 Ryan S. Tonkin1
Ryan S. Tonkin1 Yilin Mao2
Yilin Mao2 Simon J. O’Carroll3Louise F. B. Nicholson3Colin R. Green4
Simon J. O’Carroll3Louise F. B. Nicholson3Colin R. Green4 Catherine A. Gorrie2
Catherine A. Gorrie2 Gila Moalem-Taylor1*
Gila Moalem-Taylor1*
- 1School of Medical Sciences, Faculty of Medicine, University of New South Wales, Sydney, NSW, Australia
- 2School of Medical and Molecular Bioscience, Faculty of Science, University of Technology, Sydney, NSW, Australia
- 3Department of Anatomy with Radiology and Centre for Brain Research, Faculty of Medical and Health Sciences, University of Auckland, Auckland, New Zealand
- 4Department of Ophthalmology, Faculty of Medical and Health Sciences, University of Auckland, Auckland, New Zealand
Gap junctions are specialized intercellular communication channels that are formed by two hexameric connexin hemichannels, one provided by each of the two adjacent cells. Gap junctions and hemichannels play an important role in regulating cellular metabolism, signaling, and functions in both normal and pathological conditions. Following spinal cord injury (SCI), there is damage and disturbance to the neuronal elements of the spinal cord including severing of axon tracts and rapid cell death. The initial mechanical disruption is followed by multiple secondary cascades that cause further tissue loss and dysfunction. Recent studies have implicated connexin proteins as playing a critical role in the secondary phase of SCI by propagating death signals through extensive glial networks. In this review, we bring together past and current studies to outline the distribution, changes and roles of various connexins found in neurons and glial cells, before and in response to SCI. We discuss the contribution of pathologically activated connexin proteins, in particular connexin 43, to functional recovery and neuropathic pain, as well as providing an update on potential connexin specific pharmacological agents to treat SCI.
Introduction
Gap junctions are specialized intercellular connections that allow direct electrical and metabolic communication between two adjacent cells; they are therefore vital to many physiological processes. By controlling the movement of amino acids, second messengers, ions, and other metabolites, gap junctions are able to coordinate cellular signaling and propagate electrical signals (Hertzberg et al., 1981; Söhl et al., 2005). In vertebrates, gap junctions are formed by two hemichannels (or connexons), one provided by each of two cells. Each hemichannel consists of six oligomerized connexin proteins (abbreviated as Cx), which can be either homomeric or heteromeric (two or more different connexins in a connexon) and form homotypic or heterotypic (different connexons) gap junction channels (Orthmann-Murphy et al., 2008). Connexins are named according to their estimated molecular weights and have been shown to be expressed in almost all mammalian tissues (Söhl and Willecke, 2003; Söhl et al., 2005). The structure of these connexin proteins is described in Figures 1A,B.
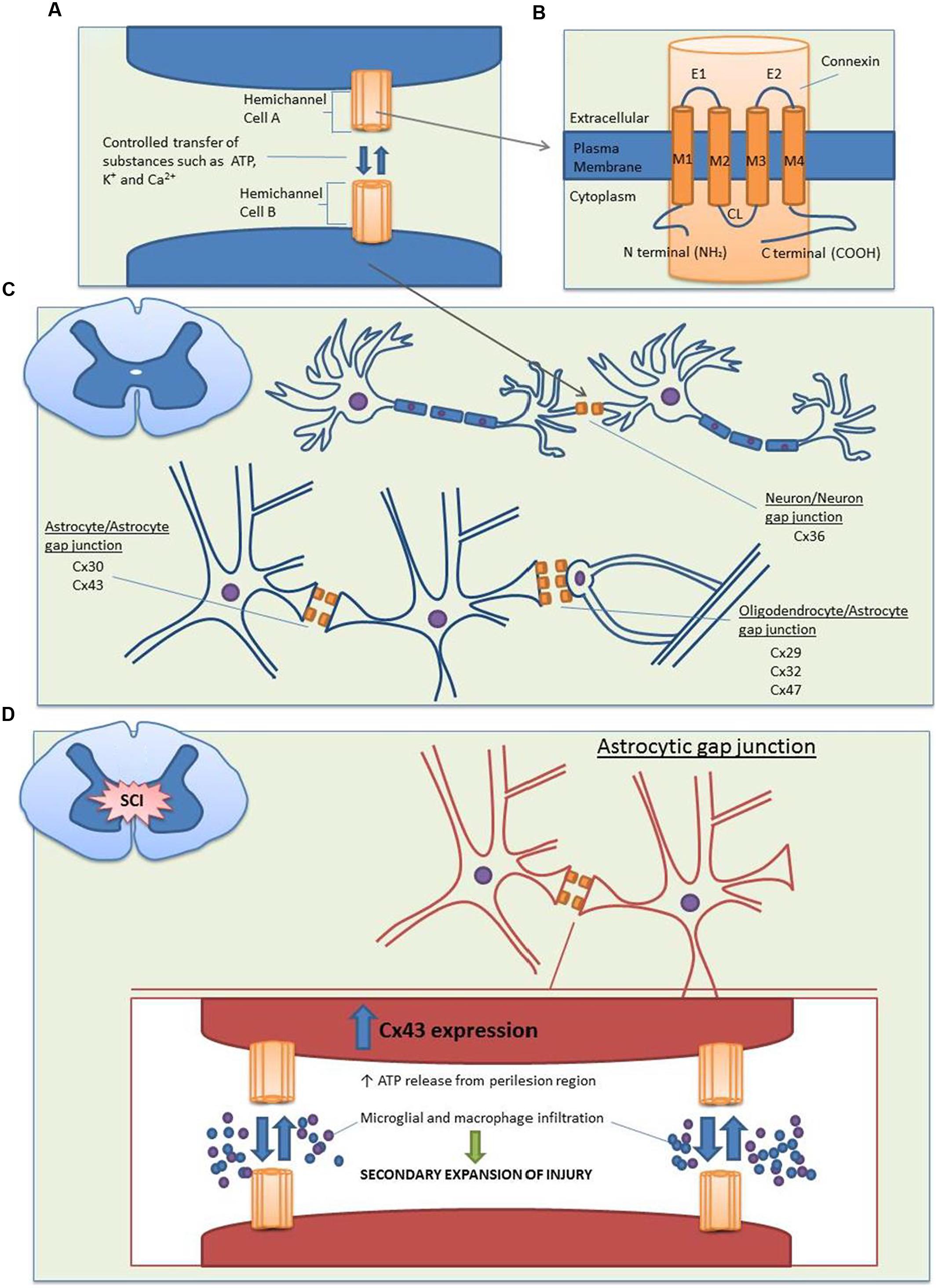
FIGURE 1. Structure and distribution of the main connexin proteins in the spinal cord before and after SCI. (A) Gap junction channels are produced by the conjoining of two hemichannels, with one hemichannel provided by each cell. Compared to the closed state (∼1.8 nm), opened channels (∼2.5 nm) allow the transfer of substances less than 1 kDa between the cytoplasms of two cells, including nutrients, metabolites, K+, ATP, cAMP, and Ca2+. (B) Each hemichannel is composed of six individual connexin proteins, which contain four transmembrane domains. The cytoplasmic loop (CL), amino (N), and carboxy (C) terminals are located on the cytoplasmic side, with multiple regulatory phosphorylation sites located on the C termini. There are two extracellular loops (E1 and E2) which contain three highly conserved cysteine bonds that play a role in the docking of hemichannels from opposing cells via intramolecular cysteine/cysteine bonds. (C) In the spinal cord, there is a large distribution of connexins. Neuron/neuron gap junctions contain Cx36, astrocyte/astrocyte junctions contain Cx30 and Cx43 and oligodendrocyte/astrocyte gap junctions contain Cx29, Cx32, and Cx47 with the major heterotypic channels composed of Cx43Cx47 and Cx30Cx32. (D) Following SCI, Cx43 expression is increased in astrocytes. This causes an uncontrolled release of the small molecule ATP from the perilesion region. ATP release leads to the activation of a destructive inflammatory response including recruitment of microglia and macrophages to the site of injury, which causes an increased secondary expansion of the lesion.
While two hemichannels form gap junctions that directly connect the interiors of two adjacent cells without facing the external milieu, increasing evidence indicates that unopposed hemichannels can open to the extracellular space in response to various physiological and pathological stimuli (Bennett et al., 2003). In such contexts, connexin hemichannels may become activated in response to various intracellular signals including changes in pH, phosphorylation status, extracellular signals such as a low Ca2+ environment, and mechanical, oxidative, metabolic, and ischemic stresses (Contreras et al., 2002; Retamal et al., 2007; Shintani-Ishida et al., 2007). Indeed, studies using dye uptake measurements, recording of hemichannel activity in single cells, and pharmacological blockade of hemichannels in transfected cells have demonstrated that connexins form unopposed hemichannels that can open to the exterior (Contreras et al., 2003; Li et al., 2004). Opened hemichannels lead to the release of molecules such as potassium, ATP, and glutamate into the extracellular space, that under physiological conditions may modulate neuronal activity and under pathological conditions may ultimately induce cell death (Evans et al., 2006; Chever et al., 2014). In addition, gap junctional communication can be likewise regulated by numerous signals, such as connexin phosphorylation and post-translational modification at the C-terminal tail, intracellular acidification, intracellular pH, and inflammatory mediators (Lampe and Lau, 2000; Faustmann et al., 2003; Hinkerohe et al., 2005; Haghikia et al., 2008; Iyyathurai et al., 2013).
In this review, we discuss the expression and function of gap junction/hemichannel proteins in the intact and injured spinal cord, their role in neuropathic pain following spinal cord injury (SCI), and the therapeutic implications.
Distribution and Roles of Connexins in the Spinal Cord
In the normal functioning central nervous system (CNS), gap junctions are formed by fully docked connexons, which play a vital physiological role in the development of communicative pathways in glial and neuronal cells. Studies examining the distribution and functional role of connexins in the spinal cord indicate that connexin-formed gap junctions undergo cell-to-cell coupling between neurons, adjacent astrocytes, and astrocytes/oligodendrocytes (Figure 1C; Chang et al., 1999; Chang and Balice-Gordon, 2000; Rash et al., 2000; Kleopa et al., 2004; Lee et al., 2005; Orthmann-Murphy et al., 2008).
In neurons, gap junctional coupling is widespread during neural development but declines during the first postnatal week. Gap junctional communication in the developing spinal cord is vital in synchronizing neuronal cells and providing electrical synapses, which act as support frameworks for neuronal activity prior to the development of chemical synapses (Chang et al., 1999; Kiehn et al., 2000; Altevogt et al., 2002; Kiehn and Tresch, 2002; Wilson et al., 2007). Developing motor neurons in the spinal cord express Cx36, Cx37, Cx40, Cx43, and Cx45 mRNA. While electrical and dye coupling decrease at about 1 week postnatal along with Cx40 and Cx45 expression, the remaining connexins persist into adulthood (Chang et al., 1999; Chang and Balice-Gordon, 2000). Although earlier studies by Chang et al. (1999) were unable to demonstrate Cx36 and Cx37 protein by immunohistochemistry, more recent studies using sensitive techniques such as in situ hybridization and freeze-fracture replica immunogold labeling (FRIL) were able to detect Cx36 mRNA and protein, respectively. Cx36 mRNA was found to be confined to the cytoplasm of spinal neurons but was more strongly expressed in developing neurons compared to adult rodent spinal cord neurons (Lee et al., 2005). A recent study using transgenic mice expressing Cx36 protein tagged with enhanced green florescent protein, showed a substantial number of florescent clusters in the white matter of the spinal cord providing further evidence for the presence of Cx36 in the adult spinal cord (Meyer et al., 2014). Using FRIL, there was no evidence for the presence of Cx26 in neuronal gap junctions in the perinatal or adult spinal cord (Nagy et al., 2001). While it may be possible that there are as yet unidentified connexins in neuronal gap junctions, more recent studies have implicated Cx36 as the major connexin in mediating electrical synapses in neurons of the spinal cord (Bautista et al., 2014). In adults, neuronal gap junction channels are proposed to contribute to a number of different cognitive processes such as perception, memory, and learning (Buzsáki and Chrobak, 1995; Fricker and Miles, 2001). These gap junction channels are able to sharpen neuronal activity by enhancing the efficacy and precision of synchronous oscillatory activity in neurons (Hormuzdi et al., 2004; Gibson et al., 2005).
In astrocytes, Cx43 and Cx30 are abundantly expressed and are found densely populated around the ependymal and leptomeningeal membranes of the neonatal rodent spinal cord, roughly 4 weeks postnatal (Dahl et al., 1996; Kunzelmann et al., 1999; Lee et al., 2005). It has also been shown using FRIL analysis that leptomeningeal cells in the rats midthoracic spinal cord are highly labeled for Cx26, and that most astrocyte gap junctions in the parenchyma of adult spinal cord are labeled for both Cx26 and Cx30 or Cx26 alone (Nagy et al., 2001). However, recent evidence has suggested a degree of uncertainty over the presence of Cx26 in astrocytes. In postnatal day 4 rats, regions of spinal leptomeningeal cells were found to be largely unlabeled for Cx26 using immunohistochemistry (Nagy et al., 2001). While it is possible that this result reflects the low labeling efficiency for Cx26 at postnatal day 4, recent evidence has shown that the Cx26 antibody may cross-react with Cx30 (Altevogt and Paul, 2004; Orthmann-Murphy et al., 2008). Furthermore, a study using mice with genetically altered Cx26 allele that allows visualization of Cx26 expression has shown that in both the embryonic and mature CNS, Cx26 was restricted to meningeal cells and could not be detected by either neurons or glia, including astrocytes (Filippov et al., 2003). The importance of these astrocytic connexins to normal physiology appears to be linked to their ability to regulate synaptic function. For example, blockade and deletion of astrocytic Cx43 has been shown to impair fear memory consolidation and cause alterations in synaptic transmission and plasticity in rats (Pannasch et al., 2011; Stehberg et al., 2012).
There are limited studies on microglial connexins in the spinal cord. A study by Lee et al. (2005) using immunohistochemistry and triple labeling of Cx43, glial fibrillary acidic protein (GFAP; a marker of astrocytes) and OX-42 (a marker of microglia) showed that 1 week following SCI, Cx43 was colocalized with GFAP, rather than OX-42, suggesting that resting (ramified) and reactive (rounded phagocytic) microglia rarely express Cx43 in the spinal cord.
In oligodendrocytes, Cx29, Cx32, and Cx47 are expressed in regions of the corticospinal tract and are localized to oligodendrocytic cell bodies as well as abaxonal membranes of myelinated fibers, and these three connexins have been shown to participate in astrocytic/oligodendritic gap junctions (Kleopa et al., 2004; Li et al., 2004; Kamasawa et al., 2005). In examining astrocytic/oligodendritic interfaces, Nagy et al. (2001) observed astrocytic Cx43 and Cx30 staining at apposed oligodendrocyte somata in wild type mice. When Cx32 in oligodendrotcyes was knocked out, Cx30 disappeared, while Cx43 levels remain active, furthering the notion of a Cx43–Cx47 and a Cx30–Cx32 heterotypic astrocytic/oligodendritic coupling (Nagy et al., 2001). Similarly, Kamasawa et al. (2005) showed that on the oligodendrocytic side, a greater abundance of Cx47 compared to Cx32 corresponded to a greater level of Cx43 on the astrocytic side, further supporting the notion of Cx43–Cx47 and Cx30–Cx32 heterotypic channels as being the major components of astrocytic/oligodendritic gap junctions.
The most likely role of coupling between glial cells is the creation of the glial syncytium, which amongst other processes such as the propagation of Ca2+ waves and transfer of metabolites, plays a crucial role in spatially buffering increases in extracellular K+. In the CNS, a low extracellular K+ concentration is required for normal neuronal activity and thus pathological changes to connexin expressions are able to have a great impact on the balance of this system (Nagy and Rash, 2003; Rash, 2010). In addition, gap junction communication in glial cells is essential for normal central myelination. Indeed, mice deficient in both Cx32 and Cx47, which are expressed by oligodendrocytes, showed severe demyelinating phenotypes (Menichella et al., 2003; Odermatt et al., 2003). These double knockout mice developed gross tremors and tonic seizures, and died by the sixth postnatal week from profound abnormalities in central myelination. Abnormal changes included axons with thinly or absent myelinated sheaths, myelinated axons with markedly enlarged extracellular spaces separating the axon from its myelin sheath and occasional myelinated axons with enlarged collars of periaxonal oligodendrocyte cytoplasm (Menichella et al., 2003).
Role of Gap Junctions in Spinal Cord Injury
Following a SCI, the initial impact leads to immediate hemorrhage and damage or disturbance to the neuronal elements of the spinal cord including severing of axon tracts and rapid cell death. The initial mechanical disruption is followed by multiple secondary cascades that cause further tissue loss and dysfunction. This secondary phase takes place during the first hours, days, or even months after the initial trauma. During this secondary phase, damage spreads to surrounding neuronal tissue, destroying gray and white matter in a process called the ‘bystander’ effect (Norenberg et al., 2004; Profyris et al., 2004). This encompasses glial cell responses such as astrogliosis and inflammatory cell activation, which can not only prevent neuronal repair, but also cause neuronal cell death. Cell death occurs via processes such as apoptosis, oxidative damage, and cytotoxic edema in astrocytes due to the buildup of glutamate and K+ (Del Bigio and Johnson, 1989; Crowe et al., 1997; Springer et al., 1999; Norenberg et al., 2004; Fitch and Silver, 2008).
In terms of specific connexins, an early study showed that following a mechanically induced SCI in rodents, alterations in astrocytic Cx43 occurred in gray and white matter of the spinal cord (Theriault et al., 1997). Reactive astrocytes displaying GFAP appeared within 1 day and reached maximum levels at 3 days in the area of injury and areas that were both adjacent to and farther away from the lesion epicenter. The authors concluded that during their transformation to reactive states, spinal cord astrocytes experience altered Cx43 expression as a direct result of traumatic SCI (Theriault et al., 1997). These results were replicated by Lee et al. (2005), who used a model of transected adult spinal injury and in situ hybridization showing that astrocytic Cx43 protein and mRNA levels were largely increased 4 h after injury, while there were no changes in Cx32 or Cx36. After 4 weeks, levels of astrocytic Cx43 were three times higher in the caudal stump than the rostral stump, especially adjacent to the injury site, suggesting that increases in Cx43 are related to glial responses to injured tissue (Lee et al., 2005). Several studies that used selective inhibition of Cx43 channels in ex vivo and in vivo models of SCI have shown that Cx43 plays a role in the spread of injury and its blockage leads to improved recovery (Cronin et al., 2008; Zhang et al., 2010).
While increases in Cx43 protein have been widely reported following SCI, this does not appear to be the case with all connexins. Following up on reports that Cx36 mRNA was down regulated at the site of injury during the first week after SCI (Lee et al., 2005), Yates et al. (2008) observed that 7 days after a complete T10 transection in rats, Cx36 protein levels decreased in the lumbar enlargement distant to the site of injury in association with decreased electrical coupling. While this downregulation lasted 14 days post-injury, Cx36 returned to control levels over 2–4 weeks. This coincided with the onset of hyperreflexia and was normalized following oral administration of the eugeroic agent, Modafinil (Yates et al., 2008). The changes in neuronal gap junction protein Cx36 below the level of the lesion suggest that disruption to electrical coupling contributes to hyperreflexia and spasticity following SCI (Yates et al., 2011).
Recently, studies have shifted attention from gap junctional activity to the role of undocked hemichannels following SCI. While it was previously thought that these unopposed hemichannels occurred in closed states, recent evidence shows that contrastingly, hemichannels are able to open independent of channel docking, in a regulated manner during specialized physiological events as well as unregulated opening during pathological events (Contreras et al., 2004; Retamal et al., 2006, 2007). These hemichannels form large cell pores in the cell membrane which, upon opening, allow the exchange of ions and small molecules between intra and extracellular environments. It was found that in addition to playing a traditional role in gap junctions, hexamers of Cx43 are inserted at astroglial membranes as large pore single hemichannels that are able to mediate direct cytosolic exchange with the extracellular space (Giaume et al., 2013).
Of the small molecule substances released by opened undocked hemichannels following SCI, particular attention has been directed at ATP (Figure 1D). ATP release following increased hemichannel expression has been demonstrated both in vitro and in vivo. The application of an acidic fibroblast growth factor-1 (FGF-1) is able to activate spinal astrocytes in culture, causing increased levels of ATP and the opening of connexin channels, in particular, Cx43 (Garré et al., 2010). FGF-1 acts on the FGF receptor, and its activation causes increased levels of cytoplasmic Ca2+ and the release of ATP from vesicles through the activation of phospholipase C (Bennett et al., 2012). When a FGF-1 inhibitor was applied following a weight drop SCI, this caused reduced ATP levels, accompanied by a reduction in inflammatory cell recruitment and lesion expansion (Bennett et al., 2012). A recent study using Cx43 KO mice specific for astrocytes showed that there was a reduction in ATP-induced inflammation, decreased macrophage and microglial recruitment, and an increased functional recovery following SCI (Huang et al., 2012). This study followed past observations that rats subjected to cord contusion have abnormally high and sustained levels of ATP release. By using bioluminescence imaging, levels of ATP in peritraumatic zones were found to be significantly higher than the center of injury, and this elevated ATP release persisted for up to 6 h following SCI. In addition, secondary enlargement of the lesion occurred with signs of inflammation that included increased expression of GFAP, and recruitment of activated microglia and macrophages (Wang et al., 2004). The link between ATP signaling and Cx43 was further strengthened in a recent study by Chever et al. (2014). It was found that when hippocampal CA1 pyramidal slices were treated with Gap26, a Cx43 hemichannel blocking peptide, there was a fivefold decrease in extracellular ATP concentration. However, when the slices were pretreated with ATP P2 receptor antagonists, Gap26 failed to induce changes in ATP concentration suggesting that basal excitatory synaptic transmissions are regulated by astroglial Cx43 hemichannels through ATP signaling (Chever et al., 2014). While there is still a need for further research to elucidate the exact mechanisms, past studies have been able to provide a framework by which Cx43 hemichannels contribute to SCI. Following SCI, increased opening of astrocytic Cx43 channels causes the uncontrolled release of ATP into the perilesion region and inflammatory cell recruitment such as macrophage and microglia, which in turn causes the increased secondary expansion of the lesion.
Role of Gap Junctions in Neuropathic Pain Following Spinal Cord Injury
As many as 80% of SCI patients will suffer some form of chronic pain; nociceptive (e.g., musculoskeletal pain, visceral pain), neuropathic pain (at-level SCI pain, below-level SCI pain), or other pain (Finnerup and Baastrup, 2012). Neuropathic pain is the more debilitating and difficult to treat. It is important to note that neuropathic pain represents a series of heterogeneous conditions including various painful peripheral neuropathies (e.g., traumatic nerve injury, chemotherapy-induced peripheral neuropathy, post-herpetic neuralgia) and central neuropathic pain, such as that occurring after SCI. However, despite the different etiologies and underlying mechanisms, the clinical manifestation of the pain is somewhat similar across the different neuropathic syndromes. Symptoms of both types of neuropathic pain include spontaneous pain presenting as burning, stinging, stabbing, or shooting sensations that is ongoing or intermittent; stimulus-evoked pain, which occurs in response to normally non-noxious (e.g., light touch) or noxious (e.g., cold, punctuate) stimuli; and abnormal sensations (e.g., paresthesia and dysesthesia). Neuropathic pain after SCI results from injury-induced neurochemical and neuroanatomical changes that contribute to maladaptive synaptic circuits and neuronal hyperexcitability in the spinal dorsal horn (Gwak and Hulsebosch, 2011). This pain has been reported as an important factor in decreased quality of life and has been shown to adversely impact function and daily routines in persons with SCI (Cairns et al., 1996; Siddall et al., 1999; Woolf and Mannion, 1999; Norenberg et al., 2004).
The conceptual basis of glial cells including astrocytes and microglia contributing to neuropathic pain is relatively new. The involvement of glial activation has been demonstrated in several chronic pain conditions, including inflammatory pain, peripheral, and central neuropathic pain (Wu et al., 2012). Activated glia can release ATP, excitatory amino acids, proinflammatory cytokines, and reactive oxygen species that in turn promote neuronal hyperexcitability in the dorsal horn of the spinal cord and contribute to neuropathic pain (Austin et al., 2012).
Glial activation has also been demonstrated in neuropathic pain following SCI (Hulsebosch et al., 2009). Several studies have demonstrated that rodents with SCI develop mechanical allodynia and thermal hyperalgesia, in association with astrocytic and microglial activation in segments that are uninjured and rostral to the initial injury site (Hains and Waxman, 2006; Peng et al., 2006; Carlton et al., 2009). Moreover, treatment with glial inhibitors, such as minocycline and propentofylline, reduced both mechanical allodynia and thermal hyperalgesia in the hind limbs of rats with below-level pain following SCI (Gwak et al., 2008; Gwak and Hulsebosch, 2009; Marchand et al., 2009).
Gap junctional activation via connexin channels has been proposed to play a role in glial cell activation, and thus neuropathic pain following SCI. Indeed, Roh et al. (2010) showed that in rats with T13 spinal cord hemisection the development of below-level neuropathic pain, including thermal hyperalgesia and mechanical allodynia, was attenuated following an intrathecal injection of carbenoxolone, a gap junction decoupler. In addition, daily treatment with carbenoxolone significantly reduced the SCI-induced increases in GFAP immunoreactivity and the phosphorylated NMDA receptor NR1 immunoreactivity in the dorsal horn of the spinal cord. These effects were observed when carbenoxolone was administered during the induction phase (0–5 days after SCI), but not during the maintenance phase (15–20 days after SCI), when pain is already established (Roh et al., 2010). This suggests there is a critical time window in which a treatment to block astrocytic gap junctions could be effective in stopping the development of SCI pain.
A recent study using a targeted deletion approach has shown that Cx43 is linked to the development of neuropathic pain following acute SCI (Chen et al., 2012). After a weight drop SCI, control wild type and Cx30 deleted mice developed persistent neuropathic pain lasting from 4 to 8 weeks, which presented as heat hyperalgesia and mechanical allodynia in the presence of increased levels of GFAP positive cells. However, in Cx43/Cx30 deleted mice, both the heat hyperalgesia and mechanical allodynia were prevented in addition to reduced levels of astrogliosis that continued up to 4 weeks. These findings indicate that Cx43 plays a significant role in the development of neuropathic pain following SCI.
Therapeutic Applications
During the past two decades, intercellular communication via gap junctions has provided a novel pharmacological target for treating SCI. The proposed mechanism of gap junction inhibitors is to induce a conformational change in the gap junction protein to control the intercellular and metabolic signaling cascades, such as ATP, glutamate, and reactive oxygen species (Takeuchi et al., 2006; Ramachandran et al., 2007; Clarke et al., 2009). Currently, there are a number of non-specific gap junction blockers such as carbenoxolone, and the antimalarial mefloquine that blocks Cx36 gap junctions at low concentrations (3 uM), while at higher concentrations (30 uM), completely inhibits Cx43 as well as Cx26 and Cx32 in neuroblastoma cells (Cruikshank et al., 2004). In addition, there are novel gap junction hemichannel blockers, such as INI-0602, which can penetrate the blood–brain barrier, and is a proven and effective treatment in mice models of neurodegenerative diseases (Takeuchi et al., 2011). It was recently shown that administration of INI-0602 in mice with SCI resulted in reduced glutamate excitotoxicity by inhibiting microglial activation. This subsequently promoted locomotor recovery and suppressed glial scar formation by reducing secondary degeneration. Further, INI-0602 treatment blocked neutrophil infiltration, stimulated increase in anti-inflammatory cytokines and elevated brain-derived neurotrophic factor levels (Umebayashi et al., 2014). Although such non-specific connexin blockers may be promising therapeutic strategies in SCI, a problem with these drugs is that it is very difficult to control their actions on specific hemichannels and thus establish effective regulation. For instance, a recent study has demonstrated that intrathecal injection of carbenoxolone up to 5 days post-hemisection SCI attenuated the induction of neuropathic pain and the astrogliosis at the lesion edge, but did not significantly improve hindlimb locomotor function (Roh et al., 2010). These global inhibitors non-specifically block all types of gap junctions, affecting physiological functions, such as thermoregulation and angiogenesis (Betageri and Rogers, 1987; Hong et al., 2009). While the global gap junction blockers have confining or questionable control of lesion spread and astrogliosis following traumatic CNS injury, other tissues containing connexin gap junctions may also be adversely affected.
A more targeted approach using antisense oligodeoxynucleotides (AS-ODN) and connexin mimetic peptides has been developed to prevent connexin signaling. Mimetic peptides are short peptides designed to mimic sequences on extracellular loops of connexin proteins. They prevent connexin hemichannel docking, thus blocking gap junctional intercellular communication (Warner et al., 1995; Kwak and Jongsma, 1999; Boitano and Evans, 2000; Martin et al., 2005). Although the mechanism of action for connexin mimetic peptides has not been fully elucidated, it has been proposed that they may induce a conformational change in connexin hemichannels via connexin phosphorylation, leading to the closure of the uncoupled hemichannels without blocking existing gap junctions (Chaytor et al., 1997). In cultured rodent spinal cords, when a Cx43-mimetic peptide, peptide 5, was given at doses between 5 and 500 μm for 24 h, swelling was reduced by 50% and neuronal loss was prevented. Furthermore, peptide 5 was able to reduce GFAP levels for up to 4 days in ex vivo spinal cord segments following SCI (O’Carroll et al., 2008). This was further supported by a study that used an AS-ODN to suppress Cx43 following a compression SCI in rodents, showing that treatment with Cx43-ODN resulted in less swelling, structural distortion and decreased GFAP positive cells in spinal cord segments (Cronin et al., 2008). In a follow-up study, peptide 5 administered after a spinal cord contusion injury in rats showed reduced levels of proinflammatory cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-1β, reduced GFAP staining in the area adjacent to the lesion epicenter, a reduction of neuronal loss 7 mm from the lesion epicenter as well as improved locomotor function (O’Carroll et al., 2013).
The application of connexin mimetic peptides has been widely studied in depressing immune responses (Neijssen et al., 2005; Matsue et al., 2006; Bopp et al., 2007), limiting cardiovascular damage (Wong et al., 2006; Shintani-Ishida et al., 2007), and inhibiting osteoclastic differentiation (Ilvesaro et al., 2001). Importantly, connexin mimetic peptides have the potential to be systemically delivered to target the lesion area of spinal cord in human patients, and offer a new hope to spinal cord-injured patients as an acute intervention. The regulation of Cx43 by AS-ODNs has been shown to not only protect neural cells after traumatic CNS injury, but also improve wound healing in many tissues, including cornea (Ratkay-Traub et al., 2001; Cursiefen et al., 2009; Grupcheva et al., 2012), skin (Coutinho et al., 2003; Qiu et al., 2003; Becker et al., 2012), skeletal muscle (Gorbe et al., 2006), cardiac muscle (Yasui et al., 2000), smooth muscle (Yeh et al., 1997; Liao and Duling, 2000), and vascular endothelium (Yeh et al., 2000; Kwak et al., 2001; Ormonde et al., 2012). Cx43 is upregulated at the wound edge of chronic wounds (Coutinho et al., 2003; Wang et al., 2007), and treatment with Cx43 AS-ODNs dampens the inflammatory response and decreases wound spread, resulting in reduced swelling and scarring and accelerated healing (Qiu et al., 2003; Coutinho et al., 2005; Mori et al., 2006). Significantly, several phase 1 or phase 2 clinical trials by CoDa Therapeutics, Inc. using the topical gel of Cx43 AS-ODNs (Nexagon®) have shown the treatment to be safe and tolerable in skin wounds (NCT00736593), venous leg ulcers (NCT00820196 and NCT01199588), diabetic foot ulcer (NCT01490879), and acute corneal wounds (NCT00654550). Thus, based on animal studies and the clinical use of connexin inhibitors in various conditions, drugs that inhibit specific connexins, such as Cx43 AS-ODNs or Cx43 mimetic peptides, hold great promise for the treatment of SCI.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from the SCI Network – “Toward Translation – Capacity Building Initiative” to Gila Moalem-Taylor, Catherine A. Gorrie, Colin R. Green, Simon J. O’Carroll, and Louise F. B. Nicholson.
References
Altevogt, B. M., Kleopa, K. A., Postma, F. R., Scherer, S. S., and Paul, D. L. (2002). Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J. Neurosci. 22, 6458–6470.
Altevogt, B. M., and Paul, D. L. (2004). Four classes of intercellular channels between glial cells in the CNS. J. Neurosci. 24, 4313–4323. doi: 10.1523/JNEUROSCI.3303-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Austin, P. J., Kim, C. F., Perera, C. J., and Moalem-Taylor, G. (2012). Regulatory T cells attenuate neuropathic pain following peripheral nerve injury and experimental autoimmune neuritis. Pain 153, 1916–1931. doi: 10.1016/j.pain.2012.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bautista, W., Mccrea, D., and Nagy, J. (2014). Connexin36 identified at morphologically mixed chemical/electrical synapses on trigeminal motoneurons and at primary afferent terminals on spinal cord neurons in adult mouse and rat. Neuroscience 236, 159–180. doi: 10.1016/j.neuroscience.2013.12.057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Becker, D. L., Thrasivoulou, C., and Phillips, A. R. (2012). Connexins in wound healing; perspectives in diabetic patients. Biochim. Biophys. Acta 1818, 2068–2075. doi: 10.1016/j.bbamem.2011.11.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bennett, M. V., Contreras, J. E., Bukauskas, F. F., and Sáez, J. C. (2003). New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 26, 610–617. doi: 10.1016/j.tins.2003.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bennett, M. V., Garré, J. M., Orellana, J. A., Bukauskas, F. F., Nedergaard, M., Giaume, C.,et al. (2012). Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res. 1487, 3–15. doi: 10.1016/j.brainres.2012.08.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Betageri, G., and Rogers, J. (1987). Thermodynamics of partitioning of β-blockers in the n-octanol-buffer and liposome systems. Int. J. Pharm. 36, 165–173. doi: 10.1016/0378-5173(87)90152-9
Boitano, S., and Evans, W. H. (2000). Connexin mimetic peptides reversibly inhibit Ca2+ signaling through gap junctions in airway cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, 623–630.
Bopp, T., Becker, C., Klein, M., Klein-Heßling, S., Palmetshofer, A., Serfling, E.,et al. (2007). Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J. Exp. Med. 204, 1303–1310. doi: 10.1084/jem.20062129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buzsáki, G., and Chrobak, J. J. (1995). Temporal structure in spatially organized neuronal ensembles: a role for interneuronal networks. Curr. Opin. Neurobiol. 5, 504–510. doi: 10.1016/0959-4388(95)80012-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cairns, D. M., Adkins, R. H., and Scott, M. D. (1996). Pain and depression in acute traumatic spinal cord injury: origins of chronic problematic pain? Arch. Phys. Med. Rehabil. 77, 329–335. doi: 10.1016/S0003-9993(96)90079-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carlton, S. M., Du, J., Tan, H. Y., Nesic, O., Hargett, G. L., Bopp, A. C.,et al. (2009). Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain 147, 265–276. doi: 10.1016/j.pain.2009.09.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, Q., and Balice-Gordon, R. J. (2000). Gap junctional communication among developing and injured motor neurons. Brain Res. Brain Res. Rev. 32, 242–249. doi: 10.1016/S0165-0173(99)00085-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, Q., Gonzalez, M., Pinter, M. J., and Balice-Gordon, R. J. (1999). Gap junctional coupling and patterns of connexin expression among neonatal rat lumbar spinal motor neurons. J. Neurosci. 19, 10813–10828.
Chaytor, A., Evans, W., Griffith, T., and Thornbury, K. (1997). Peptides homologous to extracellular loop motifs of connexin 43 reversibly abolish rhythmic contractile activity in rabbit arteries. J. Physiol. 503, 99–110. doi: 10.1111/j.1469-7793.1997.099bi.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, M. J., Kress, B., Han, X., Moll, K., Peng, W., Ji, R. R.,et al. (2012). Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia 60, 1660–1670. doi: 10.1002/glia.22384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chever, O., Lee, C.-Y., and Rouach, N. (2014). Astroglial connexin43 hemichannels tune basal excitatory synaptic transmission. J. Neurosci. 34, 11228–11232. doi: 10.1523/JNEUROSCI.0015-14.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clarke, T. C., Williams, O. J., Martin, P. E., and Evans, W. H. (2009). ATP release by cardiac myocytes in a simulated ischaemia model: inhibition by a connexin mimetic and enhancement by an antiarrhythmic peptide. Eur. J. Pharmacol. 605, 9–14. doi: 10.1016/j.ejphar.2008.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Contreras, J. E., Sáez, J. C., Bukauskas, F. F., and Bennett, M. V. (2003). Gating and regulation of connexin 43 (Cx43) hemichannels. Proc. Natl. Acad. Sci. U.S.A. 100, 11388–11393. doi: 10.1073/pnas.1434298100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Contreras, J. E., Sánchez, H. A., Eugenín, E. A., Speidel, D., Theis, M., Willecke, K.,et al. (2002). Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. U.S.A. 99, 495–500. doi: 10.1073/pnas.012589799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Contreras, J. E., Sánchez, H. A., Véliz, L. P., Bukauskas, F. F., Bennett, M. V., and Sáez, J. C. (2004). Role of connexin-based gap junction channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res. Brain Res. Rev. 47, 290–303. doi: 10.1016/j.brainresrev.2004.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coutinho, P., Qiu, C., Frank, S., Tamber, K., and Becker, D. (2003). Dynamic changes in connexin expression correlate with key events in the wound healing process. Cell Biol. Int. 27, 525–541. doi: 10.1016/S1065-6995(03)00077-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coutinho, P., Qiu, C., Frank, S., Wang, C., Brown, T., Green, C.,et al. (2005). Limiting burn extension by transient inhibition of connexin43 expression at the site of injury. Br. J. Plast. Surg. 58, 658–667. doi: 10.1016/j.bjps.2004.12.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cronin, M., Anderson, P. N., Cook, J. E., Green, C. R., and Becker, D. L. (2008). Blocking connexin43 expression reduces inflammation and improves functional recovery after spinal cord injury. Mol. Cell. Neurosci. 39, 152–160. doi: 10.1016/j.mcn.2008.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crowe, M. J., Bresnahan, J. C., Shuman, S. L., Masters, J. N., and Crowe, M. S. (1997). Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. 3, 73–76. doi: 10.1038/nm0197-73
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cruikshank, S. J., Hopperstad, M., Younger, M., Connors, B. W., Spray, D. C., and Srinivas, M. (2004). Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc. Natl. Acad. Sci. U.S.A. 101, 12364–12369. doi: 10.1073/pnas.0402044101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cursiefen, C., Bock, F., Horn, F. K., Kruse, F. E., Seitz, B., Borderie, V.,et al. (2009). GS-101 antisense oligonucleotide eye drops inhibit corneal neovascularization: interim results of a randomized phase II trial. Ophthalmology 116, 1630–1637. doi: 10.1016/j.ophtha.2009.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dahl, E., Manthey, D., Chen, Y., Schwarz, H.-J., Chang, Y. S., Lalley, P. A.,et al. (1996). Molecular cloning and functional expression of mouse connexin-30, a gap junction gene highly expressed in adult brain and skin. J. Biol. Chem. 271, 17903–17910. doi: 10.1074/jbc.271.30.17903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Bigio, M. R., and Johnson, G. E. (1989). Clinical presentation of spinal cord concussion. Spine 14, 37–40. doi: 10.1097/00007632-198901000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Evans, W., De Vuyst, E., and Leybaert, L. (2006). The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem. J. 397, 1–14. doi: 10.1042/BJ20060175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Faustmann, P. M., Haase, C. G., Romberg, S., Hinkerohe, D., Szlachta, D., Smikalla, D.,et al. (2003). Microglia activation influences dye coupling and Cx43 expression of the astrocytic network. Glia 42, 101–108. doi: 10.1002/glia.10141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Filippov, M. A., Hormuzdi, S. G., Fuchs, E. C., and Monyer, E. (2003). A reporter allele for investigating connexin 26 gene expression in the mouse brain. Eur. J. Neurosci. 18, 3183–3192. doi: 10.1111/j.1460-9568.2003.03042.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Finnerup, N. B., and Baastrup, C. (2012). Spinal cord injury pain: mechanisms and management. Curr. Pain Headache Rep. 16, 207–216. doi: 10.1007/s11916-012-0259-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fitch, M. T., and Silver, J. (2008). CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp. Neurol. 209, 294–301. doi: 10.1016/j.expneurol.2007.05.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fricker, D., and Miles, R. (2001). Interneurons, spike timing, and perception. Neuron 32, 771–774. doi: 10.1016/S0896-6273(01)00528-1
Garré, J. M., Retamal, M. A., Cassina, P., Barbeito, L., Bukauskas, F. F., Sáez, J. C.,et al. (2010). FGF-1 induces ATP release from spinal astrocytes in culture and opens pannexin and connexin hemichannels. Proc. Natl. Acad. Sci. U.S.A. 107, 22659–22664. doi: 10.1073/pnas.1013793107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Giaume, C., Leybaert, L., Naus, C. C., and Sáez, J. C. (2013). Connexin and pannexin hemichannels in brain glial cells: properties, pharmacology, and roles. Front. Pharmacol. 4:88. doi: 10.3389/fphar.2013.00088
Gibson, J. R., Beierlein, M., and Connors, B. W. (2005). Functional properties of electrical synapses between inhibitory interneurons of neocortical layer 4. J. Neurophysiol. 93, 467–480. doi: 10.1152/jn.00520.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gorbe, A., Becker, D. L., Dux, L., Krenacs, L., and Krenacs, T. (2006). In differentiating prefusion myoblasts connexin43 gap junction coupling is upregulated before myoblast alignment then reduced in post-mitotic cells. Histochem. Cell Biol. 125, 705–716. doi: 10.1007/s00418-005-0121-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grupcheva, C. N., Laux, W. T., Rupenthal, I. D., Mcghee, J., Mcghee, C. N., and Green, C. R. (2012). Improved corneal wound healing through modulation of gap junction communication using connexin43-specific antisense oligodeoxynucleotides. Invest. Ophthalmol. Vis. Sci. 53, 1130–1138. doi: 10.1167/iovs.11-8711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gwak, Y. S., Crown, E. D., Unabia, G. C., and Hulsebosch, C. E. (2008). Propentofylline attenuates allodynia, glial activation and modulates GABAergic tone after spinal cord injury in the rat. Pain 138, 410–422. doi: 10.1016/j.pain.2008.01.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gwak, Y. S., and Hulsebosch, C. E. (2009). Remote astrocytic and microglial activation modulates neuronal hyperexcitability and below-level neuropathic pain after spinal injury in rat. Neuroscience 161, 895–903. doi: 10.1016/j.neuroscience.2009.03.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gwak, Y. S., and Hulsebosch, C. E. (2011). Neuronal hyperexcitability: a substrate for central neuropathic pain after spinal cord injury. Curr. Pain Headache Rep. 15, 215–222. doi: 10.1007/s11916-011-0186-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haghikia, A., Ladage, K., Lafenetre, P., Haghikia, A., Hinkeroche, D., Smikalla, D.,et al. (2008). Intracellular application of TNF-alpha impairs cell to cell communication via gap junctions in glioma cells. J. Neurooncol. 86, 143–152. doi: 10.1007/s11060-007-9462-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hains, B. C., and Waxman, S. G. (2006). Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J. Neurosci. 26, 4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hertzberg, E. L., Lawrence, T. S., and Gilula, N. B. (1981). Gap junctional communication. Annu. Rev. Physiol. 43, 479–491. doi: 10.1146/annurev.ph.43.030181.002403
Hinkerohe, D., Smikalla, D., Haghikia, A., Heupel, K., Haase, C. G., Dermietzel, R.,et al. (2005). Effects of cytokines on microglial phenotypes and astroglial coupling in an inflammatory coculture model. Glia 52, 85–97. doi: 10.1002/glia.20223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hong, T., Wang, H., Wang, Y., and Wang, H. (2009). Effects of gap junctional blockers on cerebral vasospasm after subarachnoid hemorrhage in rabbits. Neurol. Res. 31, 238–244. doi: 10.1179/174313208X322770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hormuzdi, S. G., Filippov, M. A., Mitropoulou, G., Monyer, H., and Bruzzone, R. (2004). Electrical synapses: a dynamic signaling system that shapes the activity of neuronal networks. Biochim. Biophys. Acta 1662, 113–137. doi: 10.1016/j.bbamem.2003.10.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, C., Han, X., Li, X., Lam, E., Peng, W., Lou, N.,et al. (2012). Critical role of connexin 43 in secondary expansion of traumatic spinal cord injury. J. Neurosci. 32, 3333–3338. doi: 10.1523/JNEUROSCI.1216-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hulsebosch, C. E., Hains, B. C., Crown, E. D., and Carlton, S. M. (2009). Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res. Rev. 60, 202–213. doi: 10.1016/j.brainresrev.2008.12.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ilvesaro, J., Tavi, P., and Tukkanen, J. (2001). Connexin mimetic peptide Gap27 decreases osteoclastic activity. BMC Musculoskelet. Disord. 2:10. doi: 10.1186/1471-2474-2-10
Iyyathurai, J., D’hondt, C., Wang, N., De Bock, M., Himpens, B., Retamal, M. A.,et al. (2013). Peptides and peptide derived molecules targetting the intracellular domains of Cx43: gap junctions versus hemichannels. Neuropharmacology 75, 491–505. doi: 10.1016/j.neuropharm.2013.04.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kamasawa, N., Sik, A., Morita, M., Yasumura, T., Davidson, K., Nagy, J.,et al. (2005). Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: implications for ionic homeostasis and potassium siphoning. Neuroscience 136, 65–86. doi: 10.1016/j.neuroscience.2005.08.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiehn, O., Kjaerulff, O., Tresch, M. C., and Harris-Warrick, R. M. (2000). Contributions of intrinsic motor neuron properties to the production of rhythmic motor output in the mammalian spinal cord. Brain Res. Bull. 53, 649–659. doi: 10.1016/S0361-9230(00)00398-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiehn, O., and Tresch, M. C. (2002). Gap junctions and motor behavior. Trends Neurosci. 25, 108–115. doi: 10.1016/S0166-2236(02)02038-6
Kleopa, K. A., Orthmann, J. L., Enriquez, A., Paul, D. L., and Scherer, S. S. (2004). Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia 47, 346–357. doi: 10.1002/glia.20043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kunzelmann, P., Schröder, W., Traub, O., Steinhäuser, C., Dermietzel, R., and Willecke, K. (1999). Late onset and increasing expression of the gap junction protein connexin 30 in adult murine brain and long-term cultured astrocytes. Glia 25, 111–119. doi: 10.1002/(SICI)1098-1136(19990115)25:2<111::AID-GLIA2>3.0.CO;2-I
Kwak, B. R., and Jongsma, H. J. (1999). Selective inhibition of gap junction channel activity by synthetic peptides. J. Physiol. 516, 679–685. doi: 10.1111/j.1469-7793.1999.0679u.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kwak, B. R., Pepper, M. S., Gros, D. B., and Meda, P. (2001). Inhibition of endothelial wound repair by dominant negative connexin inhibitors. Mol. Biol. Cell 12, 831–845. doi: 10.1091/mbc.12.4.831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lampe, P. D., and Lau, A. F. (2000). Regulation of gap junctions by phosphorylation of connexins. Arch. Biochem. Biophys. 384, 205–215. doi: 10.1006/abbi.2000.2131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, I. H., Lindqvist, E., Kiehn, O., Widenfalk, J., and Olson, L. (2005). Glial and neuronal connexin expression patterns in the rat spinal cord during development and following injury. J. Comp. Neurol. 489, 1–10. doi: 10.1002/cne.20567
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, X., Ionescu, A., Lynn, B., Lu, S., Kamasawa, N., Morita, M.,et al. (2004). Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience 126, 611–630. doi: 10.1016/j.neuroscience.2004.03.063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liao, Y., and Duling, B. R. (2000). Blockade of connexin 43 expression by stable transfection of antisense cDNA in cultured vascular smooth muscle cells. Antisense Nucleic Acid Drug Dev. 10, 275–281. doi: 10.1089/108729000421457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchand, F., Tsantoulas, C., Singh, D., Grist, J., Clark, A. K., Bradbury, E. J.,et al. (2009). Effects of etanercept and minocycline in a rat model of spinal cord injury. Eur. J. Pain 13, 673–681. doi: 10.1016/j.ejpain.2008.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin, P. E., Wall, C., and Griffith, T. M. (2005). Effects of connexin-mimetic peptides on gap junction functionality and connexin expression in cultured vascular cells. Br. J. Pharmacol. 144, 617–627. doi: 10.1038/sj.bjp.0706102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsue, H., Yao, J., Matsue, K., Nagasaka, A., Sugiyama, H., Aoki, R.,et al. (2006). Gap junction-mediated intercellular communication between dendritic cells (DCs) is required for effective activation of DCs. J. Immunol. 176, 181–190. doi: 10.4049/jimmunol.176.1.181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Menichella, D. M., Goodenough, D. A., Sirkowski, E., Scherer, S. S., and Paul, D. L. (2003). Connexins are critical for normal myelination in the CNS. J. Neurosci. 23, 5963–5973.
Meyer, A., Hilgen, G., Dorgau, B., Sammler, E. M., Weiler, R., Monyer, H.,et al. (2014). All amacrine cells discriminate between heterocellular and homocellular locations when assembling connexin36 containing gap junctions. J. Cell Sci. 127, 1190–1202. doi: 10.1242/jcs.133066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mori, R., Power, K. T., Wang, C. M., Martin, P., and Becker, D. L. (2006). Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J. Cell Sci. 119, 5193–5203. doi: 10.1242/jcs.03320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagy, J. I., Li, X., Rempel, J., Stelmack, G., Patel, D., Staines, W. A.,et al. (2001). Connexin26 in adult rodent central nervous system: demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J. Comp. Neurol. 441, 302–323. doi: 10.1002/cne.1414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagy, J. I., and Rash, J. E. (2003). Astrocyte and oligodendrocyte connexins of the glial syncytium in relation to astrocyte anatomical domains and spatial buffering. Cell Commun. Adhes. 10, 401–406. doi: 10.1080/cac.10.4-6.401.406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neijssen, J., Herberts, C., Drijfhout, J. W., Reits, E., Janssen, L., and Neefjes, J. (2005). Cross-presentation by intercellular peptide transfer through gap junctions. Nature 434, 83–88. doi: 10.1038/nature03290
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Norenberg, M. D., Smith, J., and Marcillo, A. (2004). The pathology of human spinal cord injury: defining the problems. J. Neurotrauma 21, 429–440. doi: 10.1089/089771504323004575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Carroll, S. J., Alkadhi, M., Nicholson, L. F., and Green, C. R. (2008). Connexin43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes. 15, 27–42. doi: 10.1080/15419060802014164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Carroll, S. J., Gorrie, C. A., Velamoor, S., Green, C. R., and Nicholson, L. F. (2013). Connexin43 mimetic peptide is neuroprotective and improves function following spinal cord injury. Neurosci. Res. 75, 256–267. doi: 10.1016/j.neures.2013.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Odermatt, B., Wellershaus, K., Wallraff, A., Seifert, G., Degen, J., Euwens, C.,et al. (2003). Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J. Neurosci. 23, 4549–4559.
Ormonde, S., Chou, C.-Y., Goold, L., Petsoglou, C., Al-Taie, R., Sherwin, T.,et al. (2012). Regulation of connexin43 gap junction protein triggers vascular recovery and healing in human ocular persistent epithelial defect wounds. J. Membr. Biol. 245, 381–388. doi: 10.1007/s00232-012-9460-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orthmann-Murphy, J. L., Abrams, C. K., and Scherer, S. S. (2008). Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 35, 101–116. doi: 10.1007/s12031-007-9027-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pannasch, U., Vargová, L., Reingruber, J., Ezan, P., Holcman, D., Giaume, C.,et al. (2011). Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. U.S.A. 108, 8467–8472. doi: 10.1073/pnas.1016650108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peng, X. M., Zhou, Z. G., Glorioso, J. C., Fink, D. J., and Mata, M. (2006). Tumor necrosis factor–α contributes to below-level neuropathic pain after spinal cord injury. Ann. Neurol. 59, 843–851. doi: 10.1002/ana.20855
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Profyris, C., Cheema, S. S., Zang, D., Azari, M. F., Boyle, K., and Petratos, S. (2004). Degenerative and regenerative mechanisms governing spinal cord injury. Neurobiol. Dis. 15, 415–436. doi: 10.1016/j.nbd.2003.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiu, C., Coutinho, P., Frank, S., Franke, S., Law, L.-Y., Martin, P.,et al. (2003). Targeting connexin43 expression accelerates the rate of wound repair. Curr. Biol. 13, 1697–1703. doi: 10.1016/j.cub.2003.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramachandran, S., Xie, L.-H., John, S. A., Subramaniam, S., and Lal, R. (2007). A novel role for connexin hemichannel in oxidative stress and smoking-induced cell injury. PLoS ONE 2:e712. doi: 10.1371/journal.pone.0000712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rash, J., Staines, W., Yasumura, T., Patel, D., Furman, C., Stelmack, G.,et al. (2000). Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc. Natl. Acad. Sci. U.S.A. 97, 7573–7578. doi: 10.1073/pnas.97.13.7573
Rash, J. E. (2010). Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 168, 982–1008. doi: 10.1016/j.neuroscience.2009.10.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ratkay-Traub, I., Hopp, B., Bor, Z., Dux, L., Becker, D., and Krenacs, T. (2001). Regeneration of rabbit cornea following excimer laser photorefractive keratectomy: a study on gap junctions, epithelial junctions and epidermal growth factor receptor expression in correlation with cell proliferation. Exp. Eye Res. 73, 291–302. doi: 10.1006/exer.2001.1040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Retamal, M. A., Cortés, C. J., Reuss, L., Bennett, M. V., and Sáez, J. C. (2006). S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. U.S.A. 103, 4475–4480. doi: 10.1073/pnas.0511118103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Retamal, M. A., Schalper, K. A., Shoji, K. F., Bennett, M. V., and Sáez, J. C. (2007). Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc. Natl. Acad. Sci. U.S.A. 104, 8322–8327. doi: 10.1073/pnas.0702456104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roh, D. H., Yoon, S. Y., Seo, H. S., Kang, S. Y., Han, H. J., Beitz, A. J.,et al. (2010). Intrathecal injection of carbenoxolone, a gap junction decoupler, attenuates the induction of below-level neuropathic pain after spinal cord injury in rats. Exp. Neurol. 224, 123–132. doi: 10.1016/j.expneurol.2010.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shintani-Ishida, K., Uemura, K., and Yoshida, K.-I. (2007). Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 293, 1714–1720. doi: 10.1152/ajpheart.00022.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Siddall, P. J., Taylor, D. A., Mcclelland, J. M., Rutkowski, S. B., and Cousins, M. J. (1999). Pain report and the relationship of pain to physical factors in the first 6 months following spinal cord injury. Pain 81, 187–197. doi: 10.1016/S0304-3959(99)00023-8
Söhl, G., Maxeiner, S., and Willecke, K. (2005). Expression and functions of neuronal gap junctions. Nat. Rev. Neurosci. 6, 191–200. doi: 10.1038/nrn1627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Söhl, G., and Willecke, K. (2003). An update on connexin genes and their nomenclature in mouse and man. Cell Commun. Adhes. 10, 173–180. doi: 10.1080/cac.10.4-6.173.180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Springer, J. E., Azbill, R. D., and Knapp, P. E. (1999). Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat. Med. 5, 943–946. doi: 10.1038/11387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stehberg, J., Moraga-Amaro, R., Salazar, C., Becerra, A., Echeverría, C., Orellana, J. A.,et al. (2012). Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 26, 3649–3657. doi: 10.1096/fj.11-198416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takeuchi, H., Jin, S., Wang, J., Zhang, G., Kawanokuchi, J., Kuno, R.,et al. (2006). Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 281, 21362–21368. doi: 10.1074/jbc.M600504200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takeuchi, H., Mizoguchi, H., Doi, Y., Jin, S., Noda, M., Liang, J.,et al. (2011). Blockade of gap junction hemichannel suppressess disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimers disease. PLoS ONE 6:e21108. doi: 10.1371/journal.pone.0021108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Theriault, E., Frankenstein, U., Hertzberg, E., and Nagy, J. (1997). Connexin43 and astrocytic gap junctions in the rat spinal cord after acute compression injury. J. Comp. Neurol. 382, 199–214. doi: 10.1002/(SICI)1096-9861(19970602)382:2<199::AID-CNE5>3.0.CO;2-Z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Umebayashi, D., Natsume, A., Takeuchi, H., Hara, M., Nishimura, Y., Fukuyama, R.,et al. (2014). Blockade of gap junction hemichannels protects secondary spinal cord injury from activated microglia mediated glutamate exitoneurotoxicity. J. Neurotrauma 21, 1–8. doi: 10.1089/neu.2013.3223
Wang, C. M., Lincoln, J., Cook, J. E., and Becker, D. L. (2007). Abnormal connexin expression underlies delayed wound healing in diabetic skin. Diabetes 56, 2809–2817. doi: 10.2337/db07-0613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X., Arcuino, G., Takano, T., Lin, J., Peng, W. G., Wan, P.,et al. (2004). P2X7 receptor inhibition improves recovery after spinal cord injury. Nat. Med. 10, 821–827. doi: 10.1038/nm1082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Warner, A., Clements, D., Parikh, S., Evans, W., and Dehaan, R. (1995). Specific motifs in the external loops of connexin proteins can determine gap junction formation between chick heart myocytes. J. Physiol. 488, 721–728.
Wilson, J. M., Cowan, A. I., and Brownstone, R. M. (2007). Heterogeneous electrotonic coupling and synchronization of rhythmic bursting activity in mouse Hb9 interneurons. J. Neurophysiol. 98, 2370–2381. doi: 10.1152/jn.00338.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, C. W., Christen, T., Roth, I., Chadjichristos, C. E., Derouette, J.-P., Foglia, B. F.,et al. (2006). Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 12, 950–954. doi: 10.1038/nm1441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Woolf, C. J., and Mannion, R. J. (1999). Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353, 1959–1964. doi: 10.1016/S0140-6736(99)01307-0
Wu, A., Green, C. R., Rupenthal, I. D., and Moalem-Taylor, G. (2012). Role of gap junctions in chronic pain. J. Neurosci. Res. 90, 337–345. doi: 10.1002/jnr.22764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yasui, K., Kada, K., Hojo, M., Lee, J.-K., Kamiya, K., Toyama, J.,et al. (2000). Cell-to-cell interaction prevents cell death in cultured neonatal rat ventricular myocytes. Cardiovasc. Res. 48, 68–76. doi: 10.1016/S0008-6363(00)00145-0
Yates, C., Charlesworth, A., Allen, S., Reese, N., Skinner, R., and Garcia-Rill, E. (2008). The onset of hyperreflexia in the rat following complete spinal cord transection. Spinal Cord 46, 798–803. doi: 10.1038/sc.2008.49
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yates, C., Garrison, K., Reese, N. B., Charlesworth, A., and Garcia-Rill, E. (2011). Chapter 11-Novel mechanism for hyperreflexia and spasticity. Prog. Brain Res. 188, 167–180. doi: 10.1016/B978-0-444-53825-3.00016-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yeh, H. I., Lai, Y. J., Chang, H. M., Ko, Y. S., Severs, N. J., and Tsai, C. H. (2000). Multiple connexin expression in regenerating arterial endothelial gap junctions. Arterioscler. Thromb. Vasc. Biol. 20, 1753–1762. doi: 10.1161/01.ATV.20.7.1753
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yeh, H. I., Lupu, F., Dupont, E., and Severs, N. J. (1997). Upregulation of connexin43 gap junctions between smooth muscle cells after balloon catheter injury in the rat carotid artery. Arterioscler. Thromb. Vasc. Biol. 17, 3174–3184. doi: 10.1161/01.ATV.17.11.3174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, J., O’Carroll, S. J., Wu, A., Nicholson, L. F. B., and Green, C. R. (2010). A model for ex vivo spinal cord segment culture – a tool for analysis of injury repair strategies. J. Neurosci. Methods 192, 49–57. doi: 10.1016/j.jneumeth.2010.07.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: spinal cord injury, gap junctions, hemichannels, connexins, neuropathic pain
Citation: Tonkin RS, Mao Y, O’Carroll SJ, Nicholson LFB, Green CR, Gorrie CA and Moalem-Taylor G (2015) Gap junction proteins and their role in spinal cord injury. Front. Mol. Neurosci. 7:102. doi: 10.3389/fnmol.2014.00102
Received: 08 October 2014; Accepted: 12 December 2014;
Published online: 06 January 2015.
Edited by:
Nicola Maggio, The Chaim Sheba Medical Center, IsraelReviewed by:
Sheriar Hormuzdi, University of Dundee, UKMichele Papa, Seconda Università degli Studi di Napoli, Italy
Ping Liu, University of Connecticut Health Center, USA
Copyright © 2015 Tonkin, Mao, O’Carroll, Nicholson, Green, Gorrie and Moalem-Taylor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gila Moalem-Taylor, School of Medical Sciences, Faculty of Medicine, University of New South Wales, Wallace Wurth Building East, Level 3, Room 327, Sydney, NSW 2052, Australia e-mail:Z2lsYUB1bnN3LmVkdS5hdQ==