternative pathways. The canonical Wnt pathway is another important regulator of GSK3 activity. In the absence of the Wnt ligand, GSK3 forms a protein complex with Axin, β-catenin, and adenomatous polyposis coli (APC). As a result, GSK3 phosphorylates β-catenin, leading to its degradation through the ubiquitin-mediated proteasome (UPS) pathway. Upon activation of the Wnt signaling, GSK3 is dissociated from the β-catenin complex and inactivated, resulting in β-catenin stabilization and the subsequent gene expression. How the Wnt pathway inactivates GSK3 is not fully understood, but it may involve being recruited to the Wnt co-receptor low-density-lipoprotein receptor-related protein 5/6 (LRP 5/6) on the membrane (Piao et al., 2008; Wu et al., 2009) or being sequestered from the cytosol into multivesicular bodies (Taelman et al., 2010).
An important step in axon extension is the continuous addition microtubules at the nerve growth cone. Such process is tightly controlled by a battery of cytoskeletal proteins, such as the collapsin response mediator protein 2 (CRMP2), the classic microtubule-associated proteins (MAP1b and Tau), the microtubule plus end-tracking protein (+TIPs, APC, and CLASP), the microtubule-severing proteins (katanin and spastin), the microtubule-depolymerizing factors (MCAK/KIF2c and stathmin), the microtubule-based motor proteins (kinesin-5 and dynein), and the actin-based motor protein (non-muscle myosin II). These cytoskeletal proteins coordinate microtubule assembly at the growth cone to support axon extension (Kim et al., 2011). Surprisingly, many of these proteins, including CRMP2, APC, CLASP, MAP1b, Tau, stathmin, and myosin II, are validated GSK3 substrates (Hanger et al., 1992; Rubinfeld et al., 1996; Moreno and Avila, 1998; Cole et al., 2004; Trivedi et al., 2005; Kim et al., 2009; Kumar et al., 2009), whereas others, including katinin and KIF2c (Taelman et al., 2010), contain conserved GSK3 sites. Thus, GSK3 is emerging as a major regulatory molecule in the growth cone for controlling local axon assembly (Zhou et al., 2004; Zhou and Snider, 2005; Hur and Zhou, 2010; Hur et al., 2011).
There is increasing evidence that GSK3 is also an important regulator of gene expression. For instance, in a recent study, GSK3 in quiescent non-neuronal cells was directly inhibited with pharmacological inhibitors, showing that GSK3 inactivation alone is sufficient to induce gene expression in the absence of growth factor stimulation (Tullai et al., 2011). Additional analysis showed that these GSK3 inhibition-induced genes were regulated by a transcriptional network downstream of three families of transcription factors: CREB, NFκB, and AP-1. Similarly, in rat cortical neurons, knocking down GSK3α or GSK3β activates CREB, NFκB, and AP-1, along with other transcription factors such as TCF4, GRE, and Smad3/4 (Liang and Chuang, 2006). These studies provide clear evidence that GSK3 signaling can orchestrate multiple transcription factors to control a specific pattern of gene expression. To date, there is no direct evidence that GSK3-mediated gene expression is involved in regulation of axon growth. One study has shown that Wnt3a can induce axon growth from embryonic DRG neurons via its receptor Ryk independent of the neurotrophin signaling (Lu et al., 2004). Through the same Ryk receptor, Wnt3a also activates the transcription factor TCF4, which is regulated by the GSK3-β-catenin pathway. This result suggests that GSK3-mediated gene transcription via β-catenin-TCF4 may underlie Wnt3a-induced axon growth, although it was not tested in that study. Moreover, because Ryk receptor is also localized in the growth cone (Lu et al., 2004), Wnt3a may also control local axon assembly through the GSK3 pathway. Thus, GSK3 may function to coordinate gene expression in the soma and axon assembly at the growth cone during Wnt3a-induced axon growth. Inhibition of GSK3 with multiple pharmacological inhibitors, lithium and SB415286, has been shown to promote both axon sprouting and regeneration of corticospinal neurons across the lesion site (Dill et al., 2008). In that study, the promoting effect was attributed mainly to the effect of GSK3 inhibition on overcoming CNS inhibitors. However, multiple studies have indicated that overcoming CNS inhibitors alone is not sufficient to promote corticospinal axon regeneration and that enhanced intrinsic axon growth ability is required (Liu et al., 2011). Therefore, it is suggested that GSK3 inhibition promotes the intrinsic axon regeneration ability of adult corticospinal neurons, presumably via regulation of gene expression.
In summary, GSK3 signaling can regulate local axon assembly through cytoskeletal proteins and gene expression through transcription factors. Because GSK3 inhibitor lithium has been used clinically for decades and more specific inhibitors are being actively developed for treating psychological disorders, inhibition of GSK3 will be an attractive new approach for promoting axon regeneration after CNS injuries.
Potential Role of GSK3 Signaling in Regulation of Axon Growth-Associated Transcription Factors during Development and Regeneration
To date, many transcription factors have been identified for regulating axon growth during development or regeneration, including CREB, nuclear factor of activated T-cells (NFATc), protein 53 (p53), nuclear factor κB (NFκB), c-Jun, Smads, SnoN, E47, inhibitor of DNA-binding 2 (Id2), activating transcription factor 3 (ATF3), signal transducer and activator of transcription 3 (STAT3), SRF, Sox11, and Kruppel-like factors (KLFs). Many of these proteins are well-established GSK3 substrates, including CREB, NFATc, p53, NFκB, c-Jun, and Smads (see discussion below). Although some have not been identified directly as GSK3 substrates, including SRF, Sox11, Id2, and KLFs (Lasorella et al., 2006; Wickramasinghe et al., 2008; Moore et al., 2009; Jing et al., 2011; Lin et al., 2011; Yu et al., 2011), they contain multiple and well-conserved GSK3 sites (Taelman et al., 2010). In this section, we summarize the functions of these proteins in regulation of axon growth and discuss how they are regulated by the GSK3 signaling. It should be noted that the discussion below about how GSK3 signaling controls the function of these transcription factors is based mainly on studies in non-neuronal cells and that additional studies are required to determine if similar regulation occurs in neurons during axon growth.
Cyclic AMP Response Element-Binding Protein
Role of CREB in Axon Growth and Regeneration
During development, several transcription factors have been shown to control axon growth downstream of neurotrophin signaling, including CREB (Lonze et al., 2002), NFATc (Graef et al., 2003), SRF (Wickramasinghe et al., 2008), and Sox11 (Lin et al., 2011), among which CREB and NFATc are well-established GSK3 substrates. Upon neurotrophin stimulation, CREB is activated via Serine-133 phosphorylation downstream of the MAPK pathway (Bonni et al., 1995; Finkbeiner et al., 1997; Arthur et al., 2004). Inhibition of CREB activity blocks neurotrophin-mediated sympathetic neuron survival (Riccio et al., 1999). Thus, CREB knockout mice die shortly after birth with substantial apoptosis of PNS neurons (Lonze et al., 2002). In vitro, CREB null sensory and sympathetic neurons grew shorter axons in response to NGF than did those of wild-type mice when cell apoptosis was blocked by the caspase inhibitor (Lonze et al., 2002), showing that CREB-mediated gene transcription is required for neurotrophin-induced axon growth. However, in vivo data of CREB knockout mice showed that axon growth was impaired at an earlier developmental stage when axon growth was neurotrophin-independent. In another study of adult mice, overexpression of an active form of CREB in DRG neurons markedly enhanced the regeneration of the dorsal column axons after a spinal cord injury (Gao et al., 2004). These studies indicate that CREB-mediated gene expression is both necessary and sufficient for supporting axon growth.
Regulation of CREB by GSK3
The cAMP–PKA pathway activates CREB by phosphorylation of its serine-133 residue, which in turn acting as a priming site for the phosphorylation of GSK3 at serine-129. Although one study showed that phosphorylation of CREB by GSK3 at serine-129 enhanced its transcriptional activity (Fiol et al., 1994), two later studies showed that GSK3-mediated phosphorylation of CREB reduced its DNA-binding activity (Bullock and Habener, 1998; Grimes and Jope, 2001). This conclusion is consistent with the fact the growth factor-induced CREB activation is correlated with GSK3 inactivation. Moreover, the inhibitory effect of GSK3 on CREB-DNA binding was reversed by treatment with multiple GSK3 inhibitors (Grimes and Jope, 2001). Furthermore, another study found that inhibition of GSK3 in quiescent cells alone can directly induce the expression of multiple genes containing the CREB binding site (Tullai et al., 2011), providing functional evidence that GSK3 plays a negative role in regulation of CREB activity.
Nuclear Factor of Activated T-Cells
Role of NFATc in Axon Growth and Regeneration
In addition to CREB, neurotrophins also control gene expression via the transcription factor NFATc family proteins (NFATc1–4), which are activated by protein phosphatase calcineurin downstream of calcium signaling (Hogan et al., 2003). In triple NFATc knockout mice, sensory neurons show severe defects in axon growth at embryonic day 10.5 (Graef et al., 2003). In vitro cultured DRG explants from the mutant mice also have impaired axon growth in response to NGF. However, these mutant neurons extend axons normally in response to extracellular signals other than neurotrophins, indicating that NFATc specifically mediates neurotrophin-induced axon growth. Interestingly, a recent study showed that the calcium–calcineurin pathway is also required locally at the axon to mediate neurotrophin-induced axon growth by controlling neurotrophin receptor endocytosis independent of transcription (Bodmer et al., 2011). This result indicates that neurotrophins induce axon growth through the calcium–calcineurin pathway by coordinating gene transcription in the neuronal soma with local regulatory signaling in the axon.
Regulation of NFATc by GSK3
Calcineurin activates NFATc via dephosphorylating multiple serine residues in their regulatory subunits, which are located on the N-terminal to the DNA-binding domains. NFATc remains inactive in the cytoplasm, and dephosphorylation by calcineurin exposes a nuclear localization sequence while masking a nuclear export sequence, thus leading to translocation of NFATc into the nucleus and the subsequent transcriptional activation (Hogan et al., 2003). Conversely, several kinases can phosphorylate NFATc and promote their export from the nucleus. GSK3 is a key export kinase that functions to rephosphorylate NFATc and thus turns off the signaling (Beals et al., 1997). In hippocampal neurons, activity-induced NFATc translocation from cytoplasm to nucleus (and the subsequent gene transcription) is antagonized by the GSK3-mediated phosphorylation of NFATc (Graef et al., 1999). One study has shown that, in addition to negatively regulating subcellular localization, GSK3-mediated phosphorylation also negatively regulates the intrinsic DNA-binding activity of NFATc (Neal and Clipstone, 2001). Phosphorylation of NFATc by GSK3 requires prior phosphorylation by priming kinases, including PKA (Sheridan et al., 2002) and DYRK1/2 (Gwack et al., 2006), providing another level of modulation of NFATc function via GSK3.
SMADs
Role of Smad Proteins in Axon Growth and Regeneration
Smad proteins are transcription factors activated downstream of the TGFβ or the bone morphogenetic protein (BMP) signaling pathways. Different Smad isoforms mediate gene transcription downstream of different ligands, with Smad1, -5, and -8 acting downstream of BMPs and Smad2 and -3 acting downstream of the TGFβ (Schmierer and Hill, 2007). A recent study showed that Smad1 was up-regulated and activated in DRG neurons upon peripheral axotomy and that activation of Smad1 was necessary for the regenerative axon growth of DRG neurons in vitro (Zou et al., 2009). A later study using neurons from Smad1 conditional knockout mice showed that Smad1 was also required for axon growth from developing DRG neurons in vitro (Parikh et al., 2011). In addition, activation of the BMP4 signaling in DRG neurons in vivo was sufficient to promote dorsal column axon regeneration after the spinal cord injury. Although the in vivo role of Smad1 was not directly tested in these two studies, these results suggest that Smad1 is a key transcription factor controlling axon growth during both development and regeneration. In contrast to Smad1, Smad2 was shown to limit axon growth of developing cerebellar granule neurons (CGNs; Stegmuller et al., 2008). Interestingly, the negative effect of Smad2 on axon growth does not seem to depend on its transcriptional activity. Instead, Smad2 was shown to inhibit axon growth by facilitating protein degradation of another transcription factor SnoN, which functions to promote axon growth, together with the E3 ubiquitin ligase Cdh1–APC complex. Whether Smad2 also functions to limit axon regeneration in mature neurons is not known. However, knocking down Smad2 overrode the inhibitory effect of myelin on axon growth, suggesting Smad2 as a potential therapeutic target for enhancing axon regeneration (Stegmuller et al., 2008).
Regulation of Smad Proteins by GSK3
Smad1 is a well-studied GSK3 substrate. When primed with MAPK or CDK8/9, GSK3 can phosphorylate Smad1 at multiple sites in its linker region (Fuentealba et al., 2007; Aragon et al., 2011). Phosphorylation of Smad1 promotes its interaction with HECT-domain ubiquitin ligase Smurf1 and the subsequent degradation of Smad1 (Sapkota et al., 2007). Studies have shown that Smad2 and Smad3 can be phosphorylated by GSK3, which also leads to their degradation via UPS (Guo et al., 2008; Aragon et al., 2011). Unlike Smad1, degradation of Smad2/3 is mediated through a different E3 ligase, Nedd4L (Gao et al., 2009; Aragon et al., 2011). Because Smad1 and Smad2/3 are negatively regulated by GSK3-mediated protein degradation but play opposing roles in regulation of axon growth, it is unclear how modulation of GSK3 signaling can control axon growth through Smad proteins. To solve this problem, studies are needed to directly examine how GSK3 signaling regulates Smad1 or Smad2/3 in neurons during axon growth.
c-Jun
Role of C-Jun in Axon Growth and Regeneration
c-Jun is one of the early identified genes that are drastically up-regulated in DRG neurons upon axotomy (Jenkins and Hunt, 1991). Importantly, substantial up-regulation of c-Jun in DRG neurons is induced only upon peripheral axotomy, which results in successful axon regeneration but not upon central axotomy, which fails to produce regeneration (Broude et al., 1997). These results suggest that c-Jun functions to regulate axon regeneration. When c-Jun was knocked out in neural progenitors via Nestin-Cre-mediated recombination (Raivich et al., 2004), the mice developed normally with no obvious morphological or behavioral defects in the nervous system, indicating that c-Jun is not required for developmental axon growth. However, when axon regeneration was assessed in these mutant mice, regeneration of facial nerve after the injury was markedly reduced. In addition, expression of several known axon RAGs was impaired in the mutant mice upon the nerve injury, such as galanin and alpha7beta1 integrin. These results support the notion that c-Jun specifically regulates axon regeneration via controlling the expression of axon RAGs. In support, a recent study showed that knocking down c-Jun specifically in adult DRG neurons via in vivo gene electroporation blocked sciatic nerve axon regeneration, confirming a specific role of c-Jun in the regulation of axon regeneration (Saijilafu et al., 2011).
Regulation of c-Jun by GSK3
Several early studies (Boyle et al., 1991; de Groot et al., 1993; Nikolakaki et al., 1993) have shown that c-Jun and two other members of the Jun family, JunB and JunD, are all substrates of GSK3. Phosphorylation of c-Jun at sites near its DNA-binding domain reduces its DNA-binding activity and the subsequent transcription activation. Similar negative regulation of c-Jun function by GSK3 is conserved for the drosophila GSK3 homolog shaggy (de Groot et al., 1993). In addition to regulating the DNA-binding activity of c-Jun, GSK3-mediated phosphorylation of c-Jun also promotes its protein degradation through UPS (Wei et al., 2005). Lastly, there is evidence that GSK3 also negatively regulates the activity of c-Jun N-terminal kinase (JNK) downstream of the growth factor signaling (Liu et al., 2004). Together, these studies suggest that GSK3 is a major regulator of c-Jun function by controlling its protein stability, DNA-binding, and activation.
p53
Role of p53 in Axon Growth and Regeneration
p53 is well-known for its role as a tumor suppressor that functions to promote apoptosis or cell cycle arrest (Levine and Oren, 2009). In neurons, p53 has been shown to mediate cell death of sympathetic neurons upon neurotrophin withdrawn (Aloyz et al., 1998). Recent studies have revealed an important role of p53 in regulation of axon growth (Di Giovanni et al., 2006; Qin et al., 2009; Tedeschi et al., 2009). Overexpression of a dominant negative mutant of p53 (p53DN) blocks axon growth from PC12 cells and primary cultured rat cortical neurons (Di Giovanni et al., 2006). Importantly, expression of p53DN also leads to reduced expression of two axon growth-associated genes, the actin binding protein coronin1b and the GTPase Rab13, suggesting that p53 controls axon growth by controlling the expression of axon growth-associated genes. A later study has identified GAP43, which is one of the best-recognized genes associated with axon growth, as a transcriptional target of p53 (Tedeschi et al., 2009). The level of GAP43 mRNA is reduced by 60% in p53 knockout mice compared with that in wild-type mice (Tedeschi et al., 2009). As a result, axon growth of embryonic cortical neurons from the p53 null mice in vitro is substantially impaired, providing definitive evidence that p53 is required for axon growth in vitro. Whether p53 knockout mice have axon growth defects in vivo during development was not examined in that study. However, in a facial nerve axotomy model, axon regeneration of facial motor neurons was markedly reduced in comparison to that in wild-type mice, indicating that p53 function is necessary for axon regeneration in vivo. In addition to regulation of axon growth as a transcription factor, p53 has been shown to act locally at the growth cone to control growth cone motility (Qin et al., 2009). Inhibition of p53 by inhibitors, dominant negative mutants, or siRNA leads to growth cone collapse, whereas overexpression of p53 increases the growth cone size. Such an unexpected role of p53 in the growth cone was achieved by antagonizing the function of Rho kinase, a well-known negative regulator of growth cone motility. These results suggest that p53 can control axon growth via coordinating gene expression with local regulation of growth cone motility.
Regulation of p53 by GSK3
The regulation of p53 by GSK3 is mostly studied in the context of p53-induced apoptosis and cell cycle arrest in response to stress signals. However, the results are complex, with different studies showing opposing effects. Several studies have shown that GSK3 negatively regulated p53 protein levels by phosphorylation of p53, which then promoted its protein degradation via UPS (Mao et al., 2001; Qu et al., 2004; Pluquet et al., 2005; Baltzis et al., 2007; Boehme et al., 2008). In contrast, other studies have shown that GSK3 could positively regulate p53 function. One study indicated that phosphorylation of p53 by GSK3 at a different site enhanced the transcriptional activity of p53 (Turenne and Price, 2001). Two other studies found that GSK3 interacted with p53 and promoted its activity independent of phosphorylation (Watcharasit et al., 2002, 2003). Another study showed that GSK3 bound and facilitated p53 acetylation at lysine-373 and lysine-382 (Eom and Jope, 2009). Although acetylation of p53 at these two sites promotes its transcriptional activity (Olsson et al., 2007), it does not play a role in regulation of axon growth in neurons (Di Giovanni et al., 2006). Instead, acetylation of p53 at a different site, lysine-320, is specifically involved in controlling axon growth (Di Giovanni et al., 2006). These studies suggest that the complex roles of GSK3 in regulation of p53 function depend on its specific post-translational modification of p53.
NFκB
Role of NFκB in Axon Growth and Regeneration
Nuclear factor κB is a transcription factor that usually forms a dimmer with two of the five subunits, including p65, c-Rel, RelB, p50, and p52. NFκB function is negatively regulated by the inhibitor of κB (IκB) proteins, which binds to NFκB and prevents it from entering the nucleus (Chen and Greene, 2004). In PC12 cells, NGF can activate NFκB through TrkA activation, and blocking such TrkA-induced NFκB activation inhibits neurite outgrowth (Foehr et al., 2000). In primary cultured developing nodose sensory neurons, inhibition of NFκB also impairs BDNF-induced neurite outgrowth (Gutierrez et al., 2005). Importantly, this regulatory effect of NFκB on neurite outgrowth was observed only during a restricted period of development, suggesting that neurons use different transcription factors to control neurite outgrowth during different stages of development. Inhibition of NFκB activity also blocks CNTF-induced neurite outgrowth from developing nodose sensory neurons (Gallagher et al., 2007), indicating that different signaling pathways can converge on NFκB to promote neurite outgrowth. However, NFκB has also been shown to inhibit neurite outgrowth from different neuronal types. For instance, in neonatal superior cervical ganglion sympathetic neurons, enhancing NFκB transcriptional activity by multiple approaches markedly inhibits neurite outgrowth (Gutierrez et al., 2008). The molecular mechanisms by which NFκB plays opposing roles in regulation of neurite outgrowth in different neurons is unclear.
Regulation of NFκB by GSK3
Similar to reports of p53, studies have indicated opposing results about how GSK3 regulates NFκB function (Bournat et al., 2000; Hoeflich et al., 2000; Sanchez et al., 2003; Buss et al., 2004; Liang and Chuang, 2006; Tullai et al., 2011). p65 Subunit of NFκB has been shown to be a GSK3 substrate with its serine-468 phosphorylated by GSK3 (Buss et al., 2004). Overexpression of a S468A mutant of p65 in p65-deficient cells has been shown to lead to higher NFκB transcriptional activity compared with wild-type p65, indicating that GSK3-mediated phosphorylation of p65 at serine-468 negatively regulates NFκB function. Moreover, in primary cultured cortical astrocytes, overexpression of an active mutant of GSK3β (S9A) induced cell apoptosis and reduced NFκB activation (Sanchez et al., 2003), indicating that GSK3 activation inhibits NFκB function. In another study, expression of Wnt-1 in PC12 cells enhanced NFκB transcriptional activity and reduced cell apoptosis induced by serum withdrawn (Bournat et al., 2000). Importantly, the effect of Wnt-1 expression on NFκB activation can be mimicked by lithium or a dominant negative mutant of GSK3β. Taken together, these studies show that GSK3 acts to inhibit NFκB activity. In consistent with its negative role in regulation of NFκB activity, inhibition of GSK3 alone in the absence of other extracellular signals has been shown to be sufficient to activate NFκB in both neurons and non-neuronal cells (Liang and Chuang, 2006; Tullai et al., 2011). However, when GSK3β was knocked out, the mutant mice died prenatally primarily because of failure of the liver function caused by reduced NFκB activity (Hoeflich et al., 2000). Although the molecular mechanism remains unknown, this study suggests that GSK3β facilitates NFκB function and it cannot be compensated by GSK3α.
In summary, most of these transcription factors are direct GSK3 substrates. By phosphorylation of these axon growth-associated transcription factors, GSK3 plays primarily negative roles in the regulation of their function through controlling their DNA-binding activities, nuclear localization, and/or protein stability.
Concluding Remarks and Future Directions
It is well recognized that axon growth requires the coordinated regulation of gene expression in the soma and axonal assembly at the nerve growth cone. How such coordination is achieved is one of the major unanswered questions in the axon growth field. GSK3 has been shown to be a key signaling molecule that controls axonal assembly at the growth cone via regulation of multiple microtubule-binding proteins (Kim et al., 2011). Remarkably, many axon growth-associated transcription factors are also regulated by GSK3. Therefore, GSK3 could be a potential candidate for the coordinated regulation of gene expression in the soma via transcription factors and local axon assembly at the growth cone via microtubule-binding proteins (Figure 1). Compared with the regulation of microtubule-binding proteins by GSK3 signaling, we know very little about how GSK3 regulates these transcription factors in neurons. Therefore, additional studies are needed to determine if modulation of GSK3 activity in neurons regulates these transcription factors in a manner similar to that in non-neuronal cells, and whether such regulation affects axon growth. In addition to the transcription factors we have discussed, there are many other axon growth-associated transcription factors that are potential GSK3 substrates, including KLFs, SRF, Sox11, and Id2, all of which contain multiple conserved GSK3 consensus sites (Taelman et al., 2010). It will be interesting to determine if their function can be modulated by GSK3 signaling during axon growth. GSK3 could play permissive or instructive roles in the regulation of gene expression during axon growth. As discussed, direct inhibition of GSK3 alone in the absence of extracellular signals can activate several transcription factors in both non-neuronal cells and neurons (Liang and Chuang, 2006; Tullai et al., 2011), supporting the premise that GSK3 inactivation plays an instructive role in directing gene expression. It will be important to investigate if such GSK3 inactivation-induced gene expression in neurons can support axon growth.
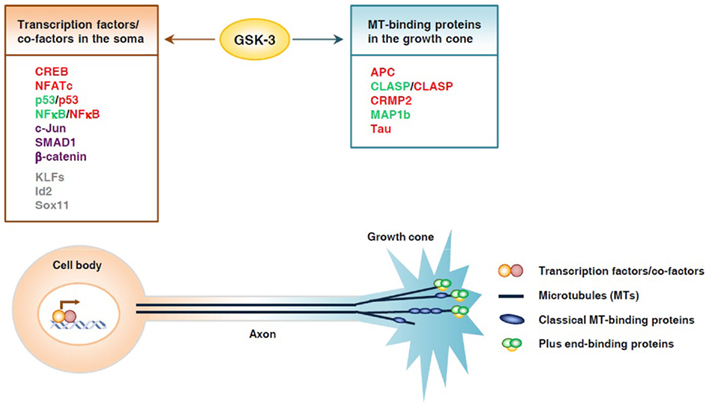
Figure 1. Glycogen synthase kinase 3 controls axon growth by coordinating gene transcription and local microtubule dynamics in the growth cone. GSK3 phosphorylates a number of transcription factors and thereby controls their function and subsequent gene expression, which may play an important part in determining the intrinsic axon growth capacity. GSK3 is also known to phosphorylate several microtubule-binding proteins, which control distinct aspects of microtubule dynamics and assembly, and thereby controls local axon assembly at the axonal growth cone. The final outcome of axon regeneration is determined by the coordination between gene expression and local cytoskeletal assembly at the growth cone. Substrates whose activities are inhibited by GSK3 are depicted in red, those that are activated by GSK3 are depicted in green. Some substrates, such as p53 and NFκB, can be either activated or inhibited upon GSK3 phosphorylation, depending on the context. Substrates that undergo proteasomal degradation when phosphorylated by GSK3 are depicted in purple. Putative substrates are shown in gray.
The loss of axon regeneration ability in adult CNS neurons is due in part to the result of reduced intrinsic axon growth capacity, which is regulated primarily by gene transcription and protein translation (Liu et al., 2011). Therefore, direct manipulation of axon growth-associated transcription factors or their upstream regulators is emerging as an effective approach for promoting axon regeneration. For instance, a recent study showed that activation of the BMP–Smad1 signaling markedly enhanced axon regeneration after the spinal cord injury (Parikh et al., 2011). Because GSK3 signaling is involved in the regulation of many transcription factors associated with axon growth, manipulation of GSK3 signaling may be a potential approach for promoting axon regeneration via enhancing the intrinsic axon growth ability by activating multiple transcription factors at the same time. Indeed, genetic deletion of Pten in adult neurons has been shown to greatly improve axon regeneration after an optic nerve crush or spinal cord injury (Park et al., 2008; Liu et al., 2010). The effect of Pten deletion is partially mediated by the mTor-mediated proteins translation. Because Pten deletion leads to GSK3 inactivation, it is very likely that GSK3-regulated gene transcription is also involved. In consistent, systemic inhibition of GSK3 activity with pharmacological inhibitors has been shown to promote corticospinal axon regeneration in vivo (Dill et al., 2008), which requires increased intrinsic axon growth ability. It is possible that the promoting effect of GSK3 inhibitors on axon regeneration in vivo is not neuron cell autonomous. Therefore, future genetic studies, in which GSK3 is specifically manipulated in neurons, are necessary to determine if inhibition of GSK3 in vivo is able to enhance the intrinsic axon growth ability of mature neurons and promotes CNS axon regeneration.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors were supported by grants from NIH (R01NS064288) and The Craig H. Neilsen Foundation (to Feng-Quan Zhou). We thank Elanie Henze for reading and editing the manuscript. We apologize to authors whose work we were unable to include because of space constraints.
References
Aloyz, R. S., Bamji, S. X., Pozniak, C. D., Toma, J. G., Atwal, J., Kaplan, D. R., and Miller, F. D. (1998). p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell Biol. 143, 1691–1703.
Aragon, E., Goerner, N., Zaromytidou, A. I., Xi, Q., Escobedo, A., Massague, J., and Macias, M. J. (2011). A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 25, 1275–1288.
Arthur, J. S., Fong, A. L., Dwyer, J. M., Davare, M., Reese, E., Obrietan, K., and Impey, S. (2004). Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J. Neurosci. 24, 4324–4332.
Atwal, J. K., Massie, B., Miller, F. D., and Kaplan, D. R. (2000). The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron 27, 265–277.
Baltzis, D., Pluquet, O., Papadakis, A. I., Kazemi, S., Qu, L. K., and Koromilas, A. E. (2007). The eIF2alpha kinases PERK and PKR activate glycogen synthase kinase 3 to promote the proteasomal degradation of p53. J. Biol. Chem. 282, 31675–31687.
Bareyre, F. M., Garzorz, N., Lang, C., Misgeld, T., Buning, H., and Kerschensteiner, M. (2011). In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 6282–6287.
Beals, C. R., Sheridan, C. M., Turck, C. W., Gardner, P., and Crabtree, G. R. (1997). Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 275, 1930–1934.
Bodmer, D., Ascano, M., and Kuruvilla, R. (2011). Isoform-specific dephosphorylation of dynamin1 by calcineurin couples neurotrophin receptor endocytosis to axonal growth. Neuron 70, 1085–1099.
Boehme, K. A., Kulikov, R., and Blattner, C. (2008). p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc. Natl. Acad. Sci. U.S.A. 105, 7785–7790.
Bonni, A., Ginty, D. D., Dudek, H., and Greenberg, M. E. (1995). Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol. Cell. Neurosci. 6, 168–183.
Bournat, J. C., Brown, A. M., and Soler, A. P. (2000). Wnt-1 dependent activation of the survival factor NF-kappaB in PC12 cells. J. Neurosci. Res. 61, 21–32.
Boyle, W. J., Smeal, T., Defize, L. H., Angel, P., Woodgett, J. R., Karin, M., and Hunter, T. (1991). Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell 64, 573–584.
Broude, E., Mcatee, M., Kelley, M. S., and Bregman, B. S. (1997). c-Jun expression in adult rat dorsal root ganglion neurons: differential response after central or peripheral axotomy. Exp. Neurol. 148, 367–377.
Bullock, B. P., and Habener, J. F. (1998). Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase A and glycogen synthase kinase-3 alters DNA-binding affinity, conformation, and increases net charge. Biochemistry 37, 3795–3809.
Buss, H., Dorrie, A., Schmitz, M. L., Frank, R., Livingstone, M., Resch, K., and Kracht, M. (2004). Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J. Biol. Chem. 279, 49571–49574.
Campbell, D. S., and Holt, C. E. (2003). Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron 37, 939–952.
Campenot, R. B. (1977). Local control of neurite development by nerve growth factor. Proc. Natl. Acad. Sci. U.S.A. 74, 4516–4519.
Campenot, R. B. (1982a). Development of sympathetic neurons in compartmentalized cultures. II. Local control of neurite survival by nerve growth factor. Dev. Biol. 93, 13–21.
Campenot, R. B. (1982b). Development of sympathetic neurons in compartmentalized cultures. Il Local control of neurite growth by nerve growth factor. Dev. Biol. 93, 1–12.
Chen, L. F., and Greene, W. C. (2004). Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5, 392–401.
Cheng, P. L., Song, A. H., Wong, Y. H., Wang, S., Zhang, X., and Poo, M. M. (2011). Self-amplifying autocrine actions of BDNF in axon development. Proc. Natl. Acad. Sci. U.S.A. 108, 18430–18435.
Cole, A. R., Knebel, A., Morrice, N. A., Robertson, L. A., Irving, A. J., Connolly, C. N., and Sutherland, C. (2004). GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 279, 50176–50180.
de Groot, R. P., Auwerx, J., Bourouis, M., and Sassone-Corsi, P. (1993). Negative regulation of Jun/AP-1: conserved function of glycogen synthase kinase 3 and the Drosophila kinase shaggy. Oncogene 8, 841–847.
Di Giovanni, S., Knights, C. D., Rao, M., Yakovlev, A., Beers, J., Catania, J., Avantaggiati, M. L., and Faden, A. I. (2006). The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 25, 4084–4096.
Dill, J., Wang, H., Zhou, F., and Li, S. (2008). Inactivation of glycogen synthase kinase 3 promotes axonal growth and recovery in the CNS. J. Neurosci. 28, 8914–8928.
Eickholt, B. J., Ahmed, A. I., Davies, M., Papakonstanti, E. A., Pearce, W., Starkey, M. L., Bilancio, A., Need, A. C., Smith, A. J., Hall, S. M., Hamers, F. P., Giese, K. P., Bradbury, E. J., and Vanhaesebroeck, B. (2007). Control of axonal growth and regeneration of sensory neurons by the p110delta PI 3-kinase. PLoS ONE 2, e869. doi: 10.1371/journal.pone.0000869
Eom, T. Y., and Jope, R. S. (2009). GSK3 beta N-terminus binding to p53 promotes its acetylation. Mol. Cancer 8, 14.
Finkbeiner, S., Tavazoie, S. F., Maloratsky, A., Jacobs, K. M., Harris, K. M., and Greenberg, M. E. (1997). CREB: a major mediator of neuronal neurotrophin responses. Neuron 19, 1031–1047.
Fiol, C. J., Williams, J. S., Chou, C. H., Wang, Q. M., Roach, P. J., and Andrisani, O. M. (1994). A secondary phosphorylation of CREB341 at Ser129 is required for the cAMP-mediated control of gene expression. A role for glycogen synthase kinase-3 in the control of gene expression. J. Biol. Chem. 269, 32187–32193.
Foehr, E. D., Lin, X., O’Mahony, A., Geleziunas, R., Bradshaw, R. A., and Greene, W. C. (2000). NF-kappa B signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J. Neurosci. 20, 7556–7563.
Fuentealba, L. C., Eivers, E., Ikeda, A., Hurtado, C., Kuroda, H., Pera, E. M., and De Robertis, E. M. (2007). Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131, 980–993.
Gallagher, D., Gutierrez, H., Gavalda, N., O’Keeffe, G., Hay, R., and Davies, A. M. (2007). Nuclear factor-kappaB activation via tyrosine phosphorylation of inhibitor kappaB-alpha is crucial for ciliary neurotrophic factor-promoted neurite growth from developing neurons. J. Neurosci. 27, 9664–9669.
Gallo, G., and Letourneau, P. C. (1998). Localized sources of neurotrophins initiate axon collateral sprouting. J. Neurosci. 18, 5403–5414.
Gao, S., Alarcon, C., Sapkota, G., Rahman, S., Chen, P. Y., Goerner, N., Macias, M. J., Erdjument-Bromage, H., Tempst, P., and Massague, J. (2009). Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell 36, 457–468.
Gao, Y., Deng, K., Hou, J., Bryson, J. B., Barco, A., Nikulina, E., Spencer, T., Mellado, W., Kandel, E. R., and Filbin, M. T. (2004). Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44, 609–621.
Goldberg, J. L., Espinosa, J. S., Xu, Y., Davidson, N., Kovacs, G. T., and Barres, B. A. (2002). Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron 33, 689–702.
Graef, I. A., Mermelstein, P. G., Stankunas, K., Neilson, J. R., Deisseroth, K., Tsien, R. W., and Crabtree, G. R. (1999). L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401, 703–708.
Graef, I. A., Wang, F., Charron, F., Chen, L., Neilson, J., Tessier-Lavigne, M., and Crabtree, G. R. (2003). Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell 113, 657–670.
Grimes, C. A., and Jope, R. S. (2001). CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J. Neurochem. 78, 1219–1232.
Guo, X., Ramirez, A., Waddell, D. S., Li, Z., Liu, X., and Wang, X. F. (2008). Axin and GSK3-control Smad3 protein stability and modulate TGF-signaling. Genes Dev. 22, 106–120.
Gutierrez, H., Hale, V. A., Dolcet, X., and Davies, A. (2005). NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. Development 132, 1713–1726.
Gutierrez, H., O’Keeffe, G. W., Gavalda, N., Gallagher, D., and Davies, A. M. (2008). Nuclear factor kappa B signaling either stimulates or inhibits neurite growth depending on the phosphorylation status of p65/RelA. J. Neurosci. 28, 8246–8256.
Gwack, Y., Sharma, S., Nardone, J., Tanasa, B., Iuga, A., Srikanth, S., Okamura, H., Bolton, D., Feske, S., Hogan, P. G., and Rao, A. (2006). A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 441, 646–650.
Hanger, D. P., Hughes, K., Woodgett, J. R., Brion, J. P., and Anderton, B. H. (1992). Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147, 58–62.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90.
Hogan, P. G., Chen, L., Nardone, J., and Rao, A. (2003). Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232.
Hur, E. M., Saijilafu, Lee, B. D., Kim, S. J., Xu, W. L., and Zhou, F. Q. (2011). GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev. 25, 1968–1981.
Hur, E. M., and Zhou, F. Q. (2010). GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551.
Jenkins, R., and Hunt, S. P. (1991). Long-term increase in the levels of c-Jun mRNA and Jun protein-like immunoreactivity in motor and sensory neurons following axon damage. Neurosci. Lett. 129, 107–110.
Jing, X., Wang, T., Huang, S., Glorioso, J. C., and Albers, K. M. (2011). The transcription factor Sox11 promotes nerve regeneration through activation of the regeneration-associated gene Sprr1a. Exp. Neurol. 233, 221–232.
Kim, N. G., Xu, C., and Gumbiner, B. M. (2009). Identification of targets of the Wnt pathway destruction complex in addition to beta-catenin. Proc. Natl. Acad. Sci. U.S.A. 106, 5165–5170.
Kim, Y. T., Hur, E. M., Snider, W. D., and Zhou, F. Q. (2011). Role of GSK3 signaling in neuronal morphogenesis. Front. Mol. Neurosci. 4:48. doi: 10.3389/fnmol.2011.00048
Kumar, P., Lyle, K. S., Gierke, S., Matov, A., Danuser, G., and Wittmann, T. (2009). GSK3beta phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J. Cell Biol. 184, 895–908.
Kuruvilla, R., Zweifel, L. S., Glebova, N. O., Lonze, B. E., Valdez, G., Ye, H., and Ginty, D. D. (2004). A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell 118, 243–255.
Labelle, C., and Leclerc, N. (2000). Exogenous BDNF, NT-3 and NT-4 differentially regulate neurite outgrowth in cultured hippocampal neurons. Brain Res. Dev. Brain Res. 123, 1–11.
Lasorella, A., Stegmuller, J., Guardavaccaro, D., Liu, G., Carro, M. S., Rothschild, G., De La Torre-Ubieta, L., Pagano, M., Bonni, A., and Iavarone, A. (2006). Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 442, 471–474.
Leibinger, M., Muller, A., Andreadaki, A., Hauk, T. G., Kirsch, M., and Fischer, D. (2009). Neuroprotective and axon growth-promoting effects following inflammatory stimulation on mature retinal ganglion cells in mice depend on ciliary neurotrophic factor and leukemia inhibitory factor. J. Neurosci. 29, 14334–14341.
Lentz, S. I., Knudson, C. M., Korsmeyer, S. J., and Snider, W. D. (1999). Neurotrophins support the development of diverse sensory axon morphologies. J. Neurosci. 19, 1038–1048.
Levine, A. J., and Oren, M. (2009). The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer 9, 749–758.
Liang, M. H., and Chuang, D. M. (2006). Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J. Biol. Chem. 281, 30479–30484.
Lin, L., Lee, V. M., Wang, Y., Lin, J. S., Sock, E., Wegner, M., and Lei, L. (2011). Sox11 regulates survival and axonal growth of embryonic sensory neurons. Dev. Dyn. 240, 52–64.
Liu, K., Lu, Y., Lee, J. K., Samara, R., Willenberg, R., Sears-Kraxberger, I., Tedeschi, A., Park, K. K., Jin, D., Cai, B., Xu, B., Connolly, L., Steward, O., Zheng, B., and He, Z. (2010). PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 13, 1075–1081.
Liu, K., Tedeschi, A., Park, K. K., and He, Z. (2011). Neuronal intrinsic mechanisms of axon regeneration. Annu. Rev. Neurosci. 34, 131–152.
Liu, R. Y., and Snider, W. D. (2001). Different signaling pathways mediate regenerative versus developmental sensory axon growth. J. Neurosci. 21, RC164.
Liu, S., Yu, S., Hasegawa, Y., Lapushin, R., Xu, H. J., Woodgett, J. R., Mills, G. B., and Fang, X. (2004). Glycogen synthase kinase 3beta is a negative regulator of growth factor-induced activation of the c-Jun N-terminal kinase. J. Biol. Chem. 279, 51075–51081.
Lonze, B. E., Riccio, A., Cohen, S., and Ginty, D. D. (2002). Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 34, 371–385.
Lowery, L. A., and Van Vactor, D. (2009). The trip of the tip: understanding the growth cone machinery. Nat. Rev. Mol. Cell Biol. 10, 332–343.
Lu, W., Yamamoto, V., Ortega, B., and Baltimore, D. (2004). Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell 119, 97–108.
Mao, C. D., Hoang, P., and Dicorleto, P. E. (2001). Lithium inhibits cell cycle progression and induces stabilization of p53 in bovine aortic endothelial cells. J. Biol. Chem. 276, 26180–26188.
Markus, A., Zhong, J., and Snider, W. D. (2002). Raf and akt mediate distinct aspects of sensory axon growth. Neuron 35, 65–76.
McManus, E. J., Sakamoto, K., Armit, L. J., Ronaldson, L., Shpiro, N., Marquez, R., and Alessi, D. R. (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 24, 1571–1583.
Ming, G., Song, H., Berninger, B., Inagaki, N., Tessier-Lavigne, M., and Poo, M. (1999). Phospholipase C-gamma and phosphoinositide 3-kinase mediate cytoplasmic signaling in nerve growth cone guidance. Neuron 23, 139–148.
Moore, D. L., Blackmore, M. G., Hu, Y., Kaestner, K. H., Bixby, J. L., Lemmon, V. P., and Goldberg, J. L. (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301.
Moreno, F. J., and Avila, J. (1998). Phosphorylation of stathmin modulates its function as a microtubule depolymerizing factor. Mol. Cell. Biochem. 183, 201–209.
Muller, A., Hauk, T. G., and Fischer, D. (2007). Astrocyte-derived CNTF switches mature RGCs to a regenerative state following inflammatory stimulation. Brain 130, 3308–3320.
Neal, J. W., and Clipstone, N. A. (2001). Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J. Biol. Chem. 276, 3666–3673.
Neumann, S., and Woolf, C. J. (1999). Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23, 83–91.
Nikolakaki, E., Coffer, P. J., Hemelsoet, R., Woodgett, J. R., and Defize, L. H. (1993). Glycogen synthase kinase 3 phosphorylates Jun family members in vitro and negatively regulates their transactivating potential in intact cells. Oncogene 8, 833–840.
O’Connor, R., and Tessier-Lavigne, M. (1999). Identification of maxillary factor, a maxillary process-derived chemoattractant for developing trigeminal sensory axons. Neuron 24, 165–178.
Olsson, A., Manzl, C., Strasser, A., and Villunger, A. (2007). How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 14, 1561–1575.
Ozdinler, P. H., and Macklis, J. D. (2006). IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat. Neurosci. 9, 1371–1381.
Parikh, P., Hao, Y., Hosseinkhani, M., Patil, S. B., Huntley, G. W., Tessier-Lavigne, M., and Zou, H. (2011). Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc. Natl. Acad. Sci. U.S.A. 108, E99–E107.
Park, K. K., Liu, K., Hu, Y., Smith, P. D., Wang, C., Cai, B., Xu, B., Connolly, L., Kramvis, I., Sahin, M., and He, Z. (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963–966.
Patel, T. D., Jackman, A., Rice, F. L., Kucera, J., and Snider, W. D. (2000). Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron 25, 345–357.
Patel, T. D., Kramer, I., Kucera, J., Niederkofler, V., Jessell, T. M., Arber, S., and Snider, W. D. (2003). Peripheral NT3 signaling is required for ETS protein expression and central patterning of proprioceptive sensory afferents. Neuron 38, 403–416.
Piao, S., Lee, S. H., Kim, H., Yum, S., Stamos, J. L., Xu, Y., Lee, S. J., Lee, J., Oh, S., Han, J. K., Park, B. J., Weis, W. I., and Ha, N. C. (2008). Direct inhibition of GSK3beta by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta-catenin signaling. PLoS ONE 3, e4046. doi: 10.1371/journal.pone.0004046
Pluquet, O., Qu, L. K., Baltzis, D., and Koromilas, A. E. (2005). Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3beta. Mol. Cell. Biol. 25, 9392–9405.
Qin, Q., Baudry, M., Liao, G., Noniyev, A., Galeano, J., and Bi, X. (2009). A novel function for p53: regulation of growth cone motility through interaction with Rho kinase. J. Neurosci. 29, 5183–5192.
Qiu, J., Cafferty, W. B., Mcmahon, S. B., and Thompson, S. W. (2005). Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. J. Neurosci. 25, 1645–1653.
Qu, L., Huang, S., Baltzis, D., Rivas-Estilla, A. M., Pluquet, O., Hatzoglou, M., Koumenis, C., Taya, Y., Yoshimura, A., and Koromilas, A. E. (2004). Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 18, 261–277.
Raivich, G., Bohatschek, M., Da Costa, C., Iwata, O., Galiano, M., Hristova, M., Nateri, A. S., Makwana, M., Riera-Sans, L., Wolfer, D. P., Lipp, H. P., Aguzzi, A., Wagner, E. F., and Behrens, A. (2004). The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43, 57–67.
Riccio, A., Ahn, S., Davenport, C. M., Blendy, J. A., and Ginty, D. D. (1999). Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286, 2358–2361.
Rubinfeld, B., Albert, I., Porfiri, E., Fiol, C., Munemitsu, S., and Polakis, P. (1996). Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 272, 1023–1026.
Saijilafu, Hur, E. M., and Zhou, F. Q. (2011). Genetic dissection of axon regeneration via in vivo electroporation of adult mouse sensory neurons. Nat. Commun. 2, 543.
Sanchez, J. F., Sniderhan, L. F., Williamson, A. L., Fan, S., Chakraborty-Sett, S., and Maggirwar, S. B. (2003). Glycogen synthase kinase 3beta-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor kappaB signaling. Mol. Cell. Biol. 23, 4649–4662.
Sapkota, G., Alarcon, C., Spagnoli, F. M., Brivanlou, A. H., and Massague, J. (2007). Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol. Cell 25, 441–454.
Schmierer, B., and Hill, C. S. (2007). TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982.
Sheridan, C. M., Heist, E. K., Beals, C. R., Crabtree, G. R., and Gardner, P. (2002). Protein kinase A negatively modulates the nuclear accumulation of NF-ATc1 by priming for subsequent phosphorylation by glycogen synthase kinase-3. J. Biol. Chem. 277, 48664–48676.
Smith, P. D., Sun, F., Park, K. K., Cai, B., Wang, C., Kuwako, K., Martinez-Carrasco, I., Connolly, L., and He, Z. (2009). SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron 64, 617–623.
Stegmuller, J., Huynh, M. A., Yuan, Z., Konishi, Y., and Bonni, A. (2008). TGFbeta-Smad2 signaling regulates the Cdh1-APC/SnoN pathway of axonal morphogenesis. J. Neurosci. 28, 1961–1969.
Taelman, V. F., Dobrowolski, R., Plouhinec, J. L., Fuentealba, L. C., Vorwald, P. P., Gumper, I., Sabatini, D. D., and De Robertis, E. M. (2010). Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148.
Tedeschi, A., Nguyen, T., Puttagunta, R., Gaub, P., and Di Giovanni, S. (2009). A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ. 16, 543–554.
Trivedi, N., Marsh, P., Goold, R. G., Wood-Kaczmar, A., and Gordon-Weeks, P. R. (2005). Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J. Cell. Sci. 118, 993–1005.
Tullai, J. W., Graham, J. R., and Cooper, G. M. (2011). A GSK-3-mediated transcriptional network maintains repression of immediate early genes in quiescent cells. Cell Cycle 10, 3072–3077.
Turenne, G. A., and Price, B. D. (2001). Glycogen synthase kinase3 beta phosphorylates serine 33 of p53 and activates p53’s transcriptional activity. BMC Cell Biol. 2, 12. doi: 10.1186/1471-2121-2-12
Watcharasit, P., Bijur, G. N., Song, L., Zhu, J., Chen, X., and Jope, R. S. (2003). Glycogen synthase kinase-3beta (GSK3beta) binds to and promotes the actions of p53. J. Biol. Chem. 278, 48872–48879.
Watcharasit, P., Bijur, G. N., Zmijewski, J. W., Song, L., Zmijewska, A., Chen, X., Johnson, G. V., and Jope, R. S. (2002). Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proc. Natl. Acad. Sci. U.S.A. 99, 7951–7955.
Wei, W., Jin, J., Schlisio, S., Harper, J. W., and Kaelin, W. G. Jr. (2005). The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 8, 25–33.
Werner, A., Willem, M., Jones, L. L., Kreutzberg, G. W., Mayer, U., and Raivich, G. (2000). Impaired axonal regeneration in alpha7 integrin-deficient mice. J. Neurosci. 20, 1822–1830.
Wickramasinghe, S. R., Alvania, R. S., Ramanan, N., Wood, J. N., Mandai, K., and Ginty, D. D. (2008). Serum response factor mediates NGF-dependent target innervation by embryonic DRG sensory neurons. Neuron 58, 532–545.
Wu, G., Huang, H., Garcia Abreu, J., and He, X. (2009). Inhibition of GSK3 phosphorylation of beta-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PLoS ONE 4, e4926. doi: 10.1371/journal.pone.0004926
Yin, Y., Cui, Q., Gilbert, H. Y., Yang, Y., Yang, Z., Berlinicke, C., Li, Z., Zaverucha-Do-Valle, C., He, H., Petkova, V., Zack, D. J., and Benowitz, L. I. (2009). Oncomodulin links inflammation to optic nerve regeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 19587–19592.
Yin, Y., Henzl, M. T., Lorber, B., Nakazawa, T., Thomas, T. T., Jiang, F., Langer, R., and Benowitz, L. I. (2006). Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat. Neurosci. 9, 843–852.
Yiu, G., and He, Z. (2006). Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 7, 617–627.
Yu, P., Zhang, Y. P., Shields, L. B., Zheng, Y., Hu, X., Hill, R., Howard, R., Gu, Z., Burke, D. A., Whittemore, S. R., Xu, X. M., and Shields, C. B. (2011). Inhibitor of DNA binding 2 promotes sensory axonal growth after SCI. Exp. Neurol. 231, 38–44.
Zhou, F. Q., and Snider, W. D. (2005). Cell biology. GSK-3beta and microtubule assembly in axons. Science 308, 211–214.
Zhou, F. Q., and Snider, W. D. (2006). Intracellular control of developmental and regenerative axon growth. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1575–1592.
Zhou, F. Q., Walzer, M., Wu, Y. H., Zhou, J., Dedhar, S., and Snider, W. D. (2006). Neurotrophins support regenerative axon assembly over CSPGs by an ECM-integrin-independent mechanism. J. Cell. Sci. 119, 2787–2796.
Zhou, F. Q., Zhou, J., Dedhar, S., Wu, Y. H., and Snider, W. D. (2004). NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron 42, 897–912.
Keywords: axon growth, axon regeneration, transcription factor, glycogen synthase kinase 3
Citation: Liu C-M, Hur E-M and Zhou F-Q (2012) Coordinating gene expression and axon assembly to control axon growth: potential role of GSK3 signaling. Front. Mol. Neurosci. 5:3. doi: 10.3389/fnmol.2012.00003
Received: 16 November 2011; Accepted: 09 January 2012;
Published online: 06 February 2012.
Edited by:
Simone Di Giovanni, University of Tuebingen, GermanyReviewed by:
Mark R. Cookson, National Institutes of Health, USAAlyson Fournier, McGill University, Canada
Copyright: © 2012 Liu, Hur and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Feng-Quan Zhou, Department of Orthopaedic Surgery, Johns Hopkins University School of Medicine, 720 Rutland Avenue, 215 Ross Research Building, Baltimore, MD 21287, USA. e-mail:Znpob3U0QGpobWkuZWR1.