 Kristina Endres
Kristina Endres Thomas Deller
Thomas Deller- 1Clinic of Psychiatry and Psychotherapy, University Medical Center Johannes Gutenberg-University Mainz, Mainz, Germany
- 2Institute of Clinical Neuroanatomy, Neuroscience Center, Goethe-University, Frankfurt/Main, Germany
ADAM10 (A Disintegrin and Metalloproteinase 10) has been identified as the major physiological alpha-secretase in neurons, responsible for cleaving APP in a non-amyloidogenic manner. This cleavage results in the production of a neuroprotective APP-derived fragment, APPs-alpha, and an attenuated production of neurotoxic A-beta peptides. An increase in ADAM10 activity shifts the balance of APP processing toward APPs-alpha and protects the brain from amyloid deposition and disease. Thus, increasing ADAM10 activity has been proposed an attractive target for the treatment of neurodegenerative diseases and it appears to be timely to investigate the physiological mechanisms regulating ADAM10 expression. Therefore, in this article, we will (1) review reports on the physiological regulation of ADAM10 at the transcriptional level, by epigenetic factors, miRNAs and/or protein interactions, (2) describe conditions, which change ADAM10 expression in vitro and in vivo, (3) report how neuronal ADAM10 expression may be regulated in humans, and (4) discuss how this knowledge on the physiological and pathophysiological regulation of ADAM10 may help to preserve or restore brain function.
ADAM10 - Portrait of a Biologically Versatile Protease
Introduction
ADAM10 (A Disintegrin and Metalloproteinase 10) was identified in vitro as a key proteinase in the processing of the amyloid precursor protein (APP) more than 15 years ago (Lammich et al., 1999). The zinc-dependent proteinase cleaves APP within the A-beta sequence, thus preventing the production of this peptide. Furthermore, APP-cleavage by ADAM10 liberates APPs-alpha, which has neuroprotective properties and is involved in the regulation of synaptic plasticity and learning and memory (reviewed in Kögel et al., 2012). In line with these findings, overexpression of ADAM10 in mice revealed elevated APPs-alpha levels and demonstrated a robust in vivo activity of ADAM10 (Postina et al., 2004). Overexpression of ADAM10 was also effective in animal mouse models of Alzheimer's disease (AD) and reduced plaque load as well as deficits in learning and memory (Postina et al., 2004; Schmitt et al., 2006). Subsequent investigations of RNAi-mediated knock-down of the enzyme in primary cortical neurons (Kuhn et al., 2010) as well as conditional knock-down in mice (Jorissen et al., 2010) consolidated the enzymes' role in APP processing in vivo. Collectively, these data point to ADAM10 as being the most important physiological alpha-secretase involved in the processing of APP in neurons.
The central role of ADAM10 in APP processing has made ADAM10 an interesting target for AD therapy. It has been proposed (e.g., Fahrenholz and Postina, 2006; Vincent and Govitrapong, 2011) that similar to the situation in intact animals (Postina et al., 2004) an increase in ADAM10 could result in decreased A-beta load and improved learning and memory in AD patients. For this approach to be effective and safe, however, the cell biology of ADAM10 and its cellular functions need to be better understood. ADAM10 is a versatile protease which cleaves not only APP but also several other proteins (see paragraph 2). Therapeutic strategies for AD focusing on ADAM10 as a target have to keep these additional substrates in mind. In the present review we will summarize the extant literature on ADAM10 and focus on what is known about its regulation in vitro and in vivo. Understanding the regulation of this enzyme may be a necessary step toward understanding its usefulness in therapeutic contexts.
Domain Structure, Cellular Synthesis, and Maturation of ADAM10
ADAM10 is a catalytically active member of the ADAM family of proteinases. The ADAMs are grouped together as a family because they share structural features with snake venom disintegrin proteases (Wolfsberg et al., 1995a,b). ADAM10 is co-translationally synthesized via the rough ER, matures and is transported via the Golgi apparatus. Maturation includes removal of the prodomain (Figure 1: 1), which keeps the enzyme in an inactive state. A cleavage site for proprotein convertases such as PC7 (Anders et al., 2001) is mandatory for production of the catalytically active enzyme as shown by analyzing mutated ADAM10. However, the prodomain has not a mere inhibitory function but is also needed as an intramolecular chaperon for correct folding (Anders et al., 2001). This is reflected by the fact that a large proportion of ADAM10 has been found to be localized in the Golgi apparatus in AR breast carcinoma cell line by confocal microscopy (Gutwein et al., 2003). The mature form of ADAM10 of about 68 kDa was found in the Golgi compartment as well as in the ER/plasma membrane-enriched fraction of postnuclear supernatant and at least cleavage of another substrate of ADAM10—the L1 adhesion molecule—seems to occur in both. Recent investigations suggested by administering the inhibitor RVKR for up to 8 h before measuring shedding activity that cleavage by proprotein convertases might be dispensable for rapid stimulation of ADAM10 (Maretzky et al., 2015). However, as the half-life time of ADAM10 is rather long (>72 h; Mezyk-Kopec et al., 2009), this result may need to be interpreted with some caution.
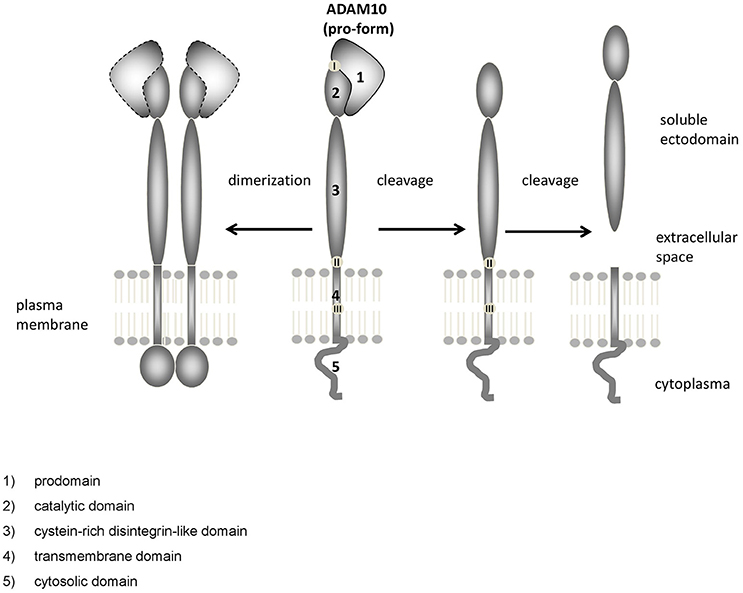
Figure 1. Domain structure of ADAM10. ADAM10 consists of several functional distinct domains: (1) prodomain, (2) catalytic domain, (3) cystein-rich disintegrin-like domain, (4) transmembrane domain, (5) cytosolic domain. Upon dimerization (left), the unstructured C-terminus converts into an ordered domain (Deng et al., 2014). Cleavage sites for proteinases such as proprotein convertases [located at the end of the prodomain, (Anders et al., 2001), I], other ADAMs [close to the membrane, (Cissé et al., 2005; Parkin and Harris, 2009; Tousseyn et al., 2009) II] or gamma-secretase [within the membrane, (Tousseyn et al., 2009), III] have been identified.
The catalytic domain of ADAM10 (Figure 1: 2) contains the characteristic zinc-binding consensus motif (HEXGHXXGXXHD) of active members of the proteinase family. A point mutation within this motif (E384A) results in a dominant negative acting protein and a decreased APPs-alpha secretion, as could be shown shown in HEK cells and mice (Fahrenholz et al., 2000; Postina et al., 2004).
The catalytic and the proximal disintegrin domain contain high-mannose as well as complex-type N-glycan attachment sites (Escrevente et al., 2008). The disintegrin domain of ADAM10 (Figure 1: 3) does not appear to be essential for ADAM10 protease activity in cell culture experiments (Fahrenholz et al., 2000). Rather, the short intracellular C-terminus seems to play an important role: Epidermal growth factor (EGF) cleavage has been reported to be partially impaired in ADAM10−/− cells overexpressing a cytoplasmic domain deletion mutant of the proteinase (Horiuchi et al., 2007). However, the cytoplasmic domain of ADAM10 has also been reported to negatively influence constitutive shedding through an ER retention motif: an ADAM10Δcyto mutant displayed increased catalytic activity compared to ADAM10 Wt with regard to Betacellulin cleavage (Maretzky et al., 2015). The cytoplasmic domain of ADAM10 contains several binding sites that may be involved in regulatory events, such as an IQ consensus binding site for calmodulin (Horiuchi et al., 2007) and two proline-rich putative Src homology 3 (SH3) binding domains. The juxtamembrane binding site affects basolateral localization of ADAM10 in epithelial cells (Wild-Bode et al., 2006), while in neurons the SH3 binding domains direct ADAM10 to the postsynaptic membrane (Marcello et al., 2007). Using a phage library analysis comprising 305 human SH3 domains, 38 candidate binding proteins for the ADAM10 C-terminus were identified, including endophilin-A2, Lck, or ZDHHC6 (Ebsen et al., 2014). Although the biological relevance of many of these putative ADAM10 binding partners needs to be determined, this finding suggests that regulatory interactions at the C-terminus could play a major role regarding the cellular localization as well as the activity of the proteinase.
Developmental and Adult Expression of ADAM10 in Mouse and Human Brain
ADAM10 is expressed in various tissues in mice (Marcinkiewicz and Seidah, 2000). Its presence in the developing as well as in the adult CNS underscores its importance for normal brain development and function. Since ADAM10 can only process a putative substrate if both, protease and substrate are expressed at the same time and in the same cellular compartments, it is important to know the temporospatial pattern of ADAM10 expression in the brain. This pattern can then be compared to corresponding data of putative substrates or binding partners.
The distribution of ADAM10 was studied in mouse cerebral cortex from embryonic day (E) 14.5 to postnatal day (P) 1 using in situ hybridization analysis. This revealed ADAM10 expression within the ventricular zone and the cortical plate from E17.5 to P1 (Ma et al., 2013; see also Figure 2). These data on ADAM10 mRNA were corroborated by immunofluorescence analyses which detected ADAM10 protein in developing cerebral cortex from E14.5 to E18.5 (Ma et al., 2013).
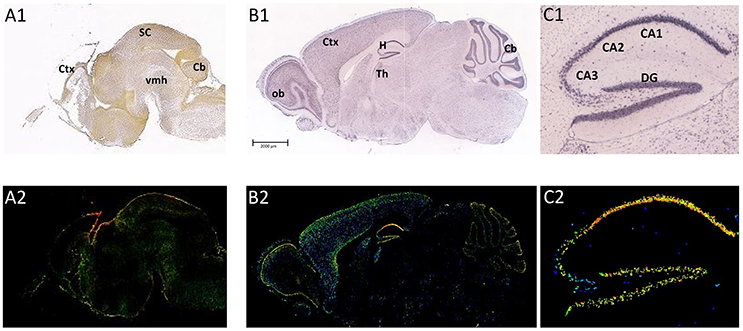
Figure 2. Distribution of ADAM10 mRNA in the murine brain. Sagittal section of C57Bl6/J mouse brain (male) at E18.5 (A1,A2; Image credit: Allen Institute; http://developingmouse.brain-map.org/experiment/show/100055949, ©2016. Allen Institute for Brain Science) and P56 (B1,B2; Image credit: Allen Institute; http://developingmouse.brain-map.org/experiment/show/69514738, ©2016. Allen Institute for Brain Science C1,C2: magnification of hippocampal area of the adult brain). ADAM10 mRNA expression is revealed by in situ hybridization [A1–C1, upper row ISH; A2–C2, lower row expression energy (cells with highest probability of gene expression)]. CA1-3, Cornu Ammonis regions; Cb, cerebellum; Ctx, cerebral cortex; DG, Dentate Gyrus; H, hippocampus; ob, olfactory bulb; SC, Superior Colliculus; Th, thalamus; vmh, ventral mid-/hindbrain
ADAM10 plays an essential role during development. Animals with a conventional ADAM10 knock-out die on E9.5 (Hartmann et al., 2002), which underlines the general importance of this protease. More recently generated conditional Nestin-Cre-ADAM10 knock-out mice with a cell-specific inactivation of ADAM10 in neural progenitor cells (NPCs), NPC-derived neurons and glial cells prolonged the life span of the mice to a perinatal time point (Jorissen et al., 2010). These mutants displayed a disrupted neocortex and a severe reduction of the ganglionic eminence. Knock-out of ADAM10 in the postnatal CNS using a CaMKII-alpha-Cre driver finally allowed investigation of adult mice (Prox et al., 2013). This conditional mutant did not show gross morphological abnormalities but exhibited synaptic dysfunction, increased early perinatal lethality, altered behavior, and epileptic seizures. Similar results were reported by another group which independently established an adult ADAM10 knock-out model (Zhuang et al., 2015). Taken together, these studies indicate that ADAM10-deficiency results in major developmental phenotypes. Lack of the protease at later stages is compatible with life but results in a number of dysfunctions.
The cellular expression pattern of ADAM10 was also investigated in some of these studies. Interestingly, ADAM10 protein expression partially correlated with both, S100β and Tuj1 expression (E16.5 to P1), which indicates a relevance of ADAM10 for glial as well as neuronal cell function during late embryonic cerebral cortex development stages (Ma et al., 2013). In the developing brain of chicken, ADAM10 shows a weak but widespread expression at E12 in most gray matter areas (Lin et al., 2008). Expression intensity decreased from E14 to E19, with the exception of the telencephalon and the cerebellum. Some ADAM10-positive non-neuronal cells may be oligodendrocytes, since they were shown to co-express galactocerebroside, which is a marker for oligodendrocytes at late stages of chicken embryogenesis (Lin et al., 2008).
ADAM10 expression has also been studied in developing human brain: Bernstein and colleagues compared the amount of ADAM10 in temporal cortex of stillborn children with those of normal aged adults and found a general increase (Bernstein et al., 2003). In a follow-up study, a weak expression of ADAM10 in the cytoplasm of pyramidal as well as non-pyramidal neurons was confirmed in pre- and perinatal human brains (Bernstein et al., 2009). In contrast to these findings, analysis of total human fetal brain RNA obtained from a commercial source (Clontech, Kaczur et al., 2007) failed to detect a prominent ADAM10 expression using microarray analysis. Differences in the stage of fetal development (which has not been reported) or technical issues, e.g., detection of ADAM10 in total mRNA preparations, may explain these discrepancies. Analysis of transcripts from a human fetal brain library, however, revealed two types of ADAM10 cDNAs: one encoding a 748 amino acid protein [designated Kuzbanian (Kuz)L] and a second one (KuzS, encoding a 568 amino acid protein), which lacks the cysteine-rich, transmembrane, and cytoplasmic domain (Yavari et al., 1998). Fetal human brain expressed substantially more of the short than of the long variant, while fetal lung predominantly contained the longer variant. In adult human brain tissue, Northern blot demonstrated the persistence of both forms in different amounts (Yavari et al., 1998). The presence of the short transcript appears to be unique to humans as the transcript could not be detected in adult and embryonic tissue of mice. Whether it is functionally relevant, i.e., whether it is translated into a biologically active protein is unclear.
The expression of ADAM10 in the adult brain has been studied in rodents and humans. By using northern blot technique it could be shown that adult human amygdala, caudate nucleus and corpus callosum contain relatively high amounts of ADAM10 transcripts whereas mRNA levels in the subthalamic nucleus and the thalamus were comparably low (Yavari et al., 1998). In the adult rodent brain, ADAM10 mRNA was reported to be moderately expressed throughout the whole brain, including the olfactory bulb, the hippocampus or the subthalamic region (mouse and rat: Kärkkäinen et al., 2000, see also Figure 2: P56). Semiquantitative evaluation of ADAM10 mRNA using ISH analysis revealed only the Pontine nuclei as a brain structure not expressing the protease (Kärkkäinen et al., 2000). These findings have been confirmed by a recent investigation that also described positive ISH-stainings for neurons of the cerebral cortex, hippocampus, thalamus, and cerebellar granular cells in the CNS of adult mice (Guo et al., 2016).
ADAM10—Physiological Substrates and Functions
Physiological Substrates of ADAM10
ADAM10 is probably best known for its ability to process APP. ADAM10 cleaves APP at the alpha-secretase cleavage site and in vitro as well as in vivo studies have implicated ADAM10 as the biologically most relevant neuronal alpha-secretase (e.g., Postina et al., 2004; Jorissen et al., 2010; Kuhn et al., 2010). Of note, the regional and cellular overlap of ADAM10 and APP, which is necessary for ADAM10 to process APP in tissues, is age-dependent: at early developmental stages the mRNA distributions of ADAM10 and APP are not fully congruent but with aging the overlap increases (Marcinkiewicz and Seidah, 2000). This finding—but also the wealth of data on other substrates of ADAM10 (see below)—suggests that ADAM10 substrates may change: During development and in the young brain ADAM10 may preferentially cleave substrates other than APP and the role of ADAM10 as alpha-secretase of APP may emerge with aging.
Presently, a rather large number of ADAM10 substrates have been identified in different experimental settings (e.g., reviewed for proteomic approaches in Müller et al., 2016). Of notice, ADAM10 substrates belong to type I as well as type II transmembrane but also Glycosylphosphatidylinisotol (GPI)-anchored proteins, indicating a considerable flexibility of the protease with regard to substrate recognition. Consensus cleavage motifs for proteases are commonly deduced from the amino acids surrounding the naturally occurring cleavage sites within protein substrates. This approach failed in the case of ADAM10 because it lacks a well-defined consensus sequence: for ADAM10 leucine was found to be preferred (and tyrosine accepted) in the P1' position (immediately downstream of the cleavage site) in an investigation using oriented peptide mixture libraries which gives evidence of a shallow or deep S1' site (John et al., 2004). ADAM10's preference for larger residues at P1' has been confirmed but acceptance of aromatic amino acids and even glutamine were also reported (Caescu et al., 2009). This tolerance for aromatic residues in P1' may be the most relevant difference in cleavage site specificities between ADAM10 and its close relative ADAM17 (Tucher et al., 2014). In the early investigation tyrosine was found to be favored at P1 (immediately upstream of the cleavage site; John et al., 2004). In contrast to this, selectivity for small residues such as alanine at the P1 was described by Caescu et al. (2009) and specificities for proline and basic residues were recently reported (Tucher et al., 2014). In sum, these reports show the methodological limitations and uncertainties involved in pinpointing cleavage site specificities from linear unmodified peptide libraries. In addition, the activity state of the cell may also influence shedding capacity, as is the case for NG2 (Sakry et al., 2014) as well as the synaptic marker neuroligin 1 (Suzuki et al., 2012), further complicating work in this direction.
At present, a wide variety of substrates has been identified for ADAM10 and some of them have been confirmed not only in primary culture but also in vivo. In line with its ubiquitous expression, ADAM10 substrates are linked to a number of biological systems and physiological as well as pathological functions (c.f. chapter 4), including the immune and nervous system but also cancerogenesis (e.g., Vincent and Checler, 2012). In their review Pruessmeyer and Ludwig reported on the “good, the bad and the ugly” ADAM10 substrates (Pruessmeyer and Ludwig, 2009). Since then, a number of additional ADAM10 substrates were identified and have resulted in a more complete, albeit even more complex picture of ADAM10 (Table 1).
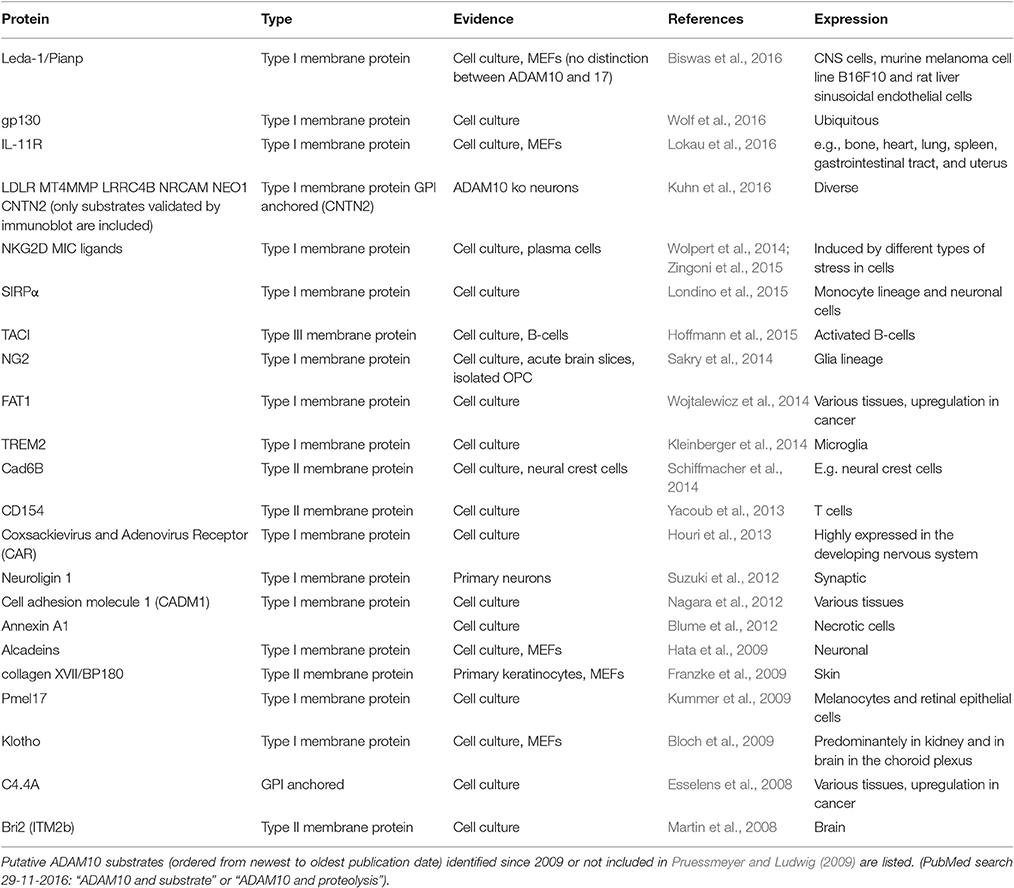
Table 1. ADAM10 substrates identified within the last years.
Functions of ADAM10 at the Synapse and in Non-neuronal Cells
ADAM10 processes other proteins and thus, influences the functions of its substrates by in-/activating them or by liberating biologically active fragments. Thereby, the biological effects of ADAM10 activity are tightly linked to the functions of the substrates and their cleavage products. Because of the large number of ADAM10 substrates identified to date, we focus in this review on those which are known to have an important impact on brain function and which are likely to co-localize with ADAM10 at the synapse or in glial cells.
The earliest study on the distribution of ADAM10 at synapses was based on immunocytochemistry and suggested that ADAM10 co-localizes with the postsynaptic scaffold protein Synapse-associated protein 97 (SAP-97) but not with the presynaptic vesicle protein synaptophysin (Marcello et al., 2007). However, a more recent study using the sensitive proximity ligation assay reported proximity of the enzyme with synaptophysin in mouse primary hippocampal neurons (Lundgren et al., 2015). This suggests that ADAM10 can be present in both parts of a synapse. One example where this could be functionally relevant is the neurexin-neuroligin-interaction: neurexins and neuroligins are cell-adhesion molecules which form transsynaptic complexes (e.g., Tsetsenis et al., 2014). They appear to be important for normal synapse specification and function (Jedlicka et al., 2011, 2015). For the postsynaptic protein Neuroligin 1, ADAM10 has been found to act as the major sheddase, as could be shown by pharmacological and genetic means in primary rat cortical neurons (Suzuki et al., 2012). NMDA receptor activation as well as prolonged epileptic seizure condition increased shedding, suggesting a role for neuronal activity in this context. Interestingly, shedding of Neuroligin 1 could be induced by soluble neurexin 1α or β derived from overexpressing HEK293 cells (Suzuki et al., 2012), indicating that ligand binding at the cell surface also regulates Neuroligin 1 shedding. Similar observations have been made for the Notch-Delta complex where Notch1 cleavage by ADAM10 is induced by Delta binding (e.g., reviewed in Van Tetering and Vooijs, 2011). Intriguingly, Notch 1 as well as its ligands - Delta or Jagged - have been found to be cleaved by ADAM10 (for example: Pan and Rubin, 1997; Lavoie and Selkoe, 2003). A recent publication regarding systemic characterization of ADAM10 substrates from neurons highlighted that ADAM10 is also in principle capable of shedding the Neuroligin ligands Neurexins 2 and 3, although deletion of the proteinase resulted only in a comparably mild reduction of the shedding (Kuhn et al., 2016). If this role for ADAM10 in the cleavage of major anchoring proteins can be verified in vivo and in human brain, interfering with ADAM10 activity may indeed be a powerful tool to influence synaptic structure and function.
ADAM10 has also been found to process substrates of non-neuronal cells. Since neurons and glial cells are highly interdependent and jointly regulate synapse functions, ADAM10 may also influence network activities through glial cells. For example, the marker transmembrane proteoglycan nerve-glia antigen 2 (NG2), commonly found on the so-called “NG2-glial cells” (Eugenín-Von Bernhardi and Dimou, 2016), has also been identified as a substrate of ADAM10 (Sakry et al., 2014). Similar to what has been reported for Neuroligin 1, shedding of NG2 is also regulated by neuronal activity. Moreover, neurons from NG2-knock-out mice exhibited diminished amplitudes of AMPA receptor-currents which could be rescued by application of the partial NG2 ectodomain (Sakry et al., 2014). This suggests that an NG2-cell derived ectodomain produced by ADAM10 processing regulates synaptic activity and plays a role in neuron-glia communication.
Another non-neuronal substrate of ADAM10 with implications for glial and neuronal function is the microglial surface protein triggering receptor expressed on myeloid cells 2 (TREM2). TREM2 has been suggested to play a role in phagocytosis and has been recently recognized as a genetic risk-factor for AD (Frank et al., 2008; Colonna and Wang, 2016). Using HEK293 Flp-In cells and enzymatic inhibitors, the release of soluble TREM2 ectodomain was demonstrated to depend on ADAM10 but not on ADAM17 or beta-secretase (beta-site amyloid precursor protein cleaving enzyme 1; BACE1) (Kleinberger et al., 2014). Reduction of cell surface TREM2 decreases the ability of microglia to phagocytose and remove cellular debris or apoptotic neurons (Kleinberger et al., 2014). Of note, TREM2 ligands were identified on Neuro2A cells and on cultured cortical and dopamine neurons (Hsieh et al., 2009), suggesting an impact of a non-neuronal shedding event on neurons.
Finally, there is growing evidence for a role of exosomes in neuron-glia communication (Frühbeis et al., 2013). In this regard it is of interest that functionally active ADAM10 has been found in exosomes from ovarian carcinoma cells where it contributes to L1 and CD44 cleavage (Stoeck et al., 2006) and in exosomes of primed B-cells (Padro et al., 2013). Whether microglial or neuronal cells also use exosomes to deliver ADAM10 or shedded substrates among themselves is currently unknown.
Regulation of ADAM10
ADAM10 is a multifunctional protease active throughout the life of an organism and its regulation is controlled at transcriptional, epigenetic, translational and post-translational levels. These different levels of regulation allow a cell to adapt ADAM10 levels rapidly to functional perturbations as well as to slower changes induced by aging and/or maturation.
Transcriptional Regulation of ADAM10
The human ADAM10 gene is localized on chromosome 15, whereas its murine homolog is found on chromosome 9 (Yamazaki et al., 1997a,b). Both genes are comprised of about 160 kb with high sequence preservation within the first 500 bp upstream of the translation initiation site (Prinzen et al., 2005). The human core promoter is positioned at −508 to −300 bp and contains no TATA box but several functional binding sites for common transcription factors such as Sp1 and USF (Prinzen et al., 2005). SNPs in the human promoter region at position −279 and −630 indicated no association with AD (Prinzen et al., 2005), whereas a SNP located at −644 was correlated with CSF APPs-alpha levels (Bekris et al., 2011). The 5′ UTR of the human gene was located 444 bp upstream of the start codon (Lammich et al., 2010), the 3′ UTR up to 1254 bp downstream of the stop codon (Augustin et al., 2012).
Even before the promoter of human ADAM10 was described, several pathways regulating the enzyme's expression had been identified: for example, in the prostate cancer cell line LNCaP insulin-like growth factor I combined with 5 alpha-dihydrotestosterone increased mature and immature ADAM10 protein amounts (McCulloch et al., 2004). Similarly, EGF led to the up-regulation of ADAM10 mRNA and protein in those cells. In addition, the Tcf/Lef-family of transcription factors which is known to interact with beta-catenin (Wisniewska, 2013) also seems to be involved: Wang et al. demonstrated in transgenic AD mice the induction of Wnt signaling by huperzine A. This was accompanied by elevated beta-catenin levels and increased ADAM10 protein levels (Wang et al., 2011). These findings were corroborated by the observation that NMDA receptor activation in primary neurons similarly increased ADAM10 via Wnt/MAPK signaling (Wan et al., 2012).
Using different cell systems Paired Box Genes (PAX) were similarly identified as putative ADAM10 regulators. In melanoma cells chromatin immunoprecipitation assay and overexpression as wells as siRNA-mediated knock-down gave evidence that PAX2 can regulate ADAM10 expression (Lee et al., 2011). Downregulation of PAX2 via siRNA in A498 (renal carcinoma), EAhy (endothelial), T98G (glioblastoma), and SKOV3ip (ovarian carcinoma) cells revealed a nearly total loss of ADAM10 protein as demonstrated by Western blot analysis (Doberstein et al., 2011). Therefore, PAX2 seems to play an important role in ADAM10 expression control—at least in cancer cells. Interestingly, the related PAX4 has been shown to regulate ADAM10 post-transcriptionally (see paragraph “Regulation of ADAM10 at the translational level”).
Another signaling pathway that increases ADAM10 amount within the cell via gene regulation requires melatonin. It has been reported that melatonin elevates ADAM10 level in HEK293 and neuronal SH-SY5Y cells via G protein-coupled receptor-induced PKC/Erk activation (Panmanee et al., 2015; Shukla et al., 2015). This effect seems to depend on human ADAM10 promoter region −1193 to −555 as a respective deletion construct failed to respond in a reporter gene assay (Shukla et al., 2015). The authors of the report discuss that the binding sites of cAMP response element-binding protein (CREB) and octamer-binding transcription factor 1 (Oct-1) which were described earlier (Prinzen et al., 2005) might contribute to the regulation or that a yet unidentified Hypoxia-inducible factor 1 (HIF-1) binding site might be responsible. The regulation of ADAM10 via the sleep hormone melatonin seems highly interesting as sleep disturbances are considered characteristic symptoms of AD (for example Sung et al., 2017).
Agonists specific for Peroxisome Proliferator-Activated Receptor alpha (PPARalpha) but not PPARbeta, delta, or gamma elevated ADAM10 protein amount in primary murine hippocampal neurons (Corbett et al., 2015). Seven PPAR responsible elements were identified by in silico analysis and the specific agonist GEM led to enrichment of PPARalpha and its heterodimer Retinoid X Receptor alpha (RXRalpha) binding partner at two direct repeat 1 PPAR responsive elements (PPRE) located in the ADAM10 promoter in wild type, but not in PPARalpha knock-out hippocampal neurons. 9-cis retinoic acid failed to synergistically increase ADAM10 amount in this context, therefore a non-permissive PPARalpha/RXRalpha heterodimer seems to regulate ADAM10 expression similar to the RARalpha/beta/RXR dimer from earlier investigations (Tippmann et al., 2009). PPARalpha is known to be involved in fatty acid metabolism. In this regard, it is of interest that lowering the cholesterol amount of cells increased ADAM10's catalytic but not transcriptional activity (Kojro et al., 2010) and that various fatty acids and lipids such as Docosahexaenoic acid (DHA) interfere with the balance of APP processing (e.g., Eckert et al., 2011; Grimm et al., 2016). Additionally, Sex-determining region Y-box 2 (Sox2), a major factor of adult tissue homeostasis and regeneration control, was recently identified to upregulate ADAM10 expression in HEK293 cells using overexpression experiments (Sarlak et al., 2016).
The retinoic acid receptor (RAR) family is particularly interesting with regard to ADAM10 regulation because of its therapeutic potential. Both, RAR alpha and beta are capable of inducing human ADAM10 promoter activity (Tippmann et al., 2009). Moreover, the commercially available drug acitretin which intracellularly liberates retinoic acid (Ortiz et al., 2013), shifts APP processing in AD model mice toward the alpha-secretase cleavage pathway (Tippmann et al., 2009). The neuroprotective property of RARalpha agonists has been shown in cortical cultures, an AD mouse model (Tg2576 mice) (Jarvis et al., 2010), as well as in hippocampal tissue of aged SAMP8 mice (Kitaoka et al., 2013). Cilostazol-stimulated N2A cells with overexpression of human mutated APP also displayed ADAM10 elevation which was significantly attenuated by a RARbeta inhibitor and RARbeta-gene silencing (Lee et al., 2014). The effect of cilostazol on ADAM10 expression could be antagonized by sirtinol and by Sirtuin 1 (SIRT1)-gene silencing, suggesting that RARbeta and this class of deacetlyase together act on the ADAM10 promoter.
For a systematic approach on transcription factors relevant to ADAM10 regulation, we performed a screening approach (Reinhardt et al., 2014). Figure 3 sums up transcription factors that showed a significant influence on ADAM10 expression in these investigations. One has to consider that the screening approach was performed in human neuronal SH-SY5Y cells and only included single expression plasmids for 704 human transcription factors. Therefore, accessory proteins for single factors might not have been present in the cell line or combinations of transcription factors might be needed for full activation. However, we identified 11% transcription factors with a comparably strong influence on promoter activity of ADAM10 with nine factors inhibiting and 74 factors increasing transcriptional activity (Figure 3). Starting from this screening we were able to further characterize regulation of ADAM10 via one of the strongest inducers—X-Box binding protein 1 (XBP-1, Calfon et al., 2002). The active transcription factor is built upon ER stress sensor Inositol requiring enzyme 1 alpha (IRE1 alpha) activation and leads to increase in ADAM10 mRNA as well as protein and subsequent release of the APP cleavage product APPs-alpha (Reinhardt et al., 2014). Interestingly, we also found the amount of XBP1-mRNA to be decreased in Alzheimer model mice at higher age and also in Alzheimer's disease patients.
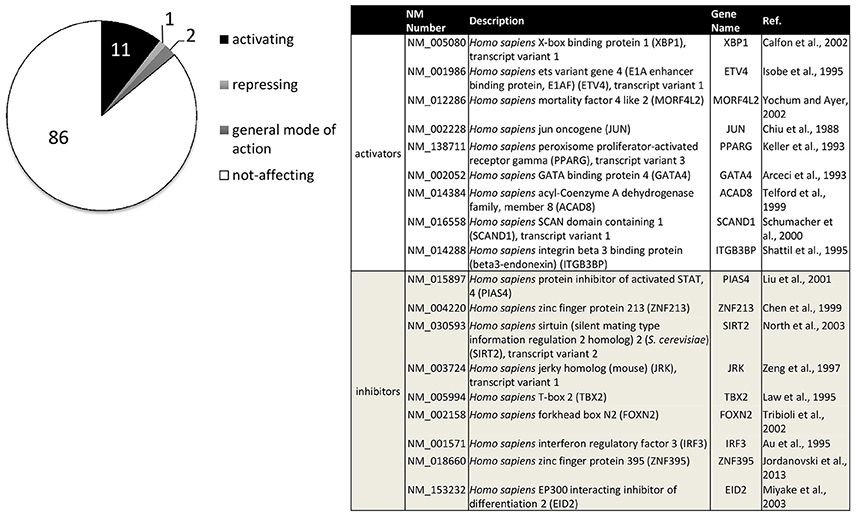
Figure 3. Transcription factors influencing human ADAM10 promoter activity in SH-SY5Y cells. Original data published in: Reinhardt et al. (2014). Factors filtered for effect size (promoter activity above 100+5xSD or below 100-2.5xSD of control) and reproducibility (SD≤ 15% of effect size in at least three independent experiments). Percentage of activating or repressing factors are indicated (left), factors with a general mode of action on transcription such as activator of basal transcription 1 (ABT1) were excluded. The table (right) shows the nine transcription factors with either strongest activating or inhibiting effects on the human ADAM10 promoter ranked from strongest to weakest.
Epigenetic Regulation of ADAM10
Currently, little is known about the epigenetic regulation of ADAM10. The 5′-untranslated region of the human ADAM10 gene contains a large GC-rich domain at −700 to +200 bp. The GC content of the first 600 bp upstream of the ATG of the human ADAM10 gene is 67% and nine CpG islands have been predicted (Prinzen et al., 2005). This abundance of CpGs suggests that cytosine methylation could play a role in regulating the proteinases' expression. SIRT1 an evolutionarily conserved NAD+-dependent deacetylase pivotal for metabolic control has been identified to increase ADAM10 expression (Lee et al., 2014). SIRT1 is involved in histone deacetylation and methylation, promoter CpG island methylation, and inactivation of non-histone transcription factors (Zhang and Kraus, 2010). Conceivably, SIRT1 is also involved in deacetylation of RAR or in chromatin modifications upon recruitment by the receptor but currently this has not yet been demonstrated. Investigations into these regulatory mechanisms are non-trivial and complicated by the fact that SIRT1 also acts on the cellular retinoid binding protein II (CRABPII) and also has a more general effect on RA signaling (Tang et al., 2014).
In transgenic AD model mice (5 × FAD) a significant increase in global DNA methylation, measured by 5-methyl cytosine, has been reported and additional changes in e.g., demethylase Dnmt3b or enzymes of histon acetylation/ deacetylation such as Hdac2, Jarid1a, or G9a (Griñán-Ferré et al., 2016). Surprisingly, no changes of ADAM10 expression were observed when using whole brain mRNA preparations. Although ADAM10 was found within the top CpG sites of an epigenomic analysis of psychiatric tic-diseases using peripheral blood samples (cg00785856, Zilhão et al., 2015), the methylation site did not reach significance at the genome-wide threshold.
Finally, as melatonin seems to be able to increase the level of deacetylase in young and aged primary neurons (Tajes et al., 2009), the observed induction of ADAM10 by melatonin (Panmanee et al., 2015; Shukla et al., 2015) might also rely on deacetylase activation.
Regulation of ADAM10 at the Translational Level
Besides regulation on the transcriptional/epigenetic level, translational modifiers can regulate the amount and availability of ADAM10: RNA structure, RNA-binding proteins (RBPs), and miRNAs have been reported to play a role.
The working group of Christian Haass explored a suppression of ADAM10 expression by its 5′UTR (Lammich et al., 2010) and identified a stable G-quadruplex structure of ADAM10 mRNA (Lammich et al., 2011). The stability of a G-quadruplex structure depends in part on binding proteins, such as fragile X mental retardation protein (FMRP; Oostra and Willems, 1995) and indeed, FMRP immunoprecipitated from cortical mouse tissue revealed bound ADAM10 mRNA (Pasciuto et al., 2015). Mice lacking FMRP displayed a shift of APP processing toward the non-amyloidogenic pathway during early stages of development, which subsequently led to synaptic and behavioral deficits (Pasciuto et al., 2015). Lack of FMRP could increase ADAM10 levels because FMRP stabilizes the G-quadruplex structure and can thus perturb translation initiation, as has been previously suggested for two other mRNAs (MAP1B and PP2A) that are FMRP targets (Lu et al., 2004; Castets et al., 2005). Another RNA-binding protein found to regulate ADAM10 is the neuronal ELAV protein: nELAV was shown by using immunoprecipitation to bind ADAM10 mRNA via an adenine- and uridine-rich element (Amadio et al., 2009). This might result in an increase in amyloidogenic APP processing. Since A-beta peptides have been found to inhibit ELAV-binding to ADAM10 mRNA (Amadio et al., 2009), this could reduce the ADAM10 amount even further, potentially leading to a vicious cycle.
miRNAs can silence cytoplasmic mRNAs either by triggering degradation or by promoting translation repression. For ADAM10 a prominent example for such a regulatory mechanism is hepatic miR-122, which decreased ADAM10 protein in human hepatic cancer cell lines (Bai et al., 2009). Using a systematic approach, i.e., a combination of different bioinformatics tools, we identified several candidate miRNAs that should act on ADAM10 and evaluated three of them via reporter gene assay—miR-103, -107, and -1306 (Augustin et al., 2012). Additionally, miR-144/451 which has been shown to be induced by A-beta peptide in SH-SY5Y cells decreased ADAM10 protein amount (Cheng et al., 2013). This regulation might be indirect and based on the transcription factor PAX4 (Zhang et al., 2015). In gastric cancer tissue miR-448 (Wu et al., 2016) and in tumor initiator cells of head and neck squamous cell carcinoma miR-494 (Chang et al., 2015) were also identified as novel regulators of ADAM10.
Post-translational Regulation of ADAM10: Maturation and Interaction Partners
After their synthesis, membrane proteins mature along the secretory pathway; they are transported to distinct compartments of the cell and finally, they locally interact with proteins and lipids of the phospholipid-bilayer. Eventually, they are degraded. Protein synthesis and removal are in homeostasis and thus determine the concentration of functional intramembranous proteins. In principle, ADAM10 can be regulated at all of these stages, offering possibilities for intervention.
The ADAM10 zymogen is cleaved by proprotein convertases within the secretory pathway to yield the active enzyme (see paragraph 1). Removal of the prodomain of ADAMs likely involves a canonical consensus site for the proprotein convertase Furin (Roebroek et al., 1994), which is located between the pro- and the catalytic domain of ADAM10 (Anders et al., 2001). More recently, a novel cleavage site upstream of the prodomain has been identified (Wong et al., 2015). ADAM10 has four potential N-glycosylation sites of which three are located in the metalloprotease domain (N267, N278, and N439) and one in the disintegrin domain (N551). In bovine ADAM10 all four have been found glycosylated and required for full in vivo activity (Escrevente et al., 2008).
Binding of ADAM10 to synapse associated protein 97 (SAP97) is required for inserting ADAM10 into the synaptic membrane (Marcello et al., 2013). Interaction of SAP97 with ADAM10 is mediated via a protein kinase C (PK C) phosphorylation site within the SAP97 SRC homology domain (Saraceno et al., 2014). Removal of ADAM10 from excitatory synapses occurs by clathrin-mediated endocytosis in human hippocampal tissue (Marcello et al., 2013). This is mediated by the clathrin adaptor protein AP2 which interacts with the ADAM10 C-terminal domain. In addition to control of surface concentrations of ADAM10 by transport mechanisms, further cleavage events may occur: the ectodomain of ADAM10 can be processed by ADAM9/15 or gamma-secretase (Cissé et al., 2005; Parkin and Harris, 2009; Tousseyn et al., 2009). Using recombinant mouse ADAM9 prodomain as a competitive inhibitor of ADAM9, Moss et al. demonstrated an increase of ADAM10-dependent APP processing in human neuronal SH-SY5Y cells (Moss et al., 2011). However, a truncated soluble ADAM10 construct was incapable of shedding cell-associated amyloid precursor protein while earlier reports described that shedded ADAM10 had the ability to cleave endogenous Prion protein in fibroblasts (Cissé et al., 2005).
The intensity of ADAM10 cleavage may further depend on the cytoskeleton: a dominant negative dynamin I mutant not only increased surface expression of both, immature, and mature ADAM10 but also strongly increased the amount of the C-terminal cleavage product of ADAM10 (Carey et al., 2011). In addition to its role as a protease acting at the cell surface it has been speculated that the soluble ADAM10 C-terminus could act as a signaling molecule, facilitating nuclear entry of other proteins (Endsley et al., 2014). A protein class which is deeply involved in for example cytoskeletal anchoring and protein-trafficking is the tetraspanin family (Charrin et al., 2014). Several tetraspanins have been identified to interact with ADAM10: tetraspanin 12 (Tspan 12) binds to ADAM10 in a palmitylation-dependent mechanism and increases non-amyloidogenic shedding of APP by increased enzymatic maturation of the protease (Xu et al., 2009). Co-immunoprecipitation experiments also identified specific ADAM10 interactions with Tspan5, Tspan10, Tspan14, Tspan15, Tspan17, and Tspan33/Penumbra (Haining et al., 2012), which all led to enhanced enzyme maturation. Interestingly, only overexpression of Tspan15 resulted in a reduction of ligand-induced Notch-1 processing by ADAM10 (Jouannet et al., 2016). This led to the assumption that the tetraspanins might differentially influence compartimentalization of ADAM10. Indeed, the apparent diffusion coefficient of ADAM10 was higher in cells overexpressing Tspan15 as compared to control cells or Tspan5 overexpressing cells and also decreased the co-immunoprecipitation of proteins of the tetraspanin web with ADAM10 (Jouannet et al., 2016). Tspan12 and 17 also seem to stabilize a high molecular weight protein complex that tethers ADAM10 to the gamma-secretase allowing rapid sequential processing of substrates (Chen et al., 2015).
ADAM10 is known to be mainly located outside of lipid rafts and alpha-secretase cleavage of APP occurs in non-raft domains (Kojro et al., 2001). Targeting ADAM10 artificially into lipid raft domains of the plasma membrane resulted in impaired enzymatic activity in human neuroblastoma cells (Harris et al., 2009; Kojro et al., 2010). Depletion of one of the constituents of lipid rafts, i.e., cholesterol, enhanced ADAM10 activity in different cellular models (Kojro et al., 2001, 2010; Matthews et al., 2003). The sigma-1 receptor contains a cholesterol recognition domain in its C-terminus and is able to remodel lipid rafts by changing the relative distribution of cholesterol between raft and non-raft fractions (Takebayashi et al., 2004). Interestingly, overexpression of sigma-1 in HEK293 or COS cells diminished Betacellulin cleavage by ADAM10 further substantiating the lipid-sensitivity of the enzyme (Li et al., 2012). Several investigations also report on influence of different lipid species such as trans fatty acids on APP processing balance (e.g., Eckert et al., 2011; Grimm et al., 2012) but in this regard it is not clear if this has a direct influence on ADAM10 or whether indirect mechanisms are involved.
Roles of ADAM10 in Neural Homeostasis and Pathology
ADAM10 has a number of physiological functions (see above) contributing to brain development or neural homeostasis. Diseases challenge this physiological state and the brain reacts to such perturbations with adaptations at the molecular, cellular, and functional level. The picture that is currently emerging from studies using animal models and human brains suggests a two-faced role of ADAM10 in diseases: beneficial as well as detrimental effects can be attributed to the protease depending on the specific setting and the substrates involved. In the following we review some of the conditions and diseases in which ADAM10 has been implicated.
ADAM10 in Aging and Alzheimer's Disease
ADAM10's role in these contexts is of particular interest because of its function as in vivo alpha secretase (Jorissen et al., 2010; Kuhn et al., 2010). Cleavage of APP along the non-amyloidogenic pathway yields APPs-alpha, which is important for neuroprotection (Kögel et al., 2012), learning and memory (Taylor et al., 2008; Hick et al., 2015; Xiong et al., 2016), and the structural integrity of neurons (Lee et al., 2010; Tyan et al., 2012; Weyer et al., 2014; Hick et al., 2015). Because cleavage of APP along the non-amyloidogenic pathway decreases with aging (Kern et al., 2006) and reduced APPs-alpha levels were found in CSF of some AD patients (Lannfelt et al., 1995; Sennvik et al., 2000), it is likely that insufficient APPs-alpha levels could contribute to the cognitive deficits of AD patients.
What is known about age-dependent changes in ADAM10 levels or activity in human brain? Unfortunately, with the notable exception of a publication from Bernstein et al. (2003) who compared still-born children with normal aged adults and found an increase in ADAM10 amount, data on ADAM10 in human brain are scarce. To study ADAM10 in humans, peripheral surrogate markers have been used, although it is unclear how comparable they are to CNS expression levels. A study aiming at comparing brain and leukocyte APP processing reported that while ADAM10 is present in brain it remains undetectable in the blood leukocyte fraction (Delvaux et al., 2013). Others, however, demonstrated ADAM10 expression in peripheral mononuclear blood cells as well as in platelets (Colciaghi et al., 2002). Using three groups of cognitively healthy subjects, we recently described an elevation of ADAM10 protein amount as well as catalytic function with cognitively healthy aging (Schuck et al., 2016). The reason why ADAM10 should be up-regulated is unclear. It is conceivable that it is a reaction to age-dependent changes in stress signatures (such as ER stress; e.g., Taylor, 2016) and thus represents a protective response. Although more data are needed, “healthy agers” show an ADAM10-increase whereas AD patients show a decrease (see below). In the former case APPs-alpha could be present in sufficient amounts protecting the brain whereas in the latter case APPs-alpha levels might be insufficient.
Using animal models of AD the role of ADAM10 as a protective protease has been demonstrated: overexpression of the protease at low level (30% above endogenous expression) was sufficient to nearly abolish plaque deposition in APP/PS1 AD model mice (Postina et al., 2004). These changes went hand-in-hand with improvements of learning and memory. In line with this gain-of-function approach, overexpression of a dominant negative ADAM10 mutant reduced alpha-secretase activity and worsened cognitive deficits (Schmitt et al., 2006). Interestingly, investigations using peripheral platelets of AD patients and healthy controls reported a decreased ADAM10 amount in AD patients (Colciaghi et al., 2002). Furthermore, ADAM10 levels in patient platelets were highly correlated with performance of the patients in psychological tests (Manzine et al., 2013, 2014). Together, these data suggest that normalizing or even increasing ADAM10 levels in AD could have a disease-modifying or at least disease-protracting effect.
Finally, it should be kept in mind that ADAM10 is multifunctional and that some effects of ADAM10 in the context of aging and AD could depend on ADAM10-mediated cleavage of other substrates than APP, such as Klotho (Chen et al., 2007; Bloch et al., 2009). This protein is linked to longevity (Kurosu et al., 2005) and soluble Klotho (s-Klotho) may be cardioprotective (Xie et al., 2012). Lower CSF s-Klotho levels have also been associated with endothelial dysfunction and neuronal damage in neuropsychiatric systemic lupus erythematosus patients (Ushigusa et al., 2016). Thus, lower ADAM10 levels in the aged brain may have detrimental effects on several levels involving APP processing as well as the processing of other ADAM10 substrates.
ADAM10, Dendritic Spines and Fragile X Syndrome
The level and/or activity of ADAM10 affect neuronal structures in the adult brain, in particular dendritic spines. This was shown using conditional ADAM10-deficient mice (Prox et al., 2013), which exhibited hippocampal neurons with fewer and abnormally shaped spines. The effect of ADAM10 on spines may depend on several substrates involved in the regulation of spine density, geometry and dynamics, including APP, N-cadherins, Neurexins, Neuroligins, and Nectin-1 (Prox et al., 2013). These substrates act as cell adhesion molecules and are known to influence spine morphology as well as synaptic transmission.
Of particular interest in this context is again the link between ADAM10 and APP. APP and in particular its cleavage product APPs-alpha have been shown to regulate dendritic complexity as well as spine numbers of hippocampal neurons (Lee et al., 2010; Tyan et al., 2012; Weyer et al., 2014). This effect appears to be age-dependent: whereas young APP-deficient mice had normal spine numbers, older APP-deficient mice showed a decrease in their spine density (Tyan et al., 2012). It may also depend on the brain region, since APP levels may show regional variations (Del Turco et al., 2016). Since APPs-alpha is generated by ADAM10 cleavage of APP, it is likely that some of the structural effects on spines seen in conditional ADAM10 knock-out mice (Prox et al., 2013) are the result of reduced APPs-alpha levels. Indeed, Prox et al. (2013) reported a reduction of APPs-alpha in brain of conditional ADAM10 knock-out mice to 5% of control levels. Since aging is also associated with reduced dendritic complexity and spine densities (Dickstein et al., 2007), it is attractive to speculate that reduced ADAM10 levels/activity and reduced APPs-alpha levels could play a role in this context (Lannfelt et al., 1995; Sennvik et al., 2000).
Whereas, reduction of ADAM10 may contribute to conditions in which fewer dendritic spines are observed, too much ADAM10 could contribute to diseases with the opposite phenotype, i.e., too many spines. Fragile X syndrome (FXS) is a good example for such a disease and is characterized by increased spine numbers and abnormally long spines. Mice with a fragile X mental retardation protein (FMRP) knock-out at an early adult age (P21, Pasciuto et al., 2015) showed a parallel increase in the expression of APP and mature ADAM10, suggesting that ADAM10 processing of APP could play a role. Indeed, primary fibroblasts obtained from adolescent and adult patients with FXS showed similar changes (Pasciuto et al., 2015), suggesting that an upregulation of ADAM10 and APP could also occur in brains of FXS-patients. In line with these findings, overexpression of APP (Lee et al., 2010) caused FXS-like spine changes in vitro.
In sum, under healthy conditions ADAM10 and its processing of cell adhesion molecules at synapses is in a homeostatic balance. Reduction of ADAM10 levels may cause a reduction in spine densities. Conversely, an increase in ADAM10 levels may increase the density of spines. Normalizing ADAM10 levels could be a potential therapeutic strategy.
Synaptic Function and Epilepsy
Dendritic spines and excitatory synaptic neurotransmission are intimately linked (Kasai et al., 2010). It is, therefore, in line with the effects of ADAM10 on dendritic spines that conditional ADAM10 knock-out mice show functional abnormalities at excitatory synapses: electrophysiological analysis of hippocampal CA1 neurons revealed almost normal basal synaptic transmission and short-term-plasticity but a grossly impaired induction of long-term-potentiation (Prox et al., 2013). These electrophysiological abnormalities were accompanied by reduction of postsynaptic density protein-95 (PSD-95) and several NMDA-receptor subunits, suggesting a severe disruption of synaptic architecture and function. Spatial learning was impaired at the behavioral level (Prox et al., 2013). Mechanistically, the impairment of synaptic plasticity and learning could be linked to several of the substrates of ADAM10 at the synapse. Again, APP is one of the more interesting candidates because its fragment APPs-alpha has been shown to be involved in synaptic plasticity, as well as learning and memory in the hippocampus (Taylor et al., 2008; Hick et al., 2015). At present it is unknown whether some of the abnormalities of the conditional ADAM10 mice could be rescued by recombinant APPs-alpha. Answering this question could help to better understand the relative importance of APP in this context.
Gain-of-function experiments resulted in an increased susceptibility of neurons for seizures: in mice overexpressing ADAM10 under the Thy1 promoter (~postnatal day 1), kainate-treatment evoked stronger and longer episodes of seizures as compared to wild type mice (Clement et al., 2008). Moreover, a dominant negative variant of ADAM10 seemed protective against this form of experimental epilepsy as shown e.g., by decreased neuronal damage score. The role of ADAM10 in epilepsy is complex, however, since conditional ADAM10 knock-out mice also showed seizures. However, these seizures may be linked to the gliosis observed in these mice (Prox et al., 2013). Thus, different disease mechanisms could play a role and additional work is needed before the role of ADAM10 in epilepsy can be assessed.
ADAM10 and Traumatic Brain Injury
ADAM10 is upregulated at injury sites (Zohar et al., 2011) and in denervated areas of the brain following brain injury (Warren et al., 2012; Del Turco et al., 2014). Reactive astroglia but not microglia has been shown to upregulate the protease following denervation (Warren et al., 2012; Del Turco et al., 2014). ADAM10's role in traumatic brain injury is still poorly understood and different modes of action have been proposed, which may not be mutually exclusive. First, the protease could be involved in the reorganization of the extracellular matrix of denervated regions (e.g., Deller et al., 2000), which may be a requirement for denervation-induced synaptic reorganization to occur (Warren et al., 2012). Secondly, ADAM10 could process synaptic adhesion molecules such as N-cadherin (Malinverno et al., 2010; Warren et al., 2012), neuroligins (Suzuki et al., 2012), or ephrins (Janes et al., 2005), which could tether degenerating terminals to their postsynaptic membranes. Cleaving the transsynaptic molecular bridge could be a necessary first step for re-innervation. Thirdly, ADAM10 could cleave APP and liberate APPs-alpha (Del Turco et al., 2014), which is neuroprotective in vitro (Kögel et al., 2012) and protects neurons in vivo following brain injury (reviewed in: Plummer et al., 2016). Of note, in these contexts upregulation of ADAM10 is associated with a short-term neural “defense”-reaction. This reaction seems to be transient and ADAM10 levels return to normal within a few days. In contrast, under lesioning conditions resulting in long-term upregulation of ADAM10 (Warren et al., 2012) synaptic reorganization failed and functional deficits persisted. Under these conditions, pharmacological blockade of ADAM10 helped to restore function, suggesting that long-term upregulation of ADAM10 is detrimental for brain rewiring. Collectively, these findings suggest the following model for ADAM10's role in brain injury: ADAM10 plays a plasticity-enhancing and neuroprotective role during the first phase following injury. It shapes the extracellular environment for sprouting fibers, clears synaptic sites, and liberates neuroprotective APP fragments. During the second phase, however, sprouting of surviving fibers occurs and new synapses form. If ADAM10 is still upregulated at this time point it could interfere with the stabilization of new synapses by cleaving the molecular bridge that binds pre- and postsynaptic structures (Figure 4).
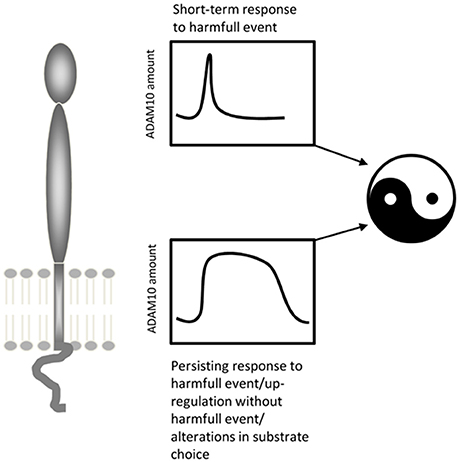
Figure 4. ADAM10's potential two-faced role under conditions of brain injury. Whereas a transiently increased activity/amount of ADAM10 seems to be part of a protective and restorative response to mild neural lesions, a persistent upregulation of ADAM10 as seen following severe lesions may be deleterious.
Stroke and Psychiatric Diseases
A positive association between the rs653765 polymorphism of ADAM10 and atherosclerotic cerebral infarction has been found in a Chinese population cohort (Li et al., 2013). Patients that carried the rs653765 C > T mutation also showed increased ADAM10 mRNA in PBMCs as did aged patients in comparison to younger patients or healthy controls (>70 years). As already mentioned, an association between CpG-site methylation in the ADAM10 locus and psychiatric tic-disorders has been identified (Zilhão et al., 2015). Beside this epigenetic association, ADAM10 has been characterized as one of the candidates within a low density GWA study for conduct disorder (Jian et al., 2011). Further associations with psychiatric disorders are conceivable, since ADAM10 processes neuroligins, which have also been identified as candidate genes in autism spectrum disorders and schizophrenia (e.g., Sun et al., 2011; Chen et al., 2014). In this regard it is of interest that Ray and colleagues recently reported alterations of APP processing and amount not only in FXS patients but also in autism spectrum disorder patients (Ray et al., 2016). However, they also reported age-dependent elevation of ADAM17 in the latter so that this protease might be due to observed changes instead of ADAM10.
Brain Tumors
ADAM10 may have deleterious effects for patients with brain tumors because it may promote the spreading of tumor cells. Reduced motility of glioblastoma cells treated with ADAM10-targeted siRNA has been observed (Kohutek et al., 2009) and invasiveness of pituitary adenomas correlated with ADAM10 expression level (Pan et al., 2012). Both publications suggests that ADAM10 may process putative barriers restricting tumor cells. With regard to cancer stem cells Bulstrode et al. reported that ADAM10 promotes the self-renewal of brain tumor sphere forming cells (Bulstrode et al., 2012). Additionally, treatment with inhibitors specific for ADAM10 or ADAM17 increased immune recognition of glioblastoma-initiating cells by natural killer cells (Wolpert et al., 2014). This seemed to be due to enhanced cell surface expression of UL16-binding protein 2 (ULBP2), which is shedded by both proteinases. In sum, stimulating ADAM10 expression, as suggested for AD patients (see below), may not be an option for oncology patients.
Conclusion and Outlook—ADAM10-Targeting Drugs as Novel Therapeutics?
ADAM10 is a biologically multifunctional protease involved in many important processes. It is expressed almost ubiquitously in the body. High amounts of ADAM10 are found in neural tissue during development, maturation and aging. Under conditions of neuronal activity and under some pathological conditions, ADAM10 expression is altered which, in turn, leads to changes in the processing of its substrates. The biological activity of these substrates and their cleavage products lead to measurable changes in function, biochemistry and even neural structures.
How can ADAM10 be considered a target for therapy in spite of the large number of substrates with multiple functions? First of all it has to be kept in mind that the majority of data on ADAM10 was obtained using in vitro systems. Although these studies can show putative interactions, such in vitro interactions require in vivo verification. ADAM10 can only cleave putative substrates if protease and substrate are in the same microcompartment at the same time. Since the availability of substrates and their distribution changes during development and aging, it is likely that changes in ADAM10 expression result in different effects depending on the age and stage of development of an organism. Of particular importance for the use of drugs targeting ADAM10 is the fact that ADAM10 shows an increasing overlap with its substrate APP with age (Marcinkiewicz and Seidah, 2000), suggesting that ADAM10-mediated APP cleavage may become more relevant at later stages in life. Regardless of these considerations, a rational approach to therapy development will take all these possibilities into account and will look at the net biological effects changes in ADAM10 expression induce in neural tissue. Complex in vitro systems, such as organotypic slice cultures (e.g., Gähwiler et al., 1997; Del Turco and Deller, 2007) and in vivo models (e.g., Postina et al., 2004) will help to address these questions.
The duration of ADAM10 expression changes may also play a critical role during the course of a disease. The enzyme can be briefly upregulated or persistently increased, depending on the specific conditions. Thus, ambivalent or even opposite outcomes can be expected for ADAM10 effects on brain structure and function, as has been shown for its role in brain injury (see Figure 4). Finally, patients may have different genetic predispositions or constitutively elevated ADAM10 levels, which might also harm the brain as has been shown for infarction and cancerogenesis (Pan et al., 2012; Li et al., 2013).
In sum, there are drug safety-issues which need to be explored before ADAM10 targeting drugs can be considered for therapy. The complex expression patterns and time courses of ADAM10 and its substrates may constrain the use of ADAM10-targeting drugs to specific situations, aged patients or some diseases. A clinical pilot study using acitretin was, however, promising (Endres et al., 2014). Acitretin (Neotigason), which increases APP processing along the non-amyloidogenic pathway in vitro, in primary cells, and in AD model mice (Tippmann et al., 2009; Reinhardt et al., 2016), was given to patients with mild to moderate AD for 4 weeks. Compared to the placebo group, treated patients showed a significant increase in their CSF APPs-alpha levels. Acitretin-treatment was well-tolerated and considered overall safe (Endres et al., 2014). Longer and larger trials will now be needed to evaluate the potential of acitretin as a novel AD-therapeutic. In any case, the pilot study raises hopes that at least for some AD patient groups ADAM10-targeting therapies may eventually prove to be useful.
Author Contributions
KE and TD contributed equally to all aspects of this review, including development of the overall concept, writing and creating the figures.
Funding
Grant sponsor: Deutsche Forschungsgemeinschaft (DFG, FOR 1332 to TD) and (NGFN, FKZ01GS08130) and the Alfons Geib-Stiftung to KE.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Amadio, M., Pascale, A., Wang, J., Ho, L., Quattrone, A., Gandy, S., et al. (2009). nELAV proteins alteration in Alzheimer's disease brain: a novel putative target for amyloid-β reverberating on AβPP processing. J. Alzheimers. Dis. 16, 409–419. doi: 10.3233/JAD-2009-0967
Anders, A., Gilbert, S., Garten, W., Postina, R., and Fahrenholz, F. (2001). Regulation of the α-secretase ADAM10 by its prodomain and proprotein convertases. FASEB J. 15, 1837–1839. doi: 10.1096/fj.01-0007fje
Arceci, R. J., King, A. A., Simon, M. C., Orkin, S. H., and Wilson, D. B. (1993). Mouse GATA-4: a retinoic acid-inducible GATA-binding transcription factor expressed in endodermally derived tissues and heart. Mol. Cell. Biol. 13, 2235–2246. doi: 10.1128/MCB.13.4.2235
Au, W. C., Moore, P. A., Lowther, W., Juang, Y. T., and Pitha, P. M. (1995). Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. U.S.A. 92, 11657–11661. doi: 10.1073/pnas.92.25.11657
Augustin, R., Endres, K., Reinhardt, S., Kuhn, P. H., Lichtenthaler, S. F., Hansen, J., et al. (2012). Computational identification and experimental validation of microRNAs binding to the Alzheimer-related gene ADAM10. BMC Med. Genet. 13:35. doi: 10.1186/1471-2350-13-35
Bai, S., Nasser, M. W., Wang, B., Hsu, S. H., Datta, J., Kutay, H., et al. (2009). MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J. Biol. Chem. 284, 32015–32027. doi: 10.1074/jbc.M109.016774
Bekris, L. M., Galloway, N. M., Millard, S., Lockhart, D., Li, G., Galasko, D. R., et al. (2011). Amyloid precursor protein (APP) processing genes and cerebrospinal fluid APP cleavage product levels in Alzheimer's disease. Neurobiol. Aging 32, 556–523. doi: 10.1016/j.neurobiolaging.2010.10.020
Bernstein, H. G., Bukowska, A., Krell, D., Bogerts, B., Ansorge, S., and Lendeckel, U. (2003). Comparative localization of ADAMs 10 and 15 in human cerebral cortex normal aging, Alzheimer disease and Down syndrome. J. Neurocytol. 32, 153–160. doi: 10.1023/B:NEUR.0000005600.61844.a6
Bernstein, H. G., Stricker, R., Lendeckel, U., Bertram, I., Dobrowolny, H., Steiner, J., et al. (2009). Reduced neuronal co-localisation of nardilysin and the putative α-secretases ADAM10 and ADAM17 in Alzheimer's disease and Down syndrome brains. Age (Dordr). 31, 11–25. doi: 10.1007/s11357-008-9076-x
Biswas, S., Adrian, M., Weber, J., Evdokimov, K., Winkler, M., and Géraud, C. (2016). Posttranslational proteolytic processing of Leda-1/Pianp involves cleavage by MMPs, ADAM10/17 and gamma-secretase. Biochem. Biophys. Res. Commun. 477, 661–666. doi: 10.1016/j.bbrc.2016.06.116
Bloch, L., Sineshchekova, O., Reichenbach, D., Reiss, K., Saftig, P., Kuro-o, M., et al. (2009). Klotho is a substrate for α-, β- and γ-secretase. FEBS Lett. 583, 3221–3224. doi: 10.1016/j.febslet.2009.09.009
Blume, K. E., Soeroes, S., Keppeler, H., Stevanovic, S., Kretschmer, D., Rautenberg, M., et al. (2012). Cleavage of annexin A1 by ADAM10 during secondary necrosis generates a monocytic “find-me” signal. J. Immunol. 188, 135–145. doi: 10.4049/jimmunol.1004073
Bulstrode, H., Jones, L. M., Siney, E. J., Sampson, J. M., Ludwig, A., Gray, W. P., et al. (2012). A-Disintegrin and Metalloprotease (ADAM) 10 and 17 promote self-renewal of brain tumor sphere forming cells. Cancer Lett. 326, 79–87. doi: 10.1016/j.canlet.2012.07.022
Caescu, C. I., Jeschke, G. R., and Turk, B. E. (2009). Active-site determinants of substrate recognition by the metalloproteinases TACE and ADAM10. Biochem. J. 424, 79–88. doi: 10.1042/BJ20090549
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. doi: 10.1038/415092a
Carey, R. M., Blusztajn, J. K., and Slack, B. E. (2011). Surface expression and limited proteolysis of ADAM10 are increased by a dominant negative inhibitor of dynamin. BMC Cell Biol. 12:20. doi: 10.1186/1471-2121-12-20
Castets, M., Schaeffer, C., Bechara, E., Schenck, A., Khandjian, E. W., Luche, S., et al. (2005). FMRP interferes with the Rac1 pathway and controls actin cytoskeleton dynamics in murine fibroblasts. Hum. Mol. Genet. 14, 835–844. doi: 10.1093/hmg/ddi077
Chang, Y. C., Jan, C. I., Peng, C. Y., Lai, Y. C., Hu, F. W., and Yu, C. C. (2015). Activation of microRNA-494-targeting Bmi1 and ADAM10 by silibinin ablates cancer stemness and predicts favourable prognostic value in head and neck squamous cell carcinomas. Oncotarget 6, 24002–24016. doi: 10.18632/oncotarget.4365
Charrin, S., Jouannet, S., Boucheix, C., and Rubinstein, E. (2014). Tetraspanins at a glance. J. Cell Sci. 127, 3641–3648. doi: 10.1242/jcs.154906
Chen, A. C., Kim, S., Shepardson, N., Patel, S., Hong, S., and Selkoe, D. J. (2015). Physical and functional interaction between the α- and γ-secretases: a new model of regulated intramembrane proteolysis. J. Cell Biol. 211, 1157–1176. doi: 10.1083/jcb.201502001
Chen, C. D., Podvin, S., Gillespie, E., Leeman, S. E., and Abraham, C. R. (2007). Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. U.S.A. 104, 19796–19801. doi: 10.1073/pnas.0709805104
Chen, J., Yu, S., Fu, Y., and Li, X. (2014). Synaptic proteins and receptors defects in autism spectrum disorders. Front. Cell. Neurosci. 8:276. doi: 10.3389/fncel.2014.00276
Chen, X., Hamon, M., Deng, Z., Centola, M., Sood, R., Taylor, K., et al. (1999). Identification and characterization of a zinc finger gene (ZNF213) from 16p13.3. Biochim. Biophys. Acta 1444, 218–230. doi: 10.1016/S0167-4781(98)00273-5
Cheng, C., Li, W., Zhang, Z., Yoshimura, S., Hao, Q., Zhang, C., et al. (2013). MicroRNA-144 is regulated by activator protein-1 (AP-1) and decreases expression of Alzheimer disease-related a disintegrin and metalloprotease 10 (ADAM10). J. Biol. Chem. 288, 13748–13761. doi: 10.1074/jbc.M112.381392
Chiu, R., Boyle, W. J., Meek, J., Smeal, T., Hunter, T., and Karin, M. (1988). The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell 54, 541–552. doi: 10.1016/0092-8674(88)90076-1
Cissé, M. A., Sunyach, C., Lefranc-Jullien, S., Postina, R., Vincent, B., and Checler, F. (2005). The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J. Biol. Chem. 280, 40624–40631. doi: 10.1074/jbc.M506069200
Clement, A. B., Hanstein, R., Schröder, A., Nagel, H., Endres, K., Fahrenholz, F., et al. (2008). Effects of neuron-specific ADAM10 modulation in an in vivo model of acute excitotoxic stress. Neuroscience 152, 459–468. doi: 10.1016/j.neuroscience.2007.10.060
Colciaghi, F., Borroni, B., Pastorino, L., Marcello, E., Zimmermann, M., Cattabeni, F., et al. (2002). [α]-Secretase ADAM10 as well as [α]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 8, 67–74.
Colonna, M., and Wang, Y. (2016). TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 17, 201–207. doi: 10.1038/nrn.2016.7
Corbett, G. T., Gonzalez, F. J., and Pahan, K. (2015). Activation of peroxisome proliferator-activated receptor α stimulates ADAM10-mediated proteolysis of APP. Proc. Natl. Acad. Sci. U.S.A. 112, 8445–8450. doi: 10.1073/pnas.1504890112
Deller, T., Haas, C. A., and Frotscher, M. (2000). Reorganization of the rat fascia dentata after a unilateral entorhinal cortex lesion. Role of the extracellular matrix. Ann. N.Y. Acad. Sci. 911, 207–220. doi: 10.1111/j.1749-6632.2000.tb06728.x
Del Turco, D., and Deller, T. (2007). Organotypic entorhino-hippocampal slice cultures–a tool to study the molecular and cellular regulation of axonal regeneration and collateral sprouting in vitro. Methods Mol. Biol. 399, 55–66. doi: 10.1007/978-1-59745-504-6_5
Del Turco, D., Paul, M. H., Schlaudraff, J., Hick, M., Endres, K., Müller, U. C., et al. (2016). Region-specific differences in amyloid precursor protein expression in the mouse hippocampus. Front. Mol. Neurosci. 9:134. doi: 10.3389/fnmol.2016.00134
Del Turco, D., Schlaudraff, J., Bonin, M., and Deller, T. (2014). Upregulation of APP, ADAM10 and ADAM17 in the denervated mouse dentate gyrus. PLoS ONE 9:e84962. doi: 10.1371/journal.pone.0084962
Delvaux, E., Bentley, K., Stubbs, V., Sabbagh, M., and Coleman, P. D. (2013). Differential processing of amyloid precursor protein in brain and in peripheral blood leukocytes. Neurobiol. Aging 34, 1680–1686. doi: 10.1016/j.neurobiolaging.2012.12.004
Deng, W., Cho, S., Su, P. C., Berger, B. W., and Li, R. (2014). Membrane-enabled dimerization of the intrinsically disordered cytoplasmic domain of ADAM10. Proc. Natl. Acad. Sci. U.S.A. 111, 15987–15992. doi: 10.1073/pnas.1409354111
Dickstein, D. L., Kabaso, D., Rocher, A. B., Luebke, J. I., Wearne, S. L., and Hof, P. R. (2007). Changes in the structural complexity of the aged brain. Aging Cell 6, 275–284. doi: 10.1111/j.1474-9726.2007.00289.x
Doberstein, K., Pfeilschifter, J., and Gutwein, P. (2011). The transcription factor PAX2 regulates ADAM10 expression in renal cell carcinoma. Carcinogenesis 32, 1713–1723. doi: 10.1093/carcin/bgr195
Ebsen, H., Lettau, M., Kabelitz, D., and Janssen, O. (2014). Identification of SH3 domain proteins interacting with the cytoplasmic tail of the a disintegrin and metalloprotease 10 (ADAM10). PLoS ONE 9:e102899. doi: 10.1371/journal.pone.0102899
Eckert, G. P., Chang, S., Eckmann, J., Copanaki, E., Hagl, S., Hener, U., et al. (2011). Liposome-incorporated DHA increases neuronal survival by enhancing non-amyloidogenic APP processing. Biochim. Biophys. Acta 1808, 236–243. doi: 10.1016/j.bbamem.2010.10.014
Endres, K., Fahrenholz, F., Lotz, J., Hiemke, C., Teipel, S., Lieb, K., et al. (2014). Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 83, 1930–1935. doi: 10.1212/WNL.0000000000001017
Endsley, M. A., Somasunderam, A. D., Li, G., Oezguen, N., Thiviyanathan, V., Murray, J. L., et al. (2014). Nuclear trafficking of the HIV-1 pre-integration complex depends on the ADAM10 intracellular domain. Virology 454–455, 60–66. doi: 10.1016/j.virol.2014.02.006
Escrevente, C., Morais, V. A., Keller, S., Soares, C. M., Altevogt, P., and Costa, J. (2008). Functional role of N-glycosylation from ADAM10 in processing, localization and activity of the enzyme. Biochim. Biophys. Acta 1780, 905–913. doi: 10.1016/j.bbagen.2008.03.004
Esselens, C. W., Malapeira, J., Colomé, N., Moss, M., Canals, F., and Arribas, J. (2008). Metastasis-associated C4.4A, a GPI-anchored protein cleaved by ADAM10 and ADAM17. Biol. Chem. 389, 1075–1084. doi: 10.1515/BC.2008.121
Eugenín-Von Bernhardi, J., and Dimou, L. (2016). NG2-glia, more than progenitor cells. Adv. Exp. Med. Biol. 949, 27–45. doi: 10.1007/978-3-319-40764-7_2
Fahrenholz, F., Gilbert, S., Kojro, E., Lammich, S., and Postina, R. (2000). α-secretase activity of the disintegrin metalloprotease ADAM 10. Influences of domain structure. Ann. N.Y. Acad. Sci. 920, 215–222. doi: 10.1111/j.1749-6632.2000.tb06925.x
Fahrenholz, F., and Postina, R. (2006). α-secretase activation–an approach to Alzheimer's disease therapy. Neurodegener. Dis. 3, 255–261. doi: 10.1159/000095264
Frank, S., Burbach, G. J., Bonin, M., Walter, M., Streit, W., Bechmann, I., et al. (2008). TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56, 1438–1447. doi: 10.1002/glia.20710
Franzke, C. W., Bruckner-Tuderman, L., and Blobel, C. P. (2009). Shedding of collagen XVII/BP180 in skin depends on both ADAM10 and ADAM9. J. Biol. Chem. 284, 23386–23396. doi: 10.1074/jbc.M109.034090
Frühbeis, C., Fröhlich, D., Kuo, W. P., and Kramer-Albers, E. M. (2013). Extracellular vesicles as mediators of neuron-glia communication. Front. Cell. Neurosci. 7:182. doi: 10.3389/fncel.2013.00182
Gähwiler, B. H., Capogna, M., Debanne, D., McKinney, R. A., and Thompson, S. M. (1997). Organotypic slice cultures: a technique has come of age. Trends Neurosci. 20, 471–477. doi: 10.1016/S0166-2236(97)01122-3
Grimm, M. O., Haupenthal, V. J., Mett, J., Stahlmann, C. P., Blümel, T., Mylonas, N. T., et al. (2016). Oxidized docosahexaenoic acid species and lipid peroxidation products increase amyloidogenic amyloid precursor protein processing. Neurodegener. Dis. 16, 44–54. doi: 10.1159/000440839
Grimm, M. O., Rothhaar, T. L., Grösgen, S., Burg, V. K., Hundsdörfer, B., Haupenthal, V. J., et al. (2012). Trans fatty acids enhance amyloidogenic processing of the Alzheimer amyloid precursor protein (APP). J. Nutr. Biochem. 23, 1214–1223. doi: 10.1016/j.jnutbio.2011.06.015
Griñán-Ferré, C., Sarroca, S., Ivanova, A., Puigoriol-Illamola, D., Aguado, F., Camins, A., et al. (2016). Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging (Albany NY). 8, 664–684. doi: 10.18632/aging.100906
Guo, Z. B., Su, Y. Y., Wang, Y. H., Wang, W., and Guo, D. Z. (2016). The expression pattern of Adam10 in the central nervous system of adult mice: detection by in situ hybridization combined with immunohistochemistry staining. Mol. Med. Rep. 14, 2038–2044. doi: 10.3892/mmr.2016.5501
Gutwein, P., Mechtersheimer, S., Riedle, S., Stoeck, A., Gast, D., Joumaa, S., et al. (2003). ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles. FASEB J. 17, 292–294. doi: 10.1096/fj.02-0430fje
Haining, E. J., Yang, J., Bailey, R. L., Khan, K., Collier, R., Tsai, S., et al. (2012). The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. J. Biol. Chem. 287, 39753–39765. doi: 10.1074/jbc.M112.416503
Harris, B., Pereira, I., and Parkin, E. (2009). Targeting ADAM10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res. 1296, 203–215. doi: 10.1016/j.brainres.2009.07.105
Hartmann, D., De Strooper, B., Serneels, L., Craessaerts, K., Herreman, A., Annaert, W., et al. (2002). The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for α-secretase activity in fibroblasts. Hum. Mol. Genet. 11, 2615–2624. doi: 10.1093/hmg/11.21.2615
Hata, S., Fujishige, S., Araki, Y., Kato, N., Araseki, M., Nishimura, M., et al. (2009). Alcadein cleavages by amyloid β-precursor protein (APP) α- and β-secretases generate small peptides, p3-Alcs, indicating Alzheimer disease-related gamma-secretase dysfunction. J. Biol. Chem. 284, 36024–36033. doi: 10.1074/jbc.M109.057497
Hick, M., Herrmann, U., Weyer, S. W., Mallm, J. P., Tschape, J. A., Borgers, M., et al. (2015). Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol. 129, 21–37. doi: 10.1007/s00401-014-1368-x
Hoffmann, F. S., Kuhn, P. H., Laurent, S. A., Hauck, S. M., Berer, K., Wendlinger, S. A., et al. (2015). The immunoregulator soluble TACI is released by ADAM10 and reflects B cell activation in autoimmunity. J. Immunol. 194, 542–552. doi: 10.4049/jimmunol.1402070
Horiuchi, K., Le, G. S., Schulte, M., Yamaguchi, T., Reiss, K., Murphy, G., et al. (2007). Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 18, 176–188. doi: 10.1091/mbc.E06-01-0014
Houri, N., Huang, K. C., and Nalbantoglu, J. (2013). The Coxsackievirus and Adenovirus Receptor (CAR) undergoes ectodomain shedding and regulated intramembrane proteolysis (RIP). PLoS ONE 8:e73296. doi: 10.1371/journal.pone.0073296
Hsieh, C. L., Koike, M., Spusta, S. C., Niemi, E. C., Yenari, M., Nakamura, M. C., et al. (2009). A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x
Isobe, M., Yamagishi, F., Yoshida, K., Higashino, F., and Fujinaga, K. (1995). Assignment of the ets-related transcription factor E1A-F gene (ETV4) to human chromosome region 17q21. Genomics 28, 357–359. doi: 10.1006/geno.1995.1158
Janes, P. W., Saha, N., Barton, W. A., Kolev, M. V., Wimmer-Kleikamp, S. H., Nievergall, E., et al. (2005). Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell 123, 291–304. doi: 10.1016/j.cell.2005.08.014
Jarvis, C. I., Goncalves, M. B., Clarke, E., Dogruel, M., Kalindjian, S. B., Thomas, S. A., et al. (2010). Retinoic acid receptor-α signalling antagonizes both intracellular and extracellular amyloid-β production and prevents neuronal cell death caused by amyloid-β. Eur. J. Neurosci. 32, 1246–1255. doi: 10.1111/j.1460-9568.2010.07426.x
Jedlicka, P., Hoon, M., Papadopoulos, T., Vlachos, A., Winkels, R., Poulopoulos, A., et al. (2011). Increased dentate gyrus excitability in neuroligin-2-deficient mice in vivo. Cereb. Cortex 21, 357–367. doi: 10.1093/cercor/bhq100
Jedlicka, P., Vnencak, M., Krueger, D. D., Jungenitz, T., Brose, N., and Schwarzacher, S. W. (2015). Neuroligin-1 regulates excitatory synaptic transmission, LTP and EPSP-spike coupling in the dentate gyrus in vivo. Brain Struct. Funct. 220, 47–58. doi: 10.1007/s00429-013-0636-1
Jian, X. Q., Wang, K. S., Wu, T. J., Hillhouse, J. J., and Mullersman, J. E. (2011). Association of ADAM10 and CAMK2A polymorphisms with conduct disorder: evidence from family-based studies. J. Abnorm. Child Psychol. 39, 773–782. doi: 10.1007/s10802-011-9524-4
John, L., Krstenansky, J. W., Gregg, R., Chenail, Matthew R., Tiffany, et al. (2004). Probing proteinase active sites using oriented peptide mixture libraries – ADAM-10. Lett. Drug Des. Discov. 1, 6–13. doi: 10.2174/1570180043485770
Jordanovski, D., Herwartz, C., Pawlowski, A., Taute, S., Frommolt, P., and Steger, G. (2013). The hypoxia-inducible transcription factor ZNF395 is controlled by IkB kinase-signaling and activates genes involved in the innate immune response and cancer. PLoS ONE 8:e74911. doi: 10.1371/journal.pone.0074911
Jorissen, E., Prox, J., Bernreuther, C., Weber, S., Schwanbeck, R., Serneels, L., et al. (2010). The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 30, 4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010
Jouannet, S., Saint-Pol, J., Fernandez, L., Nguyen, V., Charrin, S., Boucheix, C., et al. (2016). TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell. Mol. Life Sci. 73, 1895–1915. doi: 10.1007/s00018-015-2111-z
Kaczur, V., Puskas, L. G., Nagy, Z. U., Miled, N., Rebai, A., Juhasz, F., et al. (2007). Cleavage of the human thyrotropin receptor by ADAM10 is regulated by thyrotropin. J. Mol. Recognit. 20, 392–404. doi: 10.1002/jmr.851
Kärkkäinen, I., Rybnikova, E., Pelto-Huikko, M., and Huovila, A. P. (2000). Metalloprotease-disintegrin (ADAM) genes are widely and differentially expressed in the adult CNS. Mol. Cell. Neurosci. 15, 547–560. doi: 10.1006/mcne.2000.0848
Kasai, H., Fukuda, M., Watanabe, S., Hayashi-Takagi, A., and Noguchi, J. (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129. doi: 10.1016/j.tins.2010.01.001
Keller, H., Dreyer, C., Medin, J., Mahfoudi, A., Ozato, K., and Wahli, W. (1993). Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc. Natl. Acad. Sci. U.S.A. 90, 2160–2164. doi: 10.1073/pnas.90.6.2160
Kern, A., Roempp, B., Prager, K., Walter, J., and Behl, C. (2006). Down-regulation of endogenous amyloid precursor protein processing due to cellular aging. J. Biol. Chem. 281, 2405–2413. doi: 10.1074/jbc.M505625200
Kitaoka, K., Shimizu, N., Ono, K., Chikahisa, S., Nakagomi, M., Shudo, K., et al. (2013). The retinoic acid receptor agonist Am80 increases hippocampal ADAM10 in aged SAMP8 mice. Neuropharmacology 72, 58–65. doi: 10.1016/j.neuropharm.2013.04.009
Kleinberger, G., Yamanishi, Y., Suarez-Calvet, M., Czirr, E., Lohmann, E., Cuyvers, E., et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6:243ra286. doi: 10.1126/scitranslmed.3009093
Kögel, D., Deller, T., and Behl, C. (2012). Roles of amyloid precursor protein family members in neuroprotection, stress signaling and aging. Exp. Brain Res. 217, 471–479. doi: 10.1007/s00221-011-2932-4
Kohutek, Z. A., Dipierro, C. G., Redpath, G. T., and Hussaini, I. M. (2009). ADAM-10-mediated N-cadherin cleavage is protein kinase C-α dependent and promotes glioblastoma cell migration. J. Neurosci. 29, 4605–4615. doi: 10.1523/JNEUROSCI.5126-08.2009
Kojro, E., Fuger, P., Prinzen, C., Kanarek, A. M., Rat, D., Endres, K., et al. (2010). Statins and the squalene synthase inhibitor zaragozic acid stimulate the non-amyloidogenic pathway of amyloid-β protein precursor processing by suppression of cholesterol synthesis. J. Alzheimers Dis. 20, 1215–1231. doi: 10.3233/JAD-2010-091621
Kojro, E., Gimpl, G., Lammich, S., Marz, W., and Fahrenholz, F. (2001). Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc. Natl. Acad. Sci. U.S.A. 98, 5815–5820. doi: 10.1073/pnas.081612998
Kuhn, P. H., Colombo, A. V., Schusser, B., Dreymueller, D., Wetzel, S., Schepers, U., et al. (2016). Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife 5:e12748. doi: 10.7554/eLife.12748
Kuhn, P. H., Wang, H., Dislich, B., Colombo, A., Zeitschel, U., Ellwart, J. W., et al. (2010). ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032. doi: 10.1038/emboj.2010.167
Kummer, M. P., Maruyama, H., Huelsmann, C., Baches, S., Weggen, S., and Koo, E. H. (2009). Formation of Pmel17 amyloid is regulated by juxtamembrane metalloproteinase cleavage, and the resulting C-terminal fragment is a substrate for gamma-secretase. J. Biol. Chem. 284, 2296–2306. doi: 10.1074/jbc.M808904200
Kurosu, H., Yamamoto, M., Clark, J. D., Pastor, J. V., Nandi, A., Gurnani, P., et al. (2005). Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833. doi: 10.1126/science.1112766
Lammich, S., Buell, D., Zilow, S., Ludwig, A. K., Nuscher, B., Lichtenthaler, S. F., et al. (2010). Expression of the anti-amyloidogenic secretase ADAM10 is suppressed by its 5′-untranslated region. J. Biol. Chem. 285, 15753–15760. doi: 10.1074/jbc.M110.110742
Lammich, S., Kamp, F., Wagner, J., Nuscher, B., Zilow, S., Ludwig, A. K., et al. (2011). Translational repression of the disintegrin and metalloprotease ADAM10 by a stable G-quadruplex secondary structure in its 5'-untranslated region. J. Biol. Chem. 286, 45063–45072. doi: 10.1074/jbc.M111.296921
Lammich, S., Kojro, E., Postina, R., Gilbert, S., Pfeiffer, R., Jasionowski, M., et al. (1999). Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927. doi: 10.1073/pnas.96.7.3922
Lannfelt, L., Basun, H., Wahlund, L. O., Rowe, B. A., and Wagner, S. L. (1995). Decreased α-secretase-cleaved amyloid precursor protein as a diagnostic marker for Alzheimer's disease. Nat. Med. 1, 829–832. doi: 10.1038/nm0895-829
Lavoie, M. J., and Selkoe, D. J. (2003). The Notch ligands, Jagged and Delta, are sequentially processed by α-secretase and presenilin/gamma-secretase and release signaling fragments. J. Biol. Chem. 278, 34427–34437. doi: 10.1074/jbc.M302659200
Law, D. J., Gebuhr, T., Garvey, N., Agulnik, S. I., and Silver, L. M. (1995). Identification, characterization, and localization to chromosome 17q21-22 of the human TBX2 homolog, member of a conserved developmental gene family. Mamm. Genome 6, 793–797. doi: 10.1007/BF00539006
Lee, H. R., Shin, H. K., Park, S. Y., Kim, H. Y., Lee, W. S., Rhim, B. Y., et al. (2014). Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J. Neurosci. Res. 92, 1581–1590. doi: 10.1002/jnr.23421
Lee, K. J., Moussa, C. E., Lee, Y., Sung, Y., Howell, B. W., Turner, R. S., et al. (2010). β amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience 169, 344–356. doi: 10.1016/j.neuroscience.2010.04.078
Lee, S. B., Doberstein, K., Baumgarten, P., Wieland, A., Ungerer, C., Burger, C., et al. (2011). PAX2 regulates ADAM10 expression and mediates anchorage-independent cell growth of melanoma cells. PLoS ONE 6:e22312. doi: 10.1371/journal.pone.0022312
Li, J., Liu, B., Gao, X., Ma, Z., Caosong, T., Mei, Y. A., et al. (2012). Overexpression of sigma-1 receptor inhibits ADAM10 and ADAM17 mediated shedding in vitro. Protein Cell 3, 153–159. doi: 10.1007/s13238-012-2006-9
Li, Y., Liao, F., Yin, X. J., Cui, L. L., Ma, G. D., Nong, X. X., et al. (2013). An association study on ADAM10 promoter polymorphisms and atherosclerotic cerebral infarction in a Chinese population. CNS Neurosci. Ther. 19, 785–794. doi: 10.1111/cns.12136
Lin, J., Luo, J., and Redies, C. (2008). Differential expression of five members of the ADAM family in the developing chicken brain. Neuroscience 157, 360–375. doi: 10.1016/j.neuroscience.2008.08.053
Liu, B., Gross, M., Ten Hoeve, J., and Shuai, K. (2001). A transcriptional corepressor of Stat1 with an essential LXXLL signature motif. Proc. Natl. Acad. Sci. U.S.A. 98, 3203–3207. doi: 10.1073/pnas.051489598
Lokau, J., Nitz, R., Agthe, M., Monhasery, N., Aparicio-Siegmund, S., Schumacher, N., et al. (2016). Proteolytic cleavage governs interleukin-11 trans-signaling. Cell Rep. 14, 1761–1773. doi: 10.1016/j.celrep.2016.01.053
Londino, J. D., Gulick, D., Isenberg, J. S., and Mallampalli, R. K. (2015). Cleavage of Signal Regulatory Protein α(SIRPα) enhances inflammatory signaling. J. Biol. Chem. 290, 31113–31125. doi: 10.1074/jbc.M115.682914
Lu, R., Wang, H., Liang, Z., Ku, L., O'donnell, W. T., Li, W., et al. (2004). The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc. Natl. Acad. Sci. U.S.A. 101, 15201–15206. doi: 10.1073/pnas.0404995101
Lundgren, J. L., Ahmed, S., Schedin-Weiss, S., Gouras, G. K., Winblad, B., Tjernberg, L. O., et al. (2015). ADAM10 and BACE1 are localized to synaptic vesicles. J. Neurochem. 135, 606–615. doi: 10.1111/jnc.13287
Ma, Z., Li, Q., Zhang, Z., and Zheng, Y. (2013). A disintegrin and metalloprotease 10 in neuronal maturation and gliogenesis during cortex development. Neural Regen. Res. 8, 24–30. doi: 10.3969/j.issn.1673-5374.2013.01.003
Malinverno, M., Carta, M., Epis, R., Marcello, E., Verpelli, C., Cattabeni, F., et al. (2010). Synaptic localization and activity of ADAM10 regulate excitatory synapses through N-cadherin cleavage. J. Neurosci. 30, 16343–16355. doi: 10.1523/JNEUROSCI.1984-10.2010
Manzine, P. R., Barham, E. J., Vale, F. A., Selistre-De-Araujo, H. S., Pavarini, S. C., and Cominetti, M. R. (2014). Platelet a disintegrin and metallopeptidase 10 expression correlates with clock drawing test scores in Alzheimer's disease. Int. J. Geriatr. Psychiatry 29, 414–420. doi: 10.1002/gps.4020
Manzine, P. R., Barham, E. J., Vale Fde, A., Selistre-De-Araujo, H. S., Iost Pavarini, S. C., and Cominetti, M. R. (2013). Correlation between mini-mental state examination and platelet ADAM10 expression in Alzheimer's disease. J. Alzheimers Dis. 36, 253–260. doi: 10.3233/JAD-130125
Marcello, E., Gardoni, F., Mauceri, D., Romorini, S., Jeromin, A., Epis, R., et al. (2007). Synapse-associated protein-97 mediates α-secretase ADAM10 trafficking and promotes its activity. J. Neurosci. 27, 1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007
Marcello, E., Saraceno, C., Musardo, S., Vara, H., De La Fuente, A. G., Pelucchi, S., et al. (2013). Endocytosis of synaptic ADAM10 in neuronal plasticity and Alzheimer's disease. J. Clin. Invest. 123, 2523–2538. doi: 10.1172/JCI65401
Marcinkiewicz, M., and Seidah, N. G. (2000). Coordinated expression of β-amyloid precursor protein and the putative β-secretase BACE and α-secretase ADAM10 in mouse and human brain. J. Neurochem. 75, 2133–2143. doi: 10.1046/j.1471-4159.2000.0752133.x
Maretzky, T., Evers, A., Le, G. S., Alabi, R. O., Speck, N., Reiss, K., et al. (2015). The cytoplasmic domain of a disintegrin and metalloproteinase 10 (ADAM10) regulates its constitutive activity but is dispensable for stimulated ADAM10-dependent shedding. J. Biol. Chem. 290, 7416–7425. doi: 10.1074/jbc.M114.603753
Martin, L., Fluhrer, R., Reiss, K., Kremmer, E., Saftig, P., and Haass, C. (2008). Regulated intramembrane proteolysis of Bri2 (Itm2b) by ADAM10 and SPPL2a/SPPL2b. J. Biol. Chem. 283, 1644–1652. doi: 10.1074/jbc.M706661200
Matthews, V., Schuster, B., Schutze, S., Bussmeyer, I., Ludwig, A., Hundhausen, C., et al. (2003). Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J. Biol. Chem. 278, 38829–38839. doi: 10.1074/jbc.M210584200
McCulloch, D. R., Akl, P., Samaratunga, H., Herington, A. C., and Odorico, D. M. (2004). Expression of the disintegrin metalloprotease, ADAM-10, in prostate cancer and its regulation by dihydrotestosterone, insulin-like growth factor I, and epidermal growth factor in the prostate cancer cell model LNCaP. Clin. Cancer Res. 10, 314–323. doi: 10.1158/1078-0432.CCR-0846-3
Mezyk-Kopec, R., Bzowska, M., Stalinska, K., Chelmicki, T., Podkalicki, M., Jucha, J., et al. (2009). Identification of ADAM10 as a major TNF sheddase in ADAM17-deficient fibroblasts. Cytokine 46, 309–315. doi: 10.1016/j.cyto.2009.03.002
Miyake, S., Yanagisawa, Y., and Yuasa, Y. (2003). A novel EID-1 family member, EID-2, associates with histone deacetylases and inhibits muscle differentiation. J. Biol. Chem. 278, 17060–17065. doi: 10.1074/jbc.M212212200
Moss, M. L., Powell, G., Miller, M. A., Edwards, L., Qi, B., Sang, Q. X., et al. (2011). ADAM9 inhibition increases membrane activity of ADAM10 and controls α-secretase processing of amyloid precursor protein. J. Biol. Chem. 286, 40443–40451. doi: 10.1074/jbc.M111.280495
Müller, S. A., Scilabra, S. D., and Lichtenthaler, S. F. (2016). Proteomic substrate identification for membrane proteases in the brain. Front. Mol. Neurosci. 9:96. doi: 10.3389/fnmol.2016.00096
Nagara, Y., Hagiyama, M., Hatano, N., Futai, E., Suo, S., Takaoka, Y., et al. (2012). Tumor suppressor cell adhesion molecule 1 (CADM1) is cleaved by a disintegrin and metalloprotease 10 (ADAM10) and subsequently cleaved by γ-secretase complex. Biochem. Biophys. Res. Commun. 417, 462–467. doi: 10.1016/j.bbrc.2011.11.140
North, B. J., Marshall, B. L., Borra, M. T., Denu, J. M., and Verdin, E. (2003). The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444. doi: 10.1016/S1097-2765(03)00038-8
Oostra, B. A., and Willems, P. J. (1995). A fragile gene. Bioessays 17, 941–947. doi: 10.1002/bies.950171107
Ortiz, N. E., Nijhawan, R. I., and Weinberg, J. M. (2013). Acitretin. Dermatol. Ther. 26, 390–399. doi: 10.1111/dth.12086
Padro, C. J., Shawler, T. M., Gormley, M. G., and Sanders, V. M. (2013). Adrenergic regulation of IgE involves modulation of CD23 and ADAM10 expression on exosomes. J. Immunol. 191, 5383–5397. doi: 10.4049/jimmunol.1301019
Pan, D., and Rubin, G. M. (1997). Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell 90, 271–280. doi: 10.1016/S0092-8674(00)80335-9
Pan, Y., Han, C., Wang, C., Hu, G., Luo, C., Gan, X., et al. (2012). ADAM10 promotes pituitary adenoma cell migration by regulating cleavage of CD44 and L1. J. Mol. Endocrinol. 49, 21–33. doi: 10.1530/JME-11-0174
Panmanee, J., Nopparat, C., Chavanich, N., Shukla, M., Mukda, S., Song, W., et al. (2015). Melatonin regulates the transcription of βAPP-cleaving secretases mediated through melatonin receptors in human neuroblastoma SH-SY5Y cells. J. Pineal Res. 59, 308–320. doi: 10.1111/jpi.12260
Parkin, E., and Harris, B. (2009). A disintegrin and metalloproteinase (ADAM)-mediated ectodomain shedding of ADAM10. J. Neurochem. 108, 1464–1479. doi: 10.1111/j.1471-4159.2009.05907.x
Pasciuto, E., Ahmed, T., Wahle, T., Gardoni, F., D'Andrea, L., Pacini, L., et al. (2015). Dysregulated ADAM10-mediated processing of APP during a critical time window leads to synaptic deficits in fragile X syndrome. Neuron 87, 382–398. doi: 10.1016/j.neuron.2015.06.032
Plummer, S., Van Den Heuvel, C., Thornton, E., Corrigan, F., and Cappai, R. (2016). The neuroprotective properties of the amyloid precursor protein following traumatic brain injury. Aging Dis. 7, 163–179. doi: 10.14336/AD.2015.0907
Postina, R., Schroeder, A., Dewachter, I., Bohl, J., Schmitt, U., Kojro, E., et al. (2004). A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest. 113, 1456–1464. doi: 10.1172/JCI20864
Prinzen, C., Muller, U., Endres, K., Fahrenholz, F., and Postina, R. (2005). Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J. 19, 1522–1524. doi: 10.1096/fj.04-3619fje
Prox, J., Bernreuther, C., Altmeppen, H., Grendel, J., Glatzel, M., D'Hooge, R., et al. (2013). Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J. Neurosci. 33, 12915–12928, 12928a. doi: 10.1523/jneurosci.5910-12.2013
Pruessmeyer, J., and Ludwig, A. (2009). The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Biol. 20, 164–174. doi: 10.1016/j.semcdb.2008.09.005
Ray, B., Sokol, D. K., Maloney, B., and Lahiri, D. K. (2016). Finding novel distinctions between the sAPPα -mediated anabolic biochemical pathways in Autism Spectrum Disorder and Fragile X Syndrome plasma and brain tissue. Sci. Rep. 6:26052. doi: 10.1038/srep26052
Reinhardt, S. G. M., Stahlmann, C., Hartmann, T., Shudo, K., Tomita, T., and Endres, K. (2016). Rescue of Hypovitaminosis A induces non-amyloidogenic Amyloid Precursor Protein (APP) Processing. Curr. Alzheimer Res. 13, 1277–1289. doi: 10.2174/1567205013666160603002105
Reinhardt, S., Schuck, F., Grosgen, S., Riemenschneider, M., Hartmann, T., Postina, R., et al. (2014). Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer's disease. FASEB J. 28, 978–997. doi: 10.1096/fj.13-234864
Roebroek, A. J., Creemers, J. W., Ayoubi, T. A., and Van De Ven, W. J. (1994). Furin-mediated proprotein processing activity: involvement of negatively charged amino acid residues in the substrate binding region. Biochimie 76, 210–216. doi: 10.1016/0300-9084(94)90148-1
Sakry, D., Neitz, A., Singh, J., Frischknecht, R., Marongiu, D., Biname, F., et al. (2014). Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol. 12:e1001993. doi: 10.1371/journal.pbio.1001993
Saraceno, C., Marcello, E., Di Marino, D., Borroni, B., Claeysen, S., Perroy, J., et al. (2014). SAP97-mediated ADAM10 trafficking from Golgi outposts depends on PKC phosphorylation. Cell Death Dis. 5:e1547. doi: 10.1038/cddis.2014.492
Sarlak, G., Htoo, H. H., Hernandez, J. F., Iizasa, H., Checler, F., Konietzko, U., et al. (2016). Sox2 functionally interacts with βAPP, the βAPP intracellular domain and ADAM10 at a transcriptional level in human cells. Neuroscience 312, 153–164. doi: 10.1016/j.neuroscience.2015.11.022
Schiffmacher, A. T., Padmanabhan, R., Jhingory, S., and Taneyhill, L. A. (2014). Cadherin-6B is proteolytically processed during epithelial-to-mesenchymal transitions of the cranial neural crest. Mol. Biol. Cell 25, 41–54. doi: 10.1091/mbc.E13-08-0459
Schmitt, U., Hiemke, C., Fahrenholz, F., and Schroeder, A. (2006). Over-expression of two different forms of the α-secretase ADAM10 affects learning and memory in mice. Behav. Brain Res. 175, 278–284. doi: 10.1016/j.bbr.2006.08.030
Schuck, F., Wolf, D., Fellgiebel, A., and Endres, K. (2016). Increase of α-Secretase ADAM10 in Platelets Along Cognitively Healthy Aging. J. Alzheimers Dis. 50, 817–826. doi: 10.3233/JAD-150737
Schumacher, C., Wang, H., Honer, C., Ding, W., Koehn, J., Lawrence, Q., et al. (2000). The SCAN domain mediates selective oligomerization. J. Biol. Chem. 275, 17173–17179. doi: 10.1074/jbc.M000119200
Sennvik, K., Fastbom, J., Blomberg, M., Wahlund, L. O., Winblad, B., and Benedikz, E. (2000). Levels of α- and β-secretase cleaved amyloid precursor protein in the cerebrospinal fluid of Alzheimer's disease patients. Neurosci. Lett. 278, 169–172. doi: 10.1016/S0304-3940(99)00929-5
Shattil, S. J., O'toole, T., Eigenthaler, M., Thon, V., Williams, M., Babior, B. M., et al. (1995). β 3-endonexin, a novel polypeptide that interacts specifically with the cytoplasmic tail of the integrin β 3 subunit. J. Cell Biol. 131, 807–816. doi: 10.1083/jcb.131.3.807
Shukla, M., Htoo, H. H., Wintachai, P., Hernandez, J. F., Dubois, C., Postina, R., et al. (2015). Melatonin stimulates the nonamyloidogenic processing of βAPP through the positive transcriptional regulation of ADAM10 and ADAM17. J. Pineal Res. 58, 151–165. doi: 10.1111/jpi.12200
Stoeck, A., Keller, S., Riedle, S., Sanderson, M. P., Runz, S., Le, N. F., et al. (2006). A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem. J. 393, 609–618. doi: 10.1042/BJ20051013
Sun, C., Cheng, M. C., Qin, R., Liao, D. L., Chen, T. T., Koong, F. J., et al. (2011). Identification and functional characterization of rare mutations of the neuroligin-2 gene (NLGN2) associated with schizophrenia. Hum. Mol. Genet. 20, 3042–3051. doi: 10.1093/hmg/ddr208
Sung, P. S., Yeh, C. C., Wang, L. C., Hung, P. H., Muo, C. H., Sung, F. C., et al. (2017). Increased risk of dementia in patients with non-apnea sleep disorder. Curr. Alzheimer Res. 14, 309–316 doi: 10.2174/1567205013666161108104703
Suzuki, K., Hayashi, Y., Nakahara, S., Kumazaki, H., Prox, J., Horiuchi, K., et al. (2012). Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 76, 410–422. doi: 10.1016/j.neuron.2012.10.003
Tajes, M., Gutierrez-Cuesta, J., Ortuno-Sahagun, D., Camins, A., and Pallas, M. (2009). Anti-aging properties of melatonin in an in vitro murine senescence model: involvement of the sirtuin 1 pathway. J. Pineal Res. 47, 228–237. doi: 10.1111/j.1600-079X.2009.00706.x
Takebayashi, M., Hayashi, T., and Su, T. P. (2004). Sigma-1 receptors potentiate epidermal growth factor signaling towards neuritogenesis in PC12 cells: potential relation to lipid raft reconstitution. Synapse 53, 90–103. doi: 10.1002/syn.20041
Tang, S., Huang, G., Fan, W., Chen, Y., Ward, J. M., Xu, X., et al. (2014). SIRT1-mediated deacetylation of CRABPII regulates cellular retinoic acid signaling and modulates embryonic stem cell differentiation. Mol. Cell 55, 843–855. doi: 10.1016/j.molcel.2014.07.011
Taylor, C. J., Ireland, D. R., Ballagh, I., Bourne, K., Marechal, N. M., Turner, P. R., et al. (2008). Endogenous secreted amyloid precursor protein-α regulates hippocampal NMDA receptor function, long-term potentiation and spatial memory. Neurobiol. Dis. 31, 250–260. doi: 10.1016/j.nbd.2008.04.011
Taylor, R. C. (2016). Aging and the UPR(ER). Brain Res. 1648, 588–593. doi: 10.1016/j.brainres.2016.04.017
Telford, E. A., Moynihan, L. M., Markham, A. F., and Lench, N. J. (1999). Isolation and characterisation of a cDNA encoding the precursor for a novel member of the acyl-CoA dehydrogenase gene family. Biochim. Biophys. Acta 1446, 371–376. doi: 10.1016/S0167-4781(99)00102-5
Tippmann, F., Hundt, J., Schneider, A., Endres, K., and Fahrenholz, F. (2009). Up-regulation of the α-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 23, 1643–1654. doi: 10.1096/fj.08-121392
Tousseyn, T., Thathiah, A., Jorissen, E., Raemaekers, T., Konietzko, U., Reiss, K., et al. (2009). ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the γ-secretase. J. Biol. Chem. 284, 11738–11747. doi: 10.1074/jbc.M805894200
Tribioli, C., Robledo, R. F., and Lufkin, T. (2002). The murine fork head gene Foxn2 is expressed in craniofacial, limb, CNS and somitic tissues during embryogenesis. Mech. Dev. 118, 161–163. doi: 10.1016/S0925-4773(02)00220-4
Tsetsenis, T., Boucard, A. A., Araç, D., Brunger, A. T., and Sudhof, T. C. (2014). Direct visualization of trans-synaptic neurexin-neuroligin interactions during synapse formation. J. Neurosci. 34, 15083–15096. doi: 10.1523/JNEUROSCI.0348-14.2014
Tucher, J., Linke, D., Koudelka, T., Cassidy, L., Tredup, C., Wichert, R., et al. (2014). LC-MS based cleavage site profiling of the proteases ADAM10 and ADAM17 using proteome-derived peptide libraries. J. Proteome Res. 13, 2205–2214. doi: 10.1021/pr401135u
Tyan, S. H., Shih, A. Y., Walsh, J. J., Maruyama, H., Sarsoza, F., Ku, L., et al. (2012). Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell. Neurosci. 51, 43–52. doi: 10.1016/j.mcn.2012.07.009
Ushigusa, T., Ichinose, K., Sato, S., Michitsuji, T., Shimizu, T., Umeda, M., et al. (2016). Soluble α-klotho is a potential biomarker associated with neuropsychiatric systemic lupus erythematosus. Clin. Immunol. 165, 29–34. doi: 10.1016/j.clim.2016.03.001
Van Tetering, G., and Vooijs, M. (2011). Proteolytic cleavage of Notch: “HIT and RUN.” Curr. Mol. Med. 11, 255–269. doi: 10.2174/156652411795677972
Vincent, B., and Checler, F. (2012). α-Secretase in Alzheimer's disease and beyond: mechanistic, regulation and function in the shedding of membrane proteins. Curr. Alzheimer Res. 9, 140–156. doi: 10.2174/156720512799361646
Vincent, B., and Govitrapong, P. (2011). Activation of the α-secretase processing of AβPP as a therapeutic approach in Alzheimer's disease. J. Alzheimers Dis. 24(Suppl. 2), 75–94. doi: 10.3233/JAD-2011-110218
Wan, X. Z., Li, B., Li, Y. C., Yang, X. L., Zhang, W., Zhong, L., et al. (2012). Activation of NMDA receptors upregulates a disintegrin and metalloproteinase 10 via a Wnt/MAPK signaling pathway. J. Neurosci. 32, 3910–3916. doi: 10.1523/JNEUROSCI.3916-11.2012
Wang, C. Y., Zheng, W., Wang, T., Xie, J. W., Wang, S. L., Zhao, B. L., et al. (2011). Huperzine A activates Wnt/β-catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology 36, 1073–1089. doi: 10.1038/npp.2010.245
Warren, K. M., Reeves, T. M., and Phillips, L. L. (2012). MT5-MMP, ADAM-10, and N-cadherin act in concert to facilitate synapse reorganization after traumatic brain injury. J. Neurotrauma 29, 1922–1940. doi: 10.1089/neu.2012.2383
Weyer, S. W., Zagrebelsky, M., Herrmann, U., Hick, M., Ganss, L., Gobbert, J., et al. (2014). Comparative analysis of single and combined APP/APLP knockouts reveals reduced spine density in APP-KO mice that is prevented by APPsα expression. Acta Neuropathol. Commun. 2:36. doi: 10.1186/2051-5960-2-36
Wild-Bode, C., Fellerer, K., Kugler, J., Haass, C., and Capell, A. (2006). A basolateral sorting signal directs ADAM10 to adherens junctions and is required for its function in cell migration. J. Biol. Chem. 281, 23824–23829. doi: 10.1074/jbc.M601542200
Wisniewska, M. B. (2013). Physiological role of β-catenin/TCF signaling in neurons of the adult brain. Neurochem. Res. 38, 1144–1155. doi: 10.1007/s11064-013-0980-9
Wojtalewicz, N., Sadeqzadeh, E., Weiss, J. V., Tehrani, M. M., Klein-Scory, S., Hahn, S., et al. (2014). A soluble form of the giant cadherin Fat1 is released from pancreatic cancer cells by ADAM10 mediated ectodomain shedding. PLoS ONE 9:e90461. doi: 10.1371/journal.pone.0090461
Wolf, J., Waetzig, G. H., Chalaris, A., Reinheimer, T. M., Wege, H., Rose-John, S., et al. (2016). Different soluble forms of the interleukin-6 family signal transducer gp130 fine-tune the blockade of interleukin-6 trans-signaling. J. Biol. Chem. 291, 16186–16196. doi: 10.1074/jbc.M116.718551
Wolfsberg, T. G., Primakoff, P., Myles, D. G., and White, J. M. (1995a). ADAM, a novel family of membrane proteins containing A Disintegrin and Metalloprotease domain: multipotential functions in cell-cell and cell-matrix interactions. J. Cell Biol. 131, 275–278. doi: 10.1083/jcb.131.2.275
Wolfsberg, T. G., Straight, P. D., Gerena, R. L., Huovila, A. P., Primakoff, P., Myles, D. G., et al. (1995b). ADAM, a widely distributed and developmentally regulated gene family encoding membrane proteins with a disintegrin and metalloprotease domain. Dev. Biol. 169, 378–383. doi: 10.1006/dbio.1995.1152
Wolpert, F., Tritschler, I., Steinle, A., Weller, M., and Eisele, G. (2014). A disintegrin and metalloproteinases 10 and 17 modulate the immunogenicity of glioblastoma-initiating cells. Neuro Oncol. 16, 382–391. doi: 10.1093/neuonc/not232
Wong, E., Maretzky, T., Peleg, Y., Blobel, C. P., and Sagi, I. (2015). The functional maturation of A Disintegrin and Metalloproteinase (ADAM) 9, 10, and 17 requires processing at a newly identified Proprotein Convertase (PC) cleavage site. J. Biol. Chem. 290, 12135–12146. doi: 10.1074/jbc.M114.624072
Wu, X., Tang, H., Liu, G., Wang, H., Shu, J., and Sun, F. (2016). miR-448 suppressed gastric cancer proliferation and invasion by regulating ADAM10. Tumour Biol. 37, 10545–10551. doi: 10.1007/s13277-016-4942-0
Xie, J., Cha, S. K., An, S. W., Kuro, O., Birnbaumer, L., and Huang, C. L. (2012). Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 3:1238. doi: 10.1038/ncomms2240
Xiong, M., Jones, O. D., Peppercorn, K., Ohline, S. M., Tate, W. P., and Abraham, W. C. (2016). Secreted amyloid precursor protein-α can restore novel object location memory and hippocampal LTP in aged rats. Neurobiol. Learn. Mem. 138, 291–299. doi: 10.1016/j.nlm.2016.08.002
Xu, D., Sharma, C., and Hemler, M. E. (2009). Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 23, 3674–3681. doi: 10.1096/fj.09-133462
Yacoub, D., Benslimane, N., Al-Zoobi, L., Hassan, G., Nadiri, A., and Mourad, W. (2013). CD154 is released from T-cells by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM17 in a CD40 protein-dependent manner. J. Biol. Chem. 288, 36083–36093. doi: 10.1074/jbc.M113.506220
Yamazaki, K., Mizui, Y., Sagane, K., and Tanaka, I. (1997a). Assignment of a disintegrin and metalloproteinase domain 10 (Adam10) gene to mouse chromosome 9. Genomics 46, 528–529. doi: 10.1006/geno.1997.5043
Yamazaki, K., Mizui, Y., and Tanaka, I. (1997b). Radiation hybrid mapping of human ADAM10 gene to chromosome 15. Genomics 45, 457–459. doi: 10.1006/geno.1997.4910
Yavari, R., Adida, C., Bray-Ward, P., Brines, M., and Xu, T. (1998). Human metalloprotease-disintegrin Kuzbanian regulates sympathoadrenal cell fate in development and neoplasia. Hum. Mol. Genet. 7, 1161–1167. doi: 10.1093/hmg/7.7.1161
Yochum, G. S., and Ayer, D. E. (2002). Role for the mortality factors MORF4, MRGX, and MRG15 in transcriptional repression via associations with Pf1, mSin3A, and Transducin-Like Enhancer of Split. Mol. Cell. Biol. 22, 7868–7876. doi: 10.1128/MCB.22.22.7868-7876.2002
Zeng, Z., Kyaw, H., Gakenheimer, K. R., Augustus, M., Fan, P., Zhang, X., et al. (1997). Cloning, mapping, and tissue distribution of a human homologue of the mouse jerky gene product. Biochem. Biophys. Res. Commun. 236, 389–395. doi: 10.1006/bbrc.1997.6935
Zhang, J., Qin, X., Sun, Q., Guo, H., Wu, X., Xie, F., et al. (2015). Transcriptional control of PAX4-regulated miR-144/451 modulates metastasis by suppressing ADAMs expression. Oncogene 34, 3283–3295. doi: 10.1038/onc.2014.259
Zhang, T., and Kraus, W. L. (2010). SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim. Biophys. Acta 1804, 1666–1675. doi: 10.1016/j.bbapap.2009.10.022
Zhuang, J., Wei, Q., Lin, Z., and Zhou, C. (2015). Effects of ADAM10 deletion on Notch-1 signaling pathway and neuronal maintenance in adult mouse brain. Gene 555, 150–158. doi: 10.1016/j.gene.2014.10.056
Zilhão, N. R., Padmanabhuni, S. S., Pagliaroli, L., Barta, C., Smit, D. J., Cath, D., et al. (2015). Epigenome-wide association study of tic disorders. Twin Res. Hum. Genet. 18, 699–709. doi: 10.1017/thg.2015.72
Zingoni, A., Cecere, F., Vulpis, E., Fionda, C., Molfetta, R., Soriani, A., et al. (2015). Genotoxic stress induces senescence-associated ADAM10-dependent release of NKG2D MIC ligands in multiple myeloma cells. J. Immunol. 195, 736–748. doi: 10.4049/jimmunol.1402643
Keywords: ADAM10, aging, alpha-secretase, Alzheimer's disease, mouse models, promoter, transcription factors, spine
Citation: Endres K and Deller T (2017) Regulation of Alpha-Secretase ADAM10 In vitro and In vivo: Genetic, Epigenetic, and Protein-Based Mechanisms. Front. Mol. Neurosci. 10:56. doi: 10.3389/fnmol.2017.00056
Received: 21 December 2016; Accepted: 20 February 2017;
Published: 17 March 2017.
Edited by:
Ashok Hegde, Georgia College and State University, USAReviewed by:
Eckart D. Gundelfinger, Leibniz Institute for Neurobiology, GermanyBaojin Ding, University of Texas Southwestern Medical Center, USA
Copyright © 2017 Endres and Deller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristina Endres, a3Jpc3RpbmEuZW5kcmVzQHVuaW1lZGl6aW4tbWFpbnouZGU=