 Yimeng Xia
Yimeng Xia Xiaoyun Sun2
Xiaoyun Sun2 Creed M. Stary
Creed M. Stary- 1Department of Anesthesiology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China
- 2Department of Anesthesiology, Perioperative and Pain Medicine, Stanford University School of Medicine, Stanford, CA, United States
The underlying mechanisms of isoflurane neurotoxicity in the developing brain remain unclear. Ferroptosis is a recently characterized form of programmed cell death distinct from apoptosis or autophagy, characterized by iron-dependent reactive oxygen species (ROS) generation secondary to failure of glutathione-dependent antioxidant defenses. The results of the present study are the first to demonstrate in vitro that ferroptosis is a central mechanism contributing to isoflurane neurotoxicity. We observed in embryonic mouse primary cortical neuronal cultures (day-in-vitro 7) that 6 h of 2% isoflurane exposure was associated with decreased transcription and protein expression of the lipid repair enzyme glutathione peroxidase 4. In parallel, isoflurane exposure resulted in increased ROS generation, disruption in mitochondrial membrane potential, and cell death. These effects were significantly attenuated by pre-treatment with the selective ferroptosis inhibitor ferrostatin-1 (Fer-1). Collectively, these observations provide a novel mechanism for isoflurane-induced injury in the developing brain and suggest that pre-treatment with Fer-1 may be a potential clinical intervention for neuroprotection.
Introduction
Isoflurane remains a commonly used volatile anesthetic worldwide despite emerging evidence of neurotoxicity to the developing brain (Andropoulos, 2018). Extensive work in rodents has attributed neonatal cognitive dysfunction following inhalational anesthetics exposure to apoptosis, neuroinflammation, reactive oxygen species (ROS) accumulation, neurotransmitter disturbances, and/or changes in synaptic plasticity (Patel and Sun, 2009; Brambrink et al., 2012; Lei et al., 2012; Xia et al., 2017). Mechanistically, isoflurane neurotoxicity has been linked with RhoA-mediated actin depolymerization of the neuronal cytoskeletal architecture (Lemkuil et al., 2011), however inhibition of this process is not clinically protective (Schilling et al., 2017). Therefore, a definitive characterization of the central, underlying mechanisms that regulate neonatal anesthetic toxicity remain a critical knowledge gap impeding the development of interventional therapies.
Ferroptosis (Figure 1A), first described in Dixon et al. (2012), is a newly recognized mechanism of programmed cell death characterized by iron-dependent accumulation of lipid peroxides (Angeli et al., 2017; Stockwell et al., 2017; Zilka et al., 2017), driven by inactivation of glutathione peroxidase 4 (GPX4, Yang et al., 2014). Emerging evidence suggests ferroptosis contributes to neurodegenerative diseases and brain injury associated with oxidative stress (Skouta et al., 2014), including brain hemorrhage (Li et al., 2017), Alzheimer's disease (Fine et al., 2012; Guo et al., 2013), Huntington's disease (Yang et al., 2005; Niatsetskaya et al., 2010; Speer et al., 2013), stroke (Tuo et al., 2017) and traumatic brain injury (Anthonymuthu et al., 2018). Oxidative stress is associated with neonatal neuronal cell death after inhalational anesthetic exposure (Zhang et al., 2012), however whether ferroptosis contributes to this process has not been investigated. In the present study we assessed GPX4 expression, mitochondrial function and cell death in primary neonatal neuronal cultures following isoflurane exposure, with and without pre-treatment with the selective ferroptosis inhibitor ferrostatin-1 (Fer-1, Kabiraj et al., 2015).
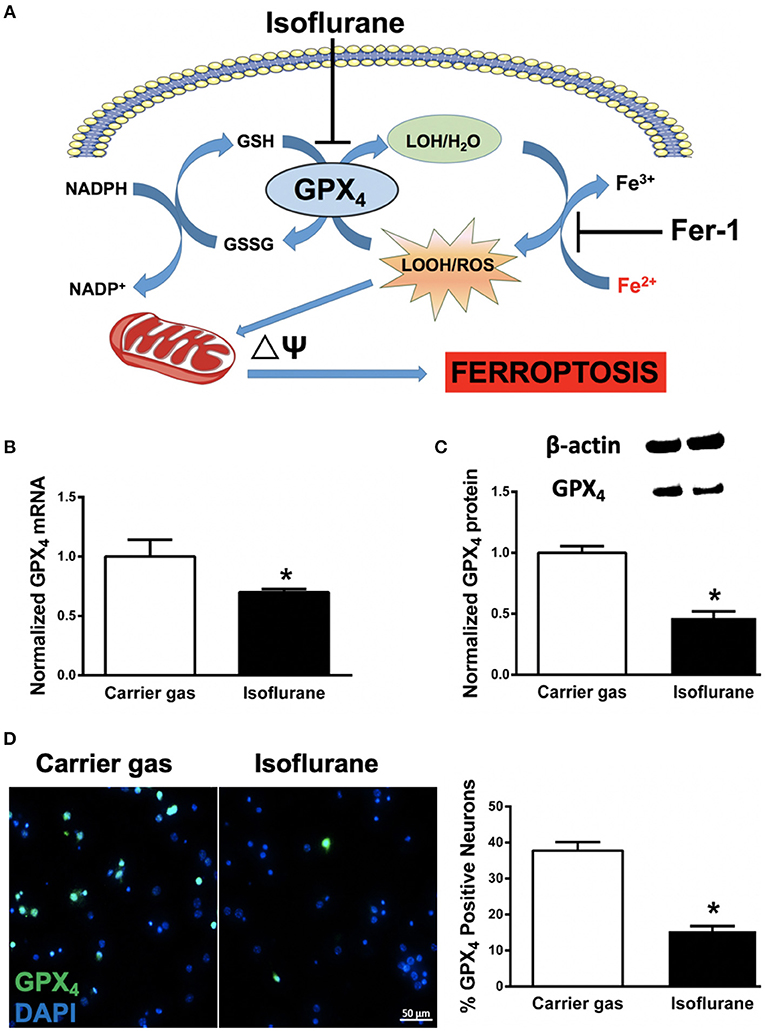
Figure 1. (A) Proposed model illustrating ferroptosis in isoflurane neurotoxicity. Exposure of developing neurons to isoflurane suppresses glutathione peroxidase 4 (GPX4) expression, leading to augmentation of ferrous- (Fe2+) driven formation of reactive lipid peroxides (LOOH) and ROS generation, impairment in mitochondrial membrane potential (ΔΨ) and ferroptosis (cell death). Inhibition by co-administration of ferrostatin-1 (Fer-1) inhibits ferrous-driven formation of LOOH. (B) Expression of GPX4 mRNA in primary neuronal cultures after isoflurane exposure, normalized to carrier gas exposure. (C) Quantification of levels of GPX4 protein assessed by immunoblot in neuronal cultures after isoflurane or carrier gas exposure, with example of immunoblot above. (D) Immunofluorescent images (left) of GPX4 (green) in primary neuronal cultures co-stained with the cell-permeant nuclear dye DAPI (blue). Quantitation of GPX4 positive cells (right) as a percentage of total nuclei. All graphs represent n = 12 pooled data from three independent experiments. Data are expressed as mean ± SEM. *p < 0.01 compared with the control group. Scale bar = 50 μm. DAPI = 4′,6-diamidino-2-phenylindole, dihydrochloride. Fe3+, ferric (iron) ion; GSSG, glutathione disulfide; GSH, glutathione; LOH, non-reactive lipid; NADH+, nicotinamide adenine dinucleotide; NADPH, reduced nicotinamide adenine dinucleotide; ROS, reactive oxygen species.
Materials and Methods
Animals and Primary Neuronal Cultures
All animal experiments were approved by Stanford University Animal Care and Use Committee (Stanford, CA, USA) and conducted according to the National Institutes of Health guidelines for animal welfare. Primary cortical neuronal cultures were prepared from embryonic/gestational day 15 or 16 Swiss Webster mice as previously described (Stary et al., 2015). Please refer to Supplementary Material for detailed methods.
Experimental Protocol
Primary neuronal cultures were pre-treated at day-in-vitro (DIV) 7 with 1μM Fer-1 (CAT#SML0583, Sigma Chemicals) a selective ferroptosis inhibitor (Kabiraj et al., 2015; Wu et al., 2018), or 1:5,000 DMSO (Sigma Chemicals) as vehicle control 1 h prior to 6 h of 2% isoflurane exposure or carrier gas (5%CO2, 21%O2, balance N2). Immediately after isoflurane or carrier gas exposure cultures were processed for either reverse transcription–quantitative real-time-PCR, immunoblot, immunocytochemical staining, or live-cell fluorescent imaging of ROS/mitochondrial membrane potential. Please refer to Supplementary Material for detailed methods.
Reverse-Quantitative Polymerase Chain Reaction (RT–qPCR)
Total RNA was extracted and reverse transcription and PCR were performed as previously described (Ouyang et al., 2012a,b). Ct-values for GPX4 were normalized to GAPDH as the internal control and comparisons calculated as the inverse log of the ΔΔCT (Livak and Schmittgen, 2001). Please refer to Supplementary Material for detailed methods.
Immunoblot
Please refer to Supplementary Material for detailed methods. After protein gel electrophoresis and transfer membranes were blocked and incubated at 4°C overnight with primary antibodies to GPX4 (1:500, #125066; Abcam) and anti-β-actin (1:20,000, #A1978; Sigma). Membranes were then incubated with 1:3,000 goat anti-rabbit (CST, #7074) for GPX4 and horse anti-mouse (CST, #7076) for β-actin. GPX4 band intensity was normalized to β-actin and the isoflurane group then normalized to the carrier gas group.
Immunocytochemistry
Please refer to Supplementary Material for detailed methods. After fixation cultures were incubated with rabbit monoclonal antibody to GPX4 (1:500; ab125066, Abcam) and secondary Alexa Fluor 488-conjugated donkey anti-rabbit (CAT#A-21206, 1:1,000; ThermoFisher Scientific). Cells were visualized at 400X on an inverted Zeiss Observer microscope (Carl Zeiss, Göttingen, Germany).
Cell Death Assay
Subsequent to isoflurane or carrier gas exposure, cultures were incubated with Hoechst 33342 (5 μM, Sigma) and propidium iodide (PI, 5 μM, Sigma). Automated fluorescent image capture was performed at 200X using a LumascopeTM 720 (Etaluma, Carlsbad, CA). The number of PI-positive and Hoechst-positive cells were quantified using Image J software (v1.49b, National Institutes of Health, USA) and expressed as percentage of total cells.
Assessment of Reactive Oxygen Species (ROS) and Mitochondrial Membrane Potential
Cultures were incubated with the ROS sensitive dye CellROXTM green (5 μM; #C10444, Life Technologies, Carlsbad, CA) or the mitochondrial membrane potential dye tetramethylrhodamine ethyl ester (TMRE, 50 nM, ThermoFisher Scientific) according to the manufacturer's instructions. Fluorescence was assessed at 200X with a LumascopeTM 720 and fluorescence intensity was quantified using Image J software (v1.49b).
Statistics
All results are expressed as mean ± standard error (SE). Statistical analysis was performed using SPSS 18.0 software. GPX4 mRNA and protein expression levels, CellROS and TMRE fluorescent values were normalized to those of the control group (carrier gas, vehicle alone). All data represent pooled data from 3 individual experiments containing n = 4 samples for each treatment group. After normality and equal variance tests, statistical differences between two groups were compared using Student's t-test. For data with non-normal distributions, Kruskal–Wallis test was used. For all measurements p < 0.05 (95% confidence interval) were considered significant. Please refer to Supplementary Material for detailed methods.
Results
Isoflurane Decreases GPX4 Expression
In order to assess whether ferroptosis contributes to isoflurane neurotoxicity, we assessed mRNA and protein levels of the central regulator of ferroptosis, GPX4 (Dixon et al., 2012; Cardoso et al., 2017). Compared to carrier gas control, GPX4 mRNA levels assessed by RT-qPCR significantly (p = 0.0035, Student's t-test) decreased after 6 h isoflurane treatment (Figure 2B). The mRNA levels of the internal control GAPDH were not statistically different between the two groups. Protein levels of GPX4 as assessed by immunoblot were concomitantly significantly (p < 0.01, Student's t-test) decreased secondary to isoflurane exposure (Figure 2C). In parallel we confirmed a significant (p < 0.01, Student's t-test) decrease in cellular expression of GPX4 by immunocytochemistry (Figure 2D) after isoflurane relative to carrier gas control levels. Together these results demonstrate that 6 h of 2% isoflurane exposure decreases expression of GPX4 in these primary neuronal cell cultures.
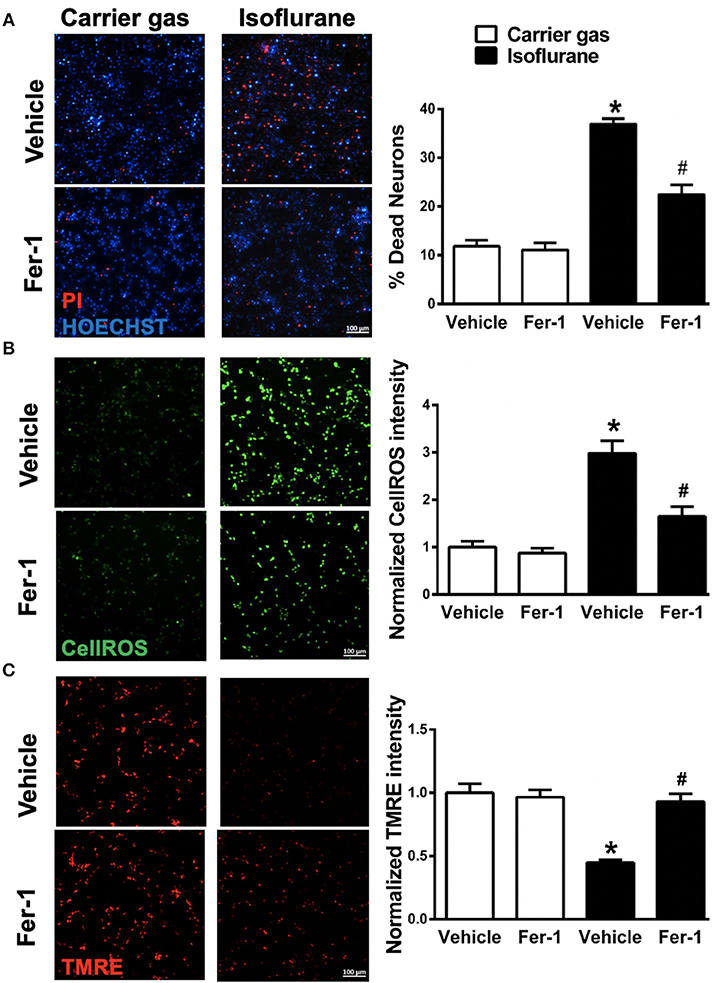
Figure 2. Effects on ferrostatin-1 (Fer-1) on neuronal cell death and mitochondrial function. (A) Representative micrographs (left) and quantification of cell death (right) in primary neuronal cultures exposed to isoflurane or carrier gas, with or without Fer-1 pre-treatment, stained with propidium iodide (dead cells, red) and Hoescht (live cells, blue). (B) Representative micrographs (left) of ROS generation (CellROX fluorescence, green) and quantitation of fluorescence intensity (right) in primary neuronal cultures exposed to isoflurane or carrier gas, with or without Fer-1 pre-treatment. (C) Representative micrographs of mitochondrial membrane potential (TMRE fluorescence, red) and quantitation of fluorescence intensity (right) in primary neuronal cultures exposed to isoflurane or carrier gas, with or without Fer-1 pre-treatment. Graphs represent n = 12, pooled data from three independent experiments. All data were expressed as mean ± SEM. *p < 0.01 compared with carrier gas control group; #p < 0.01 compared with isoflurane. Scale bar = 100 μm. PI, propidium iodide; ROS, reactive oxygen species; TMRE, tetramethylrhodamine ethyl ester.
Fer-1 Alleviates Isoflurane-Induced Cell Death
To investigate whether ferroptosis contributes to isoflurane neurotoxicity, cell death was assessed in neuronal cultures secondary to isoflurane exposure with and without pre-treatment with the ferroptosis selective inhibitor Fer-1. Isoflurane exposure induced a significant (p < 0.01, Kruskal–Wallis test) increase in cell death relative to carrier gas control (Figure 2A), assessed by automated fluorescence microscopy after double staining with Hoescht/PI. Notably, pre-treatment with Fer-1 provided substantial protection by significantly (p < 0.01, Kruskal–Wallis test) alleviating cell death by ~50%. Application of Fer-1 had no measurable toxicity in carrier gas control treated cultures (Figure 2A). These findings suggest that ferroptosis contributes to isoflurane neurotoxicity and that Fer-1 pre-treatment is protective in these neuronal cultures.
Fer-1 Attenuates Isoflurane-Induced ROS Generation
Next, in order to assess whether Fer-1 provides neuroprotection against isoflurane exposure by mitigation of ROS generation CellROXTM green was utilized for live-cell fluorescent imaging. CellROXTM fluorescence increased significantly (p < 0.01, Kruskal–Wallis test) in neuronal cultures after isoflurane exposure compared with carrier gas control treatment (Figure 2B). Fer-1 pre-treatment significantly (p < 0.01, Kruskal–Wallis test) attenuated CellROXTM fluorescence in isoflurane treated cultures (Figure 2B) but had no effect on CellROXTM fluorescence in carrier gas control treated cultures. These results demonstrate that Fer-1 pre-treatment attenuates ROS generation secondary to isoflurane exposure in these neuronal cultures, likely contributing to observations of improved cell survival.
Fer-1 Mitigates Isoflurane-Induced Mitochondrial Dysfunction
Finally, we assessed the role of ferroptosis in mitochondrial dysfunction secondary to isoflurane exposure by evaluating mitochondrial membrane potential with and without Fer-1 pre-treatment. Compared with carrier gas control treated cultures, TMRE fluorescence intensity was significantly (p < 0.01, Kruskal–Wallis test) depressed following isoflurane exposure (Figure 2C) relative to carrier gas in vehicle-control cultures. However, in Fer-1 treated cultures mitochondrial membrane potential remained largely preserved after isoflurane exposure (Figure 2C). No significant difference in TMRE fluorescence intensity was measured in carrier gas control treated cultures with Fer-1 application. These results demonstrate that Fer-1 mitigates mitochondrial dysfunction secondary to isoflurane exposure.
Discussion
Proposed mechanisms whereby volatile anesthetics induce cognitive dysfunction during neurodevelopment include GABA-A receptor-mediated Cl− ion efflux and depolarization, disruption in cell-survival signaling pathways leading to neuronal apoptosis, impaired neuronal differentiation, neurogenesis and neuronal migration, and disruption in synaptogenesis and synaptic pruning (for review, see Patel and Sun, 2009; Brambrink et al., 2012; Lei et al., 2012). In vitro models utilizing primary neuronal cell cultures have demonstrated that isoflurane only contributes to injury at DIV 4–7 cultures, and not at DIV 10, which roughly parallels the neonatal period in vivo brain characterized by active synaptogenesis. Prior studies have demonstrated that mitochondrial function is central to several critical processes for proper brain development (Arrázola et al., 2018; Devine and Kittler, 2018), including neurogenesis, neuronal differentiation, neuronal migration, synaptogenesis and synaptic pruning secondary to programmed apoptosis. Therefore, mitochondria represent a unifying target for therapies aimed at protecting or rescuing the neonatal brain following anesthetic exposure. In the present study, we utilized an isoflurane exposure protocol (2% isoflurane for 6 h) known to induce ROS generation, impair mitochondrial membrane potential and induce cell death in DIV 7 primary neuronal cell cultures (Xie et al., 2006, 2007; Zhang et al., 2010, 2012) in order to assess the potential role of ferroptosis in anesthetic neurotoxicity. We report here for the first time that pre-treatment with the selective ferroptosis inhibitor Fer-1 preserved mitochondrial function and mitigated neuronal cell death secondary to isoflurane exposure (Figure 2), suggesting that ferroptosis represents a central mechanism of anesthetic neurotoxicity in these primary neuronal cell cultures.
Ferroptosis is defined by iron-dependent accumulation of lipid peroxides, and is genetically and biochemically distinct from other forms of programmed cell death such as apoptosis, necrosis and autophagy (Yang and Stockwell, 2016). GPX4 is a lipid repair enzyme, which reduces lipid hydroperoxides to lipid alcohols and limit the initiation of ferroptosis (Cao and Dixon, 2016; Tonnus and Linkermann, 2016). Inhibition of GPX4 activity leads to rapid accumulation of lipid peroxides and cell death, and deletion of GPX4 is embryonic lethal (Seiler et al., 2008; Yang et al., 2014), therefore GPX4 expression has been used as a marker for ferroptosis (Cardoso et al., 2017; Conrad et al., 2018). In the present study we observed a significant decrease in GPX4 mRNA and GPX4 protein expression with isoflurane exposure (Figure 1), supporting our hypothesis that isoflurane induces ferroptosis. These observations, in combination with our parallel observations that mitochondrial dysfunction secondary to isoflurane exposure were largely reversed by pre-treatment with Fer-1 (Figure 2), imply a central role for ferroptosis in isoflurane neurotoxicity. Future in vivo studies selectively altering GPX4 expression are needed to more accurately assess the mechanistic contribution of decreased GPX4 to isoflurane neurotoxicity.
Whether general anesthetics have the capacity to permanently impair cognitive function remains controversial (Hudson and Hemmings, 2011; Zhou et al., 2015), and clinical trials in matched cohorts testing protective compounds may provide the only avenue for direct evidence of neurotoxicity in children. In the present study we observed that Fer-1 had no effect on ROS generation, mitochondrial membrane potential or cell death in neuronal cultures, suggesting that Fer-1 may provide a clinically safe therapeutic intervention. However, one limitation of the present study is that cell cultures were grown at atmospheric partial pressure of O2 (21%), which represents a hyperoxic state relative to physiological O2 levels in the brain which approach 1–8% (Erecinska and Silver, 2001). Our group have previously described differences in mitochondrial function between cultures grown at 21% and cultures grown at 5% O2 (Sun et al., 2015), suggesting that in vitro experiments assessing mitochondrial mechanisms should account for this. Second, although 2% isoflurane in vitro treatment represents in vivo physiologic levels of exposure (Herold et al., 2017), a 6 h duration of single agent exposure would not represent a typical pediatric anesthetic. Further studies extending these findings to clinically relevant in vivo models utilizing multiple anesthetic agents and more common exposure durations are required to advance Fer-1 as a potential neuroprotectant in pediatric patients requiring anesthesia.
Author Contributions
YX planned and executed experiments and drafted the manuscript. XS assisted with experiments. YL assisted in intellectual design and reviewed manuscript. CS assisted in methodological planning, experimental execution, data review, and manuscript preparation.
Funding
This study was supported by American Heart Association grant 14FTF-19970029 to CS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00486/full#supplementary-material
References
Andropoulos, D. B. (2018). Effect of anesthesia on the developing brain: infant and fetus. Fetal Diagn. Ther. 43, 1–11. doi: 10.1159/000475928
Angeli, J. P. F., Shah, R., Pratt, D. A., and Conrad, M. (2017). Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol. Sci. 38, 489–498. doi: 10.1016/j.tips.2017.02.005
Anthonymuthu, T. S., Kenny, E. M., Lamade, A. M., Kagan, V. E., and Bayir, H. (2018). Oxidized phospholipid signaling in traumatic brain injury. Free Radic. Biol. Med. 124, 493–503. doi: 10.1016/j.freeradbiomed.2018.06.031
Arrázola, M. S., Andraini, T., Szelechowski, M., Mouledous, L., Arnaune-Pelloquin, L., Davezac, N., et al. (2018). Mitochondria in developmental and adult neurogenesis. Neurotox Res. doi: 10.1007/s12640-018-9942-y. [Epub ahead of print].
Brambrink, A. M., Orfanakis, A., and Kirsch, J. R. (2012). Anesthetic neurotoxicity. Anesthesiol. Clin. 30, 207–228. doi: 10.1016/j.anclin.2012.06.002
Cao, J. Y., and Dixon, S. J. (2016). Mechanisms of ferroptosis. Cell Mol. Life Sci. 73, 2195–2209. doi: 10.1007/s00018-016-2194-1
Cardoso, B. R., Hare, D. J., Bush, A. I., and Roberts, B. R. (2017). Glutathione peroxidase 4: a new player in neurodegeneration? Mol. Psychiatry 22, 328–335. doi: 10.1038/mp.2016.196
Conrad, M., Kagan, V. E., Bayir, H., Pagnussat, G. C., Head, B., Traber, M. G., et al. (2018). Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 32, 602–619. doi: 10.1101/gad.314674.118
Devine, M. J., and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19, 63–80. doi: 10.1038/nrn.2017.170
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Erecinska, M., and Silver, I. A. (2001). Tissue oxygen tension and brain sensitivity to hypoxia. Respir. Physiol. 128, 263–276.
Fine, J. M., Baillargeon, A. M., Renner, D. B., Hoerster, N. S., Tokarev, J., Colton, S., et al. (2012). Intranasal deferoxamine improves performance in radial arm water maze, stabilizes HIF-1alpha, and phosphorylates GSK3beta in P301L tau transgenic mice. Exp. Brain Res. 219, 381–390. doi: 10.1007/s00221-012-3101-0
Guo, C., Wang, T., Zheng, W., Shan, Z. Y., Teng, W. P., and Wang, Z. Y. (2013). Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer's disease. Neurobiol. Aging 34, 562–575. doi: 10.1016/j.neurobiolaging.2012.05.009
Herold, K. F., Sanford, R. L., Lee, W., Andersen, O. S., and Hemmings, H. C. Jr. (2017). Clinical concentrations of chemically diverse general anesthetics minimally affect lipid bilayer properties. Proc. Natl. Acad. Sci. U.S.A. 114, 3109–3114. doi: 10.1073/pnas.1611717114
Hudson, A. E., and Hemmings, H. C. Jr. (2011). Are anaesthetics toxic to the brain? Br. J. Anaesth. 107, 30–37. doi: 10.1093/bja/aer122
Kabiraj, P., Valenzuela, C. A., Marin, J. E., Ramirez, D. A., Mendez, L., Hwang, M. S., et al. (2015). The neuroprotective role of ferrostatin-1 under rotenone-induced oxidative stress in dopaminergic neuroblastoma cells. Protein J. 34, 349–358. doi: 10.1007/s10930-015-9629-7
Lei, X., Guo, Q., and Zhang, J. (2012). Mechanistic insights into neurotoxicity induced by anesthetics in the developing brain. Int. J. Mol. Sci. 13, 6772–6799. doi: 10.3390/ijms13066772
Lemkuil, B. P., Head, B. P., Pearn, M. L., Patel, H. H., Drummond, J. C., and Patel, P. M. (2011). Isoflurane neurotoxicity is mediated by p75NTR-RhoA activation and actin depolymerization. Anesthesiology 114, 49–57. doi: 10.1097/ALN.0b013e318201dcb3
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2:e90777. doi: 10.1172/jci.insight.90777
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Niatsetskaya, Z., Basso, M., Speer, R. E., McConoughey, S. J., Coppola, G., Ma, T. C., et al. (2010). HIF prolyl hydroxylase inhibitors prevent neuronal death induced by mitochondrial toxins: therapeutic implications for Huntington's disease and Alzheimer's disease. Antioxid. Redox Signal. 12, 435–443. doi: 10.1089/ars.2009.2800
Ouyang, Y. B., Lu, Y., Yue, S., and Giffard, R. G. (2012a). miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion 12, 213–219. doi: 10.1016/j.mito.2011.09.001
Ouyang, Y. B., Lu, Y., Yue, S., Xu, L. J., Xiong, X. X., White, R. E., et al. (2012b). miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiol. Dis. 45, 555–563. doi: 10.1016/j.nbd.2011.09.012
Patel, P., and Sun, L. (2009). Update on neonatal anesthetic neurotoxicity insight into molecular mechanisms and relevance to humans. Anesthesiology 110, 703–708. doi: 10.1097/Aln.0b013e31819c42a4
Schilling, J. M., Kassan, A., Mandyam, C., Pearn, M. L., Voong, A., Grogman, G. G., et al. (2017). Inhibition of p75 neurotrophin receptor does not rescue cognitive impairment in adulthood after isoflurane exposure in neonatal mice. Br. J. Anaesth. 119, 465–471. doi: 10.1093/bja/aew299
Seiler, A., Schneider, M., Forster, H., Roth, S., Wirth, E. K., Culmsee, C., et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248. doi: 10.1016/j.cmet.2008.07.005
Skouta, R., Dixon, S. J., Wang, J., Dunn, D. E., Orman, M., Shimada, K., et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556. doi: 10.1021/ja411006a
Speer, R. E., Karuppagounder, S. S., Basso, M., Sleiman, S. F., Kumar, A., Brand, D., et al. (2013). Hypoxia-inducible factor prolyl hydroxylases as targets for neuroprotection by “antioxidant” metal chelators: from ferroptosis to stroke. Free Radic. Biol. Med. 62, 26–36. doi: 10.1016/j.freeradbiomed.2013.01.026
Stary, C. M., Sun, X., and Giffard, R. G. (2015). Astrocytes protect against isoflurane neurotoxicity by buffering pro-brain-derived neurotrophic factor. Anesthesiology 123, 810–819. doi: 10.1097/ALN.0000000000000824
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Sun, X., Voloboueva, L. A., Stary, C. M., and Giffard, R. G. (2015). Physiologically normal 5% O2 supports neuronal differentiation and resistance to inflammatory injury in neural stem cell cultures. J. Neurosci. Res. 93, 1703–1712. doi: 10.1002/jnr.23615
Tonnus, W., and Linkermann, A. (2016). “Death is my Heir”–Ferroptosis connects cancer pharmacogenomics and ischemia-reperfusion injury. Cell. Chem. Biol. 23, 202–203. doi: 10.1016/j.chembiol.2016.02.005
Tuo, Q. Z., Lei, P., Jackman, K. A., Li, X. L., Xiong, H., Li, X. L., et al. (2017). Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 22, 1520–1530. doi: 10.1038/mp.2017.171
Wu, C., Zhao, W., Yu, J., Li, S., Lin, L., and Chen, X. (2018). Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci. Rep. 8:574. doi: 10.1038/s41598-017-18935-1
Xia, Y., Xu, H., Jia, C., Hu, X., Kang, Y., Yang, X., et al. (2017). Tanshinone IIA attenuates sevoflurane neurotoxicity in neonatal mice. Anesth. Analg. 124, 1244–1252. doi: 10.1213/ANE.0000000000001942
Xie, Z., Dong, Y., Maeda, U., Alfille, P., Culley, D. J., Crosby, G., et al. (2006). The common inhalation anesthetic isoflurane induces apoptosis and increases amyloid β protein levels. Anesthesiology 104, 988–994. doi: 10.1097/00000542-200605000-00015
Xie, Z., Dong, Y., Maeda, U., Moir, R. D., Xia, W., Culley, D. J., et al. (2007). The inhalation anesthetic isoflurane induces a vicious cycle of apoptosis and amyloid -protein accumulation. J. Neurosci. 27, 1247–1254. doi: 10.1523/jneurosci.5320-06.2007
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yang, W. S., and Stockwell, B. R. (2016). Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 26, 165–176. doi: 10.1016/j.tcb.2015.10.014
Yang, Y. T., Ju, T. C., and Yang, D. I. (2005). Induction of hypoxia inducible factor-1 attenuates metabolic insults induced by 3-nitropropionic acid in rat C6 glioma cells. J. Neurochem. 93, 513–525. doi: 10.1111/j.1471-4159.2005.03032.x
Zhang, Y., Dong, Y., Wu, X., Lu, Y., Xu, Z., Knapp, A., et al. (2010). The mitochondrial pathway of anesthetic isoflurane-induced apoptosis. J. Biol. Chem. 285, 4025–4037. doi: 10.1074/jbc.M109.065664
Zhang, Y., Xu, Z., Wang, H., Dong, Y., Shi, H. N., Culley, D. J., et al. (2012). Anesthetics isoflurane and desflurane differently affect mitochondrial function, learning, and memory. Ann. Neurol. 71, 687–698. doi: 10.1002/ana.23536
Zhou, L., Wang, Z., Zhou, H., Liu, T., Lu, F., Wang, S., et al. (2015). Neonatal exposure to sevoflurane may not cause learning and memory deficits and behavioral abnormality in the childhood of Cynomolgus monkeys. Sci. Rep. 5:11145. doi: 10.1038/srep11145
Keywords: glutathione peroxidase, ferrostatin-1, iron, reactive oxygen species, mitochondria, volatile anesthetic
Citation: Xia Y, Sun X, Luo Y and Stary CM (2019) Ferroptosis Contributes to Isoflurane Neurotoxicity. Front. Mol. Neurosci. 11:486. doi: 10.3389/fnmol.2018.00486
Received: 13 September 2018; Accepted: 14 December 2018;
Published: 09 January 2019.
Edited by:
Gregg E. Homanics, University of Pittsburgh, United StatesReviewed by:
David F. Werner, Binghamton University, United StatesRalf J. Braun, University of Bayreuth, Germany
Copyright © 2019 Xia, Sun, Luo and Stary. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Creed M. Stary, Y3N0YXJ5QHN0YW5mb3JkLmVkdQ==