 Kara L. Schneider
Kara L. Schneider Thomas Nyström
Thomas Nyström Per O. Widlund
Per O. Widlund- Department of Microbiology and Immunology, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
Protein quality control (PQC) is critical to maintain a functioning proteome. Misfolded or toxic proteins are either refolded or degraded by a system of temporal quality control and can also be sequestered into aggregates or inclusions by a system of spatial quality control. Breakdown of this concerted PQC network with age leads to an increased risk for the onset of disease, particularly neurological disease. Saccharomyces cerevisiae has been used extensively to elucidate PQC pathways and general evolutionary conservation of the PQC machinery has led to the development of several useful S. cerevisiae models of human neurological diseases. Key to both of these types of studies has been the development of several different model misfolding proteins, which are used to challenge and monitor the PQC machinery. In this review, we summarize and compare the model misfolding proteins that have been used to specifically study spatial PQC in S. cerevisiae, as well as the misfolding proteins that have been shown to be subject to spatial quality control in S. cerevisiae models of human neurological diseases.
Introduction
The presence of protein inclusions is a hallmark of many age-related neurological diseases (Kaytor and Warren, 1999; Koo et al., 1999; Paulson, 1999). There is much evidence to suggest that the misfolded proteins generated during progression of these diseases are deposited into inclusions by the cell’s protein quality control (PQC) machinery to shield cellular components from their toxic properties (Saudou et al., 1998; Arrasate et al., 2004; Takahashi et al., 2008; Tyedmers et al., 2010).
Though protein deposits are common to many neurological diseases, inclusions can also be seen in aged neuronal cells of healthy animals (Fiori, 1987; Peters et al., 1991), reinforcing the idea that inclusions are a normal response of the PQC machinery to misfolded proteins. However, there are several lines of evidence for an age-related decline in the cells ability to process damaged proteins, which may explain the increased incidence of neurological disease with age (Cuervo and Dice, 2000; Koga et al., 2011; Kruegel et al., 2011; Andersson et al., 2013; Oling et al., 2014; Saez and Vilchez, 2014).
While the observed inclusions vary in form and intracellular location across species ranging from bacteria to humans, the formation of cellular inclusions in response to aberrantly folded proteins is evolutionarily conserved and is a consequence of the cell’s ongoing effort to maintain protein homeostasis, or proteostasis. Organisms across the evolutionary tree have evolved systems of temporal quality control and spatial quality control to maintain a functioning proteome (Hartl and Hayer-Hartl, 2009; Mogk and Bukau, 2017). The chaperones of the temporal quality control system ensure proper folding of newly synthesized proteins, attempt to refold misfolded proteins and promote degradation of those that cannot be effectively refolded (Hill et al., 2017; Josefson et al., 2017; Sontag et al., 2017). A system of spatial quality control runs in parallel to sort and deposit potentially harmful misfolded proteins and its action is most apparent when the temporal quality control system fails or is overloaded. When this occurs, misfolded proteins are partitioned into inclusions which shield the cell from their toxicity and can aid in their eventual clearance (Taylor et al., 2003; Escusa-Toret et al., 2013; Wolfe et al., 2013).
Due to the general conservation of the PQC machinery, several model organisms have been used to study not only the pathways involved in PQC, but also how they manage misfolded proteins that have been identified as important in the development of several human age-related neurological diseases. One of these is the proven model eukaryote, Saccharomyces cerevisiae. It is well-suited for the study of PQC, particularly in the context of aging, because aging can be studied in two major contexts: non-proliferative and proliferative cells. Yeast can be grown to stationary phase, where they eventually lose viability. This loss of viability, sometimes called chronological aging, can in many ways mimic aging experienced by differentiated cells like neuronal cells. Furthermore, proliferative cells, like stem cells, can also be modeled since yeast similarly undergo asymmetric cell divisions.
Asymmetric divisions are important in the context of spatial quality control because they allow damage, notably damaged proteins, to be segregated asymmetrically. During asymmetric cell division in yeast, the daughter cell is rejuvenated and one reason for this is that damaged or misfolded proteins are retained in the mother cell as part of the system of spatial quality control (Aguilaniu et al., 2003; Shcheprova et al., 2008). Evidence is emerging that some stem cell types also segregate damage asymmetrically to allow the stem cell lineage to propagate free of damage (Rujano et al., 2006; Fuentealba et al., 2008; Bufalino et al., 2013; Ogrodnik et al., 2014). Since this asymmetrical division of damage is limited to proliferating cells, it is possible that the higher incidence of inclusions in differentiated cells like neurons may be a consequence of the inability of the spatial quality control machinery to remove damage through division. Spatial PQC, therefore, plays a key role in both aging and age-related neurological disease and yeast has been successfully used to study the pathways involved in both contexts.
A key reason for this success has been due to the development of model proteins that either probe how the cell responds to misfolding proteins in general or how they deal with those that are thought to be the main causative agents of human disease. Model misfolding proteins are especially useful in the study of both temporal and spatial quality control as they can be used to track processing by the quality control machinery with minimal perturbation to the system itself. Fluorescently tagged substrates are indispensable, particularly in the study of spatial quality control, as they allow straightforward tracking of aggregate formation and localization by light microscopy. They also allow the study of spatial quality control in relation to temporal control by following aggregates in the cell through time. Several misfolding model proteins have been developed for these purposes. Furthermore, several human disease proteins have also been successfully used in yeast to study both the nature of the toxicity of the misfolded proteins, as well as how the PQC machinery responds to these proteins. Herein, we review the major model proteins used in S. cerevisiae to study spatial PQC pathways and the role of spatial quality control in the molecular basis of human disease.
Model Misfolding Proteins
Model misfolding proteins have helped to define the different quality control sites that have been identified in yeast (Kaganovich et al., 2008; Miller et al., 2015; Hill et al., 2017) (Figure 1). When induced, they often initially accumulate at stress foci, called CytoQs/Q-bodies/peripheral aggregates, in the cytoplasm or at the surface of organelles including the endoplasmic reticulum, mitochondria, and vacuole (Specht et al., 2011; Spokoini et al., 2012; Escusa-Toret et al., 2013; Miller et al., 2015). During prolonged stress, the aggregates coalesce into larger foci, often called inclusions, which are deposited or collected at several defined sites: the juxtanuclear quality control (JUNQ), the intranuclear quality control (INQ) and the insoluble protein deposit (IPOD) site (Kaganovich et al., 2008; Miller et al., 2015). They also can associate with, and be imported into, mitochondria (Zhou et al., 2014; Ruan et al., 2017). Other sites are likely to exist as some misfolding human disease models do not appear to localize to these defined sites (Tenreiro et al., 2014; Farrawell et al., 2015). The misfolding models are summarized in Table 1. We grouped them into three general categories: Temperature-sensitive (Ts) misfolding proteins, continuously misfolding proteins, and human disease proteins. For each category, we will first describe the development of model proteins for S. cerevisiae and then discuss how they have been used to elucidate spatial quality control pathways.
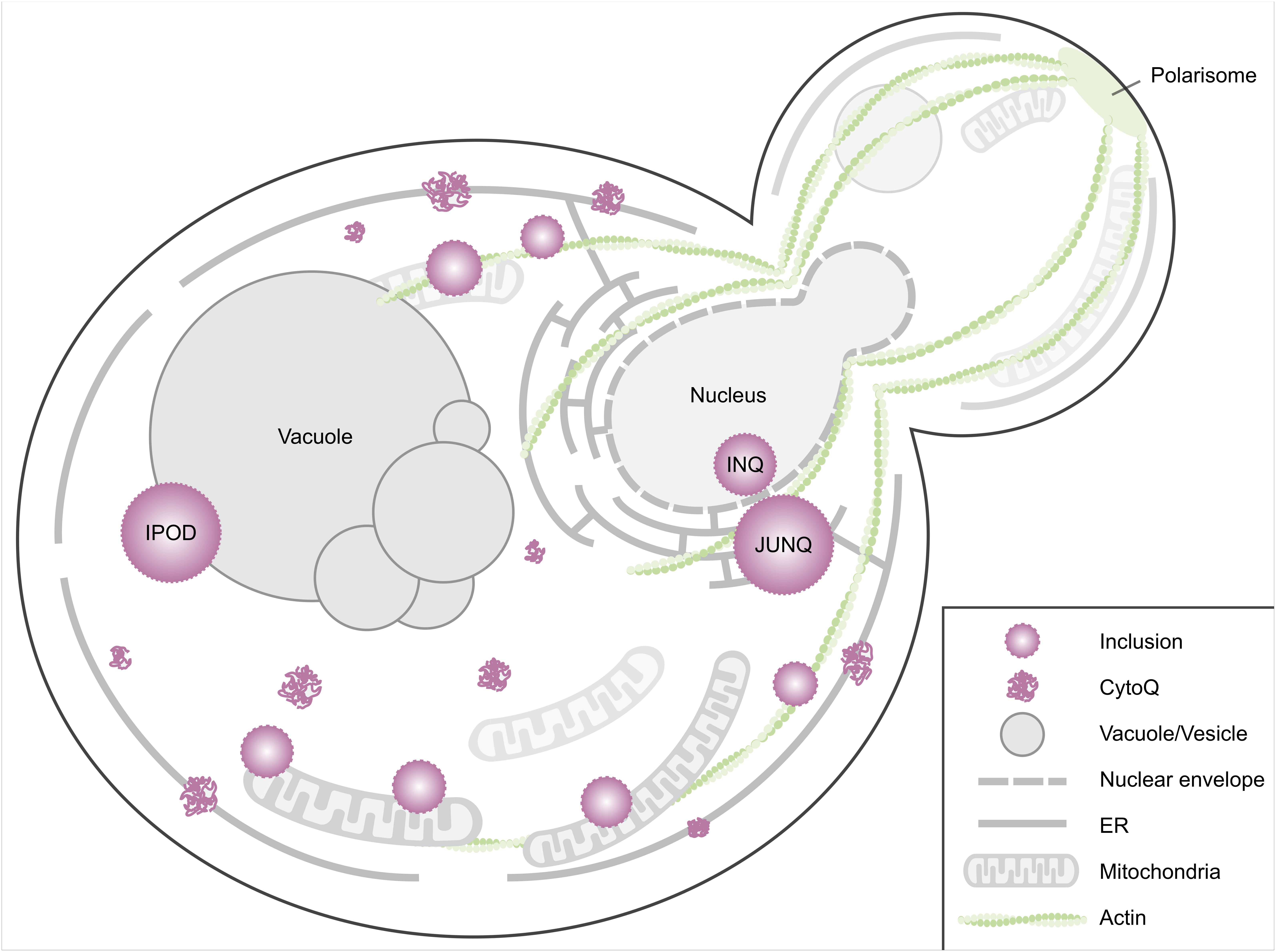
FIGURE 1. Spatial protein quality control sites in Saccharomyces cerevisiae. When induced to misfold, proteins aggregate in the cytosol and on the ER membrane (Escusa-Toret et al., 2013). These initial cytosolic aggregates are called CytoQ (Miller et al., 2015), stress foci (Spokoini et al., 2012), peripheral aggregates (Specht et al., 2011), cytosolic puncta (Kaganovich et al., 2008) or Q-bodies (Escusa-Toret et al., 2013). These coalesce into larger structures, usually referred to as inclusions. Smaller inclusions have been observed tethered to actin cables (Liu et al., 2010; Song et al., 2014) or are captured by mitochondria (Zhou et al., 2014; Böckler et al., 2017). Yeast cells have several distinct larger inclusions including, but not limited to, the intranuclear quality control site (INQ), the juxtanuclear quality control site (JUNQ), and the perivacuolar IPOD site (Kaganovich et al., 2008; Miller et al., 2015). Other IPOD-like peripheral inclusions likely exist as some aggregates of model substrates do not co-localize with the IPOD, e.g., the non-amyloidogenic disease protein OPTN (Kryndushkin et al., 2012). An additional site, the age-associated protein deposit site (APOD, not depicted here) has been identified in aged cells (Saarikangas and Barral, 2015).
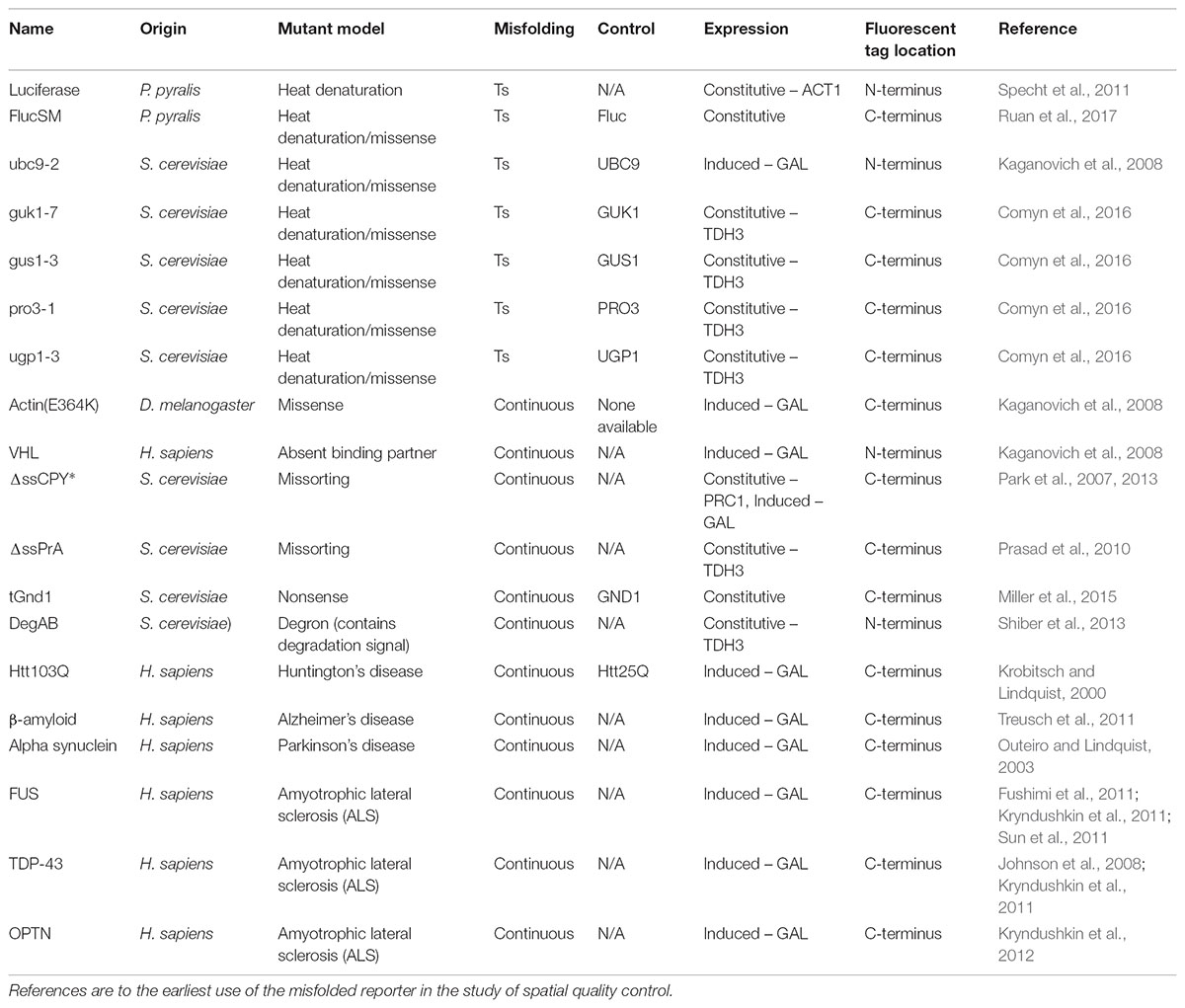
TABLE 1. Fluorescently tagged misfolding model proteins.
Temperature-Sensitive Misfolding Proteins
Luciferase, FlucSM/DM
Photinus pyralis luciferase was an early model substrate used in the study of PQC. It was selected to elucidate the cellular chaperone machinery in Escherichia coli because it was thermolabile, could be reactivated in vivo, and activity could readily be monitored by luminescence assay (Schröder et al., 1993). A fluorescently tagged version was later used to study spatial quality control in E. coli (Winkler et al., 2010) before versions were adapted for use in S. cerevisiae (Specht et al., 2011). Mutant versions of Luciferase, FlucSM/DM were developed to be more susceptible to heat denaturation (Gupta et al., 2011). S. cerevisiae compatible constructs were recently developed (Ruan et al., 2017).
Ubc9ts (ubc9-2)
Temperature-sensitive mutants of S. cerevisiae genes have been used to study gene function for decades, particularly of essential genes. Many Ts alleles behave as effective nulls and one mechanism for this was shown with the gene product of a Ts allele of the ubiquitin conjugating enzyme, Ubc9. Several mutant ubc9 proteins were shown to be short-lived at the restrictive temperature and the observed rapid breakdown could be suppressed and was dependent on proteasome activity (Betting and Seufert, 1996). A GFP tagged version of ubc9-2 containing the point mutation Y69L was then used to show that misfolding proteins partition between at least two quality control compartments, the JUNQ and IPOD (Kaganovich et al., 2008).
guk1-7, gus1-3, pro3-1, ugp1-3
A screen of a panel of 22 Ts alleles of six essential genes encoding predominantly cytoplasmic proteins showed that a significant fraction was degraded at the restrictive temperature, clearly demonstrating degradation as a major mechanism for Ts phenotypes (Khosrow-Khavar et al., 2012). Four unstable mutants were fluorescently tagged and used as model misfolding substrates: guk1-7, gus1-3, pro3-1, and ugp1-3 (Comyn et al., 2016). A Ts mutant of a guanylate kinase, guk1-7, was selected for further characterization. The temperature sensitivity of guk1-7 is a consequence of four missense mutations. It was shown to co-localize with Hsp104-mCherry and with Hsp42-mCherry foci (Comyn et al., 2016), and based on these markers, they are likely deposited into one or more of the major PQCs like Q-bodies, JUNQ/INQ or IPOD.
Quality Control of Temperature-Sensitive Proteins
Many temperature sensitive proteins are not degraded at the restrictive temperature. The conditional lethal phenotype caused by these stable variants are likely due to local perturbations in domain structure caused by the mutations. This local effect is apparent in Ts alleles that encode homomultimeric proteins where intragenic complementation is possible (Sundberg and Davis, 1997). Dominant Ts alleles have been proposed to affect protein structure locally (McMurray, 2014). Indeed, these local effects may generally help to explain the wide range of phenotypes that can be observed with different Ts mutants of the same gene.
In contrast, a significant percentage of temperature-sensitive mutants display a recessive, null phenotype at the restrictive temperature that is often due to degradation of the expressed protein (Khosrow-Khavar et al., 2012). The null phenotype of unstable Ts alleles can often be rescued by removal of one or a combination of genes involved in PQC, indicating that the mutations do not significantly affect protein function, but instead cause partial unfolding that the quality control machinery recognizes, resulting in that protein being targeted for destruction (Betting and Seufert, 1996; Gardner et al., 2005; Khosrow-Khavar et al., 2012). It is this class of Ts mutants that has been adapted for the study of PQC.
Of the Ts substrates adapted, Luciferase is the only one with several well-established assays to monitor enzymatic activity, which is one of the main reasons it was initially chosen as a model substrate (Schröder et al., 1993). When used as a spatial quality control substrate, it has a rather mild aggregation phenotype that either requires a higher 42°C heat shock or proteasomal inhibition for aggregates to be clearly visible. As a consequence, destabilized versions of luciferase, FlucSM and FlucDM were later engineered to make them more amenable to studies of proteome stress (Gupta et al., 2011). FlucSM expressed in yeast forms clear aggregates at 42°C, but experiments with milder 37°C heat stress have not yet been reported in yeast (Ruan et al., 2017). Luciferase is also the only exogenous Ts substrate used in the study of spatial quality control. This has the potential advantage that it is unlikely to specifically interact with endogenous proteins. However, it may also not be recognized by the PQC machinery in the same way as an endogenous protein, potentially limiting its use in elucidating endogenous spatial quality control pathways.
The remaining substrates are Ts versions of S. cerevisiae proteins which have been selected to be unstable at or above 37°C. Ubc9-2 has often been used as a spatial quality control substrate since it was originally used to help define the JUNQ and IPOD deposition sites (Kaganovich et al., 2008). The guk1-7, gus1-3, pro3-1, and ugp1-3 proteins expand the number of available Ts substrates significantly and allow interesting comparisons between processing of endogenous misfolded proteins since they differ in their wild-type gene function, cellular localization and pathways of degradation upon misfolding. At the permissive temperature, these Ts substrates have been reported to have the following localization patterns: ubc9-2, nucleus; guk1-7, nucleus and cytoplasm; gus1-3, cytoplasm and mitochondria; pro3-1, cytoplasm; ugp1-3, cytoplasm and plasma membrane1. It is perhaps not surprising that these model proteins show notable differences in the way they are processed by the PQC machinery. For example, degradation of pro3-1 is partially dependent on the E3 ubiquitin ligase San1 for degradation while guk1-7 is significantly less dependent. They also show different responses to deletion of prefoldin subunits. Furthermore, gus1-3 appears to be unique in that it does not appear to depend on the proteasome for degradation (Khosrow-Khavar et al., 2012). Table 2 summarizes what is known about the processing of the Ts model substrates.
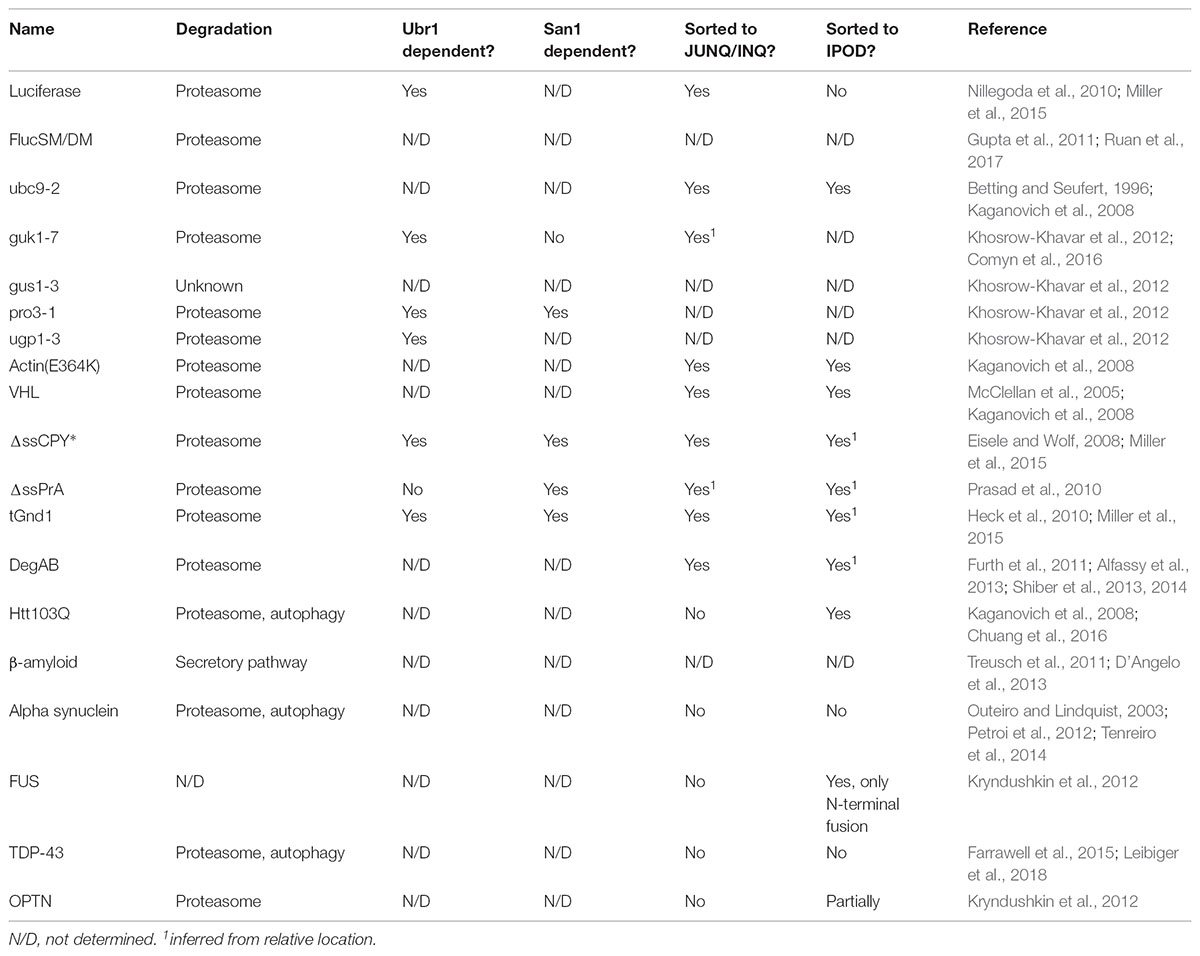
TABLE 2. Spatial quality control pathways implicated in processing of the misfolding model substrates, as well as dependence on the proteasome for degradation.
There are further subclasses of Ts proteins that are important to distinguish between, especially in the context of quality control. Some proteins are TL (thermolabile) while others are TSS (temperature sensitive synthesis) (Sadler and Novick, 1965; McMurray, 2014) (Figure 2). TL mutants are universally destabilized at the restrictive temperature, whereas TSS mutants are those that only misfold during synthesis. In yeast, a clear distinction between these phenotypes was shown with Ts mutants of Gal80 (Matsumoto et al., 1978). These unique properties can be exploited to examine whether the PQC machinery handles misfolding of newly synthesized or aged proteins differently. Of the Ts proteins used to study spatial quality control, only Ubc9ts has been characterized in this way and was shown to be TL as it could form foci during heat shock even after expression was shut off (Escusa-Toret et al., 2013). For this reason, we cannot generally distinguish between TL and TSS in this review.
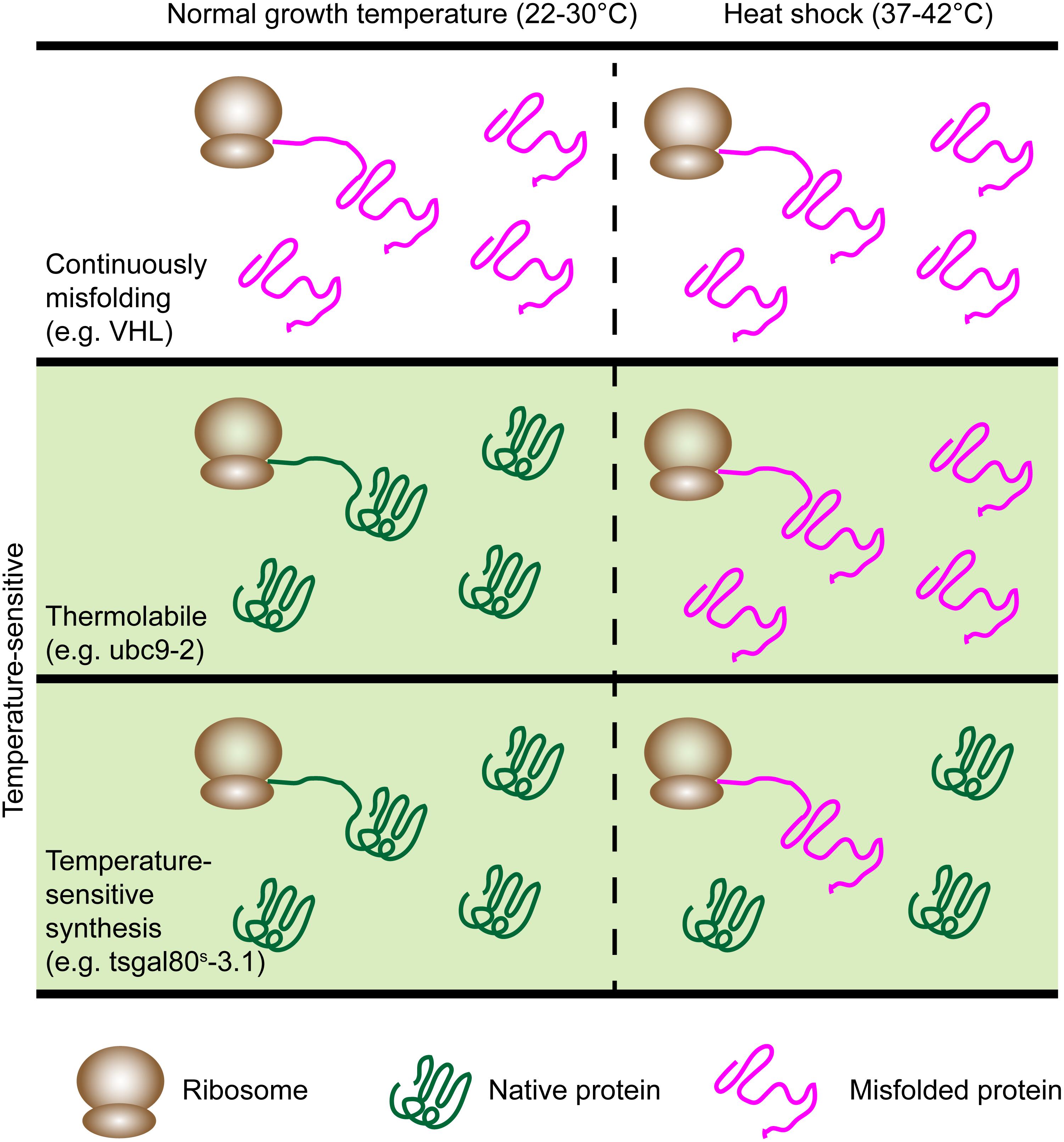
FIGURE 2. The model proteins have different patterns of misfolding. Continuously misfolding proteins misfold at all temperatures. Ts proteins (green panel) misfold during heat shock and fall into two classes: Thermolabile (TL) and temperature-sensitive synthesis (TSS). TL proteins universally misfold during heat shock while TSS proteins misfold only if synthesized during heat shock. Examples are listed for each category.
Continuously Misfolding Proteins
Actin(E364K)
The actin mutant actin(E364K) was originally isolated in Drosophila melanogaster (Drummond et al., 1991) and was later shown to be degraded by the PQC machinery (McClellan et al., 2005). A GFP tagged version localized to quality control compartments seen with ubc9-2 (Kaganovich et al., 2008).
von Hippel–Lindau (VHL)
The von Hippel–Lindau (VHL) tumor-suppressor protein is subject to chaperone mediated folding in mammalian cells (Feldman et al., 1999). Tumor-causing mutations that disrupt VHL binding to Elongin B/C leads to misfolding and degradation by the proteasome (Feldman et al., 1999; Schoenfeld et al., 2000). Wild type VHL was shown to be degraded in yeast due to absence of Elongin B/C and this was similarly dependent on the ubiquitin-proteasome pathway (McClellan et al., 2005). GFP tagged VHL was shown to be subject to spatial quality control in yeast similar to ubc9-2 and actin(E346K) (Kaganovich et al., 2008).
ΔssCPY∗
The first mutant version of the vacuolar enzyme carboxypeptidase Y (Y, yscY, CPY, PRC1) was isolated in 1975 (Wolf and Fink, 1975) in a screen with the purpose of investigating the function of the proteinase itself. However, CPY∗ did not localize to the vacuole, its regular destination, but instead was retained in the ER for proteasome-independent rapid degradation and was found to be misfolded (Finger et al., 1993).
Many derivatives of CPY have been used to study their degradation or PQC pathways, which differ depending on the domains they are fused to, e.g., the fusion to a transmembrane domain to study ER membrane proteins (Stolz and Wolf, 2012). One such derivative is ΔssCPY∗, which is a cytoplasmic misfolding substrate made by eliminating the ER-targeting signal sequence from CPY∗. Though originally used as a negative control when studying ERAD dependent degradation, (Medicherla et al., 2004) GFP tagged ΔssCPY∗ was shown to form inclusions and is subject to spatial quality control (Prasad et al., 2010; Park et al., 2013). Based on this construct, a similar cytosolic misfolding protein, ΔssPrA, was made using vacuolar proteinase A (PEP4) (Prasad et al., 2010).
tGnd1
A dosage suppressor screen using a high copy (2 μ) genomic library was performed to identify factors involved in stabilization of the misfolded model ΔssCPY∗ (Heck et al., 2010). The recovered hits were found to express truncated proteins, one of which was a truncated form of Gnd1, a phosphogluconate dehydrogenase. The truncation proved to be a competing PQC substrate. Fluorescently tagged tGnd1 was shown to localize to JUNQ/INQ and CytoQ deposits (Miller et al., 2015).
DegAB
A study of the degradation signal (degron) of the kinetochore protein, Ndc10, showed that it functions autonomously, as it leads to degradation of various other stable proteins when it is attached (Furth et al., 2011). This degron consists of two parts, DegA and DegB, leading to the collective name DegAB (Alfassy et al., 2013). This model degron differs from conventionally used terminally misfolding model substrates for PQC as it does not aggregate spontaneously, is not cytotoxic and can model mildly misfolded PQC substrates, which remain soluble and derive from an endogenous yeast protein (Shiber et al., 2013). A GFP fusion of DegAB, GFP-DegAB, can be used to study spatial PQC and generally forms two inclusions, one of which is juxtanuclear.
Quality Control of Continuously Misfolding Proteins
All non-thermolabile misfolding proteins presented here are targeted for degradation via the ubiquitin-proteasome pathway (Schoenfeld et al., 2000; McClellan et al., 2005; Park et al., 2007; Heck et al., 2010; Furth et al., 2011). Nonetheless, the factors required for degradation of substrates differ. For example, actin(E364K) and VHL have been compared in the same study and it was shown that Hsp90 is required for VHL degradation but not for actin(E364K) degradation. Additionally, VHL requires the Hsp70 chaperone Ssa1 and its co-chaperone Sti1 for degradation (McClellan et al., 2005). ΔssCPY∗ and tGnd1 share a requirement for the Ubr1 and San1 E3 ubiquitin ligases for their degradation (Eisele and Wolf, 2008; Heck et al., 2010; Miller et al., 2015). Similarly, DegAB was found to depend on Ssa1 or Ssa2 (Shiber et al., 2013).
Another similarity of all non-thermolabile substrates studied so far in this regard is their spatial sorting in the cell, as they have been observed to at least partially localize to both the JUNQ/INQ and IPOD compartment upon aggregation. Actin(E364K) and VHL both localize to JUNQ/INQ and IPOD but VHL requires heat shock for localization to the IPOD (Kaganovich et al., 2008). Proteasome inhibition alone results in a single nuclear inclusion, most likely the JUNQ/INQ (Kaganovich et al., 2008; Specht et al., 2011; Spokoini et al., 2012; Miller et al., 2015). ΔssCPY∗ and tGnd1 have been demonstrated to localize to the JUNQ/INQ in the stabilizing ubr1Δ san1Δ background in which these model proteins are no longer, or only marginally, ubiquitinated. This observation lead to the conclusion that ubiquitination is not a requirement for JUNQ/INQ targeting (Miller et al., 2015). In these experiments, tGnd1 is also targeted to a second cytoplasmic focus, which most likely corresponds to the IPOD, while ΔssCPY∗ appears exclusively targeted to INQ. However, cytoplasmic foci have also been observed for ΔssCPY∗. DegAB foci were shown to co-localize with Hsp42 and Hsp104 (Shiber et al., 2013) and detergent solubility properties of DegAB were determined to be similar to those of ΔssCPY∗ as measured by flow cytometry (Shiber et al., 2014). In summary, there are clear differences in sorting of these model proteins, which makes it reasonable to use more than one misfolding substrate when general conclusions about spatial sorting pathways are to be drawn.
Human Disease Proteins
Age-associated proteopathies have been increasingly modeled in yeast to gain more nuanced insight into the molecular bases of the diseases while circumventing potential ethical issues such as those with patient samples. The successful introduction of a range of different disease model proteins highlights the versatility and applicability of this model organism in the study of proteostasis, aging, and disease development (Braun et al., 2010). Different types of high-throughput screens with budding yeast are particularly powerful and have been used, in part, to find modifiers of cytotoxicity of disease proteins (Willingham et al., 2003; Cooper et al., 2006; Liang et al., 2008; Elden et al., 2010; Ju et al., 2011; Kayatekin et al., 2014). Importantly, fluorescently tagged versions of several proteins implicated in disease progression have been introduced to study how the spatial quality control machinery handles them and to help determine what contributes to their toxicity in diseased neurons. Here, we focus exclusively on substrates that have been shown to be subject to spatial quality control and aim to provide guidance in the selection of model substrates used to study spatial quality control pathways relevant to these proteopathies. For more thoroughly detailed descriptions of yeast models of human neurodegenerative disease, we refer the reader several excellent reviews (Braun et al., 2010; Pereira et al., 2012; Tenreiro et al., 2013; Braun, 2015; Menezes et al., 2015; Fruhmann et al., 2017). The disease models are summarized below and common constructs are listed in Table 1.
Huntington’s Disease
Htt103Q/Htt97Q
Yeast models for characterization of Huntingtin (Htt), the protein that is mutated in patients of Huntington’s disease, were established by testing variants of exon 1 of the N-terminus of human Huntingtin. These differ in the lengths of the polyglutamine expansions that modify aggregation behavior (Krobitsch and Lindquist, 2000). The Huntingtin model protein commonly used in yeast is Htt103Q, which is named after the number of glutamines in the expansion. A non-expanded variant, Htt25Q, is used as a control. Non-expanded variants, like Htt25Q, display diffuse localization in the cytoplasm, while expansions of 72 glutamines or more cause the proteins to form visible aggregates of varying size and quality (Krobitsch and Lindquist, 2000). Interestingly, it has been reported that an intermediate length huntingtin with a 47 glutamine expansion forms aggregates in yeast upon entry into stationary phase, which supports the use of stationary phase cells as a reasonable model for aged neuronal cells (Cohen et al., 2012). Models with longer expansions, such as Htt103Q, have different patterns of localization, ranging from on major focus per cell to multiple foci, that depend on expression levels and the regions flanking the polyglutamine expansion (Krobitsch and Lindquist, 2000; Duennwald et al., 2006; Wang et al., 2009; Song et al., 2014; Berglund et al., 2017).
Alzheimer’s Disease
β-Amyloid
Alzheimer’s disease (AD) models focus on one of the proteins implicated in the pathology of AD, the amyloid-β peptide (Aβ), as extracellular amyloid plaques containing Aβ appear in affected individuals. The Aβ peptides arise through cleavage of the amyloid precursor protein (APP) via the amyloidogenic pathway, resulting in different sizes of Aβ, mostly Aβ40 and Aβ42 (O’Brien and Wong, 2011). The latter is more aggregation-prone and nucleates plaques in diseased individuals and is therefore often selected as a model. A GFP-Aβ42 fusion protein was shown to form aggregates and induced a stress response (Caine et al., 2007). Toxicity models expressing the Aβ peptide were established later by targeting Aβ to the secretory pathway (Treusch et al., 2011; D’Angelo et al., 2013). Expression via an inducible promoter caused cytotoxicity without extensive cell death compared to a less toxic Aβ40 peptide control (Treusch et al., 2011).
Parkinson’s Disease
Alpha Synuclein
Alpha synuclein (aSyn) is a lipid-binding protein and the main constituent of Lewy Bodies, which are protein deposits occurring in diseased neurons that are affected by Parkinson’s Disease (PD) and other disorders. aSyn and derivatives were first investigated in mammalian cells (McLean et al., 2001) and later used to develop a PD model in yeast (Outeiro and Lindquist, 2003). While several PD models have been established in yeast [reviewed in (Menezes et al., 2015)], aSyn is the most commonly used model protein. Expression of aSyn in yeast causes dose-dependent cytotoxicity, a phenotype reminiscent of what is seen in human cells. Toxicity can therefore be modified by inducing different levels of expression by regulatable promoters or by changing plasmid copy number. As with human cells, aSyn has been reported to localize mainly to the plasma membrane and partially to the cytoplasm. However, overexpression causes visible aggregation of aSyn into cytosolic inclusions (Outeiro and Lindquist, 2003).
Amyotrophic Lateral Sclerosis (ALS)
FUS, TDP-43, OPTN
Mutations associated with amyotrophic lateral sclerosis (ALS) appear in the proteins SOD1, FUS, TDP-43, and OPTN which become misfolded and aggregate. Yeast studies have focused on FUS and especially TDP-43. Both proteins act as DNA/RNA-binding proteins and are found in neuronal inclusions of affected individuals. A TDP-43 model in yeast mimics several disease characteristics. Expression from an inducible plasmid causes it to form nuclear inclusions, while higher expression induces mislocalization to the cytoplasm where it forms visible inclusions (Johnson et al., 2008). These cytoplasmic aggregates are toxic to the cell. Similarly, FUS models have shown that expression in yeast is also cytotoxic (Fushimi et al., 2011; Kryndushkin et al., 2011; Sun et al., 2011). The model protein forms numerous cytoplasmic aggregates, which co-localize with and cause formation of RNA processing sites (P-bodies and stress granules) as observed in human cells (Kryndushkin et al., 2011). Another protein known to form inclusions in ALS-affected individuals is optineurin (OPTN). In a recently developed yeast model, wild type OPTN was shown to be toxic, as were versions with disease-causing mutations (Kryndushkin et al., 2012).
Quality Control of Human Disease Proteins
The disease proteins summarized above do not require stress conditions such as heat shock or proteasome inhibition to induce aggregate formation since they aggregate autonomously. Therefore, they are most commonly expressed using inducible promoters (Table 1). Additionally, it should be noted that none of the human misfolding proteins discussed here have orthologs in yeast.
The spatial quality control mechanisms handling several disease model proteins in yeast have not yet been extensively studied, however, it appears that amyloidogenic proteins in general are targeted to the IPOD compartment, which has been shown using Htt mutant proteins (Kaganovich et al., 2008; Escusa-Toret et al., 2013). The aggregates formed by aSyn are likely neither localized to JUNQ nor IPOD, as they did not co-localize with any of the commonly used markers for these quality control compartments (Outeiro and Lindquist, 2003; Tenreiro et al., 2014). In contrast to the other disease model proteins, TDP-43, FUS, and OPTN form non-amyloid aggregates. The spatial quality control mechanisms involved in the transport and processing of TDP-43, FUS, and OPTN have not been extensively investigated in yeast. A study using a mouse cell line concluded that the inclusions visible for TDP-43 and FUS might be distinct from several of the known quality control compartments: aggresome, JUNQ and IPOD (Farrawell et al., 2015). However, both TDP-43 and FUS have been reported to co-localize and physically interact in yeast (Kryndushkin et al., 2011), suggesting involvement of similar spatial PQC mechanisms for both model proteins. Furthermore, a GFP tagged version of FUS localizes to the IPOD while OPTN also shows only partial localization to this quality control site (Kryndushkin et al., 2012). OPTN forms non-amyloid cytoplasmic aggregates, which are distinct from FUS and TDP-43 foci, as they do not exclusively co-localize. OPTN appears to be handled in a unique way by the cell in that a single focus appears early after induction of expression, while upon later time points several small additional foci become visible. The foci only partially overlap with previously characterized model proteins reported to localize to the IPOD, further indicating that additional deposition sites may exist in the cytoplasm.
General Considerations for Model Selection
When selecting a candidate model misfolding protein, it is useful to consider what type of misfolding is to be modeled, the temperature sensitivity, whether a properly folding control is available, the promoter used for expression and the effects of the fluorescent tag. The categories are listed in Table 1 and summarized below.
Yeast vs. Non-yeast
Both mutated yeast proteins and non-yeast proteins have been used to study spatial quality control in S. cerevisiae. Use of mutated native proteins, such as the frequently used Ubc9ts, has the general advantage that it most closely mimics a typical error during protein production. The yeast spatial PQC machinery is more likely to properly recognize a misfolded endogenous protein compared to a non-native protein and it is therefore more likely to be sequestered to an “authentic” deposition site in the cell. However, mutated yeast proteins may also cause dominant negative effects, particularly when overexpressed, for example by sequestration of native binding partners. Non-native proteins, particularly those lacking yeast homologs, are less likely to bind endogenous proteins and can therefore be useful alternatives in this regard.
Mutant Model
Aberrant folding of proteins in wild-type cells can happen for several reasons. It is a major consequence of stress, such as with heat-induced denaturation. It can also be a consequence of mutations or a result of errors during protein production. Additionally, changes in expression can also result in a lack of binding partners necessary for proper folding. Model misfolding proteins have been engineered or selected to mimic the misfolding that occurs in these instances so that specific responses by the PQC machinery can be elucidated.
Temperature-Sensitive vs. Continuously Misfolding Reporters
Ts substrates are useful as unfolding can be triggered easily and rapidly by temperature shift so that the immediate response to unfolded proteins can be studied. In the case of TL substrates, it also allows decoupling from translation as one can terminate translation using cycloheximide before temperature shift. However, this temperature shift will also trigger the general heat shock response, which upregulates a range of chaperones. Non-thermolabile proteins do not require this temperature shift; therefore, while chaperones may be induced in response to the continuously misfolding substrate, temperature-induced changes in the levels of molecular chaperones are avoided.
Normally Folding Controls
Negative controls play an important role in studies on PQC as it is important to assess whether observable effects on PQC can be actually attributed to the misfolding protein itself and not to other factors such as their fluorescent tag or dominant negative effects. Negative controls exist for a subset of the human disease models. The Huntingtin models expressed in yeast, Htt103Q or other variants with expanded polyQ stretches, can be compared to a diffusely distributed cytosolic version, Htt25Q, which serves as a wild type control. Similarly, there are comparably less toxic variants of the amyloid beta peptide, which can be used as controls in some experimental setups. Alpha-synuclein aggregation depends on the level of expressed protein. While high expression levels result in aggregation, expression from a low copy number plasmid leaves the protein localized to the plasma membrane and the cytoplasm. A similar principle applies to TDP-43.
Several misfolding proteins can be directly compared to a wild-type control. For example, Ubc9 is used as a control for ubc9-2 (Kaganovich et al., 2008). Corresponding wild-type alleles are also available for guk1-7, gus1-3, pro3-1, and ugp1-3 (Khosrow-Khavar et al., 2012). Wild-type Gnd1 is used as a control for the truncated misfolding tGnd1 (Heck et al., 2010). However, the majority of the continuously misfolding proteins including VHL, actin(E364K), ΔssCPY∗, and DegAB do not have obvious normally folding controls.
Promoter
Misfolding substrates have generally been placed behind strong promoters, such as TDH3, with the intent of overloading the temporal quality control system in order to easily visualize aggregates and inclusions. If the model protein of choice is not obviously cytotoxic, constitutively active promoters are useful in that they require no change in experimental conditions (such as a change in carbon source) to induce expression.
Inducible promoters have been commonly used when working with misfolding proteins that are cytotoxic, such as alpha-synuclein (Table 1). Thereby, cytotoxic effects of the misfolding protein can be avoided during strain construction and limited to the experiment itself. Additionally, inducible promoters can be useful for misfolding proteins that show a dose-dependent cytotoxicity, as it is easy to compare cytotoxicity when the expression of the misfolding protein is tightly regulated. It is also possible to shut off translation of the misfolding protein at a specific time point without interfering with translation of other proteins with agents such as cycloheximide. This is especially important when the cellular response to any particular misfolding protein is partially or fully dependent on a functional translation machinery.
One major limitation of the constitutive and inducible promoters used for the vast majority of the models covered in this review, is that they are among the strongest promoters characterized in S. cerevisiae (Peng et al., 2015). While inclusions can easily be visualized as a result, the high concentration of protein can cause toxicity unrelated to protein misfolding. For example, it can lead to sequestration of native and non-native binding partners and it may result in transport into non-native cellular compartments or organelles. Normally folding controls may mitigate this problem, as they can be used to identify the abnormal or toxic effects of overexpression.
Fluorescent Tagging
Fluorescent tags should, ideally, minimally affect protein function and are therefore generally placed accordingly. Practically, however, the tag will always have some effect regardless of whether it is placed N-terminally, C-terminally, or at an internal site. Even though the proteasome can process N-terminally or C-terminally tagged substrates (Liu et al., 2003), tagging can stabilize certain mutants (Mikalsen et al., 2005; Khosrow-Khavar et al., 2012), perhaps due to shielding of the unfolded domain recognized by the quality control machinery. Tag location will also determine if the misfolding protein exits the ribosome before or after folding of the fluorescent tag can begin, which can affect stability of the model protein or have an effect on the fluorescent tag itself. In fact, certain misfolded proteins have been shown to significantly affect GFP chromophore formation. This effect was taken advantage of to develop a protein folding assay (Waldo et al., 1999). In this study, fluorescence of GFP tagged test substrates correlated with solubility, indicating that aggregation of their test substrates affected chromophore formation. Therefore, for any substrate, it is important to determine whether the fluorescent signal observed is proportional to the amount of protein present. Along the same lines, the fluorescent signal may not reflect the presence of a misfolded protein as the proteasome can degrade it while leaving the fluorophore intact (Liu et al., 2003). Finally, the tag itself has been shown to influence toxicity of misfolding proteins. Tagging affected toxicity of different huntingtin constructs (Jiang et al., 2017) and the location of the GFP-tag and the design of the linker on FUS and OPTN was shown to influence toxicity and the constructs’ aggregation propensities (Kryndushkin et al., 2012).
Concluding Remarks
Deposition into quality control sites is a common, if not universal, response to misfolding proteins as these sites have been described in a wide range of organisms. Model misfolding proteins have been key tools used in the identification of, and distinction between, these quality control sites. Importantly, the use and characterization of a range of different misfolded proteins in yeast has made it clear that while many are handled similarly, there are often notable differences. For example, several models vary in their dependence on the San1 and Ubr1 E3 ubiquitin ligases for proteasomal degradation, while others do not depend on the proteasome but are degraded or removed in an undefined manner (Table 2). Furthermore, human disease models appear to be even more distinct in their processing. The huntingtin model, along with other amyloid aggregates, does not obviously sort to the JUNQ deposition site and ALS model proteins appear to deposit at sites that are distinct from both the JUNQ and IPOD. Similarly, aggregates of the Parkinson’s disease model, aSyn, did not colocalize to these defined sites (Tenreiro et al., 2014). Taken together, the evidence points toward the existence of uncharacterized spatial quality control pathways or, at the very least, modifications of defined pathways that have not yet been well characterized. Continued use of model misfolding proteins under varying conditions alone, and in combination, will do much to further define these spatial quality control pathways that play such an important role in the maintenance of proteostasis and prevention of disease progression.
Author Contributions
PW designed the review outline. PW and KS wrote the manuscript with guidance and input from TN.
Funding
This work was supported by grants from the Swedish Natural Science Research Council (VR) (TN), the Knut and Alice Wallenberg Foundation (Wallenberg Scholar) (TN), and the ERC (Advanced Grant; QualiAge) (TN).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Lisa Larsson Berglund for critical reading of the manuscript and all members of the Nyström lab for useful discussions.
Footnotes
References
Aguilaniu, H., Gustafsson, L., Rigoulet, M., and Nyström, T. (2003). Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753. doi: 10.1126/science.1080418
Alfassy, O. S., Cohen, I., Reiss, Y., Tirosh, B., and Ravid, T. (2013). Placing a disrupted degradation motif at the C terminus of proteasome substrates attenuates degradation without impairing ubiquitylation. J. Biol. Chem. 288, 12645–12653. doi: 10.1074/jbc.M410234200
Andersson, V., Hanzén, S., Liu, B., Molin, M., and Nyström, T. (2013). Enhancing protein disaggregation restores proteasome activity in aged cells. Aging 5, 802–812. doi: 10.18632/aging.100613
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. doi: 10.1038/nature02998
Berglund, L. L., Hao, X., Liu, B., Grantham, J., and Nyström, T. (2017). Differential effects of soluble and aggregating polyQ proteins on cytotoxicity and type-1 myosin-dependent endocytosis in yeast. Sci. Rep. 7:11328. doi: 10.1038/s41598-017-11102-6
Betting, J., and Seufert, W. (1996). A yeast Ubc9 mutant protein with temperature-sensitive in vivo function is subject to conditional proteolysis by a ubiquitin- and proteasome-dependent pathway. J. Biol. Chem. 271, 25790–25796. doi: 10.1074/jbc.271.42.25790
Böckler, S., Chelius, X., Hock, N., Klecker, T., Wolter, M., Weiss, M., et al. (2017). Fusion, fission, and transport control asymmetric inheritance of mitochondria and protein aggregates. J. Cell Biol. 216, 2481–2498. doi: 10.1038/ng1341
Braun, R. J. (2015). Ubiquitin-dependent proteolysis in yeast cells expressing neurotoxic proteins. Front. Mol. Neurosci. 8:8. doi: 10.3389/fnmol.2015.00008
Braun, R. J., Büttner, S., Ring, J., Kroemer, G., and Madeo, F. (2010). Nervous yeast: modeling neurotoxic cell death. Trends Biochem. Sci. 35, 135–144. doi: 10.1016/j.tibs.2009.10.005
Bufalino, M. R., DeVeale, B., and van der Kooy, D. (2013). The asymmetric segregation of damaged proteins is stem cell-type dependent. J. Cell Biol. 201, 523–530. doi: 10.1083/jcb.201207052
Caine, J., Sankovich, S., Antony, H., Waddington, L., Macreadie, P., Varghese, J., et al. (2007). Alzheimer’s Abeta fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Res. 7, 1230–1236. doi: 10.1111/j.1567-1364.2007.00285.x
Chuang, K.-H., Liang, F., Higgins, R., and Wang, Y. (2016). Ubiquilin/Dsk2 promotes inclusion body formation and vacuole (lysosome)-mediated disposal of mutated huntingtin. Mol. Biol. Cell 27, 2025–2036. doi: 10.1091/mbc.E16-01-0026
Cohen, A., Ross, L., Nachman, I., and Bar-Nun, S. (2012). Aggregation of polyQ proteins is increased upon yeast aging and affected by Sir2 and Hsf1: novel quantitative biochemical and microscopic assays. PLoS One 7:e44785. doi: 10.1371/journal.pone.0044785
Comyn, S. A., Young, B. P., Loewen, C. J., and Mayor, T. (2016). Prefoldin promotes proteasomal degradation of cytosolic proteins with missense mutations by maintaining substrate solubility. PLoS Genet. 12:e1006184. doi: 10.1371/journal.pgen.1006184
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328. doi: 10.1126/science.1129462
Cuervo, A. M., and Dice, J. F. (2000). Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 275, 31505–31513. doi: 10.1074/jbc.M002102200
D’Angelo, F., Vignaud, H., Di Martino, J., Salin, B., Devin, A., Cullin, C., et al. (2013). A yeast model for amyloid-β aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity. Dis. Model. Mech. 6, 206–216. doi: 10.1242/dmm.010108
Drummond, D. R., Hennessey, E. S., and Sparrow, J. C. (1991). Characterisation of missense mutations in the Act88F gene of Drosophila melanogaster. Mol. Gen. Genet. 226, 70–80. doi: 10.1007/BF00273589
Duennwald, M. L., Jagadish, S., Muchowski, P. J., and Lindquist, S. (2006). Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. U.S.A. 103, 11045–11050. doi: 10.1073/pnas.0604547103
Eisele, F., and Wolf, D. H. (2008). Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett. 582, 4143–4146. doi: 10.1016/j.febslet.2008.11.015
Elden, A. C., Kim, H.-J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
Escusa-Toret, S., Vonk, W. I. M., and Frydman, J. (2013). Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol. 15, 1231–1243. doi: 10.1038/ncb2838
Farrawell, N. E., Lambert-Smith, I. A., Warraich, S. T., Blair, I. P., Saunders, D. N., Hatters, D. M., et al. (2015). Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Nature 5:13416. doi: 10.1038/srep13416
Feldman, D. E., Thulasiraman, V., Ferreyra, R. G., and Frydman, J. (1999). Formation of the VHL-elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol. Cell 4, 1051–1061. doi: 10.1016/S1097-2765(00)80233-6
Finger, A., Knop, M., and Wolf, D. H. (1993). Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur. J. Biochem. 218, 565–574. doi: 10.1111/j.1432-1033.1993.tb18410.x
Fiori, M. G. (1987). Intranuclear inclusions in Schwann cells of aged fowl ciliary ganglia. J. Anat. 154, 201–214. doi: 10.1111/(ISSN)1469-7580
Fruhmann, G., Seynnaeve, D., Zheng, J., Ven, K., Molenberghs, S., Wilms, T., et al. (2017). Yeast buddies helping to unravel the complexity of neurodegenerative disorders. Mech. Ageing Dev. 161, 288–305. doi: 10.1016/j.mad.2016.05.002
Fuentealba, L. C., Eivers, E., Geissert, D., Taelman, V., and De Robertis, E. M. (2008). Asymmetric mitosis: unequal segregation of proteins destined for degradation. Proc. Natl. Acad. Sci. U.S.A. 105, 7732–7737. doi: 10.1073/pnas.0803027105
Furth, N., Gertman, O., Shiber, A., Alfassy, O. S., Cohen, I., Rosenberg, M. M., et al. (2011). Exposure of bipartite hydrophobic signal triggers nuclear quality control of Ndc10 at the endoplasmic reticulum/nuclear envelope. Mol. Biol. Cell 22, 4726–4739. doi: 10.1091/mbc.E11-05-0463
Fushimi, K., Long, C., Jayaram, N., Chen, X., Li, L., and Wu, J. Y. (2011). Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell 2, 141–149. doi: 10.1007/s13238-011-1014-5
Gardner, R. G., Nelson, Z. W., and Gottschling, D. E. (2005). Degradation-mediated protein quality control in the nucleus. Cell 120, 803–815. doi: 10.1016/j.cell.2005.01.016
Gupta, R., Kasturi, P., Bracher, A., Loew, C., Zheng, M., Villella, A., et al. (2011). Firefly luciferase mutants as sensors of proteome stress. Nat. Meth. 8, 879–884. doi: 10.1016/j.molcel.2006.08.017
Hartl, F. U., and Hayer-Hartl, M. (2009). Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16, 574–581. doi: 10.1038/nsmb.1591
Heck, J. W., Cheung, S. K., and Hampton, R. Y. (2010). Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. U.S.A. 107, 1106–1111. doi: 10.1038/35050524
Hill, S. M., Hanzén, S., and Nyström, T. (2017). Restricted access: spatial sequestration of damaged proteins during stress and aging. EMBO Rep. 18, 377–391. doi: 10.15252/embr.201643458
Jiang, Y., Di Gregorio, S. E., Duennwald, M. L., and Lajoie, P. (2017). Polyglutamine toxicity in yeast uncovers phenotypic variations between different fluorescent protein fusions. Traffic 18, 58–70. doi: 10.1111/tra.12453
Johnson, B. S., McCaffery, J. M., Lindquist, S., and Gitler, A. D. (2008). A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 6439–6444. doi: 10.1073/pnas.0802082105
Josefson, R., Andersson, R., and Nyström, T. (2017). How and why do toxic conformers of aberrant proteins accumulate during ageing? Essays Biochem. 61, 317–324. doi: 10.1042/EBC20160085
Ju, S., Tardiff, D. F., Han, H., Divya, K., Zhong, Q., Maquat, L. E., et al. (2011). A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 9:e1001052. doi: 10.1371/journal.pbio.1001052
Kaganovich, D., Kopito, R., and Frydman, J. (2008). Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095. doi: 10.4161/auto.2.2.2388
Kayatekin, C., Matlack, K. E. S., Hesse, W. R., Guan, Y., Chakrabortee, S., Russ, J., et al. (2014). Prion-like proteins sequester and suppress the toxicity of huntingtin exon 1. Proc. Natl. Acad. Sci. U.S.A. 111, 12085–12090. doi: 10.1073/pnas.1412504111
Kaytor, M. D., and Warren, S. T. (1999). Aberrant protein deposition and neurological disease. J. Biol. Chem. 274, 37507–37510. doi: 10.1074/jbc.274.53.37507
Khosrow-Khavar, F., Fang, N. N., Ng, A. H. M., Winget, J. M., Comyn, S. A., and Mayor, T. (2012). The yeast ubr1 ubiquitin ligase participates in a prominent pathway that targets cytosolic thermosensitive mutants for degradation. G3 2, 619–628. doi: 10.1534/g3.111.001933
Koga, H., Kaushik, S., and Cuervo, A. M. (2011). Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res. Rev. 10, 205–215. doi: 10.1016/j.arr.2010.02.001
Koo, E. H., Lansbury, P. T., and Kelly, J. W. (1999). Amyloid diseases: abnormal protein aggregation in neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 96, 9989–9990. doi: 10.1073/pnas.96.18.9989
Krobitsch, S., and Lindquist, S. (2000). Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1589–1594. doi: 10.1073/pnas.97.4.1589
Kruegel, U., Robison, B., Dange, T., Kahlert, G., Delaney, J. R., Kotireddy, S., et al. (2011). Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 7:e1002253. doi: 10.1371/journal.pgen.1002253
Kryndushkin, D., Ihrke, G., Piermartiri, T. C., and Shewmaker, F. (2012). A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol. Microbiol. 86, 1531–1547. doi: 10.1111/mmi.12075
Kryndushkin, D., Wickner, R. B., and Shewmaker, F. (2011). FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell 2, 223–236. doi: 10.1007/s13238-011-1525-0
Leibiger, C., Deisel, J., Aufschnaiter, A., Ambros, S., Tereshchenko, M., Verheijen, B. M., et al. (2018). TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum. Mol. Genet. 27, 1593–1607. doi: 10.1093/hmg/ddy066
Liang, J., Clark-Dixon, C., Wang, S., Flower, T. R., Williams-Hart, T., Zweig, R., et al. (2008). Novel suppressors of alpha-synuclein toxicity identified using yeast. Hum. Mol. Genet. 17, 3784–3795. doi: 10.1093/hmg/ddn276
Liu, B., Larsson, L., Caballero, A., Hao, X., Öling, D., Grantham, J., et al. (2010). The polarisome is required for segregation and retrograde transport of protein aggregates. Cell 140, 257–267. doi: 10.1016/j.cell.2009.12.031
Liu, C.-W., Corboy, M. J., DeMartino, G. N., and Thomas, P. J. (2003). Endoproteolytic activity of the proteasome. Science 299, 408–411. doi: 10.1126/science.1079293
Matsumoto, K., Toh-e, A., and Oshima, Y. (1978). Genetic control of galactokinase synthesis in Saccharomyces cerevisiae: evidence for constitutive expression of the positive regulatory gene gal4. J. Bacteriol. 134, 446–457.
McClellan, A. J., Scott, M. D., and Frydman, J. (2005). Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 121, 739–748. doi: 10.1016/j.cell.2005.03.024
McLean, P. J., Kawamata, H., and Hyman, B. T. (2001). Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 104, 901–912. doi: 10.1016/S0306-4522(01)00113-0
McMurray, M. (2014). Lean forward: Genetic analysis of temperature-sensitive mutants unfolds the secrets of oligomeric protein complex assembly. Bioessays 36, 836–846. doi: 10.1074/jbc.M112.406710
Medicherla, B., Kostova, Z., Schaefer, A., and Wolf, D. H. (2004). A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 5, 692–697. doi: 10.1038/414652a
Menezes, R., Tenreiro, S., Macedo, D., Santos, C. N., and Outeiro, T. F. (2015). From the baker to the bedside: yeast models of Parkinson’s disease. Microb. Cell 2, 262–279. doi: 10.15698/mic2015.08.219
Mikalsen, T., Johannessen, M., and Moens, U. (2005). Sequence- and position-dependent tagging protects extracellular-regulated kinase 3 protein from 26S proteasome-mediated degradation. Int. J. Biochem. Cell Biol. 37, 2513–2520. doi: 10.1016/j.biocel.2005.06.007
Miller, S. B., Ho, C. T., Winkler, J., Khokhrina, M., Neuner, A., Mohamed, M. Y., et al. (2015). Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 34, 778–797. doi: 10.15252/embj.201489524
Mogk, A., and Bukau, B. (2017). Role of sHsps in organizing cytosolic protein aggregation and disaggregation. Cell Stress Chaperones 22, 493–502. doi: 10.1007/s12192-017-0762-4
Nillegoda, N. B., Theodoraki, M. A., Mandal, A. K., Mayo, K. J., Ren, H. Y., Sultana, R., et al. (2010). Ubr1 and Ubr2 function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol. Biol. Cell 21, 2102–2116. doi: 10.1091/mbc.E10-02-0098
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Ogrodnik, M., Salmonowicz, H., Brown, R., Turkowska, J., Średniawa, W., Pattabiraman, S., et al. (2014). Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin. Proc. Natl. Acad. Sci. U.S.A. 111, 8049–8054. doi: 10.1073/pnas.1324035111
Oling, D., Eisele, F., Kvint, K., and Nystrom, T. (2014). Opposing roles of Ubp3-dependent deubiquitination regulate replicative life span and heat resistance. EMBO J. 33, 747–761. doi: 10.1002/embj.201386822
Outeiro, T. F., and Lindquist, S. (2003). Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775. doi: 10.1126/science.1090439
Park, S.-H., Bolender, N., Eisele, F., Kostova, Z., Takeuchi, J., Coffino, P., et al. (2007). The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin-proteasome system. Mol. Biol. Cell 18, 153–165. doi: 10.1091/mbc.E06-04-0338
Park, S.-H., Kukushkin, Y., Gupta, R., Chen, T., Konagai, A., Hipp, M. S., et al. (2013). PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154, 134–145. doi: 10.1016/j.cell.2013.06.003
Paulson, H. L. (1999). Protein fate in neurodegenerative proteinopathies: polyglutamine diseases join the (mis)fold. Am. J. Hum. Genet. 64, 339–345. doi: 10.1086/302269
Peng, B., Williams, T. C., Henry, M., Nielsen, L. K., and Vickers, C. E. (2015). Controlling heterologous geneexpression in yeast cell factories on di erent carbon substrates and across the diauxic shift: a comparison of yeast promoter activities. Microb. Cell Fact. 14:91. doi: 10.1186/s12934-015-0278-5
Pereira, C., Bessa, C., Soares, J., Leão, M., and Saraiva, L. (2012). Contribution of yeast models to neurodegeneration research. J. Biomed. Biotechnol. 2012:941232. doi: 10.1155/2012/941232
Peters, A., Josephson, K., and Vincent, S. L. (1991). Effects of aging on the neuroglial cells and pericytes within area 17 of the rhesus monkey cerebral cortex. Anat. Rec. 229, 384–398. doi: 10.1002/ar.1092290311
Petroi, D., Popova, B., Taheri-Talesh, N., Irniger, S., Shahpasandzadeh, H., Zweckstetter, M., et al. (2012). Aggregate clearance of α-synuclein in Saccharomyces cerevisiae depends more on autophagosome and vacuole function than on the proteasome. J. Biol. Chem. 287, 27567–27579. doi: 10.1371/journal.pone.0005515
Prasad, R., Kawaguchi, S., and Ng, D. T. W. (2010). A nucleus-based quality control mechanism for cytosolic proteins. Mol. Biol. Cell 21, 2117–2127. doi: 10.1091/mbc.E10-02-0111
Ruan, L., Zhou, C., Jin, E., Kucharavy, A., Zhang, Y., Wen, Z., et al. (2017). Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543, 443–446. doi: 10.1038/nature21695
Rujano, M. A., Bosveld, F., Salomons, F. A., Dijk, F., van Waarde, M. A. W. H., van der Want, J. J. L., et al. (2006). Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 4:e417. doi: 10.1371/journal.pbio.0040417
Saarikangas, J., and Barral, Y. (2015). Protein aggregates are associated with replicative aging without compromising protein quality control. eLife 4:e06197. doi: 10.7554/eLife.06197
Sadler, J. R., and Novick, A. (1965). The properties of repressor and the kinetics of its action. J. Mol. Biol. 12, 305–327. doi: 10.1016/S0022-2836(65)80255-8
Saez, I., and Vilchez, D. (2014). The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genomics 15, 38–51. doi: 10.2174/138920291501140306113344
Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M. E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66. doi: 10.1016/S0092-8674(00)81782-1
Schoenfeld, A. R., Davidowitz, E. J., and Burk, R. D. (2000). Elongin BC complex prevents degradation of von Hippel-Lindau tumor suppressor gene products. Proc. Natl. Acad. Sci. U.S.A. 97, 8507–8512. doi: 10.1073/pnas.97.15.8507
Schröder, H., Langer, T., Hartl, F. U., and Bukau, B. (1993). DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 12, 4137–4144.
Shcheprova, Z., Baldi, S., Frei, S. B., Gonnet, G., and Barral, Y. (2008). A mechanism for asymmetric segregation of age during yeast budding. Nature 454, 728–734. doi: 10.1038/nature07212
Shiber, A., Breuer, W., Brandeis, M., and Ravid, T. (2013). Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Mol. Biol. Cell 24, 2076–2087. doi: 10.1091/mbc.E13-01-0010
Shiber, A., Breuer, W., and Ravid, T. (2014). Flow cytometric quantification and characterization of intracellular protein aggregates in yeast. Prion 8, 276–284. doi: 10.4161/19336896.2014.968445
Song, J., Yang, Q., Yang, J., Larsson, L., Hao, X., Zhu, X., et al. (2014). Essential genetic interactors of SIR2 required for spatial sequestration and asymmetrical inheritance of protein aggregates. PLoS Genet. 10:e1004539. doi: 10.1371/journal.pgen.1004539.s009
Sontag, E. M., Samant, R. S., and Frydman, J. (2017). Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 86, 97–122. doi: 10.1146/annurev-biochem-060815-14616
Specht, S., Miller, S. B. M., Mogk, A., and Bukau, B. (2011). Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J. Cell Biol. 195, 617–629. doi: 10.1083/jcb.136.1.111
Spokoini, R., Moldavski, O., Nahmias, Y., England, J. L., Schuldiner, M., and Kaganovich, D. (2012). Confinement to organelle-associated inclusion structures mediates asymmetric inheritance of aggregated protein in budding yeast. Cell Rep. 2, 738–747. doi: 10.1016/j.celrep.2012.08.024
Stolz, A., and Wolf, D. H. (2012). Use of CPY and its derivatives to study protein quality control in various cell compartments. Methods Mol. Biol. 832, 489–504. doi: 10.1007/978-1-61779-474-2_35
Sun, Z., Diaz, Z., Fang, X., Hart, M. P., Chesi, A., Shorter, J., et al. (2011). Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 9:e1000614. doi: 10.1371/journal.pbio.1000614
Sundberg, H. A., and Davis, T. N. (1997). A mutational analysis identifies three functional regions of the spindle pole component Spc110p in Saccharomyces cerevisiae. Mol. Biol. Cell 8, 2575–2590. doi: 10.1091/mbc.8.12.2575
Takahashi, T., Kikuchi, S., Katada, S., Nagai, Y., Nishizawa, M., and Onodera, O. (2008). Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 17, 345–356. doi: 10.1093/hmg/ddm311
Taylor, J. P., Tanaka, F., Robitschek, J., Sandoval, C. M., Taye, A., Markovic-Plese, S., et al. (2003). Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 12, 749–757. doi: 10.1093/hmg/ddg074
Tenreiro, S., Munder, M. C., Alberti, S., and Outeiro, T. F. (2013). Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. J. Neurochem. 127, 438–452. doi: 10.1111/jnc.12271
Tenreiro, S., Reimão-Pinto, M. M., Antas, P., Rino, J., Wawrzycka, D., Macedo, D., et al. (2014). Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet. 10:e1004302. doi: 10.1371/journal.pgen.1004302
Treusch, S., Hamamichi, S., Goodman, J. L., Matlack, K. E. S., Chung, C. Y., Baru, V., et al. (2011). Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 334, 1241–1245. doi: 10.1126/science.1213210
Tyedmers, J., Mogk, A., and Bukau, B. (2010). Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 11, 777–788. doi: 10.1038/nrm2993
Waldo, G. S., Standish, B. M., Berendzen, J., and Terwilliger, T. C. (1999). Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 17, 691–695. doi: 10.1038/10904
Wang, Y., Meriin, A. B., Zaarur, N., Romanova, N. V., Chernoff, Y. O., Costello, C. E., et al. (2009). Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J. 23, 451–463. doi: 10.1096/fj.08-117614
Willingham, S., Outeiro, T. F., DeVit, M. J., Lindquist, S. L., and Muchowski, P. J. (2003). Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 302, 1769–1772. doi: 10.1126/science.1090389
Winkler, J., Seybert, A., König, L., Pruggnaller, S., Haselmann, U., Sourjik, V., et al. (2010). Quantitative and spatio-temporal featuresof protein aggregation in Escherichia coli and consequences on protein quality control and cellular ageing. EMBO J 29, 910–923. doi: 10.1038/emboj.2009.412
Wolf, D. H., and Fink, G. R. (1975). Proteinase C (carboxypeptidase Y) mutant of yeast. J. Bacteriol. 123, 1150–1156.
Wolfe, K. J., Ren, H. Y., Trepte, P., and Cyr, D. M. (2013). The Hsp70/90 cochaperone, Sti1, suppresses proteotoxicity by regulating spatial quality control of amyloid-like proteins. Mol. Biol. Cell 24, 3588–3602. doi: 10.1091/mbc.E13-06-0315
Keywords: protein quality control, spatial protein quality control, protein misfolding, misfolding model, inclusions, aging, cell stress, temperature-sensitive
Citation: Schneider KL, Nyström T and Widlund PO (2018) Studying Spatial Protein Quality Control, Proteopathies, and Aging Using Different Model Misfolding Proteins in S. cerevisiae. Front. Mol. Neurosci. 11:249. doi: 10.3389/fnmol.2018.00249
Received: 15 April 2018; Accepted: 02 July 2018;
Published: 23 July 2018.
Edited by:
Ralf J. Braun, University of Bayreuth, GermanyReviewed by:
Jens Tyedmers, Universitätsklinikum Heidelberg, GermanyMartin Lothar Duennwald, University of Western Ontario, Canada
Copyright © 2018 Schneider, Nyström and Widlund. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Per O. Widlund, cGVyLndpZGx1bmRAZ3Uuc2U=