

- 1Department of Neuroscience, Rosalind Franklin University of Medicine and Science, North Chicago, IL, USA
- 2Department of Cellular and Molecular Pharmacology, Rosalind Franklin University of Medicine and Science, North Chicago, IL, USA
In animal models of drug addiction, cocaine exposure has been shown to increase levels of calcium-permeable AMPA receptors (CP-AMPARs) in two brain regions that are critical for motivation and reward—the ventral tegmental area (VTA) and the nucleus accumbens (NAc). This review compares CP-AMPAR plasticity in the two brain regions and addresses its functional significance. In VTA dopamine neurons, cocaine exposure results in synaptic insertion of high conductance CP-AMPARs in exchange for lower conductance calcium-impermeable AMPARs (CI-AMPARs). This plasticity is rapid in onset (hours), GluA2-dependent, and can be observed with a single cocaine injection. Whereas it is short-lived after experimenter-administered cocaine, it persists for months after cocaine self-administration. In addition to strengthening synapses and altering Ca2+ signaling, CP-AMPAR insertion alters subsequent induction of plasticity at VTA synapses. However, CP-AMPAR insertion is unlikely to mediate the increased DA cell activity that occurs during early withdrawal from cocaine exposure. Metabotropic glutamate receptor 1 (mGluR1) exerts a negative influence on CP-AMPAR accumulation in the VTA. Acutely, mGluR1 stimulation elicits a form of LTD resulting from CP-AMPAR removal and CI-AMPAR insertion. In medium spiny neurons (MSNs) of the NAc, extended access cocaine self-administration is required to increase CP-AMPAR levels. This is first detected after approximately a month of withdrawal and then persists. Once present in NAc synapses, CP-AMPARs mediate the expression of incubation of cue-induced cocaine craving. The mechanism of their accumulation may be GluA1-dependent, which differs from that observed in the VTA. However, similar to VTA, mGluR1 stimulation removes CP-AMPARs from MSN synapses. Loss of mGluR1 tone during cocaine withdrawal may contribute to CP-AMPAR accumulation in the NAc. Thus, results in both brain regions point to the possibility of using positive modulators of mGluR1 as treatments for cocaine addiction.
Introduction
One way to classify AMPA receptors is according to whether or not they contain the GluA2 subunit. Those lacking GluA2 differ from the more common GluA2-containing receptors in that they are Ca2+-permeable, exhibit larger single channel conductance and faster kinetics, and display voltage-dependent block by intracellular polyamines. These Ca2+-permeable AMPARs (CP-AMPARs) have emerged as a highly regulated AMPAR subtype that mediates diverse types of neuronal plasticity (Cull-Candy et al., 2006; Isaac et al., 2007; Liu and Zukin, 2007; Lee, 2012). Here we will review results implicating CP-AMPARs in rodent models of cocaine addiction, focusing on the ventral tegmental area (VTA) and the nucleus accumbens (NAc). We will contrast the two brain regions with respect to the type of cocaine exposure required to increase synaptic CP-AMPAR levels, the time-course of CP-AMPAR plasticity, and its underlying mechanisms. Commonalities between the two regions will also be discussed. Most notably, metabotropic glutamate receptor 1 (mGluR1) plays a negative modulatory role in both the VTA and the NAc, promoting removal of CP-AMPARs from synapses and their replacement by GluA2-containing Ca2+-impermeable AMPARs (CI-AMPARs). Finally, we will critically evaluate the significance of CP-AMPARs for cellular and behavioral adaptations that occur in animal models of cocaine addiction. For a broader view of cocaine-induced AMPAR plasticity in the NAc, see previous reviews (Wolf, 2010a; Wolf and Ferrario, 2010).
To help the reader determine which studies can reasonably be compared and integrated into hypotheses, we have provided fairly detailed information about features of experimental design that may influence the neuroadaptations produced by drug exposure. The type of cocaine regimen utilized is particularly important because it is well established that different regimens produce different behavioral and cellular plasticity. As discussed in detail in this review, this applies to CP-AMPAR plasticity as well (see Table 1 for a summary). An important variable is whether cocaine exposure is contingent or non-contingent. When cocaine is administered by the experimenter, drug administration is not dependent upon the animal's behavior and is therefore described as non-contingent. When cocaine is self-administered, drug administration is dependent on the animal's response, such as a nose poke or lever press, and is therefore contingent. Contingent and non-contingent cocaine exposure differently affect glutamate transmission (e.g., Chen et al., 2008; McCutcheon et al., 2011a). Cocaine self-administration regimens are commonly classified as limited access procedures, in which drug is generally available for 1–2 h per session for ~2 weeks, or extended-access procedures, in which drug is available for longer periods of time (e.g., 6 h/day) or for more days. Extended access to cocaine, but not limited access, leads to behaviors that model the compulsive drug-seeking and drug-taking characteristic of addiction, including escalation of intake, increased motivation for drug, and pursuit of cocaine despite adverse consequences (Ahmed, 2011). The effect of cocaine exposure on synaptic plasticity is also influenced by the duration of withdrawal (e.g., Conrad et al., 2008) and whether extinction training occurs during withdrawal (e.g., Knackstedt et al., 2010). Finally, age is particularly important in studies of synaptic plasticity (McCutcheon and Marinelli, 2009), so the age (and species) of animals used in each cocaine-related primary paper cited herein is listed in Table 2.
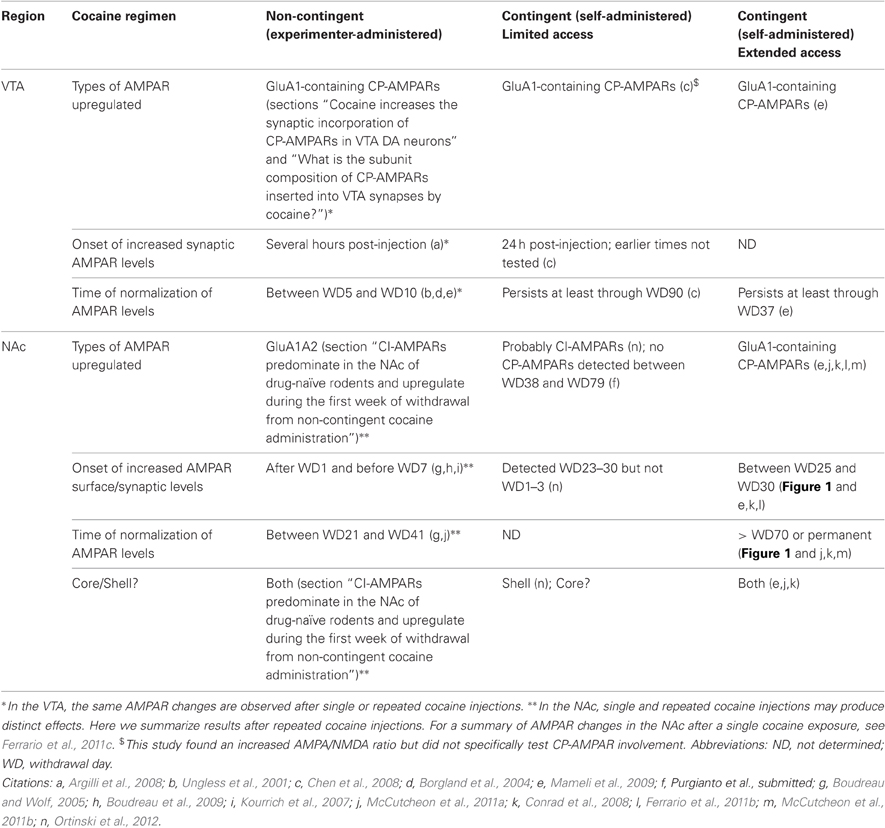
Table 1. Summary of AMPAR changes in VTA and NAc after different cocaine regimens.
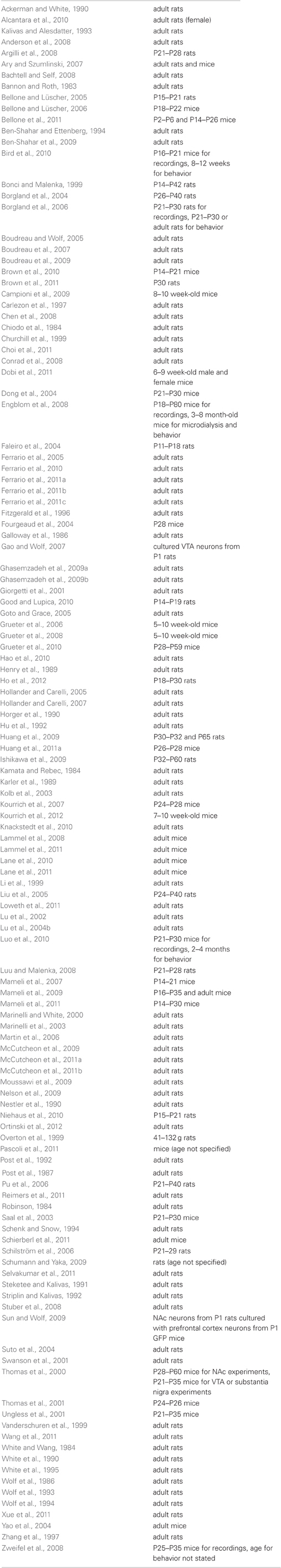
Table 2. Species and age of experimental subjects in cocaine-related primary papers cited in this review (male animals unless noted otherwise; adult rat = >300 g at time of testing or so described by authors; adult mouse = so described by authors).
CP-AMPARs in the VTA
Drug-Induced Changes in Synaptic Strength in the VTA: Background
Most studies of CP-AMPARs in the VTA have been performed in the context of behavioral sensitization to cocaine. Therefore, in order to understand the functional significance of CP-AMPARs in the VTA, it is necessary to discuss behavioral sensitization and the early findings that led to studies of synaptic plasticity in the VTA.
Behavioral sensitization refers to the progressive augmentation of behavioral responses to a drug that occurs during repeated drug exposure and then persists for many weeks after drug exposure is discontinued (Vezina, 2004; Robinson and Berridge, 2008; Vanderschuren and Pierce, 2010). It can be produced by non-contingent or contingent cocaine administration. Both the locomotor activating effects of cocaine and its incentive motivational properties (properties that make it “wanted”) undergo sensitization. In rodents, incentive sensitization can be detected as the ability to more rapidly learn to self-administer a low dose of cocaine or greater motivation to work for cocaine in progressive ratio experiments.
By far the most commonly used experimental approach to study behavioral sensitization is to administer repeated i.p. cocaine injections and monitor progressive increases in locomotor activity. Even a single injection of cocaine is sufficient to produce locomotor sensitization (detected as an enhanced response to a second injection and sometimes termed “one-shot sensitization”). There are many similarities between behavioral sensitization produced by one versus multiple injections of psychostimulants (e.g., Vanderschuren et al., 1999), although it is clear that the total amount of drug exposure (which is greater with multiple injections) is proportional to the robustness of locomotor sensitization (e.g., Robinson, 1984) and to the robustness of at least some sensitization-related adaptations (e.g., Kolb et al., 2003). However, direct comparisons are few and far between. Some caution may be warranted by early reports that behavioral sensitization produced by a single cocaine injection is completely context-dependent (i.e., rats express a sensitized response only when tested in the same environment in which drug was first administered), whereas sensitization produced by multiple injections has a context-independent component (Post et al., 1987, 1992). Furthermore, two types of adaptations in the VTA, both of which facilitate LTP, have been observed after repeated cocaine injections that are not observed after a single injection (Liu et al., 2005; Pu et al., 2006; see section “Evidence for increased AMPA/NMDA ratios and facilitation of LTP in VTA DA neurons from cocaine-exposed rodents” for more discussion).
Despite the widespread use of locomotor sensitization to study neuroadaptations produced by cocaine, incentive sensitization obviously has greater face validity as a model for motivational changes underlying intensification of drug seeking after prior exposure. In section “CI-AMPARs predominate in the NAc of drug-naïve rodents and upregulate during the first week of withdrawal from non-contingent cocaine administration”, we will consider the possibility that AMPAR upregulation in the NAc, which is dissociable from locomotor sensitization, is more directly related to incentive sensitization.
Significance of Increased DA Cell Firing for Behavioral Sensitization
It has long been known that behavioral sensitization to psychostimulants is initiated by psychostimulant actions within the VTA whereas, once it has developed, it is expressed in response to a challenge injection of psychostimulant into the NAc but not the VTA (Kalivas and Stewart, 1991; Vezina, 2004). Anatomical separation of sites for the initiation and expression of sensitization implies a mechanism to “transfer” sensitization from the VTA to the NAc and other forebrain regions. Supporting this idea, adaptations in the VTA occur early in withdrawal and are often transient, whereas adaptations in the NAc may require a period of withdrawal to become evident but then exhibit greater persistence (e.g., Wolf et al., 1993).
Considerable evidence indicates that the “transfer” mechanism involves a transient increase in the firing rate and bursting of VTA DA neurons that is detected during the first few days after discontinuing repeated non-contingent cocaine or amphetamine injections. This effect is most pronounced on the first day of withdrawal and dissipates completely by withdrawal day (WD) 10–14 (Kamata and Rebec, 1984; White and Wang, 1984; Henry et al., 1989; Wolf et al., 1993). Increased DA cell firing and bursting, with the same transient time-course, are also observed after discontinuing cocaine self-administration (Marinelli et al., 2003). This increase in DA neuronal firing during withdrawal is opposite to the decreased activity of VTA DA cells observed while psychostimulants are “on board”. This decrease occurs because psychostimulants elevate extracellular DA levels by blocking DA transporters, leading to DA autoreceptor activation in the VTA as well as activation of inhibitory postsynaptic feedback pathways (Marinelli, 2007)1.
The transient increase in DA cell activity observed during early withdrawal from psychostimulant exposure has been closely linked to the development of behavioral sensitization by many lines of evidence (White, 1996; Wolf, 1998). For example, following non-drug treatments that are expected to increase DA cell firing, drug-naïve rats appear “sensitized” when subsequently exposed to an addictive drug (Steketee and Kalivas, 1991; Ben-Shahar and Ettenberg, 1994; Schenk and Snow, 1994; Carlezon et al., 1997). Several other findings link a faster firing rate of DA neurons to addiction vulnerability: (1) Rats that exhibit faster firing and more bursting activity of VTA DA cells exhibit greater propensity to acquire psychostimulant self-administration (Marinelli and White, 2000). (2) Rats that exhibit a high locomotor response to novelty, which predicts greater addiction liability, exhibit more persistent increases in firing and bursting of VTA DA cells during withdrawal from limited access cocaine self-administration (McCutcheon et al., 2009). (3) Decreasing DA cell firing rate by stimulating D2 DA autoreceptors reduces cocaine-seeking behavior (Marinelli et al., 2003; Xue et al., 2011).
What mechanism underlies the increased DA cell activity observed during early withdrawal from psychostimulant exposure? This question led to early theories about LTP-like effects and ultimately to the discovery that drugs of abuse do in fact increase the strength of excitatory synapses onto VTA DA neurons. Ironically, however, recent results indicate that drug-induced increases in the AMPA/NMDA ratio can be dissociated from increased DA cell activity, as discussed below (section “CP-AMPARs in the VTA: Functional significance”) and previously (McCutcheon et al., 2009).
Evidence for Increased AMPA/NMDA Ratios and Facilitation of LTP in VTA DA Neurons from Cocaine-Exposed Rodents
Several lines of evidence led investigators to hypothesize, in the late 1980's and early 1990's, that NMDAR-dependent synaptic potentiation was involved in the activation of VTA DA neurons observed during early psychostimulant withdrawal (Wolf, 1998). Most importantly, the induction of behavioral sensitization was shown to require NMDAR transmission in the VTA (e.g., Karler et al., 1989; Kalivas and Alesdatter, 1993). Furthermore, interfering with NMDAR transmission not only prevented the induction of sensitization but also prevented adaptations in the VTA during early withdrawal that are associated with induction of sensitization as well as more delayed NAc adaptations that are associated with persistence of sensitization (e.g., Wolf et al., 1994; Li et al., 1999; Mameli et al., 2009).
The first support for the synaptic potentiation hypothesis came from the demonstration that VTA DA neurons showed enhanced sensitivity to the excitatory effects of iontophoretic glutamate when recorded three days after discontinuing repeated i.p. cocaine or amphetamine injections (White et al., 1995). Subsequently, we found enhanced sensitivity to iontophoretic AMPA in recordings conducted three but not 14 days after discontinuing repeated cocaine or amphetamine injections (Zhang et al., 1997; see also Giorgetti et al., 2001). These results were consistent with the LTP hypothesis because LTP can be expressed as a selective increase in the AMPAR component of the EPSP (Kauer et al., 1988).
A few years later, a landmark paper directly linked behavioral sensitization with the induction of LTP in VTA DA neurons (Ungless et al., 2001). Using in vitro recordings, the authors compared excitatory transmission in DA neurons from control mice and mice that had received a single cocaine injection 24 h earlier. In VTA DA neurons from cocaine-treated mice, a facilitation of AMPAR-mediated synaptic transmission relative to the NMDAR-mediated response (the AMPA/NMDA ratio) was observed. This facilitation was NMDAR-dependent (i.e., it was blocked when an NMDAR antagonist was administered along with cocaine), linking it to NMDAR-dependence of the induction of behavioral sensitization (see first paragraph of this section). Subsequent studies showed that the magnitude of the increase in the AMPA/NMDA ratio was similar regardless of whether single or multiple i.p. cocaine injections were administered (Borgland et al., 2004). The AMPA/NMDA ratio was also increased 24 h after systemic administration of amphetamine, nicotine, morphine, and ethanol, as well as after stress (Saal et al., 2003). These treatments also exhibit cross-sensitization with each other (e.g., Marinelli and Piazza, 2002), further suggesting a relationship between synaptic potentiation in the VTA and the development of behavioral sensitization.
The increased AMPA/NMDA ratio observed after cocaine injection was interpreted as a form of LTP because electrically evoked LTP could not be elicited in the cocaine-exposed mice, whereas LTD was enhanced (Ungless et al., 2001). Similarly, spike timing-dependent LTP could be induced in VTA DA neurons from control animals but not those previously treated with single or multiple cocaine injections (Argilli et al., 2008; Luu and Malenka, 2008; Ho et al., 2012; Mameli et al., 2011). These results indicate occlusion of LTP by prior cocaine exposure (see Argilli et al., 2008). Supporting this, NMDAR transmission is required for both the cocaine-induced increase in the AMPA/NMDA ratio (Ungless et al., 2001) and electrically-induced LTP in midbrain DA neurons (Bonci and Malenka, 1999; Overton et al., 1999). Interestingly, maintenance of cocaine-induced LTP requires the activity of protein kinase Mζ (PKMζ; Ho et al., 2012), an autonomously active protein kinase C (PKC) isoform, whereas spike timing-dependent LTP in VTA DA neurons of drug-naïve mice depends on conventional PKC isoforms (Luu and Malenka, 2008).
Cocaine acts locally within the VTA to increase the AMPA/NMDA ratio, since in vitro incubation with cocaine was sufficient to elicit the increase (Argilli et al., 2008). Cocaine also acts rapidly—whether injected systemically or applied in vitro, increased AMPA/NMDA ratios were observed within 3–5 h of cocaine exposure (Argilli et al., 2008). However, cocaine produces an even more rapid enhancement of NMDAR transmission that may be important in enabling the subsequent NMDAR-dependent increase in the AMPA/NMDA ratio. Thus, within minutes of cocaine exposure, NMDAR transmission is potentiated by insertion of NR2B-containing NMDARs into synapses through a mechanism that requires activation of D5 receptors, protein kinase A, and new protein synthesis (Schilström et al., 2006; Argilli et al., 2008; also see Dong et al., 2004). Endogenous orexin transmission is also required for the rapid cocaine-induced insertion of NR2B-containing receptors, as well as the increased AMPA/NMDA ratio and the induction of behavioral sensitization (Borgland et al., 2006).
The increased AMPA/NMDA ratio is not the only cocaine-induced adaptation observed in VTA DA neurons. Twenty-four hours after repeated cocaine injections (5–7 days) but not a single injection of cocaine, facilitation of spike-timing dependent LTP was observed that was attributable to reduced GABAA receptor-mediated inhibition. Interestingly, rats treated in the same manner showed an increased AMPA/NMDA ratio, indicating that the increased ratio can co-exist with GABAA receptor-mediated plasticity (Liu et al., 2005). At longer withdrawal times (10–15 days) from repeated cocaine injections, but not a single cocaine injection, a distinct form of enhanced susceptibility to LTP induction was observed that was attributable to elevated BDNF signaling in the VTA (Pu et al., 2006). Finally, drugs of abuse (morphine, cocaine, and nicotine) or stress cause a loss of LTP of GABAergic synapses onto VTA DA neurons; this could act in concert with the increased AMPA/NMDA ratio to enhance the excitability of VTA DA neurons (Niehaus et al., 2010). To understand the functional significance of these various changes, it will be helpful to examine how they track with different behavioral adaptations produced after limited or extended access cocaine self-administration.
Recent Findings have Clarified the Mechanism, Duration, and Cell Specificity of Cocaine-Induced Synaptic Potentiation in the VTA
Three findings are particularly significant in this regard. The first relates to CP-AMPAR and NMDAR involvement. Initially, it was proposed that the increased AMPA/NMDA ratio observed after drug exposure reflected an increase in AMPAR insertion into synapses in the absence of changes in NMDAR transmission, since bath application of AMPA to brain slices from sensitized animals elicited increased currents relative to controls whereas bath application of NMDA did not (Ungless et al., 2001). More recent results have indicated that the increased AMPA/NMDA ratio is due to replacement of lower conductance CI-AMPARs with higher conductance CP-AMPARs (increasing the numerator of the ratio; Bellone and Lüscher, 2006; Mameli et al., 2007; Argilli et al., 2008; Ho et al., 2012; see section “Cocaine increases the synaptic incorporation of CP-AMPARs in VTA DA neurons” for more discussion) combined with a decrease in synaptic NMDAR currents (decreasing the denominator; Mameli et al., 2011). The latter effect was detected by uncaging glutamate at single putative glutamate synapses on VTA DA neurons 24 h after a single injection of cocaine (Mameli et al., 2011). These authors speculated that decreased NMDAR currents were not detected by Ungless et al. (2001) because bath applied NMDA activated both synaptic and extrasynaptic NMDARs. The observed decrease in NMDAR synaptic currents may help to explain the difficulty in inducing LTP after cocaine exposure (Mameli et al., 2011). It should be noted that decreased NMDAR transmission 24 h after cocaine (Mameli et al., 2011) is not necessarily at odds with the very rapid increase in NMDAR transmission observed within minutes of cocaine exposure (Schilström et al., 2006; Argilli et al., 2008; see section “Evidence for increased AMPA/NMDA ratios and facilitation of LTP in VTA DA neurons from cocaine-exposed rodents”).
The second extension concerns the duration of the effect. Initially, the increased AMPA/NMDA ratio was considered a transient effect that enabled more persistent forms of plasticity in target regions of VTA DA neurons such as the NAc. This was based on studies in which cocaine was administered non-contingently (i.p.) and the AMPA/NMDA ratio was elevated five days but not 10 days after the last cocaine injection (Ungless et al., 2001; Borgland et al., 2004). This idea was revised after Chen et al. (2008) found a much more persistent elevation of the AMPA/NMDA ratio in rats that had self-administered cocaine (2 h/day for 14–19 days). In these animals, the magnitude of the elevation was similar to that produced by non-contingent cocaine, but the ratio remained at this elevated level regardless of whether VTA neurons were recorded seven days, 21 days, or three months after the last i.v. cocaine infusion. Indeed, the ratio remained elevated even after extinction training. Persistent elevation of the AMPA/NMDA ratio in VTA DA neurons after cocaine self-administration was also observed by Mameli et al. (2009) and shown to result from CP-AMPAR synaptic incorporation (section “Cocaine increases the synaptic incorporation of CP-AMPARs in VTA DA neurons”). In contrast, food or sucrose self-administration produced only a transient elevation of the AMPA/NMDA ratio, detected after seven but not 21 days of withdrawal (Chen et al., 2008). Importantly, yoked controls (animals that received non-signaled i.v. cocaine infusions in a pattern and dosage similar to cocaine self-administering animals) did not exhibit an increased AMPA/NMDA ratio, even when recordings were performed on WD1, a time when the AMPA/NMDA ratio is increased after a single i.p. cocaine injection. These results suggest that this plasticity requires learning about cues that signal drug delivery (see Chen et al., 2008 for more discussion). Furthermore, they suggest that cocaine self-administration, compared to non-contingent administration, produces a much longer temporal window in which synaptic potentiation in the VTA may lead to altered neurotransmission or plasticity. The effect of different cocaine regimens on AMPAR plasticity in the VTA is summarized in Table 1.
The third extension is that subpopulations of VTA DA neurons differ in their ability to undergo cocaine-induced LTP (Lammel et al., 2011). Thus, cocaine increased the AMPA/NMDA ratio in VTA DA neurons projecting to the NAc medial shell but not those projecting to the medial prefrontal cortex, while an aversive stimulus selectively strengthened synapses on DA neurons projecting to prefrontal cortex. Both rewarding and aversive stimuli strengthened synapses on DA neurons projecting to the lateral shell of the NAc. The NAc core was not evaluated. These findings extend prior studies demonstrating that DA neurons with different projection targets show different properties. For example, unique characteristics of mesoprefrontal DA neurons are due in part to lack of DA autoreceptor modulation of firing rate and DA synthesis (Bannon and Roth, 1983; Chiodo et al., 1984; Galloway et al., 1986) although nerve terminal DA autoreceptors do modulate DA release in prefrontal cortex (Galloway et al., 1986; Wolf et al., 1986) (for review, see Wolf and Roth, 1987; for a more recent demonstration of these unique properties, see Lammel et al., 2008). Even at the level of a single DA neuron, different synapses may exhibit different plasticity depending on the origin of the presynaptic input (Good and Lupica, 2010; see section “Are CP-AMPARs present in VTA DA neurons of drug-naïve rodents?” for more discussion).
Are CP-AMPARs Present in VTA DA Neurons of Drug-Naïve Rodents?
Many principal neurons express CP-AMPARs shortly after birth but undergo a developmental switch during the first weeks of postnatal development in which CP-AMPARs are replaced by CI-AMPARs (Kumar et al., 2002; Eybalin et al., 2004; Ho et al., 2007; Brill and Huguenard, 2008). Consistent with these results, CP-AMPARs were found in excitatory synapses on VTA DA neurons in P2–6 but not P14–26 mice (Bellone et al., 2011). Previous studies had failed to detect CP-AMPARs in VTA DA neurons of P14–35 drug-naïve mice (Thomas et al., 2000; Bellone and Lüscher, 2006; Mameli et al., 2007, 2009; Brown et al., 2010). The situation is more complex in rats. Bellone and Lüscher (2005) detected a substantial complement of CP-AMPARs in P15–P21 drug-naïve rats. More recently, Good and Lupica (2010) found that VTA DA neurons may express CP-AMPARs at some of their synapses but not others. They reached this conclusion after observing that Joro spider toxin (JST), which selectively blocks CP-AMPARs, had no effect on EPSCs elicited by stimulation of the pedunculopontine nucleus but inhibited a subpopulation of EPSCs (64%) elicited by intra-VTA stimulation. Two studies did not detect CP-AMPARs in VTA DA neurons of drug-naïve rats (Faleiro et al., 2004; Argilli et al., 2008). However, Faleiro et al. (2004) did not include a polyamine such as spermine in the recording pipette. This could prevent detection of inward rectification, a hallmark of CP-AMPARs, since rectification is a direct reflection of CP-AMPAR block by intracellular polyamines. Other methodological differences such as location of the stimulating electrode might also be important. Finally, different levels of stress could affect results, since stress influences AMPA/NMDA ratios (Saal et al., 2003).
Cocaine Increases the Synaptic Incorporation of CP-AMPARs in VTA DA Neurons
Initial studies of the cocaine-induced increase in the AMPA/NMDA ratio did not attempt to distinguish between increased synaptic strength due to incorporation of CP-AMPARs versus CI-AMPARs (e.g., Ungless et al., 2001). However, biochemical and behavioral evidence pointed to the possibility that homomeric GluA1 receptors were elevated in the VTA of cocaine-sensitized rats (Carlezon and Nestler, 2002). Subsequent electrophysiological studies demonstrated that CP-AMPAR levels in VTA DA neurons increase after non-contingent cocaine administration, both in mice (Bellone and Lüscher, 2006; Mameli et al., 2007, 2009, 2011) and rats (Argilli et al., 2008; Good and Lupica, 2010; Ho et al., 2012). This plasticity depends on NMDAR transmission in VTA DA neurons since it is not observed in NR1DATCreERT2 mice in which NMDARs are lacking in DA neurons (Engblom et al., 2008; Mameli et al., 2009). While most of these studies were conducted 24 h after a single cocaine injection, CP-AMPARs are also detected 24 h after repeated cocaine injections (Mameli et al., 2009) and as early as 3 h after a single injection (Argilli et al., 2008). A single cocaine injection increased CP-AMPAR levels in VTA DA neurons regardless of whether the population of synapses sampled possessed CP-AMPARs prior to cocaine treatment (Good and Lupica, 2010). After cocaine self-administration, CP-AMPAR incorporation in VTA DA neurons was much more persistent, detectable even on WD35 (Mameli et al., 2009), corresponding to the more persistent increase in the AMPA/NMDA ratio (Chen et al., 2008; section “Recent findings have clarified the mechanism, duration, and cell specificity of cocaine-induced synaptic potentiation in the VTA”).
In the first study showing that cocaine increased CP-AMPAR synaptic incorporation, Bellone and Lüscher (2006) recorded from mouse VTA DA neurons 24 h after a single i.p. cocaine injection. They found that the current-voltage relationship, previously linear, now showed inward rectification. They also observed sensitivity to the CP-AMPAR blocker JST. The cocaine-induced inward rectification was paralleled by an increased AMPA/NMDA ratio. A significant correlation between these measures indicated that the increased AMPA/NMDA ratio was due, at least in part, to synaptic incorporation of CP-AMPARs (Bellone and Lüscher, 2006), which have higher conductance than CI-AMPARs (Isaac et al., 2007). A subsequent study showed that cocaine no longer elevated the AMPA/NMDA ratio in the presence of intracellular spermine, which blocks CP-AMPARs, arguing that CP-AMPAR insertion accounts fully for the increased AMPA/NMDA ratio (Argilli et al., 2008). However, as noted in section “Recent findings have clarified the mechanism, duration, and cell specificity of cocaine-induced synaptic potentiation in the VTA”, a recent study indicated that the situation is more complex by demonstrating that cocaine-induced CP-AMPAR synaptic incorporation is accompanied by a reduction of unitary NMDAR currents (Mameli et al., 2011). Thus, in addition to the insertion of CP-AMPARs, a decrease in NMDAR transmission also contributes to the increase in the AMPA/NMDA ratio observed in the VTA following cocaine. These recent findings add to intriguing evidence that synapses containing CP-AMPARs tend to exhibit lower NMDAR transmission (Lamsa et al., 2007; Wang and Gao, 2010), although a more complex relationship between CP-AMPARs and NMDARs has been found during postnatal development of the VTA (Bellone et al., 2011).
Nicotine, Morphine, and Amphetamine: Increased CP-AMPAR Levels in the VTA?
Similar to results with cocaine, synaptic insertion of CP-AMPARs was detected in VTA DA neurons 24 h after a single injection of nicotine or morphine, or after optogenetic activation of DA neurons (Brown et al., 2010). Together with other results (Gao and Wolf, 2007; Brown et al., 2010), these findings indicate that drugs of abuse produce CP-AMPAR insertion due to their common ability to increase extracellular DA levels. If so, amphetamine would be expected to produce the same effect. However, some results raise questions about this. First, although Faleiro et al. (2004) detected a robust increase in the AMPA/NMDA ratio 24 h after amphetamine injection, the current-voltage relationship remained linear. As noted in section “Are CP-AMPARs present in VTA DA neurons of drug-naïve rodents?” failure to observe inward rectification could be explained by omission of spermine from the recording pipette. However, whereas increased GluA1 surface expression was detected in VTA 24 h after cocaine injection (Boudreau and Wolf, 2005), no such change was found 24 h after amphetamine injection (Faleiro et al., 2004). These results suggest that cocaine and amphetamine may differ in the mechanism by which they increase the AMPA/NMDA ratio. This is reminiscent of the markedly different effects of cocaine and amphetamine on AMPAR surface expression in the NAc (Nelson et al., 2009). In addition, although both drugs increase the AMPA/NMDA ratio within 2–3 h of systemic injection (Faleiro et al., 2004; Argilli et al., 2008), in vitro incubation of VTA slices with cocaine reproduces this effect (Argilli et al., 2008) while in vitro amphetamine incubation does not (Faleiro et al., 2004). The difference could suggest a distinct site of action for amphetamine, although it could also reflect the fact that AMPA/NMDA ratios were measured during amphetamine perfusion but after cocaine washout. Overall, it remains unclear whether cocaine and amphetamine produce qualitatively different effects on AMPAR transmission in the VTA.
What is the Subunit Composition of CP-AMPARs Inserted into VTA Synapses by Cocaine?
Many findings implicate GluA1-containing CP-AMPARs. The first evidence came from immunoblotting studies of GluA1 in VTA homogenates combined with studies in which VTA GluA1 levels were manipulated using viral vectors; these results led Carlezon and Nestler (2002) to argue that formation of homomeric GluA1 receptors and a resultant increase in Ca2+ signaling in the VTA were responsible for triggering behavioral sensitization. Subsequent biochemical studies supported a role for GluA1-containing CP-AMPARs. For example, we detected increased GluA1 surface expression in the VTA 24 h after a single i.p. cocaine injection (Boudreau and Wolf, 2005) whereas GluA2 was unchanged (Boudreau and Wolf, unpublished findings). Whether increased surface and/or synaptic GluA1 is reflected by increased total GluA1 protein appears to depend on how GluA1 is measured (Fitzgerald et al., 1996; Churchill et al., 1999; Lu et al., 2002). While all of the above studies utilized non-contingent cocaine treatment, a more recent study found increased total GluA1 and P-845 GluA1 protein in the VTA 24 h after discontinuing cocaine self-administration, although GluA2 protein was also increased (Choi et al., 2011).
Electrophysiological results have also supported a role for GluA1. AMPAR currents, measured 3–5 h after in vitro cocaine exposure, were sensitive to PhTx-74, a compound that selectively blocks GluA1-containing AMPARs (Argilli et al., 2008). Furthermore, a single cocaine injection was able to increase the AMPA/NMDA ratio in mice lacking GluA2 in DA neurons (Engblom et al., 2008) but not in mice lacking GluA1 (Dong et al., 2004; Engblom et al., 2008; for more discussion of these studies, see section “CP-AMPARs in the VTA: Functional significance”). Finally, electronic microscopy studies found increased plasma membrane and synaptic GluA1 levels in the VTA after discontinuing cocaine exposure, although this depended on the number of injections, the duration of withdrawal, and the VTA subregion sampled; furthermore, increased GluA1 was detected in non-DA dendrites as well as in DA neurons (Lane et al., 2010, 2011).
Despite strong evidence for GluA1 involvement, a role for GluA3 cannot be ruled out. After incubating cultured VTA neurons with DA for 1 h (to mimic the duration of elevated DA levels produced by systemic cocaine injection), increases were observed in cell surface levels of GluA1, GluA3, and the area of GluA1–GluA3 colocalization, while surface GluA2 was unaltered; these results suggest that DA transmission might regulate homomeric GluA1 and GluA1A3-containing CP-AMPAR levels (Gao and Wolf, 2007). GluA3 levels in the VTA have not been measured after in vivo cocaine exposure.
mGLuR1 Negatively Regulates CP-AMPAR Levels in VTA DA Neurons
Group I mGluR-dependent long-term depression (mGluR-LTD) is recognized as playing an important role in diseases including drug addiction (Gerdeman et al., 2003; Grueter et al., 2007; Lüscher and Huber, 2010). The discovery that mGluR1 stimulation removes CP-AMPARs from VTA synapses was an important advance in the field (Bellone and Lüscher, 2005). Subsequent studies found the same effect in cerebellar stellate cells in drug-naïve rats (Kelly et al., 2009) and NAc neurons from cocaine-exposed rats (McCutcheon et al., 2011b), suggesting a special relationship between mGluR1 and CP-AMPARs (see sections “mGluR1 negatively regulates synaptic levels of CP-AMPARs in the NAc” and “Removal of CP-AMPARs by mGluR1 stimulation and its therapeutic implications” for more discussion).
In their initial report, Bellone and Lüscher (2005) studied CP-AMPAR containing synapses in the VTA of drug-naïve rats. They found that short bursts of 66 Hz stimulation or application of the non-selective group I mGluR agonist dihydroxyphenylglycine (DHPG) led to a form of mGluR-LTD that was blocked by an antagonist of mGluR1 but not mGluR5. After mGluR-LTD, inward rectification was no longer evident and the depressed EPSCs were no longer sensitive to JST, indicating that mGluR-LTD involved removal of CP-AMPARs from synapses. As will be discussed in section “Cocaine-induced synaptic incorporation of CP-AMPARs and reversal of this plasticity by mGluR1 stimulation are GluA2-dependent processes,” this is accompanied by insertion of CI-AMPARs but the net effect is LTD because high conductance CP-AMPARs are being replaced by lower conductance CI-AMPARs. In agreement with these results, mGluR-LTD was detected in VTA DA neurons from drug-naïve rats only at synapses that express CP-AMPARs but not at synapses expressing only CI-AMPARs; however, it could be detected at all synapses after cocaine exposure (Good and Lupica, 2010).
Following up on their results in drug-naïve rats, Bellone and Lüscher (2006) found that mGluR-LTD reversed cocaine-induced accumulation of CP-AMPARs in mouse VTA DA neurons. Thus, CP-AMPARs were removed from synapses by a positive modulator of mGluR1 (Ro 67-7476), either applied directly to slices from cocaine-treated mice or injected 24 h after i.p. cocaine (recordings performed a total of 48 h after cocaine injection, i.e., 24 h after Ro 67-7476). CP-AMPAR removal was later shown to require dynamin-dependent internalization (Mameli et al., 2007). Lüscher and colleagues then demonstrated that mGluR1 activation in vivo normally limits the duration of elevated CP-AMPAR levels in VTA synapses of cocaine treated animals. Thus, in control rats, inward rectification (a hallmark of CP-AMPARs) was detected on WD1 but not WD7 after a single cocaine injection. However, if mGluR1 function in the VTA was reduced 24 h before cocaine injection (via intra-VTA injection of a TAT-conjugated peptide that disrupts the interaction between group I mGluRs and Homer1b/c), then the cocaine-induced inward rectification persisted on WD7 (Mameli et al., 2009). The cocaine-increase in synaptic CP-AMPARs was similarly prolonged if mGluR1 function in the VTA was disrupted by daily i.p. injections of an mGluR1 antagonist (1-aminoindan-1,5-dicarboxylic acid; AIDA). Conversely, administering cocaine along with the mGluR1 positive modulator Ro 67-7476 (for 7 days) prevented the increased inward rectification normally produced by this repeated cocaine regimen. Activation of mGluR1 in the VTA also exerts a braking effect on cocaine-induced plasticity in the NAc (Mameli et al., 2009; see section “mGluR1 negatively regulates synaptic levels of CP-AMPARs in the NAc”). This negative regulatory role for mGluR1 in both VTA and NAc suggests mGluR1 as a potential therapeutic target in addiction (section “Removal of CP-AMPARs by mGluR1 stimulation and its therapeutic implications”).
Overall, these results indicate that tonic activation of mGluR1 in the VTA normally exerts a braking influence on the cocaine-induced accumulation of CP-AMPARs; decreasing mGluR1 tone in the VTA permits elevation of CP-AMPARs to persist for a longer time (Mameli et al., 2009). Given that CP-AMPARs contribute substantially to the increased AMPA/NMDA ratio after cocaine exposure (section “Cocaine increases the synaptic incorporation of CP-AMPARs in VTA DA neurons”), these results suggest a potential explanation for the longer duration of the elevated AMPA/NMDA ratio after contingent versus non-contingent cocaine (Chen et al., 2008; section “Recent findings have clarified the mechanism, duration, and cell specificity of cocaine-induced synaptic potentiation in the VTA”), namely that cocaine self-administration persistently depresses mGluR1 signaling in the VTA. However, this has yet to be tested.
Cocaine-Induced Synaptic Incorporation of CP-AMPARs and Reversal of this Plasticity by mGLuR1 Stimulation are GLuA2-Dependent Processes
As noted by He et al. (2009), there appear to be two general mechanisms for regulating the synaptic content of CP-AMPARs; in some sitiuations, relative levels of CP-AMPARs and CI-AMPARs are altered via a GluA2-dependent mechanism requiring GluA2-PICK1 interactions, but in others, the accumulation of CP-AMPARs is associated with increased GluA1 but very little change in GluA2. Cocaine-induced CP-AMPAR incorporation in the VTA falls into the former category, although this is not so clear for the NAc (see section “Accumulation of CP-AMPARs after cocaine exposure”).
PICK1 is a protein that interacts with the C-terminus of GluA2 (and GluA3) but not GluA1 (Xia et al., 1999). It plays complex roles in regulating AMPAR trafficking during different types of plasticity (Anggono and Huganir, 2012) and, most relevant to this review, is required for exchanging CP-AMPARs for CI-AMPARs. This was first demonstrated in cerebellar stellate cells (Liu and Cull-Candy, 2005). Subsequently, Bellone and Lüscher (2006) found that interfering with the interaction between GluA2 and PICK1, using a TAT-conjugated peptide, prevented cocaine-induced insertion of CP-AMPARs. This suggested that CI-AMPARs are removed from synapses when CP-AMPARs are inserted (i.e., there is an exchange between the two AMPAR subtypes), as opposed to insertion of CP-AMPARs on top of existing synaptic CI-AMPARs. Direct evidence for an exchange was provided by electron microscopy studies showing that GluA2 immunolabeling in VTA DA neurons of control mice is mainly synaptic, whereas cocaine reduces synaptic GluA2 labeling and increases cytoplasmic labeling (Mameli et al., 2007). Similar results were found after nicotine or morphine exposure (Brown et al., 2010).
The GluA2 subunit also plays a key role in the reversal of this exchange during mGluR-LTD in VTA DA neurons. Whereas cocaine caused a translocation of GluA2 immunolabeling from synapse to cytoplasm, activation of mGluR1 with DHPG reversed this effect (Mameli et al., 2007), indicating that CP-AMPAR removal during mGluR-LTD occurs in tandem with their replacement by lower-conductance CI-AMPARs. Further supporting an exchange, non-stationary fluctuation analysis indicated that the average number of AMPARs open remained constant during mGluR-LTD. Additional experiments showed that the CI-AMPARs inserted into synapses during mGluR-LTD are synthesized de novo, as the mGluR-LTD was inhibited by antisense oligonucleotide or siRNA to GluA2. This is likely to reflect local translation of GluA2 mRNA based on sensitivity of mGluR-LTD to translational inhibitors or disruption of mTOR signaling (Mameli et al., 2007).
CP-AMPARs in the VTA: Functional Significance
As described in section “Significance of increased DA cell firing for behavioral sensitization,” VTA DA cell firing rate and bursting are increased on the day after discontinuing cocaine or amphetamine treatment, an effect that dissipates gradually and is no longer observed after 10–14 days of withdrawal. This transient increase in DA cell activity is believed to be crucial for enabling downstream neuronal adaptations to occur in the NAc that correlate with persistent expression of sensitization, i.e., “transferring” sensitization from the VTA to the NAc (White, 1996; Wolf, 1998). Several adaptations have been described that could theoretically contribute to increased DA cell activity during the early withdrawal period, including decreased VTA levels of inhibitory G-protein subunits (Nestler et al., 1990; Striplin and Kalivas, 1992), increased VTA levels of glutamate receptors (Fitzgerald et al., 1996; see Wolf, 2002 for review), the development of functional subsensitivity of D2 autoreceptors which normally exert inhibitory control over DA cell activity (Kamata and Rebec, 1984; White and Wang, 1984; Ackerman and White, 1990; Wolf et al., 1993) and, most recently, the elevated AMPA/NMDA ratio discussed herein. However, which of these actually mediates the increased DA cell activity?
Considerable evidence points to a primary role for DA autoreceptor subsensitivity. First, the time-course of increased DA cell activity exactly parallels the time-course of DA autoreceptor subsensitivity during withdrawal from either repeated i.p. injections of cocaine or amphetamine (White and Wang, 1984; Henry et al., 1989) or cocaine self-administration (Marinelli et al., 2003)2. Second, DA autoreceptor subsensitivity is required for the induction of lasting behavioral sensitization. If DA autoreceptor subsensitivity is prevented, more slowly developing adaptations in the NAc, linked to expression of behavioral sensitization and its persistence, do not occur (Wolf et al., 1994; Li et al., 1999). Furthermore, repeated treatment with a D1 receptor agonist, which does not produce DA autoreceptor sensitivity, fails to produce lasting sensitization (White et al., 1990; Hu et al., 1992). Third, cocaine, amphetamine, and stress, which produce cross-sensitization, all produce transient DA autoreceptor subsensitivity in the VTA (White, 1996; Marinelli, 2007). Thus, there is strong evidence that DA autoreceptor subsensitivity and increased DA cell firing rate are related, and that together they represent early causal steps leading to induction of behavioral sensitization and to more persistent sensitization-related adaptations in the NAc (White, 1996). Even though DA autoreceptors are subsensitive shortly after discontinuing psychostimulant treatment, they are not completely “off-line.” DA autoreceptor stimulation during the early withdrawal period can reduce DA cell activity and cocaine self-administration, although, as expected, their stimulation is more efficacious at eliciting both effects on WD10, when autoreceptor subsensitivity has subsided (Marinelli et al., 2003).
Does the increased AMPA/NMDA ratio also contribute to increased DA cell firing during psychostimulant withdrawal? In experiments in which behavioral sensitization is elicited by repeated i.p. injections of cocaine, the increased AMPA/NMDA ratio (Ungless et al., 2001; Borgland et al., 2004) and increased DA cell firing rate (Henry et al., 1989) have similar time courses; both are transient. However, after withdrawal from cocaine self-administration, the increased AMPA/NMDA ratio persists for three months (Chen et al., 2008; Mameli et al., 2009), whereas the increase in DA cell activity normalizes within a week (Marinelli et al., 2003; McCutcheon et al., 2009). As noted by McCutcheon et al. (2009), this indicates that increased strength of excitatory synapses onto VTA DA neurons “does not translate into the integrated output of these cells as measured by firing activity.” In contrast, the time course of DA autoreceptor subsensitivity parallels increased DA cell activity during cocaine withdrawal, regardless of how cocaine is administered (see previous paragraph).
If the cocaine-induced increase in the AMPA/NMDA ratio is dissociable from the increase in DA cell firing rate, what is the functional significance of the increased AMPA/NMDA ratio? There are clues, but no clear answers. The demonstration that cue-reward learning also increases the AMPA/NMDA ratio in VTA DA neurons (Stuber et al., 2008) suggests that this increase may be associated with or facilitate learning about the rewarding properties of drugs. Other results suggest that it may alter aspects of such learning that depend on subsequent synaptic plasticity in the VTA. The incorporation of CP-AMPARs into VTA DA neurons is particularly significant in this regard, as Mameli et al. (2011) showed that it inverts the rules for induction of synaptic plasticity. Thus, in control animals, they found that pairing of presynaptic activity and postsynaptic depolarization (to activate NMDARs) elicited LTP, as expected. However, in the VTA of cocaine-treated mice, LTP relied on Ca2+ entry through CP-AMPARs and was independent of NMDARs. This type of LTP, also reported in other synapses that contain CP-AMPARs (Kullmann and Lamsa, 2011), can be induced when presynaptic activity is paired with a slight postsynaptic hyperpolarization. As a result of this change, the pattern of activity which potentiates DA cell excitation will be different in cocaine-exposed animals, perhaps setting the stage for a cascade of altered responsiveness that may ultimately contribute to addiction (Mameli et al., 2011). It remains to be determined if the increased AMPA/NMDA ratio after cue-reward learning, which is more transient than after drug exposure (Stuber et al., 2008), involves CP-AMPAR synaptic incorporation and is therefore associated with altered rules for induction of synaptic plasticity in the VTA.
The dissociation between the increased AMPA/NMDA ratio and increased DA cell firing rate may explain a puzzle in the literature. In several transgenic mouse lines in which glutamate receptor expression was disrupted, cocaine no longer elicited an increased AMPA/NMDA ratio in VTA DA neurons but mice were nevertheless able to develop locomotor sensitization (Dong et al., 2004; Engblom et al., 2008; Zweifel et al., 2008; Bird et al., 2010; see also Luo et al., 2010, which assessed electrically induced LTP rather than cocaine-induced LTP). We suggest that cocaine, despite its inability to increase the AMPA/NMDA ratio, elicited other adaptations in the VTA (such as DA autoreceptor subsensitivity) that are more directly related to the increased DA cell firing rate, which in turn is strongly linked to induction of behavioral sensitization. Another factor to keep in mind is that cocaine also alters AMPAR subunit distribution in non-DA cells of the VTA (Lane et al., 2010, 2011). In fact, the ability of intra-VTA NMDAR antagonist injection to prevent locomotor sensitization (e.g., Kalivas and Alesdatter, 1993) may reflect blockade of NMDARs in non-DA neurons, since this effect is still observed in mice that lack NR1 in VTA DA neurons (Luo et al., 2010).
Do results obtained in transgenic mice (see previous paragraph) rule out any role for the increased AMPA/NMDA ratio in the induction of behavioral sensitization? Two arguments suggest that this would be premature. First, parallels between locomotor sensitization and the AMPA/NMDA ratio cannot be discounted, i.e., both are induced by drug actions in the VTA, both require NMDAR transmission in the VTA, and cross-sensitization occurs between treatments (different drugs and stress) that also increase the AMPA/NMDA ratio (see section “Evidence for increased AMPA/NMDA ratios and facilitation of LTP in VTA DA neurons from cocaine-exposed rodents”). Second, there are caveats associated with all of the transgenic studies cited above. These include compensatory changes in glutamate transmission in the VTA (Dong et al., 2004; Engblom et al., 2008; Bird et al., 2010) and, in some cases, the possibility of changes in glutamate receptor expression in other brain regions (Dong et al., 2004; Bird et al., 2010). More importantly, synaptic potentiation was typically measured in much younger animals than were used for assessing sensitization of locomotor activity (Engblom et al., 2008; Bird et al., 2010; Luo et al., 2010) and, in all cases, the AMPA/NMDA ratio was assessed after a single cocaine exposure whereas locomotor sensitization was assessed after repeated cocaine exposure (Dong et al., 2004; Engblom et al., 2008; Zweifel et al., 2008; Bird et al., 2010). Although not addressed here, there are also interesting differences in the development of cocaine-induced conditioned place preference (CPP) in these mouse strains (see Luo et al., 2010 for discussion).
Summing up, the neuroadaptation most convincingly linked to the development of locomotor sensitization is a transient increase in VTA DA cell firing during early withdrawal from psychostimulant exposure. This increase in DA cell activity results, at least in part, from transient subsensitivity of impulse-modulating DA autoreceptors. However, more work is needed to precisely delineate the relationship between specific neuroadaptations in the VTA (e.g., DA autoreceptor subsensitivity vs. increased AMPA/NMDA ratio) and specific behavioral changes. It may be illuminating to more closely examine behaviors related to drug seeking/incentive sensitization rather than limiting the focus to locomotor sensitization. For example, viral vector-mediated GluA1 upregulation in the VTA increased the motivation of rats to work for cocaine in progressive ratio experiments, suggesting a potential link between synaptic potentiation and incentive sensitization (Choi et al., 2011).
Finally, despite differences in the persistence of VTA plasticity after different cocaine regimens (section “Recent findings have clarified the mechanism, duration, and cell specificity of cocaine-induced synaptic potentiation in the VTA”; Table 1), it is important to recognize that the AMPA/NMDA ratio is increased to a similar degree after single and repeated i.p. cocaine injections (Borgland et al., 2004), as well as limited access (Chen et al., 2008) and extended access cocaine self-administration (Mameli et al., 2009). Similarly, a comparable increase in the rectification index (indicating CP-AMPAR incorporation) is observed following a single i.p. cocaine injection, repeated i.p. cocaine injections, and extended access cocaine self-administration (e.g., Bellone and Lüscher, 2006; Mameli et al., 2009). This may indicate that synaptic potentiation in the VTA is permissive rather than causal with respect to behavioral changes most directly related to drug addiction (which generally emerge only after extended access cocaine self-administration; Ahmed, 2011). This does not diminish its functional significance, as an understanding of factors that predispose to drug-taking is a major goal of addiction research.
Summary: CP-AMPARs in the VTA
In drug-naïve young rats (P14–21), most evidence indicates that VTA DA neurons express CP-AMPARs at some of their synapses. In mice, VTA DA neurons contain abundant CP-AMPARs early in development (P2–6) but these are replaced with CI-AMPARs by P14–26. Exposure to drugs of abuse or stress (treatments that produce behavioral sensitization and cross-sensitization) leads to an increased AMPA/NMDA ratio at excitatory synapses onto VTA DA neurons. In the case of cocaine, which has been most thoroughly studied, the AMPA/NMDA ratio increases within hours of drug exposure. This increase may be triggered by a more rapid cocaine-induced increase in NMDAR transmission, occurring within minutes of cocaine injection. The persistence of the increased AMPA/NMDA ratio during withdrawal depends on whether cocaine is experimenter-administered or self-administered. The AMPA/NMDA ratio remains elevated for about a week in the first case and for months in the latter case. However, the magnitude of the increase in the AMPA/NMDA ratio is similar regardless of whether cocaine exposure is contingent or non-contingent (see final paragraph of section “CP-AMPARs in the VTA: Functional significance” for a summary). The AMPA/NMDA ratio increases after cocaine exposure because high conductance CP-AMPARs are inserted into synapses and lower conductance CI-AMPARs are removed. The insertion of CP-AMPARs into synapses may be accompanied by decreased NMDAR transmission, further contributing to elevation of the AMPA/NMDA ratio. The cocaine-induced switch in AMPAR subtypes (CP-AMPAR inserted, CI-AMPAR removed) depends on GluA2-PICK1 interactions. Stimulation of mGluR1 leading to mGluR-LTD reverses this process: CP-AMPARs internalize and are replaced with CI-AMPARs through a mechanism that requires locally translated GluA2. In vivo, tonic activation of mGluR1 receptors in the VTA limits the duration of cocaine-induced CP-AMPAR synaptic incorporation, helping to restore these synapses to the pre-cocaine state. The functional significance of the increased AMPA/NMDA ratio may be related to the fact that CP-AMPAR incorporation alters the rules for subsequent induction of LTP, although the behavioral correlates of this alteration remain to be worked out. In contrast, subsensitivity of impulse-modulating DA autoreceptors is more likely to explain the increase in DA cell firing rate that occurs during early psychomotor stimulant withdrawal and is closely linked to the development of behavioral sensitization. A primary role for DA autoreceptor subsensitivity, rather than the increased AMPA/NMDA ratio, in driving increased DA firing rate and the development of sensitization may help explain reports that mice lacking GluA1 do not show cocaine-induced synaptic potentiation but do develop locomotor sensitization. See Table 1 for a summary of CP-AMPAR plasticity in the VTA after different cocaine regimens.
CP-AMPARs in the NAc
NAc: Background
The NAc, comprised primarily (90–95%) of GABAergic medium spiny projection neurons (MSNs), plays a key role in goal-directed behaviors including those related to drugs of abuse. MSNs integrate excitatory inputs from regions that process information related to motivation and behavioral control (the prefrontal cortex, hippocampus, basolateral amygdala, and thalamus) and project to regions important for motor output (including the ventral pallidum, substantia nigra, and VTA). Based on this connectivity, the NAc has been described as a limbic-motor interface, a concept supported by functional studies (Mogenson et al., 1980; Everitt et al., 1999; Groenewegen et al., 1999; Meredith et al., 2008; Sesack and Grace, 2010).
Disrupted processing of reward-related information within the NAc has been implicated in drug addiction, focusing interest on excitatory synaptic transmission onto MSNs. This led to the demonstration that cocaine seeking behavior, measured in a number of different animal models, requires activation of AMPARs on NAc MSNs (Kalivas and Volkow, 2005), although neuronal circuitry involving the dorsal striatum becomes important as drug use becomes habitual (Everitt and Robbins, 2005). Over the last 10 years, evidence has accumulated to show that cell surface and synaptic levels of AMPARs are upregulated in the NAc after withdrawal from cocaine exposure (Wolf, 2010a,b; Wolf and Ferrario, 2010). It is logical to presume that this enhances behavioral output related to cocaine and cocaine-associated stimuli. In some cases this has been demonstrated (e.g., Anderson et al., 2008; Conrad et al., 2008). However, despite considerable recent interest in drug-induced synaptic plasticity in the NAc (Kauer and Malenka, 2007; Bowers et al., 2010; Lüscher and Malenka, 2011), the relationship between particular types of plasticity and particular behavioral changes remains unclear (Wolf, 2010a,b; Wolf and Ferrario, 2010). Here we will distinguish between cocaine regimens leading to upregulation of CI-AMPARs vs. CP-AMPARs and then focus on the mechanisms and functional consequences of CP-AMPAR upregulation.
CI-AMPARs Predominate in the NAc of Drug-Naïve Rodents and Upregulate During the First Week of Withdrawal from Non-Contingent Cocaine Administration
Biochemical data indicate that nearly all AMPARs in the NAc of adult drug-naïve rats contain the GluA2 subunit; most are GluA1A2, although GluA2A3 receptors are also present (Boudreau et al., 2007; Conrad et al., 2008; Reimers et al., 2011). Electrophysiological studies in younger mice generally support the same conclusion, based on linear current-voltage relationships (Kourrich et al., 2007; Mameli et al., 2009; Grueter et al., 2010; but see Campioni et al., 2009). However, there is both biochemical and electrophysiological evidence for a small (~5–10%) contribution of CP-AMPARs to NAc glutamatergic synaptic transmission in adult drug-naïve rats (Conrad et al., 2008; Reimers et al., 2011). This CP-AMPAR population probably includes homomeric GluA1 and GluA1A3 receptors, but homomeric GluA3 receptors cannot be excluded.
In the first study to investigate cocaine's effects on AMPAR surface expression in the NAc, Boudreau and Wolf (2005) administered repeated i.p. cocaine injections using a regimen that produced locomotor sensitization in about 50% of the cocaine-treated rats (see section “Drug-induced changes in synaptic strength in the VTA: Background” for background on behavioral sensitization). Only the sensitized rats exhibited increased GluA1 and GluA2 surface expression in the NAc (combined core/shell dissection). If a more robust regimen was used, locomotor sensitization and AMPAR upregulation occurred in all cocaine-treated rats (Ferrario et al., 2010). This AMPAR upregulation was observed on WD7, WD14, and WD21, but not on WD1 or WD41, indicating that surface expression of GluA1A2 receptors increases during the first week of withdrawal, remains high for several weeks, and then normalizes (Boudreau and Wolf, 2005; Boudreau et al., 2007, 2009; Ferrario et al., 2010; McCutcheon et al., 2011a). Biochemical results from other groups support this time-course of AMPAR upregulation during withdrawal from repeated cocaine injections, the involvement of GluA1 and GluA2, and its occurrence in both core and shell subregions (Ghasemzadeh et al., 2009a; Schumann and Yaka, 2009; Schierberl et al., 2011). Mechanisms implicated in AMPAR upregulation in the NAc of cocaine-sensitized animals include ERK activation (Boudreau et al., 2007, 2009; Schumann and Yaka, 2009; Pascoli et al., 2011), phosphorylation of GluA1 at the PKA site (S845) (Schierberl et al., 2011), and nitrosylation of stargazin (Selvakumar et al., 2011).
Electrophysiological correlates of increased AMPAR surface expression are the increased AMPA/NMDA ratio observed in the NAc shell of cocaine-sensitized mice on WD10–14 (Kourrich et al., 2007) and potentiation of AMPAR-mediated synaptic transmission in D1R-expressing MSN in both core and shell subregions on WD7–10 in cocaine-sensitized mice (Pascoli et al., 2011). In contrast, experiments performed on WD1 found that the AMPA/NMDA ratio was decreased in the NAc shell of cocaine-sensitized mice and electrically evoked LTD was occluded (Kourrich et al., 2007; Mameli et al., 2009). If AMPAR surface expression is unchanged on WD1 (Boudreau and Wolf, 2005), why is the AMPA/NMDA ratio depressed? Huang et al. (2009) provided a possible answer by showing that NR2B-containing silent synapses are generated in the NAc shell during the course of repeated i.p. cocaine injections3. Thus, the decreased AMPA/NMDA ratio on WD1 may reflect increased NMDAR-mediated transmission whereas the AMPAR upregulation later in withdrawal may result from insertion of new AMPARs into these silent synapses (Huang et al., 2009; Brown et al., 2011; Huang et al., 2011b; Lee and Dong, 2011). Indeed, the cocaine-induced increase in silent synapses wanes during the first week of withdrawal (Huang et al., 2009), just as increases in GluA1 and GluA2 surface expression are detected (Boudreau and Wolf, 2005; Boudreau et al., 2009). Interestingly, an increased AMPA/NMDA ratio was observed on WD1 following 28 injections of cocaine (Dobi et al., 2011). Perhaps during the longer regimen there is sufficient time for silent synapses to be formed and “filled” with AMPARs, whereas these processes spill over into early withdrawal when the period of cocaine exposure is shorter.
In VTA DA neurons, single and repeated i.p. injections of cocaine similarly increase the magnitude of the AMPA/NMDA ratio (Borgland et al., 2004), although its duration and other types of plasticity in VTA depend on the regimen (sections “Drug-induced changes in synaptic strength in the VTA: Background”, “Evidence for increased AMPA/NMDA ratios and facilitation of LTP in VTA DA neurons from cocaine-exposed rodents”, and “Recent findings have clarified the mechanism, duration, and cell specificity of cocaine-induced synaptic potentiation in the VTA”). It is less clear to what extent a single i.p. cocaine injection reproduces the effects of multiple injections on AMPAR transmission in the NAc. In some cases, effects are the same (Pascoli et al., 2011) but other results suggest a complex picture (Kourrich et al., 2007; Wolf and Ferrario, 2010; Ferrario et al., 2011c). Therefore, in Table 1, the summary information provided on the effects of non-contingent (i.p.) cocaine injections in the VTA applies to both single and repeated injections, whereas the summary information for non-contingent cocaine in the NAc refers only to results obtained after repeated injections.
Overall, results summarized in this section provide strong evidence for increased levels of CI-AMPARs (GluA1A2) in NAc synapses during the first three weeks of withdrawal from repeated non-contingent (i.p.) cocaine exposure. In contrast, CP-AMPARs accumulate in NAc synapses after a month of withdrawal from extended access cocaine self-administration, as described in the next section. This raises the question of whether AMPAR upregulation “shifts” from CI-AMPARs to CP-AMPARs as withdrawal from non-contingent cocaine treatment progresses. We addressed this issue and found that this was not the case. Using biochemical and electrophysiological measures, we found that non-contingent cocaine exposure does not lead to CP-AMPAR incorporation in the NAc of adult rats regardless of the withdrawal period (McCutcheon et al., 2011a). More recently, we failed to find significant increases in CP-AMPAR levels, assessed based on sensitivity to naspm, after prolonged withdrawal from limited access cocaine self-administration (Purgianto et al., submitted; see section “CP-AMPARs in the NAc: Functional significance”). However, even though CP-AMPARs are not involved, there does appear to be AMPAR upregulation occurring during withdrawal from limited access cocaine self-administration. This is based on a recent study showing increased mEPSC amplitude and AMPA/NMDA ratios in the NAc shell 3–4 weeks after discontinuing limited access cocaine self-administration (2 h/day for 14 d) (Ortinski et al., 2012; core was not examined), as well as prior indirect evidence in the core (Knackstedt et al., 2010). Together, these results indicate that, in adult rats, CP-AMPAR accumulation occurs only after extended access cocaine self-administration, where CI-AMPAR upregulation occurs after non-contingent cocaine or limited access cocaine self-administration. Interestingly, non-contingent cocaine exposure is sufficient to produce CP-AMPAR accumulation in the NAc shell of young mice (Mameli et al., 2009), perhaps reflecting age-related differences in synaptic plasticity (McCutcheon and Marinelli, 2009). It has been shown that CP-AMPARs are prevalent in cultured NAc neurons derived from P1 rats (Sun and Wolf, 2009) but the time-course of their decline during development is not known.
What is the functional significance of CI-AMPAR upregulation after withdrawal from non-contingent cocaine exposure? Although correlations have been reported between upregulated AMPAR transmission and locomotor sensitization (Boudreau and Wolf, 2005; Pascoli et al., 2011), the two phenomenon are dissociable under some conditions (reviewed by Wolf and Ferrario, 2010). For example, locomotor sensitization is expressed on WD1, prior to AMPAR upregulation (Boudreau and Wolf, 2005); 24 h after cocaine challenge, when AMPAR levels and AMPAR-mediated synaptic transmission fall below basal levels (Thomas et al., 2001; Boudreau et al., 2007; Kourrich et al., 2007; Ferrario et al., 2010; and on WD41, when AMPAR upregulation has waned (McCutcheon et al., 2011a). Studies in mutant mice also show a dissociation between AMPAR upregulation and another adaptation strongly linked to locomotor sensitization, namely decreased intrinsic excitability of MSN (Kourrich et al., 2012). Perhaps AMPAR upregulation is dissociable from the expression of locomotor sensitization because expression depends more on the massive increase in extracellular DA levels produced by a challenge injection of cocaine than on enhanced excitatory synaptic transmission (but see Pierce et al., 1996). If so, AMPAR upregulation may be more important for those addiction-related behaviors that can be manifest in the absence of cocaine challenge and rely more directly on excitatory transmission.
Based on this rationale, we have proposed that CI-AMPAR upregulation is critical for incentive sensitization (sensitization of drug “wanting”) (Ferrario et al., 2010; Wolf and Ferrario, 2010). Incentive sensitization can be illustrated by treating rats with non-contingent psychostimulant injections (leading to locomotor sensitization) and then giving them the opportunity to self-administer cocaine. Previously sensitized rats show greater motivation for psychostimulants, that is, they more rapidly learn to self-administer low doses of drug and they will work harder for drug in progressive ratio experiments (e.g., Horger et al., 1990; Suto et al., 2004; reviewed by Vezina, 2004). We speculate that motivation increases because AMPAR upregulation in the NAc strengthens synaptic connections between MSN and the cortico-limbic glutamate inputs that trigger cocaine seeking. Supporting this theory, our preliminary studies suggest that prevention of AMPAR upregulation reduces incentive sensitization whereas locomotor sensitization still develops (Wang et al., 2011). Other results support a relationship between incentive sensitization and changes in AMPAR responsiveness in the NAc (Suto et al., 2004). It should be noted that cocaine CPP provides a measure of the acute rewarding effects of cocaine, and not of incentive sensitization, so dissociation between CPP and AMPAR upregulation (e.g., Kourrich et al., 2012) does not contradict our hypothesis.
What happens to upregulated CI-AMPAR levels if animals are re-exposed to cocaine (“cocaine challenge”) during withdrawal? Biochemical, electrophysiological, and behavioral studies have found that AMPAR upregulation is temporarily reversed, that is, surface and synaptic CI-AMPAR levels are decreased 24 h after the injection but then recover to upregulated levels within a week (Boudreau et al., 2007; Kourrich et al., 2007; Bachtell and Self, 2008). It is less clear what happens immediately after the challenge injection, during the behavioral response to cocaine. We did not detect any change in AMPAR surface expression 30 min after administering an i.p. cocaine challenge to sensitized rats (i.e., toward the end of the period in which locomotor sensitization is maximally expressed) (Ferrario et al., 2010). However, another study found that cocaine challenge increased cell surface GluA1 but not GluA2 at the 30 min time-point, suggesting that trafficking of homomeric GluA1 receptors to the surface immediately after cocaine challenge may serve to further boost excitation of MSN (Schierberl et al., 2011). This is reminiscent of an earlier study in which i.p. cocaine challenge elicited reinstatement of cocaine self-administration accompanied by a rapid increase in GluA1 surface expression (Anderson et al., 2008; for more discussion, see section “CP-AMPARs in the NAc: Functional significance”). In both studies, D1 receptors, L-type calcium channels, CaMKII and increased GluA1 phosphorylation at S831 were implicated in cocaine's effect on GluA1 (Anderson et al., 2008; Schierberl et al., 2011). These in vivo results may be related to the ability of D1 receptor stimulation (which would be enhanced after cocaine injection) to rapidly increase expression of GluA1-containing AMPARs in cultured MSNs (Sun et al., 2008; Wolf, 2010b).
Future studies are needed to fully understand the effects of cocaine re-exposure on AMPAR transmission in the NAc. Overall, however, these results—which show no change or an increase in AMPAR surface expression after cocaine challenge—as well as other evidence previously considered in detail (Wolf and Ferrario, 2010), argue against the theory that the expression of locomotor sensitization depends on AMPAR internalization, a theory based primarily on studies of amphetamine and on effects of cocaine in the shell (Brebner et al., 2005; Kourrich et al., 2012).
CP-AMPARs Accumulate in the NAc During Withdrawal from Extended Access Cocaine Self-Administration
Whereas non-contingent cocaine exposure or limited access cocaine self-administration lead to increased surface and synaptic levels of CI-AMPARs (section “CI-AMPARs predominate in the NAc of drug-naïve rodents and upregulate during the first week of withdrawal from non-contingent cocaine administration”), CP-AMPARs accumulate in the NAc of adult rodents after prolonged withdrawal from extended access cocaine self-administration (Conrad et al., 2008; Mameli et al., 2009; Ferrario et al., 2011b; McCutcheon et al., 2011a,b). In the first of these studies, Conrad et al. (2008) tested the hypothesis that increased AMPAR levels in the NAc underlie the incubation of cue-induced cocaine seeking. Incubation refers to the progressive intensification of cue-induced cocaine craving that occurs during withdrawal from extended access cocaine self-administration (Lu et al., 2004a; Pickens et al., 2011). To test this hypothesis, Conrad et al. (2008) trained rats to self-administer saline or cocaine for 6 h/day for 10 days, an extended access cocaine self-administration regimen (see “Introduction”). Each infusion was paired with a light cue. After 42–47 days of withdrawal, current-voltage relationships were linear in MSN from the saline group, as expected, whereas an inward rectification of AMPAR-mediated transmission was observed in MSN recorded from the cocaine group. Accordingly, bath application of the CP-AMPAR antagonist Naspm produced only a small reduction in electrically-evoked EPSC amplitude in the saline group (~5%), whereas a more robust reduction (~30%) was observed in the cocaine group (Conrad et al., 2008). While these original studies were performed in the NAc core, we have found very similar results in the NAc shell, albeit with greater variability (McCutcheon et al., 2011a). To test the behavioral significance of CP-AMPAR synaptic incorporation, Conrad et al. (2008) administered bilateral injections of Naspm into the NAc core 15 min before a test for cue-induced cocaine seeking on WD45. Naspm markedly reduced the magnitude of cue-induced cocaine seeking, demonstrating that CP-AMPARs in the NAc core mediate the expression of incubated cue-induced cocaine seeking at this late withdrawal time (Conrad et al., 2008). We propose that the synaptic incorporation of CP-AMPARs enhances the responsiveness of NAc MSN to glutamate inputs from cortical and limbic regions. Thus, when cocaine-associated cues are presented after prolonged withdrawal from cocaine and glutamate is released in the NAc, MSN respond more robustly, leading to enhanced cocaine-seeking.
Similar conclusions about the functional significance of CP-AMPAR synaptic incorporation were reached using NR1DATCreERT2 mice in which NMDARs are lacking in DA neurons (Mameli et al., 2009). A single i.p. cocaine injection does not elicit CP-AMPAR synaptic incorporation in VTA DA neurons from these mice (Engblom et al., 2008; see section “CP-AMPARs in the VTA: Functional significance”). Following cocaine self-administration (4 h/day for eight days; infusions were paired with a light cue), Mameli et al. (2009) found that control mice showed significantly greater cue-induced seeking than NR1DATCreERT2 mice on WD35. Recordings performed within 48 h of the behavioral test revealed CP-AMPARs in VTA DA neurons and NAc shell MSN in the control cocaine-exposed mice, but not in the NR1DATCreERT2 mice. A significant correlation between the number of lever presses during the test and the rectification index in MSN (a measure of the level of CP-AMPARs) was observed (Mameli et al., 2009). These results suggest that NMDAR-dependent activation of DA neurons in the VTA is a prerequisite for CP-AMPAR synaptic incorporation in both VTA DA neurons and MSN of the NAc shell, and for robust cocaine seeking after prolonged withdrawal.
Biochemical results have provided further evidence for CP-AMPAR accumulation in the NAc after prolonged withdrawal from extended access cocaine self-administration, as well as clues to their subunit composition (Conrad et al., 2008). Rats were killed on WD1 or WD45 after discontinuing cocaine or saline self-administration, and AMPAR subunit surface expression was determined. On WD1, when cocaine seeking was low, cocaine rats showed modest decreases in surface, intracellular, and total GluA1 compared to saline controls. This could represent synaptic scaling (scaling down) in response to high levels of synaptic activity during the 10 days of cocaine self-administration training. On WD45, when cocaine seeking was high (incubated), cocaine rats exhibited significant increases in all of these GluA1 measures relative to saline controls, while GluA2 measures were not significantly altered, consistent with an increase in CP-AMPARs. Overall, our analysis of surface, intracellular and total AMPAR subunit levels suggest that, after prolonged withdrawal from cocaine, the normal complement of GluA2-containing AMPARs is supplemented by the addition of GluA1-containing CP-AMPARs (see section “Accumulation of CP-AMPARs after cocaine exposure” for more discussion). Supporting this conclusion, co-immunoprecipitation (co-IP) studies detected an increase in the portion of GluA1 that was not physically associated with GluA2 or any other AMPAR subunit, indicating formation of homomeric GluA1 receptors in the NAc during incubation. However, a role for GluA3-containing CP-AMPARs cannot be ruled out in light of increased cell surface GluA3 in cocaine exposed rats on both WD1 and WD45 (Conrad et al., 2008). For a detailed review of studies that measured GluA1 in NAc homogenates or membrane fractions after extended access cocaine self-administration, see section 5.3 of Wolf and Ferrario (2010).
By measuring the AMPAR rectification index at different withdrawals from the same extended access cocaine self-administration regimen (6 h/day × 10 days), we have shown that CP-AMPARs are first detected between WD25 and WD35; once present, they persist through at least WD70 (Figure 1). Biochemical results also support a delay in the CP-AMPAR increase. Compared to saline controls, surface GluA1 in the NAc was decreased on WD1 after cocaine self-administration, unchanged on WD3, and slightly increased on WD21, although total GluA1 was not yet increased on WD21 (on WD45, both surface and total GluA1 were increased markedly) (Conrad et al., 2008). Interestingly, behavioral studies have shown that incubation of cue-induced cocaine craving is already evident by WD7 (Lu et al., 2004a) and therefore occurs more rapidly than CP-AMPAR accumulation in the NAc. Thus, different mechanisms must be responsible for the expression of incubation of cocaine craving at earlier withdrawal times. This is not surprising in light of the involvement of multiple brain regions in this phenomenon (Pickens et al., 2011).
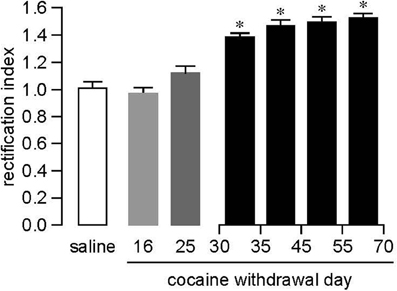
Figure 1. Changes in AMPAR-mediated synaptic transmission rectification index (RI) measured in NAc core MSNs following withdrawal from extended access cocaine self-administration. Compared to saline-treated animals (n = 17, pooled from withdrawal days [WD] 35–70), no apparent changes in RI were observed during the first month of withdrawal from cocaine self-administration (WD16, n = 5; WD25, n = 5). However, a significant increase in RI was observed after a month of withdrawal (WD30–35, n = 7). The increased RI persisted through WD70 (WD35–45, n = 10; WD45–55, n = 13; WD55–70, n = 7), the latest time-point measured. *P < 0.01 vs. saline, Tukey post-hoc test after significant ANOVA.
The delay in accumulation of CP-AMPARs suggests that homeostatic plasticity might underlie AMPAR adaptations during incubation (Boudreau and Wolf, 2005; Conrad et al., 2008; Wolf, 2010a; Huang et al., 2011b). For example, decreased metabolic activity in regions sending glutamate projections to the NAc during cocaine withdrawal could lead to a synaptic scaling-mediated increase in CP-AMPAR levels on MSN (see Sun and Wolf, 2009 for discussion). However, the time required for CP-AMPAR accumulation during incubation is considerably longer than that required for known forms of homeostatic plasticity in NAc neurons (Ishikawa et al., 2009; Sun and Wolf, 2009), possibly implying a cascade of changes in excitatory activity in different brain regions that could result in sprouting of new synaptic connections in the NAc.
mGLuR1 Negatively Regulates Synaptic Levels of CP-AMPARs in the NAc
Prompted by evidence that mGluR1 removes CP-AMPARs from synapses in other brain regions (see section “mGluR1 negatively regulates CP-AMPAR levels in VTA DA neurons”), we recently investigated the effect of the group I mGluR agonist DHPG on AMPAR transmission in the NAc of rats that underwent >45 days of withdrawal from the same extended access cocaine self-administration regimen used in Conrad et al. (2008). We found that DHPG rapidly normalized the elevated rectification index elicited by this regimen (McCutcheon et al., 2011b). This normalization remains present for at least 30 min post-DHPG washout (data not shown). These results indicate DHPG-induced removal of CP-AMPARs via either internalization or inhibition of their function (McCutcheon et al., 2011b). Supporting the former possibility, dynamin-dependent internalization is required for analogous effects of DHPG in VTA DA neurons (Mameli et al., 2007) and our preliminary results indicate that DHPG rapidly internalizes GluA1-containing CP-AMPARs in cultured NAc neurons (Loweth et al., 2010; see also Mangiavacchi and Wolf, 2004).
The DHPG-induced normalization of the rectification index in NAc MSN of cocaine-exposed rats was associated with two concurrent events: facilitation of EPSCs recorded at +40 mV (EPSC+40 mV) and attenuation of EPSCs recorded at −70 mV (EPSC−70 mV). Since CP-AMPARs contribute to synaptic transmission at −70 mV but not +40 mV due to voltage-dependent block by polyamines, we suggest that attenuation of EPSC−70 mV reflects removal of CP-AMPARs whereas EPSC+40 mV facilitation reflects enhancement of CI-AMPAR transmission (McCutcheon et al., 2011b). Thus, DHPG may promote an exchange between CP-AMPARs and CI-AMPARs in the NAc after prolonged cocaine withdrawal, analogous to its effects in VTA DA neurons from cocaine-treated mice (Bellone and Lüscher, 2006; Mameli et al., 2007) and in cerebellar stellate neurons of drug-naïve rats (Kelly et al., 2009). Supporting an exchange in MSN, our results indicate mechanistic links between DHPG-induced CP-AMPAR removal and CI-AMPAR facilitation in the NAc MSN from the cocaine group. Thus, both effects were dependent on postsynaptic Ca2+ elevation and PKC activation. Furthermore, if Naspm was present prior to DHPG application, both effects were blocked, suggesting that CP-AMPARs must be activated in order for CI-AMPAR transmission to be facilitated (McCutcheon et al., 2011b). In the VTA, DHPG-induced exchange of CP-AMPARs and CI-AMPARs requires local translation of new GluA2 subunits and is associated with GluA2 redistribution from cytoplasm back to synapses (Mameli et al., 2007). It remains to be determined if a similar mechanism operates in the NAc.
Whereas DHPG attenuated EPSC−70 mV and facilitated EPSC+40 mV in cocaine rats, markedly different effects were observed in NAc MSN from saline control rats. In this group, DHPG did not alter EPSC+40 mV but it did attenuate EPSC−70 mV. This synaptic attenuation was associated with facilitation of the paired-pulse ratio, completely prevented by bath application of the CB1R antagonist AM-251, and blocked by the mGluR5 antagonist MTEP (McCutcheon et al., 2011b). In contrast, DHPG attenuation of EPSC−70 mV in MSNs from cocaine rats was blocked by the mGluR1 antagonist LY367385 but not by AM-251, and did not involve a change in the paired-pulse ratio. Lack of effect of AM-251 was not due to impaired CB1R function, since bath application of the CB1R agonist WIN-55,212-2 was able to elicit synaptic inhibition in MSNs from the cocaine group; in fact, this inhibition was more robust in cocaine rats than saline controls (McCutcheon et al., 2011b). Together, these results indicate a dramatic shift in the mechanism of DHPG-dependent regulation of synaptic transmission. In saline controls, the effect of DHPG resembles the well-studied striatal mGluR-LTD that involves mGluR5, postsynaptic endocannabinoid release, presynaptic CB1R activation and reduction of glutamate release (Robbe et al., 2002; Lovinger, 2008)4. This mechanism is apparently disabled after extended access cocaine self-administration and prolonged withdrawal. Instead, a novel mechanism emerges that relies on mGluR1, postsynaptic PKC signaling, and CP-AMPAR removal (McCutcheon et al., 2011b).
Based on our finding that mGluR1 stimulation removed CP-AMPARs from MSN synapses, we hypothesized that a persistent decrease in mGluR1 signaling in the NAc during cocaine withdrawal might contribute to synaptic accumulation of CP-AMPARs. Supporting this hypothesis, mGluR1 surface expression was similar in saline and cocaine groups on WD14, but was decreased in the cocaine group on WD25, just before CP-AMPARs are detected, and remained low on WD48, after the increase in CP-AMPAR levels has stabilized (Loweth et al., 2011)5. The Homer family of postsynaptic scaffolding proteins is critical for group I mGluR signaling (Fagni et al., 2002; Szumlinski et al., 2008; Bockaert et al., 2010), so decreased coupling of mGluR1 to Homer proteins could also contribute to decreased mGluR1 tone during incubation. Indeed, there is evidence for decreased Homer levels in the NAc after cocaine exposure (Szumlinski et al., 2008) including extended access cocaine self-administration (Ben-Shahar et al., 2009). However, in preliminary co-IP experiments, we have not detected significant changes in association between mGluR1 and Homer proteins in the NAc during incubation, suggesting that the decrease in mGluR1 surface expression is the primary reason for altered mGluR1 signaling (Loweth et al., 2011).
In contrast, co-IP experiments did reveal decreased coupling between mGluR5 and Homer proteins (1b/c and 2) in cocaine rats on WD45 (Loweth et al., 2011), which could help explain why NAc MSN from cocaine rats do not exhibit mGluR5-mediated synaptic depression (McCutcheon et al., 2011b). Blunting of mGluR5-mediated LTD in the NAc has been reported after various types of cocaine exposure, including a single injection (Fourgeaud et al., 2004; Grueter et al., 2010), repeated i.p. injections (Huang et al., 2011a), and cocaine self-administration followed by extinction training (Moussawi et al., 2009), so it is unlikely that this effect is specific to incubation or related to the presence of CP-AMPARs in synapses6. It should be noted that there are many discrepancies, too complex to be reviewed here, about whether all cocaine regimens reduce mGluR5 levels or function, whether this occurs in core or shell, whether it occurs in D1R or D2R expressing cells, and whether it occurs after abstinence or only after extinction training (Swanson et al., 2001; Fourgeaud et al., 2004; Ary and Szumlinski, 2007; Szumlinski et al., 2008; Ben-Shahar et al., 2009; Ghasemzadeh et al., 2009b; Moussawi et al., 2009; Grueter et al., 2010; Hao et al., 2010; Knackstedt et al., 2010; Huang et al., 2011a; McCutcheon et al., 2011b).
While this section has focused on regulatory effects of “local” mGluR1 receptors within the NAc, CP-AMPAR levels in the NAc are also regulated by mGluR1 receptors in the VTA (Mameli et al., 2009). This was demonstrated with a TAT-conjugated dominant negative peptide that interferes with the binding of Homer1b/c to the C-terminus of group I mGluRs. While 10 i.p. cocaine injections, but not a single injection, led to CP-AMPAR accumulation in the NAc shell of young mice on WD35, a single cocaine injection became sufficient when preceded by intra-VTA injection of the dominant negative peptide. Together with other results (see section “mGluR1 negatively regulates CP-AMPAR levels in VTA DA neurons”), these findings demonstrate that mGluR1 receptors in the VTA normally exert a “braking effect” on plasticity in both the VTA and the NAc. Interrupting mGluR1 signaling renders animals more vulnerable to cocaine-induced plasticity, while potentiating mGluR1 signaling opposes this plasticity. Our results show a similar negative regulatory role for mGluR1 receptors located in the NAc (McCutcheon et al., 2011b). Together, these findings point to the potential therapeutic utility of mGluR1 positive allosteric modulators for the treatment of addiction (see section “Removal of CP-AMPARs by mGluR1 stimulation and its therapeutic implications”).
Heterogeneity of MSN in the NAc
There has been considerable recent interest in whether synaptic plasticity, including drug-related plasticity, is differently expressed in subpopulations of NAc neurons. Recent technical advances have shed light on this issue. Studies using BAC transgenic mice have shown that D1R and D2R expressing populations of MSN in both dorsal striatum and ventral striatum (NAc) can exhibit different types of synaptic plasticity and distinct functional roles, and optogenetic techniques offer the potential to selectively monitor plasticity at synapses between MSN and specific afferent pathways (e.g., afferents originating in the prefrontal cortex) (Lüscher and Malenka, 2011). However, it is worth raising four points related to defining MSN based on DA receptor expression: (1) In the dorsal striatum, D1Rs are preferentially expressed in MSNs from the direct pathway whereas D2Rs are preferentially found in MSNs from the indirect pathway. In the NAc, however, the segregation is less complete. D2R-expressing MSNs project to the ventral pallidum (equivalent to the indirect pathway from the dorsal striatum), whereas D1R-expressing MSNs project to both the ventral pallidum and the output nuclei of the direct pathway (Humphries and Prescott, 2010; Sesack and Grace, 2010). (2) There are many instances in which D1- and D2-class receptors exert cooperative effects on NAc firing and related behaviors (e.g., Clark and White, 1987; Seif et al., 2011). (3) Although DA receptor expression is an important characteristic of MSN, many anatomical distinctions and gradients (not limited to core versus shell) contribute to MSN diversity and function (Voorn et al., 2004; Humphries and Prescott, 2010; Sesack and Grace, 2010). (4) The role of DA receptor subtypes other than D1 and D2 in cocaine's effects (e.g., Beninger and Banasikowski, 2008), and their potential overlap with D1 and D2 receptors in MSNs, should not be disregarded (Surmeier et al., 1996; Humphries and Prescott, 2010).
Given that MSNs show heterogeneity with respect to certain types of plasticity (Lüscher and Malenka, 2011), the question arises as to whether this applies to CI- or CP-AMPAR upregulation during cocaine withdrawal. As reviewed previously (Wolf and Ferrario, 2010), CI-AMPAR upregulation after withdrawal from repeated i.p. cocaine injections appears to be quite global. Likewise, silent synapse formation seems to be widespread rather than confined to a subpopulation of MSNs (Brown et al., 2011), consistent with the idea that silent synapses are the substrate for CI-AMPAR upregulation (Huang et al., 2009). Most of these studies were performed in rats. On the other hand, synaptic potentiation in the NAc of cocaine-sensitized BAC transgenic mice is restricted to D1R-expressing MSN in both core and shell (Pascoli et al., 2011). It will be important to understand the reasons for these different results. What about CP-AMPAR accumulation during incubation? In the core subregion, we find that CP-AMPARs are present in nearly all NAc MSN recorded after more than a month of withdrawal from extended access cocaine self-administration (Figure 1; McCutcheon et al., 2011a). However, more variability is evident in the shell (McCutcheon et al., 2011a).
How can such widespread plasticity be reconciled with selective strengthening of addiction-related behaviors after cocaine exposure or, more generally, with evidence that MSN participate in functionally distinct neuronal ensembles? An important point is that single MSNs receive convergent inputs from multiple brain regions (O'Donnell and Grace, 1995; Finch, 1996). Thus, even if every MSN gains CP-AMPARs during incubation, they could be added selectively to spines innervated by a subpopulation of glutamate afferents. For example, selective addition of CP-AMPARs to spines innervated by the basolateral amygdala, a structure important for associative learning, could help explain incubation of cue-induced cocaine craving. It is also possible that CP-AMPARs are selectively added to new synapses generated by cocaine exposure (see section “CP-AMPARs, spine plasticity and silent synapses”). Alternatively, CP-AMPARs may be added more broadly, that is, across synapses innervated by multiple brain regions. In this case, selectivity may be conferred through plasticity upstream of the NAc that promotes activation of particular afferent pathways to the NAc; this could occur through a variety of mechanisms and might even involve sprouting of new presynaptic terminals. Such cocaine-induced plasticity could affect the ability of different inputs to compete for influence over MSN output (e.g., Goto and Grace, 2005). Once a particular afferent pathway is activated, global postsynaptic adaptations such as AMPAR upregulation would help to determine the strength of ensemble activation. For discussion of how different types of cocaine-induced neuroadaptations may interact to influence MSN output, see previous reviews (Wheeler and Carelli, 2009; Wolf, 2010a; Huang et al., 2011b; Lee and Dong, 2011).
CP-AMPARs, Spine Plasticity, and Silent Synapses
A major complicating factor in understanding cocaine-induced AMPAR plasticity is that it is superimposed upon structural plasticity. New spines are observed in the NAc after both non-contingent and contingent cocaine exposure (Robinson and Kolb, 2004; Russo et al., 2010) and may participate in functional synapses (Alcantara et al., 2010; Dobi et al., 2011). For example, extended access cocaine self-administration followed by prolonged withdrawal leads not only to CP-AMPAR accumulation (section “CP-AMPARs accumulate in the NAc during withdrawal from extended access cocaine self-administration”), but also to increased spine density in the NAc (Ferrario et al., 2005). Are the CP-AMPARs added to the new spines, to pre-existing spines, or both? An intriguing possibility is that new spines, populated with CP-AMPARs, synapse with new presynaptic terminals that have sprouted during withdrawal. This would represent a very profound rewiring of NAc circuits that could help explain the complexity of addiction-related behavioral changes.
The question of how synaptic and structural plasticity are integrated after cocaine exposure is very broad. A more specific question is how silent synapses may be involved. As noted above, NR2B-containing silent synapses are generated in the NAc during repeated i.p. cocaine injections and are proposed to house CI-AMPARs added during withdrawal (see section “CI-AMPARs predominate in the NAc of drug-naïve rodents and upregulate during the first week of withdrawal from non-contingent cocaine administration”). Could the same scenario (formation of NR2B-containing silent synapses followed by AMPAR incorporation) explain the accumulation of CP-AMPARs in the incubation model? This seems unlikely if silent synapse formation occurs during cocaine exposure, since weeks pass between the last cocaine self-administration session and the detection of CP-AMPARs (Figure 1). Furthermore, given evidence for a mechanistic link between mGluR1-mediated CP-AMPAR removal and CI-AMPAR insertion (McCutcheon et al., 2011b), it seems likely that CP-AMPARs accumulate at synapses that either express CI-AMPARs or have the potential to rapidly produce them in response to mGluR1 stimulation. The main theoretical support for possible CP-AMPAR incorporation into silent synapses comes from evidence that NR2B-containing NMDARs, silent synapses, and CP-AMPARs are all prevalent early in development. For example, in the VTA, NR2B-containing NMDARs and CP-AMPARs undergo coordinated regulation during early postnatal development (Bellone et al., 2011). Based on these and other results, it has been suggested that cocaine exposure recapitulates the highly plastic synapses characteristic of early development, thereby facilitating learning about drugs (e.g., Bellone et al., 2011; Lee and Dong, 2011), although this idea must be reconciled with the behavioral inflexibility exhibited once addiction is established.
While these theories are intriguing, the role of silent synapses in synapse maturation during normal development is still not fully understood (Kerchner and Nicoll, 2008). Furthermore, cocaine may not faithfully recapitulate normal development or normal plasticity. Perhaps CP-AMPARs are inserted into new spines, but the new spines do not correspond to silent synapses generated during cocaine exposure; instead, they might result from weeks of withdrawal leading to multiple rounds of interacting synaptic and structural plasticity. It is very difficult to predict how relationships between new spines, their complement of NMDARs, and AMPAR synaptic insertion evolve during withdrawal from extended access cocaine self-administration. However, in comparing cocaine and saline groups on WD45, we have found that a smaller proportion of spines from the NAc of cocaine rats exhibit Ca2+ influx in response to NMDAR activation (Ferrario et al., 2012). We speculate that spines containing CP-AMPARs may have low NMDAR levels, similar to what has been observed in the VTA of cocaine-exposed mice (Mameli et al., 2011) and in other CP-AMPAR-containing synapses (Lamsa et al., 2007; Wang and Gao, 2010).
CP-AMPARs in the NAc: Functional Significance
As noted in section “NAc: Background,” cocaine seeking requires AMPAR transmission in the NAc. Therefore, addition of AMPARs to excitatory synapses onto MSNs of the NAc is expected to enhance cocaine seeking. Due to the higher conductance of CP-AMPARs compared to CI-AMPARs (Cull-Candy et al., 2006; Isaac et al., 2007), excitatory synaptic transmission onto MSN should be increased more robustly after regimens leading to CP-AMPAR incorporation (extended access cocaine self-administration followed by prolonged withdrawal) compared to regimens leading to increased CI-AMPAR levels (non-contingent cocaine administration or limited access cocaine self-administration). This may explain greater (incubated) cue-induced cocaine craving in the former situation. Of course, the presence of CP-AMPARs also provides a new source of Ca2+ influx into dendritic spines of MSN. Which attribute of CP-AMPARs (higher conductance, Ca2+ permeability, or different kinetics and voltage-dependence) is most important for drug seeking (or how they may differently contribute) is unknown. In addition, as noted in the previous section, a thorough understanding of altered NAc function after cocaine exposure will require integration of information on presynaptic plasticity, postsynaptic plasticity, and the associated structural plasticity. Although beyond the scope of this review, additional important factors are cocaine-induced alterations in voltage-gated conductances and the subsequent changes in intrinsic excitability in MSNs; such changes interact in a homeostatic fashion with the regulation of synaptic strength (Ishikawa et al., 2009; for review see Wolf, 2010a; Huang et al., 2011b). By focusing on postsynaptic glutamate receptors, we may be examining only a very tiny portion of the elephant.
Many questions remain about the circumstances in which NAc CP-AMPARs are functionally important for drug seeking. To date, CP-AMPAR synaptic incorporation has been definitively linked to incubation of cue-induced craving, i.e., blocking CP-AMPARs in the NAc core prevents the expression of incubated craving (Conrad et al., 2008; section “CP-AMPARs accumulate in the NAc during withdrawal from extended access cocaine self-administration”). Interestingly, electrophysiological recordings in the NAc core of awake rats indicate that incubation is associated with an increase in the number of MSN that encode information related to cocaine or cocaine-associated cues, as well as the strength of neuronal activation during cocaine self-administration (Hollander and Carelli, 2005, 2007)7. Synaptic strengthening via CP-AMPAR incorporation could account for these observations. Are CP-AMPARs important beyond the incubation model? One issue is that the extended access cocaine regimen typically used to produce incubation (6 h/day × 10 days) is shorter than the “classical” extended access regimen developed by Ahmed and colleagues (e.g., 10 or more days of limited access training followed by 10 or more additional days at 6 h/day; Ahmed, 2011). Recently, we found that this “classical” extended access regimen also resulted in CP-AMPAR accumulation in the NAc after prolonged withdrawal, whereas significant changes in CP-AMPAR levels were not observed after prolonged withdrawal from limited access cocaine self-administration (Purgianto et al., submitted; section “CI-AMPARs predominate in the NAc of drug-naïve rodents and upregulate during the first week of withdrawal from non-contingent cocaine administration”). This raises the possibility that CP-AMPARs contribute to addiction-related behaviors observed exclusively after extended access cocaine self-administration, such as escalation of drug intake, willingness to seek drug despite punishment, and greater motivation to work for drug (Ahmed, 2011).
Do CP-AMPARs contribute to reinstatement of cocaine seeking evoked by stress or a priming injection of cocaine? This is also unknown. We suggest that CP-AMPAR accumulation during cocaine withdrawal is not important for cocaine-primed reinstatement because this behavioral response does not incubate (Lu et al., 2004b). Furthermore, the state of MSN during cocaine-primed reinstatement is certainly very different than during cue-induced seeking or other drug-free tests. In the first situation, the dominant effect may be the cocaine-induced elevation of extracellular DA levels. In the latter situations, the major consequence may be cue-induced activation of excitatory afferents onto MSNs—in this case, the presence of CP-AMPARs would be of greatest significance. This is analogous to our speculation that AMPAR plasticity is more relevant to incentive sensitization (glutamate-dominated) than to locomotor sensitization (DA-dominated) (see section “CI-AMPARs predominate in the NAc of drug-naïve rodents and upregulate during the first week of withdrawal from non-contingent cocaine administration”). However, as noted for cocaine challenge after non-contingent exposure (section “CI-AMPARs predominate in the NAc of drug-naïve rodents and upregulate during the first week of withdrawal from non-contingent cocaine administration”), it is possible that cocaine re-exposure after cocaine self-administration rapidly increases CP-AMPAR levels. This is based on a study of cocaine-primed reinstatement after limited access cocaine self-administration (Anderson et al., 2008). These authors showed that the priming injection of cocaine led to a rapid increase (30 min) in GluA1 surface expression, and that reinstatement was attenuated by intra-NAc shell injection of a viral vector expressing a peptide (GluA1-C99) which impairs the transport of GluA1-containing AMPARs. Unfortunately GluA2 was not measured in this study, so it remains unknown whether reinstatement involves increased surface expression of GluA1A2-containing AMPARs or homomeric GluA1 receptors, although indirect evidence argues that GluA1 and GluA2 are differently regulated in this model (Famous et al., 2008). If cocaine-primed reinstatement does elicit synaptic incorporation of CP-AMPARs, they most likely originate from the small pool normally present in MSN, because CP-AMPAR levels do not increase significantly after withdrawal from limited access cocaine self-administration (see previous paragraph).
A potential functional consequence of CP-AMPAR insertion into MSN synapses could be altered rules for induction of synaptic plasticity, as demonstrated in the VTA (Mameli et al., 2011; see section “CP-AMPARs in the VTA: Functional significance”), but this has not been tested in the NAc. The work that has been done has primarily examined the effect of non-contingent or limited access cocaine self-administration on subsequent synaptic plasticity. The main value of such studies is that they provide insight into the mechanisms by which drugs of abuse alter the properties of a particular synapse, which may shed light on potential therapeutic strategies. However, describing the consequences of a particular cocaine regimen as a simple function of LTP or LTD impairment or facilitation is less informative because LTP and LTD are exogenously induced phenomena that are very dependent on experimental conditions. Nevertheless, it is perceived as a problem in the literature that very discrepant reports have appeared regarding the ability to induce LTP or LTD in the NAc of cocaine-exposed animals (e.g., Thomas et al., 2001; Yao et al., 2004; Goto and Grace, 2005; Martin et al., 2006; Mameli et al., 2009; Moussawi et al., 2009; Kasanetz et al., 2010; Knackstedt et al., 2010; Pascoli et al., 2011). The differences can usually be explained based on regimen or withdrawal time (Wolf and Ferrario, 2010). For example, after a week or so of withdrawal from non-contingent cocaine, when CI-AMPAR upregulation has occurred, occlusion of LTP would be predicted and has been reported (Pascoli et al., 2011). However, in studies conducted at early withdrawal times or shortly after cocaine challenge (Thomas et al., 2001; Yao et al., 2004; Goto and Grace, 2005; Kasanetz et al., 2010), it is likely that synaptic AMPAR levels are in flux, so occlusion of LTP due to AMPAR upregulation would not necessarily be expected8. Other aspects of the treatment regimen can also be critical in determining the nature of plasticity. For example, the same limited access cocaine self-administration regimen leads to impairment of both LTP and LTD if followed by extinction training, but impairment of only LTP if followed by home-cage withdrawal (Moussawi et al., 2009; Knackstedt et al., 2010). Perhaps the most important point is that useful hypotheses will not be developed by lumping together studies that differ in critical aspects of their experimental design.
Summary: CP-AMPARs in the NAc
CI-AMPARs predominate in the NAc of drug-naïve rodents, although CP-AMPARs make a small contribution to synaptic transmission. Repeated i.p. cocaine injections, leading to behavioral sensitization, result in increased cell surface and synaptic levels of CI-AMPARs in the NAc core and shell. This occurs during the first week of withdrawal, persists for at least 2 more weeks, but ultimately wanes. Despite correlations between the development of AMPAR upregulation and the development of locomotor sensitization, they can be dissociated, prompting us to propose that elevated AMPAR levels in the NAc may be more directly related to incentive sensitization. Limited access cocaine self-administration also increases synaptic AMPAR levels in a withdrawal-dependent manner in the NAc shell (core has not been directly tested); these appear to be CI-AMPARs rather than CP-AMPARs. In contrast, extended access cocaine self-administration (leading to incubation of cocaine craving) results in CP-AMPAR accumulation. This occurs in both core and shell subregions of the NAc. The increase in CP-AMPAR levels is quite delayed—a significant increase in the rectification index is first observed approximately a month after the last cocaine self-administration session—but endures through at least WD70 and perhaps much longer. Once CP-AMPARs are present, they can be rapidly removed from NAc synapses by acute pharmacological stimulation of mGluR1 receptors in the NAc. This is accompanied by strengthening of CI-AMPAR transmission, although the net effect is synaptic depression since high conductance CP-AMPAR transmission is eliminated and low conductance CI-AMPAR transmission is enhanced. These and other results suggest that mGluR1-mediated CP-AMPAR removal is coupled to CI-AMPAR insertion into NAc synapses. In studies designed to understand the delayed accumulation of CP-AMPARs during withdrawal, we found that mGluR1 surface expression in the NAc is decreased on WD25, prior to the detection of increased CP-AMPAR levels, suggesting that loss of inhibitory mGluR1 tone may be permissive for CP-AMPAR accumulation. Overall, the delay in CP-AMPAR accumulation suggests that it represents homeostatic plasticity perhaps involving slowly developing structural changes. Functionally, CP-AMPARs in the NAc mediate the expression of incubated cue-induced cocaine seeking at long withdrawal times, most likely by strengthening synaptic connections between MSN and forebrain regions that trigger drug seeking. A better understanding of how CP-AMPARs promote addiction-related behaviors will require experiments to determine if CP-AMPARs are inserted into new or pre-existing spines, which afferents contact these spines, and whether newly formed afferents are involved.
Comparison of Mechanisms Regulating CP-AMPAR Levels in the VTA and the NAc
Accumulation of CP-AMPARs After Cocaine Exposure
NMDAR transmission in VTA DA neurons during cocaine exposure is a common requirement for CP-AMPAR insertion in both VTA DA neurons and NAc MSN (Engblom et al., 2008; Mameli et al., 2009; see sections “Cocaine increases the synaptic incorporation of CP-AMPARs in VTA DA neurons” and “CP-AMPARs accumulate in the NAc during withdrawal from extended access cocaine self-administration”). However, beyond this, marked differences in the conditions required to elicit CP-AMPAR plasticity in the VTA and NAc suggest divergent underlying mechanisms (see Table 1 for a summary).
A major difference is that CP-AMPARs can be detected in VTA synapses within hours of cocaine exposure, whereas CP-AMPARs require approximately a month of withdrawal to accumulate in NAc synapses (Table 1). The role of GluA2 in CP-AMPAR accumulation may also differ between the two regions. As noted in section “Cocaine-induced synaptic incorporation of CP-AMPARs and reversal of this plasticity by mGluR1 stimulation are GluA2-dependent processes,” the mechanisms regulating CP-AMPAR levels in synapses can be divided into two types, one dependent on GluA2, and the other involving increased GluA1 with very little change in GluA2. In VTA DA neurons, cocaine-induced CP-AMPAR insertion is GluA2-dependent. It requires a GluA2-PICK interaction (Bellone and Lüscher, 2006) and is associated with redistribution of GluA2 labeling from synapses to the cytoplasm (Mameli et al., 2007; Brown et al., 2010). Thus, there is an exchange of CP-AMPARs and CI-AMPARs, apparently driven by greater CI-AMPAR availability. In the NAc, however, the accumulation of CP-AMPARs was originally detected as a robust increase in surface, intracellular and total GluA1 on WD45 in the absence of significant changes in GluA2 (Conrad et al., 2008). However, small changes in GluA2 were observed. Thus, cocaine rats on WD1 had slightly higher surface GluA2 than saline controls, whereas cocaine rats on WD45 had slightly lower surface GluA2 than saline controls (neither change was statistically significant). The same pattern was observed for the surface/intracellular (S/I) ratio, but in this case the GluA2 S/I ratio was significantly increased in cocaine rats on WD1 compared to all other groups (Conrad et al., 2008). Overall, given that GluA1 changes were many-fold more robust than GluA2 changes and that GluA2 changes were most evident on WD1, our results argue that the main effect after prolonged withdrawal is an addition of CP-AMPARs on top of existing CI-AMPARs, rather than an exchange between the two subtypes. Even if some exchange does occur, aspects of the exchange process or its triggering must differ between VTA and NAc in light of the markedly different time-courses of CP-AMPAR accumulation in these regions. One difference may be the role of mGluR1. As discussed in section “mGluR1 negatively regulates synaptic levels of CP-AMPARs in the NAc,” CP-AMPAR accumulation in the NAc may be attributable in part to a delayed but persistent decrease in mGluR1 surface expression which removes the normal mGluR1-mediated braking effect on CP-AMPAR levels (Loweth et al., 2011). In the VTA, mGluR1 similarly exerts a negative regulatory influence on CP-AMPAR levels (Bellone and Lüscher, 2005, 2006; Mameli et al., 2009; see section “mGluR1 negatively regulates CP-AMPAR levels in VTA DA neurons”), but there is no evidence to indicate that a cocaine-induced reduction in mGluR1 signaling contributes to cocaine-induced increase in CP-AMPAR levels in the VTA.
Accumulation of CP-AMPARs in the NAc during incubation may result not only from impaired mGluR1-mediated removal but also from an increased rate of synaptic insertion. This idea is based on increased levels of GluA1 phosphorylated on serine 845 (pS845 GluA1) in extrasynaptic membrane fractions prepared from the NAc of rats after extended access cocaine self-administration and 45 days of withdrawal (conditions leading to incubation and CP-AMPAR synaptic incorporation), compared to saline controls (Ferrario et al., 2011b). How might this be linked to CP-AMPAR synaptic insertion? We showed previously, in cultured NAc neurons, that GluA1 phosphorylation at S845 accelerates AMPAR externalization onto extrasynaptic regions of the MSN plasma membrane, increasing the size of the extrasynaptic AMPAR pool; this in turn primes AMPARs for NMDAR-dependent synaptic insertion (Sun et al., 2008; Wolf, 2010b). Thus, the observed increase in extrasynaptic pS845 GluA1 after incubation could help explain increased CP-AMPAR levels in NAc synapses (Ferrario et al., 2011b). GluA1 phosphorylation at S845 is also associated with increased GluA1 and GluA2 surface expression in the NAc after non-contingent cocaine exposure leading to locomotor sensitization (Schierberl et al., 2011), so this mechanism may be relevant for cocaine-induced increases in both CI-AMPARs and CP-AMPARs.
The observation that CP-AMPARs are largely excluded from NAc synapses under normal conditions but are permitted to accumulate after incubation suggests the existence of mechanisms that selectively regulate CP-AMPAR association with the PSD. Although patch clamp recordings have established that CP-AMPARs participate in synaptic transmission once they accumulate during cocaine withdrawal (Conrad et al., 2008; Ferrario et al., 2011b; McCutcheon et al., 2011a,b), biochemical results suggest that these CP-AMPARs are loosely tethered to the PSD. The latter conclusion was based on surprising findings in a detergent insoluble PSD fraction. Although we had expected that GluA1 levels would be increased in PSD fractions prepared from the NAc after prolonged withdrawal, we actually observed a decrease. However, the expected increase in GluA1 was detected in an LP1 fraction from identically treated rats (the LP1 fraction is a less purified synaptic membrane fraction that has not been exposed to detergent). These and other results suggested that CP-AMPARs are weakly anchored to the PSD, resulting in their loss during preparation of the PSD fraction (Ferrario et al., 2011b). Other results are indicative of “un-anchored” homomeric GluA1 receptors under some conditions (Goel et al., 2011). We speculate that this unique relationship between CP-AMPARs and the PSD may contribute to their selective regulation. For example, if loose anchoring of CP-AMPARs indicates a perisynaptic location, this could result in physical proximity to perisynaptic mGluR1 receptors, explaining the ability of mGluR1 stimulation to selective remove CP-AMPARs from synapses.
Interactions of CP-AMPARs with transmembrane AMPAR regulatory proteins (TARPs) may also be important during incubation. In the NAc of drug-naïve animals, the TARP γ 2 is enriched in synaptic membranes, whereas γ 4 is mainly found in an extrasynaptic membrane-enriched fraction (Ferrario et al., 2011a). Results obtained after extended access cocaine self-administration (WD35–45), by measuring TARPs and GluA1 in the same subcellular fractions and in biotinylation experiments, suggest that γ 4 is associated with CP-AMPARs added to extrasynaptic membranes during incubation, whereas synaptic CP-AMPARs may be associated with γ 2 (Ferrario et al., 2011b). These findings raise the possibility that TARPs contribute to cocaine-induced neuroadaptations.
Removal of CP-AMPARs by mGLuR1 Stimulation and its Therapeutic Implications
In both VTA and NAc, mGluR1 stimulation results in coordinated removal of high conductance CP-AMPARs and their replacement by lower conductance CI-AMPARs, resulting in synaptic depression (sections “mGluR1 negatively regulates CP-AMPAR levels in VTA DA neurons,” “Cocaine-induced synaptic incorporation of CP-AMPARs and reversal of this plasticity by mGluR1 stimulation are GluA2-dependent processes,” and “mGluR1 negatively regulates synaptic levels of CP-AMPARs in the NAc”). Considerably more is known about mechanisms involved in this process in the VTA, particularly with respect to CI-AMPAR insertion, which requires local translation of GluR2 (Mameli et al., 2007). Whether a similar mechanism underlies mGluR1-dependent CI-AMPAR insertion in the NAc is unknown (see section “mGluR1 negatively regulates synaptic levels of CP-AMPARs in the NAc”).
One difference between NAc and VTA concerns the properties of mGluR-LTD in the control versus the cocaine-exposed state. In the NAc of drug-naïve rats, DHPG normally acts on mGluR5 receptors to elicit a presynaptically expressed CB1R-dependent form of mGluR-LTD. After extended access cocaine self-administration, this pathway is disabled in concert with the withdrawal-dependent emergence of CP-AMPARs and postsynaptically expressed mGluR1-dependent LTD (McCutcheon et al., 2011b; see section “mGluR1 negatively regulates synaptic levels of CP-AMPARs in the NAc” for more discussion of mGluR-LTD after other cocaine regimens). In contrast, mGluR-LTD is not observed in the VTA of control mice, but only emerges after cocaine exposure (Bellone and Lüscher, 2006; Mameli et al., 2007).
Stimulation of mGluR1 also removes CP-AMPARs from synapses in cerebellar stellate cells of drug-naïve rats (Kelly et al., 2009), indicating a general relationship between mGluR1 and CP-AMPARs that extends across different brain regions. Interestingly, CP-AMPARs in stellate cells are probably homomeric GluA3 (Keinanen et al., 1990; Sato et al., 1993; Liu and Cull-Candy, 2002), whereas in the VTA and NAc of cocaine-exposed rodents they appear to be GluA1-containing (sections “What is the subunit composition of CP-AMPARs inserted into VTA synapses by cocaine?” and “CP-AMPARs accumulate in the NAc during withdrawal from extended access cocaine self-administration”). Thus, if a common mechanism is operating in all brain regions, it may not be selective for GluA1 vs. GluA3 despite marked differences in their C-terminals and thus in their regulation by phosphorylation and protein–protein interactions (Shepherd and Huganir, 2007).
The negative regulatory role for mGluR1 with regard to CP-AMPAR levels in both VTA and NAc suggests that mGluR1 positive allosteric modulators might be useful in the treatment of addiction. It remains to be determined how long CP-AMPARs remain down-regulated in VTA or NAc neurons of cocaine-exposed animals following mGluR1 stimulation. However, in a study of developmental consequences of in utero cocaine exposure, a single i.p. injection of the mGluR1 positive allosteric modulator Ro 67-7476 produced effects lasting 10 days (Bellone et al., 2011). Even a very transient down-regulation of CP-AMPARs might be therapeutically useful given that CP-AMPARs mediate expression of the incubation of cue-induced craving (Conrad et al., 2008). Perhaps treatment with an mGluR1 agonist prior to an anticipated encounter with cocaine-related cues could thereby maintain abstinence.
At first glance, the suggestion that mGluR1 stimulation has potential therapeutic utility seems to conflict with a large body of work showing anti-craving effects of systemic and intra-NAc blockade of group I mGluRs (in particular mGluR5) (Olive, 2009). However, none of these studies have tested cocaine seeking in animals that have experienced both extended access cocaine self-administration and a prolonged period of withdrawal—conditions that lead to CP-AMPAR accumulation in NAc synapses. Once CP-AMPARs are present in synapses, stimulating mGluR1, rather than inhibiting group I mGluRs, may be a more effective way to decrease excitatory transmission in the NAc and reduce craving. It is not surprising that different mGluR-based pharmacological strategies may be optimal for reducing cocaine seeking depending on the mode of cocaine exposure and the duration of withdrawal. Both of these variables can significantly influence the effect of cocaine on group I mGluR expression and signaling in the NAc (e.g., Ben-Shahar et al., 2009; Hao et al., 2010).
Summary of Comparison
The NAc is similar to the VTA in that cocaine exposure increases CP-AMPAR levels and mGluR1 stimulation reverses this effect. However, CP-AMPAR insertion in VTA is rapid (hours), GluA2-dependent, and produced by even a single non-contingent cocaine injection. In contrast, CP-AMPAR accumulation in the NAc occurs only after extended access cocaine self-administration and prolonged withdrawal (~30 days), and may be GluA1-dependent. Once CP-AMPARs are present in VTA or NAc synapses, they can be rapidly removed by mGluR1 stimulation. Given that CP-AMPARs in the NAc mediate the intensified cue-induced cocaine craving observed after prolonged withdrawal, drugs that stimulate mGluR1 may prove useful in reducing craving and extending abstinence.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge their support from the NIH (DA009621 to Marina E. Wolf and Kuei Y. Tseng, DA015835 and DA029099 to Marina E. Wolf, and MH086507 to Kuei Y. Tseng) and Rosalind Franklin University of Medicine and Science. We thank Dr. Michela Marinelli for her insightful comments on this manuscript.
Footnotes
- ^In vivo, all addictive drugs acutely activate DA transmission in brain regions that are the targets of midbrain dopamine neurons (e.g., the NAc). Psychostimulants do this by acting within these target regions to block the DA transporter and increase extracellular DA levels; thus, they activate DA transmission in the target regions despite the fact that they inhibit DA cell firing at the level of the midbrain (where they elevate DA levels and thus activate inhibitory somatodendritic DA autoreceptors). In contrast, nicotine, opiates, ethanol, and cannabinoids act within the midbrain to increase DA cell firing rate and thereby activate DA transmission in dopaminergic target regions (Pierce and Kumaresan, 2006; Wolf, 2012). Thus, although activation of DA transmission in target regions is a common effect of all drugs of abuse, it is important to keep in mind that the firing rate of midbrain DA neurons is differently affected by different classes of abused drugs (see White, 1996; Marinelli, 2007).
- ^It should be acknowledged that the relationship between DA autoreceptor sensitivity and DA cell activity depends on how the latter is assessed. On WD1 from repeated cocaine injections, when autoreceptors are subsensitive, increases in both firing rate and the number of spontaneously active cells were reported (Henry et al., 1989). On WD7, a time when DA autoreceptor subsensitivity has dissipated, firing rate was normalized but the number of spontaneously active VTA DA cells continued to be higher than control levels (Gao et al., 1998). Together, these studies suggest that different mechanisms regulate the number of spontaneously active cells and the firing rate of active cells during cocaine withdrawal.
- ^Silent synapses are generally defined as synapses that express an NMDAR-mediated response but not an AMPAR-mediated response (Kerchner and Nicoll, 2008).
- ^In D2R-positive MSNs of D2-EGFP heterozygotic BAC transgenic mice, mGluR5-stimulated endocannabinoid formation also triggers a postsynaptically expressed form of LTD-mediated via TRPV1 channel activation and internalization of CI-AMPARs (Grueter et al., 2010).
- ^Relatively few prior studies have measured mGluR1 in the NAc after cocaine exposure, and results are difficult to compare to our own due to procedural differences (see Swanson et al., 2001; Ary and Szumlinski, 2007; Ben-Shahar et al., 2009).
- ^Blunting of mGluR5-CB1R-mediated LTD has also been reported in the bed nucleus of the stria terminalis following cocaine exposure (Grueter et al., 2006, 2008).
- ^Interestingly, these changes are not seen in the NAc shell (Hollander and Carelli, 2005, 2007), despite the presence of CP-AMPARs in the mouse NAc shell at long withdrawal times associated with high cue-induced cocaine seeking (Mameli et al., 2009; McCutcheon et al., 2011b), as discussed in more detail previously (Wolf and Ferrario, 2010).
- ^At short withdrawal times from non-contingent cocaine injections, CI-AMPAR upregulation may be absent or just developing. In the days following cocaine challenge, AMPAR levels may be transitioning between a cocaine-induced decrease and a withdrawal-dependent upregulation. For details, see section “CI-AMPARs predominate in the NAc of drug-naïve rodents and upregulate during the first week of withdrawal from non-contingent cocaine administration” and Wolf and Ferrario (2010).
References
Ackerman, J. M., and White, R. J. (1990). A10 somatodendritic dopamine autoreceptor sensitivity following withdrawal from repeated cocaine treatment. Neurosci. Lett. 117, 181–187.
Ahmed, S. H. (2011). “Escalation of drug use,” in Animal Models of Drug Addiction, Neuromethods, Vol. 53, Part 2, ed M. C. Olmsted (New York, NY: Springer Science+Business Media, LLC 2011), 267–292.
Alcantara, A. A., Lim, H. L., Floyd, C. E., Garces, J., Mendenhall, J. M., Lyons, C. L., and Berlanga, M. L. (2010). Cocaine- and morphine-induced synaptic plasticity in the nucleus accumbens. Synapse 65, 309–320.
Anderson, S. M., Famous, K. R., Sadri-Vakili, G., Kumaresan, V., Schmidt, H. D., Bass, C. E., Terwilliger, E. F., Cha, J. H., and Pierce, R. C. (2008). CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat. Neurosci. 11, 344–353.
Anggono, V., and Huganir, R. L. (2012). Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. doi: 10.1016/j.conb.2011.12.006. [Epub ahead of print].
Argilli, E., Sibley, D. R., Malenka, R. C., England, P. M., and Bonci, A. (2008). Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J. Neurosci. 28, 9092–9100.
Ary, A. W., and Szumlinski, K. K. (2007). Regional differences in the effects of withdrawal from repeated cocaine upon Homer and glutamate receptor expression: a two-species comparison. Brain Res. 1184, 295–305.
Bachtell, R. K., and Self, D. W. (2008). Renewed cocaine exposure produces transient alterations in nucleus accumbens AMPA receptor-mediated behavior. J. Neurosci. 28, 12808–12814.
Bannon, M. J., and Roth, R. H. (1983). Pharmacology of mesocortical dopamine neurons. Pharmacol. Rev. 35, 53–68.
Bellone, C., and Lüscher, C. (2005). mGluRs induce a long-term depression in the ventral tegmental area that involves a switch of the subunit composition of AMPA receptors. Eur. J. Neurosci. 21, 1280–1288.
Bellone, C., and Lüscher, C. (2006). Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat. Neurosci. 9, 636–641.
Bellone, C., Mameli, M., and Lüscher, C. (2011). In utero exposure to cocaine delays postnatal synaptic maturation of glutamatergic transmission in the VTA. Nat. Neurosci. 14, 1439–1446.
Beninger, R. J., and Banasikowski, T. J. (2008). Dopaminergic mechanism of reward-related incentive learning: focus on the dopamine D(3) receptor. Neurotox. Res. 14, 57–70.
Ben-Shahar, O., and Ettenberg, A. (1994). Repeated stimulation of the ventral tegmental area sensitizes the hyperlocomotor response to amphetamine. Pharmacol. Biochem. Behav. 48, 1005–1009.
Ben-Shahar, O., Obara, I., Ary, A. W., Ma, N., Mangiardi, M. A., Medina, R. L., and Szumlinski, K. K. (2009). Extended daily access to cocaine results in distinct alterations in homer 1b/c and NMDA receptor subunit expression within the medial prefrontal cortex. Synapse 63, 598–609.
Bird, M. K., Reid, C. A., Chen, F., Tan, H. O., Petrou, S., and Lawrence, A. J. (2010). Cocaine-mediated synaptic potentiation is absent in VTA neurons form mGlu5-deficient mice. Int. J. Neuropsychopharmacol. 13, 133–1341.
Bockaert, J., Perroy, J., Bécamel, C., Marin, P., and Fagni, L. (2010). GPCR interacting proteins (GIPs) in the nervous system: roles in physiology and pathologies. Annu. Rev. Pharmacol. Toxicol. 50, 89–109.
Bonci, A., and Malenka, R. C. (1999). Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J. Neurosci. 19, 3723–3730.
Borgland, S. L., Malenka, R. C., and Bonci, A. (2004). Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J. Neurosci. 24, 7482–7490.
Borgland, S. L., Taha, S., Sarti, F., Fields, H. L., and Bonci, A. (2006). Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron 49, 589–601.
Boudreau, A. C., Ferrario, C. R., Glucksman, M. J., and Wolf, M. E. (2009). Signaling pathway adaptations and novel protein kinase A substrates related to behavioral sensitization to cocaine. J. Neurochem. 110, 363–377.
Boudreau, A. C., Reimers, J. M., Milovanovic, M., and Wolf, M. E. (2007). Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J. Neurosci. 27, 10621–10635.
Boudreau, A. C., and Wolf, M. E. (2005). Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J. Neurosci. 25, 9144–9151.
Bowers, M. S., Chen, B. T., and Bonci, A. (2010). AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron 67, 11–24.
Brebner, K., Wong, T. P., Liu, L., Liu, Y., Campsall, P., Gray, S., Phelps, L., Phillips, A. G., and Wang, Y. T. (2005). Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 310, 1340–1343.
Brill, J., and Huguenard, J. R. (2008). Sequential changes in AMPA receptor targeting in the developing neocortical excitatory circuit. J. Neurosci. 28, 13918–13928.
Brown, M. T., Bellone, C., Mameli, M., Labouèbe, G., Bocklisch, C., Balland, B., Dahan, L., Luján, R., Deisseroth, K., and Lüscher, C. (2010). Drug-driven AMPA receptor redistribution mimicked by selective dopamine neuron stimulation. PLoS ONE 5:e15870. doi: 10.1371/journal.pone.0015870
Brown, T. E., Lee, B. R., Mu, P., Ferguson, D., Dietz, D., Ohnishi, Y. N., Lin, Y., Suska, A., Ishikawa, M., Huang, Y. H., Shen, H., Kalivas, P. W., Sorg, B. A., Zukin, R. S., Nestler, E. J., Dong, Y., and Schlüter, O. M. (2011). A silent synapse-based mechanism for cocaine-induced locomotor sensitization. J. Neurosci. 31, 8163–8174.
Campioni, M. R., Xu, M., and McGehee, D. S. (2009). Stress-induced changes in nucleus accumbens glutamate synaptic plasticity. J. Neurophysiol. 101, 3192–3198.
Carlezon, W. A. Jr., Boundy, V. A., Haile, C. N., Lane, S. B., Kalb, R. G., Neve, R. L., and Nestler, E. J. (1997). Sensitization to morphine induced by viral-mediated gene transfer. Science 277, 812–814.
Carlezon, W. A. Jr., and Nestler, E. J. (2002). Elevated levels of GluR1 in the midbrain: a trigger for sensitization to drugs of abuse? Trends Neurosci. 25, 610–615.
Chen, B. T., Bowers, M. S., Martin, M., Hopf, F. W., Guillory, A. M., Carelli, R. M., Chou, J. K., and Bonci, A. (2008). Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 59, 288–297.
Chiodo, L. A., Bannon, M. J., Grace, A. A., Roth, R. H., and Bunney, B. S. (1984). Evidence for the absence of impulse-regulating somatodendritic and synthesis-modulating nerve terminal autoreceptors on subpopulations of mesocortical dopamine neurons. Neuroscience 12, 1–16.
Choi, K. H., Edwards, S., Graham, D. L., Larson, E. B., Whisler, K. N., Simmons, D., Friedman, A. K., Walsh, J. J., Rahman, Z., Monteggia, L. M., Eisch, A. J., Neve, R. L., Nestler, E. J., Han, M. H., and Self, D. W. (2011). Reinforcement-related regulation of AMPA glutamate receptor subunits in the ventral tegmental area enhances motivation for cocaine. J. Neurosci. 31, 7927–7937.
Churchill, L., Swanson, C. J., Urbina, M., and Kalivas, P. W. (1999). Repeated cocaine alters glutamate receptor subunit levels in the nucleus accumbens and ventral tegmental area of rats that develop behavioral sensitization. J. Neurochem. 72, 2397–2403.
Clark, D., and White, F. J. (1987). D1 dopamine receptor–the search for a function: a critical evaluation of the D1/D2 dopamine receptor classification and its functional implications. Synapse 1, 347–388.
Conrad, K. L., Tseng, K. Y., Uejima, J. L., Reimers, J. M., Heng, L. J., Shaham, Y., Marinelli, M., and Wolf, M. E. (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121.
Cull-Candy, S., Kelly, L., and Farrant, M. (2006). Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr. Opin. Neurobiol. 16, 288–297.
Dobi, A., Seabold, G. K., Christensen, C. H., Bock, R., and Alvarez, V. A. (2011). Cocaine-induced plasticity in the nucleus accumbens is cell specific and develops without prolonged withdrawal. J. Neurosci. 31, 1895–1904.
Dong, Y., Saal, D., Thomas, M., Faust, R., Bonci, A., Robinson, T., and Malenka, R. C. (2004). Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(-/-) mice. Proc. Natl. Acad. Sci. U.S.A. 101, 14282–14287.
Engblom, D., Bilbao, A., Sanchis-Segura, C., Dahan, L., Perreau-Lenz, S., Balland, B., Parkitna, J. R., Luján, R., Halbout, B., Mameli, M., Parlato, R., Sprengel, R., Lüscher, C., Schütz, G., and Spanagel, R. (2008). Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron 59, 497–508.
Everitt, B. J., Parkinson, J. A., Olmstead, M. C., Arroyo, M., Robledo, P., and Robbins, T. W. (1999). Associative processes in addiction and reward. The role of amygdala-ventral striatal subsystems. Ann. N.Y. Acad. Sci. 877, 412–438.
Everitt, B. J., and Robbins, T. W. (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat. Neurosci. 8, 1481–1489.
Eybalin, M., Caicedo, A., Renard, N., Ruel, J., and Puel, J. L. (2004). Transient Ca2+-permeable AMPA receptors in postnatal rat primary auditory neurons. Eur. J. Neurosci. 20, 2981–2989.
Fagni, L., Worley, P. F., and Ango, F. (2002). Homer as both a scaffold and transduction molecule. Sci. STKE 137:re8.
Faleiro, L. J., Jones, S., and Kauer, J. A. (2004). Rapid synaptic plasticity of glutamatergic synapses on dopamine neurons in the ventral tegmental area in response to acute amphetamine injection. Neuropsychopharmacology 29, 2115–2125.
Famous, K. R., Kumaresan, V., Sadri-Vakili, G., Schmidt, H. D., Mierke, D. F., Cha, J. H., and Pierce, R. C. (2008). Phosphorylation-dependent trafficking of GluR2-containing AMPA receptors in the nucleus accumbens plays a critical role in the reinstatement of cocaine seeking. J. Neurosci. 28, 11061–11070.
Ferrario, C. R., Gorny, G., Crombag, H. S., Li, Y., Kolb, B., and Robinson, T. E. (2005). Neural and behavioral plasticity associated with the transition from controlled to escalated cocaine use. Biol. Psychiatry 58, 751–759.
Ferrario, C. R., Goussakov, I., Stutzmann, G. E., and Wolf, M. E. (2012). Withdrawal from cocaine self-administration alters NMDA receptor-mediated Ca2+ entry in nucleus accumbens dendritic spines. PLoS ONE, in press.
Ferrario, C. R., Li, X., Wang, X., Reimers, J. M., Uejima, J. L., and Wolf, M. E. (2010). The role of glutamate receptor redistribution in locomotor sensitization to cocaine. Neuropsychopharmacology 35, 818–833.
Ferrario, C. R., Loweth, J. A., Milovanovic, M., Wang, X., and Wolf, M. E. (2011a). Distribution of AMPA receptor subunits and TARPs in synaptic and extrasynaptic membranes of the adult rat nucleus accumbens. Neurosci. Lett. 490, 180–184.
Ferrario, C. R., Loweth, J. A., Milovanovic, M., Ford, K. A., Galiñanes, G. L., Heng, L. J., Tseng, K. Y., and Wolf, M. E. (2011b). Alterations in AMPA receptor subunits and TARPs in the rat nucleus accumbens related to the formation of Ca2+-permeable AMPA receptors during the incubation of cocaine craving. Neuropharmacology 61, 1141–1151.
Ferrario, C. R., Li, X., and Wolf, M. E. (2011c). Effects of acute cocaine or dopamine receptor agonists on AMPA receptor distribution in the nucleus accumbens. Synapse 65, 54–63.
Finch, D. M. (1996). Neurophysiology of converging synaptic inputs from the rat prefrontal cortex, amygdala, midline thalamus, and hippocampal formation onto single neurons of the caudate/putamen and nucleus accumbens. Hippocampus 6, 495–512.
Fitzgerald, L. W., Ortiz, J., Hamedani, A. G., and Nestler, E. J. (1996). Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J. Neurosci. 16, 274–282.
Fourgeaud, L., Mato, S., Bouchet, D., Hémar, A., Worley, P. F., and Manzoni, O. J. (2004). A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J. Neurosci. 24, 6939–6945.
Galloway, M. P., Wolf, M. E., and Roth, R. H. (1986). Regulation of dopamine synthesis in the medial prefrontal cortex is mediated by release modulating autoreceptors: studies in vivo. J. Pharmacol. Exp. Ther. 236, 689–698.
Gao, C., and Wolf, M. E. (2007). Dopamine alters AMPA receptor synaptic expression and subunit composition in dopamine neurons of the ventral tegmental area cultured with prefrontal cortex neurons. J. Neurosci. 27, 14725–14785.
Gao, W. Y., Lee, T. H., King, G. R., and Ellinwood, E. H. (1998). Alterations in baseline activity and quinpirole sensitivity in putative dopamine neurons in the substantia nigra and ventral tegmental area after withdrawal from cocaine pretreatment. Neuropsychopharmacology 18, 222–232.
Gerdeman, G. L., Partridge, J. G., Lupica, C. R., and Lovinger, D. M. (2003). It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 26, 184–192.
Ghasemzadeh, M. B., Mueller, C., and Vasudevan, P. (2009a). Behavioral sensitization to cocaine is associated with increased glutamate receptor trafficking to the postsynaptic density after extended withdrawal period. Neuroscience 159, 414–426.
Ghasemzadeh, M. B., Vasudevan, P., Mueller, C., Seubert, C., and Mantsch, J. R. (2009b). Neuroadaptations in the cellular and postsynaptic group I metabotropic glutamate receptor mGluR5 and Homer proteins following extinction of cocaine self-administration. Neurosci. Lett. 452, 167–171.
Giorgetti, M., Hotsenpiller, G., Ward, P., Teppen, T., and Wolf, M. E. (2001). Amphetamine-induced plasticity of AMPA receptors in the ventral tegmental area: effects on extracellular levels of dopamine and glutamate in freely moving rats. J. Neurosci. 21, 6362–6369.
Goel, A., Xu, L. W., Snyder, K. P., Song, L., Goenaga-Vazquez, Y., Megill, A., Takamiya, K., Huganir, R. L., and Lee, H. K. (2011). Phosphorylation of AMPA receptors is required for sensory eprivation-induced homeostatic synaptic plasticity. PLoS ONE 6:e18264. doi: 10.1371/journal.pone.0018264
Good, C. H., and Lupica, C. R. (2010). Afferent-specific AMPA receptor subunit composition and regulation of synaptic plasticity in midbrain dopamine neurons by abused drugs. J. Neurosci. 30, 7900–7909.
Goto, Y., and Grace, A. A. (2005). Dopamine-dependent interactions between limbic and prefrontal cortical plasticity in the nucleus accumbens: disruption by cocaine sensitization. Neuron 47, 255–266.
Groenewegen, H. J., Wright, C. I., Beijer, A. V., and Voorn, P. (1999). Convergence and segregation of ventral striatal inputs and outputs. Ann. N.Y. Acad. Sci. 877, 49–63.
Grueter, B. A., Brasnjo, G., and Malenka, R. C. (2010). Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat. Neurosci. 13, 1519–1525.
Grueter, B. A., Gosnell, H. B., Olsen, C. M., Schramm-Sapyta, N. L., Nekrasova, T., Landreth, G. E., and Winder, D. G. (2006). Extracellular-signal regulated kinase 1-dependent metabotropic glutamate receptor 5-induced long-term depression in the bed nucleus of the stria terminalis is disrupted by cocaine administration. J. Neurosci. 26, 3210–3219.
Grueter, B. A., McElligott, Z. A., and Winder, D. G. (2007). Group I mGluRs and long-term depression: potential roles in addiction? Mol. Neurobiol. 36, 232–244.
Grueter, B. A., McElligott, Z. A., Robison, A. J., Mathews, G. C., and Winder, D. G. (2008). In vivo metabotropic glutamate receptor 5 (mGluR5) antagonism prevents cocaine-induced disruption of postsynaptically maintained mGluR5-dependent long-term depression. J. Neurosci. 28, 9261–9270.
Hao, Y., Martin-Fardon, R., and Weiss, F. (2010). Behavioral and functional evidence of metabotropic glutamate receptor 2/3 and metabotropic glutamate receptor 5 dysregulation in cocaine-escalated rats: factor in the transition to dependence. Biol. Psychiatry 68, 240–248.
He, K., Song, L., Cummings, L. W., Goldman, J., Huganir, R. L., and Lee, H. K. (2009). Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 20033–20038.
Henry, D. J., Greene, M. A., and White, F. J. (1989). Electrophysiological effects of cocaine in the mesoaccumbens dopamine system: repeated administration. J. Pharmacol. Exp. Ther. 251, 833–839.
Ho, M. T., Pelkey, K. A., Topolnik, L., Petralia, R. S., Takamiya, K., Xia, J., Huganir, R. L., Lacaille, J. C., and McBain, C. J. (2007). Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J. Neurosci. 27, 11651–11662.
Ho, S. Y., Chen, C. H., Liu, T. H., Chang, H. F., and Liou, J. C. (2012). Protein kinase Mζ is necessary for cocaine-induced synaptic potentiation in the ventral tegmental area. Biol. Psychiatry 71, 706–713.
Hollander, J. A., and Carelli, R. M. (2005). Abstinence from cocaine self-administration heightens neural encoding of goal-directed behaviors in the accumbens. Neuropsychopharmacology 30, 1464–1474.
Hollander, J. A., and Carelli, R. M. (2007). Cocaine-associated stimuli increase cocaine seeking and activate accumbens core neurons after abstinence. J. Neurosci. 27, 3535–3539.
Horger, B. A., Shelton, K., and Schenk, S. (1990). Pre-exposure sensitizes rats to the rewarding effects of cocaine. Pharmacol. Biochem. Behav. 37, 707–711.
Hu, X. T., Brooderson, R. J., and White, F. J. (1992). Repeated stimulation of D1 dopamine receptors causes time-dependent alterations in the sensitivity of both D1 and D2 dopamine receptors within the rat striatum. Neuroscience 50, 137–147.
Huang, C. C., Yeh, C. M., Wu, M. Y., Chang, A. Y., Chan, J. Y., Chan, S. H., and Hsu, K. S. (2011a). Cocaine withdrawal impairs metabotropic glutamate receptor-dependent long-term depression in the nucleus accumbens. J. Neurosci. 31, 4194–4203.
Huang, Y. H., Schlüter, O. M., and Dong, Y. (2011b). Cocaine-induced homeostatic regulation and dysregulation of nucleus accumbens neurons. Behav. Brain Res. 216, 9–18.
Huang, Y. H., Lin, Y., Mu, P., Lee, B. R., Brown, T. E., Wayman, G., Marie, H., Liu, W., Yan, Z., Sorg, B. A., Schluter, O. M., Zukin, R. S., and Dong, Y. (2009). In vivo cocaine experience generates silent synapses. Neuron 63, 40–47.
Humphries, M. D., and Prescott, T. J. (2010). The ventral basal ganglia, a selection mechanism at the crossroads of space, strategy, and reward. Prog. Neurobiol. 90, 385–417.
Isaac, J. T., Ashby, M. C., and McBain, C. J. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871.
Ishikawa, M., Mu, P., Moyer, J. T., Wolf, J. A., Quock, R. M., Davies, N. M., Hu, X. T., Schlüter, O. M., and Dong, Y. (2009). Homeostatic synapse-driven membrane plasticity in nucleus accumbens neurons. J. Neurosci. 29, 5820–5831.
Kalivas, P. W., and Alesdatter, J. E. (1993). Involvement of N-methyl-D-aspartate receptor stimulation in the ventral tegmental area and amygdala in behavioral sensitization to cocaine. J. Pharmacol. Exp. Ther. 267, 486–495.
Kalivas, P. W., and Stewart, J. (1991). Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res. Rev. 16, 223–244.
Kalivas, P. W., and Volkow, N. D. (2005). The neural basis of addiction: a pathology of motivation and choice. Am. J. Psychiatry 162, 1403–1413.
Kamata, K., and Rebec, G. V. (1984). Long-term amphetamine treatment attenuates or reverses the depression of neuronal activity produced by dopamine agonists in the ventral tegmental area. Life Sci. 34, 2419–2427.
Karler, R., Calder, L. D., Chaudhry, I. A., and Turkanis, S. A. (1989). Blockade of ‘reverse tolerance’ to cocaine and amphetamine by MK-801. Life Sci. 45, 599–606.
Kasanetz, F., Deroche-Gamonet, V., Berson, N., Balado, E., Lafourcade, M., Manzoni, O., and Piazza, P. V. (2010). Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science 328, 1709–1712.
Kauer, J. A., and Malenka, R. C. (2007). Synaptic plasticity in addiction. Nat. Rev. Neurosci. 8, 844–858.
Kauer, J. A., Malenka, R. C., and Nicoll, R. A. (1988). A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron 1, 911–917.
Keinanen, K., Wisden, W., Sommer, B., Werner, P., Herb, A., Verdoorn, T. A., Sakmann, B., and Seeberg, P. H. (1990). A family of AMPA-selective glutamate receptors. Science 249, 556–560.
Kelly, L., Farrant, M., and Cull-Candy, S. G. (2009). Synaptic mGluR activation drives plasticity of calcium-permeable AMPA receptors. Nat. Neurosci. 12, 593–601.
Kerchner, G. A., and Nicoll, R. A. (2008). Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci. 9, 813–825.
Knackstedt, L. A., Moussawi, K., Lalumiere, R., Schwendt, M., Klugmann, M., and Kalivas, P. W. (2010). Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J. Neurosci. 30, 7984–7992.
Kolb, B., Gorny, G., Li, Y., Samaha, A. N., and Robinson, T. E. (2003). Amphetamine or cocaine limits the ability of later experience to promote structural plasticity in the neocortex and nucleus accumbens. Proc. Natl. Acad. Sci. U.S.A. 100, 10523–10528.
Kourrich, S., Klug, J. R., Mayford, M., and Thomas, M. J. (2012). AMPAR-independent effect of striatal αCaMKII promotes the sensitization of cocaine reward. J. Neurosci. 32, 6578–6586.
Kourrich, S., Rothwell, P. E., Klug, J. R., and Thomas, M. J. (2007). Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J. Neurosci. 27, 7921–7928.
Kullmann, D. M., and Lamsa, K. P. (2011). LTP and LTD in cortical GABAergic interneurons: emerging rules and roles. Neuropharmacology 60, 712–719.
Kumar, S. S., Bacci, A., Kharazia, V., and Huguenard, J. R. (2002). A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J. Neurosci. 22, 3005–3015.
Lammel, S., Hetzel, A., Häckel, O., Jones, I., Liss, B., and Roeper, J. (2008). Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron 57, 760–773.
Lammel, S., Ion, D. I., Roeper, J., and Malenka, R. C. (2011). Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron 70, 855–862.
Lamsa, K. P., Heeroma, J. H., Somogyi, P., Rusakov, D. A., and Kullmann, D. M. (2007). Anti-Hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science 315, 1262–1266.
Lane, D. A., Jaferi, A., Kreek, M. J., and Pickel, V. M. (2010). Acute and chronic cocaine differentially alter the subcellular distribution of AMPA GluR1 subunits in region-specific neurons within the mouse ventral tegmental area. Neuroscience 169, 559–573.
Lane, D. A., Reed, B., Kreek, M. J., and Pickel, V. M. (2011). Differential glutamate AMPA-receptor plasticity in subpopulations of VTA neurons in the presence or absence of residual cocaine: implications for the development of addiction. Neuropharmacology 61, 1129–1140.
Lee, H. K. (2012). Ca2+-permeable AMPA receptors in homeostatic synaptic plasticity. Front. Mol. Neurosci. 5:17. doi: 10.3389/fnmol.2012.00017. [Epub ahead of print].
Lee, B. R., and Dong, Y. (2011). Cocaine-induced metaplasticity in the nucleus accumbens: silent synapse and beyond. Neuropharmacology 61, 1060–1069.
Li, Y., Hu, X. T., Berney, T. G., Vartanian, A. J., Stine, C., Wolf, M. E., and White, F. J. (1999). Both glutamate receptor antagonists and prefrontal cortex lesions prevent the induction of cocaine sensitization and associated neuroadaptations. Synapse 34, 169–180.
Liu, S. J., and Cull-Candy, S. G. (2002). Activity-dependent change in AMPA receptor properties in cerebellar stellate cells. J. Neurosci. 22, 3881–3889.
Liu, S. J., and Cull-Candy, S. G. (2005). Subunit interaction with PICK and GRIP controls Ca2+ permeability of AMPARs at cerebellar synapses. Nat. Neurosci. 8, 768–775.
Liu, Q. S., Pu, L., and Poo, M. M. (2005). Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature 437, 1027–1031.
Liu, S. J., and Zukin, R. S. (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134.
Lovinger, D. M. (2008). Presynaptic modulation by endocannabinoids. Handb. Exp. Pharmacol. 184, 435–477.
Loweth, J. A., McCutcheon, J. E., Milovanovic, M., Ford, K. A., Scheyer, A. F., Marinelli, M., Tseng, K. Y., and Wolf, M. E. (2011). Cocaine-induced AMPA receptor plasticity: modulation by metabotropic glutamate receptors. Soc. Neurosci. Abstr. 37, 67.08.
Loweth, J. A., Reimers, J. M., Milovanovic, M., Ford, K. A., Ferrario, C. R., McCutcheon, J. E., Marinelli, M., Tseng, K. Y., and Wolf, M. E. (2010). Group I metabotropic glutamate receptors regulate calcium-permeable AMPA receptors in the rat nucleus accumbens. Soc. Neurosci. Abstr. 36, 366.18.
Lu, L., Grimm, J. W., Hope, B. T., and Shaham, Y. (2004a). Incubation of cocaine craving after withdrawal: a review of preclinical data. Neuropharmacology 47(Suppl. 1) 214–226.
Lu, L., Grimm, J. W., Dempsey, J., and Shaham, Y. (2004b). Cocaine seeking over extended withdrawal periods in rats: different time courses of responding induced by cocaine cues versus cocaine priming over the first 6 months. Psychopharmacology 176, 101–108.
Lu, W., Monteggia, L. M., and Wolf, M. E. (2002). Repeated administration of amphetamine or cocaine does not alter AMPA receptor subunit expression in the rat midbrain. Neuropsychopharmacology 26, 1–13.
Luo, Y., Good, C. H., Diaz-Ruiz, O., Zhang, Y., Hoffman, A. F., Shan, L., Kuang, S. Y., Malik, N., Chefer, V. I., Tomac, A. C., Lupica, C. R., and Bäckman, C. M. (2010). NMDA receptors on non-dopaminergic neurons in the VTA support cocaine sensitization. PLoS ONE 5:e12141. doi: 10.1371/journal.pone.0012141
Lüscher, C., and Huber, K. M. (2010). Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459.
Lüscher, C., and Malenka, R. C. (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663.
Luu, P., and Malenka, R. C. (2008). Spike timing-dependent long-term potentiation in ventral tegmental area dopamine cells requires PKC. J. Neurophysiol. 100, 533–538.
Mameli, M., Balland, B., Lujan, R., and Lüscher, C. (2007). Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317, 530–533.
Mameli, M., Bellone, C., Brown, M. T., and Lüscher, C. (2011). Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat. Neurosci. 14, 414–416.
Mameli, M., Halbout, B., Creton, C., Engblom, D., Parkitna, J. R., Spanagel, R., and Lüscher, C. (2009). Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat. Neurosci. 12, 1036–1041.
Mangiavacchi, S., and Wolf, M. E. (2004). Stimulation of N-methyl-D-aspartate receptors, AMPA receptors or metabotropic glutamate receptors leads to rapid internalization of AMPA receptors in cultured nucleus accumbens neurons. Eur. J. Neurosci. 20, 649–657.
Marinelli, M. (2007). “Dopaminergic reward pathways and effects of stress,” in Stress and Addiction: Biological and Psychological Mechanisms, ed M. al'Absi (Amsterdam, The Netherlands: Elsevier Science), 41–83.
Marinelli, M., and White, F. J. (2000). Enhanced vulnerability to cocaine self-administration is associated with elevated impulse activity of midbrain dopamine neurons. J. Neurosci. 20, 8876–8885.
Marinelli, M., Cooper, D. C., Baker, L. K., and White, F. J. (2003). Impulse activity of midbrain dopamine neurons modulates drug-seeking behavior. Psychopharmacology (Berl.) 168, 84–98.
Marinelli, M., and Piazza, P. V. (2002). Interaction between glucocorticoid hormones, stress and psychostimulant drugs. Eur. J. Neurosci. 16, 387–394.
Martin, M., Chen, B. T., Hopf, F. W., Bowers, M. S., and Bonci, A. (2006). Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat. Neurosci. 9, 868–869.
McCutcheon, J. E., Wang, X., Tseng, K. Y., Wolf, M. E., and Marinelli, M. (2011a). Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. J. Neurosci. 31, 5737–5743.
McCutcheon, J. E., Loweth, J. A., Ford, K. A., Marinelli, M., Wolf, M. E., and Tseng, K. Y. (2011b). Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J. Neurosci. 31, 14536–14541.
McCutcheon, J. E., White, F. J., and Marinelli, M. (2009). Individual differences in dopamine cell neuroadaptations following cocaine self-administration. Biol. Psychiatry 66, 801–803.
Meredith, G. E., Baldo, B. A., Andrezjewski, M. E., and Kelley, A. E. (2008). The structural basis for mapping behavior onto the ventral striatum and its subdivisions. Brain Struct. Funct. 213, 17–27.
Mogenson, G. J., Jones, D. L., and Yim, C. Y. (1980). From motivation to action: functional interface between the limbic system and the motor system. Prog. Neurobiol. 14, 69–97.
Moussawi, K., Pacchioni, A., Moran, M., Olive, M. F., Gass, J. T., Lavin, A., and Kalivas, P. W. (2009). N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat. Neurosci. 12, 182–189.
Nelson, C. L., Milovanovic, M., Wetter, J. B., Ford, K. A., and Wolf, M. E. (2009). Behavioral sensitization to amphetamine is not accompanied by changes in glutamate receptor surface expression in the rat nucleus accumbens. J. Neurochem. 109, 35–51.
Nestler, E. J., Terwilliger, R. Z., Walker, J. R., Sevarino, K. A., and Duman, R. S. (1990). Chronic cocaine treatment decreases levels of the G protein subunits Giα and Goα in discrete regions of rat brain. J. Neurochem. 55, 1079–1082.
Niehaus, J. L., Murali, M., and Kauer, A. J. (2010). Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur. J. Neurosci. 32, 108–117.
O'Donnell, P., and Grace, A. A. (1995). Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J. Neurosci. 15, 3622–3639.
Olive, M. F. (2009). Metabotropic glutamate receptor ligands as potential therapeutics for addiction. Curr. Drug Abuse Rev. 2, 83–98.
Ortinski, P. I., Vassoler, F. M., Carlson, G. C., and Pierce, R. C. (2012). Temporally dependent changes in cocaine-induced synaptic plasticity in the nucleus accumbens shell are reversed by D1-Like dopamine receptor stimulation. Neuropsychopharmacology 37, 1671–1682.
Overton, P. G., Richards, C. D., Berry, M. S., and Clark, D. (1999). Long-term potentiation at excitatory amino acid synapses on midbrain dopamine neurons. Neuroreport 10, 221–226.
Pascoli, V., Turiault, M., and Lüscher, C. (2011). Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behavior. Nature 481, 71–75.
Pickens, C. L., Airavaara, M., Theberge, F., Fanous, S., Hope, B. T., and Shaham, Y. (2011). Neurobiology of the incubation of drug craving. Trends Neurosci. 34, 411–420.
Pierce, R. C., Bell, K., Duffy, P., and Kalivas, P. W. (1996). Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J. Neurosci. 16, 1550–1560.
Pierce, R. C., and Kumaresan, V. (2006). The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci. Biobehav. Rev. 30, 215–238.
Post, R. M., Weiss, S. R. B., Fontana, D., and Pert, A. (1992). “Conditioned sensitization to the psychomotor stimulant cocaine,” in The Neurobiology of Drug and Alcohol Addiction, eds P. W. Kalivas and H. H. Samson (New York, NY: The New York Academy of Sciences), 386–399.
Post, R. M., Weiss, S. R. B., and Pert, A. (1987). The role of context and conditioning in behavioral sensitization to cocaine. Psychopharmacol. Bull. 23, 425–429.
Pu, L., Liu, Q. S., and Poo, M. M. (2006). BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat. Neurosci. 9, 605–607.
Reimers, J. M., Milovanovic, M., and Wolf, M. E. (2011). Quantitative analysis of AMPA receptor subunit composition in addiction-related brain regions. Brain Res. 1367, 223–233.
Robbe, D., Kopf, M., Remaury, A., Bockaert, J., and Manzoni, O. J. (2002). Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc. Natl. Acad. Sci. U.S.A. 99, 8384–8388.
Robinson, T. E. (1984). Behavioral sensitization: characterization of enduring changes in rotational behavior produced by intermittent injections of amphetamine in male and female rats. Psychopharmacology 84, 466–475.
Robinson, T. E., and Berridge, K. C. (2008). Review. The incentive sensitization theory of addiction: some current issues. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 3137–3146.
Robinson, T. E., and Kolb, B. (2004). Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 47(Suppl. 1), 33–46.
Russo, S. J., Dietz, D. M., Dumitriu, D., Morrison, J. H., Malenka, R. C., and Nestler, E. J. (2010). The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 33, 267–276.
Saal, D., Dong, Y., Bonci, A., and Malenka, R. C. (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37, 577–582.
Sato, K., Kiyama, H., and Tohyama, M. (1993). The differential expression of patterns of messenger RNAs encoding non-N-methyl-D-aspartate glutamate receptor subunits (GluR1–4) in the rat brain. Neuroscience 52, 515–539.
Schenk, S., and Snow, S. (1994). Sensitization to cocaine's motor activating properties produced by electrical kindling of the medial prefrontal cortex but not of the hippocampus. Brain Res. 659, 17–22.
Schierberl, K., Hao, J., Tropea, T. F., Ra, S., Giordano, T. P., Xu, Q., Garraway, S. M., Hofmann, F., Moosmang, S., Striessnig, J., Inturrisi, C. E., and Rajadhyaksha, A. M. (2011). Cav1.2 L-type Ca 2+ channels mediate cocaine-induced GluA1 trafficking in the nucleus accumbens, a long-term adaptation dependent on ventral tegmental area Cav1.3 channels. J. Neurosci. 31, 13562–13575.
Schilström, B., Yaka, R., Argilli, E., Suvarna, N., Schumann, J., Chen, B. T., Carman, M., Singh, V., Mailliard, W. S., Ron, D., and Bonci, A. (2006). Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J. Neurosci. 26, 8549–8558.
Schumann, J., and Yaka, R. (2009). Prolonged withdrawal from repeated noncontingent cocaine exposure increases NMDA receptor expression and ERK activity in the nucleus accumbens. J. Neurosci. 29, 6955–6963.
Seif, T., Makriyannis, A., Kunos, G., Bonci, A., and Hopf, F. W. (2011). The endocannabinoid 2-arachidonoylglycerol mediates D1 and D2 receptor cooperative enhancement of rat nucleus accumbens core neuron firing. Neuroscience 193, 21–33.
Selvakumar, B., Campbell, P. W., Milovanovic, M., Park, D. J., West, A. R., Snyder, S. H., and Wolf, M. E. (2011). S-nitrosylation of stargazin mediates cocaine sensitization-linked AMPA receptor externalization in nucleus accumbens. Soc. Neurosci. Abstr. 37, 40.10.
Sesack, S. R., and Grace, A. A. (2010). Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology 35, 27–47.
Shepherd, J. D., and Huganir, R. L. (2007). The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 23, 613–643.
Steketee, J. D., and Kalivas, P. W. (1991). Sensitization to psychostimulants and stress after injection of pertussis toxin into the A10 dopamine region. J. Pharmacol. Exp. Ther. 259, 916–924.
Striplin, C. D., and Kalivas, P. W. (1992). Correlation between behavioral sensitization to cocaine and G protein ADP-ribosylation in the ventral tegmental area. Brain Res. 579, 181–186.
Stuber, G. D., Klanker, M., de Ridder, B., Bowers, M. S., Joosten, R. N., Feenstra, M. G., and Bonci, A. (2008). Reward-predictive cues enhance excitatory synaptic strength onto midbrain dopamine neurons. Science 321, 1690–1692.
Sun, X., Milovanovic, M., Zhao, Y., and Wolf, M. E. (2008). Acute and chronic dopamine receptor stimulation modulates AMPA receptor trafficking in nucleus accumbens neurons cocultured with prefrontal cortex neurons. J. Neurosci. 28, 4216–4230.
Sun, X., and Wolf, M. E. (2009). Nucleus accumbens neurons exhibit synaptic scaling that is occluded by repeated dopamine pre-exposure. Eur. J. Neurosci. 30, 539–550.
Surmeier, D. J., Song, W. J., and Yan, Z. (1996). Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J. Neurosci. 16, 6579–6591.
Suto, N., Tanabe, L. M., Austin, J. D., Creekmore, E., Pham, C. T., and Vezina, P. (2004). Previous exposure to psychostimulants enhances the reinstatement of cocaine seeking by nucleus accumbens AMPA. Neuropsychopharmacology 29, 2149–2159.
Swanson, C. J., Baker, D. A., Carson, D., Worley, P. F., and Kalivas, P. W. (2001). Repeated cocaine administration attenuates group I metabotropic glutamate receptor-mediated glutamate release and behavioral activation: a potential role for Homer. J. Neurosci. 21, 9043–9052.
Szumlinski, K. K., Ary, A. W., and Lominac, K. D. (2008). Homers regulate drug-induced neuroplasticity: implications for addiction. Biochem. Pharmacol. 75, 112–133.
Thomas, M. J., Beurrier, C., Bonci, A., and Malenka, R. C. (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat. Neurosci. 4, 1217–1223.
Thomas, M. J., Malenka, R. C., and Bonci, A. (2000). Modulation of long-term depression by dopamine in the mesolimbic system. J. Neurosci. 20, 5581–5586.
Ungless, M. A., Whistler, J. L., Malenka, R. C., and Bonci, A. (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411, 583–587.
Vanderschuren, L. J., and Pierce, R. C. (2010). Sensitization processes in drug addiction. Curr. Top Behav. Neurosci. 3, 179–195.
Vanderschuren, L. J., Schmidt, E. D., De Vries, T. J., Van Moorsel, C. A., Tilders, F. J., and Schoffelmeer, A. N. (1999). A single exposure to amphetamine is sufficient to induce long-term behavioral, neuroendocrine, and neurochemical sensitization in rats. J. Neurosci. 19, 9579–9586.
Vezina, P. (2004). Sensitization of midbrain dopamine neuron reactivity and the self-administration of psychomotor stimulant drugs. Neurosci. Biobehav. Rev. 27, 827–839.
Voorn, P., Vanderschuren, L. J. M. J., Groenewegen, H. J., Robbins, T. W., and Pennartz, C. M. A. (2004). Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci. 27, 468–474.
Yao, W. D., Gainetdinov, R. R., Arbuckle, M. I., Sotnikova, T. D., Cyr, M., Beaulieu, J. M., Torres, G. E., Grant, S. G., and Caron, M. G. (2004). Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41, 625–638.
Wang, H. X., and Gao, W. J. (2010). Development of calcium-permeable AMPA receptors and their correlation with NMDA receptors in fast-spiking interneurons of rat prefrontal cortex. J. Physiol. (Lond.) 588, 2823–2838.
Wang, X., Cahill, M. E., Xie, Z., Loweth, J. A., Werner, C. T., Russo, S. J., Penzes, P., and Wolf, M. E. (2011). Kalirin-7 contributes to the increased AMPA receptor surface expression and spine density observed in the nucleus accumbens of cocaine-sensitized rats. Soc. Neurosci. Abstr. 37, 67.09.
Wheeler, R. A., and Carelli, R. M. (2009). Dissecting motivational circuitry to understand substance abuse. Neuropharmacology 56(Suppl. 1), 149–159.
White, F. J. (1996). Synaptic regulation of mesocorticolimbic dopamine neurons. Annu. Rev. Neurosci. 19, 405–436.
White, F. J., Hu, X. T., and Brooderson, R. J. (1990). Repeated stimulation of dopamine D1 receptors enhances the effects of dopamine receptor agonists. Eur. J. Pharmacol. 191, 497–499.
White, F. J., Hu, X. T., Zhang, X. F., and Wolf, M. E. (1995). Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J. Pharmacol. Exp. Ther. 273, 445–454.
White, F. J., and Wang, R. Y. (1984). Electrophysiological evidence for A10 dopamine autoreceptor subsensitivity following chronic D-amphetamine treatment. Brain Res. 309, 283–292.
Wolf, M. E. (1998). The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog. Neurobiol. 54, 679–720.
Wolf, M. E. (2002). “Effects of psychomotor stimulants on glutamate receptor expression,” in Methods in Molecular Medicine, Vol. 79 : Drugs of Abuse: Analysis of Neurological Effects, ed J. Q. Wang (Totowa, NJ: Humana Press), 13–31.
Wolf, M. E. (2010a). The Bermuda triangle of cocaine-induced neuroadaptations. Trends Neurosci. 33, 391–398.
Wolf, M. E. (2010b). Regulation of AMPA receptor trafficking in the nucleus accumbens by dopamine and cocaine. Neurotoxicol. Res. 18, 393–409.
Wolf, M. E. (2012). “Addiction,” in Basic Neurochemistry, 8th edition, eds S. T. Brady, G. J. Siegel, R. W. Albers., and D. L. Price (Waltham, MA: Elsevier Academic Press), 1037–1055.
Wolf, M. E., and Ferrario, C. R. (2010). AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neurosci. Biobehav. Rev. 35, 185–211.
Wolf, M. E., Galloway, M. P., and Roth, R. H. (1986). Regulation of dopamine synthesis in the medial prefrontal cortex: studies in brain slices. J. Pharmacol. Exp. Ther. 236, 699–707.
Wolf, M. E., and Roth, R. H. (1987). “Dopamine autoreceptors,” in Structure and Function of Dopamine Receptors, Receptor Biochemistry and Methodology, Vol. 9, eds I. Creese and C. M. Fraser, (New York, NY: Alan R. Liss, Inc.), 45–96.
Wolf, M. E., White, F. J., and Hu, X. T. (1994). MK-801 prevents alterations in the mesoaccumbens dopamine system associated with behavioral sensitization to amphetamine. J. Neurosci. 14, 1735–1745.
Wolf, M. E., White, F. J., Nassar, R., Brooderson, R. J., and Khansa, M. R. (1993). Differential development of autoreceptor subsensitivity and enhanced dopamine release during amphetamine sensitization. J. Pharmacol. Exp. Ther. 264, 249–255.
Xia, J., Zhang, X., Staudinger, J., and Huganir, R. L. (1999). Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron 22, 179–187.
Xue, Y., Steketee, J. D., Rebec, G. V., and Sun, W. (2011). Activation of D2-like receptors in rat ventral tegmental area inhibits cocaine-reinstated drug-seeking behavior. Eur. J. Neurosci. 33, 1291–1298.
Yao, W. D., Gainetdinov, R. R., Arbuckle, M. I., Sotnikova, T. D., Cyr, M., Beaulieu, J. M., Torres, G. E., Grant, S. G., and Caron, M. G. (2004). Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41, 625–638.
Zhang, X. F., Hu, X. T., White, F. J., and Wolf, M. E. (1997). Increased responsiveness of ventral tegmental area dopamine neurons to glutamate after repeated administration of cocaine or amphetamine is transient and selectively involves non-NMDA receptors. J. Pharmacol. Exp. Ther. 281, 699–706.
Keywords: addiction, calcium-permeable AMPA receptor, cocaine, nucleus accumbens, ventral tegmental area
Citation: Wolf ME and Tseng KY (2012) Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why?. Front. Mol. Neurosci. 5:72. doi: 10.3389/fnmol.2012.00072
Received: 19 January 2012; Paper pending published: 20 February 2012;
Accepted: 21 May 2012; Published online: 27 June 2012.
Edited by:
R. Suzanne Zukin, Albert Einstein College of Medicine, USAReviewed by:
Lorna Role, SUNY Stony Brook, USAMiou Zhou, University of California, Los Angeles, USA
Copyright: © 2012 Wolf and Tseng. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Marina E. Wolf, Department of Neuroscience, Rosalind Franklin University of Medicine and Science, 3333 Green Bay Road, North Chicago, IL 60048, USA. e-mail:bWFyaW5hLndvbGZAcm9zYWxpbmRmcmFua2xpbi5lZHU=