 Andrea Bednářová1,2*
Andrea Bednářová1,2* Marley Hanna3Isabella Durham4Tara VanCleave2Alexis England2Anathbandhu Chaudhuri5
Marley Hanna3Isabella Durham4Tara VanCleave2Alexis England2Anathbandhu Chaudhuri5 Natraj Krishnan2
Natraj Krishnan2- 1Department of Biochemistry and Physiology, Institute of Entomology, Biology Centre, Academy of Sciences, České Budějovice, Czechia
- 2Laboratory of Molecular Biology and Biochemistry, Department of Biochemistry, Molecular Biology, Entomology and Plant Pathology, Mississippi State University, Mississippi State, MS, USA
- 3Molecular Biosciences Program, Arkansas State University, Jonesboro, AR, USA
- 4Department of Wildlife, Fisheries and Aquaculture, Mississippi State University, Mississippi State, MS, USA
- 5Department of Natural Sciences, Stillman College, Tuscaloosa, AL, USA
Transfer RNAs (tRNAs) are key molecules participating in protein synthesis. To augment their functionality they undergo extensive post-transcriptional modifications and, as such, are subject to regulation at multiple levels including transcription, transcript processing, localization and ribonucleoside base modification. Post-transcriptional enzyme-catalyzed modification of tRNA occurs at a number of base and sugar positions and influences specific anticodon–codon interactions and regulates translation, its efficiency and fidelity. This phenomenon of nucleoside modification is most remarkable and results in a rich structural diversity of tRNA of which over 100 modified nucleosides have been characterized. Most often these hypermodified nucleosides are found in the wobble position of tRNAs, where they play a direct role in codon recognition as well as in maintaining translational efficiency and fidelity, etc. Several recent studies have pointed to a link between defects in tRNA modifications and human diseases including neurological disorders. Therefore, defects in tRNA modifications in humans need intensive characterization at the enzymatic and mechanistic level in order to pave the way to understand how lack of such modifications are associated with neurological disorders with the ultimate goal of gaining insights into therapeutic interventions.
Chemical modifications of RNA is a ubiquitous phenomenon in all kingdoms of life (Grosjean, 2009) and more than 100 chemically distinct modifications in cellular RNA have been identified to date. Initially, studies on such modifications were restricted to the ones that were the most abundant such as: peudouridine (ψ), N1-methyladenosine (m1A) and 2′-O-methylation (2′OMe) in ribosomal RNA (rRNA) and transfer RNA (tRNA), and 2′OMe, N6-methyladenosine (m6A) in messenger RNA (mRNA) and viral RNA (Desrosiers et al., 1974; Perry and Kelley, 1974; Dubin and Taylor, 1975). With technological advances in high throughput sequencing and mass spectrometry, additional less prevalent modifications on less abundant RNA species are being discovered (Nachtergaele and He, 2017; Song and Yi, 2017) and their biological significance uncovered.
tRNA Modifications
tRNA molecules translate the genetic code by recognizing cognate mRNA codons during the process of translation and protein synthesis. This ribosome-mediated interaction of the mRNA codons with the anticodon of the tRNA results in the discrimination of cognate vs. near-cognate and non-cognate codons (Ogle et al., 2001; Yusupov et al., 2001). The tRNAs undergo extensive post-transcriptional modifications during the process of maturation. In eukaryotes, around 100 different chemical modifications have been described occurring at different positions on the tRNA (Jackman and Alfonzo, 2013; tRNA modification database1 and MODOMICS2). Among all the modified nucleosides observed in tRNA, eight are present at the same position and the same subpopulation of tRNA isoacceptors in all domains of life, suggesting that tRNA modifications have highly conserved origins and functions (Björk, 1986; Björk et al., 2001). The largest diversity of the post-transcriptional nucleoside modifications occurs at the wobble position 34 in the anticodon or immediately 3′ adjacent to the anticodon triplet at position 37 (Rozenski et al., 1999; Dunin-Horkawicz et al., 2006).
Elusive Biological Functions of tRNA Modifications
A major limitation in the study of the biological significance and function of tRNA base modifications has been the lack of a phenotype associated with such modifications under defined settings. However, with technological advances, several modification defects and their phenotypes have now been described which provides insights into their biological roles (Hopper and Phizicky, 2003). In general, the modifications of bases at the wobble position in the anticodon or immediately adjacent to the anticodon triplet often impact the decoding abilities of tRNA’s by restriction and/or improvement of the codon-anticodon interactions, which may affect maintenance of the reading frame (Agris, 1991; Lim, 1994; Björk, 1995; Yokoyama and Nishimura, 1995; Johansson et al., 2008). Some modified bases present in the anticodon loop may be required for aminoacylation (Giege et al., 1998), while other modifications outside the anticodon loop, e.g., m1A58 of tRNAiMet, may be important for the structure or stability of the tRNA (Anderson et al., 1998; Calvo et al., 1999; Kadaba et al., 2004). Some of these tRNA base modifications may also regulate the speed and fidelity of translation (Hori, 2014; Manickam et al., 2016). There is also evidence that the loss of certain single modifications can be compensated by the presence of others, which suggests the existence of some redundancy in the system (Alexandrov et al., 2006). This indicates that modifications in tRNA affect translational function in very subtle ways and their main goal is to maintain tRNA functionality in the cell.
The cell utilizes the tRNA nucleoside modifications which are located in or near the anticodon loop to alter the repertoire of proteins that are preferentially synthesized. The affinity of codon-anticodon interaction can also regulate the biased subsets of mRNA, and so by adjusting the frequency of modifications in tRNA, the cells are capable of rapidly reacting to environmental challenges and thus focus their synthesis capacity on the proteins needed the most during these stress situations. For example, in Escherichia coli, stress resistance was significantly altered when heat shock genes were targeted by closely associated mutations (Krisko et al., 2014).
A feature of wobble modifications is their frequent tRNA specificity. It has been shown in yeast (Saccharomyces cerevisiae) that tRNA methyltransferase 4 (Trm4), which is multi-site specific, catalyzes 5-methylcytosine (m5C) formation in over 34 species of tRNA and this occurs at position 48 (between variable arm and T stem loop) most frequently (Czerwoniec et al., 2009). However, the only tRNA with m5C at the wobble position is tRNALeu(CAA) and this uniqueness has a role in the regulation of translation in response to oxidative stress in cells (Chan et al., 2012; Gu et al., 2014). This has given rise to the concept of tRNA modification tunable transcripts (MoTTs; Endres et al., 2015). This concept is defined as tRNA modifications that lead to: (a) transcripts that use specific degenerate codons and codon biases to encode critical stress response proteins; and (b) transcripts whose translation is influenced by changes in wobble base tRNA modification.
Defects in tRNA Modifications Associated with Neurological Disorders
Although the role of tRNA in the process of translation has been known since the late 1950s it is only in 1990, a mutation in tRNA was linked to a human disease (Kobayashi et al., 1990). The range of disorders now associated with defects in tRNA modifications range from metabolic (Type II Diabetes), respiratory defects and myopathies, mitochondrial disorders such as mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) and myoclonus epilepsy associated with ragged-red fibers (MERRF), to X-linked intellectual disability and familial dysautonomia (Duechler et al., 2016).
In S. cerevisiae, many of the tRNA modifications and the enzymes involved have been well characterized (Phizicky and Hopper, 2010). However, the homologs for many of these enzymes and their biological significance in humans have only recently started to emerge (Towns and Begley, 2012). This has led to a new appreciation of the link between tRNA modifications and human diseases (Torres et al., 2014; Hou et al., 2015).
The human brain is particularly sensitive to defects in tRNA modifications and several neurological disorders can be attributed to mutations in genes that affect the post-transcriptional modifications that occur in certain residues of the tRNA (Figure 1A; Table 1).
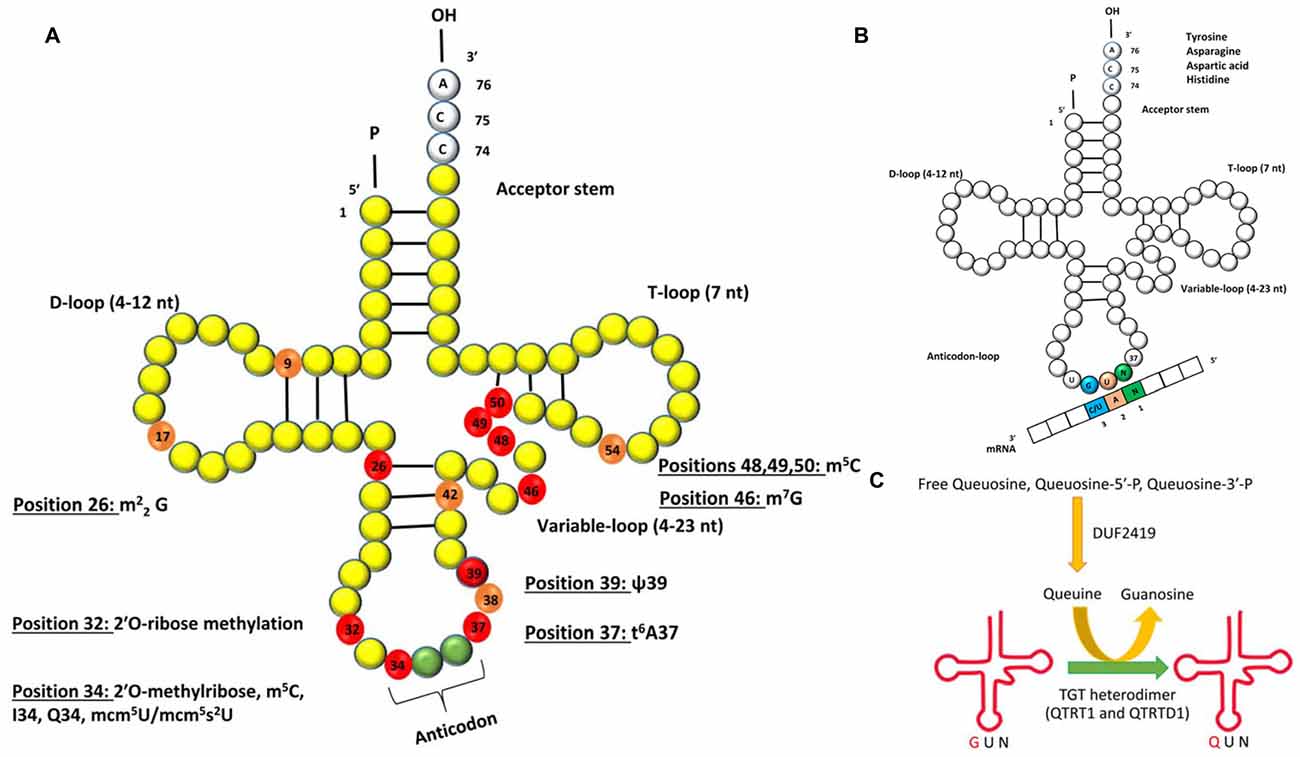
Figure 1. (A) Schematic representation of the secondary structure of transfer RNA (tRNA) with post-transcriptionally modified residues in light orange and red (numbered). The residues marked in red are of relevance to human neurological disorders. The abbreviations for the modifications are as follows: m22G: N2, N2-dimethyl guanosine; m5C: 5-methylcytosine; I34: inosine at position 34; Q34: queuosine at position 34, mcm5U: 5-methoxycarbonylmethyluridine; mcm5s2U: 5-methoxycarbonylmethyl-2-thiouridine; ψ: pseudouridine; t6A37: N6-threonyl-carbamoyl-adenosine at position 37. (B) Queuosine modification of tRNA. The G34U35N36 anticodon sequence of tRNA isoacceptors for amino acids tyrosine, asparagine, aspartic acid and histidine will base pair with a N1A2C/U3 codon of mRNA. G = guanine, U = uridine, A = adenine, N = any base. (C) Single step of Q modification of tRNAs in eukaryotes showing replacement of G34 by Q in the anticodon triplet by the enzyme heterodimeric TGT enzyme complex. Free Queuosine, Queuosine-5′-P, Queuosine-3′-P are obtained from degradation of Q-tRNA. Salvage of Q is accomplished by protein DUF2419.
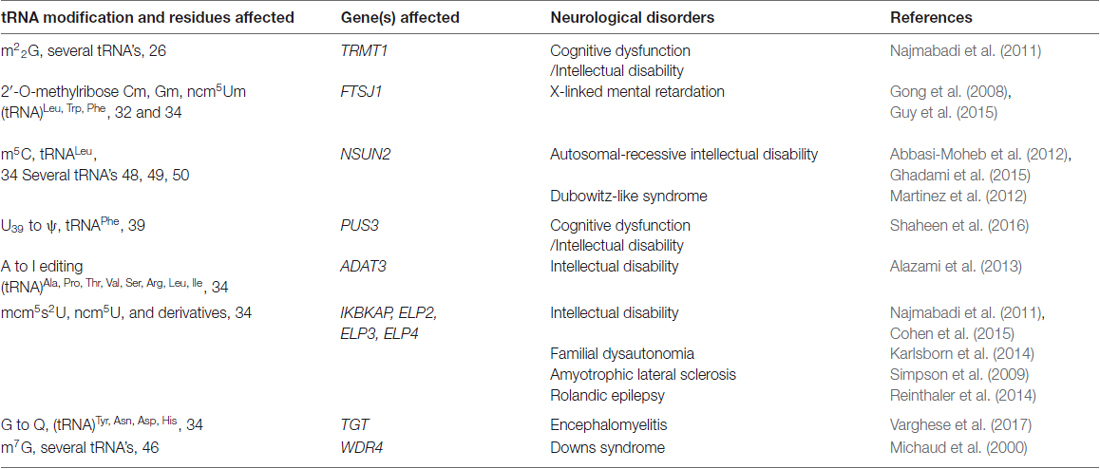
Table 1. Transfer RNA (tRNA) modifications, genes affected and their associated neurological disorders.
Cognitive Disorders and Intellectual Disability
Dimethylation of guanosines (m22G) occurs at position 26 of tRNAs and this is catalyzed by the human tRNA methyltransferase 1 (Liu and Straby, 2000; Figure 1A). Inactivation of this gene by a homozygous frameshift mutation is a biomarker for recessive cognitive disorders (Najmabadi et al., 2011). In human tRNAs so far characterized, pseudouridine is located at 13 different positions and different pseudouridylases (Pus) catalyze the modification at each of the site(s). It was recently reported that a nonsense mutation in PUS3 gene known for its role in isomerizing uracil to pseudouridine via Pus3 at position 39 in human tRNA was significantly reduced in patients with intellectual disability (Shaheen et al., 2016, Table 1).
Positions 32 and 34 on tRNALeu, tRNATrp and tRNAPhe are methylated (Figure 1A) by the gene encoding a methyltransferase—the FtsJ methyltransferase homolog 1 (FTSJ1), homologous to the yeast methyltransferase 7 (TRM7). Non-syndromic X linked mental retardation and intellectual disability are associated with mutations in this gene (Gong et al., 2008; Guy et al., 2015). At position 34 of tRNALeu and also at positions 48–50 on several tRNAs, m5C formation occurs (Brzezicha et al., 2006; Hussain et al., 2013; Khoddami and Cairns, 2013; Figure 1A). In higher eukaryotes, the only known m5C RNA methyltransferases are NSun2 and Dnmt2 and both enzymes are confirmed to target tRNA (Brzezicha et al., 2006). Deficiency in Nsun2 has been linked to intellectual disability (Abbasi-Moheb et al., 2012; Ghadami et al., 2015; Table 1). Interestingly, deletion of the ortholog of Nsun2 in fly results in severe short-term memory deficits (Abbasi-Moheb et al., 2012). Dubowitz-like syndrome associated with intellectual disability is also linked to mutations in Nsun2 (Martinez et al., 2012). Adenosine deaminase catalyze the conversion of adenosine-to-inosine at position 34 (I34) of tRNAs (Figure 1), and is encoded by heterodimeric adenosine deaminase (hetADAT). A single missense mutation in ADAT3 encoding for one of the subunits of hetADAT is present in families with individuals affected with intellectual disability (Alazami et al., 2013). 7-methylguanosine (m7G) modification of tRNA occuring in eukaryotes and bacteria, is nearly always found at position 46 (Figure 1), and is a modification that confers a positive charge to the base. The human WD repeat domain 4 (WDR4) is the closest homolog to the yeast TRM82 protein complex responsible for formation of m7G. WDR4 has been identified in a search for candidate genes of Down’s syndrome phenotypes (Michaud et al., 2000), but a direct association has not yet been demonstrated.
Neurodevelopmental Disabilities
Elongator is a highly conserved multi-subunit protein complex (ELP 1–6) that is essential to transcription elongation, histone acetylation as well as tRNA modification. The association between elongator complex and translational fidelity via regulation of tRNA modifications has gathered accumulating evidence in the last decade (Kojic and Wainwright, 2016). In eukaryotes, uridine at position 34 are modified to 5-carbamoyl-methyl-uridine (ncm5U), 5-methoxy-carbonyl-methyl-uridine (mcm5U), or 5-methoxy-carbonyl-methyl-2-thio-uridine (mcm52U) in the anticodons of tRNALys, tRNAGlu and tRNAGln. These modifications require the elongator complex (Esberg et al., 2006; Johansson et al., 2008; Bauer and Hermand, 2012). The Elongator mediated transfer of methyl-group to tRNA U34 involves a SAM-mediated mechanism coupled with an electron transfer from Kti11/Kti13, a cofactor complex (Boal et al., 2011; Kolaj-Robin et al., 2015). Recently, Glatt et al. (2016) showed the structural basis for such a modification of tRNA by ELP3. Moreover, it has been shown that a missense variant of the gene encoding for ELP2 is linked to neurodevelopmental disabilities (Najmabadi et al., 2011; Cohen et al., 2015).
Rolandic Epilepsy
Rare and deleterious variants of ELP4 have been associated with atypical rolandic epilepsy (RE; Reinthaler et al., 2014, Table 1). ELP4 has a putative role in neuronal migration and variants in the gene confer susceptibility to parts of the RE disease spectrum.
Motor Neuron Diseases
Allelic variants of ELP3 were associated with amyotrophic lateral sclerosis (ALS; a spontaneous, progressive motor neuron disease), in three human populations (Simpson et al., 2009), and, in the same study, two different loss of function mutations in ELP3 and genes important for neuronal communication and survival were identified in a mutagenesis screen in Drosophila. Moreover, dose-dependent motor axonal abnormalities were observed in zebrafish embryos when ELP3 protein levels were knocked down using antisense morpholinos. (Simpson et al., 2009). Mutations in ELPC1 and ELPC3 in Caenorhabditis elegans are associated with neurological and developmental dysfunctions (Chen et al., 2009). These studies implicate the importance of the elongator complex which are essential for tRNA modifications.
Interestingly, phenotypes associated with defective Elongator are due to lack of formation of mcm5s2U at position 34 of tRNAs (Phizicky and Hopper, 2010; Figure 1A). Mutation in the inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase complex-associated protein (IKBKAP) gene leads to reduced levels of the modified nucleoside mcm5s2U in tRNA and is associated with familial dysautonomia, a recessive neurodegenerative genetic disease (Karlsborn et al., 2014). Since Elongator influences acetylation of α-tubulin in neurons, there is a strong possibility that it may also play a significant role in neurological disorders such as Huntington’s disease, Alzheimer’s disease, Parkinson’s disease and ALS (Nguyen et al., 2010).
Neurodegeneration
The N6-threonyl-carbamoyl-adenosine (t6A) modification, is a complex modification of adenosine located at position 37 (t6A37) next to the anticodon stem loop of many tRNA’s that decode the ANN codons (Figure 1A). A recent report on a biosynthetic defect of the t6A molecule as a result of a mutation to kinase-associated endopeptidase (KAE1) gene of the highly conserved Kinase, Endopeptidase and Other Proteins of small Size (KEOPS) complex manifested in a neurodegenerative phenotype in two patients (Edvardson et al., 2017). This adds to the growing list of defects in cytoplasmic tRNA modification enzymes associated with neurological disorders.
Significance of Queuosine Modification of Trna and Its Role in Multiple Sclerosis and Other Neurological Disorders
Queuosine (Q) is among the most elaborate of the known tRNA modifications occurring in the wobble base (position 34) of tRNAs with G34U35N36 anticodons (tRNAGUN; where N = any base) that incorporate tyrosine, asparagine, aspartic acid or histidine amino acids (Katze et al., 1982; Nishimura, 1983; El Yacoubi et al., 2012; Figure 1B). This modification is widely distributed in most prokaryotic and eukaryotic phyla with the exception of yeast and mycoplasma (Katze et al., 1982). Structurally, Q comprises a 7-deazaguanosine core (Iwata-Reuyl, 2003). Q base is known to exist in four forms in biological systems: free nucleoside, free nucleotide and nucleoside incorporated into tRNA and free queuine base. tRNA-guanine transglycosylase (TGTase; EC 2.4.2.29) which was renamed later as tRNA-guanine ribosyltransferase (TGRase) by the Enzyme Commission, is the enzyme that catalyzes the formation of this modified tRNA (Farkas et al., 1984). Even though the Q modification is widely distributed in Bacteria and Eukarya, it is synthesized de novo by most bacteria, whereas all eukaryotes solely rely on salvage from environment to incorporate this complex modification. In eukaryotes, queuosine production is from the fully formed queuine base obtained from diet or microflora in intestinal, circulatory or membrane transport systems and incorporated post-transcriptionally by the eukaryotic analog of the prokaryotic TGTase. The completely modified base queuine is utilized by eukaryotic TGTase as a substrate and is irreversibly incorporated by a base-exchange reaction at position 34 of specific tRNAs (Kersten and Kersten, 1990; Figure 1C). Eukaryotic TGTase was shown to be a heterodimeric complex of 100–104 kDa protein, comprising a putative 60–66 kDa (QTRTD1) regulatory subunit and a 34–45 kDa (QTRT1) catalytic subunit (Morris et al., 1995; Slany and Müller, 1995; Deshpande et al., 1996). The former is probably involved in tRNA binding (Chen et al., 2010). The identification of DUF2419 as a potential member of the Q salvage protein family involved in Q-recycling was recently reported from phylogenetic analysis of plant, animal and fungal micronutrient Q salvage systems (Zallot et al., 2014). Queuosine modification system in mammalian cells is dependent on: (a) the uptake of queuine base into the cells by a queuine-specific membrane transport system (Elliott and Crane, 1990; Morris et al., 1996, 1999); (b) enzymatic incorporation of queuine into the first position of the anticodon loop of tRNA by TGTase (Morris et al., 1995, 1996, 1999; Slany and Müller, 1995); and (c) salvage of queuine by DUF2419 from queuosine 5′ monophosphate, free queuosine or queuosine-3′ monophosphate, resulting from tRNA degradation (Gündüz and Katze, 1982, 1984; Morris et al., 1999; Zallot et al., 2014).
The physiological significance of Q-tRNA and role of queuine has not been comprehensively established in eukaryotes. The absence of Q leads to no obvious developmental phenotypes in C. elegans (Gaur et al., 2007), in contrast, Q-deficient Drosophila are more sensitive to cadmium stresses (Siard et al., 1991). In mammals, the absence of both Q and tyrosine cause severe symptoms leading to death (Marks and Farkas, 1997). This suggests that queuine, the Q precursor is an essential micronutrient and also plays an important role in biosynthesis of Q to form Q-tRNA (Nishimura, 1983; Kersten, 1988). Queuine has been shown to promote the activity of antioxidant enzymes and thus may have a role in improving the antioxidant defense system (Pathak et al., 2008). Queuine has been shown to induce cell differentiation in cancerous cells (Chen and Wu, 1994). Lack of Q in first position of anticodons in tRNAs of Q-family is reported in various tumor cells (Dirheimer et al., 1995). The histopathological grade of malignancy is strongly correlated to amount of Q-deficient tRNA, and increased deficiency is observed in metastatic ovarian malignant tumors compared with primary malignancies (Baranowski et al., 1994). In short, Q modification of tRNA has been shown to play a divergent role in cellular machinery (Vinayak and Pathak, 2010).
Autoimmune diseases, including multiple sclerosis, are characterized by the rapid expansion of T cells directed to self-antigens. The potential medicinal relevance of targeting the hypomodification of Q-tRNA was recently demonstrated in the treatment of a chronic multiple sclerosis model—murine experimental autoimmune encephalomyelitis (Varghese et al., 2017). The administration of a de novo designed eukaryotic TGT substrate (NPPDAG) led to an unprecedented complete reversal of clinical symptoms and a dramatic reduction of markers associated with immune hyperactivation and neuronal damage. Thus, TGT is essential for the therapeutic effect, since animals deficient in TGT activity were refractory to therapy. The data suggests that exploitation of the eukaryotic TGT enzyme is a promising approach for the treatment of multiple sclerosis.
It has also been shown that deficiency in queuine in human HepG2 cells and mice made deficient in Q-tRNA, by disrupting the tRNA TGTase, have impaired ability to produce tyrosine from phenylalanine. This is because of a decrease in supply of the cofactor BH4 by increased oxidation of tetrahydrobiopterin leading to elevation of dihydrobiopterin (BH2) by an as yet undetermined mechanism (Rakovich et al., 2011). This has significant implications for dopamine (DA) biosynthesis which depends on a robust supply of BH4 (Nagatsu and Ichinose, 1996). It is thus interesting to speculate if such a deficiency of Q-tRNA could mimic Parkinsonian disorders.
All these defects in tRNA modifications and the consequent neurological disorders strongly indicate a functional association between aberrant tRNA modification and development of neurological disease. This raises a question of how many more defective proteins lead to lack of tRNA modifications and could these also lead to human pathologies, particularly neurological and/or neurodegenerative disorders?
Conclusions
Complex clinical pathologies arise as a result of mutations in tRNA genes and tRNA processing enzymes. Central to these tRNA modifications is the precise biological roles played by such changes or lack thereof. Once these roles are defined, strategies eventually will emerge to develop therapeutics directed towards correcting hypomodified tRNA or modulation of the expression of tRNA modification enzymes. In this context, recent advances in the field of tRNA biology, detection methods of modified tRNA, advances in genomics and proteomics together with studies on animal models are promising for a better understanding of the complex mechanisms in tRNA modifications leading to neuronal disorders and/or neuroprotection. Such advances will stimulate research in novel tRNA modification based therapeutics.
Author Contributions
AB, AC and NK conceived the content, AB and NK provided the ideas, supervised the work and wrote the critical review. MH, ID, TVC, AE and AC contributed to the content. All authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported in part by Undergraduate Research Scholars Program and the Office for Research and Development funding of the Mississippi State University (grant nos. URSP 2016 and ORED 2016) to NK. AB acknowledges Fellowship No. L200961701 from the Program of Support of Promising Human Resources, awarded by The Czech Academy of Sciences (Akademie Věd České Republiky).
Footnotes
References
Abbasi-Moheb, L., Mertel, S., Gonsior, M., Nouri-Vahid, L., Kahrizi, K., Cirak, S., et al. (2012). Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 90, 847–855. doi: 10.1016/j.ajhg.2012.03.021
Agris, P. F. (1991). Wobble position modified nucleosides evolved to select transfer RNA codon recognition: a modified-wobble hypothesis. Biochimie 73, 1345–1349. doi: 10.1016/0300-9084(91)90163-u
Alazami, A. M., Hijazi, H., Al-Dosari, M. S., Shaheen, R., Hashem, A., Aldahmesh, M. A., et al. (2013). Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J. Med. Genet. 50, 425–430. doi: 10.1136/jmedgenet-2012-101378
Alexandrov, A., Chernyakov, I., Gu, W., Hiley, S. L., Hughes, H. R., Grayhack, E. J., et al. (2006). Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21, 87–96. doi: 10.1016/j.molcel.2005.10.036
Anderson, J., Phan, L., Cuesta, R., Carlson, B. A., Pak, M., Asano, K., et al. (1998). The essential Gcd10p-Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev. 12, 3650–3662. doi: 10.1101/gad.12.23.3650
Baranowski, W., Dirheimer, G., Jankowiski, J. A., and Keith, G. (1994). Significant deficiency of queuine, a highly modified base purine base, in transfer ribonucleic acids from primary and metastatic ovarian malignant tumor in women. Cancer Res. 54, 4468–4471.
Bauer, F., and Hermand, D. (2012). A corordinated codon-dependent regulation of translation by Elongator. Cell Cycle 11, 4524–4529. doi: 10.4161/cc.22689
Björk, G. R. (1995). “Biosynthesis and function of modified nucleosides,” in tRNA: Structure, Biosynthesis and Function, eds D. Söll and U. L. RajBhandary (Washington, DC: ASM Press), 165–205.
Björk, G. R., Jacobsson, K., Nilsson, K., Johansson, M. J., Byström, A. S., and Persson, O. P. (2001). A primordial tRNA modification required for the evolution of life? EMBO J. 20, 231–239. doi: 10.1093/emboj/20.1.231
Boal, A. K., Grove, T. L., McLaughlin, M. I., Yennawar, N. H., Booker, S. J., and Rosenzweig, A. C. (2011). Structural basis for methyl transfer by a radical SAM enzyme. Science 332, 1089–1092. doi: 10.1126/science.1205358
Brzezicha, B., Schmidt, M., Makalowska, I., Jarmolowski, A., Pienkowska, J., and Szweykowska-Kulinska, Z. (2006). Identification of human tRNA: m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNA Leu (CAA). Nucleic Acids Res. 34, 6034–6043. doi: 10.1093/nar/gkl765
Calvo, O., Cuesta, R., Anderson, J., Gutierrez, N., Garcia-Barrio, M. T., Hinnebusch, A. G., et al. (1999). GCD14p, a repressor of GCN4 translation, cooperates with Gcd10p and Lhp1p in the maturation of initiator methionyl-tRNA in Saccharomyces cerevisiae. Mol. Cell Biol. 19, 4167–4181. doi: 10.1128/mcb.19.6.4167
Chan, C. T., Pang, Y. L., Deng, W., Babu, I. R., Dyavaiah, M., Begley, T. J., et al. (2012). Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 3:937. doi: 10.1038/ncomms1938
Chen, C., Tuck, S., and Byström, A. S. (2009). Defects in tRNA modification associated with neurological and developmental dysfunctions in Caenorhabditis elegans elongator mutants. PLoS Genet. 5:e1000561. doi: 10.1371/journal.pgen.1000561
Chen, Y.-C., Kelley, V. P., Stachura, S. V., and Garcia, G. A. (2010). Characterization of the human tRNA-guanine transglycosylase: confirmation of the heterodimeric subunit structure. RNA 16, 958–968. doi: 10.1261/rna.1997610
Chen, Y. L., and Wu, R. T. (1994). Altered queuine modification of tRNA involved in the differentiation of human K562 erythroleukemia cells in the presence of distinct differentiation inducers. Cancer Res. 54, 2192–2198.
Cohen, J. S., Srivastava, S., Farwell, K. D., Lu, H. M., Zeng, W., Lu, H., et al. (2015). ELP2 is a novel gene implicated in neurodevelopmental disabilities. Am. J. Med. Genet. A. 167, 1391–1395. doi: 10.1002/ajmg.a.36935
Czerwoniec, A., Dunin-Horkawicz, S., Purta, E., Kaminska, K. H., Kasprzak, J. M., Bujmicki, J. M., et al. (2009). MODOMICS: a database of RNA modification pathways. 2008 update. Nucleic Acids Res. 37, D118–D121. doi: 10.1093/nar/gkn710
Deshpande, K. L., Seubert, P. H., Tillman, D. M., Farkas, W. R., and Katze, J. R. (1996). Cloning and characterization of cDNA encoding the rabbit tRNA-guanine transglycosylase 60-kDa subunit. Arch. Biochem. Biophys. 326, 1–7. doi: 10.1006/abbi.1996.0039
Desrosiers, R., Friderici, K., and Rottman, F. (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. U S A 71, 3971–3975. doi: 10.1073/pnas.71.10.3971
Dirheimer, G., Baranowski, W., and Keith, G. (1995). Variation in tRNA modifications, particularly of their queuine content in higher eukaryotes. Its relation to malignancy grading. Biochimie 77, 99–103. doi: 10.1016/0300-9084(96)88111-9
Dubin, D. T., and Taylor, R. H. (1975). The methylation state of polyA–containing–messenger RNA from cultured hamster cells. Nucleic Acids Res. 2, 1653–1658. doi: 10.1093/nar/2.10.1653
Duechler, M., Leszczyńlska, G., Sochacka, E., and Nawrot, B. (2016). Nucleoside modfications in the regulation of gene expression: focus on tRNA. Cell Mol. Life Sci. 73, 3075–3095. doi: 10.1007/s00018-016-2217-y
Dunin-Horkawicz, S., Czerwoniec, A., Gajda, M. J., Feder, M., Grosjean, H., and Bujnicki, J. M. (2006). MODOMICS: a database of RNA modification pathways. Nucleic Acids Res. 34, D145–D149. doi: 10.1093/nar/gkj084
Edvardson, S., Prunetti, L., Arraf, A., Haas, D., Bacusmo, J. M., Hu, J. F., et al. (2017). tRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and renal tubulopathy. Eur. J. Hum. Genet. 25, 545–551. doi: 10.1038/ejhg.2017.30
El Yacoubi, B., Bailly, M., and de Crécy-Lagard, V. (2012). Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 46, 69–95. doi: 10.1146/annurev-genet-110711-155641
Elliott, M. S., and Crane, D. L. (1990). Protein kinase C modulation of queuine uptake in cultured human fibroblasts. Biochem. Biophys. Res. Commun. 171, 393–400. doi: 10.1016/0006-291x(90)91406-i
Endres, L., Dedon, P. C., and Begley, T. J. (2015). Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 12, 603–614. doi: 10.1080/15476286.2015.1031947
Esberg, A., Huang, B., Johansson, M. J. O., and Byström, A. S. (2006). Elevated levels of two tRNA species bypass the requirement of elongator complex in transcription and exocytosis. Mol. Cell 24, 139–148. doi: 10.1016/j.molcel.2006.07.031
Farkas, W. R., Jacobson, K. B., and Katzeöm, J. R. (1984). Substrate and inhibitor specificity of tRNA-guanine ribosyltransferase. Biochim. Biophys. Acta 781, 64–75. doi: 10.1016/0167-4781(84)90124-6
Gaur, R., Björk, G., Tuck, S., and Varshney, U. (2007). Diet dependent depletion of queuosine in tRNAs in Caenorhabditis elegans does not lead to a developmental block. J. Biosci. 32, 747–754. doi: 10.1007/s12038-007-0074-4
Ghadami, S., Mohammadi, H. M., Malbin, J., Masoodifard, M., Sarhaddi, A. B., Tavakkoly-Bazzaz, J., et al. (2015). Frequencies of six (five novel) STR markers linked to TUSC3 (MRT7) or NSUN2 (MRT5) genes used for homozygosity mapping of recessive intellectual disability. Clin. Lab. 61, 925–932. doi: 10.7754/clin.lab.2015.150101
Giege, R., Sissler, M., and Florentz, C. (1998). Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 26, 5017–5035. doi: 10.1093/nar/26.22.5017
Glatt, S., Zabel, R., Kolaj-Robin, O., Onuma, O. F., Baudin, F., Graziadei, A., et al. (2016). Structural basis for tRNA modification by Elp3 from Dehalococcoides mccartyi. Nat. Struct. Mol. Biol. 23, 794–802. doi: 10.1038/nsmb.3265
Gong, P., Li, J., Dai, L., Zhang, K., Zheng, Z., Gao, X., et al. (2008). Genetic variations in FTSJ1 influence cognitive ability in young males in the Chinese Han population. J. Neurogenet. 22, 277–287. doi: 10.1080/01677060802337299
Grosjean, H. (2009). “Nucleic acids are not boring long polymers of only four types of nucleotides: a guided tour,” in DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution, ed. H. Grosjean (Austin, TX: Landes Bioscience), 1–18.
Gu, C., Begley, T. J., and Dedon, P. C. (2014). tRNA modifications regulate translation during cellular stress. FEBS Lett. 588, 4287–4296. doi: 10.1016/j.febslet.2014.09.038
Gündüz, U., and Katze, J. R. (1982). Salvage of the nucleic acid base queuine from queuine-containing tRNA by animal cells. Biochem. Biophys. Res. Commun. 109, 159–167. doi: 10.1016/0006-291x(82)91579-0
Gündüz, U., and Katze, J. R. (1984). Queuine salvage in mammalian cells. Evidence that queuine is generated from queuosine 5′-phosphate. J. Biol. Chem. 259, 1110–1113.
Guy, M. P., Shaw, M., Weiner, C. L., Hobson, L., Stark, Z., Rose, K., et al. (2015). Defects in tRNA anticodon loop 2′-O-methylation are implicated in nonsyndromic X-linked intellectual disability due to mutations in FTSJ1. Hum. Mutat. 36, 1176–1187. doi: 10.1002/humu.22897
Hopper, A. K., and Phizicky, E. M. (2003). tRNA transfers to the limelight. Genes Dev. 17, 162–180. doi: 10.1101/gad.1049103
Hori, H. (2014). Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 5:144. doi: 10.3389/fgene.2014.00144
Hou, Y.-M., Gamper, H., and Yang, W. (2015). Post-transcriptional modifications to tRNA—a response to the genetic code degeneracy. RNA 21, 642–644. doi: 10.1261/rna.049825.115
Hussain, S., Sajini, A. A., Blanco, S., Dietmann, S., Lombard, P., Sugimoto, Y., et al. (2013). NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 4, 255–261. doi: 10.1016/j.celrep.2013.06.029
Iwata-Reuyl, D. (2003). Biosynthesis of the 7-deazaguanosine hypermodified nucleosides of transfer RNA. Bioorg. Chem. 31, 24–43. doi: 10.1016/s0045-2068(02)00513-8
Jackman, J. E., and Alfonzo, J. D. (2013). Transfer RNA modifications: nature’s combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA 4, 35–48. doi: 10.1002/wrna.1144
Johansson, M. J., Esberg, A., Huang, B., Björk, G. R., and Bystrom, A. S. (2008). Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell Biol. 28, 3301–3312. doi: 10.1128/MCB.01542-07
Kadaba, S., Krueger, A., Trice, T., Krecic, A. M., Hinnebusch, A. G., and Anderson, J. (2004). Nuclear surveillance and degradation of hypo-modified initiator tRNAMet in S. cerevisiae. Genes Dev. 18, 1227–1240. doi: 10.1101/gad.1183804
Karlsborn, T., Tükenmez, H., Chen, C., and Byström, A. S. (2014). Familial dysautonomia (FD) patients have reduced levels of the modified wobble nucleoside mcm(5)s(2)U in tRNA. Biochem. Biophys. Res. Commun. 454, 441–445. doi: 10.1016/j.bbrc.2014.10.116
Katze, J. R., Basile, B., and McCloskey, J. A. (1982). Queuine, a modified base incorporated posttranscriptionally into eukaryotic transfer RNA: wide distribution in nature. Science 216, 55–56. doi: 10.1126/science.7063869
Kersten, H. (1988). The nutrient factor queuine: biosynthesis, occurrence in transfer RNA and function. Biofactors 1, 27–29. doi: 10.1016/b978-012417762-8.50088-0
Kersten, H., and Kersten, W. (1990). “Biosynthesis and function of queuine and queosine tRNAs,” in Chromatography and Modification of Nucleosides. Part B. Biological Roles and Function of Modification, eds C. W. Gehrke and K. C. T. Kuo (Amsterdam: Elsevier), B69–B108.
Khoddami, V., and Cairns, B. R. (2013). Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 31, 458–464. doi: 10.1038/nbt.2566
Kobayashi, Y., Momoi, M. Y., Tominaga, K., Momoi, T., Nihei, K., Yanagisawa, M., et al. (1990). A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem. Biophys. Res. Commun. 173, 816–822. doi: 10.1016/S0006-291X(05)80860-5
Kojic, M., and Wainwright, B. (2016). The many faces of elongator in neurodevelopment and disease. Front. Mol. Neurosci. 9:115. doi: 10.3389/fnmol.2016.00115
Kolaj-Robin, O., McEwen, A. G., Cavarelli, J., and Séraphin, B. (2015). Structure of the Elongator cofactor complex Kti11/Kti13 provides insight into the role of Kti13 in Elongator-dependent tRNA modification. FEBS J. 282, 819–833. doi: 10.1111/febs.13199
Krisko, A., Copic, T., Gabaldón, T., Lehner, B., and Supek, F. (2014). Inferring gene function from evolutionary change in signatures of translation efficiency. Genome Biol. 15:R44. doi: 10.1186/gb-2014-15-3-r44
Lim, V. I. (1994). Analysis of action of wobble nucleoside modifications on codon-anticodon pairing within the ribosome. J. Mol. Biol 240, 8–19. doi: 10.1006/jmbi.1994.1413
Liu, J., and Straby, K. B. (2000). The human tRNA(m22G26)dimethyltransferase: functional expression and characterization of a cloned hTRM1 gene. Nucleic Acids Res. 28, 3445–3451. doi: 10.1093/nar/28.18.3445
Manickam, N., Joshi, K., Bhatt, M. J., and Farabaugh, P. J. (2016). Effects of tRNA modification on translational accuracy depend on intrinsic codon-anticodon strength. Nucleic Acids Res. 44, 1871–1881. doi: 10.1093/nar/gkv1506
Marks, T., and Farkas, W. R. (1997). Effects of a diet deficient in tyrosine and queuine on germfree mice. Biochem. Biophys. Res. Commun. 230, 233–237. doi: 10.1006/bbrc.1996.5768
Martinez, F. J., Lee, J. H., Lee, J. E., Blanco, S., Nickerson, E., Gabriel, S., et al. (2012). Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med. Genet. 49, 380–385. doi: 10.1136/jmedgenet-2011-100686
Michaud, J., Kudoh, J., Berry, A., Bonne-Tamir, B., Lalioti, M. D., Rossier, C., et al. (2000). Isolation and characterization of a human chromosome 21q22.3 gene (WDR4) and its mouse homologue that code for a WD-repeat protein. Genomics. 68, 71–79. doi: 10.1006/geno.2000.6258
Morris, R. C., Brooks, B. J., Eriotou, P., Kelly, D. F., Sagar, S., Hart, K. L., et al. (1995). Activation of transfer RNA-guanine ribosyltransferase by protein kinase C. Nucleic Acids Res. 23, 2492–2498. doi: 10.1093/nar/23.13.2492
Morris, R. C., Brooks, B. J., Hart, K. L., and Elliott, M. S. (1996). Modulation of queuine uptake and incorporation into tRNA by protein kinase C and protein phosphatase. Biochim. Biophys. Acta. 1311, 124–132. doi: 10.1016/0167-4889(95)00184-0
Morris, R. C., Galicia, M. C., Clase, K. L., and Elliott, M. S. (1999). Determination of queuosine modification system deficiencies in cultured human cells. Mol. Genet. Metab. 68, 56–67. doi: 10.1006/mgme.1999.2889
Nachtergaele, S., and He, C. (2017). The emerging biology of RNA post-transcriptional modifications. RNA Biol. 14, 156–163. doi: 10.1080/15476286.2016.1267096
Nagatsu, T., and Ichinose, H. (1996). GTP cyclohydrolase I gene, tetrahydrobiopterin and tyrosine hydroxylase gene: their relations to dystonia and parkinsonism. Neurochem. Res. 21, 245–250. doi: 10.1007/bf02529141
Najmabadi, H., Hu, H., Garshasbi, M., Zemojtel, T., Abedini, S. S., Chen, W., et al. (2011). Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478, 57–63. doi: 10.1038/nature10423
Nguyen, L., Humbert, S., Saudou, F., and Chariot, A. (2010). Elongator—an emerging role in neurological disorders. Trends Mol. Med. 16, 1–6. doi: 10.1016/j.molmed.2009.11.002
Nishimura, S. (1983). Structure, biosynthesis, and function of queuosine in transfer RNA. Prog. Nucleic Acid Res. Mol. Biol. 28, 49–73. doi: 10.1016/s0079-6603(08)60082-3
Ogle, J. M., Brodersen, D. E., Clemons, W. M. Jr., Tarry, M. J., Carter, A. P., and Ramakrishnan, V. (2001). Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292, 897–902. doi: 10.1126/science.1060612
Pathak, C., Jaiswal, Y. K., and Vinayak, M. (2008). Modified base queuine promotes cellular antioxidant defense system in cancer. Biosci. Rep. 28, 73–81. doi: 10.1042/BSR20070011
Perry, R. P., and Kelley, D. E. (1974). Existence of methylated messenger RNA in mouse L cells. Cell 1, 37–42. doi: 10.1016/0092-8674(74)90153-6
Phizicky, E. M., and Hopper, A. K. (2010). tRNA biology charges to the front. Genes Dev. 24, 1832–1860. doi: 10.1101/gad.1956510
Rakovich, T., Boland, C., Bernstein, I., Chikwana, V. M., Iwata-Reuyl, D., and Kelly, V. P. (2011). Queuosine deficiency in eukaryotes compromises tyrosine production through increased tetrahydrobiopterin oxidation. J. Biol. Chem. 286, 19354–19363. doi: 10.1074/jbc.M111.219576
Reinthaler, E. M., Lal, D., Jurkowski, W., Feucht, M., Steinböck, H., Gruber-Sedlmayr, U., et al. (2014). Analysis of ELP4, SRPX2, and interacting genes in typical and atypical rolandic epilepsy. Epilepsia 55, e89–e93. doi: 10.1111/epi.12712
Rozenski, J., Crain, P. F., and McCloskey, J. A. (1999). The RNA modification database: 1999 update. Nucleic Acids Res. 27, 196–197. doi: 10.1093/nar/27.1.196
Shaheen, R., Han, L., Faqeih, E., Ewida, N., Alobeid, E., Pizicky, E. M., et al. (2016). A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum. Genet. 135, 707–713. doi: 10.1007/s00439-016-1665-7
Siard, T. J., Jacobson, K. B., and Farkas, W. R. (1991). Queuine metabolism and cadmium toxicity in Drosophila melanogaster. Biofactors 3, 41–47.
Simpson, C. L., Lemmens, R., Miskiewicz, K., Broom, W. J., Hansen, V. K., van Vught, P. W., et al. (2009). Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum. Mol. Genet. 18, 472–481. doi: 10.1093/hmg/ddn375
Slany, R. K., and Müller, S. O. (1995). tRNA-guanine transglycosylase from bovine liver. Purification of the enzyme to homogeneity and biochemical characterization. Eur. J. Biochem. 230, 221–228. doi: 10.1111/j.1432-1033.1995.tb20554.x
Song, J., and Yi, C. (2017). Chemical modifications to RNA: a new layer of gene expression regulation. ACS Chem. Biol. 12, 316–325. doi: 10.1021/acschembio.6b00960
Torres, A. G., Batlle, E., and Ribas de Pouplana, L. (2014). Role of tRNA modifications in human diseases. Trends Mol. Med. 20, 306–314. doi: 10.1016/j.molmed.2014.01.008
Towns, W. L., and Begley, T. J. (2012). Transfer RNA methytransferases and their corresponding modifications in budding yeast and humans: activities, predications, and potential roles in human health. DNA Cell Biol. 31, 434–454. doi: 10.1089/dna.2011.1437
Varghese, S., Cotter, M., Chevot, F., Fergus, C., Cunningham, C., Mills, K. H., et al. (2017). In vivo modification of tRNA with an artificial nucleobase leads to full disease remission in an animal model of multiple sclerosis. Nucleic Acids Res. 45, 2029–2030. doi: 10.1093/nar/gkw847
Vinayak, M., and Pathak, C. (2010). Queuosine modification of tRNA: its divergent role in cellular machinery. Biosci. Rep. 30, 135–148. doi: 10.1042/BSR20090057
Yokoyama, S., and Nishimura, S. (1995). “Modified nucleosides and codon recognition,” in tRNA: Structure, Biosynthesis, and Function, eds D. Söll and U. L. RajBhandary (Washington, DC: ASM Press), 207–223.
Yusupov, M. M., Yusupova, G. Z., Baucom, A., Lieberman, K., Earnest, T. N., Cate, J. H., et al. (2001). Crystal structure of the ribosome at 5.5 A resolution. Science 292, 883–896. doi: 10.1126/science.1060089
Keywords: transfer RNA modifications, modified nucleosides, neurological disease, queuosine, Q-tRNA
Citation: Bednářová A, Hanna M, Durham I, VanCleave T, England A, Chaudhuri A and Krishnan N (2017) Lost in Translation: Defects in Transfer RNA Modifications and Neurological Disorders. Front. Mol. Neurosci. 10:135. doi: 10.3389/fnmol.2017.00135
Received: 27 February 2017; Accepted: 20 April 2017;
Published: 09 May 2017.
Edited by:
Hermona Soreq, Hebrew University of Jerusalem, IsraelReviewed by:
Baojin Ding, University of Texas Southwestern Medical Center, USAHong Qing, Beijing Institute of Technology, China
Copyright © 2017 Bednářová, Hanna, Durham, VanCleave, England, Chaudhuri and Krishnan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Bednářová, YS5iZWRuYXJvdmFAZW50dS5jYXMuY3o=