 Padinjat Raghu
Padinjat Raghu Annu Joseph
Annu Joseph Harini Krishnan
Harini Krishnan- National Centre for Biological Sciences-TIFR, Bengaluru, India
Phosphoinositides, the seven phosphorylated derivatives of phosphatidylinositol have emerged as regulators of key sub-cellular processes such as membrane transport, cytoskeletal function and plasma membrane signaling in eukaryotic cells. All of these processes are also present in the cells that constitute the nervous system of animals and in this setting too, these are likely to tune key aspects of cell biology in relation to the unique structure and function of neurons. Phosphoinositides metabolism and function are mediated by enzymes and proteins that are conserved in evolution, and analysis of knockouts of these in animal models implicate this signaling system in neural function. Most recently, with the advent of human genome analysis, mutations in genes encoding components of the phosphoinositide signaling pathway have been implicated in human diseases although the cell biological basis of disease phenotypes in many cases remains unclear. In this review we evaluate existing evidence for the involvement of phosphoinositide signaling in human nervous system diseases and discuss ways of enhancing our understanding of the role of this pathway in the human nervous system’s function in health and disease.
Historical Perspective
Phosphoinositides are low abundance cellular membrane lipids generated by phosphorylation on the inositol headgroup of phosphatidylinositol. Historically, studies on brain tissue played an important part in the discovery of these molecules. Biochemical fractionation studies by Jordi Folch-Pi on brain extracts lead to the discovery of a mixture (which he named diphosphoinositide) that contained mainly what we now know to be phosphatidylinositol (PI), phosphatidylinositol 4 phosphate (PI4P) and phosphatidylinositol 4,5 bisphosphate [PI(4,5)P2] (Folch and Wolleey, 1942; Folch, 1949a, b). The signaling functions of phosphoinositides originated from observations by Hokin and Hokin (1955) that stimulation of pancreatic slices led to the incorporation of phosphate in lipids. It was also reported that stimulation of brain tissue led to the incorporation of phosphate into phosphatidic acid and phosphoinositides (Dawson, 1954a, b); the Hokins’ also discovered that stimulation of nervous system tissue (brain slices or dorsal root ganglion) with acetylcholine, resulted in a similar incorporation of phosphate into lipid fractions (Hokin et al., 1960). Subsequently, it became apparent that in cells, the activity of a phosphoinositide phospholipase C (Rhee and Choi, 1992) results in the hydrolysis of phosphoinositides leading to generation the products, soluble inositol 1,4,5 triphosphate (IP3) and diacyl glycerol (DAG) that act as second messengers (Berridge and Irvine, 1984). In the subsequent years, numerous studies have demonstrated the ability of phosphoinositides to regulate key cellular functions through non-PLC mediated mechanisms in a range of cell types. These include actin dynamics, vesicular transport and nuclear function which have all been extensively reviewed (Divecha et al., 1993; Martin, 1998; Simonsen et al., 2001; Haucke, 2005; Di Paolo and De Camilli, 2006; Wu et al., 2014; Posor et al., 2015). Although phosphoinositides are present in every cell type in eukaryota, given the long-established observation that inositol lipids are enriched in the brain, these lipids are likely to support key cellular functions in the human brain and alterations in these could lead to diseases of the nervous system. In this review, we provide a review of the major known functions of phosphoinositides in controlling neural cell function and human disease.
Overview of Phosphoinositide Signaling
The myo-inositol head group of the lipid phosphatidylinositol (PI) (Figure 1A) can be selectively phosphorylated at positions 3, 4 and 5 to generate seven unique species. These include three monophosphates -phosphatidylinositol 3 phosphate (PI3P), phosphatidylinositol 4 phosphate (PI4P) and phosphatidylinositol 5 phosphate (PI5P); three bisphosphates-phosphatidylinositol 4,5 bisphosphate [PI(4,5)P2], phosphatidylinositol 3,5 bisphosphate [PI(3,5)P2] and phosphatidylinositol 3,4 bisphosphate [PI(3,4)P2] and a single trisphosphate-phosphatidylinositol 3,4,5 trisphosphate [PI(3,4,5)P3] (Figure 1B). Biochemical studies across multiple cell types have revealed that phosphatidylinositol and the seven phosphoinositides are present in defined proportions (Figures 1C,D) and in many cases their cellular levels change in a precise and reversible manner during the response to specific cellular changes or environmental stimuli; these observations underscore their definition as important mediators of information transfer in cells. From a cell biological perspective, it is important to bear in mind that being lipids, phosphoinositides are not freely diffusible in the aqueous cytoplasm; therefore they are spatially restricted to the membrane at which they are produced and can move between intracellular compartments either by vesicular transport or through the action of lipid transport proteins (Cockcroft and Raghu, 2018). As a result of these constraints, each of the phosphoinositides has a distinct spatial pattern of distribution among organelles; most phosphoinositides are primarily enriched at one or two organelle membranes although minor pools of each lipid may also be found at other organelle membranes (Figure 2A) [reviewed in Di Paolo and De Camilli (2006), Balla (2013)]. In this review, we focus on the cellular functions of phosphoinositides in the nervous system. A comprehensive description of the cellular functions of these lipids has been published elsewhere and readers are referred to here for a detailed description of specific topics (Balla, 2013).
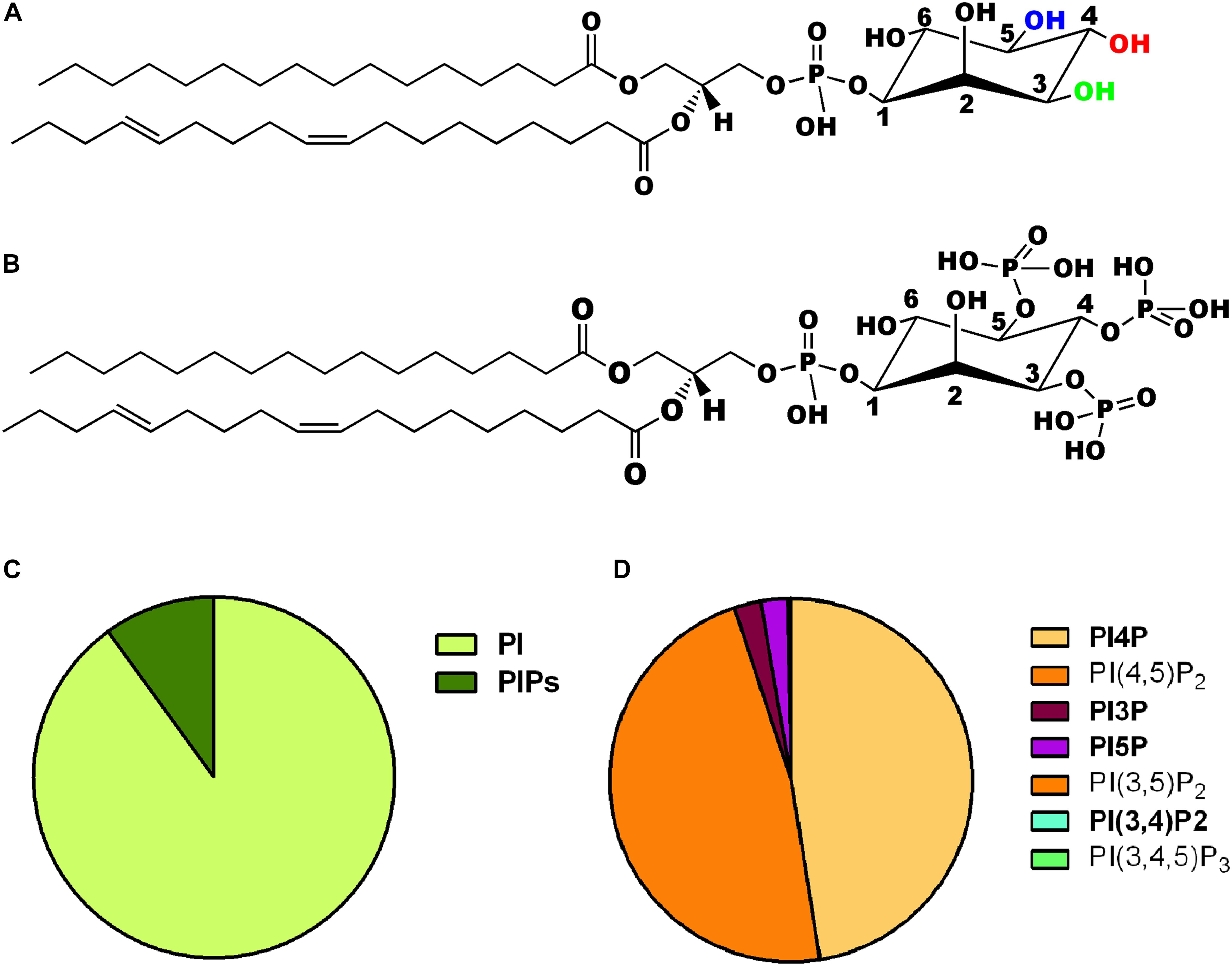
Figure 1. (A) Chemical structure of phosphatidylinositol. The three hydroxyl groups at positions 3, 4, and 5 on the inositol ring are shown in green, red, and blue, respectively. (B) Chemical structure of one of the seven phosphoinositides, phosphatidylinositol 3,4,5 trisphosphate is shown where the hydroxyls are position 3,4,5 of the inositol headgroup are all phosphorylated. (C) Pie chart representing the relative abundance of phosphatidylinositol (PI) and its phosphorylated derivatives (PIPs) in animal cells. (D) Pie chart representing the relative abundance of the seven PIPs in animal cells. The two most abundant forms PI4P and PI(4,5)P2 are shown individually along with the other five low abundance derivatives [PI3P, PI5P, PI(3,5)P2, PI(3,4)P2, and PI(3,4,5)P3].
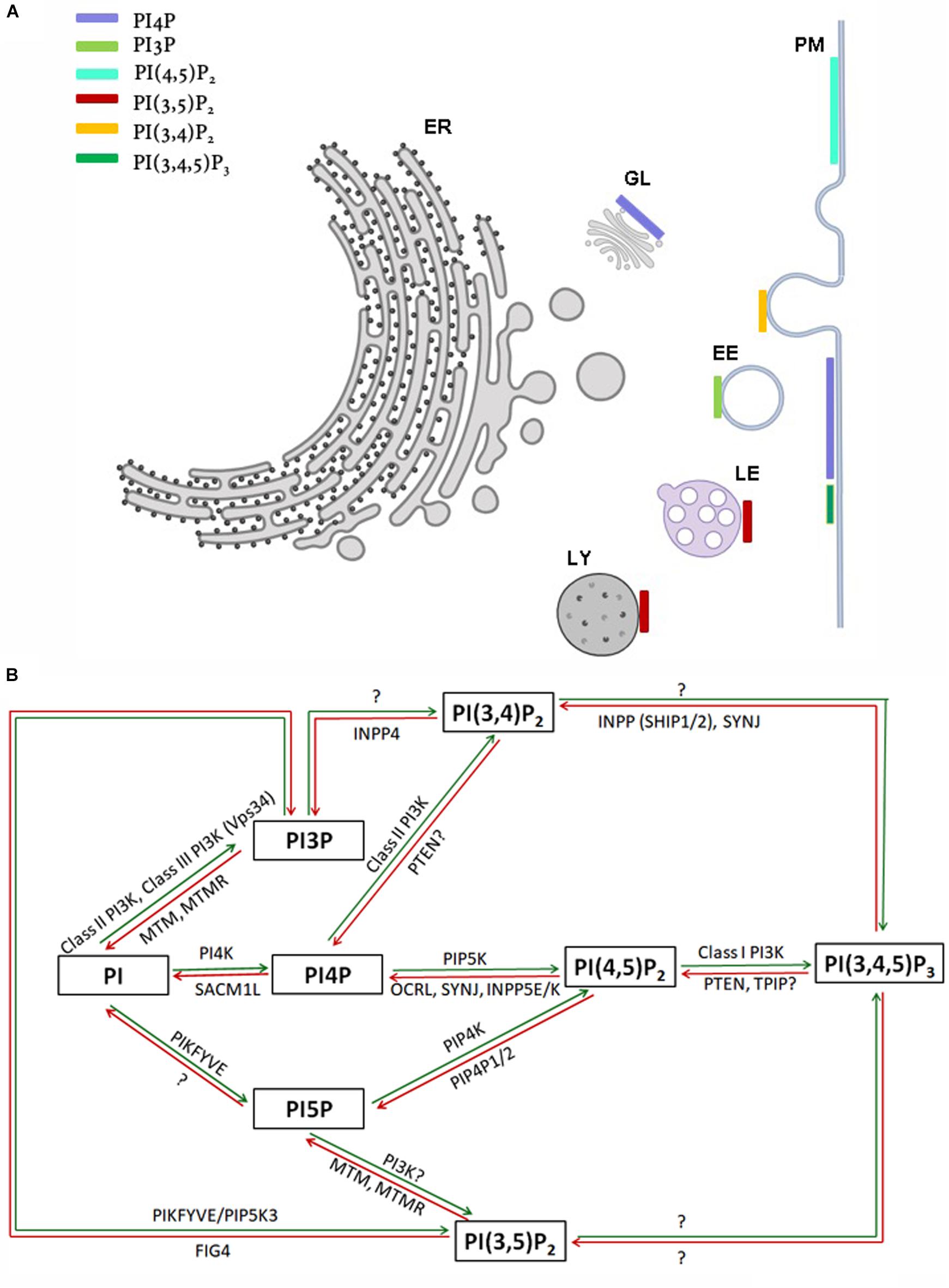
Figure 2. (A) Sub-cellular distribution of phosphoinositides in a eukaryotic cell. Major organelles are shown: PM-plasma membrane, ER-endoplasmic reticulum, EE-early endosome, LE-late endosome, LY-lysosome, GL-Golgi. The presence of a major pool of a specific phosphoinositide at a particular organelle membrane is indicated by a colored bar. Minor pools of each species at organelles though reported are not represented. (B) Schematic representation of kinases and phosphatases regulating phosphoinositide metabolism. Green and red arrows represent phosphorylation and dephosphorylation reactions, respectively. Proteins separated by commas catalyze the same reaction. The steps in which the existence of an enzymatic reaction is uncertain are indicated by question marks.
Control of Cellular Phosphoinositide Levels
Within cells, phosphoinositides are generated through the selective addition or removal of phosphate groups from the myo-inositol headgroup. In general, the addition of phosphates is performed by an evolutionarily conserved group of enzymes called the phosphoinositide kinases. These enzymes are selective in two respects (i) They are specific for the substrate molecule on which they will act: for example a lipid kinase may act only on PI5P but not PI4P (ii) They are specific for the OH group on the myo-inositol ring at which they will add the phosphate group: for example an enzyme that will only add a phosphate at position 4 but not position 5. A large and evolutionarily conserved family of kinases has been described that show selectivity based on the above criteria (Table 1, Supplementary Table 1, and Figure 2B). The properties of these enzymes and the huge body of experimental analysis of these have been covered in an excellent and detailed review (Sasaki et al., 2009). Most enzymes of this family are conserved across all of eukaryota although some are seen only in metazoan genomes (e.g., Class I PI3K, PIP4K). In addition to the phosphoinositide kinases, lipid phosphatases that are able to selectively remove a phosphate group from the myo-inositol headgroup have been described. These enzymes also show substrate specificity and defined catalytic activity just like the phosphoinositide kinases (Table 1, Supplementary Figure 1, and Figure 2B) and (Sasaki et al., 2009) and are evolutionarily well conserved.
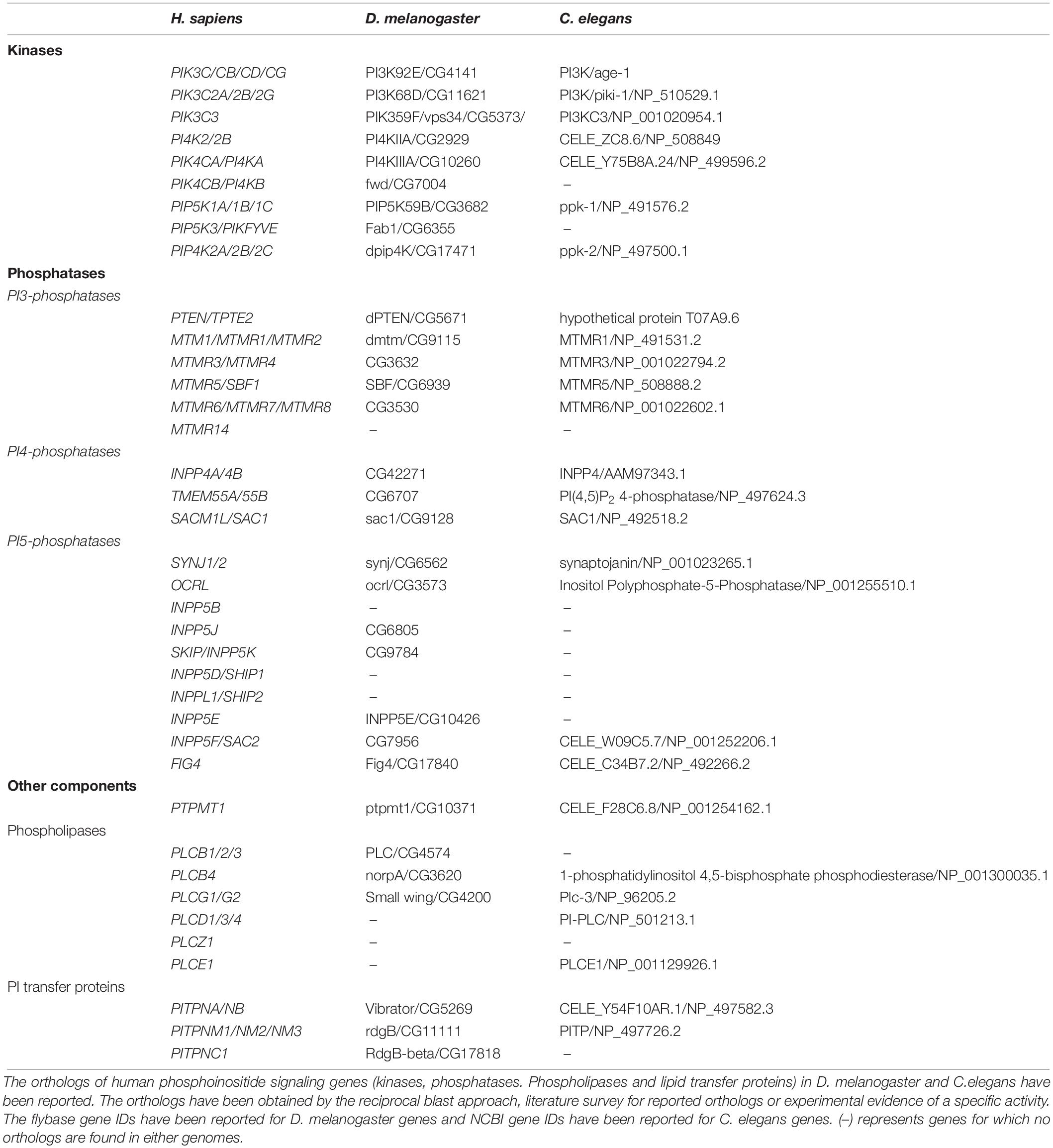
Table 1. Phosphoinositide kinase and phosphatase orthologs in Drosophila melanogaster and Caenorhabditis elegans.
Phospholipases are enzymes which belong to the class hydrolases, i.e., those that use a molecule of water to degrade substrates; they catalyze the breakdown of phospholipids into fatty acids and other constituent molecules (Dennis, 2015). They are named based on the position they hydrolyze on the backbone of phospholipids. Phospholipases are known to be involved in phospholipid turnover, membrane remodeling and neurotransmitter release in brain. Under pathological conditions they result in altered membrane permeability, ion homeostasis and accumulation of lipid peroxidases. The phospholipase of most relevance to phosphoinositide signaling is phospholipase C (EC 3.1.4.11) that is able to hydrolyze PI(4,5)P2 to generate inositol 1,4,5 trisphosphate (IP3) and diacylglycerol (DAG) (Rhee, 2001). The expression of phospholipase C genes varies across different brain compartments and tissues as detailed in this review. The phospholipases genes are well conserved across evolution (Table 1).
The phospholipid transfer proteins (LTPs) are molecules that facilitate the transfer of phospholipids between the membranes of sub-cellular compartments thus contributing to lipid homeostasis of cellular organelles. There are many classes of lipid transfer proteins; in the context of phosphoinositide signaling, the most important are the phosphatidylinositol transfer protein family that supports the transfer of PI from its site of synthesis, the endoplasmic reticulum, to the plasma membrane (Hsuan and Cockcroft, 2001). More recently additional classes of proteins such as the OSH/OSBP family have been discovered that appear to be required for PI4P transfer between organelle membranes (Cockcroft and Raghu, 2018). These lipid transfer proteins are distributed across a range of organisms (Table 1). Collectively the molecules described above are core components in the regulation of phosphoinositide signaling in eukaryotic cells (Balakrishnan et al., 2015) including those in the brain. The metabolism of each phosphoinositide and its cellular functions with respect to neurons are presented below.
Cellular Functions of Phosphoinositides in the Nervous System
PI3P
PI3P can be produced from PI by Class III PI3K (Vps34) in endosomes (Schu et al., 1993) or by class II phosphatidylinositol 3-phosphate kinase (PI3K) at the plasma membrane (Vanhaesebroeck et al., 2010). In addition, PI3P can be generated by dephosphorylation of PI(3,4)P2 by INPP4a (Sasaki et al., 2010) or PI(3,5)P2 by FIG4 (Chow et al., 2007). PI3P is mainly found localized at early endosomes and is involved in endosomal trafficking (Gaidarov et al., 2001) but is also generated at the autophagosomal membrane thus regulating autophagy (Noda et al., 2010).
With respect to neural cell function, PI3P may be particularly important in regulating the levels of cell surface receptors for neurotransmitters, or for the control of autophagy which is believed to be a key ongoing process in neurons that is relevant to neurodegeneration (Menzies et al., 2017). In hippocampal neurons of mice, PI3P was found to be localized at dendrites, axons and partially at synapses (Wang et al., 2011). The early endosomal pool of PI3P is involved in the post synaptic clustering of GABA receptors, thus regulating the strength of inhibitory post synapses in cultured hippocampal neurons (Papadopoulos et al., 2017). Depletion of Vps34, the key enzyme in PI3P generation, in selected brain regions results in neuronal degeneration and reactive gliosis that appear to be associated with defects in the endosomal system but not changes in autophagy (Zhou et al., 2010; Wang et al., 2011). Depletion of Vps34 in Schwann cells, the glia of the peripheral nervous system results in defective myelination associated with altered endo-lysosomal system and autophagy (Logan et al., 2017). Finally, PI3P has been found to be selectively deficient in the brains of human patients with Alzheimer’s disease (Morel et al., 2013) and mouse models of Alzheimer’s disease with associated defects in endosomal-lysosomal network [reviewed in Nixon (2017)].
PI4P
PI4P is found in at least two major subcellular locations in cells, the Golgi complex and the plasma membrane. PI4P at Golgi coordinates several functions including vesicle trafficking, membrane biogenesis and lipid homeostasis (D’Angelo et al., 2008). At the Golgi apparatus, PI4P recruits proteins that bind this lipid and mediate its functions at this location thus regulating processes such as sphingolipid biosynthesis (Kawano et al., 2006) and cargo sorting. At the plasma membrane, PI4P is required for maintaining PI(4,5)P2 levels during receptor activated PLC signaling; PI4P can directly regulate the function of plasma membrane proteins such as KCNQ2/3 channels (Dickson et al., 2014) and smoothens the receptor for hedgehog signaling (Jiang et al., 2016). PI4P also binds to clathrin adaptors such as epsin R (Hirst et al., 2003) and AP-1 (Wang et al., 2003) thus regulating endosome trafficking. PI4P is generated by the phosphatidylinositol 4-kinase (PI4K) family of enzymes (Balla and Balla, 2006). At the plasma membrane PI4P is produced by the PI4KIIIα class of enzymes and regulates PLC dependent functions (Nakatsu et al., 2012; Balakrishnan et al., 2018). By contrast PI4P at the Golgi is generated by the PI4KIIIβ family (Godi et al., 1999; Walch-Solimena and Novick, 1999). PI4P can also be produced by dephosphorylation of PI(4,5)P2 by 5′ lipid phosphatases such as oculocerebrorenal syndrome of Lowe (OCRL) and Synaptojanin. These enzymes are likely to be particularly important in the context of neural cell function (see below). PI4P can also be degraded by the lipid phosphatase SACM1L (Sac1 in yeast), an ER resident enzyme that at plasma membrane endoplasmic reticulum contact sites (Stefan et al., 2011; Zewe et al., 2018).
In the nervous system, very little information is available on the direct effect of perturbations in PI4P levels, and more on the enzymes regulating it. The presence of PI4K2α at high concentration in synaptic vesicles indicates a direct or indirect role of PI4P in neuronal function, as a precursor of PI(4,5)P2 (Guo et al., 2003), which in turn has been identified to be crucial for clathrin mediated endocytosis (Wenk et al., 2001). Pi4k2a mutant adult mice lacking kinase activity exhibit progressive neurological disorders, including substantial degeneration of spinal axons (Simons et al., 2009). In Drosophila models, depletion of a protein complex that includes PI4KIIIα that generates PI4P at the plasma membrane has been shown to affect the neuronal accumulation Aβ42 oligomers (Zhang X. et al., 2017).
PI5P
PI5P is the most recently discovered phosphoinositide (<10% of total cellular phosphatidylinositol monophosphates) (Rameh et al., 1997). Biochemical fractionation studies indicate that PI5P is distributed across multiple cellular compartments including the plasma membrane, nucleus, endo-lysosomal system and the Golgi (Sarkes and Rameh, 2010). A number of studies have suggested a role for PI5P in regulating chromatin function and transcriptional regulation in the nucleus [reviewed in Fiume et al. (2015)]. Ectopic expression of the Shigella phosphatase IpgD, that generates PI5P is known to cause endosomal sorting defects of the epidermal growth factor receptor (EGFR) (Ramel et al., 2011) and PI5P binding proteins have been identified in early endosomal factions (Boal et al., 2015). In vitro, PI5P is known to stimulate myotubularin (Schaletzky et al., 2003), proteins that play an important role in early endosomal sorting (Naughtin et al., 2010). Collectively these observations suggest that PI5P might regulate endosomal trafficking, but the mechanism remains unclear. The mechanism by which PI5P is generated remains unresolved. It has been argued that the enzyme PIKFYVE/Fab1 might synthesize PI5P from PI (Shisheva et al., 2015). An alternative model is that PI5P can be generated by the lipid phosphatase activity of the myotubularin family of enzymes acting on PI(3,5)P2 to generate PI5P in yeast (Walker et al., 2001) or mammalian cells (Vaccari et al., 2011). PI5P concentration can also be regulated by the phospholipid kinase phosphatidylinositol 5 phosphate 4-kinase (PIP4K) that is able to convert PI5P into PI(4,5)P2 (Gupta et al., 2013) and reviewed in Kolay et al. (2016).
There is limited information available on the role of PI5P in neurons. In vitro knock down or pharmacological inhibition of the PIP4K isoform PIP4K2C is reported to reduce mutant huntingtin protein aggregates by increasing basal autophagy, suggesting that the kinase could be a potential target for treatment of the progressive neurodegenerative Huntington’s disease (Al-Ramahi et al., 2017). Drosophila mutants with elevated PI5P levels have been reported to have defects in the early endosomal compartment of neurons (Kamalesh et al., 2017). It has also recently been reported that depletion of PIP4K2C results in defects in the recycling of Notch to the cell surface (Zheng and Conner, 2018); since Notch is a key regulator of neurogenesis, these findings may imply a role for PI5P, the substrate of PIP4K2C in brain development. Collectively these observations suggest a role for PI5P in regulating membrane transport in neural cells that remains to be fully understood.
PI(4,5)P2
PI(4,5)P2 is mainly localized at the plasma membrane of cells although other minor pools of this lipid have been described on the membranes of internal organelles such as endosomes and lysosomes (van den Bout and Divecha, 2009). In this location it controls a number of cellular processes. Historically the oldest known function of PI(4,5)P2 is to serve as the substrate for receptor activated phospholipase C which generates inositol 1,4,5 trisphosphate (IP3) and diacylglycerol (DAG) which themselves serve as second messengers. However, it is now apparent that a major function of PI(4,5)P2 is to interact with and modulate the activity of numerous proteins that regulate key sub-cellular processes such as vesicular transport (endocytosis and exocytosis), cytoskeletal reorganization and the regulation of ion channel and transporter activity [reviewed in Kolay et al. (2016)]. PI(4,5)P2 is mainly produced by the phosphorylation of PI4P by phosphatidylinositol 4-phosphate 5-kinase (PIP5K) (Funakoshi et al., 2011). In humans, three PIP5K isozymes are expressed namely PIP5Kα, β, and γ (Funakoshi et al., 2011). A minor pool of PI(4,5)P2 is also synthesized by phosphatidylinositol 5-phosphate 4-kinase (PIP4K) using PI5P (Rameh et al., 1997) and PTEN using PIP3 as substrates (Rahdar et al., 2009). PI(4,5)P2 is consumed by the activity of PLC, Class I PI3K that converts it into PI(3,4,5)P3 and also by the activities of the 5’ phosphatases synaptojanin (SYNJ) and OCRL.
PI(4,5)P2 has a number of critical functions in the nervous system. Many receptors for neurotransmitters in the brain use G-protein coupled, PLC mediated hydrolysis of PI(4,5)P2 as a key step in signal transduction [reviewed in Dickson and Hille (2019)] and numerous ion channels in the nervous system require PI(4,5)P2 for their normal function [reviewed in Hille et al. (2015)]. In addition, PI(4,5)P2 regulates multiple steps of the synaptic vesicle cycle. The lipid kinase PIP5Kγ is enriched at nerve terminals and plays a key role in regulating PI(4,5)P2 levels (Wenk et al., 2001); PIP5Kγ–/– mice show decreased PI(4,5)P2 in the brain and defects in synaptic transmission (Di Paolo et al., 2004). Dephosphorylation of PI(4,5)P2 is also important for the recycling synaptic vesicles that are endocytosed at the presynaptic terminal. The PI(4,5)P2 5-phosphatase SYNJ1 plays a key role in the uncoating of clathrin-coated synaptic vesicles (Mani et al., 2007). Increased PI(4,5)P2 levels and clustering of clathrin-coated vesicles at nerve endings were observed in the neurons of SYNJ1-deficient mice (Cremona et al., 1999), and the internalization of postsynaptic AMPA receptor, involved in fast excitatory synaptic transmission, was inhibited in cultured Synj1 knockout mouse hippocampal neurons (Gong and De Camilli, 2008). A role for PI(4,5)P2 has also been proposed for regulating cytoskeletal function in neurons; the kinesin motor Unc104 can bind PI(4,5)P2 (Klopfenstein et al., 2002) and this binding plays a key role in kinesin based transport in neurons (Klopfenstein and Vale, 2004; Kumar et al., 2010). Since PI(4,5)P2 performs multiple functions in neurons, an interesting question arises on how these are controlled independently. Recent studies have proposed a role for functional pools of PI(4,5)P2 generated in neurons by specific lipid kinases [(Chakrabarti et al., 2015) and reviewed in Kolay et al. (2016)].
PI(3,5)P2
PI(3,5)P2 is a lipid that is primarily found on late endosomes and lysosomal membranes. Its levels rise in response to changes in extracellular stimuli including osmotic stress in yeast or stimulation with growth factors and phagocytosis in mammalian cells [reviewed in Hasegawa et al. (2017)]. A number of studies implicate PI(3,5)P2 in late endosomal dynamics and it has also been proposed to regulate the function of TPC ion channels on the lysosomal membrane (Jha et al., 2014). Tsuruta et al. (2009), Seebohm et al. (2012). PI(3,5)P2 is produced in cells by the activity of a PI3P-5 kinase (Fab1 in yeast and PIKFYVE in mammals). PI(3,5)P2 can be dephosphorylated in vitro by the activity of the phosphatase FIG4 to generate PI3P; members of the MTMR family have also been proposed to regulate PI(3,5)P2 levels although the relevance of these mechanisms in vivo remains unclear [reviewed in Hasegawa et al. (2017)].
In neurons decreased levels of PI(3,5)P2 directly affect NMDA-induced voltage-gated Ca2+ channel internalization which causes neuronal excitotoxicity (Tsuruta et al., 2009) and in the hippocampus neurons of new-born rats, defects in postsynaptic GluA1 receptor turnover are seen when PI(3,5)P2 levels are altered (Seebohm et al., 2012). PI(3,5)P2 levels have also been shown to increase during homeostatic downscaling, where neurons reduce their postsynaptic strength to prevent chronic neuronal hyperactivity (McCartney et al., 2014). A number of studies have shown a role for PI(3,5)P2 in regulating lysosomal function and autophagy in the nervous system. Analysis of spontaneous mouse mutants of Fig4 and Vac14, members of the PIKFYVE-FIG4 enzyme complex, has shown that although these proteins are widely expressed across the body, the nervous system that is particularly susceptible to the loss of these enzymes, presumably through altered PI(3,5)P2 levels. These effects include neuronal degeneration, myelination defects and accumulation of inclusions in astrocytes [reviewed in Lenk and Meisler (2014)]. These findings have led to a mechanistic explanation for a number of human neurological syndromes in which proteins involved in PI(3,5)P2 metabolism are affected (see below).
PI(3,4)P2
PI(3,4)P2 is a lipid that is primarily found at the plasma membrane and the early endosomal system. The function of this lipid is unclear but it has been proposed to regulate early endosomal dynamics and also alter the gain of the Class I PI3kinase signaling pathway (Zhang S. et al., 2017). The best characterized route of PI(3,4)P2 synthesis is the dephosphorylation of PI(3,4,5)P3 by 5′ phosphatases [reviewed in Hawkins and Stephens (2016)] although it has also been proposed to be generated in the early endosomal system during clathrin mediated endocytosis by Class II PI3K activity on PI4P (Posor et al., 2013; He et al., 2017). The myotubularin family of lipid phosphatases may control the levels of this lipid by degrading it to PI4P.
The available literature points to the role of PI(3,4)P2 in neurite initiation and dendrite morphogenesis (Zhang S. et al., 2017) by promoting actin aggregation at the site of initiation, leading to the cytoskeletal reorganization for forming the cylindrical neurite. PI(3,4)P2 was also shown to be present in the postmitotic multipolar neurons derived from radial glia. The actin remodeling protein lamellipodin binds to PI(3,4)P2 and recruits Ena/vasodilator-stimulated phosphoprotein (VASP) to positively regulate the number of primary processes in the multipolar cells (Yoshinaga et al., 2012).
PI(3,4,5)P3
PI(3,4,5)P3 is a very low abundance lipid found at the plasma membrane of cells following activation of plasma membrane receptors (Ming et al., 1999; Henle et al., 2011; Arendt et al., 2014). The principal mechanism of synthesis is the activity of Class I PI3K enzymes that phosphorylate PI(4,5)P2 to generate PI(3,4,5)P3. PI(3,4,5)P3 can be degraded by the activity of 3′ phosphatases such as PTEN or by the action of 5′ phosphatases such as SHIP.
As in all other tissues and cell types, the levels of PI(3,4,5)P3 play a key role in growth through its ability to control cell division and size through activation of a number of intracellular signaling pathways (Engelman et al., 2006). PI(3,4,5)P3 has been found enriched in the growth cones of developing neurites (Ménager et al., 2004), and has been implicated in the control of numerous processes in both developing and mature neurons. These include remodeling of the actin cytoskeleton during neurite outgrowth and dendrite morphogenesis (Ming et al., 1999; Henle et al., 2011). Synthesis and availability of PI(3,4,5)P3 is also required for maintaining AMPA-type glutamate receptors at synaptic membrane, and this in turn regulates synaptic function in hippocampal neurons (Arendt et al., 2014). PTEN, that regulates PI(3,4,5)P3 levels is expressed and plays an important role during neuronal morphogenesis and differentiation (Lachyankar et al., 2000; van Diepen and Eickholt, 2008). Knockout of PTEN and resulting elevated PI(3,4,5)P3 level leads to an increase in the diameter of parallel fiber axons of granule cells, an increase in oligodendrocyte differentiation and de novo myelination of normally unmyelinated parallel fibers (Goebbels et al., 2017). Inhibition of PTEN by bisperoxovanadium has also shown to promote oligodendrocyte proliferation and myelination of dorsal root ganglion neurons (De Paula et al., 2014). At the same time, PTEN loss does not show an improvement in remyelination following brain injury (Harrington et al., 2010).
Effectors of Phosphoinositide Signaling
The oldest known function of phosphoinositides is the regulation of calcium signaling. This process is triggered by the metabolic conversion of PI(4,5)P2 into IP3 and DAG by phospholipase C leading the activation of intracellular calcium release channels and plasma membrane calcium influx channels (Berridge and Irvine, 1989). However, we now know that phosphoinositides function through multiple mechanisms including their ability to bind to cellular proteins and regulate their activity. Given their negative charge, phosphoinositides may bind to proteins by interacting with single or clusters of positively charged residues (as in the case of KiR channels) (Hansen, 2015) or via well-defined protein domains (e.g., PH, PX, FYVE domains) [reviewed in Hammond and Balla (2015)]. A specific domain may be found in the context of multiple proteins that bind a specific phosphoinositide (for example identification of an FYVE domain may be an indication of a protein that binds PI3P and the context for its cellular function). More recently an unbiased quantitative mass spectrometry analysis has described a large set of more than 400 proteins that bind phosphoinositides (Jungmichel et al., 2014) and are likely to be key mediators of the effects of phosphoinositides in cells.
The well-characterized phosphoinositide-binding domains in effector proteins are the PH (pleckstrin homology), PX (phox homology), FYVE (Fab1, YOTB, Vac1 and EEA1), ENTH (epsin amino-terminal homology) and FERM (Four-point-one, ezrin, radixin and moesin) domains (Balla, 2005). Through their interactions with specific phosphoinositides, these effector proteins are recruited onto membrane surfaces and participate in various cellular processes (Di Paolo and De Camilli, 2006).
Expression of Phosphoinositide Signaling Genes in the Human Nervous System
Gene expression in human tissues and cell types is differentially regulated depending on the developmental stage, tissue type, cell type and in some cases is altered in specific disease conditions. In addition to core genetic mechanisms, gene expression is also controlled epigenetically. Post-transcriptional processes can change the levels of transcripts and control of translation and protein stability can also affect the levels of proteins in the nervous system. The spatial and temporal pattern of gene expression can often give an insight into the potential function of a gene. Several studies have documented patterns of gene expression in human cells and tissues and an analysis of data on expression in brain regions, cell types and temporal patterns from such studies can provide an insight into the function of specific genes in the nervous system.
Several large-scale studies have documented gene expression in the human nervous system. Gene expression studies in the brain are mostly done from post-mortem samples. Studies have shown that death results in significant expression change in only 10% of genes with varied functional categorization, thus justifying the use of post-mortem brain samples for expression analysis (Franz et al., 2005). Such studies have included (i) the analysis of gene expression in specific cell types of the nervous system1, (ii) expression in distinct regions of the normal human brain and also a profile of the expression of each gene as a function of time starting with fetal development through to adult human brains and subsequently in the ageing brain2 and (iii) gene expression analysis in the diseased brain3. Likewise the Human Protein Atlas provides information on the sub-cellular localization and expression of more than 50% of human protein coding genes in major human tissues and organs (Bae et al., 2015). We have mined such databases for information on the expression of genes encoding components of the phosphoinositide signaling system in the human nervous system. The expression of genes in all these datasets is represented as TPM (Transcripts Per Million base pairs) values which is normalized for the dataset under study and cannot be compared across different datasets. These TPM values have been used to plot the heat maps and tables discussed below. The expression of genes in brain regions, cell types and their temporal expression profile is provided with a view to identifying those with potentially important roles in the structure and function of the nervous system.
Spatial Expression Pattern of Phosphoinositide Signaling Genes in the Nervous System
Approximately 80–90% of protein-coding genes are expressed in some part of the brain during development and adult human life, though there are a large number of genes with a specific expression and alternate splicing patterns (Bae et al., 2015). Human specific expression (as compared to other primates) when noted is usually associated with enrichment in the neocortex and hippocampus. The expression values (TPM values) of 214 genes (phosphoinositide signaling genes listed in Table 1) in different brain regions was extracted from the GTEx database (see footnote 2) (Lonsdale et al., 2013). The expression of each gene was normalized to its expression in whole blood (TPMbrain/TPMwhole_blood) and is represented in the form of a heatmap that has been clustered across different regions of the brain (along the columns of the heatmap) (Figures 3A,B). This analysis reveals that most genes enriched in the nervous system are usually expressed in the cerebral cortex, cerebellum and tibial nerve. We then calculated the number of phosphoinositide signaling genes with TPMbrain/TPMwhole_blood > 1.5 or TPMbrain/TPMwhole_blood < 0.6 in each brain region in order to identify genes that are either highly expressed or minimally expressed in specific regions of the brain (Figure 4). This analysis revealed that the maximum number of genes with high TPMbrain/TPMwhole_blood values were observed in peripheral nerves (represented by the tibial nerve), the cerebral and cerebellar hemispheres. In most of the other brain regions, the phosphoinositide signaling genes seem to be expressed at very low levels, with basal ganglia, hippocampus and amygdala having slightly higher numbers of such genes.
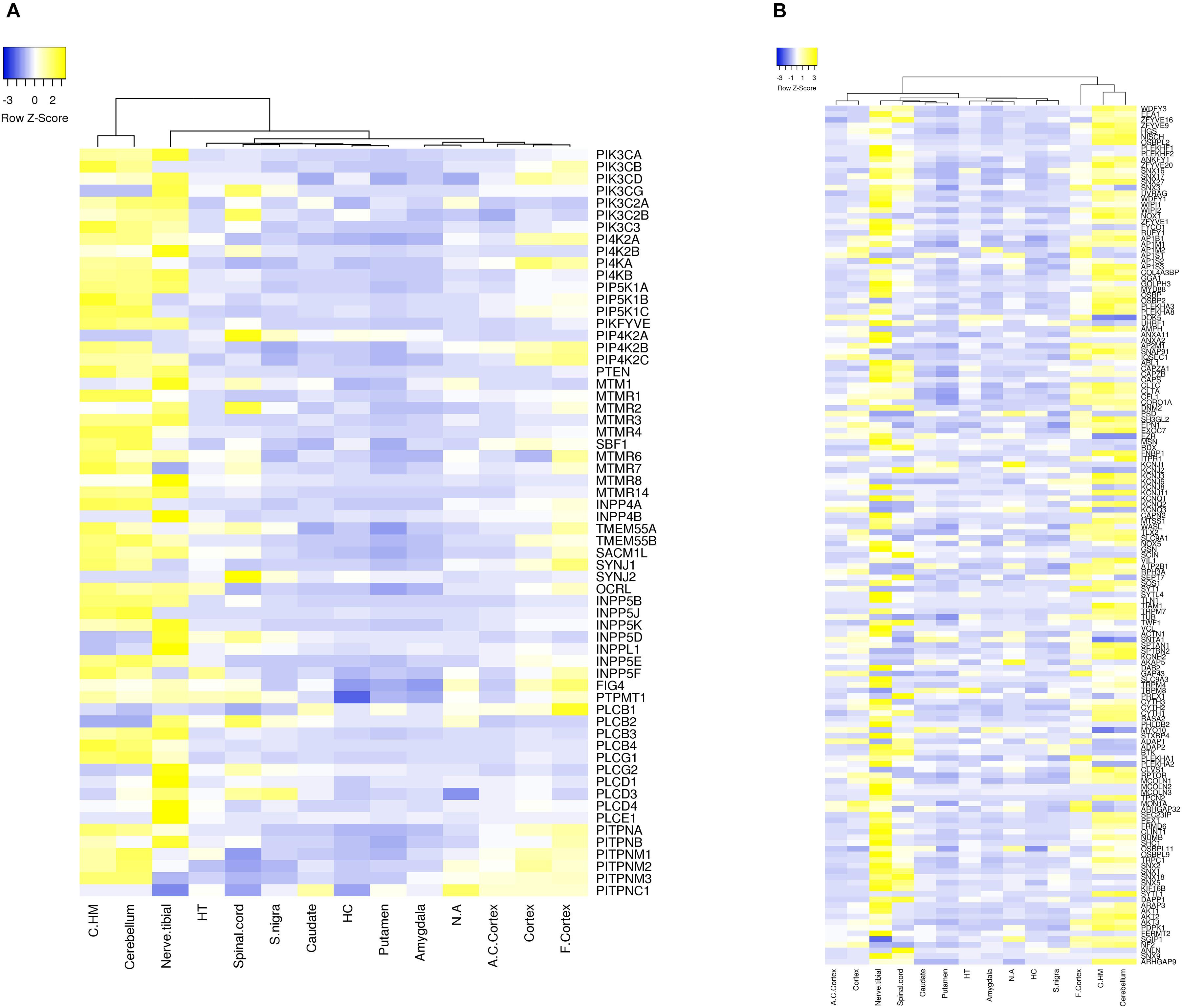
Figure 3. Expression of phosphoinositide signaling genes across human brain regions. A heat map showing the expression patterns. Data were extracted from the GTEx database. The expression values of genes for each of the brain region is normalized to the expression value of the gene in whole blood and the matrix thus obtained is represented as a heatmap. The normalized expression values range from –3 to 3 (Blue to Yellow). The map is clustered based on expression of genes in different regions of the Brain (across the column). The various brain regions represented are Cerebellar Hemisphere (C.HM), Cerebellum, Nerve tibial (N. tibial), Hypothalamus (HT), Spinal cord, Substantia nigra (S. nigra), Caudate, Hippocampus (HC), Putamen, Amygdala, Nucleus accumbens (N. accumbens), Anterior cingulate cortex (A. C. cortex), Cortex and Frontal cortex (F. cortex). Panel (A) shows the expression of kinases, phosphatases, phospholipases and lipid transfer protein while (B) shows the expression of phosphoinositide binding proteins. Individual proteins are indicated using HUGO nomenclature.
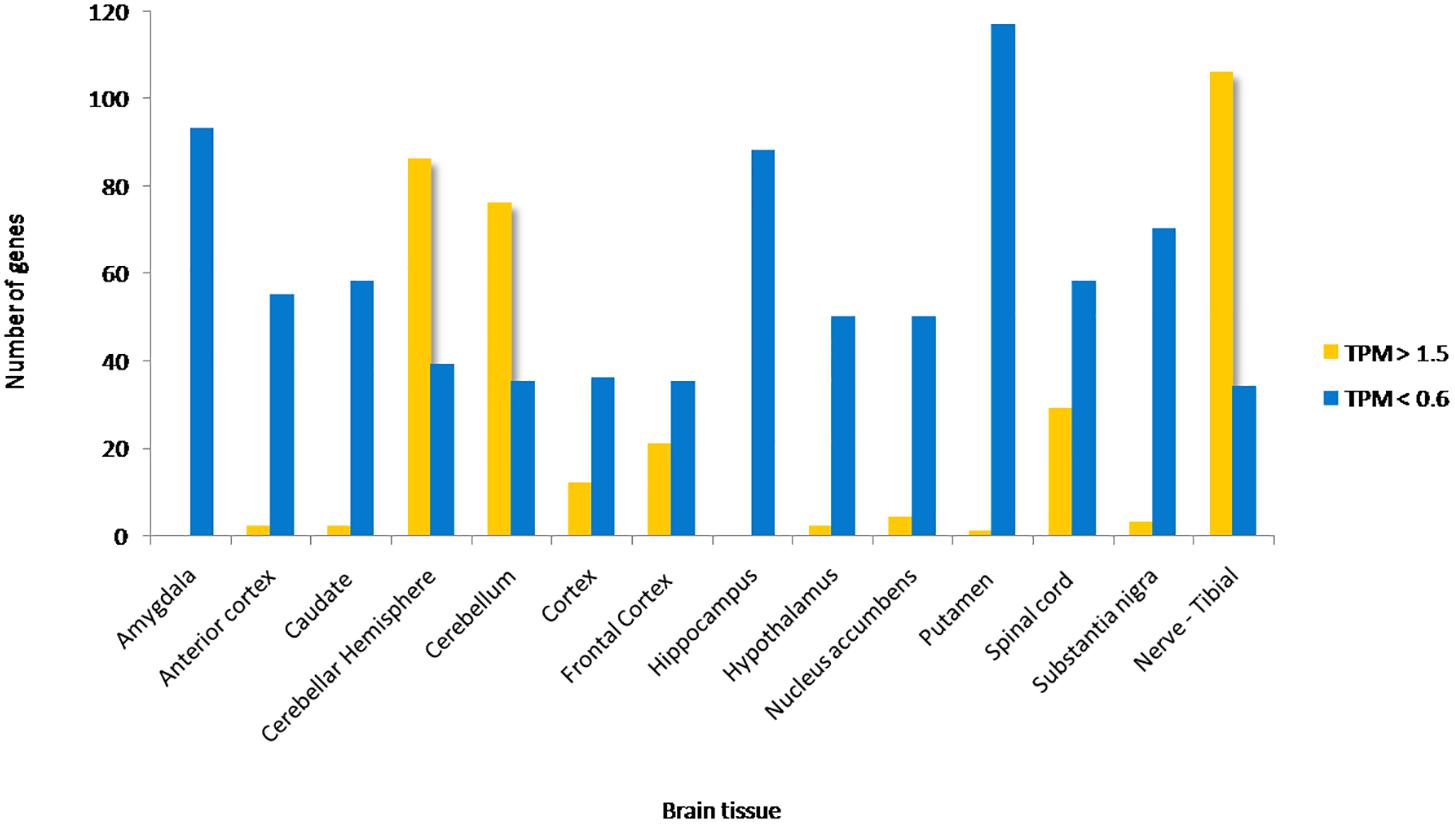
Figure 4. Proportion of phosphoinositide signaling genes with high and low expression in brain regions. The proportion of genes encoding members of the phosphoinositide signaling pathway with TPM values greater than 1.5 and less than 0.6 in specific brain regions are represented as a bar graph. Yellow bars represent TPM > 1.5, i.e., high expression genes and Blue bars represent TPM < 0.6, i.e., low expression genes.
Of all the highly enriched genes in the brain, the 10 genes that have highest TPM values across various brain regions were noted. Of these, seven genes are highly expressed in at least 10 of the 15 major regions of the brain. These genes include GAP43, KCNQ2, SNAP91, DOK 5, SH3GL2 and SYT1. GAP43 a growth-associated protein that is highly expressed during neuronal growth and axonal regeneration (Holahan, 2017); KCNQ2 a potassium channel which plays a key role in neuronal excitability (Jentsch, 2000); SNAP91, Synaptosome Associated Protein 91 a regulator of clathrin dependent endocytosis (Hill et al., 2001); DOK5 which interacts with phosphorylated receptor tyrosine kinases, activates MAP kinase signaling and is essential for neurite outgrowth (Grimm et al., 2001); SH3GL2 (SH3 Domain Containing GRB2 Like 2/, Endophilin A1) a known regulator of synaptic vesicle endocytosis (Vehlow et al., 2013) and SYT1(synaptotagmin 1) a regulator of neurotransmitter release at the synapse (18). There were also a number of highly expressed genes code for phosphoinositide binding proteins unique to the tibial nerve, i.e., not expressed in other parts of the nervous system (FERMT2, FRMD6, SLC9A3, PLCE1, TRPC1, KCNJ8, and MCOLN3) and may indicate a significant role for these in the function of peripheral nerves. It is interesting to note that most genes that show such enrichment patterns are phosphoinositide binding proteins or effectors, whereas genes encoding phosphoinositide metabolizing enzymes (kinases, phosphatases, lipases and transport proteins) appear to not show enrichment in a specific brain region, These findings are consistent with a broad role for phosphoinositide signaling in the nervous system with downstream effects being mediated in specific regions by proteins with more restricted expression patterns. While the data sets generated in such large scale gene expression analyses can be influenced by many factors, these data suggest the choices of tissue types for analyses as well as the potential nervous system compartments in which this signaling pathway could play a particularly important functional role in the context of normal physiology as well as diseases of the nervous system.
Expression of Phosphoinositide Signaling Genes in Neural Cell Types
Every region of the nervous system includes multiple cell types including neurons, glial cells (astrocytes, microglia, oligodendrocytes) and endothelial cells of the vasculature. Transcriptomes of all these cell types have been generated and are available at http://www.brainrnaseq.org. More than 2000 genes seem to be differentially enriched in specific cell types (Zhang et al., 2016). Single cell transcriptome analysis shows that neurons express a higher number of genes as compared to other cell types and microglia and endothelial cells express fewest genes (Darmanis et al., 2015). Neuronal cells can be further grouped into seven sub-populations with differential gene expression patterns and gene-expression in distinct cell types has also shown to vary with age and under neuropathological conditions (Hagenauer et al., 2018).
We extracted expression values (TPM values) for phosphoinositide signaling genes from Brain RNA-Seq data for various cell-types. The cell type specific expression values for 214 genes have been represented as a heatmap that has been clustered across different cell types in the brain (column-wise clustering in the heatmap) (Figures 5A,B). The probable connection between differential expression of genes and the disease condition has been discussed at the end of this section. The fraction of genes with TPM > 1.5 and TPM < 0.6 in each cell-type is presented (Figure 6). This analysis suggests that among all cell types, fetal astrocytes express the largest number of highly expressed genes (TPM > 1.5) and are also the cell type that show the smallest number of downregulated genes (TPM < 0.6). Among the various cell types, endothelial cells express the fewest number of differentially expressed phosphoinositide signaling genes. The top ten highly expressed genes for each cell-type were identified and compared for expression in multiple cell types. Of these AKT3, a serine/threonine protein kinase is highly expressed in all six cell types; AKT3, a mediator of growth factor signaling is implicated in a variety of biological processes such as cell proliferation, differentiation, apoptosis, tumorigenesis and glucose uptake. AKT3 is important for brain development and autophagy in neural cells (Howell et al., 2017; Soutar et al., 2018). Cofilin 1 (CFL1) is highly expressed in four of the cell-types (Fetal astrocytes, oligodendrocytes, microglia and endothelial cells). This protein is involved in polymerization and depolymerization of F-actin and G-actin in a pH dependent manner (Kanellos and Frame, 2016). CFL1 is required for neural tube morphogenesis and neural crest cell migration (Gehler et al., 2004). Clathrin heavy chain (CLTC) is also expressed in four of the cell types (neurons, oligodendrocytes, microglia and mature astrocytes). Clathrin is a major protein present in the cytoplasm that plays a role in vesicular transport and has been associated with neurological disorders (Nahorski et al., 2015). Spectrin alpha (SPTAN1), an essential cytoskeletal protein is expressed in the astrocytes, neurons and endothelial cells in the brain, and mutations in this gene is known to result in epileptic encephalopathy (Tohyama et al., 2015). Interestingly, among the most highly expressed genes, SNAP91, PLCB1, SYNJ1 and AP1S2 are all specific to neurons. Such expression patterns highlight the most useful cell type for the analysis of the cellular function of a specific gene. They could also suggest the cellular basis of a brain disorder when mutations are found in a given gene in human patients with brain disorders.
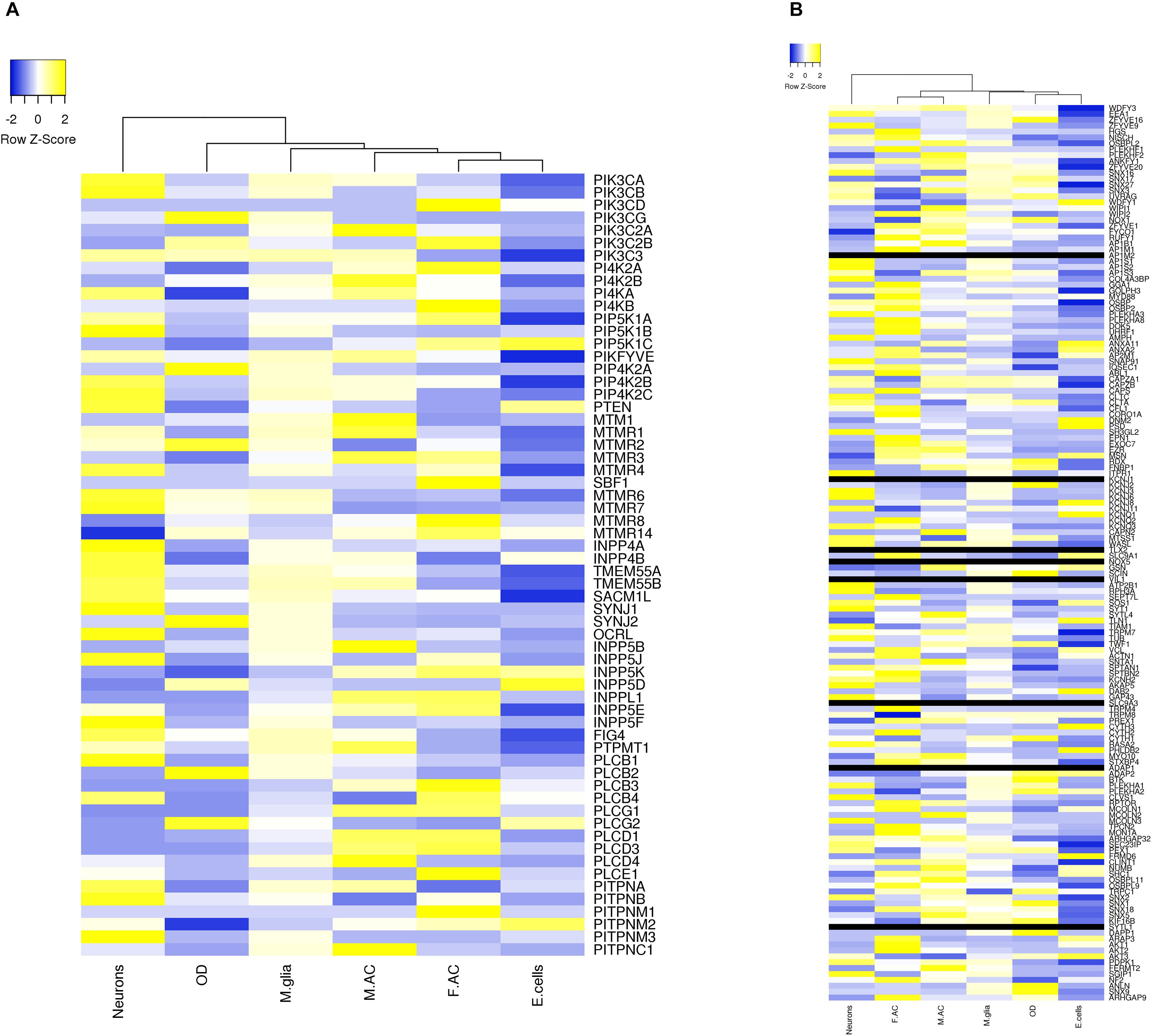
Figure 5. Expression level of phosphoinositide signaling genes in various brain cell types. The expression of genes involved in phosphoinositide signaling in specific neural cell types in human brain is shown. The data has been compiled from BrainRNAseq. The expression values range from –2 to 2 (Blue to Yellow). Black bar represents genes whose expression values were not available in the data set. The different cell types represented are Neurons, Oligodendrocytes (OD), Microglia (M.glia), Mature astrocytes (M.AC), Fetal astrocytes (F.AC) and Endothelial cells (E.cells). (A) Represents expression values of kinases, phosphatases, phospholipases and lipid transfer protein and (B) represents expression values of lipid binding proteins.
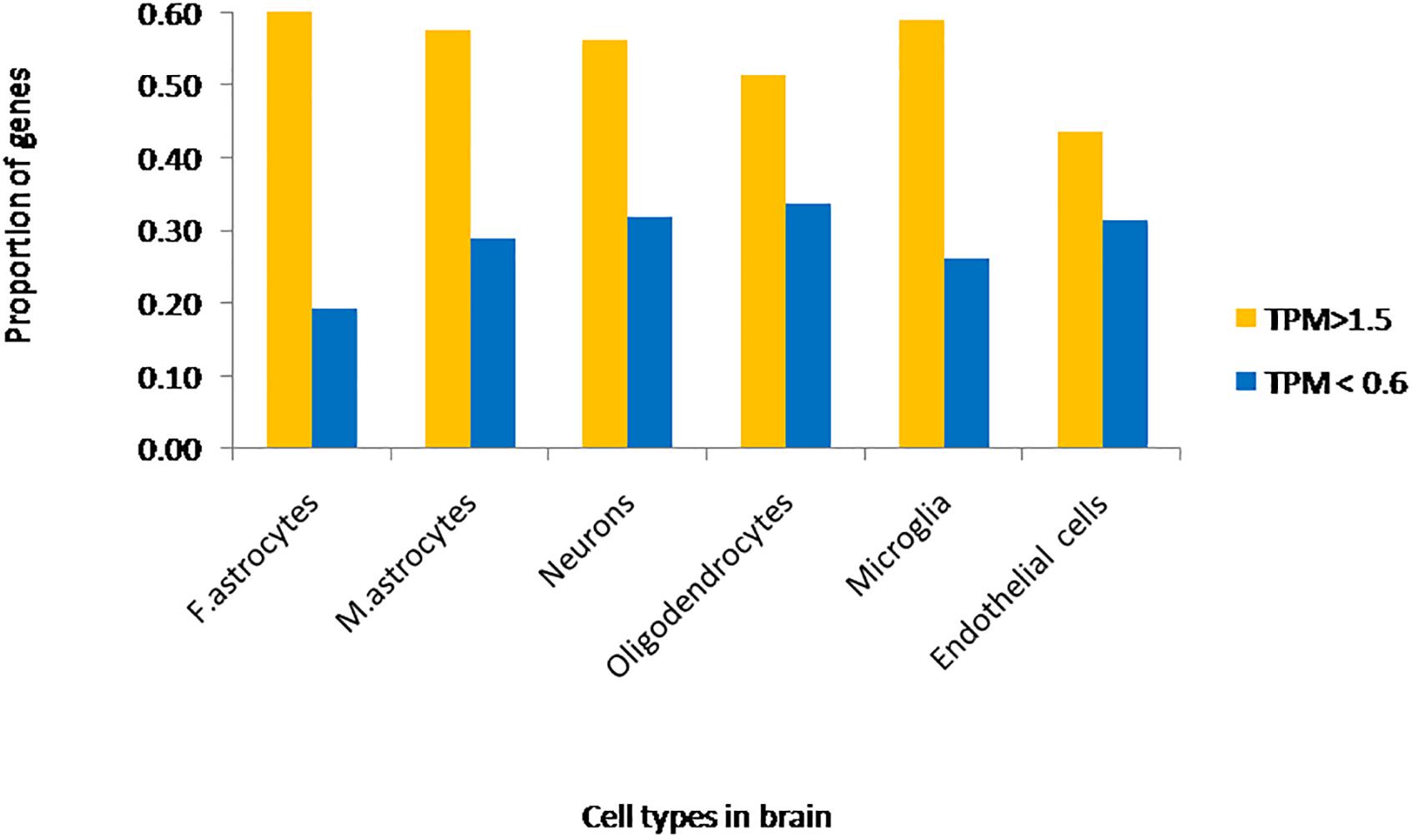
Figure 6. Fraction of phosphoinositide signaling genes with high and low expression in various cell types of the brain. The fraction of genes with TPM values greater than 1.5 and less than 0.6 in different neural cell types are represented in the graph. Yellow bars represent TPM > 1.5, i.e., high expression genes and Blue bars represent TPM < 0.6, i.e., low expression genes.
Temporal Expression of Phosphoinositide Signaling Pathway Genes in the Brain
The BrainCloud application (see footnote 3) was used to obtain the expression (TPM values) of phosphoinositide signaling genes in the brain cortex of individuals with no neuropathological diagnosis during development (from fetal development to 80 years of age) (Colantuoni et al., 2011). Expression values were arranged as a function of increasing order of age and binned at every 5 years. As observed for all genes in the study (Colantuoni et al., 2011), significant changes in the expression of phosphoinositide signaling genes is seen immediately after birth, at adolescence and then at 70 years of age. Most enzymes in the phosphoinositide signaling cascade show limited temporal variation in expression. However, 20% of phosphoinositide metabolizing enzymes show deviations with expression values ranging from (0 to +1.5 or 0 to −1.5). A similar analysis on small set of genes has shown stable transcriptional networks and gene colocalization control PI metabolism in brain cortex during development (Rapoport et al., 2015). Thus, expression levels of such enzymes could be used as an indication of their potential role in the context of human disease.
Gene Expression Data and Disease Relevance
Specific patterning of gene expression across different tissues and cell types in the brain is a feature observed in the expression data. Such expression patterns can be correlated well to some of the disease conditions of the brain as mentioned below. This data on expression could also be used to connect genes involved in PI signaling and their function in nervous system for a disease condition under study.
Phosphoinositide Kinases
Mutations in the genes PIK3CA and PI4KA that encode phosphoinositide kinases are associated with polymicrogyria and cortical dysplasia, disorders that result in altered cerebral cortex morphology and cellular composition (Mirzaa et al., 2012; Pagnamenta et al., 2015). Expression data shows that PIK3CA and PI4KA are highly expressed in the cerebral hemisphere compared to other brain regions and also enriched in the neurons as compared to other cell types in the brain. The high expression of these genes in the cerebral cortex tissue and a clear cortical development phenotype resulting from mutations in these genes underscores the potential value of using gene expression data to link gene function to disease phenotype in human brain disorders. A similar example is the PIK3C3 gene that shows high expression in neurons; loss of this gene is associated with neuronal apoptosis and neurodegeneration in a mouse knockout model (Zhou et al., 2010). Loss of PI4K2A results in a progressive neurological motor disorder associated cerebellar and spinal cord degeneration (Simons et al., 2009) and this is well correlated with high expression of this gene in the cerebellum and cerebral cortex. Loss of PIKFYVE is associated with abnormal brain morphology and decreased brain weight (Zolov et al., 2012). This gene is highly expressed in mature astrocytes as compared to other cell types. Astrocytes are known to provide metabolic support to neurons and maintain brain morphology (Jha and Morrison, 2018) thus correlating the expression of this gene with phenotypes resulting from its depletion.
Phosphoinositide Phosphatases
Expression data shows that the MTMR2 gene is upregulated in oligodendrocytes (cells required for myelination of axons in the CNS) and this is well-correlated with the finding that mutations in this gene can result in Charcot-Marie-tooth disease, type 4B1 (due to demyelination of the axons in the brain) (Bolino et al., 2000). Parkinson’s disease involves the slow degeneration of neurons in the substantia nigra area of the midbrain. The INPP5F gene that is linked by GWAS studies to Parkinson’s disease condition is differentially expressed in neurons (Nalls et al., 2014). Similarly the PLCB1 gene, implicated in Alzheimer’s disease involving nerve cell damage and atrophy in the pre-frontal cortex seems to be enriched in this region (Bakkour et al., 2013). Overall these examples illustrate the value of expression data in trying to link human brain disease phenotypes with specific genes by studying their expression pattern in the brain.
Phosphoinositide Signaling and Diseases of the Human Nervous System
Conceptually, disorders of the nervous system may arise from defects in processes that impact the development of the brain, the maintenance of normal function in developed brain cells and those that result in an inability to maintain the normal structure and function of the nervous system leading to neurodegenerative disease. There may of course be some overlap between these categories since it is now postulated that adult neurodegenerative diseases may arise from defects in brain development (Kovacs et al., 2014). Brain disorders can result from mutations in a single gene [Online Mendelian Inheritance in Man (OMIM)4 ] or DNA sequence variants in genes can alter the severity and clinical spectrum of a disorder. Defects in phosphoinositide signaling implicated in nervous system disorders of both these categories are discussed below. We include a brief description on the key clinical features of each disease; comprehensive descriptions on each neurological disorder can be found in Brain’s Diseases of the Nervous system (Donaghy, 2011).
Monogenic Disorders
Brain Development Disorders
Dominant or recessive single-gene mutations in proteins regulating phosphoinositide signaling have been implicated in neurodevelopmental disorders. The oldest and most well-known, that impacts neurodevelopment, is the X-linked monogenic disorder OCRL, or Lowe syndrome. Lowes syndrome results from mutations in the OCRL gene, that encodes one of the inositol polyphosphate 5-phosphatase enzymes that dephosphorylates PI(4,5)P2 to generate PI4P [(Olivos-glander et al., 1995) and reviewed in Mehta et al. (2014)]. The disease affects the central nervous system along with the eyes and kidneys, although the extent to which each organ is affected is variable. With regard to the nervous system, most patients show varying degrees of developmental delay, intellectual disability, hypotonia, absence of deep tendon reflexes and convulsions. Motor development is affected, and many patients show moderate mental retardation. Maladaptive behaviors including stubbornness and temper tantrums have been reported (Kenworthy et al., 1993; Bökenkamp and Ludwig, 2016). PI(4,5)P2 is known to control a number of key cellular processes in developing neural cells particularly in relation to endocytosis and the control of plasma membrane receptor composition and alterations in these may result in abnormal brain function. However, the basis for nervous system defects remains unknown and the cellular mechanism by which OCRL deficiency results in neural cell dysfunction also remains to be investigated.
Monogenic mutations in members of the Class I PI3K pathway has been found to cause multiple disorders affecting brain development in addition to non-neural consequences. Many of these mutations occur as somatic mosaics and the extent of defects in brain development depend on the stage of embryonic development at which the mutation occurs (Madsen et al., 2018). A comprehensive study on 33 children with pediatric epilepsy identified germline mutations (PTEN) or mosaic activating mutations (PIK3CA and AKT3) in PI3K pathway genes which dramatically manifests as different forms of brain malformations like megalencephaly and cortical dysplasia (Poduri et al., 2012; Jansen et al., 2015). Phosphatase and tensin homolog (PTEN) gene codes for a phosphatidylinositol 3,4,5 trisphosphate 3-phosphatase which negatively regulates PI3K/AKT pathway (Sansal and Sellers, 2014). Dominant mutations in PTEN are associated with macrocephaly autism syndrome, extreme macrocephaly ranging from >2.5 to 8.0 SD above the mean (Butler et al., 2005; Varga et al., 2009; Mcbride et al., 2010), poorly developed white matter and reduced cognitive abilities (Frazier et al., 2015). Autosomal mutations in PIK3CA, coding for p110α subunit of Class I phosphatidylinositol 3-kinase (PI3K), results in megalencephaly-capillary malformation-polymicrogyria syndrome which is characterized by brain overgrowth (megalencephaly), capillary malformations, and thick cerebral cortex due to development of excessive, unusually small folds on brain surface (polymicrogyria) (Mirzaa et al., 2012). And finally, mosaic mutation at p.Glu17Lys in pleckstrin homology (PH) domain of AKT3, a predominant effector of PI3K signaling, causes an elevation in binding to phosphatidylinositol-3,4-bisphosphate, with patients exhibiting asymmetric cortical dysplasia, while constitutive mutation in other domains showed a range of brain malformations (Poduri et al., 2012; Alcantara et al., 2017). Such overgrowth phenotypes in the brain are likely to arise from the key role that PI(3,4,5)P3 plays in the control of cell proliferation and cell growth.
Polymicrogyria is also a symptom in disorders resulting from PIK4CA (phosphatidylinositol 4-kinase), FIG4 (phosphoinositide 5-phosphatase) and AKT3 mutations (Baulac et al., 2014; Pagnamenta et al., 2015), while mutations in the 5-phosphatase domain of polyphosphate 5-phosphatase INPP5E manifests as the ciliopathy Joubert Syndrome 1 (Bielas et al., 2009) characterized by cerebellar hypoplasia/aplasia (incomplete development of cerebellum), thickened cerebellar peduncles and abnormally large interpeduncular fossa (‘molar tooth sign’) (Travaglini et al., 2013; Shetty et al., 2017). These observations illustrate the function phosphoinositides in brain development; a key biochemical mechanism is likely to be the role of PI(4,5)P2 and PI(3,45)P3 in cell division and growth.
Neurodegeneration
Phosphoinositides play an active role in membrane trafficking and cellular signaling. Since neurons are terminally differentiated cells, they are sensitive to cellular stress and susceptible to cell death. Any perturbation in the tightly regulated levels of phosphoinositides affects intracellular vesicular trafficking, membrane turnover and may result in the accumulation of cellular components that should have been degraded resulting ultimately in neuronal degeneration. Such alterations in phosphoinositide levels in the brain can result from mutations in enzymes that regulate their levels or drugs that inhibit the activity of enzymes involved in PI signaling. Important examples of such mutations are discussed below.
Synaptojanin 1 (SYJN1), a polyphosphoinositide phosphatase whose Sac1 domain dephosphorylates PI(4,5)P2 and PI(3,4,5)P3 (Cremona et al., 1999), is highly concentrated at nerve terminals (McPherson et al., 1996). Two distinct disorders have been associated with mutations in SYNJ1 gene. Alterations in SYNJ1 likely alter PI(4,5)P2 at the synapse, influence the synaptic vesicle cycle and hence contribute to the brain phenotypes described in human patients. Studies by independent groups identified homozygous missense mutation at R258Q of SYNJ1 in patients with early onset Parkinson disease-20 that affect the phosphatase activity of Sac1 domain (Krebs et al., 2013; Quadri et al., 2013; Olgiati et al., 2014). The patients exhibited tremor and bradykinesia with mild cerebral cortical atrophy. At the same time, mutations resulting in complete loss of SYNJ1 function have been identified in patients with early infantile epileptic encephalopathy 53, a severe neurodegenerative disorder characterized by epileptic seizures, severe intellectual disability and spastic quadriplegia (Hardies et al., 2016; Al Zaabi et al., 2018). A homozygous truncating mutation in SYNJ1 identified in a patient with intractable seizures also showed neurofibrillary degeneration and presence of tau protein in substantia nigra region of brain (Dyment et al., 2015). In addition to specific mutations in SYNJ1, trisomy at the locus 21q22.11 containing SYNJ1 gene has been identified in multiple lymphoblastoid cell lines developed from individuals with Down syndrome; these cells show enlarged endosomes as a result of overexpression of SYNJ1 in these cells (Cossec et al., 2012), indicating a link of SYNJ1 to Down syndrome. This result was substantiated from previous observations of increased SYNJ1 expression in brains of patients with Down syndrome (Arai et al., 2002), as well in Ts65Dn mice, a mouse model for Down syndrome exhibiting altered PI(4,5)P2 levels (Voronov et al., 2008). It is noted that patients with Down syndrome show early onset Alzheimer’s disease; this could result from a combination of overexpression of APP, the precursor of the Aβ peptide and overexpression of SYNJ1, which results in decreased levels of PI(4,5)P2, altered cellular handling of the Aβ peptide and hence early onset disease. Overall these findings imply that the specific phenotype in the patient may be impact by the type of mutation in a given gene and its effect on the activity of the enzyme it encodes as well as interactions with other genetic changes in the patient’s genome leading to altered disease phenotype.
FIG4 is another Sac domain-containing phosphoinositide 5′-phosphatase that dephosphorylates PI(3,5)P2 to PI3P. ‘Pale tremor mouse’ that carries a mutation in the Fig4 gene shows enlarged late-endosomes/lysosomes with severe neurodegeneration of dorsal root ganglion cells and large myelinated axons. The mice exhibit severe tremor and impaired motor coordination resembling Charcot-Marie-Tooth disorder in humans (Chow et al., 2007) and has been widely used as a model to study neurodegenerative disorders associated with FIG4 mutations. Compound-heterozygous loss-of-function mutation and I41T/R183X missense mutation have been identified in the autosomal recessive Charcot-Marie-Tooth type 4J (CMT4J) syndrome (Chow et al., 2007) with patients exhibiting progressive, asymmetric motor neuron degeneration, severe demyelination and axonal loss (Zhang et al., 2008; Nicholson et al., 2011). Individuals carrying one FIG4 null allele or missense mutation in one allele, and a normal allele, exhibit one form of autosomal dominant amyotrophic lateral sclerosis (Chow et al., 2009), with less severe symptoms to CMT4J. In contrast, null mutations in FIG4 resulting in complete loss of function is the cause of Yunis-Varon syndrome, which is characterized by severe neurological impairment affecting central nervous system with enlarged vacuoles in neurons, muscle and cartilage, in addition to skeletal abnormalities (Campeau et al., 2013; Nakajima et al., 2013). Around 30 individuals have thus far been identified with this syndrome (Campeau et al., 2013). PI3P is a lipid that controls endosomal trafficking and autophagy. Thus, phenotypes resulting from FIG4 and MTMR mutations likely affect the brain through alterations in membrane turnover in neural cells.
Charcot-Marie-Tooth type 4B is a group of autosomal recessive demyelinating neuropathic disorders that are typically characterized by irregular thickness of myelin sheaths due to abnormal outfolding (Othmane et al., 1999). Mutations in myotubularin related protein 2 (MTMR2), which dephosphorylates PI3P and PI(3,5)P2 and the catalytically inactive pseudophosphatase MTMR13/SBF2 (SET binding factor 2) genes are responsible for CMT4B1 and CMT4B2, respectively (Bolino et al., 2000; Azzedine et al., 2003). Even though MTMR13 is catalytically inactive, it has been suggested that the protein associates with MTMR2 to form a membrane-associated complex to regulate phosphoinositide levels (Robinson and Dixon, 2005). Since Both FIG2 and MTMR2 regulate the PI(3,5)P2 pool in the cells; the myelin sheath folding abnormalities could be due to the vesicle trafficking defects in neurons or Schwann cells as a result of PI(3,5)P2 dysregulation.
Mutations in other phosphoinositide signaling genes have been implicated in less severe forms of neuropathy and intellectual disability. In addition to the developmental disorder Joubet Syndrome, mutations in 5′-phosphatase inositol polyphosphate 5-phosphatase E (INPP5E) causes moderate mental retardation, truncal obesity, retinal dystrophy, and micropenis (MORM) syndrome (Hampshire et al., 2006). Similarly, mutations in 5’-phosphatase inositol polyphosphate 5-phosphatase K (INPP5K) result in the autosomal recessive MDCCAID (Muscular Dystrophy, Congenital, Cataracts And Intellectual Disability) disorder, where the patients suffer from mild intellectual disability in addition to the main symptoms of muscular dystrophy and early-onset cataract (Osborn et al., 2017; Wiessner et al., 2017). Table 2 lists nervous system disorders associated with mutations in phosphoinositide signaling genes. Since a monogenic disease cannot be studied experimentally in humans, several mouse strains with specific gene knockouts have been generated. Mouse knockouts of genes involved in phosphoinositide pathway with their phenotypes collated from Mouse Genome Informatics database5 are summarized in Supplementary Table 1.
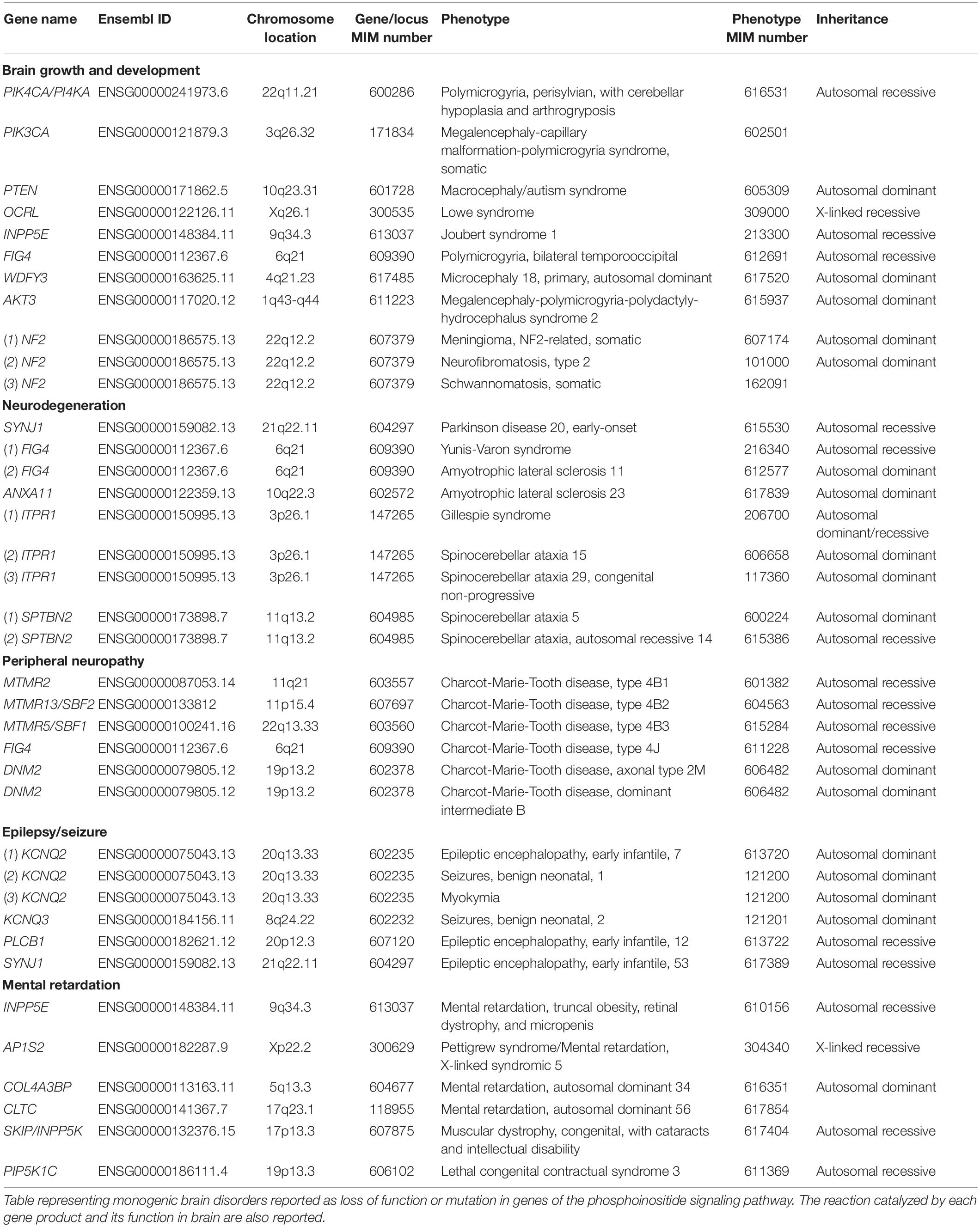
Table 2. Monogenic disorders due to mutations in phosphoinositide signaling genes.
Phosphoinositide Signaling in Bipolar Disorder
Bipolar disorder (BD) is a psychiatric illness characterized by disruptive mood swings resulting in alterations between mania and depression (Simon, 2003; Tighe et al., 2011). Studies in the early 1990s showed elevated levels of phosphatidylinositol-4, 5-bisphosphate (PIP2) in platelets from BD patients compared to controls (Soares and Mallinger, 1995) and that Protein kinase C (PKC) activity is enhanced in BD patients. These observations are consistent with a hyperactive PI cycle (Friedman et al., 1993) in BD patients. Although it was John Cade who first had treated manic depression with Lithium bromide (Cade, 1949) an understanding of the therapeutic action of lithium (Li+) came from the studies of Allison and Stewart who discovered that Li+ decreases the concentration of myo-inositol in the cerebral cortex of rats (Berridge et al., 1982, 1989) and that the decrease in myo-inositol concentration upon Li+ treatment is accompanied by an increase in the concentration of inositol monophosphate suggesting that Li+ might inhibit the enzyme inositol monophosphatase (IMPase) (Berridge et al., 1982; Harwood, 2005). Elevation of the intracellular inositol monophosphate level upon Li+ treatment was also observed in the salivary glands in insects (Berridge et al., 1982). IMPase dephosphorylates inositol 1-monophosphate to generate free myo-inositol and thereby replenish intracellular myo-inositol pool (Ohnishi et al., 2007; Agam et al., 2009). Myo-inositol is a precursor of phosphatidylinositol and when condensed with CDP-diacylglycerol by the enzyme phosphatidylinositol synthase generates phosphatidylinositol. Thus, IMPase could be integral to the maintenance of the phosphoinositide pools in mammalian cells. Such observations led Berridge to propose the “inositol depletion hypothesis” which suggests that Li+ prevents the recycling of myo-inositol by inhibiting the enzyme IMPase thereby preventing re-synthesis of phosphatidylinositol and phosphatidylinositol-4, 5-bisphosphate (PIP2) synthesis (Berridge et al., 1989). Purified IMPase was subsequently shown to be inhibited by Li+ (Hallcher and Sherman, 1980) via a non-competitive mechanism. Li+ binds to the enzyme-substrate complex, displacing Mg2+ from the active site thereby preventing the hydrolysis of the phosphate from inositol monophosphate (Harwood, 2005). However, decisive evidence that inositol depletion and alteration in PIP2 levels underlie the therapeutic action of Li+ in BD patients remains to be established and it has also recently been proposed that elevated inositol monophosphate levels may underlie the mechanism of action of Li+ (Saiardi and Mudge, 2018). Although a recent elegant biochemical analysis has once again established the ability of Li+ to regulate phosphatidylinositol turnover in neurons, the mechanism by which it does so and the relevance to therapeutic effects in BD remains a topic of active investigation (Mertens et al., 2015). Apart from IMPase, Li+ has multiple additional targets including the inositol transporter SMIT1, cyclooxygenase (COX), beta-arrestin 2 (βArr2) and glycogen synthase kinase 3 (GSK-3) (Freland and Beaulieu, 2012). The relative importance of these additional targets and their relation to PI turnover in the brain remains to be investigated.
Phosphoinositide Signaling Gene Loci Linked to Neurological Disorders
Identification of genetic variants associated with specific complex phenotypes for the brain have been made possible through the use of genome-wide association studies (GWAS) (Ripke et al., 2014; Ikeda et al., 2018). This approach does away with the bias of candidate gene approach where genes to be analyzed are selected based on pre-existing knowledge regarding their biological relevance. GWAS have generated huge amount of information spanning the entire genome to identify single nucleotide polymorphisms (SNPs) and other genomic variants associated with brain disorders. A catalog of such variants is curated and maintained by NHGRI-EBI6 (MacArthur et al., 2017) and can be mined to identify variants associated with genes involved in phosphoinositide signaling.
Genes encoding a number of phosphoinositide kinases and phosphoinositide binding proteins have been linked in GWAS studies with brain disorders. For example, variants in the kinase PIK3C2A, and the PI binding proteins UHRF1 (PI5P), SNAP91 and CAPN2 [PI(4,5)P2], RPTOR [PI(3,5)P2], AKT3 [PI(3,4)P2 and PI(3,4,5)P3] have been linked to schizophrenia patients (Goes et al., 2015) and another study identified PIK3C2A linked to schizophrenia and bipolar disorder (Ruderfer et al., 2014). In other studies, associations have been proposed between PIK4CA and PIP4K2A (previously named PIP5K2A) and schizophrenia (Schwab et al., 2006; Jungerius et al., 2008; Thiselton et al., 2009). Association studies have also suggested a link between PIP4K2A and a protective effect in tardive dyskinesia, which occurs as a complication of long-term anti-psychotic treatment (Fedorenko et al., 2015), while the diplotype ATTGCT/ATTGCT in PIP4K2A results in poor antipsychotic response in schizophrenia patients (Kaur et al., 2014). Another GWAS study has associated a PI4K2B SNP with cannabis dependence (Sherva et al., 2016), which in turn is associated with increased risk of schizophrenia (Vaucher et al., 2017), while PI4K2B has also been identified as a candidate gene in schizophrenia on a study on Scottish population cohort (Houlihan et al., 2009).
Mirroring the numerous monogenic disorders that result from mutations in phosphoinositide phosphatases, GWAS studies have linked mutations in the 5-phosphatase gene SYNJ1 causing PARK20 to Alzheimer’s disease (Herold et al., 2016), while another study has directly correlated SYNJ1 polymorphisms to age of onset in familial Alzheimer’s disease (Miranda et al., 2018). In a large study conducted in 74,026 individuals, a new locus was also identified with SNPs in INPP5D/SHIP1, the enzyme dephosphorylating PI(3,4,5)P3 into PI(3,4)P2, linking it to Alzheimer’s disease (Lambert et al., 2013). SNPs in genes for the 4-phosphatase INPP4B and the 5-phosphatase INPP5B have been linked to sporadic amyotrophic lateral sclerosis (Xie et al., 2014). An intronic variant was identified in the genetic locus of myotubularin related protein 7 (MTMR7), a 3-phosphatase dephosphorylating PI3P and inositol 1,3-bisphosphate and linked to variant Creutzfeldt-Jakob disease susceptibility (Sanchez-Juan et al., 2012).
Although the detection of a disease susceptibility locus is by no means a validated proof implicating a gene in a brain disorder, it can provide a starting point for validation of the mechanistic role of that gene through other forms of genetic analysis. Table 3 depicts the genetic loci in phosphoinositide signaling genes that are linked to brain disorders in humans.
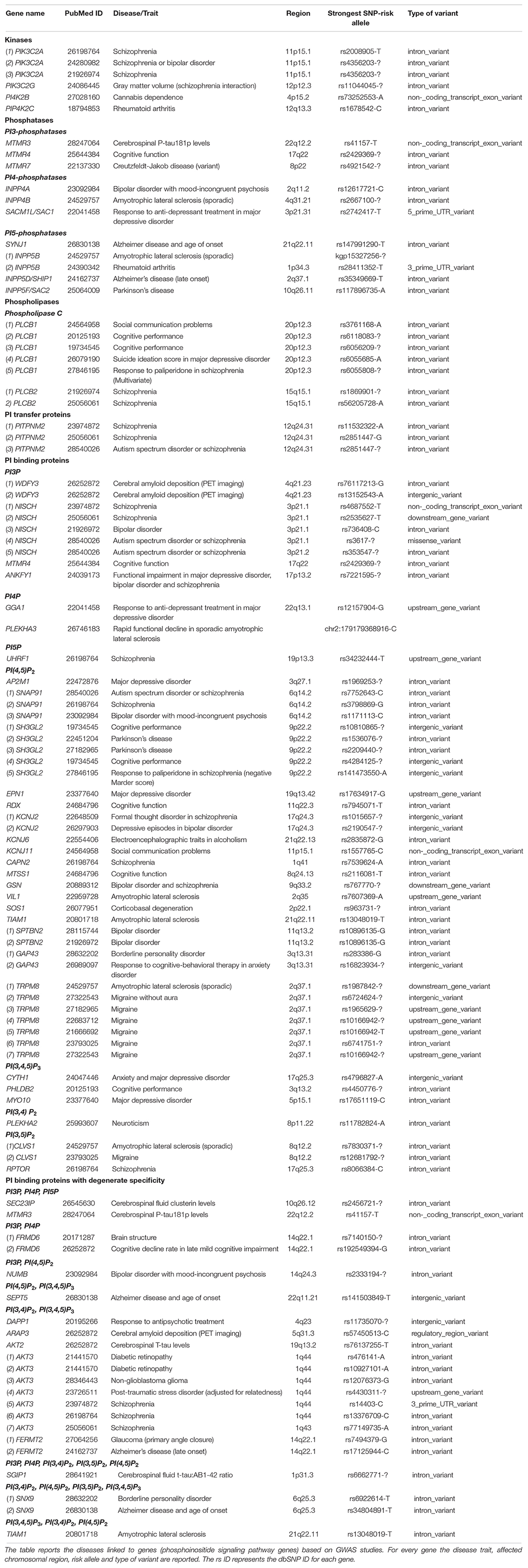
Table 3. GWAS and other association studies linking phosphoinositide signaling genes to brain disorders.
Conclusion
In the past few years, our understanding of phosphoinositide function in cell biology and physiology has rapidly advanced. Although these are quantitatively minor lipids, their impact on neuronal cell biology and function is clearly widespread. Most of these studies were done in experimentally tractable model organisms with a complex nervous system such as Drosophila, Caenorhabditis elegans or rodents and indicate a crucial role of phosphoinositides in orchestrating major subcellular pathways. Over the same period, advances in next generation DNA sequencing technology and related techniques in human genetic analysis have thrown up a large number of DNA sequence variants and suggested links between these variants and human diseases (Guerreiro et al., 2014; Splinter et al., 2018; Ganapathy et al., 2019); such observations are also true for genes involved in phosphoinositide signaling and their potential function in the context of diseases of the human nervous system. However, there have been two major challenges in this area of science (i) the ability to observe and measure aspects of the cell biology of human brain cells and to test the significance of the observations experimentally (ii) evaluating the functional significance of genetic variants reported in the context of brain disorders in genes related to phosphoinositide signaling. Recent technological advances offer the promise of rapid advances and progress in this area. The difficulty in obtaining live biopsy samples of the human brain has been a major deterrent in studying cellular mechanisms underlying neurological disorders. One possible solution to the inability to observe and experimentally manipulate human brain cells has emerged over the last 10 years in the form of induced pluripotent stem (iPS) cell technology. Using this approach one can derive pluripotent stem cells from somatic human tissue through reprograming (Shi et al., 2017). These reprogrammed stem cells can then be differentiated into neural cell types and used to study neural cell biology and physiology.
To date, several neurological diseases have been modeled based on patient-derived iPS cell technology and subsequent neural differentiation to study cellular mechanisms in disease (Russo et al., 2015; Wen et al., 2016). A recent study by Mertens et al., using iPSC technology, has revealed mitochondrial abnormalities and neuronal hyper-excitability in young neurons derived from BD patients; this hyperexcitability can be reversed by Li+ treatment only in neurons derived from Li+ sensitive patients (Mertens et al., 2015). Another recent study on iPSC derived neurons from Parkinson’s disease patients has revealed defects in the regulation of ER Ca2+ stores, which can be linked to the PI(4,5)P2 cycle since it actively regulates Ca2+ homeostasis (Korecka et al., 2019). An iPSC model for Lowe Syndrome has also been developed by Barnes et al., that suggests abnormalities in F-actin polymerization, WAVE-1 expression and altered PI(4,5)P2 levels in patient specific iPSC derived neurons, giving a further insight about the disease pathology at the cellular level (Barnes et al., 2018).
A second major development in recent times is the development of genome engineering technologies such as Zn2+ finger nucleases, TALEN and CRISPR/Cas9 that allow DNA sequence changes to be introduced into many human cell types including iPS cells. By adopting this approach, one can experimentally test the contributions of DNA sequence variants described in patient samples to the development of disease phenotypes, as demonstrated by Barnes et al. (2018). Together, these approaches are likely to accelerate our understanding of the role of phosphoinositide signaling in human disease biology in relation to the nervous system.
Author Contributions
PR conceptualized and wrote the review. AJ, HK, PS, and SS wrote the review. HK and AJ mined public databases and collated gene expression data and disease data.
Funding
Work in the authors’ laboratory is supported by the National Centre for Biological Sciences-TIFR and a Wellcome-DBT India Alliance Senior Fellowship to PR (IA/S/14/2/501540). PR is supported by the Accelerator Program for Discovery in Brain Disorders using Stem Cells, Department of Biotechnology, Government of India and the Pratiksha Trust.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research Associateship from DBT India to AJ is also gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2019.00208/full#supplementary-material
Footnotes
- ^ http://www.brainrnaseq.org
- ^ https://gtexportal.org/home/
- ^ https://www.libd.org/brain-cloud/
- ^ https://www.omim.org
- ^ http://www.informatics.jax.org
- ^ http://www.ebi.ac.uk/gwas
References
Agam, G., Bersudsky, Y., Berry, G. T., Moechars, D., Lavi-Avnon, Y., and Belmaker, R. H. (2009). Knockout mice in understanding the mechanism of action of lithium. Biochem. Soc. Trans. 37, 1121–1125. doi: 10.1042/BST0371121
Al Zaabi, N., Noora, Z., and Al-Jasmi, F. (2018). SYNJ1 gene associated with neonatal onset of neurodegenerative disorder and intractable seizure. Mol. Genet. Genomic Med. 6, 109–113. doi: 10.1002/mgg3.341
Alcantara, D., Timms, A. E., Gripp, K., Baker, L., Park, K., Collins, S., et al. (2017). Mutations of AKT3 are associated with a wide spectrum of developmental disorders including extreme megalencephaly. Brain 140, 2610–2622. doi: 10.1093/brain/awx203
Al-Ramahi, I., Giridharan, S. S. P., Chen, Y.-C., Patnaik, S., Safren, N., Hasegawa, J., et al. (2017). Inhibition of PIP4Kg ameliorates the pathological effects of mutant huntingtin protein. eLife 6:e29123. doi: 10.7554/eLife.29123
Arai, Y., Ijuin, T., Takenawa, T., Becker, L. E., and Takashima, S. (2002). Excessive expression of synaptojanin in brains with Down syndrome. Brain Dev. 24, 67–72. doi: 10.1016/S0387-7604(01)00405-3
Arendt, K. L., Benoist, M., Lario, A., Draffin, J. E., Munoz, M., and Esteban, J. A. (2014). PTEN counteracts PIP3 upregulation in spines during NMDA-receptor-dependent long-term depression. J. Cell Sci. 127, 5253–5260. doi: 10.1242/jcs.156554
Azzedine, H., Bolino, A., Taïeb, T., Birouk, N., Di Duca, M., Bouhouche, A., et al. (2003). Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of charcot-marie-tooth disease associated with early-onset glaucoma. Am. J. Hum. Genet. 72, 1141–1153. doi: 10.1086/375034
Bae, B.-I., Jayaraman, D., and Walsh, C. A. (2015). Genetic changes shaping the human brain. Dev. Cell 32, 423–434. doi: 10.1016/j.devcel.2015.01.035
Bakkour, A., Morris, J. C., Wolk, D. A., and Dickerson, B. C. (2013). The effects of aging and Alzheimer’s disease on cerebral cortical anatomy: specificity and differential relationships with cognition. Neuroimage 76, 332–344. doi: 10.1016/j.neuroimage.2013.02.059.The
Balakrishnan, S. S., Basu, U., and Raghu, P. (2015). Phosphoinositide signalling in Drosophila. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1851, 770–784. doi: 10.1016/j.bbalip.2014.10.010
Balakrishnan, S. S., Basu, U., Shinde, D., Thakur, R., Jaiswal, M., and Raghu, P. (2018). Regulation of PI4P levels by PI4KIIIα during G-protein-coupled PLC signaling in Drosophila photoreceptors. J. Cell Sci. 131:jcs217257. doi: 10.1242/jcs.217257
Balla, A., and Balla, T. (2006). Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 16, 351–361. doi: 10.1016/j.tcb.2006.05.003
Balla, T. (2005). Inositol-lipid binding motifs: signal integrators through protein-lipid and protein-protein interactions. J. Cell. Sci. 118, 2093–2104. doi: 10.1242/jcs.02387
Balla, T. (2013). Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 93, 1019–1137. doi: 10.1152/physrev.00028.2012
Barnes, J., Salas, F., Mokhtari, R., Dolstra, H., Pedrosa, E., and Lachman, H. M. (2018). Modeling the neuropsychiatric manifestations of Lowe syndrome using induced pluripotent stem cells: defective F-actin polymerization and WAVE-1 expression in neuronal cells. Mol. Autism 9, 1–16. doi: 10.1186/s13229-018-0227-3
Baulac, S., Lenk, G. M., Couarch, P., Larson, P. A., Ferguson, C. J., Noé, E., et al. (2014). Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria. Neurology 82, 1068–1075. doi: 10.1212/WNL.0000000000000241
Berridge, M. J., Downes, C. P., and Hanley, M. R. (1989). Neural and developmental actions of lithium: a unifying hypothesis. Cell 59, 411–419. doi: 10.1016/0092-8674(89)90026-3
Berridge, M. J., Downest, C. P., and Hanleyt, M. R. (1982). Lithium amplifies agonist-dependent phosphatidylinositol responses in brain and salivary glands. Biochem. J. 206, 587–595. doi: 10.1042/bj2060587
Berridge, M. J., and Irvine, R. F. (1984). Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature 312, 315–321. doi: 10.1038/312315a0
Berridge, M. J., and Irvine, R. F. (1989). Inositol phosphates and cell signalling. Nature 341, 197–205. doi: 10.1038/341197a0
Bielas, S. L., Silhavy, J. L., Brancati, F., Kisseleva, M. V., Al-gazali, L., Sztriha, L., et al. (2009). Mutations in INPP5E, encoding inositol polyphosphate- 5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 41, 1032–1036. doi: 10.1038/ng.423
Boal, F., Mansour, R., Gayral, M., Saland, E., Chicanne, G., Xuereb, J.-M., et al. (2015). TOM1 is a PI5P effector involved in the regulation of endosomal maturation. J. Cell Sci. 128, 815–827. doi: 10.1242/jcs.166314
Bökenkamp, A., and Ludwig, M. (2016). The oculocerebrorenal syndrome of Lowe: an update. Pediatr. Nephrol. 31, 2201–2212. doi: 10.1007/s00467-016-3343-3
Bolino, A., Muglia, M., Conforti, F. L., LeGuern, E., Salih, M. A. M., Georgiou, D.-M., et al. (2000). Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat. Genet. 25, 17–19. doi: 10.1038/75542
Butler, M. G., Dasouki, M. J., Zhou, X.-P., Talebizadeh, Z., Brown, M., Takahashi, T. N., et al. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 42, 318–321. doi: 10.1136/jmg.2004.024646
Cade, J. F. (1949). Lithium salts in the treatment of psychotic excitement. Med. J. Aust. 2, 349–352. doi: 10.3109/00048678209159969
Campeau, P. M., Lenk, G. M., Lu, J. T., Bae, Y., Burrage, L., Morandi, L., et al. (2013). Yunis-Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am. J. Hum. Genet. 92, 781–791. doi: 10.1016/j.ajhg.2013.03.020
Chakrabarti, P., Kolay, S., Yadav, S., Kumari, K., Nair, A., Trivedi, D., et al. (2015). A dPIP5K dependent pool of phosphatidylinositol 4,5 bisphosphate (PIP2) is required for G-protein coupled signal transduction in Drosophila photoreceptors. PLoS Genet. 11:e1004948. doi: 10.1371/journal.pgen.1004948
Chow, C. Y., Landers, J. E., Bergren, S. K., Sapp, P. C., Grant, A. E., Jones, J. M., et al. (2009). Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 84, 85–88. doi: 10.1016/j.ajhg.2008.12.010
Chow, C. Y., Zhang, Y., Dowling, J. J., Jin, N., Adamska, M., Shiga, K., et al. (2007). Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448, 68–72. doi: 10.1038/nature05876
Cockcroft, S., and Raghu, P. (2018). Phospholipid transport protein function at organelle contact sites. Curr. Opin. Cell Biol. 53, 52–60. doi: 10.1016/j.ceb.2018.04.011
Colantuoni, C., Lipska, B. K., Ye, T., Hyde, T. M., Tao, R., Leek, J. T., et al. (2011). Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523. doi: 10.1038/nature10524
Cossec, J.-C., Lavaur, J., Berman, D. E., Rivals, I., Hoischen, A., Stora, S., et al. (2012). Trisomy for Synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum. Mol. Genet. 21, 3156–3172. doi: 10.1093/hmg/dds142
Cremona, O., Di Paolo, G., Wenk, M. R., Lüthi, A., Kim, W. T., Takei, K., et al. (1999). Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179–188. doi: 10.1016/s0092-8674(00)81649-9
D’Angelo, G., Vicinanza, M., Di Campli, A., and De Matteis, M. A. (2008). The multiple roles of PtdIns(4)P – not just the precursor of PtdIns(4,5)P2. J. Cell Sci. 121, 1955–1963. doi: 10.1242/jcs.023630
Darmanis, S., Sloan, S. A., Zhang, Y., Enge, M., Caneda, C., Shuer, L. M., et al. (2015). A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. U.S.A. 112, 7285–7290. doi: 10.1073/pnas.1507125112
Dawson, R. M. (1954a). Studies on the labelling of brain phospholipids with radioactive phosphorus. Biochem. J. 57, 237–245. doi: 10.1042/bj0570237
Dawson, R. M. (1954b). The measurement of 32P labelling of individual kephalins and lecithin in a small sample of tissue. Biochim. Biophys. Acta 14, 374–379. doi: 10.1016/0006-3002(54)90195-x
De Paula, M. L., Cui, Q.-L., Hossain, S., Antel, J., and Almazan, G. (2014). The PTEN inhibitor bisperoxovanadium enhances myelination by amplifying IGF-1 signaling in rat and human oligodendrocyte progenitors.pdf. Glia 62, 64–77. doi: 10.1002/glia.22584
Dennis, E. A. (2015). Introduction to thematic review series: phospholipases: central role in lipid signaling and disease. J. Lipid Res. 56, 1245–1247. doi: 10.1194/jlr.E061101
Di Paolo, G., and De Camilli, P. (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. doi: 10.1038/nature05185
Di Paolo, G., Moskowitz, H. S., Gipson, K., Wenk, M. R., Voronov, S., Obayashi, M., et al. (2004). Impaired PtdIns (4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431, 415–422. doi: 10.1038/nature02896
Dickson, E. J., and Hille, B. (2019). Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem. J. 476, 1–23. doi: 10.1042/BCJ20180022
Dickson, E. J., Jensen, J. B., and Hille, B. (2014). Golgi and plasma membrane pools of PI(4)P contribute to plasma membrane PI(4,5)P2 and maintenance of KCNQ2/3 ion channel current. Proc. Natl. Acad. Sci. U.S.A. 111, E2281–E2290. doi: 10.1073/pnas.1407133111
Divecha, N., Banfić, H., and Irvine, R. F. (1993). Inositides and the nucleus and inositides in the nucleus. Cell 74, 405–407. doi: 10.1016/0092-8674(93)80041-c
Donaghy, M. (ed.) (2011). Brain’s Diseases of the Nervous System, 12th Edn. Oxford: Oxford University Press, doi: 10.1093/med/9780198569381.001.0001
Dyment, D. A., Smith, A. C., Humphreys, P., Schwartzentruber, J., Beaulieu, C. L., Consortium, F. C., et al. (2015). Neurobiology of Aging Homozygous nonsense mutation in SYNJ1 associated with intractable epilepsy and tau pathology. Neurobiol. Aging 36:1222.e1-.e5. doi: 10.1016/j.neurobiolaging.2014.09.005
Engelman, J. A., Luo, J., and Cantley, L. C. (2006). The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619. doi: 10.1038/nrg1879
Fedorenko, O. Y., Loonen, A. J. M., Lang, F., Toshchakova, V. A., Boyarko, E. G., Semke, A. V., et al. (2015). Association study indicates a protective role of phosphatidylinositol-4-phosphate-5-kinase against Tardive Dyskinesia. Int. J. Neuropsychopharmacol. 18:pyu098. doi: 10.1093/ijnp/pyu098
Fiume, R., Stijf-Bultsma, Y., Shah, Z. H., Keune, W. J., Jones, D. R., Jude, J. G., et al. (2015). PIP4K and the role of nuclear phosphoinositides in tumour suppression. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1851, 898–910. doi: 10.1016/j.bbalip.2015.02.014
Folch, J. (1949a). Brain diphosphoninositide, a new phosphatide having inositol metadiphosphate as a constituent. J. Biol. Chem. 177, 505–519.
Folch, J. (1949b). Complete fractionation of brain cephalin; isolation from it of phosphatidyl serine, phosphatidyl ethanolamine, and diphosphoinositide. J. Biol. Chem. 177, 497–504.
Folch, J., and Wolleey, D. W. (1942). Inositol, a constituent of brain phosphatidate. J. Biol. Chem 142, 963–964.
Franz, H., Ullmann, C., Becker, A., Ryan, M., Bahn, S., Arendt, T., et al. (2005). Systematic analysis of gene expression in human brains before and after death. Genome Biol. 6:R112. doi: 10.1186/gb-2005-6-13-r112
Frazier, T. W., Embacher, R., Tilot, A. K., Koenig, K., Mester, J., and Eng, C. (2015). Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol. Psychiatry 20, 1132–1138. doi: 10.1038/mp.2014.125
Freland, L., and Beaulieu, J.-M. (2012). Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front. Mol. Neurosci. 5:14. doi: 10.3389/fnmol.2012.00014
Friedman, E., Hoau-Yan-Wang, Levinson, D., Connell, T. A., and Singh, H. (1993). Altered platelet protein kinase C activity in bipolar affective disorder, manic episode. Biol. Psychiatry 33, 520–525. doi: 10.1016/0006-3223(93)90006-Y
Funakoshi, Y., Hasegawa, H., and Kanaho, Y. (2011). Regulation of PIP5K activity by Arf6 and its physiological significance. J. Cell. Physiol. 226, 888–895. doi: 10.1002/jcp.22482
Gaidarov, I., Smith, M. E., Domin, J., and Keen, J. H. (2001). The class II phosphoinositide 3-kinase C2alpha is activated by clathrin and regulates clathrin-mediated membrane trafficking. Mol. Cell 7, 443–449. doi: 10.1016/s1097-2765(01)00191-5
Ganapathy, A., Mishra, A., Soni, M. R., Kumar, P., Sadagopan, M., Kanthi, A. V., et al. (2019). Multi-gene testing in neurological disorders showed an improved diagnostic yield: data from over 1000 Indian patients. J. Neurol. 266, 1919–1926. doi: 10.1007/s00415-019-09358-1
Gehler, S., Shaw, A. E., Sarmiere, P. D., Bamburg, J. R., and Letourneau, P. C. (2004). Brain-derived neurotrophic factor regulation of retinal growth cone filopodial dynamics is mediated through actin depolymerizing factor/cofilin. J. Neurosci. 24, 10741–10749. doi: 10.1523/JNEUROSCI.2836-04.2004
Godi, A., Pertile, P., Meyers, R., Marra, P., Di Tullio, G., Iurisci, C., et al. (1999). ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol. 1, 280–287. doi: 10.1038/12993
Goebbels, S., Wieser, G. L., Pieper, A., Spitzer, S., Weege, B., Yan, K., et al. (2017). A neuronal PI(3,4,5)P3-dependent program of oligodendrocyte precursor recruitment and myelination. Nat. Neurosci. 20, 10–15. doi: 10.1038/nn.4425
Goes, F. S., Mcgrath, J., Avramopoulos, D., Wolyniec, P., Pirooznia, M., Ruczinski, I., et al. (2015). Genome-wide association study of schizophrenia in Ashkenazi Jews. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 168, 649–659. doi: 10.1002/ajmg.b.32349
Gong, L.-W., and De Camilli, P. (2008). Regulation of postsynaptic AMPA responses by synaptojanin 1. Proc. Natl. Acad. Sci. U.S.A. 105, 17561–17566. doi: 10.1073/pnas.0809221105
Grimm, J., Sachs, M., Britsch, S., Di Cesare, S., Schwarz-Romond, T., Alitalo, K., et al. (2001). Novel p62dok family members, dok-4 and dok-5, are substrates of the c-Ret receptor tyrosine kinase and mediate neuronal differentiation. J. Cell Biol. 154, 345–354. doi: 10.1083/jcb.200102032
Guerreiro, R., Bras, J., Hardy, J., and Singleton, A. (2014). Next generation sequencing techniques in neurological diseases: redefining clinical and molecular associations. Hum. Mol. Genet. 23, R47–R53. doi: 10.1093/hmg/ddu203
Guo, J., Wenk, M. R., Pellegrini, L., Onofri, F., Benfenati, F., and De Camilli, P. (2003). Phosphatidylinositol 4-kinase type IIalpha is responsible for the phosphatidylinositol 4-kinase activity associated with synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 100, 3995–4000. doi: 10.1073/pnas.0230488100
Gupta, A., Toscano, S., Trivedi, D., Jones, D. R., Mathre, S., Clarke, J. H., et al. (2013). Phosphatidylinositol 5-phosphate 4-kinase (PIP4K) regulates TOR signaling and cell growth during Drosophila development. Proc. Natl. Acad. Sci. U.S.A. 110, 5963–5968. doi: 10.1073/pnas.1219333110
Hagenauer, M. H., Schulmann, A., Li, J. Z., Vawter, M. P., Walsh, D. M., Thompson, R. C., et al. (2018). Inference of cell type content from human brain transcriptomic datasets illuminates the effects of age, manner of death, dissection, and psychiatric diagnosis. PLoS One 13:e0200003. doi: 10.1371/journal.pone.0200003
Hallcher, L. M., and Sherman, W. R. (1980). The effects of lithium ion and other agents on the activity of myo- inositol-1-phosphatase from bovine brain. J. Biol. Chem. 255, 10896–10901.
Hammond, G. R. V., and Balla, T. (2015). Polyphosphoinositide binding domains: key to inositol lipid biology. Biochim. Biophys. Acta 1851, 746–758. doi: 10.1016/j.bbalip.2015.02.013
Hampshire, D. J., Ayub, M., Springell, K., Roberts, E., Jafri, H., Rashid, Y., et al. (2006). MORM syndrome (mental retardation, truncal obesity, retinal dystrophy and micropenis), a new autosomal recessive disorder, links to 9q34. Eur. J. Hum. Genet. 14, 543–548. doi: 10.1038/sj.ejhg.5201577
Hansen, S. B. (2015). Lipid agonism: the PIP2 paradigm of ligand-gated ion channels. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1851, 620–628. doi: 10.1016/j.bbalip.2015.01.011
Hardies, K., Cai, Y., Jardel, C., Jansen, A. C., Cao, M., May, P., et al. (2016). Loss of SYNJ1 dual phosphatase activity leads to early onset refractory seizures and progressive neurological decline. Brain 139(Pt 9), 2420–2430. doi: 10.1093/brain/aww180
Harrington, E. P., Zhao, C., Fancy, S. P. J., Franklin, R. J. M., and Rowitch, D. H. (2010). Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann. Neurol. 68, 703–716. doi: 10.1002/ana.22090.Oligodendrocyte
Harwood, A. J. (2005). Lithium and bipolar mood disorder: the inositol-depletion hypothesis revisited. Mol. Psychiatry 10, 117–126. doi: 10.1038/sj.mp.4001618
Hasegawa, J., Strunk, B. S., and Weisman, L. S. (2017). PI5P and PI(3,5)P2: minor, but essential phosphoinositides. Cell Struct. Funct. 42, 49–60. doi: 10.1247/csf.17003
Haucke, V. (2005). Phosphoinositide regulation of clathrin-mediated endocytosis. Biochem. Soc. Trans. 33, 1285–1289. doi: 10.1042/BST20051285
Hawkins, P. T., and Stephens, L. R. (2016). Emerging evidence of signalling roles for PI(3,4)P2 in Class I and II PI3K-regulated pathways. Biochem. Soc. Trans. 44, 307–314. doi: 10.1042/BST20150248
He, K., Marsland, R. III, Upadhyayula, S., Song, E., Dang, S., Capraro, B. R., et al. (2017). Dynamics of phosphoinositide conversion in clathrin-mediated endocytic traffic. Nature 552, 410–414. doi: 10.1038/nature25146
Henle, S. J., Wang, G., Liang, E., Wu, M., Poo, M., and Henley, J. R. (2011). Asymmetric PI(3,4,5)P3 and Akt signaling mediates chemotaxis of axonal growth cones. J. Neurosci. 31, 7016–7027. doi: 10.1523/JNEUROSCI.0216-11.2011
Herold, C., Hooli, B. V., Mullin, K., Liu, T., Roehr, J. T., Mattheisen, M., et al. (2016). Family-based association analyses of imputed genotypes reveal genome-wide significant association of Alzheimer’s disease with OSBPL6, PTPRG, and PDCL3. Mol. Psychiatry 21, 1608–1612. doi: 10.1038/mp.2015.218
Hill, E., van Der Kaay, J., Downes, C. P., and Smythe, E. (2001). The role of dynamin and its binding partners in coated pit invagination and scission. J. Cell Biol. 152, 309–323.
Hille, B., Dickson, E. J., Kruse, M., Vivas, O., and Suh, B.-C. (2015). Phosphoinositides regulate ion channels. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1851, 844–856. doi: 10.1016/j.bbalip.2014.09.010
Hirst, J., Motley, A., Harasaki, K., Peak Chew, S. Y., and Robinson, M. S. (2003). EpsinR: an ENTH domain-containing protein that interacts with AP-1. Mol. Biol. Cell 14, 625–641. doi: 10.1091/mbc.E02-09-0552
Hokin, L. E., and Hokin, M. R. (1955). Effects of acetylcholine on the turnover of phosphoryl units in individual phospholipids of pancreas slices and brain cortex slices. Biochim. Biophys. Acta 18, 102–110. doi: 10.1016/0006-3002(55)90013-5
Hokin, M. R., Hokin, L. E., and Shelp, W. D. (1960). The effects of acetylcholine on the turnover of phosphatidic acid and phosphoinositide in sympathetic ganglia, and in various parts of the central nervous system in vitro. J. Gen. Physiol. 44, 217–226. doi: 10.1085/jgp.44.2.217
Holahan, M. R. (2017). A shift from a pivotal to supporting role for the growth-associated protein (GAP-43) in the coordination of axonal structural and functional plasticity. Front. Cell. Neurosci. 11:266. doi: 10.3389/fncel.2017.00266
Houlihan, L. M., Christoforou, A., Arbuckle, M. I., Torrance, H. S., Anderson, S. M., Muir, W. J., et al. (2009). A case-control association study and family-based expression analysis of the bipolar disorder candidate gene PI4K2B. J. Psychiatr. Res. 43, 1272–1277. doi: 10.1016/j.jpsychires.2009.05.004
Howell, K. R., Floyd, K., and Law, A. J. (2017). PKBγ/AKT3 loss-of-function causes learning and memory deficits and deregulation of AKT/mTORC2 signaling: relevance for schizophrenia. PLoS One 12:e0175993. doi: 10.1371/journal.pone.0175993
Hsuan, J., and Cockcroft, S. (2001). The PITP family of phosphatidylinositol transfer proteins. Genome Biol. 2:REVIEWS3011. doi: 10.1186/gb-2001-2-9-reviews3011
Ikeda, M., Saito, T., Kondo, K., and Iwata, N. (2018). Genome-wide association studies of bipolar disorder: a systematic review of recent findings and their clinical implications. Psychiatry Clin. Neurosci. 72, 52–63. doi: 10.1111/pcn.12611
Jansen, L. A., Mirzaa, G. M., Ishak, G. E., Roak, B. J. O., Hiatt, J. B., Roden, W. H., et al. (2015). PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 138, 1613–1628. doi: 10.1093/brain/awv045
Jentsch, T. J. (2000). Neuronal KCNQ potassium channels:physislogy and role in disease. Nat. Rev. Neurosci. 1, 21–30. doi: 10.1038/35036198
Jha, A., Ahuja, M., Patel, S., Brailoiu, E., and Muallem, S. (2014). Convergent regulation of the lysosomal two-pore channel-2 by Mg2+, NAADP, PI(3,5)P2 and multiple protein kinases. EMBO J. 33, 501–511. doi: 10.1002/embj.201387035
Jha, M. K., and Morrison, B. M. (2018). Glia-neuron energy metabolism in health and diseases: new insights into the role of nervous system metabolic transporters. Exp. Neurol. 309, 23–31. doi: 10.1016/j.expneurol.2018.07.009
Jiang, K., Liu, Y., Fan, J., Zhang, J., Li, X.-A., Evers, B. M., et al. (2016). PI(4)P promotes phosphorylation and conformational change of smoothened through interaction with its C-terminal Tail. PLoS Biol. 14:e1002375. doi: 10.1371/journal.pbio.1002375
Jungerius, B. J., Hoogendoorn, M. L. C., Bakker, S. C., Slot, R., Van Bardoel, A. F., and Ophoff, R. A. (2008). An association screen of myelin-related genes implicates the chromosome 22q11 PIK4CA gene in schizophrenia. Mol. Psychiatry 13, 1060–1068. doi: 10.1038/sj.mp.4002080
Jungmichel, S., Sylvestersen, K. B., Choudhary, C., Nguyen, S., Mann, M., and Nielsen, M. L. (2014). Specificity and commonality of the phosphoinositide-binding proteome analyzed by quantitative mass spectrometry. Cell Rep. 6, 578–591. doi: 10.1016/j.celrep.2013.12.038
Kamalesh, K., Trivedi, D., Toscano, S., Sharma, S., Kolay, S., and Raghu, P. (2017). Phosphatidylinositol 5-phosphate 4-kinase regulates early endosomal dynamics during clathrin-mediated endocytosis. J. Cell Sci. 130, 2119–2133. doi: 10.1242/jcs.202259
Kanellos, G., and Frame, M. C. (2016). Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci. 129, 3211–3218. doi: 10.1242/jcs.187849
Kaur, H., Jajodia, A., Grover, S., Baghel, R., Gupta, M., Jain, S., et al. (2014). Genetic variations of PIP4K2A confer vulnerability to poor antipsychotic response in severely Ill Schizophrenia patients. PLoS One 9:e102556. doi: 10.1371/journal.pone.0102556
Kawano, M., Kumagai, K., Nishijima, M., and Hanada, K. (2006). Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 281, 30279–30288. doi: 10.1074/jbc.M605032200
Kenworthy, L., Park, T., and Charnas, L. R. (1993). Cognitive and behavioral profile of the oculocerebrorenal syndrome of Lowe. Am. J. Med. Genet. 46, 297–303. doi: 10.1002/ajmg.1320460312
Klopfenstein, D. R., Tomishige, M., Stuurman, N., and Vale, R. D. (2002). Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell 109, 347–358. doi: 10.1016/s0092-8674(02)00708-0
Klopfenstein, D. R., and Vale, R. D. (2004). The lipid binding pleckstrin homology domain in UNC-104 kinesin is necessary for synaptic vesicle transport in Caenorhabditis elegans. Mol. Biol. Cell 15, 3729–3739. doi: 10.1091/mbc.e04-04-0326
Kolay, S., Basu, U., and Raghu, P. (2016). Control of diverse subcellular processes by a single multi-functional lipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2]. Biochem. J. 473, 1681–1692. doi: 10.1042/BCJ20160069
Korecka, J. A., Talbot, S., Osborn, T. M., Leeuw, S. M., De Levy, S. A., Ferrari, E. J., et al. (2019). Neurite collapse and altered ER Ca2+ control in human parkinson disease patient iPSC-derived neurons with LRRK2 G2019S mutation. Stem Cell Rep. 12, 29–41. doi: 10.1016/j.stemcr.2018.11.021
Kovacs, G. G., Adle-Biassette, H., Milenkovic, I., Cipriani, S., van Scheppingen, J., and Aronica, E. (2014). Linking pathways in the developing and aging brain with neurodegeneration. Neuroscience 269, 152–172. doi: 10.1016/j.neuroscience.2014.03.045
Krebs, C. E., Karkheiran, S., Powell, J. C., Cao, M., Makarov, V., Darvish, H., et al. (2013). The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum. Mutat. 34, 1200–1207. doi: 10.1002/humu.22372
Kumar, J., Choudhary, B. C., Metpally, R., Zheng, Q., Nonet, M. L., Ramanathan, S., et al. (2010). The Caenorhabditis elegans Kinesin-3 motor UNC-104/KIF1A is degraded upon loss of specific binding to cargo. PLoS Genet. 6:e1001200. doi: 10.1371/journal.pgen.1001200
Lachyankar, M. B., Sultana, N., Schonhoff, C. M., Mitra, P., Poluha, W., Lambert, S., et al. (2000). A role for nuclear PTEN in neuronal differentiation. J. Neurosci. 20, 1404–1413. doi: 10.1523/jneurosci.20-04-01404.2000
Lambert, J.-C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74, 046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1460. doi: 10.1038/ng.2802
Lenk, G. M., and Meisler, M. H. (2014). Mouse models of PI(3,5)P2 deficiency with impaired lysosome function. Methods Enzymol. 534, 245–260. doi: 10.1016/B978-0-12-397926-1.00014-17
Logan, A. M., Mammel, A. E., Robinson, D. C., Chin, A. L., Condon, A. F., and Robinson, F. L. (2017). Schwann cell-specific deletion of the endosomal PI 3-kinase Vps34 leads to delayed radial sorting of axons, arrested myelination, and abnormal ErbB2-ErbB3 tyrosine kinase signaling. Glia 65, 1452–1470. doi: 10.1002/glia.23173
Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., et al. (2013). The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585. doi: 10.1038/ng.2653
MacArthur, J., Bowler, E., Cerezo, M., Gil, L., Hall, P., Hastings, E., et al. (2017). The new NHGRI-EBI catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45, D896–D901. doi: 10.1093/nar/gkw1133
Madsen, R. R., Vanhaesebroeck, B., and Semple, R. K. (2018). Cancer-associated PIK3CA mutations in overgrowth disorders. Trends Mol. Med. 24, 856–870. doi: 10.1016/j.molmed.2018.08.003
Mani, M., Lee, S. Y., Lucast, L., Cremona, O., Di Paolo, G., De Camilli, P., et al. (2007). The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron 56, 1004–1018. doi: 10.1016/j.neuron.2007.10.032
Martin, T. F. J. (1998). Phosphoinositide lipids as signalling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu. Rev. Cell Dev. Biol. 14, 231–264. doi: 10.1146/annurev.cellbio.14.1.231
Mcbride, K. L., Varga, E. A., Pastore, M. T., Prior, T. W., Manickam, K., Atkin, J. F., et al. (2010). Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 3, 137–141. doi: 10.1002/aur.132
McCartney, A. J., Zolov, S. N., Kauffman, E. J., Zhang, Y., Strunk, B. S., Weisman, L. S., et al. (2014). Activity-dependent PI(3,5)P2 synthesis controls AMPA receptor trafficking during synaptic depression. Proc. Natl. Acad. Sci. U.S.A. 111, E4896–E4905. doi: 10.1073/pnas.1411117111
McPherson, P. S., Garcia, E. P., Slepnev, V. I., David, C., Zhang, X., Grabs, D., et al. (1996). A presynaptic inositol-5-phosphatase. Nature 379, 353–357. doi: 10.1038/379353a0
Mehta, Z. B., Pietka, G., and Lowe, M. (2014). The cellular and physiological functions of the Lowe syndrome protein OCRL1. Traffic 15, 471–487. doi: 10.1111/tra.12160
Ménager, C., Arimura, N., Fukata, Y., and Kaibuchi, K. (2004). PIP 3 is involved in neuronal polarization and axon formation. J. Neurochem. 89, 109–118. doi: 10.1046/j.1471-4159.2004.02302.x
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F., Andrews, S. P., Ashkenazi, A., et al. (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034. doi: 10.1016/j.neuron.2017.01.022
Mertens, J., Wang, Q.-W., Kim, Y., Yu, D. X., Pham, S., Yang, B., et al. (2015). Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 527, 95–99. doi: 10.1038/nature15526
Ming, G., Song, H., Berninger, B., Inagaki, N., Tessier-Lavigne, M., and Poo, M. (1999). Phospholipase C-gamma and phosphoinositide 3-kinase mediate cytoplasmic signaling in nerve growth cone guidance. Neuron 23, 139–148. doi: 10.1016/s0896-6273(00)80760-6
Miranda, A. M., Herman, M., Cheng, R., Lee, J. H., Hussaini, S. A., Marquer, C., et al. (2018). Excess synaptojanin 1 contributes to place cell dysfunction and memory deficits in the aging hippocampus in three types of Alzheimer’s disease. Cell Rep. 23, 2967–2975. doi: 10.1016/j.celrep.2018.05.011
Mirzaa, G. M., Conway, R. L., Gripp, K. W., Lerman-sagie, T., Siegel, D. H., Linda, S., et al. (2012). Megalencephaly-capillary malformation (MCAP) and (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am. J. Med. Genet. A 158, 269–291. doi: 10.1002/ajmg.a.34402
Morel, E., Chamoun, Z., Lasiecka, Z. M., Chan, R. B., Williamson, R. L., Vetanovetz, C., et al. (2013). Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 4:2250. doi: 10.1038/ncomms3250
Nahorski, M. S., Al-Gazali, L., Hertecant, J., Owen, D. J., Borner, G. H. H., Chen, Y.-C., et al. (2015). A novel disorder reveals clathrin heavy chain-22 is essential for human pain and touch development. Brain 138, 2147–2160. doi: 10.1093/brain/awv149
Nakajima, J., Okamoto, N., Shiraishi, J., Nishimura, G., Nakashima, M., Tsurusaki, Y., et al. (2013). Novel FIG4 mutations in Yunis – Varon syndrome. J. Hum. Genet. 58, 822–824. doi: 10.1038/jhg.2013.104
Nakatsu, F., Baskin, J. M., Chung, J., Tanner, L. B., Shui, G., Lee, S. Y., et al. (2012). PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J. Cell Biol. 199, 1003–1016. doi: 10.1083/jcb.201206095
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993. doi: 10.1038/ng.3043
Naughtin, M. J., Sheffield, D. A., Rahman, P., Hughes, W. E., Gurung, R., Stow, J. L., et al. (2010). The myotubularin phosphatase MTMR4 regulates sorting from early endosomes. J. Cell Sci. 123, 3071–3083. doi: 10.1242/jcs.060103
Nicholson, G., Lenk, G. M., Reddel, S. W., Grant, A. E., Towne, C. F., Ferguson, C. J., et al. (2011). Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P2 phosphatase FIG4. Brain 134, 1959–1971. doi: 10.1093/brain/awr148
Nixon, R. A. (2017). Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J. 31, 2729–2743. doi: 10.1096/fj.201700359
Noda, T., Matsunaga, K., Taguchi-Atarashi, N., and Yoshimori, T. (2010). Regulation of membrane biogenesis in autophagy via PI3P dynamics. Semin. Cell Dev. Biol. 21, 671–676. doi: 10.1016/j.semcdb.2010.04.002
Ohnishi, T., Ohba, H., Seo, K. C., Im, J., Sato, Y., Iwayama, Y., et al. (2007). Spatial expression patterns and biochemical properties distinguish a second myo-inositol monophosphatase IMPA2 from IMPA1. J. Biol. Chem. 282, 637–646. doi: 10.1074/jbc.M604474200
Olgiati, S., Rosa, A., De Quadri, M., Criscuolo, C., Breedveld, G. J., Picillo, M., et al. (2014). PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics 15, 183–188. doi: 10.1007/s10048-014-0406-0
Olivos-glander, I. M., Janne, P. A., and Nussbaum, R. L. (1995). The oculocerebrorenal syndrome gene product is protein localized to the golgi complex. Am. J. Hum. Genet. 57, 817–823.
Osborn, D. P. S., Pond, H. L., Mazaheri, N., Dejardin, J., Munn, C. J., Mushref, K., et al. (2017). Mutations in INPP5K cause a form of congenital muscular dystrophy overlapping marinesco-sjögren syndrome and dystroglycanopathy. Am. J. Hum. Genet. 100, 537–545. doi: 10.1016/j.ajhg.2017.01.019
Othmane, K. B., Johnson, E., Menold, M., Graham, F. L., Hamida, M., Ben, et al. (1999). Identification of a new locus for autosomal recessive Charcot – Marie – tooth disease with focally folded myelin on chromosome 11p15. Genomics 62, 344–349. doi: 10.1006/geno.1999.6028
Pagnamenta, A. T., Howard, M. F., Wisniewski, E., Popitsch, N., Knight, S. J. L., Keays, D. A., et al. (2015). Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis. Hum. Mol. Genet. 24, 3732–3741. doi: 10.1093/hmg/ddv117
Papadopoulos, T., Rhee, H. J., Subramanian, D., Paraskevopoulou, F., Mueller, R., Schultz, C., et al. (2017). Endosomal phosphatidylinositol 3-phosphate promotes gephyrin clustering and GABAergic neurotransmission at inhibitory postsynapses. J. Biol. Chem. 292, 1160–1177. doi: 10.1074/jbc.M116.771592
Poduri, A., Evrony, G. D., Cai, X., Elhosary, P. C., Beroukhim, R., Lehtinen, M. K., et al. (2012). Report somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74, 41–48. doi: 10.1016/j.neuron.2012.03.010
Posor, Y., Eichhorn-Gruenig, M., Puchkov, D., Schöneberg, J., Ullrich, A., Lampe, A., et al. (2013). Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 499, 233–237. doi: 10.1038/nature12360
Posor, Y., Eichhorn-Grünig, M., and Haucke, V. (2015). Phosphoinositides in endocytosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1851, 794–804. doi: 10.1016/j.bbalip.2014.09.014
Quadri, M., Fang, M., Picillo, M., Olgiati, S., Breedveld, G. J., Graafland, J., et al. (2013). Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum. Mutat. 34, 1208–1215. doi: 10.1002/humu.22373
Rahdar, M., Inoue, T., Meyer, T., Zhang, J., Vazquez, F., and Devreotes, P. N. (2009). A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc. Natl. Acad. Sci. U.S.A. 106, 480–485. doi: 10.1073/pnas.0811212106
Rameh, L. E., Tolias, K. F., Duckworth, B. C., and Cantley, L. C. (1997). A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature 390, 192–196. doi: 10.1038/36621
Ramel, D., Lagarrigue, F., Pons, V., Mounier, J., Dupuis-Coronas, S., Chicanne, G., et al. (2011). Shigella flexneri infection generates the lipid PI5P to alter endocytosis and prevent termination of EGFR signaling. Sci. Signal. 4:ra61. doi: 10.1126/scisignal.2001619
Rapoport, S. I., Primiani, C. T., Chen, C. T., Ahn, K., and Ryan, V. H. (2015). Coordinated expression of phosphoinositide metabolic genes during development and aging of human dorsolateral prefrontal cortex. PLoS One 10:e0132675. doi: 10.1371/journal.pone.0132675
Rhee, S. G. (2001). Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 70, 281–312. doi: 10.1146/annurev.biochem.70.1.281
Rhee, S. G., and Choi, K. D. (1992). Regulation of inositol phospholipid-specific phospholipase C isozymes. J. Biol. Chem. 267, 12393–12396.
Ripke, S., Neale, B. M., Corvin, A., Walters, J. T. R., Farh, K.-H., Holmans, P. A., et al. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Robinson, F. L., and Dixon, J. E. (2005). The phosphoinositide-3-phosphatase MTMR2 associates with MTMR13, a membrane-associated pseudophosphatase also mutated in type 4B Charcot-Marie-tooth disease. J. Biol. Chem. 280, 31699–31707. doi: 10.1074/jbc.M505159200
Ruderfer, D. M., Fanous, A. H., Ripke, S., Mcquillin, A., Amdur, R. L., and Working, S. (2014). Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and Schizophrenia. Mol. Psychiatry 19, 1017–1024. doi: 10.1038/mp.2013.138
Russo, F. B., Cugola, F. R., Fernandes, I. R., Pignatari, G. C., and Beltrão-Braga, P. C. B. (2015). Induced pluripotent stem cells for modeling neurological disorders. World J. Transplant. 5, 209–221. doi: 10.5500/wjt.v5.i4.209
Saiardi, A., and Mudge, A. W. (2018). Lithium and fluoxetine regulate the rate of phosphoinositide synthesis in neurons: a new view of their mechanisms of action in bipolar disorder. Transl. Psychiatry 8:175. doi: 10.1038/s41398-018-0235-2
Sanchez-Juan, P., Bishop, M. T., Aulchenko, Y. S., Brandel, J.-P., Rivadeneira, F., Struchalin, M., et al. (2012). Genome-wide study links MTMR7 gene to variant Creutzfeldt-Jakob risk. Neurobiol. Aging 33:1487.e21-.e28. doi: 10.1016/j.neurobiolaging.2011.10.011
Sansal, I., and Sellers, W. R. (2014). The biology and clinical relevance of the PTEN tumor suppressor pathway. J. Clin. Oncol. 22, 2954–2963. doi: 10.1200/JCO.2004.02.141
Sarkes, D., and Rameh, L. E. (2010). A novel HPLC-based approach makes possible the spatial characterization of cellular PtdIns5P and other phosphoinositides. Biochem. J. 428, 375–384. doi: 10.1042/BJ20100129
Sasaki, J., Kofuji, S., Itoh, R., Momiyama, T., Takayama, K., Murakami, H., et al. (2010). The PtdIns(3,4)P(2) phosphatase INPP4A is a suppressor of excitotoxic neuronal death. Nature 465, 497–501. doi: 10.1038/nature09023
Sasaki, T., Takasuga, S., Sasaki, J., Kofuji, S., Eguchi, S., Yamazaki, M., et al. (2009). Mammalian phosphoinositide kinases and phosphatases. Prog. Lipid Res. 48, 307–343. doi: 10.1016/j.plipres.2009.06.001
Schaletzky, J., Dove, S. K., Short, B., Lorenzo, O., Clague, M. J., and Barr, F. A. (2003). Phosphatidylinositol-5-phosphate activation and conserved substrate specificity of the myotubularin phosphatidylinositol 3-phosphatases. Curr. Biol. 13, 504–509. doi: 10.1016/S0960-9822(03)00132-5
Schu, P. V., Takegawa, K., Fry, M. J., Stack, J. H., Waterfield, M. D., and Emr, S. D. (1993). Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science 260, 88–91. doi: 10.1126/science.8385367
Schwab, S. G., Knapp, M., Sklar, P., Eckstein, G. N., Sewekow, C., Borrmann-Hassenbach, M., et al. (2006). Evidence for association of DNA sequence variants in the phosphatidylinositol-4-phosphate 5-kinase IIalpha gene (PIP5K2A) with schizophrenia. Mol. Psychiatry 11, 837–846. doi: 10.1038/sj.mp.4001864
Seebohm, G., Neumann, S., Theiss, C., Novkovic, T., Hill, E. V., Tavaré, J. M., et al. (2012). Identification of a novel signaling pathway and its relevance for GluA1 recycling. PLoS One 7:e33889. doi: 10.1371/journal.pone.0033889
Sherva, R., Wang, Q., Kranzler, H., Zhao, H., Koesterer, R., Herman, A., et al. (2016). Genome-wide association study of cannabis dependence severity, novel risk variants, and shared genetic risks. JAMA Psychiatry 73, 472–480. doi: 10.1001/jamapsychiatry.2016.0036
Shetty, M., Nimmy, R., Shubhi, S., Mullapudi, N., and Hegde, S. (2017). A homozygous missense variant in INPP5E associated with joubert syndrome and related disorders. Mol. Syndromol. 8, 313–317. doi: 10.1159/000479673
Shi, Y., Inoue, H., Wu, J. C., and Yamanaka, S. (2017). Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 16, 115–130. doi: 10.1038/nrd.2016.245
Shisheva, A., Sbrissa, D., and Ikonomov, O. (2015). Plentiful PtdIns5P from scanty PtdIns(3,5)P 2 or from ample PtdIns? PIKfyve-dependent models: evidence and speculation (response to: DOI 10.1002/bies.201300012). BioEssays 37, 267–277. doi: 10.1002/bies.201400129
Simon, G. E. (2003). Social and economic burden of mood disorders. Biol. Psychiatry 54, 208–215. doi: 10.1016/S0006-3223(03)00420-7
Simons, J. P., Al-Shawi, R., Minogue, S., Waugh, M. G., Wiedemann, C., Evangelou, S., et al. (2009). Loss of phosphatidylinositol 4-kinase 2α activity causes late onset degeneration of spinal cord axons. Proc. Natl. Acad. Sci. U.S.A. 106, 11535–11539. doi: 10.1073/pnas.0903011106
Simonsen, A., Wurmser, A. E., Emr, S. D., and Stenmark, H. (2001). The role of phosphoinositides in membrane transport. Curr. Opin. Cell Biol. 13, 485–492. doi: 10.1016/S0955-0674(00)00240-4
Soares, J. C., and Mallinger, A. G. (1995). Abnormal phosphatidylinositol (PI)-signalling in bipolar disorde. Biol. Psychiatry 3223, 1995–1996.
Soutar, M. P. M., Kempthorne, L., Miyakawa, S., Annuario, E., Melandri, D., Harley, J., et al. (2018). AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci. Rep. 8:8855. doi: 10.1038/s41598-018-26949-6
Splinter, K., Adams, D. R., Bacino, C. A., Bellen, H. J., Bernstein, J. A., Cheatle-Jarvela, A. M., et al. (2018). Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med. 379, 2131–2139. doi: 10.1056/NEJMoa1714458
Stefan, C. J., Manford, A. G., Baird, D., Yamada-Hanff, J., Mao, Y., and Emr, S. D. (2011). Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 144, 389–401. doi: 10.1016/j.cell.2010.12.034
Thiselton, D. L., Maher, B. S., Webb, B. T., Bigdeli, T. B., O’Neill, F. A., Walsh, D., et al. (2009). Association analysis of the PIP4K2A gene on chromosome 10p12 and Schizophrenia in the Irish study of high density schizophrenia families (ISHDSF) and the Irish case-control study of schizophrenia (ICCSS). Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 153B, 323–331. doi: 10.1002/ajmg.b.30982
Tighe, S. K., Mahon, P. B., and Potash, J. B. (2011). Predictors of lithium response in bipolar disorder. Ther. Adv. Chronic Dis. 2, 209–226. doi: 10.1177/2040622311399173
Tohyama, J., Nakashima, M., Nabatame, S., Gaik-Siew, C., Miyata, R., Rener-Primec, Z., et al. (2015). SPTAN1 encephalopathy: distinct phenotypes and genotypes. J. Hum. Genet. 60, 167–173. doi: 10.1038/jhg.2015.5
Travaglini, L., Brancati, F., Silhavy, J., Iannicelli, M., Nickerson, E., Elkhartoufi, N., et al. (2013). Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders. Eur. J. Hum. Genet. 21, 1074–1078. doi: 10.1038/ejhg.2012.305
Tsuruta, F., Green, E. M., Rousset, M., and Dolmetsch, R. E. (2009). PIKfyve regulates CaV1.2 degradation and prevents excitotoxic cell death. J. Cell Biol. 187, 279–294. doi: 10.1083/jcb.200903028
Vaccari, I., Dina, G., Tronchère, H., Kaufman, E., Chicanne, G., Cerri, F., et al. (2011). Genetic interaction between MTMR2 and FIG4 phospholipid phosphatases involved in Charcot-Marie-Tooth neuropathies. PLoS Genet. 7:e1002319. doi: 10.1371/journal.pgen.1002319
van den Bout, I., and Divecha, N. (2009). PIP5K-driven PtdIns(4,5)P2 synthesis: regulation and cellular functions. J. Cell Sci. 122, 3837–3850. doi: 10.1242/jcs.056127
van Diepen, M. T., and Eickholt, B. J. (2008). Function of PTEN during the formation and maintenance of neuronal circuits in the brain. Dev. Neurosci. 30, 59–64. doi: 10.1159/000109852
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M., and Bilanges, B. (2010). The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341. doi: 10.1038/nrm2882
Varga, E. A., Pastore, M., Prior, T., and Herman, G. E. (2009). The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet. Med. 11, 111–117. doi: 10.1097/GIM.0b013e31818fd762
Vaucher, J., Keating, B. J., Lasserre, A. M., Gan, W., Lyall, D. M., Ward, J., et al. (2017). Cannabis use and risk of schizophrenia: a Mendelian randomization study. Nat. Publ. Gr. 23, 1287–1292. doi: 10.1038/mp.2016.252
Vehlow, A., Soong, D., Vizcay-Barrena, G., Bodo, C., Law, A.-L., Perera, U., et al. (2013). Endophilin, Lamellipodin, and Mena cooperate to regulate F-actin-dependent EGF-receptor endocytosis. EMBO J. 32, 2722–2734. doi: 10.1038/emboj.2013.212
Voronov, S. V., Frere, S. G., Giovedi, S., Pollina, E. A., Borel, C., Zhang, H., et al. (2008). Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc. Natl. Acad. Sci. U.S.A. 105, 9415–9420. doi: 10.1073/pnas.0803756105
Walch-Solimena, C., and Novick, P. (1999). The yeast phosphatidylinositol-4-OH kinase pik1 regulates secretion at the Golgi. Nat. Cell Biol. 1, 523–525. doi: 10.1038/70319
Walker, D. M., Urbé, S., Dove, S. K., Tenza, D., Raposo, G., and Clague, M. J. (2001). Characterization of MTMR3. an inositol lipid 3-phosphatase with novel substrate specificity. Curr. Biol. 11, 1600–1605. doi: 10.1016/S0960-9822(01)00501-2
Wang, L., Budolfson, K., and Wang, F. (2011). Pik3c3 deletion in pyramidal neurons results in loss of synapses, extensive gliosis and progressive neurodegeneration. Neuroscience 172, 427–442. doi: 10.1016/j.neuroscience.2010.10.035
Wang, Y. J., Wang, J., Sun, H. Q., Martinez, M., Sun, Y. X., Macia, E., et al. (2003). Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell 114, 299–310. doi: 10.1016/s0092-8674(03)00603-2
Wen, Z., Christian, K. M., Song, H., and Ming, G. (2016). Modeling psychiatric disorders with patient-derived iPSCs. Curr. Opin. Neurobiol. 36, 118–127. doi: 10.1016/j.conb.2015.11.003
Wenk, M. R., Pellegrini, L., Klenchin, V. A., Di Paolo, G., Chang, S., Daniell, L., et al. (2001). PIP kinase Igamma is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron 32, 79–88. doi: 10.1016/s0896-6273(01)00456-1
Wiessner, M., Roos, A., Munn, C. J., Viswanathan, R., Whyte, T., Cox, D., et al. (2017). Mutations in INPP5K, encoding a phosphoinositide 5-phosphatase, cause congenital muscular dystrophy with cataracts and mild cognitive impairment. Am. J. Hum. Genet. 100, 523–536. doi: 10.1016/j.ajhg.2017.01.024
Wu, C. Y., Lin, M. W., Wu, D. C., Huang, Y. B., Huang, H. T., and Chen, C. L. (2014). The role of phosphoinositide-regulated actin reorganization in chemotaxis and cell migration. Br. J. Pharmacol. 171, 5541–5554. doi: 10.1111/bph.12777
Xie, T., Deng, L., Mei, P., Zhou, Y., Wang, B., Zhang, J., et al. (2014). A genome-wide association study combining pathway analysis for typical sporadic amyotrophic lateral sclerosis in Chinese Han populations. Neurobiol. Aging 35:1778.e9-e23. doi: 10.1016/j.neurobiolaging.2014.01.014
Yoshinaga, S., Ohkubo, T., Sasaki, S., Nuriya, M., Ogawa, Y., Yasui, M., et al. (2012). A phosphatidylinositol lipids system, lamellipodin, and Ena/VASP regulate dynamic morphology of multipolar migrating cells in the developing cerebral cortex. J. Neurosci. 32, 11643–11656. doi: 10.1523/JNEUROSCI.0738-12.2012
Zewe, J. P., Wills, R. C., Sangappa, S., Goulden, B. D., and Hammond, G. R. (2018). SAC1 degrades its lipid substrate PtdIns4P in the endoplasmic reticulum to maintain a steep chemical gradient with donor membranes. eLife 7:e35588. doi: 10.7554/eLife.35588
Zhang, S., Duan, L., He, S., Zhuang, G., and Yu, X. (2017). Phosphatidylinositol 3, 4-bisphosphate regulates neurite initiation and dendrite morphogenesis via actin aggregation. Nat. Publ. Gr. 27, 253–273. doi: 10.1038/cr.2017.13
Zhang, X., Wang, W.-A., Jiang, L.-X., Liu, H.-Y., Zhang, B.-Z., Lim, N., et al. (2017). Downregulation of RBO-PI4KIIIα facilitates Aβ 42 secretion and ameliorates neural deficits in Aβ 42 -expressing Drosophila. J. Neurosci. 37, 4928–4941. doi: 10.1523/JNEUROSCI.3567-16.2017
Zhang, X., Clement, Y., Sahenk, Z., Shy, M. E., Meisler, M. H., and Li, J. (2008). Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain 131, 1990–2001. doi: 10.1093/brain/awn114
Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. doi: 10.1016/j.neuron.2015.11.013
Zheng, L., and Conner, S. D. (2018). PI5P4Kγ functions in DTX1-mediated Notch signaling. Proc. Natl. Acad. Sci. U.S.A. 115, E1983–E1990. doi: 10.1073/pnas.1712142115
Zhou, X., Wang, L., Hasegawa, H., Amin, P., Han, B., Kaneko, S., et al. (2010). Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 9424–9429. doi: 10.1073/pnas.0914725107
Keywords: phosphoinositides, brain disease, genetics, cellular organelles, inherited disorders, human genetics and genomics, neurological disorders
Citation: Raghu P, Joseph A, Krishnan H, Singh P and Saha S (2019) Phosphoinositides: Regulators of Nervous System Function in Health and Disease. Front. Mol. Neurosci. 12:208. doi: 10.3389/fnmol.2019.00208
Received: 23 March 2019; Accepted: 07 August 2019;
Published: 23 August 2019.
Edited by:
Michele Papa, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Olof Idevall-Hagren, Uppsala University, SwedenMarco Falasca, Curtin University, Australia
Copyright © 2019 Raghu, Joseph, Krishnan, Singh and Saha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Padinjat Raghu, cHJhZ2h1QG5jYnMucmVzLmlu