 José J. Miguel-Hidalgo
José J. Miguel-Hidalgo- Department of Psychiatry and Human Behavior, University of Mississippi Medical Center, Jackson, MS, United States
Postmortem studies reveal structural and molecular alterations of astrocytes and oligodendrocytes in both the gray and white matter (GM and WM) of the prefrontal cortex (PFC) in human subjects with chronic alcohol abuse or dependence. These glial cellular changes appear to parallel and may largely explain structural and functional alterations detected using neuroimaging techniques in subjects with alcohol use disorders (AUDs). Moreover, due to the crucial roles of astrocytes and oligodendrocytes in neurotransmission and signal conduction, these cells are very likely major players in the molecular mechanisms underpinning alcoholism-related connectivity disturbances between the PFC and relevant interconnecting brain regions. The glia-mediated etiology of alcohol-related brain damage is likely multifactorial since metabolic, hormonal, hepatic and hemodynamic factors as well as direct actions of ethanol or its metabolites have the potential to disrupt distinct aspects of glial neurobiology. Studies in animal models of alcoholism and postmortem human brains have identified astrocyte markers altered in response to significant exposures to ethanol or during alcohol withdrawal, such as gap-junction proteins, glutamate transporters or enzymes related to glutamate and gamma-aminobutyric acid (GABA) metabolism. Changes in these proteins and their regulatory pathways would not only cause GM neuronal dysfunction, but also disturbances in the ability of WM axons to convey impulses. In addition, alcoholism alters the expression of astrocyte and myelin proteins and of oligodendrocyte transcription factors important for the maintenance and plasticity of myelin sheaths in WM and GM. These changes are concomitant with epigenetic DNA and histone modifications as well as alterations in regulatory microRNAs (miRNAs) that likely cause profound disturbances of gene expression and protein translation. Knowledge is also available about interactions between astrocytes and oligodendrocytes not only at the Nodes of Ranvier (NR), but also in gap junction-based astrocyte-oligodendrocyte contacts and other forms of cell-to-cell communication now understood to be critical for the maintenance and formation of myelin. Close interactions between astrocytes and oligodendrocytes also suggest that therapies for alcoholism based on a specific glial cell type pathology will require a better understanding of molecular interactions between different cell types, as well as considering the possibility of using combined molecular approaches for more effective therapies.
Introduction
Alcohol use disorder (AUD) is defined in the webpage of the National Institute of Alcohol Abuse and Alcoholism as a chronic relapsing brain disease involving compulsive alcohol use, loss of control over alcohol intake, and a negative emotional state when not using. This disorder results in severe behavioral, neurological and other medical pathologies that eventually depend on disturbances of cellular function and metabolism (Abrahao et al., 2017). In the nervous system, these disturbances cause abnormal exchanges of information between brain centers, including cortical and subcortical regions that control the intake of rewarding substances such as alcohol itself, or regions involved in emotional and cognitive regulation (Moselhy et al., 2001; Schulte et al., 2010). In many AUD patients, alcohol-induced brain alterations are also reflected in structural damage in the gray and white matter (GM and WM; Rosenbloom et al., 2003; Zahr and Pfefferbaum, 2017).
It is well known that a large proportion of alcoholics, despite serious behavioral and emotional pathology, show only minor or inconspicuous neurological deficits of the kind mentioned above, and thus these subjects are dubbed as “uncomplicated” alcoholics. In these subjects, AUDs do not necessarily result in catastrophic or global loss of brain tissue of neuronal or glial cell numbers (Jensen and Pakkenberg, 1993). However, application of conventional MRI imaging techniques has shown that even in these AUD subjects atrophy of the cerebral GM volume at relatively younger ages, while later in life the reduction in volume extends to the WM and cerebral ventricles (Pfefferbaum et al., 1988, 1992). This progressive decline in GM volume was first described as affecting the brain globally, but further neuroimaging investigations have demonstrated that significant age-dependent volume reduction is particularly noticeable in the prefrontal cortex (PFC; Pfefferbaum et al., 1997). Furthermore, while only macroscopic volumetric variations can be safely assessed in brain tissue with conventional MRI, diffusion tensor imaging (DTI) studies, a more recent development of MRI with increased resolution to visualize fiber bundle structure in WM, have shown that WM volume changes parallel structural alterations in specific WM axon bundles connecting PFC to brain circuits involved in reward and emotion regulation (Schulte et al., 2010) or that, even when macrostructural changes are not patent, there could be significant microstructural disturbances of axon bundles (Pfefferbaum and Sullivan, 2002). MRI- and DTI-based detection of volumetric and fiber bundle alterations most likely betray disturbed signal processing in specific brain regions such as the PFC and the hippocampus and anomalous connectivity between those brain regions. In fact, uncomplicated subjects are not entirely free of neuropathological alterations at the cellular level either because they show regionally selective degeneration of pyramidal neurons or their dendrites in some brain regions such as the PFC (Kril et al., 1997).
Role of Glial Cells in AUDs
Since neurons are the main conveyors of information between brain regions, much attention has been placed on the role of neuronal pathology in the various disorders caused by AUDs in human subjects and experimental animals (Abrahao et al., 2017). However, it is well-known that maintenance, survival and normal activity of neurons are fully dependent on the interaction with several types of glial cells (Barres, 2008). These cells assist critically in the support of neurotransmission, propagation of action potentials, survival (of neurons and other glial cells), supply of metabolites, brain injury repair, neuroprotection and synapse formation and removal. In the central nervous system, the main classes of glial cells are astrocytes, oligodendrocytes, NG2 cells and microglia. Each of these cell types identifies mainly with one or two of the support functions mentioned (for instance, astrocytes with neurotransmitter reuptake and metabolic support, oligodendrocytes with myelin formation around axons, microglia with responses to injury and repair), but some functions are performed cooperatively by two or more cell types. Thus, if prolonged alcohol exposure damages glial cells or disrupts their activity, grave disturbances of neuronal function are to be expected. Conversely, due to the existence of neuronal signals that regulate glial physiology (Haydon, 2000; Simons and Trajkovic, 2006), indirect actions of alcohol, mediated by neuronal pathology, are to be expected on the structural and functional integrity of glial cells. Many studies have shown that alcohol exposure in vivo and in vitro profoundly affects the development, morphology, physiology and gene expression of astrocytes, oligodendrocytes, microglia and NG2 cells. The effects of AUDs on oligodendrocytes were some of the first to receive attention from clinicians and investigators because alcoholism leads to severe neurological and cognitive disorders associated with myelin pathology (Sun et al., 1979; Gallucci et al., 1989; Harper, 2009). Later developments have also shown that the development, physiology, gene expression and morphology of astrocytes are profoundly affected by alcohol abuse (Kennedy and Mukerji, 1986; Renau-Piqueras et al., 1989; Cullen and Halliday, 1994; Franke, 1995).
Many reviews and original research articles have dealt with specific morphology, molecular markers and functions that characterize classes and types of astrocytes (Rajan and McKay, 1998; Laming et al., 2000; Nedergaard et al., 2003; Oberheim et al., 2006; Takano et al., 2006; Zhang and Barres, 2010; Lovatt et al., 2012; Parpura et al., 2012; Kettenmann et al., 2013), oligodendrocytes (Cahoy et al., 2008; Wegner, 2008; Emery and Lu, 2015; Fitzpatrick et al., 2015; Simons and Nave, 2015; Purger et al., 2016; Snaidero and Simons, 2017) and the other glial cell classes in the brain of vertebrates, including the human brain. In this review, I will concentrate on astrocytes and oligodendrocytes in the brain WM and cortical GM mainly because they are the primary glial cell types implicated in the integration (astrocytes) and propagation (oligodendrocytes) of neural signals originating from and arriving in the cortex and the most extensively studied regarding AUDs. I refer to the cited reviews and original articles for more detailed information on normal development, structure, molecular biology and physiology of astrocytes, oligodendrocytes and related cell subtypes. Likewise, the present review is about the glial molecular pathology in AUDs in the context of postmortem and neuroimaging studies, and does not include a detailed discussion of glial pathology in fetal alcohol spectrum disorders (FASD), although occasional reference to FASD is made to illustrate some general points about the pathology of astrocytes or oligodendrocytes in alcohol abuse disorders.
Astrocytes
Diversity of Astrocytes
Since the earliest structural and developmental studies astrocytes and related cells have been classified into several subtypes according to their localization and morphology (Reichenbach and Wolburg, 2012). In fact, astrocytes that reside in specific neural niches tend to substantially differ from those in other niches. For instance, astrocytes in WM are considered to be mostly of the fibrillary type while most astrocytes in GM display a distinctive morphology and are classified as protoplasmic astrocytes (Reichenbach and Wolburg, 2012). In turn, in some WM tracts, such as the optic nerve, astrocytes are further subdivided into type 1 and type 2 astrocytes, while GM astrocytes in contact with the meningeal pia mater or those adjacent to the ventricular surfaces are also morphologically distinct from the protoplasmic astrocytes (Reichenbach and Wolburg, 2012). In the cerebellum, astrocytes take on a distinctive morphology that parallels the structure of Purkinje cell’s dendrites and are identified as Bergman glia, while retinal astrocytes radially span across the retinal layers and are called Mueller cells (Kettenmann et al., 2013).
In recent years, it has become evident that the morphological variety is matched by an even richer molecular differentiation of astrocytes (Verkhratsky and Nedergaard, 2018), and that even astrocytes considered as a morphologically homogenous type (Cui et al., 2001; Oberheim et al., 2006; Khakh and Sofroniew, 2015; Hu et al., 2016), for example, cerebral cortex and WM astrocytes, can be further subdivided according to their molecular markers or their ability to divide (Zhang and Barres, 2010). These subtype-specific molecular markers ultimately betray a significant degree of physiological differentiation among astrocytes as they adapt to specific neural niches and functions.
Roles of Astrocytes in Gray and White Matter
The differential features of astrocytes in the cortical GM reflect a multitude of regulatory roles in support of cortical neuronal function (Verkhratsky and Nedergaard, 2018). These roles can be significantly disturbed either by direct actions of ethanol on receptors, transporters or metabolic enzymes of astrocytes, or through indirect actions mediated by the effects of ethanol on neurotransmission and neuronal metabolism (Verkhratsky and Parpura, 2010; Adermark and Bowers, 2016). Prominent among the critical roles of astrocytes are the exchange with blood vessels of energy-rich metabolites to support neuronal metabolism, the buffering of extracellular ions exchanged during synaptic neurotransmission/propagation of action potentials, the reuptake of synaptically-released glutamate and gamma-aminobutyric acid (GABA), the recycling of these and other transmitters for reutilization or metabolism, the release of small molecular cofactors, such as serine or glycine required for activation of the N-methyl-D-aspartate-type (NMDA-type) glutamate receptors, the release of gliotransmitters or cytokine-like molecules to communicate with other neural cells, and the expression of immune-like activities within the nervous system (Cali et al., 2008). This variety of neurophysiological roles is made possible by abundant expression of specific proteins and molecules and their associated intracellular pathways, which are in many cases exclusive to or predominantly expressed in astrocytes (Verkhratsky and Nedergaard, 2018).
Many of their characteristic intracellular pathways are found in all astrocyte subtypes. However, a division of labor also exists among astrocytes such that, for instance, some astrocytes express high levels of excitatory amino acid transporter 1 (EAAT1), other astrocytes are richer in EAAT2, or others, such as Bergmann glia express EAAT5. Most WM astrocytes and those adjacent to the pia mater express high amounts of the cytoskeletal protein glial fibrillary acidic protein (GFAP). In contrast, astrocytes in the middle layers of the cortex, while rich in glutamate transporters have very low levels of GFAP, even if dramatic increases of GFAP can occur in astrocytes after brain injury, toxicity or ischemia.
In addition to their distinctive morphology, WM astrocytes constitutionally express high levels of GFAP (levels that are significantly lower in many GM astrocytes). Even if neuron to neuron synapses are very low in WM, WM astrocytes express glutamate and glutamine transporters (Banner et al., 2002; Miguel-Hidalgo et al., 2010; Roberts et al., 2014). Unlike most GM astrocytes, WM astrocytes and astrocytes in myelinated parts of the GM, are in close contact with oligodendrocyte processes and the outermost layer of myelin, and often form gap junctions with oligodendrocyte cell membranes to allow for direct communication between the astrocyte and oligodendrocyte cytoplasms (Nualart-Marti et al., 2013). The molecular composition of these junctions is connexin subtype-specific, because connexin 43 (Cx43) and Cx30, which are contained on the astrocyte side of the gap junction, are respectively matched with Cx47 and Cx32 contained in the oligodendrocyte cell membrane to form Cx43–Cx47 and Cx30–Cx32 gap junctions (Nagy and Rash, 2000; Orthmann-Murphy et al., 2008). The importance of these astrocyte-oligodendrocyte gap junctions to establishment of myelin has been confirmed in recent studies showing that absence or downregulation of astrocytic Cx43 and Cx30 (or oligodendrocyte Cx47 and Cx32) in transgenic mice models results in major disruption of myelin formation and maintenance, as well as behavioral deficits (Lutz et al., 2009; Magnotti et al., 2011; Wasseff and Scherer, 2011). In addition, the end-feet of some processes of astrocytes reach to most of the nodes of Ranvier (NR) in the CNS. Despite the close and frequent association of these processes with NR, their function is still unknown, although some researchers have proposed that astrocytes and their perinodal processes play an important role in potassium buffering or in the stabilization and organization of myelin formation at the nodes (Black and Waxman, 1988; Kalsi et al., 2004; Serwanski et al., 2017).
End-feet of astrocytic processes also closely abut the basal lamina surrounding endothelial cells of small blood vessels. These contacts around blood vessels are important in the regulation of water movements from the blood circulation and the maintenance and function of the brain blood barrier (Paemeleire, 2002; Simard et al., 2003; Abbott et al., 2006).
Molecular Pathology of Astrocytes in Alcohol Use Disorders
Alcohol-Related Neuropathology of Astrocytes
Chronic alcohol exposure induces atrophic features in glial cells, including astrocytes and their precursors in GM (Miguel-Hidalgo and Rajkowska, 2003). Furthermore, alcoholism associates with downregulation of astrocyte specific genes, particularly in subjects with hepatic pathology (Liu et al., 2006). The atrophic changes in astrocytes appear to preferentially occur in certain brain regions, such as PFC and the hippocampus, where alcohol exposure results in reduced numbers or packing density in the general population of glial cells and in astrocytes (as identified by morphological features or GFAP labeling; Korbo, 1999; Miguel-Hidalgo et al., 2002, 2006). In addition, the expression of astrocyte-related genes is significantly downregulated (Liu et al., 2006). In some subjects, chronic AUDs are associated with persistent nutritional deficiencies or hepatic damage, resulting in serious neurological disorders such as Wernicke’s encephalopathy, hepatic encephalopathy or the demyelinating disorders central pontine myelinolysis and Marchiafava-Bignami syndrome (de la Monte and Kril, 2014; Verkhratsky et al., 2014). In these cases, severe neurological symptoms correlate with macroscopically apparent, substantial degeneration of GM or WM in cerebellum, thalamus, mammillary bodies or cerebral cortex (Phillips et al., 1990; Kril and Harper, 2012). Interestingly, although neurons and oligodendrocytes are considered major targets of nutritional and metabolic disturbances, such as thiamine deficiency, astrocytes can be also critically affected by the same disturbances (Hazell, 2009). In addition, acute ethanol exposure of cultured astrocytes causes extensive gene expression changes that resemble the heat shock response (Pignataro et al., 2013).
As it could be expected, the response of astrocytes to pathogenic alcohol exposure is not limited to alterations in their number, morphology or development, but affects many of the roles played by astrocytes in the nervous system. These roles include the regulation of neuroinflammatory processes, calcium signaling, balance of excitatory and inhibitory neurotransmission, water balance/cell volume regulation, as well as the regulation of dopamine-dependent behavioral processes in brain reward circuits (Adermark and Bowers, 2016). In addition, acute or prolonged exposure of astrocytes to alcohol may substantially modify the efficacy of connections between brain areas by disturbing the maintenance of myelin (Hazell, 2009) and the buffering of ions in the proximity of nodes Ranvier. Altering ion buffering and the osmotic regulation that results from astrocyte interactions with oligodendrocytes around NR causes abnormal action potential propagation in WM and myelinated portions of GM (Gankam Kengne et al., 2011).
The alcoholism-related changes in astrocyte numbers and their markers may be due, at least partly, to direct inhibitory effects of ethanol on astrocyte proliferation or turnover. Both in astrocytes cultured from neonatal rodents (Davies and Cox, 1991; Guerri and Renau-Piqueras, 1997; Guerri, 1998) or from postmortem human brain tissue autopsies (Kane et al., 1996) ethanol exposure causes substantial inhibition of astrocyte proliferation and synthesis of DNA and protein, including reduced expression of the major astrocyte marker GFAP. After prolonged exposure, this inhibition may lead to decreased astrocyte numbers, as well as impaired ability to perform the critical functions enumerated earlier in this review. The responses of astrocytes to chronic alcohol, although mostly inhibitory, may also lead to secondary activation of gliosis-like astrocyte responses when AUDs prolong sufficiently into senescence (Miguel-Hidalgo and Rajkowska, 2003; Miguel-Hidalgo, 2009). In approaching late-age, accumulation of ethanol-related deficits in astrocyte structure and function may contribute to neuronal degeneration, and this degeneration would trigger a secondary activation of astrocyte reactivity or gliosis, which would be reflected in increased GFAP production and other gliotic changes, although it is still unclear how other markers of astrocytes respond to aging-associated neuronal degeneration.
Alcohol Effects on Glutamate Receptors and Astrocyte Components of the Cycle for Release and Reuptake of Glutamate
NMDA-Type Glutamate Receptors in AUDs
In patients with AUDs there is evidence, some of it controversial, of alcohol-related dysfunction in some aspects of glutamatergic neurotransmission such as an increase in the expression of NMDA-type glutamate receptors and a decrease in GABA receptors, particularly in chronic alcoholism (Davis and Wu, 2001). Ethanol acts antagonistically at NMDA receptors by reducing their activation by glutamate. In animal models, chronic alcohol consumption increases expression of the NR2A, NR2B and NR1 subunits of NMDA receptors in the neocortex and the hippocampus (Gass and Olive, 2008), and the increases would appear to explain the neuronal hyperexcitability found in animal models after alcohol withdrawal. Although less consistently, human studies in chronic alcoholics also show increases in ligand binding to NMDA receptors (Tsai, 1998; Tsai et al., 1998) mainly in the PFC, but not in other cortical or brain regions such as the cingulate cortex, hippocampus or cerebellar vermis (Freund and Anderson, 1996, 1999). mRNA levels for the same NMDAR subunits as above do not differ in uncomplicated alcoholics as compared to controls, but are reduced in cirrhotic alcoholics (Ridge et al., 2008).
Involvement of Astrocyte Glutamate Transporters and Glutamine Synthetase in AUDs
Regardless of how persistent glutamate receptor changes are in human chronic alcoholism, the antagonism of NMDA receptors and the potentiation of GABA receptors by ethanol are likely to alter glutamate release and the concentrations of glutamate in the extracellular space. These neurotransmitter alterations would cause the involvement of glutamate transporters, glutamate converting enzymes and glutamate receptors, which are highly expressed by astrocytes. In the cerebral cortex, the glutamate transporters of astrocytes are crucial to the synaptic reuptake of glutamate. In addition, astrocytic GABA transporters make a very important contribution to GABA reuptake. These transporters, together with the enzyme glutamine synthetase (GS) in astrocytes, are essential components of the cycle that terminates the actions of released glutamate or GABA at many synapses, and allows for further recycling and synaptic release of glutamate and GABA. Thus, some studies have determined the cortical expression of astrocytic glutamate transporter as well as GS mRNA and protein in AUD subjects (Miguel-Hidalgo et al., 2010; Ayers-Ringler et al., 2016). Interestingly, these studies have not detected significant variations in the protein levels of either EAAT1, EAAT2 or GS in alcohol dependence, and some studies in vitro actually show that ethanol exposure may cause an increase in the rate of glutamate transport per astrocyte (Smith and Zsigo, 1996; Smith, 1997; Zink et al., 2004), although in some rat brain areas such the nucleus accumbens there appears to be an ethanol-induced decrease in glutamate transport that still is not linked to a reduced expression of glutamate transporters (Melendez et al., 2005).
It is possible that homeostatic mechanisms result in unaltered or increased expression of glutamate receptors or transporters in many brain regions of human alcoholics. However, it must be kept in mind that the unchanged transporter levels we found in the orbitofrontal cortex in alcohol-dependent subjects occurred only in uncomplicated alcoholism, while in subjects with comorbid major depression there were reduced levels of glutamate transporters and GS, raising the possibility that the severity of alcohol-related pathology resulting in depression involves a decrease in the astrocytic components of the glutamate cycle (Miguel-Hidalgo et al., 2010). The loss of glutamate transporters in subjects with Wernicke’s encephalopathy, often associated with severe cases of chronic alcoholism (Hazell et al., 2010) may be considered further evidence for the view that the severity of pathology in some brain areas may depend on profound changes in the glutamate cycle components of astrocytes.
Animal studies have shown that different regimes of chronic alcohol intake, withdrawal, and reinstatement have diverse effects on the expression of glutamate cycle components. This diversity raises the possibility that different trajectories in the timing, length and frequency of withdrawal periods, or the comorbidity with other disorders cause ample variation in glutamate-related mRNA and protein markers in human alcoholics at the time of death. This variety would prevent finding statistically significant differences in AUD patients as compared to non-alcoholic subjects. In fact, as mentioned above, we found that among subjects with alcohol-dependence only those with a comorbid diagnosis of depression had significantly lower levels of glutamate transporters EAAT1 (and a tendency for lower EAAT2 transporters) than controls, while in alcoholics without other psychiatric diagnoses (Miguel-Hidalgo et al., 2010) there was no change, suggesting that uncomplicated AUDs may involve compensatory regulation of glutamate transport. In line with this suggestion, some animal models of alcoholism show increase in astrocytic glutamate transporter levels (Wu et al., 2011), even if alcohol itself can disrupt the function of those transporters (Mulholland et al., 2009). Interestingly, restoration of EAAT2-based glutamate transport with ceftriaxone actually reduces alcohol drinking (Lee et al., 2013), while the reduction of astrocytic EAAT1 resulting from deletion of the circadian period gene (Per2) in mice is accompanied by increased alcohol intake (Spanagel et al., 2005).
Astrocyte-Released NMDA Receptor Co-Agonists in AUDs
Regulation of glutamatergic transmission at NMDA receptors is also dependent on glycine, which acts as co-agonist at those receptors. At the same binding-site, astrocyte-released D-serine is also an active co-agonist. Both glycine and D-serine have a permissive role in NMDA receptor activation when binding to the glycine-binding site. Ethanol can compete with D-serine for the occupancy of that site, although the dependence of behavioral sensitivity on ethanol binding is related to the exact subunit composition of the NMDA receptors and thus differs across brain regions (Tsai, 1998). On the other hand, reduced affinity for glycine at the glycine site is positively associated with attenuated sensitivity to the behavioral effects of alcohol (Kiefer et al., 2003) in mice, while tolerance to partial agonists of that site appears to develop in alcohol dependent subjects (Krystal et al., 2011), pointing to an important role of astrocyte-produced D-serine in the effects of ethanol in chronic AUD patients.
Astrocyte Thrombospondin in AUD-Related Synaptic Alterations
Alcohol-related neuronal dysfunction may also depend on regressive changes at synaptic contacts that result from intermittent or prolonged alcohol exposure. Those changes may be due, at least partly, to impairments in the ability of astrocytes to provide factors involved in synapse maintenance such as thrombospondins and their receptors (Ullian et al., 2004). In animal models, alcohol exposure results in significant reduction of thrombospondin release that can persist for 24 days, in parallel with disturbed matching of presynaptic and postsynaptic structures (Risher et al., 2015). Hepatic damage caused by alcoholism in some subjects may also result in synaptic dysfunction indirectly mediated by astrocytes, because increased ammonia levels that follow liver dysfunction diminish thrombospondin secretion by astrocytes and reduce the levels of synaptic proteins (Jayakumar et al., 2014). Alcohol exposure during early or prenatal stages of development, and maybe later too, may cause persistent changes in synapse formation involving thrombospondin as well (Trindade et al., 2016). These changes are further accompanied by marked reductions in astrocyte-secreted extracellular matrix (ECM) proteins, such as laminin or heparan-sulfate proteoglycan (Lasek, 2016; Trindade et al., 2016). Thus, repeated alcohol exposure at different stages of prenatal and postnatal development would result in abnormal regulation of astrocyte-derived factors involved in synaptogenesis.
Astrocyte Processes at the Blood-Brain Barrier and the Involvement of Aquaporins in AUD-Related Neuropathology
Astrocytes processes abut the basal lamina surrounding the endothelial cells of small blood vessels, where they contribute to blood-brain barrier (BBB) maintenance (Prat et al., 2001). In addition, those processes are essential to the exchange of metabolic substrates with the blood circulation, and to the regulation of blood flow (Koehler et al., 2009). Chronic alcoholism disturbs components of the BBB at endothelial cells (Haorah et al., 2005; Rubio-Araiz et al., 2017), impairing to varying degrees the exchange of energy and neurotrophic metabolites such as glucose, that directly impinge on neuronal and glial function (Abdul Muneer et al., 2011a). In addition, alcohol causes direct inhibitory effects on glucose uptake by astrocytes processes (Abdul Muneer et al., 2011b).
Effects of alcohol mediated by astrocytes very likely involve changes in aquaporins as well (Kong et al., 2013), in particular aquaporin 4 (AQ4), a membrane protein highly expressed in astrocytes processes at the BBB that allows effective passage of water through the cell membrane (Badaut et al., 2002; Rajkowska et al., 2013). Repeated alcohol intake in binging rat models results in significant increases in aquaporin, astrocyte swelling (linked to brain edema) and activation of neuroinflammatory cascades (Collins and Neafsey, 2012; Collins et al., 2013). Anti-inflammatory treatments can prevent the effects of the AQ4 elevation that is concomitant with increases in neuroinflammatory markers (Tajuddin et al., 2014). On the other hand, serious consequences of alcoholism, such as the loss of myelin in central pontine myelinolysis, might be associated with reduction in astrocytic aquaporins, although more research seems to be needed to fully ascertain this possibility (Popescu et al., 2013).
In summary, multifaceted actions of ethanol on astrocytic markers involved in the formation and regulation of the BBB, amplified by their reciprocal interactions with the BBB, may be important determinants of the cellular and functional pathology of alcoholism.
Oligodendrocytes
Role of Oligodendrocytes in Gray and White Matter
The main function of oligodendrocytes is the formation and maintenance of myelin, which consists of tightly piled layers of oligodendrocyte cell membrane wrapped around the axons of neurons (Baumann and Pham-Dinh, 2001; Butt, 2005). The layers of myelin act as insulation against the dissipation of ionic gradients, allowing for fast, self-regenerating saltatory conduction of action potentials between consecutive NR along the axons to reach synaptic terminals, and thus for efficient exchange of neural impulses between brain centers (Nave and Werner, 2014). Despite this unity of purpose, most oligodendrocytes and their myelin sheaths are finely tuned and sensitive to the physiological and gene expression changes in the neurons whose axons they wrap and the astrocytes that surround them (Simons and Trajkovic, 2006; Nave and Werner, 2014). Conversely, oligodendrocytes produce signals and growth factors that support the axons they enwrap and the neurons and astrocytes in their vicinity (Du and Dreyfus, 2002; Nave and Trapp, 2008; Simons and Nave, 2015). Some oligodendrocytes in GM regions are intimately associated with neuronal cell bodies, but their functions remain so far unclear.
Molecular Pathology of Oligodendrocytes in Alcohol Use Disorders
Myelin Components
Postmortem cellular and in vivo neuroimaging studies in human subjects have revealed that prolonged and repeated alcohol intake results in various degrees of damage or adaptations in the myelin that sheaths axons in the WM and GM, as well as in the oligodendrocytes that form the myelin. In some subjects, macroscopic damage to the WM caused by alcoholism is apparent, and can be identified with loss of myelin both in neuroimaging and postmortem histochemical studies. WM and GM damage produces different neurological syndromes that can be distinguished according to the specific anatomical location and the nature of the neurological disturbances. Myelin disorders such as Marchiafava-Bignami disease, Wernicke-Korsakoff syndrome, hepatic encephalopathy, central pontine myelinolysis or alcohol cerebellar degeneration involve myelin losses in WM and GM of cortical and subcortical brain regions (Zahr and Pfefferbaum, 2017). In these disorders, BBB disruption or nutritional deficits, such as thiamine deficiency, alone or most likely in interaction with direct effects of alcohol on oligodendrocytes, are considered main culprits for myelin disturbances in chronic alcoholism (Lewohl et al., 2005; Alexander-Kaufman et al., 2007; He et al., 2007). On the other hand, given the frequent co-abuse of ethanol and tobacco, part of the deleterious effects of ethanol on myelin proteins might result from an interaction of ethanol with specific components of tobacco, such as nicotine-specific nitrosamine ketone (NNK; Tong et al., 2015; Zabala et al., 2015; Papp-Peka et al., 2017). However, even in subjects without such obvious neurological and anatomical complications (the “uncomplicated cases”), the expression of myelin and oligodendrocyte-related proteins, or their mRNAs, can be significantly altered in various brain regions, being particularly prominent in the PFC (Mayfield et al., 2002; Alexander-Kaufman et al., 2006; Liu et al., 2006), which are reflected in low levels of the main structural myelin proteins such as myelin basic protein (MBP) and possibly proteolipid protein (PLP), their companions, myelin associated-glycoprotein (MAG) and oligodendrocyte-myelin glycoprotein (Omgp; Okamoto et al., 2006), and related transcription factors (Miguel-Hidalgo et al., 2017). Reduced expression of major myelin proteins such as MBP has been detected in models of prenatal alcohol exposure (Ozer et al., 2000; Bichenkov and Ellingson, 2001), while in vitro studies have shown that the effects of ethanol on the expression of myelin components could be mediated by direct regulation of PKC-like enzymes rather than by altering their expression (Bichenkov and Ellingson, 2002).
Beside disturbing the expression of myelin proteins, alcohol can induce oligodendrocyte apoptosis during prenatal development in primates such as the macaque (Creeley et al., 2013), while during early postnatal mice development (roughly equivalent to the human third gestation trimester) there is a dramatic reduction in differentiated oligodendrocytes and oligodendrocyte progenitor cells in the corpus callosum. These cell populations recover after ceasing alcohol exposure, but deficits in MBP levels or in the structure of corpus callosum fibers persist in young adult mice (Saito et al., 2016; Newville et al., 2017). In adult mice, chronic intermittent ethanol (CIE) exposure causes also significant reduction in the levels of MBP, PLP and 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase) in several brain regions (Samantaray et al., 2015). These degenerative effects appear to be mediated by the calcium-activated protease calpain, because calpain inhibitors prevent the reductions of myelin-related proteins in the mice CIE model (Samantaray et al., 2015).
Gene Expression in Oligodendrocytes
In human postmortem brain, gene-expression studies of chronic alcoholism that involve the aggregate of GM and WM from frontal cerebral regions, have found significantly decreased mRNAs of myelin-related proteins in chronic alcoholics (Lewohl et al., 2000, 2001; Liu et al., 2006; Farris et al., 2015a). Low myelin-related mRNAs include those of the major structural myelin proteins MBP, PLP, myelin oligodendrocyte glycoprotein (MOG) and MAG. These reductions are particularly significant in chronic alcoholics with cirrhosis as compared to non-alcoholic controls or to non-cirrhotic alcoholics (Lewohl et al., 2005), suggesting that nutritional deficiencies or metabolic toxicity, possibly interacting with direct ethanol effects, strongly deplete the expression of myelin components in alcoholics, at least as assessed in studies that include GM in the probed tissue. In contrast, in a recent study from our laboratory, we used samples only from the WM adjacent to cortical area 47 (orbitofrontal cortex) in chronic alcoholics, and observed that mRNA levels for myelin proteins PLP, MAG and MOG and other oligodendrocyte markers were markedly lower than in controls, although the levels of MBP mRNA were not changed (Miguel-Hidalgo et al., 2017). Since cirrhosis had not been diagnosed in most subjects of our study, these results suggest that the effects of prolonged alcohol abuse in some regions of WM may occur without cirrhosis and be different from those when GM is included. However, the degree of hepatic compromise was not exactly quantified in our samples, so that it was not yet possible to separately assess indirect from direct effects of ethanol on the strong decreases in the expression of mRNAs for some myelin proteins. Despite the highly localized nature of our WM study (all samples were from WM adjacent to Brodmann’s cortical area 47), it is also important to note that factors related to hepatic pathology, GM contamination of samples, or RNA quality may influence changes detected in previous studies of myelin-related markers in alcoholism (Sutherland et al., 2014) and thus replication studies with well-defined locales, or in other brain areas, appear to be necessary to draw the right conclusions regarding effects of chronic alcoholism on gene expression of glial cells in the human brain. A recent study in the hippocampus of human chronic alcoholics has in fact revealed significant decreases in several genes related to myelination in addition to alterations in specific proteins of stress-related pathways that operate in astrocytes (McClintick et al., 2013).
Studies with oligodendrocyte cultures indicate that ethanol-induced degeneration or impairment of myelin maintenance may be mediated by the ethanol metabolite acetaldehyde (Coutts and Harrison, 2015), suggesting that the ability to degrade or eliminate this metabolite may influence the effects of alcohol on myelin composition. Moreover, human subjects with alcoholism most probably differ in their drinking schedules and exposure to binge or withdrawal periods. These periods, according to the results of animal experiments, can significantly alter myelin protein expression (for example causing a recovery of MBP expression; Kipp et al., 2012; Navarro and Mandyam, 2015) and strongly influence mRNA levels at the time of death. Summarizing, despite complex interactions that probably determine the individual levels of myelin-related mRNAs and proteins at the time of death, the available animal experimentation and human postmortem evidence indicates significant effects of ethanol abuse on the expression of myelin components, and the plasticity of myelin itself. These changes would significantly affect the role of myelin maintenance in action potential propagation.
Oligodendrocyte Survival and Proliferation
In addition to the effects on myelin components and structure, ethanol exposure very likely causes damage to oligodendrocyte precursors by reducing their proliferation (Newville et al., 2017) or disrupting the expression of transcription factors, such as c-fos (Bichenkov and Ellingson, 2009), that regulate the differentiation of those precursors into mature, myelin-forming oligodendrocytes. The effects on differentiation may also include abnormal acceleration of oligodendrocyte differentiation (Aspberg and Tottmar, 1994). However, at a difference with animal models of prolonged alcohol exposure, chronic alcoholism in humans may not necessarily cause an overall reduction in neuro- or glio-genesis in well-known neurogenic niches (Sutherland et al., 2013). On the other hand, a direct role of ethanol in promoting myelin pathology and the possibility of recovery from that pathology are strongly suggested by increased MBP levels in the medial PFC of rats after prolonged periods of abstinence from ethanol (Navarro and Mandyam, 2015). Potential for recovery appears to be a consequence of the involvement of oligodendrocyte precursor cells in remyelination processes (Mi et al., 2009) during abstinence from alcohol drinking.
Glia-Related Extracellular Matrix Components in AUDs
In WM, several proteoglycans and other ECM proteins are produced by astrocytes, oligodendrocytes and neurons to form adhesion complexes at the NR and axon initial segments, where they are implicated in the aggregation of voltage-gated sodium channels and other components involved in action potential generation and propagation (Zimmermann and Dours-Zimmermann, 2008; Nelson and Jenkins, 2017). Those complexes include brevican, versicans, neurocan, tenascins and neurofascins, among others, form mutual attachments involving interactions between specific protein domains. Interestingly, repetitive binge alcohol intake in adolescent rats significantly increases the levels of several of those proteins in the WM of brain areas such as the orbitofrontal cortex that participate in the pathophysiology of addictive behaviors (Coleman et al., 2014). Alcohol exposure also significantly alters the production by astrocytes and oligodendrocytes of some ECM proteins involved in the formation of perineuronal nets and synapses in the GM, and in the assembly of the basal lamina around blood vessels (Lasek, 2016), although the exact role of those changes in the mechanisms leading to behavioral and functional disturbances in AUDs is not fully understood. The importance of an involvement of ECM components in the mechanisms of alcohol addiction is suggested by the ability of ECM disturbances to modulate the seeking for alcohol and other drugs in knock-out mice lacking matrix metalloproteinase 9 (MMP-9), a protease that regulates the integrity of the ECM (Smith, 2017). These mice have reduced motivation for alcohol drinking, but rescuing MMP-9 activity in the brain’s amygdala restores normal alcohol-seeking behavior (Stefaniuk et al., 2017).
Epigenetic Changes in Oligodendrocytes and Astrocytes in AUDs
Regulation of DNA transcription into mRNA is greatly dependent on epigenetic mechanisms such as DNA methylation and acetylation, and methylation of chromatin histones (Gräff et al., 2011). In addition, at the translational level, gene expression is regulated by the activity of microRNAs (miRNAs), small forms of non-coding RNA (about 22 nucleotides long) that interfere with translation into proteins by binding to specific sequences of coding mRNA (Liu and Casaccia, 2010; Li and Yao, 2012; Emery and Lu, 2015).
In recent years, several reviews have compiled studies showing that the development of astrocytes, oligodendrocytes as well as plastic changes in myelin maintenance involve complex epigenetic pathways (MacDonald and Roskams, 2009; Kim and Rosenfeld, 2010; Yu et al., 2010; Bian et al., 2013; Namihira and Nakashima, 2013; Emery and Lu, 2015). These pathways can be significantly altered by prolonged exposure to alcohol (Aspberg and Tottmar, 1994; Bichenkov and Ellingson, 2009; Alfonso-Loeches et al., 2012; Creeley et al., 2013; Coutts and Harrison, 2015; Newville et al., 2017). Methylation of DNA at specific nucleotides, and acetylation and methylation of DNA-associated histones are known to critically determine the fate and differentiation of precursors into mature astrocytes and oligodendrocytes as well as the formation of myelin (Moyon et al., 2016).
Epigenetic and miRNA-mediated mechanisms in the central nervous system play also relevant roles in the pathophysiology of neurological, neurodegenerative and psychiatric disorders (Meza-Sosa et al., 2012). The clinical relevance of increasing our knowledge about epigenetic disturbances in oligodendrocytes and astrocytes stems from the demonstration of significant epigenetic anomalies in demyelinating disorders, and the possibility of reversing them with experimental treatments targeted to epigenetic alterations (Li and Yao, 2012; Liu et al., 2016). In addition, several miRNAs have been found to regulate directly (by impairing translation) or indirectly (through other miRNAs or transcription factors suppressed by miRNAs) the production of transcription factors and proteins during development (Bian et al., 2013).
Glial Epigenetic Markers
AUDs are associated with profound brain alterations in epigenetic markers (Zhou et al., 2011; Farris et al., 2015b; Weng et al., 2015; Legastelois et al., 2017) and significant increases in miRNAs regulating the expression of many proteins (Lewohl et al., 2011). Alcohol-related epigenetic changes have been found in DNA methylation patterns and in methylation and acetylation of histones in the human PFC, hippocampus and amygdala (Ponomarev, 2013; Farris et al., 2015b) as well as in cultured astrocytes (Zhang et al., 2014). Some studies suggest that acute alcohol intake leads to histone deacetylase (HDAC) inhibition in the amygdala that would be the basis for increased histone acetylation and anxiolysis, while withdrawal, anxiety or adolescent alcohol exposure seems to be associated with increased HDAC activity (Pandey et al., 2017) and decreased acetylation (Pandey et al., 2008). This HDAC increase would lead to reduced expression of genes involved in synaptic plasticity, and, consequently, HDAC inhibitors have been suggested as a therapeutic option to reduce anxiety and alcohol intake (Pandey et al., 2017). Other researchers have demonstrated DNA methylation disturbances, mostly reductions, in humans with AUDs, although a more recent study with different methodology points to a relatively higher percentage of hypermethylated methylation sites in brain DNA of subjects with alcoholism (Tulisiak et al., 2017).
Methylation changes in the human brain appear to be accompanied by reduction in the mRNA expression of DNA methyltransferases (DNMTs), while in animal models repeated alcohol exposure results in DNMT upregulation (Tulisiak et al., 2017). Actually, the effects of alcoholism on DNA methylation patterns involve both hypo- and hypermethylation in promoters for specific genes, producing a rather complex picture of methylation effects (Tulisiak et al., 2017). However, given that alcohol exposure in animal models increases DNMTs’ levels some researchers have explored DNMT inhibition as a therapeutic approach against excessive alcohol drinking, finding that 5-aza or decitabine, both DNMT inhibitors, acutely reduce excessive alcohol drinking in rats or mice under certain conditions (Ponomarev et al., 2017; Tulisiak et al., 2017). Likewise, developmental models of alcoholism in rodents show disruption of DNA methylation and other epigenetic markers in various regions of the central nervous system (Laufer et al., 2017; Mahnke et al., 2017; Öztürk et al., 2017).
Recent studies on the effects of HDAC and DNMT inhibitors in AUD mouse models have shown that treatment with the DNMT inhibitor decitabine results in decreased ethanol drinking and upregulated expression of genes highly represented in oligodendrocytes and astrocytes in the ventral tegmental area, key region in the brain reward pathways (Ponomarev et al., 2017). In addition, exposure of cultured astrocytes to ethanol results in decreased methylation of the tissue plasminogen activator (TPA) gene promoter (Tulisiak et al., 2017). TPA is involved in ECM degradation, and has been reported to be increased in animal models of AUDs (Zhang et al., 2014). In rats, prenatal ethanol exposure leads to hypermethylation of the promoter for the astrocyte GFAP gene and to reduction in GFAP expression during early postnatal development (Vallés et al., 1997). These findings suggest that alteration of DNA or histone epigenetic markers in astrocytes and oligodendrocytes may play an important role in mediating behavioral disturbances in AUDs.
Glial miRNA Expression Changes
Some studies have also targeted putative miRNA changes in oligodendrocytes following repeated or prolonged alcohol exposure and the possibility that such alterations may bring about disturbances in myelination. In fact, alcoholism has been found associated with upregulation in the expression of miRNAs from a gene cluster in chromosome 14q32 that have for targets the mRNAs of several proteins involved in processes of oligodendrocyte proliferation and myelination (Manzardo et al., 2013). It is also unsurprising that genome-wide examination of miRNA-protein gene co-expression networks in the brain of alcohol-dependent human subjects has identified abundant epigenetic modifications in molecular networks that operate within oligodendrocytes and astrocytes (Ponomarev et al., 2012).
Studies have also shown significant increases of PFC miRNAs in subjects with AUDs (Lewohl et al., 2011), although in WM some miRNAs would be decreased (Miguel-Hidalgo et al., 2017). Increased miRNAs include some targeting major myelin proteins such as PLP1 and CNPase as well as transcription factor C11ORF9, a regulator of myelin formation. In our recent work, we found that miR-21, high in oligodendrocytes, was strongly and positively correlated with decreased PLP1 miRNA (which is not a direct target of miR-21; Miguel-Hidalgo et al., 2017). The networks and pathways regulated by differentially expressed miRNAs in human alcoholics and mice are very highly enriched in oligodendrocytes and astrocytes, some of them exclusively for each cell type (Nunez et al., 2013). Thus, miRNA increases are consistent with downregulated expression of myelin components and other oligodendrocyte pathology in AUD patients.
Concluding Remarks
Ethanol exposure in AUDs results in disturbances in the structure of astrocytes and oligodendrocytes as well as in the expression and function of specific astrocytes and myelin proteins. The disturbances are likely to impair diverse aspects of neuronal function including regulation of synaptic transmission, synapse formation, metabolism, interactions with the brain blood supply, propagation of action potentials and neuroprotection. Moreover, the complex interaction between astrocytes and oligodendrocytes involves proteins that are affected by ethanol exposure, such as specific connexins at gap junctions, glutamate transporters, or ECM proteins produced by astrocytes, oligodendrocytes and neurons that are crucial for saltatory conduction at NR (Figure 1). However, much more research is needed to determine the mechanisms by which AUDs acting on the components that support the interactions between astrocytes and oligodendrocytes lead to failures in connectivity between brain regions, either by affecting myelin structure or the ability to regenerate action potentials between NR. In addition, chronic alcoholism causes disturbances in the expression of miRNAs and other epigenetic markers that directly influence protein expression. These regulatory changes very likely underpin alterations of proteins and functional pathways in astrocytes and oligodendrocytes observed in earlier studies. However, with a few exceptions, it is still unclear how protein expression changes and the functional pathways they serve in astrocytes and oligodendrocytes depend on non-coding RNA and epigenetic alterations and what is the contribution of these glial processes to the neuronal pathophysiology of alcoholism. In conclusion, much additional work is needed to understand at molecular and neurophysiological levels the mechanisms of alcohol-related neural damage that depend on the molecular pathology of astrocytes, oligodendrocytes and their interactions.
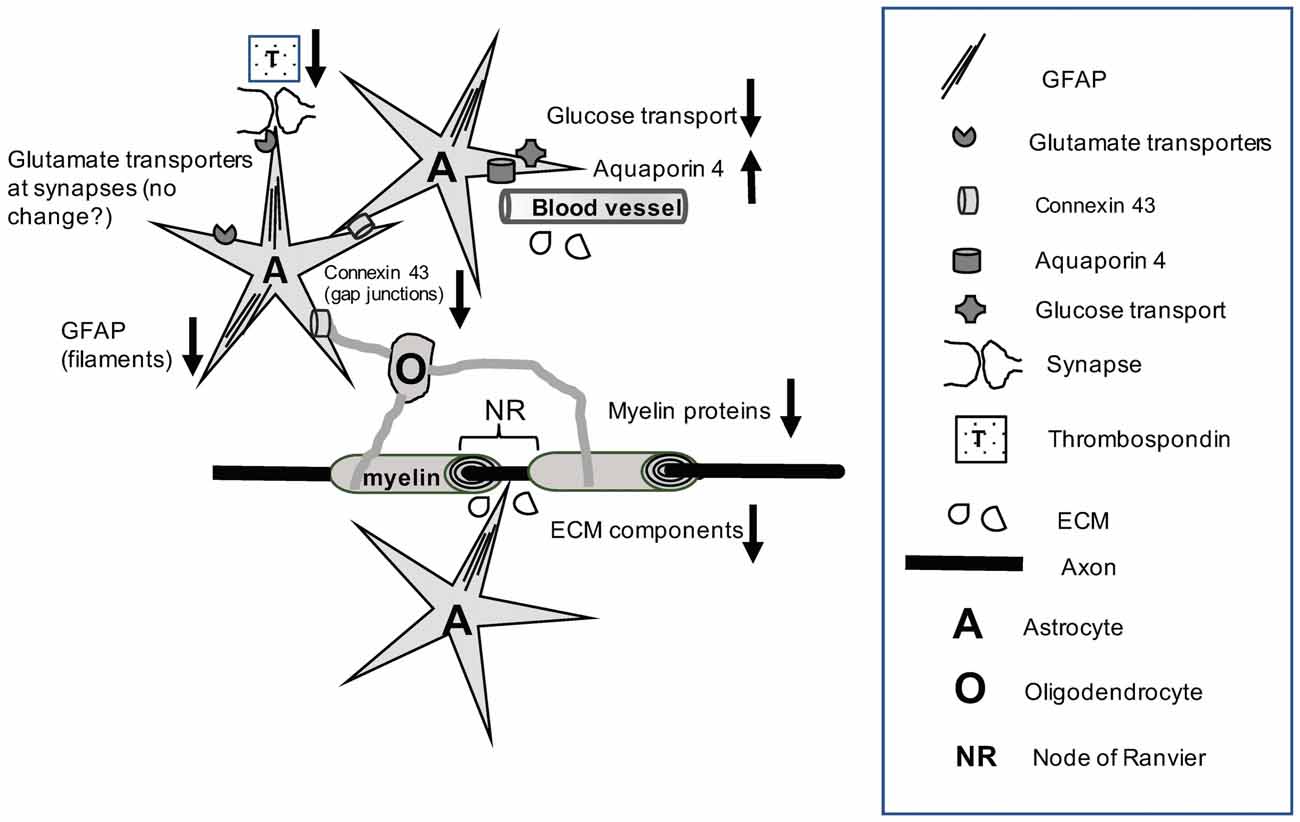
Figure 1. The cartoon illustrates that chronic alcohol exposure results in a variety of molecular disturbances that affect the reciprocal interactions between astrocytes and oligodendrocytes (for example, through reduction of connexin levels), as well as between these glial cells and neurons at synapses, NR or in the wrapping of axons by myelin. Alcohol exposure also results in pathological alterations of astrocyte-derived components at the blood brain barrier (BBB) and the ECM. More recent research work has revealed epigenetic abnormalities or changed levels of specific non-coding RNAs in alcohol use disorders (AUDs). Nevertheless, further research is needed to understand the mechanisms by which alcohol-related functional pathology at the cell and protein expression levels is linked to epigenetic and non-coding RNA markers. Abbreviations: A, astrocytes; ECM, Extracellular matrix; GFAP, glial fibrillary acidic protein; NR, node of Ranvier; O, Oligodendrocyte; T, Thrombospondin; ↓, Down-regulation; ↑, Up-regulation.
Author Contributions
JJM-H conceived and wrote the review.
Funding
The research work in bibliographic references involving the author of this article has been partly funded by National Institutes of Health (NIH) grants R21MH82297, P30GM103328 and R56MH113828 as well as the ABMRF/The Foundation for Alcohol Research.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbott, N. J., Rönnbäck, L., and Hansson, E. (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53. doi: 10.1038/nrn1824
Abdul Muneer, P. M., Alikunju, S., Szlachetka, A. M., and Haorah, J. (2011a). Inhibitory effects of alcohol on glucose transport across the blood-brain barrier leads to neurodegeneration: preventive role of acetyl-L-carnitine. Psychopharmacology 214, 707–718. doi: 10.1007/s00213-010-2076-4
Abdul Muneer, P. M., Alikunju, S., Szlachetka, A. M., Mercer, A. J., and Haorah, J. (2011b). Ethanol impairs glucose uptake by human astrocytes and neurons: protective effects of acetyl-L-carnitine. Int. J. Physiol. Pathophysiol. Pharmacol. 3, 48–56.
Abrahao, K. P., Salinas, A. G., and Lovinger, D. M. (2017). Alcohol and the brain: neuronal molecular targets, synapses, and circuits. Neuron 96, 1223–1238. doi: 10.1016/j.neuron.2017.10.032
Adermark, L., and Bowers, M. S. (2016). Disentangling the role of astrocytes in alcohol use disorder. Alcohol. Clin. Exp. Res. 40, 1802–1816. doi: 10.1111/acer.13168
Alexander-Kaufman, K., Cordwell, S., Harper, C., and Matsumoto, I. (2007). A proteome analysis of the dorsolateral prefrontal cortex in human alcoholic patients. Proteomics Clin. Appl. 1, 62–72. doi: 10.1002/prca.200600417
Alexander-Kaufman, K., James, G., Sheedy, D., Harper, C., and Matsumoto, I. (2006). Differential protein expression in the prefrontal white matter of human alcoholics: a proteomics study. Mol. Psychiatry 11, 56–65. doi: 10.1038/sj.mp.4001741
Alfonso-Loeches, S., Pascual, M., Gómez-Pinedo, U., Pascual-Lucas, M., Renau-Piqueras, J., and Guerri, C. (2012). Toll-like receptor 4 participates in the myelin disruptions associated with chronic alcohol abuse. Glia 60, 948–964. doi: 10.1002/glia.22327
Aspberg, A., and Tottmar, O. (1994). Ethanol-induced increase in catalase activity in reaggregation cultures of rat brain cells is due to increased oligodendrocyte differentiation. Alcohol. Clin. Exp. Res. 18, 620–624. doi: 10.1111/j.1530-0277.1994.tb00920.x
Ayers-Ringler, J. R., Jia, Y. F., Qiu, Y. Y., and Choi, D. S. (2016). Role of astrocytic glutamate transporter in alcohol use disorder. World J. Psychiatry 6, 31–42. doi: 10.5498/wjp.v6.i1.31
Badaut, J., Lasbennes, F., Magistretti, P. J., and Regli, L. (2002). Aquaporins in brain: distribution, physiology, and pathophysiology. J. Cereb. Blood Flow Metab. 22, 367–378. doi: 10.1097/00004647-200204000-00001
Banner, S. J., Fray, A. E., Ince, P. G., Steward, M., Cookson, M. R., and Shaw, P. J. (2002). The expression of the glutamate re-uptake transporter excitatory amino acid transporter 1 (EAAT1) in the normal human CNS and in motor neurone disease: an immunohistochemical study. Neuroscience 109, 27–44. doi: 10.1016/s0306-4522(01)00437-7
Barres, B. A. (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. doi: 10.1016/j.neuron.2008.10.013
Baumann, N., and Pham-Dinh, D. (2001). Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 81, 871–927. doi: 10.1152/physrev.2001.81.2.871
Bian, S., Xu, T. L., and Sun, T. (2013). Tuning the cell fate of neurons and glia by microRNAs. Curr. Opin. Neurobiol. 23, 928–934. doi: 10.1016/j.conb.2013.08.002
Bichenkov, E., and Ellingson, J. S. (2001). Ethanol exerts different effects on myelin basic protein and 2′,3′-cyclic nucleotide 3′-phosphodiesterase expression in differentiating CG-4 oligodendrocytes. Dev. Brain Res. 128, 9–16. doi: 10.1016/s0165-3806(01)00142-0
Bichenkov, E., and Ellingson, J. S. (2002). Protein kinase C inhibitors counteract the ethanol effects on myelin basic protein expression in differentiating CG-4 oligodendrocytes. Dev. Brain Res. 139, 29–38. doi: 10.1016/s0165-3806(02)00512-6
Bichenkov, E., and Ellingson, J. S. (2009). Ethanol alters the expressions of c-Fos and myelin basic protein in differentiating oligodendrocytes. Alcohol 43, 627–634. doi: 10.1016/j.alcohol.2009.09.026
Black, J. A., and Waxman, S. G. (1988). The perinodal astrocyte. Glia 1, 169–183. doi: 10.1002/glia.440010302
Butt, A. M. (2005). “Structure and function of oligodendrocytes,” in Neuroglia, 2nd Edn. eds H. Kettenmann and B. Ransom (New York, NY: Oxford University Press), 36–47.
Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., et al. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278. doi: 10.1523/JNEUROSCI.4178-07.2008
Cali, C., Marchaland, J., Regazzi, R., and Bezzi, P. (2008). SDF 1-α (CXCL12) triggers glutamate exocytosis from astrocytes on a millisecond time scale: imaging analysis at the single-vesicle level with TIRF microscopy. J. Neuroimmunol. 198, 82–91. doi: 10.1016/j.jneuroim.2008.04.015
Coleman, L. G. Jr., Liu, W., Oguz, I., Styner, M., and Crews, F. T. (2014). Adolescent binge ethanol treatment alters adult brain regional volumes, cortical extracellular matrix protein and behavioral flexibility. Pharmacol. Biochem. Behav. 116, 142–151. doi: 10.1016/j.pbb.2013.11.021
Collins, M. A., Moon, K. H., Tajuddin, N., Neafsey, E. J., and Kim, H. Y. (2013). Docosahexaenoic acid (DHA) prevents binge ethanol-dependent aquaporin-4 elevations while inhibiting neurodegeneration: experiments in rat adult-age entorhino-hippocampal slice cultures. Neurotox. Res. 23, 105–110. doi: 10.1007/s12640-012-9360-5
Collins, M. A., and Neafsey, E. J. (2012). Neuroinflammatory pathways in binge alcohol-induced neuronal degeneration: oxidative stress cascade involving aquaporin, brain edema, and phospholipase A2 activation. Neurotox. Res. 21, 70–78. doi: 10.1007/s12640-011-9276-5
Coutts, D. J., and Harrison, N. L. (2015). Acetaldehyde, not ethanol, impairs myelin formation and viability in primary mouse oligodendrocytes. Alcohol. Clin. Exp. Res. 39, 455–462. doi: 10.1111/acer.12642
Creeley, C. E., Dikranian, K. T., Johnson, S. A., Farber, N. B., and Olney, J. W. (2013). Alcohol-induced apoptosis of oligodendrocytes in the fetal macaque brain. Acta Neuropathol. Commun. 1:23. doi: 10.1186/2051-5960-1-23
Cui, W., Allen, N. D., Skynner, M., Gusterson, B., and Clark, A. J. (2001). Inducible ablation of astrocytes shows that these cells are required for neuronal survival in the adult brain. Glia 34, 272–282. doi: 10.1002/glia.1061
Cullen, K. M., and Halliday, G. M. (1994). Chronic alcoholics have substantial glial pathology in the forebrain and diencephalon. Alcohol Alcohol. 2, 253–257.
Davies, D. L., and Cox, W. E. (1991). Delayed growth and maturation of astrocytic cultures following exposure to ethanol: electron microscopic observations. Brain Res. 547, 62–68. doi: 10.1016/0006-8993(91)90573-E
Davis, K. M., and Wu, J. Y. (2001). Role of glutamatergic and GABAergic systems in alcoholism. J. Biomed. Sci. 8, 7–19. doi: 10.1159/000054008
de la Monte, S. M., and Kril, J. J. (2014). Human alcohol-related neuropathology. Acta Neuropathol. 127, 71–90. doi: 10.1007/s00401-013-1233-3
Du, Y., and Dreyfus, C. F. (2002). Oligodendrocytes as providers of growth factors. J. Neurosci. Res. 68, 647–654. doi: 10.1002/jnr.10245
Emery, B., and Lu, Q. R. (2015). Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harb. Perspect. Biol. 7:a020461. doi: 10.1101/cshperspect.a020461
Farris, S. P., Arasappan, D., Hunicke-Smith, S., Harris, R. A., and Mayfield, R. D. (2015a). Transcriptome organization for chronic alcohol abuse in human brain. Mol. Psychiatry 20, 1438–1447. doi: 10.1038/mp.2014.159
Farris, S. P., Harris, R. A., and Ponomarev, I. (2015b). Epigenetic modulation of brain gene networks for cocaine and alcohol abuse. Front. Neurosci. 9:176. doi: 10.3389/fnins.2015.00176
Fitzpatrick, J. M., Anderson, R. C., and McDermott, K. W. (2015). MicroRNA: key regulators of oligodendrocyte development and pathobiology. Int. J. Biochem. Cell Biol. 65, 134–138. doi: 10.1016/j.biocel.2015.05.021
Franke, H. (1995). Influence of chronic alcohol treatment on the GFAP-immunoreactivity in astrocytes of the hippocampus in rats. Acta Histochem. 97, 263–271. doi: 10.1016/s0065-1281(11)80187-x
Freund, G., and Anderson, K. J. (1996). Glutamate receptors in the frontal cortex of alcoholics. Alcohol. Clin. Exp. Res. 20, 1165–1172. doi: 10.1111/j.1530-0277.1996.tb01106.x
Freund, G., and Anderson, K. J. (1999). Glutamate receptors in the cingulate cortex, hippocampus, and cerebellar vermis of alcoholics. Alcohol. Clin. Exp. Res. 23, 1–6. doi: 10.1097/00000374-199901000-00001
Gallucci, M., Amicarelli, I., Rossi, A., Stratta, P., Masciocchi, C., Zobel, B. B., et al. (1989). MR imaging of white matter lesions in uncomplicated chronic alcoholism. J. Comput. Assist. Tomogr. 13, 395–398. doi: 10.1097/00004728-198905000-00004
Gankam Kengne, F., Nicaise, C., Soupart, A., Boom, A., Schiettecatte, J., Pochet, R., et al. (2011). Astrocytes are an early target in osmotic demyelination syndrome. J. Am. Soc. Nephrol. 22, 1834–1845. doi: 10.1681/ASN.2010111127
Gass, J. T., and Olive, M. F. (2008). Glutamatergic substrates of drug addiction and alcoholism. Biochem. Pharmacol. 75, 218–265. doi: 10.1016/j.bcp.2007.06.039
Gräff, J., Kim, D., Dobbin, M. M., and Tsai, L. H. (2011). Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 91, 603–649. doi: 10.1152/physrev.00012.2010
Guerri, C. (1998). Neuroanatomical and neurophysiological mechanisms involved in central nervous system dysfunctions induced by prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 22, 304–312. doi: 10.1097/00000374-199804000-00003
Guerri, C., and Renau-Piqueras, J. (1997). Alcohol, astroglia, and brain development. Mol. Neurobiol. 15, 65–81. doi: 10.1007/bf02740616
Haorah, J., Knipe, B., Leibhart, J., Ghorpade, A., and Persidsky, Y. (2005). Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 78, 1223–1232. doi: 10.1189/jlb.0605340
Harper, C. (2009). The neuropathology of alcohol-related brain damage. Alcohol Alcohol. 44, 136–140. doi: 10.1093/alcalc/agn102
Haydon, P. G. (2000). Neuroglial networks: neurons and glia talk to each other. Curr. Biol. 10, R712–R714. doi: 10.1016/s0960-9822(00)00708-9
Hazell, A. S. (2009). Astrocytes are a major target in thiamine deficiency and Wernicke’s encephalopathy. Neurochem. Int. 55, 129–135. doi: 10.1016/j.neuint.2009.02.020
Hazell, A. S., Sheedy, D., Oanea, R., Aghourian, M., Sun, S., Jung, J. Y., et al. (2010). Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia 58, 148–156. doi: 10.1002/glia.20908
He, X., Sullivan, E. V., Stankovic, R. K., Harper, C. G., and Pfefferbaum, A. (2007). Interaction of thiamine deficiency and voluntary alcohol consumption disrupts rat corpus callosum ultrastructure. Neuropsychopharmacology 32, 2207–2216. doi: 10.1038/sj.npp.1301332
Hu, X., Yuan, Y., Wang, D., and Su, Z. (2016). Heterogeneous astrocytes: active players in CNS. Brain Res. Bull. 125, 1–18. doi: 10.1016/j.brainresbull.2016.03.017
Jayakumar, A. R., Tong, X. Y., Curtis, K. M., Ruiz-Cordero, R., Shamaladevi, N., Abuzamel, M., et al. (2014). Decreased astrocytic thrombospondin-1 secretion after chronic ammonia treatment reduces the level of synaptic proteins: in vitro and in vivo studies. J. Neurochem. 131, 333–347. doi: 10.1111/jnc.12810
Jensen, G. B., and Pakkenberg, B. (1993). Do alcoholics drink their neurons away? Lancet 342, 1201–1204. doi: 10.1016/0140-6736(93)92185-v
Kalsi, A. S., Greenwood, K., Wilkin, G., and Butt, A. M. (2004). Kir4.1 expression by astrocytes and oligodendrocytes in CNS white matter: a developmental study in the rat optic nerve. J. Anat. 204, 475–485. doi: 10.1111/j.0021-8782.2004.00288.x
Kane, C. J., Berry, A., Boop, F. A., and Davies, D. L. (1996). Proliferation of astroglia from the adult human cerebrum is inhibited by ethanol in vitro. Brain Res. 731, 39–44. doi: 10.1016/0006-8993(96)00456-8
Kennedy, L. A., and Mukerji, S. (1986). Ethanol neurotoxicity. 1. Direct effects on replicating astrocytes. Neurobehav. Toxicol. Teratol. 8, 11–15.
Kettenmann, H., Kettenmann, H., and Ransom, B. R. (2013). Neuroglia. New York, NY: Oxford University Press.
Khakh, B. S., and Sofroniew, M. V. (2015). Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 18, 942–952. doi: 10.1038/nn.4043
Kiefer, F., Jahn, H., Koester, A., Montkowski, A., Reinscheid, R. K., and Wiedemann, K. (2003). Involvement of NMDA receptors in alcohol-mediated behavior: mice with reduced affinity of the NMDA R1 glycine binding site display an attenuated sensitivity to ethanol. Biol. Psychiatry 53, 345–351. doi: 10.1016/s0006-3223(02)01486-5
Kim, H. J., and Rosenfeld, M. G. (2010). Epigenetic control of stem cell fate to neurons and glia. Arch. Pharm. Res. 33, 1467–1473. doi: 10.1007/s12272-010-1001-z
Kipp, M., Victor, M., Martino, G., and Franklin, R. J. (2012). Endogeneous remyelination: findings in human studies. CNS Neurol. Disord. Drug Targets 11, 598–609. doi: 10.2174/187152712801661257
Koehler, R. C., Roman, R. J., and Harder, D. R. (2009). Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 32, 160–169. doi: 10.1016/j.tins.2008.11.005
Kong, L., Lian, G., Zheng, W., Liu, H., Zhang, H., and Chen, R. (2013). Effect of alcohol on diffuse axonal injury in rat brainstem: diffusion tensor imaging and aquaporin-4 expression study. Biomed Res. Int. 2013:798261. doi: 10.1155/2013/798261
Korbo, L. (1999). Glial cell loss in the hippocampus of alcoholics. Alcohol. Clin. Exp. Res. 23, 164–168. doi: 10.1097/00000374-199901000-00024
Kril, J. J., and Harper, C. G. (2012). Neuroanatomy and neuropathology associated with Korsakoff’s syndrome. Neuropsychol. Rev. 22, 72–80. doi: 10.1007/s11065-012-9195-0
Kril, J. J., Halliday, G. M., Svoboda, M. D., and Cartwright, H. (1997). The cerebral cortex is damaged in chronic alcoholics. Neuroscience 79, 983–998. doi: 10.1016/s0306-4522(97)00083-3
Krystal, J. H., Petrakis, I. L., Limoncelli, D., Nappi, S. K., Trevisan, L., Pittman, B., et al. (2011). Characterization of the interactive effects of glycine and D-cycloserine in men: further evidence for enhanced NMDA receptor function associated with human alcohol dependence. Neuropsychopharmacology 36, 701–710. doi: 10.1038/npp.2010.203
Laming, P. R., Kimelberg, H., Robinson, S., Salm, A., Hawrylak, N., Müller, C., et al. (2000). Neuronal-glial interactions and behaviour. Neurosci. Biobehav. Rev. 24, 295–340. doi: 10.1016/S0149-7634(99)00080-9
Lasek, A. W. (2016). Effects of ethanol on brain extracellular matrix: implications for alcohol use disorder. Alcohol. Clin. Exp. Res. 40, 2030–2042. doi: 10.1111/acer.13200
Laufer, B. I., Chater-Diehl, E. J., Kapalanga, J., and Singh, S. M. (2017). Long-term alterations to DNA methylation as a biomarker of prenatal alcohol exposure: From mouse models to human children with fetal alcohol spectrum disorders. Alcohol 60, 67–75. doi: 10.1016/j.alcohol.2016.11.009
Lee, M. R., Ruby, C. L., Hinton, D. J., Choi, S., Adams, C. A., Young Kang, N., et al. (2013). Striatal adenosine signaling regulates EAAT2 and astrocytic AQP4 expression and alcohol drinking in mice. Neuropsychopharmacology 38, 437–445. doi: 10.1038/npp.2012.198
Legastelois, R., Jeanblanc, J., Vilpoux, C., Bourguet, E., and Naassila, M. (2017). Epigenetic mechanisms and alcohol use disorders: a potential therapeutic target. Biol. Aujourdhui 211, 83–91. doi: 10.1051/jbio/2017014
Lewohl, J. M., Dodd, P. R., Mayfield, R. D., and Harris, R. A. (2001). Application of DNA microarrays to study human alcoholism. J. Biomed. Sci. 8, 28–36. doi: 10.1159/000054010
Lewohl, J. M., Nunez, Y. O., Dodd, P. R., Tiwari, G. R., Harris, R. A., and Mayfield, R. D. (2011). Up-regulation of microRNAs in brain of human alcoholics. Alcohol. Clin. Exp. Res. 35, 1928–1937. doi: 10.1111/j.1530-0277.2011.01544.x
Lewohl, J. M., Wang, L., Miles, M. F., Zhang, L., Dodd, P. R., and Harris, R. A. (2000). Gene expression in human alcoholism: microarray analysis of frontal cortex. Alcohol. Clin. Exp. Res. 24, 1873–1882. doi: 10.1097/00000374-200012000-00018
Lewohl, J. M., Wixey, J., Harper, C. G., and Dodd, P. R. (2005). Expression of MBP, PLP, MAG, CNP, and GFAP in the human alcoholic brain. Alcohol. Clin. Exp. Res. 29, 1698–1705. doi: 10.1097/01.alc.0000179406.98868.59
Li, J. S., and Yao, Z. X. (2012). MicroRNAs: novel regulators of oligodendrocyte differentiation and potential therapeutic targets in demyelination-related diseases. Mol. Neurobiol. 45, 200–212. doi: 10.1007/s12035-011-8231-z
Liu, J., and Casaccia, P. (2010). Epigenetic regulation of oligodendrocyte identity. Trends Neurosci. 33, 193–201. doi: 10.1016/j.tins.2010.01.007
Liu, J., Lewohl, J. M., Harris, R. A., Iyer, V. R., Dodd, P. R., Randall, P. K., et al. (2006). Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology 31, 1574–1582. doi: 10.1038/sj.npp.1300947
Liu, J., Moyon, S., Hernandez, M., and Casaccia, P. (2016). Epigenetic control of oligodendrocyte development: adding new players to old keepers. Curr. Opin. Neurobiol. 39, 133–138. doi: 10.1016/j.conb.2016.06.002
Lovatt, D., Nedergaard, M., Kettenmann, H., and Ransom, B. (2012). The astrocyte transcriptome Neuroglia, eds H. Kettenmann and B. R. Ransom (New York, NY: Oxford University Press), 347–357.
Lutz, S. E., Zhao, Y., Gulinello, M., Lee, S. C., Raine, C. S., and Brosnan, C. F. (2009). Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J. Neurosci. 29, 7743–7752. doi: 10.1523/JNEUROSCI.0341-09.2009
MacDonald, J. L., and Roskams, A. J. (2009). Epigenetic regulation of nervous system development by DNA methylation and histone deacetylation. Prog. Neurobiol. 88, 170–183. doi: 10.1016/j.pneurobio.2009.04.002
Magnotti, L. M., Goodenough, D. A., and Paul, D. L. (2011). Deletion of oligodendrocyte Cx32 and astrocyte Cx43 causes white matter vacuolation, astrocyte loss and early mortality. Glia 59, 1064–1074. doi: 10.1002/glia.21179
Mahnke, A. H., Miranda, R. C., and Homanics, G. E. (2017). Epigenetic mediators and consequences of excessive alcohol consumption. Alcohol 60, 1–6. doi: 10.1016/j.alcohol.2017.02.357
Manzardo, A. M., Gunewardena, S., and Butler, M. G. (2013). Over-expression of the miRNA cluster at chromosome 14q32 in the alcoholic brain correlates with suppression of predicted target mRNA required for oligodendrocyte proliferation. Gene 526, 356–363. doi: 10.1016/j.gene.2013.05.052
Mayfield, R. D., Lewohl, J. M., Dodd, P. R., Herlihy, A., Liu, J., and Harris, R. A. (2002). Patterns of gene expression are altered in the frontal and motor cortices of human alcoholics. J. Neurochem. 81, 802–813. doi: 10.1046/j.1471-4159.2002.00860.x
McClintick, J. N., Xuei, X., Tischfield, J. A., Goate, A., Foroud, T., Wetherill, L., et al. (2013). Stress-response pathways are altered in the hippocampus of chronic alcoholics. Alcohol 47, 505–515. doi: 10.1016/j.alcohol.2013.07.002
Melendez, R. I., Hicks, M. P., Cagle, S. S., and Kalivas, P. W. (2005). Ethanol exposure decreases glutamate uptake in the nucleus accumbens. Alcohol. Clin. Exp. Res. 29, 326–333. doi: 10.1097/01.alc.0000156086.65665.4d
Meza-Sosa, K. F., Valle-Garcia, D., Pedraza-Alva, G., and Perez-Martinez, L. (2012). Role of microRNAs in central nervous system development and pathology. J. Neurosci. Res. 90, 1–12. doi: 10.1002/jnr.22701
Mi, S., Miller, R. H., Tang, W., Lee, X., Hu, B., Wu, W., et al. (2009). Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann. Neurol. 65, 304–315. doi: 10.1002/ana.21581
Miguel-Hidalgo, J. J. (2009). “Neuropathology of depression, alcoholism, and their comorbidity in the human prefrontal cortex,” in Comorbidity of Depression and Alcohol Use Disorders, ed. L. Sher (New York, NY: Nova Science Publishers, Inc.), 171–179.
Miguel-Hidalgo, J. J., Hall, K. O., Bonner, H., Roller, A. M., Syed, M., Park, C. J., et al. (2017). MicroRNA-21: expression in oligodendrocytes and correlation with low myelin mRNAs in depression and alcoholism. Prog. Neuropsychopharmacol. Biol. Psychiatry 79, 503–514. doi: 10.1016/j.pnpbp.2017.08.009
Miguel-Hidalgo, J. J., Overholser, J. C., Meltzer, H. Y., Stockmeier, C. A., and Rajkowska, G. (2006). Reduced glial and neuronal packing density in the orbitofrontal cortex in alcohol dependence and its relationship with suicide and duration of alcohol dependence. Alcohol. Clin. Exp. Res. 30, 1845–1855. doi: 10.1111/j.1530-0277.2006.00221.x
Miguel-Hidalgo, J. J., and Rajkowska, G. (2003). Comparison of prefrontal cell pathology between depression and alcohol dependence. J. Psychiatr. Res. 37, 411–420. doi: 10.1016/s0022-3956(03)00049-9
Miguel-Hidalgo, J. J., Waltzer, R., Whittom, A. A., Austin, M. C., Rajkowska, G., and Stockmeier, C. A. (2010). Glial and glutamatergic markers in depression, alcoholism, and their comorbidity. J. Affect. Disord. 127, 230–240. doi: 10.1016/j.jad.2010.06.003
Miguel-Hidalgo, J. J., Wei, J. R., Andrew, M., Overholser, J. C., Jurjus, G., Stockmeier, C. A., et al. (2002). Glia pathology in the prefrontal cortex in alcohol dependence with and without depressive symptoms. Biol. Psychiatry 52, 1121–1133. doi: 10.1016/s0006-3223(02)01439-7
Moselhy, H. F., Georgiou, G., and Kahn, A. (2001). Frontal lobe changes in alcoholism: a review of the literature. Alcohol Alcohol. 36, 357–368. doi: 10.1093/alcalc/36.5.357
Moyon, S., Liang, J., and Casaccia, P. (2016). Epigenetics in NG2 glia cells. Brain Res. 1638, 183–198. doi: 10.1016/j.brainres.2015.06.009
Mulholland, P. J., Carpenter-Hyland, E. P., Woodward, J. J., and Chandler, L. J. (2009). Ethanol disrupts NMDA receptor and astroglial EAAT2 modulation of Kv2.1 potassium channels in hippocampus. Alcohol 43, 45–50. doi: 10.1016/j.alcohol.2008.10.001
Nagy, J. I., and Rash, J. E. (2000). Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res. Rev. 32, 29–44. doi: 10.1016/s0165-0173(99)00066-1
Namihira, M., and Nakashima, K. (2013). Mechanisms of astrocytogenesis in the mammalian brain. Curr. Opin. Neurobiol. 23, 921–927. doi: 10.1016/j.conb.2013.06.002
Navarro, A. I., and Mandyam, C. D. (2015). Protracted abstinence from chronic ethanol exposure alters the structure of neurons and expression of oligodendrocytes and myelin in the medial prefrontal cortex. Neuroscience 293, 35–44. doi: 10.1016/j.neuroscience.2015.02.043
Nave, K. A., and Trapp, B. D. (2008). Axon-glial signaling and the glial support of axon function. Annu. Rev. Neurosci. 31, 535–561. doi: 10.1146/annurev.neuro.30.051606.094309
Nave, K. A., and Werner, H. B. (2014). Myelination of the nervous system: mechanisms and functions. Annu. Rev. Cell Dev. Biol. 30, 503–533. doi: 10.1146/annurev-cellbio-100913-013101
Nedergaard, M., Ransom, B., and Goldman, S. A. (2003). New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 26, 523–530. doi: 10.1016/j.tins.2003.08.008
Nelson, A. D., and Jenkins, P. M. (2017). Axonal membranes and their domains: assembly and function of the axon initial segment and node of ranvier. Front. Cell. Neurosci. 11:136. doi: 10.3389/fncel.2017.00136
Newville, J., Valenzuela, C. F., Li, L., Jantzie, L. L., and Cunningham, L. A. (2017). Acute oligodendrocyte loss with persistent white matter injury in a third trimester equivalent mouse model of fetal alcohol spectrum disorder. Glia 65, 1317–1332. doi: 10.1002/glia.23164
Nualart-Marti, A., Solsona, C., and Fields, R. D. (2013). Gap junction communication in myelinating glia. Biochim. Biophys. Acta 1828, 69–78. doi: 10.1016/j.bbamem.2012.01.024
Nunez, Y. O., Truitt, J. M., Gorini, G., Ponomareva, O. N., Blednov, Y. A., Harris, R. A., et al. (2013). Positively correlated miRNA-mRNA regulatory networks in mouse frontal cortex during early stages of alcohol dependence. BMC Genomics 14:725. doi: 10.1186/1471-2164-14-725
Oberheim, N. A., Wang, X., Goldman, S., and Nedergaard, M. (2006). Astrocytic complexity distinguishes the human brain. Trends Neurosci. 29, 547–553. doi: 10.1016/j.tins.2006.08.004
Okamoto, H., Miki, T., Lee, K. Y., Yokoyama, T., Kuma, H., Wang, Z. Y., et al. (2006). Oligodendrocyte myelin glycoprotein (OMgp) in rat hippocampus is depleted by chronic ethanol consumption. Neurosci. Lett. 406, 76–80. doi: 10.1016/j.neulet.2006.07.023
Orthmann-Murphy, J. L., Abrams, C. K., and Scherer, S. S. (2008). Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 35, 101–116. doi: 10.1007/s12031-007-9027-5
Ozer, E., Sarioglu, S., and Gure, A. (2000). Effects of prenatal ethanol exposure on neuronal migration, neuronogenesis and brain myelination in the mice brain. Clin. Neuropathol. 19, 21–25.
Öztürk, N. C., Resendiz, M., Öztürk, H., and Zhou, F. C. (2017). DNA Methylation program in normal and alcohol-induced thinning cortex. Alcohol 60, 135–147. doi: 10.1016/j.alcohol.2017.01.006
Paemeleire, K. (2002). The cellular basis of neurovascular metabolic coupling. Acta Neurol. Belg. 102, 153–157.
Pandey, S. C., Kyzar, E. J., and Zhang, H. (2017). Epigenetic basis of the dark side of alcohol addiction. Neuropharmacology 122, 74–84. doi: 10.1016/j.neuropharm.2017.02.002
Pandey, S. C., Ugale, R., Zhang, H., Tang, L., and Prakash, A. (2008). Brain chromatin remodeling: a novel mechanism of alcoholism. J. Neurosci. 28, 3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008
Papp-Peka, A., Tong, M., Kril, J. J., De La Monte, S. M., and Sutherland, G. T. (2017). The differential effects of alcohol and nicotine-specific nitrosamine ketone on white matter ultrastructure. Alcohol Alcohol. 52, 165–171. doi: 10.1093/alcalc/agw067
Parpura, V., Heneka, M. T., Montana, V., Oliet, S. H., Schousboe, A., Haydon, P. G., et al. (2012). Glial cells in (patho)physiology. J. Neurochem. 121, 4–27. doi: 10.1111/j.1471-4159.2012.07664.x
Pfefferbaum, A., Lim, K. O., Zipursky, R. B., Mathalon, D. H., Rosenbloom, M. J., Lane, B., et al. (1992). Brain gray and white matter volume loss accelerates with aging in chronic alcoholics: a quantitative MRI study. Alcohol. Clin. Exp. Res. 16, 1078–1089. doi: 10.1111/j.1530-0277.1992.tb00702.x
Pfefferbaum, A., and Sullivan, E. V. (2002). Microstructural but not macrostructural disruption of white matter in women with chronic alcoholism. Neuroimage 15, 708–718. doi: 10.1006/nimg.2001.1018
Pfefferbaum, A., Sullivan, E. V., Mathalon, D. H., and Lim, K. O. (1997). Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcohol. Clin. Exp. Res. 21, 521–529. doi: 10.1111/j.1530-0277.1997.tb03798.x
Pfefferbaum, A., Rosenbloom, M., Crusan, K., and Jernigan, T. L. (1988). Brain CT changes in alcoholics: effects of age and alcohol consumption. Alcohol. Clin. Exp. Res. 12, 81–87. doi: 10.1111/j.1530-0277.1988.tb00137.x
Phillips, S. C., Harper, C. G., and Kril, J. J. (1990). The contribution of Wernicke’s encephalopathy to alcohol-related cerebellar damage. Drug Alcohol Rev. 9, 53–60. doi: 10.1080/09595239000185071
Pignataro, L., Varodayan, F. P., Tannenholz, L. E., Protiva, P., and Harrison, N. L. (2013). Brief alcohol exposure alters transcription in astrocytes via the heat shock pathway. Brain Behav. 3, 114–133. doi: 10.1002/brb3.125
Ponomarev, I. (2013). Epigenetic control of gene expression in the alcoholic brain. Alcohol Res. 35, 69–76.
Ponomarev, I., Stelly, C. E., Morikawa, H., Blednov, Y. A., Mayfield, R. D., and Harris, R. A. (2017). Mechanistic insights into epigenetic modulation of ethanol consumption. Alcohol 60, 95–101. doi: 10.1016/j.alcohol.2017.01.016
Ponomarev, I., Wang, S., Zhang, L., Harris, R. A., and Mayfield, R. D. (2012). Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J. Neurosci. 32, 1884–1897. doi: 10.1523/jneurosci.3136-11.2012
Popescu, B. F., Bunyan, R. F., Guo, Y., Parisi, J. E., Lennon, V. A., and Lucchinetti, C. F. (2013). Evidence of aquaporin involvement in human central pontine myelinolysis. Acta Neuropathol. Commun. 1:40. doi: 10.1186/2051-5960-1-40
Prat, A., Biernacki, K., Wosik, K., and Antel, J. P. (2001). Glial cell influence on the human blood-brain barrier. Glia 36, 145–155. doi: 10.1002/glia.1104
Purger, D., Gibson, E. M., and Monje, M. (2016). Myelin plasticity in the central nervous system. Neuropharmacology 110, 563–573. doi: 10.1016/j.neuropharm.2015.08.001
Rajan, P., and McKay, R. D. (1998). Multiple routes to astrocytic differentiation in the CNS. J. Neurosci. 18, 3620–3629.
Rajkowska, G., Hughes, J., Stockmeier, C. A., Javier Miguel-Hidalgo, J., and Maciag, D. (2013). Coverage of blood vessels by astrocytic endfeet is reduced in major depressive disorder. Biol. Psychiatry 73, 613–621. doi: 10.1016/j.biopsych.2012.09.024
Reichenbach, A., and Wolburg, H. (2012). “Astrocytes and ependymal glia,” in Neuroglia, 3rd Edn. eds H. Kettenmann and B. R. Ransom (Oxford, UK: Oxford University Press), 35–49.
Renau-Piqueras, J., Zaragoza, R., De Paz, P., Baguena-Cervellera, R., Megias, L., and Guerri, C. (1989). Effects of prolonged ethanol exposure on the glial fibrillary acidic protein-containing intermediate filaments of astrocytes in primary culture: a quantitative immunofluorescence and immunogold electron microscopic study. J. Histochem. Cytochem. 37, 229–240. doi: 10.1177/37.2.2642942
Ridge, J. P., Ho, A. M., Innes, D. J., and Dodd, P. R. (2008). The expression of NMDA receptor subunit mRNA in human chronic alcoholics. Ann. N Y Acad. Sci. 1139, 10–19. doi: 10.1196/annals.1432.053
Risher, M. L., Sexton, H. G., Risher, W. C., Wilson, W. A., Fleming, R. L., Madison, R. D., et al. (2015). Adolescent intermittent alcohol exposure: dysregulation of thrombospondins and synapse formation are associated with decreased neuronal density in the adult hippocampus. Alcohol. Clin. Exp. Res. 39, 2403–2413. doi: 10.1111/acer.12913
Roberts, R. C., Roche, J. K., and Mccullumsmith, R. E. (2014). Localization of excitatory amino acid transporters EAAT1 and EAAT2 in human postmortem cortex: a light and electron microscopic study. Neuroscience 277, 522–540. doi: 10.1016/j.neuroscience.2014.07.019
Rosenbloom, M., Sullivan, E. V., and Pfefferbaum, A. (2003). Using magnetic resonance imaging and diffusion tensor imaging to assess brain damage in alcoholics. Alcohol Res. Health 27, 146–152.
Rubio-Araiz, A., Porcu, F., Pérez-Hernández, M., García-Gutiérrez, M. S., Aracil-Fernández, M. A., Gutierrez-López, M. D., et al. (2017). Disruption of blood-brain barrier integrity in postmortem alcoholic brain: preclinical evidence of TLR4 involvement from a binge-like drinking model. Addict. Biol. 22, 1103–1116. doi: 10.1111/adb.12376
Saito, M., Chakraborty, G., Hui, M., Masiello, K., and Saito, M. (2016). Ethanol-induced neurodegeneration and glial activation in the developing brain. Brain Sci. 6:E31. doi: 10.3390/brainsci6030031
Samantaray, S., Knaryan, V. H., Patel, K. S., Mulholland, P. J., Becker, H. C., and Banik, N. L. (2015). Chronic intermittent ethanol induced axon and myelin degeneration is attenuated by calpain inhibition. Brain Res. 1622, 7–21. doi: 10.1016/j.brainres.2015.06.014
Schulte, T., Muller-Oehring, E. M., Pfefferbaum, A., and Sullivan, E. V. (2010). Neurocircuitry of emotion and cognition in alcoholism: contributions from white matter fiber tractography. Dialogues Clin. Neurosci. 12, 554–560.
Serwanski, D. R., Jukkola, P., and Nishiyama, A. (2017). Heterogeneity of astrocyte and NG2 cell insertion at the node of ranvier. J. Comp. Neurol. 525, 535–552. doi: 10.1002/cne.24083
Simard, M., Arcuino, G., Takano, T., Liu, Q. S., and Nedergaard, M. (2003). Signaling at the gliovascular interface. J. Neurosci. 23, 9254–9262.
Simons, M., and Nave, K. A. (2015). Oligodendrocytes: myelination and axonal support. Cold Spring Harb. Perspect. Biol. 8:a020479. doi: 10.1101/cshperspect.a020479
Simons, M., and Trajkovic, K. (2006). Neuron-glia communication in the control of oligodendrocyte function and myelin biogenesis. J. Cell Sci. 119, 4381–4389. doi: 10.1242/jcs.03242
Smith, T. L. (1997). Regulation of glutamate uptake in astrocytes continuously exposed to ethanol. Life Sci. 61, 2499–2505. doi: 10.1016/s0024-3205(97)00985-5
Smith, A. C. W. (2017). A glitch in the matrix: aberrant extracellular matrix proteolysis contributes to alcohol seeking. Biol. Psychiatry 81, 900–902. doi: 10.1016/j.biopsych.2017.02.1181
Smith, T. L., and Zsigo, A. (1996). Increased Na+-dependent high affinity uptake of glutamate in astrocytes chronically exposed to ethanol. Neurosci. Lett. 218, 142–144. doi: 10.1016/s0304-3940(96)13123-2
Snaidero, N., and Simons, M. (2017). The logistics of myelin biogenesis in the central nervous system. Glia 65, 1021–1031. doi: 10.1002/glia.23116
Spanagel, R., Pendyala, G., Abarca, C., Zghoul, T., Sanchis-Segura, C., Magnone, M. C., et al. (2005). The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nat. Med. 11, 35–42. doi: 10.1038/nm1163
Stefaniuk, M., Beroun, A., Lebitko, T., Markina, O., Leski, S., Meyza, K., et al. (2017). Matrix Metalloproteinase-9 and synaptic plasticity in the central amygdala in control of alcohol-seeking behavior. Biol. Psychiatry 81, 907–917. doi: 10.1016/j.biopsych.2016.12.026
Sun, G. Y., Danopoulos, V., and Sun, A. Y. (1979). The effect of chronic ethanol administration on myelin lipids. Curr. Alcohol. 7, 83–91.
Sutherland, G. T., Sheahan, P. J., Matthews, J., Dennis, C. V., Sheedy, D. S., Mccrossin, T., et al. (2013). The effects of chronic alcoholism on cell proliferation in the human brain. Exp. Neurol. 247, 9–18. doi: 10.1016/j.expneurol.2013.03.020
Sutherland, G. T., Sheedy, D., Sheahan, P. J., Kaplan, W., and Kril, J. J. (2014). Comorbidities, confounders, and the white matter transcriptome in chronic alcoholism. Alcohol. Clin. Exp. Res. 38, 994–1001. doi: 10.1111/acer.12341
Tajuddin, N., Moon, K. H., Marshall, S. A., Nixon, K., Neafsey, E. J., Kim, H. Y., et al. (2014). Neuroinflammation and neurodegeneration in adult rat brain from binge ethanol exposure: abrogation by docosahexaenoic acid. PLoS One 9:e101223. doi: 10.1371/journal.pone.0101223
Takano, T., Tian, G. F., Peng, W., Lou, N., Libionka, W., Han, X., et al. (2006). Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 9, 260–267. doi: 10.1038/nn1623
Tong, M., Yu, R., Silbermann, E., Zabala, V., Deochand, C., and De La Monte, S. M. (2015). Differential contributions of alcohol and nicotine-derived nitrosamine ketone (NNK) to white matter pathology in the adolescent rat brain. Alcohol Alcohol. 50, 680–689. doi: 10.1093/alcalc/agv102
Trindade, P., Hampton, B., Manhães, A. C., and Medina, A. E. (2016). Developmental alcohol exposure leads to a persistent change on astrocyte secretome. J. Neurochem. 137, 730–743. doi: 10.1111/jnc.13542
Tsai, G. (1998). Glutamatergic neurotransmission in alcoholism. J. Biomed. Sci. 5, 309–320. doi: 10.1159/000025345
Tsai, G. E., Ragan, P., Chang, R., Chen, S., Linnoila, V. M., and Coyle, J. T. (1998). Increased glutamatergic neurotransmission and oxidative stress after alcohol withdrawal. Am. J. Psychiatry 155, 726–732. doi: 10.1176/ajp.155.6.726
Tulisiak, C. T., Harris, R. A., and Ponomarev, I. (2017). DNA modifications in models of alcohol use disorders. Alcohol 60, 19–30. doi: 10.1016/j.alcohol.2016.11.004
Ullian, E. M., Christopherson, K. S., and Barres, B. A. (2004). Role for glia in synaptogenesis. Glia 47, 209–216. doi: 10.1002/glia.20082
Vallés, S., Pitarch, J., Renau-Piqueras, J., and Guerri, C. (1997). Ethanol exposure affects glial fibrillary acidic protein gene expression and transcription during rat brain development. J. Neurochem. 69, 2484–2493. doi: 10.1046/j.1471-4159.1997.69062484.x
Verkhratsky, A., and Nedergaard, M. (2018). Physiology of astroglia. Physiol. Rev. 98, 239–389. doi: 10.1152/physrev.00042.2016
Verkhratsky, A., and Parpura, V. (2010). Recent advances in (patho)physiology of astroglia. Acta Pharmacol. Sin. 31, 1044–1054. doi: 10.1038/aps.2010.108
Verkhratsky, A., Rodriguez, J. J., and Steardo, L. (2014). Astrogliopathology: a central element of neuropsychiatric diseases? Neuroscientist 20, 576–588. doi: 10.1177/1073858413510208
Wasseff, S. K., and Scherer, S. S. (2011). Cx32 and Cx47 mediate oligodendrocyte:astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol. Dis. 42, 506–513. doi: 10.1016/j.nbd.2011.03.003
Wegner, M. (2008). A matter of identity: transcriptional control in oligodendrocytes. J. Mol. Neurosci. 35, 3–12. doi: 10.1007/s12031-007-9008-8
Weng, J. T., Wu, L. S., Lee, C. S., Hsu, P. W., and Cheng, A. T. (2015). Integrative epigenetic profiling analysis identifies DNA methylation changes associated with chronic alcohol consumption. Comput. Biol. Med. 64, 299–306. doi: 10.1016/j.compbiomed.2014.12.003
Wu, J., Lee, M. R., Kim, T., Johng, S., Rohrback, S., Kang, N., et al. (2011). Regulation of ethanol-sensitive EAAT2 expression through adenosine A1 receptor in astrocytes. Biochem. Biophys. Res. Commun. 406, 47–52. doi: 10.1016/j.bbrc.2011.01.104
Yu, Y., Casaccia, P., and Lu, Q. R. (2010). Shaping the oligodendrocyte identity by epigenetic control. Epigenetics 5, 124–128. doi: 10.4161/epi.5.2.11160
Zabala, V., Tong, M., Yu, R., Ramirez, T., Yalcin, E. B., Balbo, S., et al. (2015). Potential contributions of the tobacco nicotine-derived nitrosamine ketone (NNK) in the pathogenesis of steatohepatitis in a chronic plus binge rat model of alcoholic liver disease. Alcohol Alcohol. 50, 118–131. doi: 10.1093/alcalc/agu083
Zahr, N. M., and Pfefferbaum, A. (2017). Alcohol’s effects on the brain: neuroimaging results in humans and animal models. Alcohol Res. 38, 183–206.
Zhang, Y., and Barres, B. A. (2010). Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 20, 588–594. doi: 10.1016/j.conb.2010.06.005
Zhang, X., Kusumo, H., Sakharkar, A. J., Pandey, S. C., and Guizzetti, M. (2014). Regulation of DNA methylation by ethanol induces tissue plasminogen activator expression in astrocytes. J. Neurochem. 128, 344–349. doi: 10.1111/jnc.12465
Zhou, Z., Yuan, Q., Mash, D. C., and Goldman, D. (2011). Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc. Natl. Acad. Sci. U S A 108, 6626–6631. doi: 10.1073/pnas.1018514108
Zimmermann, D. R., and Dours-Zimmermann, M. T. (2008). Extracellular matrix of the central nervous system: from neglect to challenge. Histochem. Cell Biol. 130, 635–653. doi: 10.1007/s00418-008-0485-9
Keywords: glia, alcoholism, postmortem, human, animal models, molecular markers, myelin, epigenetic
Citation: Miguel-Hidalgo JJ (2018) Molecular Neuropathology of Astrocytes and Oligodendrocytes in Alcohol Use Disorders. Front. Mol. Neurosci. 11:78. doi: 10.3389/fnmol.2018.00078
Received: 06 October 2017; Accepted: 28 February 2018;
Published: 20 March 2018.
Edited by:
Andrei Surguchov, University of Kansas Medical Center, United StatesReviewed by:
Susanna Amadio, Fondazione Santa Lucia (IRCCS), ItalyMark Z. Kos, University of Texas Rio Grande Valley Edinburg, United States
Copyright © 2018 Miguel-Hidalgo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José J. Miguel-Hidalgo, am1pZ3VlbC1oaWRhbGdvQHVtYy5lZHU=