 Madeline L. Pfau1,2
Madeline L. Pfau1,2 Immanuel Purushothaman1Jian Feng1
Immanuel Purushothaman1Jian Feng1 Sam A. Golden1,2Hossein Aleyasin1
Sam A. Golden1,2Hossein Aleyasin1 Zachary S. Lorsch1,2
Zachary S. Lorsch1,2 Hannah M. Cates1,2Meghan E. Flanigan1,2
Hannah M. Cates1,2Meghan E. Flanigan1,2 Caroline Menard1Mitra Heshmati1,2
Caroline Menard1Mitra Heshmati1,2 Zichen Wang2,3
Zichen Wang2,3 Avi Ma'ayan3Li Shen1Georgia E. Hodes1Scott J. Russo1*
Avi Ma'ayan3Li Shen1Georgia E. Hodes1Scott J. Russo1*- 1Fishberg Department of Neuroscience and Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 2Graduate School of Biomedical Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 3Department of Pharmacology and Systems Therapeutics, BD2K-LINCS Data Coordination and Integration Center, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Adult women are twice as likely as men to suffer from affective and anxiety disorders, although the mechanisms underlying heightened female stress susceptibility are incompletely understood. Recent findings in mouse Nucleus Accumbens (NAc) suggest a role for DNA methylation-driven sex differences in genome-wide transcriptional profiles. However, the role of another epigenetic process—microRNA (miR) regulation—has yet to be explored. We exposed male and female mice to Subchronic Variable Stress (SCVS), a stress paradigm that produces depression-like behavior in female, but not male, mice, and performed next generation mRNA and miR sequencing on NAc tissue. We applied a combination of differential expression, miR-mRNA network and functional enrichment analyses to characterize the transcriptional and post-transcriptional landscape of sex differences in NAc stress response. We find that male and female mice exhibit largely non-overlapping miR and mRNA profiles following SCVS. The two sexes also show enrichment of different molecular pathways and functions. Collectively, our results suggest that males and females mount fundamentally different transcriptional and post-transcriptional responses to SCVS and engage sex-specific molecular processes following stress. These findings have implications for the pathophysiology and treatment of stress-related disorders in women.
Introduction
Across cultures, adult women are twice as likely as men to suffer from affective and anxiety disorders including Major Depressive Disorder (MDD, Kessler et al., 1994, 2005; Bebbington, 1998). This enhanced female susceptibility begins in adolescence, declines post menopause, and is most pronounced during periods of great hormonal flux (perimenopause, postpartum period, late luteal phase), suggesting a role for female sex hormones in depression (Deecher et al., 2008; Gobinath et al., 2014). Once symptomatic, men and women experience depression differently—men are more likely to report anger, substance abuse, and risk-taking behavior; whereas women are more likely to experience somatic symptoms including irritability, fatigue, anhedonia, and sleep and appetite disturbances (Silverstein, 2002; Martin et al., 2013). Furthermore, women display enhanced risk of comorbid anxiety disorders and pain (Silverstein, 2002; Gobinath et al., 2014). Several studies suggest that sex differences may extend to treatment response, with women responding preferentially to selective serotonin reuptake inhibitors (SSRIs) over tricyclic antidepressants (TCAs), and men showing either no preference or better response to TCAs vs. SSRIs (Kornstein et al., 2000; Martenyi et al., 2001; Baca et al., 2004; Keers and Aitchison, 2010). Despite sex differences in MDD prevalence, symptoms, and treatment response, female subjects remain underrepresented in basic research, particularly in neuroscience (Beery and Zucker, 2011; Clayton and Collins, 2014).
Nevertheless, findings in animal models have yielded insight into the sexual dimorphism of MDD (Pfau and Russo, 2015). Stress is a key precipitating factor in the development of depressed mood and anxiety in human patients (Hammen, 2016; Pemberton and Fuller Tyszkiewicz, 2016). While adult female rodents often display greater cognitive resilience to chronic stress than males (Bowman et al., 2001; Luine, 2002; Conrad et al., 2003; Kitraki et al., 2004), they show enhanced emotional susceptibility to numerous stress paradigms (Westenbroek et al., 2003; Dalla et al., 2008; LaPlant et al., 2009; Sachs et al., 2014; Hodes et al., 2015a). Sex differences in the ventral striatum are increasingly implicated in adult female emotional vulnerability. The nucleus accumbens (NAc), a component structure of the ventral striatum, is essential for reward and emotion processing, and receives dense innervation from mood-related structures including the ventral tegmental area, amygdala, hippocampus, prefrontal cortex, and hypothalamus (Russo and Nestler, 2013). Recent studies highlight a role for NAc transcriptional and epigenetic processes in sex-specific stress responses. Following exposure to Subchronic Variable Stress (SCVS)—a stress paradigm that is sufficient to induce depression-like behavior in female mice but not in male mice—males and females exhibit remarkably different NAc transcriptional profiles that have been linked to both Nuclear factor κB (NfκB) transcription factor signaling (LaPlant et al., 2009) and epigenetic regulation by DNA methyltransferase 3a (Dnmt3a) (Hodes et al., 2015a).
Another potential epigenetic mechanism contributing to marked sex differences in stress-induced transcriptional patterns is post-transcriptional regulation by microRNAs (miRs). miRs are small, endogenous RNAs approximately 22 nucleotides in length. miRs do not encode proteins, but instead act to post-transcriptionally regulate the expression of target mRNA through sequence-specific binding, leading to subsequent mRNA destabilization or translational repression (Bartel, 2004; O'Carroll and Schaefer, 2013; Eichhorn et al., 2014). The evolutionary potency of miRs underscores their importance, and indeed, bioinformatic predictions estimate that 30% of mammalian protein-coding mRNAs are subject to miR regulation (O'Connor et al., 2012). Although the effect on protein expression of a single miR-mRNA interaction is modest, miRs can have hundreds of targets, and multiple miRs can target the same mRNA simultaneously (O'Carroll and Schaefer, 2013). miRs are highly enriched in the brain—50% of known mammalian miRs are expressed in the brain—and have been implicated in numerous neural processes including circuit formation and plasticity; synaptic function; and neuronal survival, differentiation, and diversity (O'Carroll and Schaefer, 2013). Dysregulated miRs have been reported in postmortem brain (Smalheiser et al., 2012; Issler et al., 2014; Lopez et al., 2014a,b; Maheu et al., 2015), blood (Belzeaux et al., 2012; Li et al., 2013; Fan et al., 2014; Issler et al., 2014; Lopez et al., 2014a; Camkurt et al., 2015), and dermal fibroblasts (Garbett et al., 2015) of depressed patients. In preclinical models, miRs have been implicated in depression-related processes ranging from glucocorticoid resistance (Uchida et al., 2008; Vreugdenhil et al., 2009) and corticotropin releasing factor sensitivity (Haramati et al., 2011), to behavioral resilience (Smalheiser et al., 2011; Dias et al., 2014; Issler et al., 2014) and antidepressant efficacy (Baudry et al., 2010; Issler et al., 2014).
In the present study, we aimed to characterize the NAc transcriptional and post-transcriptional landscape associated with sex differences in behavioral response to SCVS. Using RNA Sequencing (RNA-Seq), we performed an unbiased, genome-wide bioinformatic analysis of sex-specific miR and mRNA transcriptome profiles in mouse NAc following exposure to SCVS. We created miR-mRNA networks to probe and illustrate the complexity of stress-induced miR regulation in males and females. We find that male and female mice initiate fundamentally different transcriptional and post-transcriptional responses to stress. Furthermore, we report that male mice are not insensitive to SCVS, but rather mount a consistently robust transcriptional and post-transcriptional response to stress. This study provides new insight into the multiple levels of transcriptional processes that inform sex differences in stress susceptibility.
Materials and Methods
Animals
C57BL/6J male and female mice (Jackson Laboratory, Bar Harbor, ME) aged 8 weeks were used for all RNA-Seq and quantitative real-time PCR (qPCR) validation studies. All mice were shipped to the Icahn School of Medicine at Mount Sinai (ISMMS) animal facility at 7 weeks of age and were acclimated to the facility for 1 week prior to SCVS. Mice were group housed and maintained on a 12-h light/dark cycle in which lights were on between 7 A.M. and 7 P.M. with ad libitum access to food and water. All mouse procedures were approved by and performed in accordance with the National Institute of Health Guide for Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee at the Icahn School of Medicine at Mount Sinai. Different mouse cohorts were used for RNA-Seq and qPCR validation studies.
Subchronic Variable Stress (SCVS)
SCVS, which consists of three stressors administered over the course of 6 days, was performed as described previously (LaPlant et al., 2009; Hodes et al., 2015a). One unpredictable stressor was administered for an hour each day, and the stressors were alternated during the 6 days to prevent habituation. The three stressors included, in order: foot shock, tail suspension, and physical restraint. For foot shock stress, 10 same sex mice were placed in a fear conditioning chamber with electric grid flooring (Med Associates, St. Albans, VT) and administered 100 random 0.45 mA, 2 s foot shocks. For tail suspension stress, mouse tails were adhered to a bar, leaving the mice suspended for 1 h in an inverted position. For physical restraint stress, mice were placed in a well-ventilated, 50-mL centrifuge tube in the home cage for 1 h. The stressors were repeated in the same order on days 4–6, such that mice received foot shock stress on days 1 and 4, tail suspension stress on days 2 and 5, and restraint stress on days 3 and 6. After each stressor, mice were returned to the home cage until the next stressor or sacrifice. Unstressed controls were group housed in the home cage until sacrifice. SCVS-induced depression-like behavior was not assayed prior to sacrifice to avoid the introduction of testing-induced transcriptional changes. Furthermore, post-SCVS behavioral phenotypes have been well documented previously (LaPlant et al., 2009; Hodes et al., 2015a). The behavioral endpoints of SCVS in female mice include decreased sucrose preference (Hodes et al., 2015a), increased latency to eat in the novelty suppressed feeding test (Hodes et al., 2015a), increased immobility in the forced swim test (LaPlant et al., 2009; Hodes et al., 2015a), and increased latency to groom in the splash test (Hodes et al., 2015a). In contrast, male mice behave similarly to unstressed controls across these behavioral measures (LaPlant et al., 2009; Hodes et al., 2015a).
Estrous Cycle Monitoring
Vaginal lavage was performed on all female animals at sacrifice to determine estrous cycle stage. A sterile cotton swab moistened with 0.9% saline was used to collect a sample of vaginal cells from each animal. The sample was smeared onto a slide, left to dry overnight, and stained with 1% Toluidine Blue (Sigma-Aldrich, St. Louis, MO). Cycle stage was determined by visual inspection of cell morphology under a light microscope and recorded for each animal. Lavage was only performed at the point of sacrifice to best facilitate direct comparison of the sexes. Repeated lavage affects reward-related processes such as cocaine-stimulated motor behavior, and lavaged rats develop a conditioned place preference to a lavage-paired compartment (Walker et al., 2002). Therefore, daily lavage across all 6 days of the stress and at sacrifice could be potentially confounding. Furthermore, in an independent cohort of mice, we found that female mice cycled normally throughout exposure to the stress protocol (Figure S1) and did not demonstrate a stress-induced shift in length and duration of the estrous cycle that has been reported for other chronic stress paradigms (Konkle et al., 2003; Tou et al., 2004).
Tissue Collection, RNA Extraction, and cDNA Synthesis
NAc samples were collected from each animal as described previously (Golden et al., 2013; Hodes et al., 2015a). Twenty-four hours after the last stressor, animals were rapidly decapitated and bilateral 14-gauge ventral striatum punches were collected on ice. Punches were flash frozen on dry ice and stored at −80°C until RNA extraction. RNA was isolated using homogenization in Qiazol (Qiagen, Hilden, Germany) followed by chloroform layer separation. For sequencing, bilateral NAc punches from 5 animals (10 NAc punches) were pooled for each library at the point of homogenization. Pooling was necessary to yield sufficient input for miR sequencing, and was controlled for estrous cycle stage such that all female samples were derived from mice in estrus and proestrus. Mice in diestrus and metestrus were excluded from analysis. The aqueous RNA layer was further processed via miRNeasy mini kit (Qiagen, Hilden, Germany) to yield separate fractions enriched for small RNA (<200 bp; for small RNA sequencing) and large RNA (>200 bp; for mRNA sequencing). RNA and miR quality was assessed by Bioanalyzer (Agilent, Santa Clara, California). For qPCR validations, total RNA was extracted and purified (miRNeasy micro kit, Qiagen, Hilden, Germany) from bilateral NAc punches from individual animals. The RNA was then analyzed by Nanodrop (Thermo Fisher Scientific, Waltham, MA), and 500 ng of RNA was reverse transcribed to cDNA (qScript cDNA Supermix, Quanta Biosciences, Beverly, MA). cDNA was diluted in nuclease-free water to 500 uL (1 ng/uL).
Small RNA Sequencing and Differential Analysis
Small RNA-Seq libraries were generated from enriched small RNA fractions (<200 bp) according to the instructions of the Scriptminer Small RNA-Seq Library Preparation Kit (Epicentre, Madison, WI) with modifications as described in Dias et al. (2014). Libraries were constructed from pooled RNA samples (n = 5 mice/library) and run in independent biological replicates of 3 libraries per sex/stress condition for a total of 12 libraries. Library size and concentration were confirmed by Bioanalyzer (Agilent, Santa Clara, CA) prior to sequencing. Multiplexed libraries were pooled 12 to a lane and sequenced on a single lane of a HiSeq 2000 sequencer (Illumina, San Diego, CA) with 100 bp single-end reads at the ISMMS Genomics Core. Raw sequencing reads were processed by fastx trimmer (http://hannonlab.cshl.edu/fastx_toolkit) to remove the 3′ adaptor sequence, and sequences shorter than 16 nucleotides were subsequently discarded. Sequencing quality was assessed by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Sequencing yielded 192 million reads, with approximately 16 million high quality reads per sample. Reads were aligned to the mm9 mouse genome using Bowtie short read aligner (Langmead et al., 2009) and quantified against mature miRBase release 21 annotation using the HT-Seq Python package (Anders et al., 2015). Differential analysis was performed using the voom-limma R package (Law et al., 2014; Ritchie et al., 2015). A multifactorial, gene-wise linear model with stress and sex as main factors was fit to expression data to determine the effect of stress in males and females separately and to control for baseline variation in miR expression. An uncorrected p < 0.05 and a fold change threshold of 1.3 were used to determine differential miR expression, in accordance with the statistical cutoff used for NAc small RNA-Seq in Dias et al. (2014). Small RNA-Seq data has been made publicly available in the Gene Expression Omnibus (GEO) repository under the accession number GSE90962.
mRNA Sequencing and Differential Analysis
RNA-Seq libraries were generated from large RNA fractions (>200 bp) with RIN values > 8.8 as described in Feng et al. (2014). As for small RNA sequencing, libraries were constructed from pooled RNA samples (n = 5 mice/library) and run in independent biological replicates of 3 libraries per sex/stress condition for a total of 12 libraries. RNA (1 ug) was used for mRNA library construction according to the instructions of the TruSeq RNA Library Preparation Kit v2 (Illumina, San Diego, CA) as described in Hodes et al. (2015a). Libraries were evaluated by Bioanalyzer (Agilent, Santa Clara, CA) to verify size and concentration before pooling 12 to a lane and sequencing on 3 lanes of a HiSeq 2000 machine (Illumina, San Diego, CA) with 50 bp paired-end reads at the ISMMS Genomics Core. Sequencing yielded >30 million high quality reads per sample. Short reads were aligned to the Ensembl mus musculus NCBIM37 version 67 reference genome using TopHat2 (Kim et al., 2013). Quality assessment revealed a >90% mapping rate, with ~10% of those reads mapping to mitochondrial RNA and <1% aligning to ribosomal RNA. Individual gene counts for each sample were obtained using the HTSeq Python package (Anders et al., 2015). Only genes with >5 reads in at least 80% of samples in any one of the experimental conditions were included in differential analysis. Differential analysis was performed using the voom-limma R package (Law et al., 2014; Ritchie et al., 2015). A multifactorial, gene-wise linear model with stress and sex as main factors was fit to expression data to determine the effect of stress in males and females separately and to control for baseline variation in gene expression. An uncorrected p < 0.05 and a fold change threshold of 1.3 were used to determine differential gene expression, in accordance with the statistical cutoff used for NAc RNA-Seq in previously published reports (Hodes et al., 2015a; Bagot et al., 2016). RNA-Seq data has been made publicly available in the Gene Expression Omnibus (GEO) repository under the accession number GSE90962.
Quantitative Overlap
Quantitative overlap analysis was performed as described in Hodes et al. (2015a) using the SuperExactTest R software package (Wang et al., 2015). Differentially expressed mRNAs or miRs were divided into upregulated and downregulated lists. SuperExactTest R software was utilized to compare lists pairwise, generating 22 unique comparisons. mRNA/miR identifiers from each list were extracted for each comparison, and the quantitative overlap for each list was determined and statistically analyzed via a Fisher's Exact test. For all comparisons, significance was set at p < 0.05 and the number of background mRNAs/miRs was kept constant to reflect the total number of candidate mRNAs/miRs analyzed in the differential expression analysis (19,827 mRNAs, 781 miRs).
miR-mRNA Network Analysis
Predicted targets of stress-regulated miRs in males and females were identified using miRWalk 2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/), an online archive of predicted and experimentally validated miR-target interactions (Dweep et al., 2011; Dweep and Gretz, 2015). miRWalk searches were customized to yield only target mRNAs predicted by both the TargetScan and miRWalk algorithms for subsequent network analysis. Significant association of predicted targets of stress-regulated miRs within mRNA differential lists was assessed using chi-squared tests. The frequencies of upregulated, downregulated, and neutral mRNAs in stressed vs. control groups were computed from mRNA lists for each sex and were used as background. Then, for each differentially regulated miR, the frequencies of upregulated, downregulated and neutral target genes were computed from the mRNA differential list referencing the miRWalk predicted target list. A chi-squared test was then performed for each miR, comparing the frequencies of up, down and neutral target mRNAs versus that of the background. Significant miRs surviving this analysis were identified using a Benjamini-Hochberg adjusted p-value cut-off of p < 0.05. miR-mRNA relationships were then classified as having a positive association (both the miR and target mRNA are regulated by stress in the same direction) or a negative association (the miR and target mRNA are regulated by stress in opposite directions). miR-target network visualizations were constructed using Cytoscape v3.3.0 (cytoscape.org).
Functional Annotation and Pathway Analysis
Canonical Pathways and Diseases and Functions analyses were performed on differentially regulated genes and miR-targeted genes using Qiagen's Ingenuity Pathway Analysis (IPA, Qiagen Redwood City, CA, http://www.qiagen.com/ingenuity). Gene Ontology (GO) analysis was performed on differentially regulated and miR-targeted genes with Database for Annotation, Visualization and Integration Discovery (DAVID) Bioinformatics Resources (https://david.ncifcrf.gov/) using the DAVID mus musculus background (Huang da et al., 2009a, 2009b). IPA pathways/functions and GO terms were considered significantly enriched when Fisher's Exact –log(p-value) ≥ 1.3.
Quantitative Real-Time PCR
Quantitative real-time PCR (qPCR) biological validations of RNA-Seq targets were performed on NAc samples from individual animals. The qPCR reaction mixture consisted of 5 uL PerfeCta SYBR Green SuperMix, ROX (Quanta Biosciences, Beverly, MA), 1 uL primer (PrimeTime predesigned qPCR primers, Integrated DNA Technologies, Coralville, IA), 1 uL nuclease-free water, and 3 uL cDNA template. Samples were heated to 95°C for 2 min followed by 40 cycles of 95°C for 15 s, 60°C for 33 s, and 72°C for 33 s on an Applied Biosystems 7900 RT PCR System (Foster City, CA). Data were analyzed using the 2−ΔΔ(Ct) method (Livak and Schmittgen, 2001). Expression values were normalized to stably expressed reference genes (Lsm4, Kcnc2) that were selected from RNA-Seq gene lists based on their lack of regulation by SCVS. Expression values were normalized such that control groups show no fold change.
Statistical Analysis
For qPCR validations, differences between stressed and unstressed groups were compared using unpaired t-tests (two-tailed) or Wilcoxon Mann-Whitney (two-tailed) nonparametric U-tests when group variances were unequal. All statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA). Statistical significance was set at p < 0.05. Grubbs outlier test was performed and samples that varied more than two standard deviations from the mean were removed.
Results
SCVS Induces Largely Non-overlapping miR Transcriptional Profiles in Male and Female Mice
Gonadally intact, adult male and female mice were exposed to SCVS, consisting of 6 consecutive days of 3 alternating, unpredictable stressors: repeated foot shock, tail suspension, and physical restraint (Figure 1A). To profile stress-induced changes to the global NAc miRnome, we performed small RNA-Seq on NAc tissue collected 24 h after the last stressor. We compared stressed groups to same sex, unstressed controls in order to identify sex-specific patterns. Importantly, we controlled for female estrous cycle by including only females in estrus or proestrus so as to capture a period of great hormonal flux in estrogen and progesterone levels (Scharfman and MacLusky, 2008), and therefore recapitulate the peak sensitivity of female rodents to stress (Viau and Meaney, 1991; Carey et al., 1995; Bangasser and Valentino, 2014) and women to depression (Hendrick et al., 1998; Cohen et al., 2006; Gobinath et al., 2014). In males, 42 miRs were regulated by SCVS compared to unstressed male controls: 25 miRs were upregulated, and 17 miRs were downregulated (Figure 1B, Table S2). In females, 20% fewer miRs—28 miRs—were regulated by SCVS: 18 miRs were upregulated, and 10 miRs were downregulated (Figure 1B, Table S2). As illustrated by union maps (Figure 1B) and heatmap visualizations (Figure 1C), miR transcriptional profiles for males and females were largely non-overlapping. We submitted male and female miR differential lists to quantitative overlap analysis, and found that male and female miRnome profiles showed significant quantitative overlap only in upregulated miRs (mmu-miR-7224-3p, mmu-let-7d-3p, mmu-miR-1912-3p; p < 0.02) and oppositely regulated miRs (mmu-miR-5099, mmu-miR-206-3p; p < 0.04).

Figure 1. Male and female mice show different NAc miR profiles following Subchronic Variable Stress. (A) Schematic of the Subchronic Variable Stress (SCVS) paradigm. (B) Union maps of significantly upregulated (Left) and downregulated (Right) microRNAs (miRs) in males and females following SCVS. (C) (Left) Heatmap of miRs differentially regulated by SCVS in females compared to the same miRs in males, (Right) Heatmap of miRs differentially regulated by SCVS in males compared to the same miRs in females. Yellow indicates upregulation, blue indicates downregulation, and black indicates no change.
Males Mount a Robust, Sex-Specific Transcriptional Response to SCVS
We next sought to interrogate the effect of SCVS on gene expression in the same animals by performing RNA-Seq. RNA-Seq allowed us to quantify the expression levels of all polyA-containing transcripts in stressed mice and controls. In stressed males compared to unstressed control males, 1349 genes were regulated by SCVS (Figure 2A, See Table S3 for top 20 up and downregulated genes and Data Sheet 2 for full list). The majority of stress-regulated genes in males were downregulated (~76% or 1025 genes, vs. 324 upregulated genes). Strikingly, despite the insufficiency of SCVS to induce behavioral and neuroendocrine changes in male mice, males demonstrated a very robust transcriptional response to SCVS, potentially indicating active resilience mechanisms. In stressed females compared to unstressed control females, 348 genes were regulated by SCVS (Figure 2A, See Table S3 for top 20 up and downregulated genes and Data Sheet 2 for full list). As in males, the majority of stress-regulated genes in females were downregulated (~67% or 232 genes, vs. 116 upregulated genes). Similar to what we observed for miRs, male and female SCVS-induced genome profiles showed little overlap—approximately 2% of all upregulated genes (9 genes, p = 1.23E−4) and ~3% of all downregulated genes (39 genes, p = 6.58E−11) (Figures 2A,B). In total, 21 genes were regulated in opposite directions. Twelve genes were upregulated in males and downregulated in females (p = 4.58E−4): Plekha2, Etl4, Chrna4, AC154308.1, St14, Fhl2, Alas2, Arhgap15, Adam18, AC133502.1, Rhox4a, and Chrnb3. Nine genes were downregulated in males and upregulated in females (p = 0.15): AL671140.1, Tex16, Gm12494, Nts, U6, Arhgap36, Ropn1l, Tnfsf9, and Fas. As shown in Figures 2C,D, we performed biological validations of several genes by qPCR: Mstn [t(17) = 2.86, p = 0.01], Tlr4 [t(18) = 4.93, p < 0.001], Fmo2 (U = 3.00, p < 0.001), Tnfaip8l2 [t(19) = 3.65, p < 0.01], Sgk1 [t(19) = 3.46, p < 0.01], Npy2r (U = 10.00, p < 0.01), Crh [t(18) = 2.25, p = 0.04], Fosb [t(18) = 2.68, p = 0.02], Lynx1 (U = 24.00, p = 0.09), Drd4 (U = 13.00, p < 0.01), Adcy1 (U = 23.00, p = 0.048), and Reln (U = 17.00, p = 0.02). Genes for validation were chosen from IPA output for their involvement in significantly enriched functional networks and pathways.
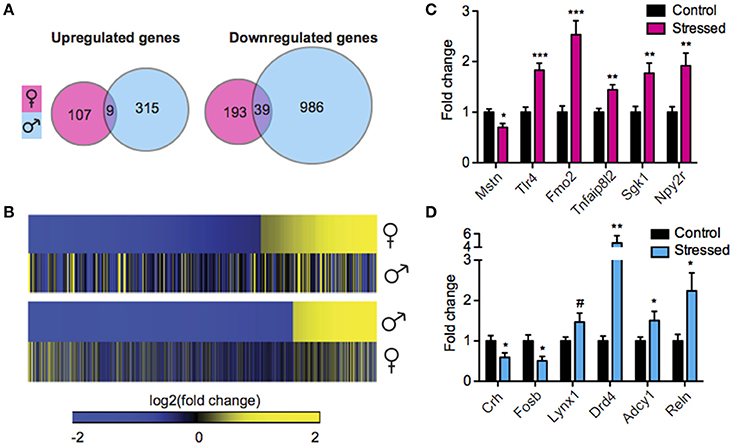
Figure 2. Male and female mice show different NAc mRNA profiles following Subchronic Variable Stress. (A) Union maps of significantly upregulated (Left) and downregulated (Right) genes in males and females following Subchronic Variable Stress (SCVS). (B) (Top) Heatmap of genes differentially regulated by SCVS in females compared with the same genes in males, (Bottom) Heatmap of genes differentially regulated by SCVS in males compared with the same genes in females. Yellow indicates upregulation, blue indicates downregulation, and black indicates no change. (C) Validation by quantitative real-time PCR (qPCR) of a subset of genes regulated by SCVS in females (n = 9–11 per group). (D) Validation by qPCR of a subset of genes regulated by SCVS in males (n = 9–11 per group). *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.1.
Functional Ontology and Pathway Analysis Reveals Sex Differences in Stress-Induced Functional Processes
We performed pathway and gene ontology (GO) analysis on our RNA-Seq mRNA differential lists using IPA and DAVID tools, respectively (Figure 3, Table S4). We utilized both tools to capitalize on their respective strengths—IPA draws from a curated, proprietary knowledgebase of published, experimentally validated information, while DAVID utilizes a comprehensive knowledgebase compiled from dozens of publicly available bio-databases (BioCarta, KEGG, GO, etc.) to maximize analytic power. Reflecting our finding of minimally overlapping SCVS-induced genes, we observed enrichment of different functional processes in males and females. Only one canonical pathway—Antigen presentation pathway, which encompasses antigen recognition and processing essential for innate and adaptive immunity—is represented among significantly enriched canonical pathways in both males and females (Figure 3A, Table S3). Among significantly enriched DAVID GO molecular function terms for males and females, we observed overlap of five functions—Neurotransmitter receptor activity and binding, peptide receptor activity and binding, and G-protein coupled peptide receptor activity—however, different or, in some cases, oppositely regulated genes were enriched within these pathways in the two sexes (Figure 3B, Table S3). Overall, we observed enrichment of several processes relevant to stress and depression, including inflammatory processes and cytokine activity (Hodes et al., 2014, 2015b), cAMP and GPCR-mediated signaling (Duman et al., 1999; Terzi et al., 2009) and ion channel activity (Friedman et al., 2014). Importantly, we observed overlap of numerous functional categories between our IPA and DAVID analyses, with terms including ion homeostasis/channel activity and GPCR signaling appearing in both analyses for males, and inflammatory processes appearing in both analyses for females.
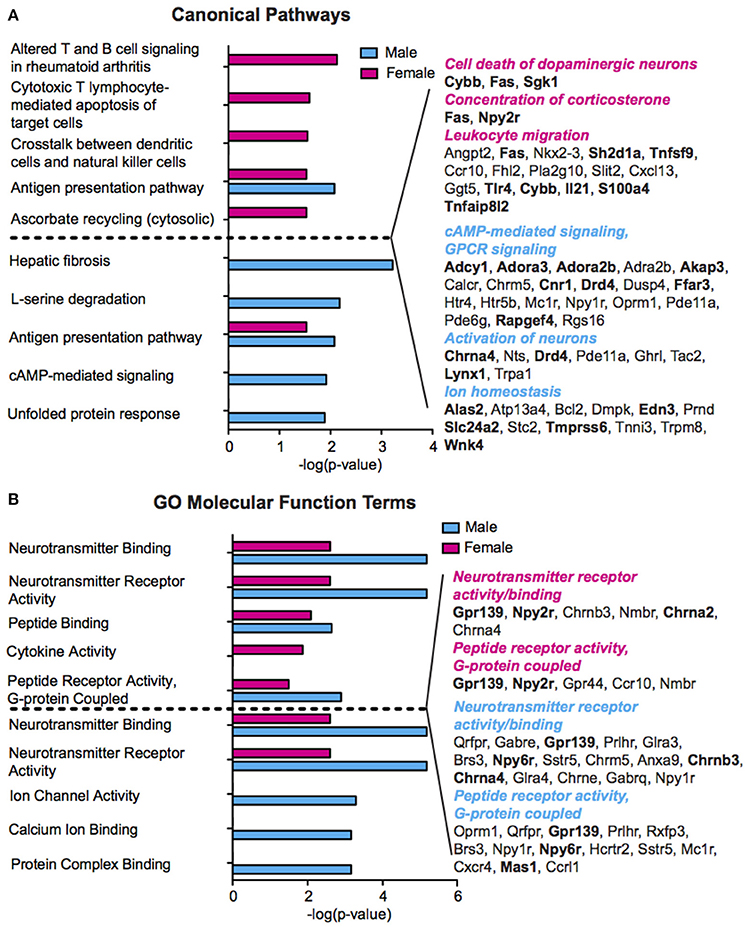
Figure 3. Sex differences in pathway enrichment and gene ontology of genes regulated by Subchronic Variable Stress. (A) (Left) The top five significantly enriched (p < 0.05) IPA canonical pathways in females (top, magenta) and males (bottom, blue), shown as –log(p-value). For pathways significantly enriched in both sexes, the degree of enrichment of the pathway in the opposite sex is shown in a separate, immediately adjacent bar. (Right) Hand curated list of genes relevant to depression pathology chosen from significantly enriched IPA Canonical Pathways or from within the top 50 significantly enriched IPA Diseases and Functions. Upregulated genes are shown in bold font. Enriched pathways/functions in females are colored magenta whereas those for males are colored blue. (B) (Left) The top five significantly enriched (p < 0.05) GO Molecular Functions in females (top, magenta) and males (bottom, blue) as identified by DAVID, shown as–log(p-value). For GO terms significantly enriched in both sexes, the degree of enrichment of the term in the opposite sex is shown in a separate, immediately adjacent bar. (Right) Genes corresponding to the neurotransmitter receptor activity and binding, and peptide receptor activity, G-protein coupled, molecular functions in females (top) and males (bottom), with upregulated genes shown in bold font.
SCVS Induces Sex Dependent miR-mRNA Networks
We next sought to overlay miR and mRNA differential lists to probe the nature and complexity of miR-mRNA interactions in males and females. In order to accomplish this, we identified all mRNA targets of SCVS-regulated miRs predicted by both TargetScan and miRWalk online databases (Lewis et al., 2005). We then applied a novel chi-squared analysis to measure enrichment of predicted miR target genes in our RNA-Seq mRNA differential lists. In females, 7 miRs (25%) survived the analysis, two downregulated miRs and 5 upregulated miRs (Figures 4A,B, Table S5). Eighty-three genes, or ~24%, were miR-targeted (Figure 4C). For simplicity, we illustrated only miR target genes that satisfy the canonical negative miR-mRNA target association in network diagrams, however, ~29% of genes showed only a positive association with targeting miRs, and ~33% of genes showed both positive and negative associations (Figure 4C). The majority of target genes—48 genes or ~58%—were targeted by more than one miR, whereas 35 genes (~42%) were targeted by only one miR (Figure 4C). We performed IPA (Figure 4D, Table S8) and DAVID (Figure 4E, Table S8) analyses on miR-targeted genes, finding enrichment of several processes relevant to depression, including Wnt/ß-Catenin signaling (Dias et al., 2014), calcium signaling and immune processes (Hodes et al., 2014, 2015b). Interestingly, we observed overlap of significantly regulated canonical pathways (Altered T and B Cell Signaling in Rheumatoid Arthritis) and GO molecular function terms (Neurotransmitter receptor activity, binding; Cytokine activity) between analyses of all SCVS-regulated genes and miR-targeted genes, indicating an involvement of miRs in prominent stress-induced molecular processes. In males, 23 miRs (~55%) survived the analysis, 12 downregulated miRs and 11 upregulated miRs (Tables S6, S7). Due to the number and size of miR-mRNA networks in males, we illustrated only the networks corresponding to the top 3 miRs most strongly downregulated by SCVS and one of the top 3 miRs most strongly upregulated by SCVS (Figures 5A,B). The remaining miRs and targeted mRNAs are listed in Tables S6, S7. Again, we only included miR target genes with a negative association to the miR of interest in network illustrations. Six hundred sixty-three genes (~49%) were miR-targeted (Figure 6A). Of these, 532 genes (~80%) showed a canonical negative association with at least 1 miR, whereas 131 genes (~20%) showed only a positive association (Figure 6A). The majority of miR-targeted genes (468 genes, or ~71%) were targeted by more than 1 miR, whereas 195 genes (~29%) were targeted by only 1 miR (Figure 6A). IPA analysis on miR-targeted genes revealed that, of the 13 significantly enriched canonical pathways for the male miR-targeted gene list, 7 overlapped with those enriched in the SCVS male differential gene list (the top 5 enriched pathways are represented in Figure 6B, the full list is represented in Table S8). DAVID analysis revealed that 24 of the 35 significantly enriched GO molecular function terms in the miR-targeted gene list overlapped with those from the full SCVS list, further illustrating the potential involvement of miR regulation in stress responsive molecular processes (the top 5 enriched GO terms are represented in Figure 6C, the full list is represented in Table S8).
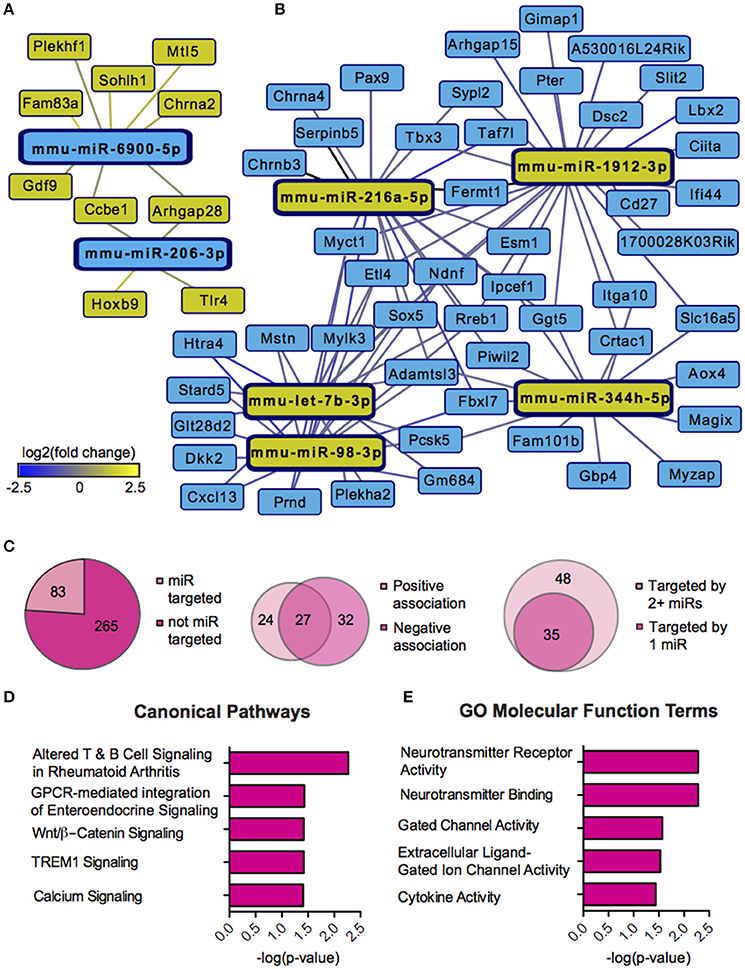
Figure 4. Female miR-mRNA networks regulated by Subchronic Variable Stress. (A) Networks of downregulated microRNAs (miRs, blue nodes) surviving network analysis and their negatively associated, upregulated target genes (yellow nodes). (B) Networks of upregulated miRs (yellow nodes) surviving network analysis and their negatively associated, downregulated target genes (blue nodes). Blue lines indicate target gene downregulation whereas yellow lines indicate target gene upregulation. Line brightness illustrates the degree of up/downregulation, with brighter lines connecting to more strongly regulated genes. (C) Pie charts and union maps illustrating genes targeted and not targeted by miRs (Left), genes showing a negative and/or positive association with targeting miRs (Middle), and genes targeted by 1 miR or 2+ miRs (Right). (D) The top five significantly enriched (p < 0.05) IPA canonical pathways for female miR-targeted genes. (E) The top five significantly enriched (p < 0.05) GO Molecular Functions for female miR-targeted genes as identified by DAVID.
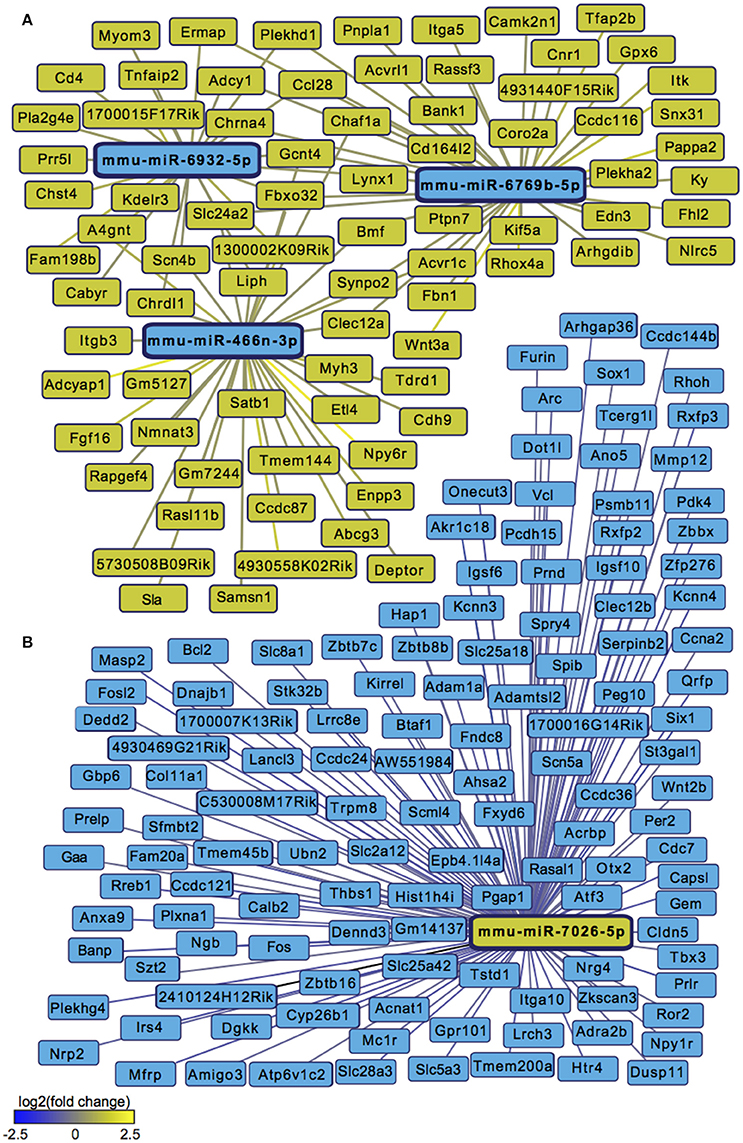
Figure 5. Male miR-mRNA networks regulated by Subchronic Variable Stress. (A) Networks of the top three most strongly downregulated miRs (blue nodes) surviving network analysis and their negatively associated, upregulated target genes (yellow nodes). (B) Network of one of the top three most strongly upregulated miRs (yellow node) surviving network analysis and its negatively associated, downregulated target genes (blue nodes). Blue lines indicate target gene downregulation whereas yellow lines indicate target gene upregulation. Line brightness illustrates the degree of up/downregulation, with brighter lines connecting to more strongly regulated genes.
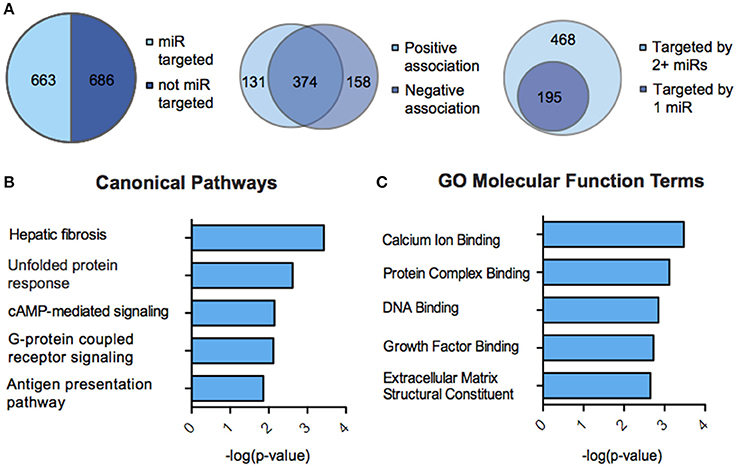
Figure 6. miR-mRNA regulatory landscape and functional enrichment of miR-targeted genes in males. (A) Pie charts and union maps illustrating genes targeted and not targeted by microRNAs (miRs, Left), genes showing a negative and/or positive association with targeting miRs (Middle), and genes targeted by 1 miR or 2+ miRs (Right). (B) The top five significantly enriched (p < 0.05) IPA canonical pathways for male miR-targeted genes. (C) The top five significantly enriched (p < 0.05) GO Molecular Functions for male miR-targeted genes as identified by DAVID.
Discussion
We find that SCVS produces striking sex differences in NAc transcriptional and post-transcriptional profiles. We observe male/female overlap of only ~3% of similarly regulated mRNAs and ~4% of similarly regulated miRs following SCVS. We also find minimal overlap between sexes in enriched functional processes. Rather than displaying a resistance to SCVS-induced transcriptional changes that mirrors the male behavioral response, males mount a robust transcriptional response to stress. This “active” response extends to male post-transcriptional profiles and miR-mRNA networks. These findings provide unique insight into the molecular underpinnings of sexual dimorphism in stress response.
The majority of studies investigating miR regulation of target mRNAs in psychiatric disease patients and animal models have used qPCR (Garbett et al., 2015), microarray (Hollander et al., 2010; Haramati et al., 2011; Miller et al., 2012; Tapocik et al., 2013; Issler et al., 2014; Lopez et al., 2014a), or candidate-based approaches (Uchida et al., 2008; Baudry et al., 2010; Im and Kenny, 2012; Maheu et al., 2015; Zhang et al., 2015). We chose to use RNA-Seq as it is a powerful technique with several key advantages: single-base resolution, broad dynamic range, low background, and high reproducibility (Finotello and Di Camillo, 2015). We performed small RNA and mRNA sequencing on separate size-enriched fractions of RNA from the same NAc dissections so as to capture the simultaneous expression patterns of miRs and genes. We then applied an in silico approach to identify miR-mRNA target interactions.
Our pipeline has several strengths. First, it provides a complete, unbiased snapshot of NAc transcription and post-transcriptional regulation in stressed males and females. Second, we implemented a requirement that miR targets be predicted by both the TargetScan and miRWalk algorithms to increase our confidence in miR-target relationships, as target prediction algorithms can produce false positives (Darnell, 2010). Furthermore, as tissue miR detection does not necessarily denote appreciable repressive activity (Mullokandov et al., 2012; La Rocca et al., 2015), we included an additional layer of anti-correlated miR-mRNA expression networks to support a functional role for identified miRs. Interestingly, we observed a non-canonical positive correlation in a substantial proportion of significant miR-mRNA relationships—61% of miR-targeted genes in females, and 76% of miR-targeted genes in males had a positive correlation with at least one targeting miR. miR activation of targeted genes has been reported (Vasudevan et al., 2007; Henke et al., 2008; Ørom et al., 2008) but is uncommon, and therefore likely represents alternative and indirect regulatory mechanisms not captured by our analysis. Alternatively, as the effect of a single miR on target expression is modest, the net effect of all targeting miRs on a gene may be negative despite some positive miR-mRNA relationships. This observation, coupled with the finding that a majority of miR-targeted genes in males and females were targeted by multiple miRs, highlights the complexity of miR regulation and the utility of coordinated miR activity in fine-tuning gene expression in the brain.
The genome-wide profiles we report corroborate our previous mRNA findings (Hodes et al., 2015a) and extend them to the level of post-transcriptional regulation. Collectively, our results suggest that male mice may be mounting an active, adaptive NAc transcriptional and post-transcriptional response that is unique to the male sex. Several recent studies suggest that a robust transcriptional response contributes to behavioral resilience. Bagot et al. (2016) report that mice resilient to chronic social defeat stress display more differentially expressed genes in NAc compared to controls than do susceptible mice at 48 h post-defeat. We previously found that following SCVS, females show a greater induction of the repressive methyltransferase gene Dnmt3a in NAc (Hodes et al., 2015a). Viral knockdown of Dnmt3a promoted behavioral resilience, and interestingly produced a greater percent overlap with male transcriptional patterns. Smalheiser et al. (2011) reported evidence that robust miR responses may also contribute to resilience. They observed that rats resilient to learned helplessness show significant regulation of 48% more miRs in cortex than do susceptible rats. Interestingly, the greater proportion of SCVS-regulated genes in males than in females was starker in this analysis than in our previously published RNA-Seq study (60% more genes in males versus 17% in our previous study, Hodes et al., 2015a). This difference may be attributable to the fact that we sequenced only females in estrus and proestrus, and may indicate that females show an especially dampened NAc transcriptional stress response during this period of heightened stress sensitivity.
We chose to sequence samples derived from reproductively intact females in estrus and proestrus due to the hormonal dynamics of the estrous cycle. The rodent estrous cycle consists of four phases: proestrus, estrus, metestrus, and diestrus. Estrogen and progesterone reach peak plasma concentrations during proestrus, decline in concentration throughout estrus, and are present at low concentrations in diestrus (Butcher et al., 1974; Bangasser and Valentino, 2014). Therefore, by studying females in estrus and proestrus, we intended to best capture a period characterized by both hormonal fluctuation and high levels of estrogen and progesterone. Adult women are most susceptible to depression during periods of great hormonal fluctuation, including the postpartum period (Hendrick et al., 1998) and during perimenopause (Cohen et al., 2006; Toffol et al., 2015). Comparatively, female rodents are most susceptible to stress during proestrus—when estrogen and progesterone are high—and exhibit higher plasma corticosterone levels at baseline and in response to stress in proestrus compared to other cycle stages (Viau and Meaney, 1991; Carey et al., 1995; Bangasser and Valentino, 2014). A recent meta-analysis of 311 neuroscience articles found that female rats at any stage of the estrous cycle are not more variable than male rats across behavioral, electrophysiological, neurochemical, and histological measures (Becker et al., 2016), further supporting our rationale for combining females in estrus and proestrus.
Our finding of minimal overlap in genes, miRs, and functional processes between the sexes suggests that the female transcriptional and post-transcriptional stress response is not a blunted male response, but is rather a fundamentally different, sex-specific response. Females engage different NAc molecular processes in response to stress than do males, and this likely has important consequences for behavioral outcome. The sex-specific patterns we observed mirror and greatly expand upon those we have previously reported, now highlighting a role for novel post-transcriptional networks in sex differences in stress responses (Hodes et al., 2015a). We also observed more overlapping enriched pathways and GO terms between total male SCVS and miR-targeted male SCVS gene lists than we did for female counterparts, indicating that miRs may play a more prominent role in stress-responsive molecular processes in males than in females. However, it is important to note that our female functional enrichment analyses were lower powered than those for males as our female gene lists were smaller, potentially exaggerating this effect. Nevertheless, the pronounced sex differences we observed in these complex miR-gene networks may shed light on important mechanistic differences governing sex differences in fundamental NAc neural processes, such as synaptic plasticity (Forlano and Woolley, 2010; Wissman et al., 2012) and neuroactive steroid signaling (Becker, 1999; Almey et al., 2015). Furthermore, our analysis revealed enrichment of pathways in miR-targeted gene lists that may yield future insights into enhanced female stress vulnerability. For instance, Wnt/ß-catenin signaling was an enriched IPA canonical pathway for female miR-targeted genes, and component genes Dkk2, Wnt7b, and Sox5 were downregulated in the female SCVS gene list. As Wnt/ß-catenin signaling is necessary for behavioral resilience to chronic social defeat stress (Dias et al., 2014), downregulation of this pathway may contribute to enhanced female vulnerability to SCVS. Our network analysis also highlights miRs for downstream functional analysis. For example, miR-206-3p—a miR that survived our network analysis in females—was oppositely regulated in males and females following SCVS. Recent studies indicate involvement of miR-206-3p—a known regulator of BDNF signaling—in rat hippocampal response to the rapidly acting antidepressant ketamine (Yang et al., 2014), medial prefrontal cortex-mediated alcohol consumption in rats (Tapocik et al., 2014) and susceptibility to bipolar disorder in human subjects (Wang et al., 2014).
In conclusion, we report that male and female mice exhibit fundamentally different NAc genome-wide responses to SCVS. Differences span both transcriptional and post-transcriptional levels, and highlight a potential role for coordinated mRNA and miR activity in behavioral outcome. These findings inform our understanding of the enhanced susceptibility of women to depression and suggest that, as stress response is strikingly different in males and females, treatment protocols and target identification strategies should take sex specificity into account.
Author Contributions
MP, GH, IP, LS, and SR designed the experiments and pipeline analyses. MP, GH, JF, SG, HA, HC, MF, CM, and MH performed the experiments and acquired data. MP, IP, ZL, ZW, and AM analyzed and interpreted the data. MP and SR wrote the article. All authors read and approved the manuscript.
Funding
This work was supported by US National Institute of Mental Health grants R01 MH090264 (SR), R01 MH104559 (SR), R01 MH104559 (SR), P50 MHo96890 (SR), R21 MH099562 (SR), F31 MH105217 (MP), T32 MH087004 (MP), T32 MH096678 (MP), and by US National Center for Complementary and Integrative Health grant P50 MH096890 (SR).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MF and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2016.00144/full#supplementary-material
References
Almey, A., Milner, T. A., and Brake, W. G. (2015). Estrogen receptors in the central nervous system and their implication for dopamine-dependent cognition in females. Horm. Behav. 74, 125–138. doi: 10.1007/s12017-014-8312-z
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1038/srep16923
Baca, E., Garcia-Garcia, M., and Porras-Chavarino, A. (2004). Gender differences in treatment response to sertraline versus imipramine in patients with nonmelancholic depressive disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 57–65. doi: 10.1016/S0278-5846(03)00177-5
Bagot, R. C., Cates, H. M., Purushothaman, I., Lorsch, Z. S., Walker, D. M., Wang, J., et al. (2016). Circuit-wide transcriptional profiling reveals brain region-specific gene networks regulating depression susceptibility. Neuron 90, 969–983. doi: 10.1006/meth.2001.1262
Bangasser, D. A., and Valentino, R. J. (2014). Sex differences in stress-related psychiatric disorders: neurobiological perspectives. Front. Neuroendocrinol. 35, 303–319. doi: 10.1016/S1877-1173(09)86010-9
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Baudry, A., Mouillet-Richard, S., Schneider, B., Launay, J. M., and Kellermann, O. (2010). miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science 329, 1537–1541. doi: 10.1126/science.1193692
Becker, J. B. (1999). Gender differences in dopaminergic function in striatum and nucleus accumbens. Pharmacol. Biochem. Behav. 64, 803–812. doi: 10.1523/JNEUROSCI.0445-14.2014
Becker, J. B., Prendergast, B. J., and Liang, J. W. (2016). Female rats are not more variable than male rats: a meta-analysis of neuroscience studies. Biol. Sex Differ. 7:34. doi: 10.1186/s13293-016-0087-5
Beery, A. K., and Zucker, I. (2011). Sex bias in neuroscience and biomedical research. Neurosci. Biobehav. Rev. 35, 565–572. doi: 10.1016/j.neubiorev.2010.07.002
Belzeaux, R., Bergon, A., Jeanjean, V., Loriod, B., Formisano-Treziny, C., Verrier, L., et al. (2012). Responder and nonresponder patients exhibit different peripheral transcriptional signatures during major depressive episode. Transl. Psychiatry 2, e185. doi: 10.1016/j.jpsychires.2014.08.007
Bowman, R. E., Zrull, M. C., and Luine, V. N. (2001). Chronic restraint stress enhances radial arm maze performance in female rats. Brain Res. 904, 279–289. doi: 10.1016/S0006-8993(01)02474-X
Butcher, R. L., Collins, W. E., and Fugo, N. W. (1974). Plasma concentration of LH, FSH, prolactin, progesterone and estradiol-17β throughout 4-day estrous-cycle of rat. Endocrinology 94, 1704–1708.
Camkurt, M. A., Acar, S., Coskun, S., Günes, M., Günes, S., Yilmaz, M. F., et al. (2015). Comparison of plasma MicroRNA levels in drug naive, first episode depressed patients and healthy controls. J. Psychiatr. Res. 69, 67–71. doi: 10.1016/j.jpsychires.2015.07.023
Carey, M. P., Deterd, C. H., de Koning, J., Helmerhorst, F., and de Kloet, E. R. (1995). The influence of ovarian steroids on hypothalamic-pituitary-adrenal regulation in the female rat. J. Endocrinol. 144, 311–321. doi: 10.1038/nn.4113
Clayton, J. A., and Collins, F. S. (2014). Policy: NIH to balance sex in cell and animal studies. Nature 509, 282–283. doi: 10.1038/509282a
Cohen, L. S., Soares, C. N., Vitonis, A. F., Otto, M. W., and Harlow, B. L. (2006). Risk for new onset of depression during the menopausal transition: the Harvard study of moods and cycles. Arch. Gen. Psychiatry 63, 385–390. doi: 10.1016/j.cell.2004.12.035
Conrad, C. D., Grote, K. A., Hobbs, R. J., and Ferayorni, A. (2003). Sex differences in spatial and non-spatial Y-maze performance after chronic stress. Neurobiol. Learn. Mem. 79, 32–40. doi: 10.1016/S1074-7427(02)00018-7
Dalla, C., Antoniou, K., Kokras, N., Drossopoulou, G., Papathanasiou, G., Bekris, S., et al. (2008). Sex differences in the effects of two stress paradigms on dopaminergic neurotransmission. Physiol. Behav. 93, 595–605. doi: 10.1016/j.physbeh.2007.10.020
Darnell, R. B. (2010). HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 1, 266–286. doi: 10.1002/wrna.31
Deecher, D., Andree, T. H., Sloan, D., and Schechter, L. E. (2008). From menarche to menopause: exploring the underlying biology of depression in women experiencing hormonal changes. Psychoneuroendocrinology 33, 3–17. doi: 10.1016/j.psyneuen.2007.10.006
Dias, C., Feng, J., Sun, H., Shao, N. Y., Mazei-Robison, M. S., Damez-Werno, D., et al. (2014). β-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature 516, 51–55. doi: 10.1038/nature13976
Duman, R. S., Malberg, J., and Thome, J. (1999). Neural plasticity to stress and antidepressant treatment. Biol. Psychiatry 46, 1181–1191. doi: 10.1038/nature09202
Dweep, H., and Gretz, N. (2015). miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat. Methods 12, 697. doi: 10.1210/endo-129-5-2503
Dweep, H., Sticht, C., Pandey, P., and Gretz, N. (2011). miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 44, 839–847. doi: 10.1677/joe.0.1440311
Eichhorn, S. W., Guo, H., McGeary, S. E., Rodriguez-Mias, R. A., Shin, C., Baek, D., et al. (2014). mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell 56, 104–115. doi: 10.1038/mp.2011.162
Fan, H. M., Sun, X. Y., Guo, W., Zhong, A. F., Niu, W., Zhao, L., et al. (2014). Differential expression of microRNA in peripheral blood mononuclear cells as specific biomarker for major depressive disorder patients. J. Psychiatr. Res. 59, 45–52. doi: 10.1016/j.jpsychires.2014.08.007
Feng, J., Wilkinson, M., Liu, X., Purushothaman, I., Ferguson, D., Vialou, V., et al. (2014). Chronic cocaine-regulated epigenomic changes in mouse nucleus accumbens. Genome Biol. 15:R65. doi: 10.1186/gb-2014-15-4-r65
Finotello, F., and Di Camillo, B. (2015). Measuring differential gene expression with RNA-seq: challenges and strategies for data analysis. Brief. Funct. Genomics 14, 130–142. doi: 10.1093/bfgp/elu035
Forlano, P. M., and Woolley, C. S. (2010). Quantitative analysis of pre- and postsynaptic sex differences in the nucleus accumbens. J. Comp. Neurol. 518, 1330–1348. doi: 10.1016/S0091-3057(99)00168-9
Friedman, A. K., Walsh, J. J., Juarez, B., Ku, S. M., Chaudhury, D., Wang, J., et al. (2014). Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science 344, 313–319. doi: 10.1126/science.1249240
Garbett, K. A., Vereczkei, A., Kalman, S., Brown, J. A., Taylor, W. D., Faludi, G., et al. (2015). Coordinated messenger RNA/microRNA changes in fibroblasts of patients with major depression. Biol. Psychiatry 77, 256–265. doi: 10.1016/j.biopsych.2014.05.015
Gobinath, A. R., Mahmoud, R., and Galea, L. A. (2014). Influence of sex and stress exposure across the lifespan on endophenotypes of depression: focus on behavior, glucocorticoids, and hippocampus. Front. Neurosci. 8:420. doi: 10.3389/fnins.2014.00420
Golden, S. A., Christoffel, D. J., Heshmati, M., Hodes, G. E., Magida, J., Davis, K., et al. (2013). Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat. Med. 19, 337–344. doi: 10.1038/nm.3090
Hammen, C. (2016). Depression and stressful environments: identifying gaps in conceptualization and measurement. Anxiety Stress Coping 29, 335–351. doi: 10.1080/10615806.2015.1134788
Haramati, S., Navon, I., Issler, O., Ezra-Nevo, G., Gil, S., Zwang, R., et al. (2011). MicroRNA as repressors of stress-induced anxiety: the case of amygdalar miR-34. J. Neurosci. 31, 14191–14203. doi: 10.1523/JNEUROSCI.1673-11.2011
Hendrick, V., Altshuler, L. L., and Suri, R. (1998). Hormonal changes in the postpartum and implications for postpartum depression. Psychosomatics 39, 93–101. doi: 10.1126/science.1249240
Henke, J. I., Goergen, D., Zheng, J., Song, Y., Schüttler, C. G., Fehr, C., et al. (2008). microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 27, 3300–3310. doi: 10.1016/j.molcel.2008.05.001
Hodes, G. E., Kana, V., Menard, C., Merad, M., and Russo, S. J. (2015b). Neuroimmune mechanisms of depression. Nat. Neurosci. 18, 1386–1393. doi: 10.1073/pnas.1113793109
Hodes, G. E., Pfau, M. L., Leboeuf, M., Golden, S. A., Christoffel, D. J., Bregman, D., et al. (2014). Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc. Natl. Acad. Sci. U.S.A. 111, 16136–16141. doi: 10.1038/tpj.2012.17
Hodes, G. E., Pfau, M. L., Purushothaman, I., Ahn, H. F., Golden, S. A., Christoffel, D. J., et al. (2015a). Sex differences in nucleus accumbens transcriptome profiles associated with susceptibility versus resilience to subchronic variable stress. J. Neurosci. 35, 16362–16376. doi: 10.1523/JNEUROSCI.1392-15.2015
Hollander, J. A., Im, H. I., Amelio, A. L., Kocerha, J., Bali, P., Lu, Q., et al. (2010). Striatal microRNA controls cocaine intake through CREB signalling. Nature 466, 197–202. doi: 10.1038/nature09202
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009a). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1016/j.yfrne.2014.03.008
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009b). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. doi: 10.1016/S0033-3182(98)71355-6
Im, H. I., and Kenny, P. J. (2012). MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 35, 325–334. doi: 10.1016/j.tins.2012.01.004
Issler, O., Haramati, S., Paul, E. D., Maeno, H., Navon, I., Zwang, R., et al. (2014). MicroRNA 135 is essential for chronic stress resiliency, antidepressant efficacy, and intact serotonergic activity. Neuron 83, 344–360. doi: 10.1371/journal.pone.0063648
Keers, R., and Aitchison, K. J. (2010). Gender differences in antidepressant drug response. Int. Rev. Psychiatry 22, 485–500. doi: 10.3109/09540261.2010.496448
Kessler, R. C., Berglund, P., Demler, O., Jin, R., Merikangas, K. R., and Walters, E. E. (2005). Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the national comorbidity survey replication. Arch. Gen. Psychiatry 62, 593–602. doi: 10.1001/archpsyc.62.6.593
Kessler, R. C., McGonagle, K. A., Zhao, S., Nelson, C. B., Hughes, M., Eshleman, S., et al. (1994). Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the national comorbidity survey. Arch. Gen. Psychiatry 51, 8–19. doi: 10.1001/archpsyc.1994.03950010008002
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. doi: 10.1093/nar/gkn923
Kitraki, E., Kremmyda, O., Youlatos, D., Alexis, M., and Kittas, C. (2004). Spatial performance and corticosteroid receptor status in the 21-day restraint stress paradigm. Ann. N.Y. Acad. Sci. 1018, 323–327. doi: 10.1196/annals.1296.039
Konkle, A. T., Baker, S. L., Kentner, A. C., Barbagallo, L. S., Merali, Z., and Bielajew, C. (2003). Evaluation of the effects of chronic mild stressors on hedonic and physiological responses: sex and strain compared. Brain Res. 992, 227–238. doi: 10.1186/gb-2014-15-2-r29
Kornstein, S. G., Schatzberg, A. F., Thase, M. E., Yonkers, K. A., McCullough, J. P., Keitner, G. I., et al. (2000). Gender differences in treatment response to sertraline versus imipramine in chronic depression. Am. J. Psychiatry 157, 1445–1452. doi: 10.1176/appi.ajp.157.9.1445
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi: 10.1016/j.neuron.2016.04.015
LaPlant, Q., Chakravarty, S., Vialou, V., Mukherjee, S., Koo, J. W., Kalahasti, G., et al. (2009). Role of nuclear factor kappaB in ovarian hormone-mediated stress hypersensitivity in female mice. Biol. Psychiatry 65, 874–880. doi: 10.1016/j.biopsych.2009.01.024
La Rocca, G., Olejniczak, S. H., Gonzalez, A. J., Briskin, D., Vidigal, J. A., Spraggon, L., et al. (2015). In vivo, Argonaute-bound microRNAs exist predominantly in a reservoir of low molecular weight complexes not associated with mRNA. Proc. Natl. Acad. Sci. U.S.A. 112, 767–772. doi: 10.1073/pnas.1424217112
Law, C. W., Chen, Y., Shi, W., and Smyth, G. K. (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. doi: 10.1016/j.jbi.2011.05.002
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi: 10.1016/j.cell.2004.12.035
Li, Y. J., Xu, M., Gao, Z. H., Wang, Y. Q., Yue, Z., Zhang, Y. X., et al. (2013). Alterations of serum levels of BDNF-related miRNAs in patients with depression. PLoS ONE 8:e63648. doi: 10.1371/journal.pone.0063648
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1001/archpsyc.63.4.385
Lopez, J. P., Fiori, L. M., Gross, J. A., Labonte, B., Yerko, V., Mechawar, N., et al. (2014b). Regulatory role of miRNAs in polyamine gene expression in the prefrontal cortex of depressed suicide completers. Int. J. Neuropsychopharmacol. 17, 23–32. doi: 10.1038/tp.2012.112
Lopez, J. P., Lim, R., Cruceanu, C., Crapper, L., Fasano, C., Labonte, B., et al. (2014a). miR-1202 is a primate-specific and brain-enriched microRNA involved in major depression and antidepressant treatment. Nat. Med. 20, 764–768. doi: 10.1017/S1461145713000941
Luine, V. (2002). Sex differences in chronic stress effects on memory in rats. Stress 5, 205–216. doi: 10.1080/1025389021000010549
Maheu, M., Lopez, J. P., Crapper, L., Davoli, M. A., Turecki, G., and Mechawar, N. (2015). MicroRNA regulation of central glial cell line-derived neurotrophic factor (GDNF) signalling in depression. Transl. Psychiatry 5, e511. doi: 10.1016/j.neuron.2014.05.042
Martenyi, F., Dossenbach, M., Mraz, K., and Metcalfe, S. (2001). Gender differences in the efficacy of fluoxetine and maprotiline in depressed patients: a double-blind trial of antidepressants with serotonergic or norepinephrinergic reuptake inhibition profile. Eur. Neuropsychopharmacol. 11, 227–232. doi: 10.1016/S0924-977X(01)00089-X
Martin, L. A., Neighbors, H. W., and Griffith, D. M. (2013). The experience of symptoms of depression in men vs women: analysis of the national comorbidity survey replication. JAMA Psychiatry 70, 1100–1106. doi: 10.1001/jamapsychiatry.2013.1985
Miller, B. H., Zeier, Z., Xi, L., Lanz, T. A., Deng, S., Strathmann, J., et al. (2012). MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc. Natl. Acad. Sci. U.S.A. 109, 3125–3130. doi: 10.1073/pnas.1113793109
Mullokandov, G., Baccarini, A., Ruzo, A., Jayaprakash, A. D., Tung, N., Israelow, B., et al. (2012). High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat. Methods 9, 840–846. doi: 10.1038/nmeth.2078
O'Carroll, D., and Schaefer, A. (2013). General principals of miRNA biogenesis and regulation in the brain. Neuropsychopharmacology 38, 39–54. doi: 10.1038/npp.2012.87
O'Connor, R. M., Dinan, T. G., and Cryan, J. F. (2012). Little things on which happiness depends: microRNAs as novel therapeutic targets for the treatment of anxiety and depression. Mol. Psychiatry 17, 359–376. doi: 10.1038/mp.2011.162
Ørom, U. A., Nielsen, F. C., and Lund, A. H. (2008). MicroRNA-10a binds the 5'UTR of ibosomal protein mRNAs and enhances their translation. Mol. Cell 30, 460–471. doi: 10.1016/j.molcel.2008.05.001
Pemberton, R., and Fuller Tyszkiewicz, M. D. (2016). Factors contributing to depressive mood states in everyday life: a systematic review. J. Affect. Disord. 200, 103–110. doi: 10.1016/j.jad.2016.04.023
Pfau, M. L., and Russo, S. J. (2015). Peripheral and central mechanisms of stress resilience. Neurobiol. Stress 1, 66–79. doi: 10.1016/j.ynstr.2014.09.004
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. doi: 10.1038/nmeth.3485
Russo, S. J., and Nestler, E. J. (2013). The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 14, 609–625. doi: 10.1038/nrn3381
Sachs, B. D., Ni, J. R., and Caron, M. G. (2014). Sex differences in response to chronic mild stress and congenital serotonin deficiency. Psychoneuroendocrinology 40, 123–129. doi: 10.1016/j.psyneuen.2013.11.008
Scharfman, H. E., and MacLusky, N. J. (2008). Estrogen-growth factor interactions and their contributions to neurological disorders. Headache 48(Suppl. 2), S77–S89. doi: 10.1073/pnas.1415191111
Silverstein, B. (2002). Gender differences in the prevalence of somatic versus pure depression: a replication. Am. J. Psychiatry 159, 1051–1052. doi: 10.1176/appi.ajp.159.6.1051
Smalheiser, N. R., Lugli, G., Rizavi, H. S., Torvik, V. I., Turecki, G., and Dwivedi, Y. (2012). MicroRNA expression is down-regulated and reorganized in prefrontal cortex of depressed suicide subjects. PLoS ONE 7:e33201. doi: 10.1371/journal.pone.0033201
Smalheiser, N. R., Lugli, G., Rizavi, H. S., Zhang, H., Torvik, V. I., Pandey, G. N., et al. (2011). MicroRNA expression in rat brain exposed to repeated inescapable shock: differential alterations in learned helplessness vs. non-learned helplessness. Int. J. Neuropsychopharmacol. 14, 1315–1325. doi: 10.1017/S1461145710001628
Tapocik, J. D., Barbier, E., Flanigan, M., Solomon, M., Pincus, A., Pilling, A., et al. (2014). microRNA-206 in rat medial prefrontal cortex regulates BDNF expression and alcohol drinking. J. Neurosci. 34, 4581–4588. doi: 10.1523/Jneurosci.0445-14.2014
Tapocik, J. D., Solomon, M., Flanigan, M., Meinhardt, M., Barbier, E., Schank, J. R., et al. (2013). Coordinated dysregulation of mRNAs and microRNAs in the rat medial prefrontal cortex following a history of alcohol dependence. Pharmacogenomics J. 13, 286–296. doi: 10.1038/tpj.2012.17
Terzi, D., Stergiou, E., King, S. L., and Zachariou, V. (2009). Regulators of G protein signaling in neuropsychiatric disorders. Prog. Mol. Biol. Transl. Sci. 86, 299–333. doi: 10.1093/ijnp/pyv025
Toffol, E., Heikinheimo, O., and Partonen, T. (2015). Hormone therapy and mood in perimenopausal and postmenopausal women: a narrative review. Menopause 22, 564–578. doi: 10.1097/GME.0000000000000323
Tou, J. C., Grindeland, R. E., and Wade, C. E. (2004). Effects of diet and exposure to hindlimb suspension on estrous cycling in Sprague-Dawley rats. Am. J. Physiol. Endocrinol. Metab. 286, E425–E33. doi: 10.1093/bioinformatics/btu638
Uchida, S., Nishida, A., Hara, K., Kamemoto, T., Suetsugi, M., Fujimoto, M., et al. (2008). Characterization of the vulnerability to repeated stress in Fischer 344 rats: possible involvement of microRNA-mediated down-regulation of the glucocorticoid receptor. Eur. J. Neurosci. 27, 2250–2261. doi: 10.1111/j.1460-9568.2008.06218.x
Vasudevan, S., Tong, Y., and Steitz, J. A. (2007). Switching from repression to activation: microRNAs can up-regulate translation. Science 318, 1931–1934. doi: 10.1126/science.1149460
Viau, V., and Meaney, M. J. (1991). Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology 129, 2503–2511. doi: 10.1016/S0006-3223(99)00177-8
Vreugdenhil, E., Verissimo, C. S., Mariman, R., Kamphorst, J. T., Barbosa, J. S., Zweers, T., et al. (2009). MicroRNA 18 and 124a down-regulate the glucocorticoid receptor: implications for glucocorticoid responsiveness in the brain. Endocrinology 150, 2220–2228. doi: 10.1210/en.2008-1335
Walker, Q. D., Nelson, C. J., Smith, D., and Kuhn, C. M. (2002). Vaginal lavage attenuates cocaine-stimulated activity and establishes place preference in rats. Pharmacol. Biochem. Behav. 73, 743–752. doi: 10.1186/gb-2009-10-3-r25
Wang, M., Zhao, Y., and Zhang, B. (2015). Efficient test and visualization of multi-set intersections. Sci. Rep. 5, 16923. doi: 10.1111/j.1526-4610.2008.01200.x
Wang, Z., Zhang, C., Huang, J., Yuan, C., Hong, W., Chen, J., et al. (2014). MiRNA-206 and BDNF genes interacted in bipolar I disorder. J. Affect. Disord. 162, 116–119. doi: 10.1016/j.jad.2014.03.047
Westenbroek, C., Ter Horst, G. J., Roos, M. H., Kuipers, S. D., Trentani, A., and den Boer, J. A. (2003). Gender-specific effects of social housing in rats after chronic mild stress exposure. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 21–30. doi: 10.1016/S0278-5846(02)00310-X
Wissman, A. M., May, R. M., and Woolley, C. S. (2012). Ultrastructural analysis of sex differences in nucleus accumbens synaptic connectivity. Brain Struct. Funct. 217, 181–190. doi: 10.1016/j.yhbeh.2015.06.010
Yang, X., Yang, Q., Wang, X., Luo, C., Wan, Y., Li, J., et al. (2014). MicroRNA expression profile and functional analysis reveal that miR-206 is a critical novel gene for the expression of BDNF induced by ketamine. Neuromol. Med. 16, 594–605. doi: 10.1016/j.jad.2014.03.047
Keywords: depression, stress, sex differences, RNA-Seq, microRNA, nucleus accumbens
Citation: Pfau ML, Purushothaman I, Feng J, Golden SA, Aleyasin H, Lorsch ZS, Cates HM, Flanigan ME, Menard C, Heshmati M, Wang Z, Ma'ayan A, Shen L, Hodes GE and Russo SJ (2016) Integrative Analysis of Sex-Specific microRNA Networks Following Stress in Mouse Nucleus Accumbens. Front. Mol. Neurosci. 9:144. doi: 10.3389/fnmol.2016.00144
Received: 28 September 2016; Accepted: 28 November 2016;
Published: 23 December 2016.
Edited by:
Oliver Stork, Otto-von-Guericke University Magdeburg, GermanyReviewed by:
Mary M. Torregrossa, University of Pittsburgh, USAMarkus Fendt, Otto-von-Guericke University Magdeburg, Germany
Copyright © 2016 Pfau, Purushothaman, Feng, Golden, Aleyasin, Lorsch, Cates, Flanigan, Menard, Heshmati, Wang, Ma'ayan, Shen, Hodes and Russo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott J. Russo, c2NvdHQucnVzc29AbXNzbS5lZHU=