 Matthieu Colom
Matthieu Colom Benjamin Vidal
Benjamin Vidal Luc Zimmer
Luc Zimmer- 1Lyon Neuroscience Research Center, INSERM, CNRS, Université de Lyon, Lyon, France
- 2CERMEP, Hospices Civils de Lyon, Bron, France
- 3Institut National des Sciences et Techniques Nucléaires, CEA Saclay, Gif-sur-Yvette, France
Positron emission tomography (PET) is a molecular imaging modality that enables in vivo exploration of metabolic processes and especially the pharmacology of neuroreceptors. G protein-coupled receptors (GPCRs) play an important role in numerous pathophysiologic disorders of the central nervous system. Thus, they are targets of choice in PET imaging to bring proof concept of change in density in pathological conditions or in pharmacological challenge. At present, most radiotracers are antagonist ligands. In vitro data suggest that properties differ between GPCR agonists and antagonists: antagonists bind to receptors with a single affinity, whereas agonists are characterized by two different affinities: high affinity for receptors that undergo functional coupling to G-proteins, and low affinity for those that are not coupled. In this context, agonist radiotracers may be useful tools to give functional images of GPCRs in the brain, with high sensitivity to neurotransmitter release. Here, we review all existing PET radiotracers used from animals to humans and their role for understanding the ligand-receptor paradigm of GPCR in comparison with corresponding antagonist radiotracers.
Introduction
From Agonist Molecules to PET Radiopharmaceuticals
Positron emission tomography is a molecular imaging modality that enables exploration of metabolic processes in vivo. It uses specific radiotracers for specific molecular targets (Van de Bittner et al., 2014). The radiotracer must have several characteristics (Halldin et al., 2001; Pike, 2009; Honer et al., 2014): i.e., a specific binding to the target of interest with an acceptable signal-to-noise ratio, a passage through the BBB, and limited radiometabolites in the brain. The most common molecular targets in PET neuroimaging are neurotransmitter receptors or transporters. PET imaging visualizes various neuroreceptors that can be located in vivo on presynaptic and/or post-synaptic sites, using a microdose of radioligand (i.e., tracer dose). PET imaging is therefore a powerful tool to demonstrate changes in neurotransmission in various CNS disorders, and can be used translationally in both animal models and humans. In addition to its contribution to the understanding of pathophysiological processes, PET imaging plays an important role in CNS drug development. It enables measurement of the proportion of receptors occupied by pharmacological doses of drugs of interest, in competition with a suitable radioligand specific to the same target. It can thus demonstrate brain penetration and in vivo binding to the target, which can be correlated to plasma concentrations to predict the effective dose range for clinical studies. They collect important information about the bioavailability of the drug candidate and contribute to the demonstration of brain penetration. Microdosing and drug occupancy studies have been shown to be very valuable for optimizing the development of drugs targeting the CNS. Another important application of PET neuroimaging is to measure in vivo fluctuations in endogenous neurotransmitter release. According to the occupancy model, the binding potential of a given radiotracer is modulated by the local concentration of the endogenous neurotransmitter in competition for the same receptors when the affinities of radioligand and neurotransmitter are in the same order of magnitude (Laruelle, 2000). All these applications rely on the development and full characterization of specific radiotracers. Consequently, although PET imaging provides interesting in vivo approaches to understanding neuropharmacology, it is currently limited by a lack of specific radiotracers for many known brain receptors. Moreover, the large majority of available radiotracers are antagonists and, as will be explained below, may not provide information about GPCR functional status in vivo; this fact, often disregarded, may be an important limitation for the interpretation of numerous clinical PET studies and probably explains certain controversies still ongoing in the field. The present review will therefore focus on the few agonist radiotracers that are currently available, highlighting their potential interest in PET neuroimaging, especially in humans in the form of radiopharmaceuticals.
The Pharmacological Paradigm of G Protein-Coupled Receptor Agonism
In vitro studies on membrane homogenates distinguished different properties in GPCR agonists and antagonists. While antagonists bind to receptors with a single affinity, agonists show two different affinities: high affinity for receptors coupled to G-proteins, and low affinity for uncoupled receptors (Figure 1). As reviewed recently (Shalgunov et al., 2019), these findings were demonstrated for various GPCRs: adrenergic (Hoffman and Lefkowitz, 1980), dopaminergic (Sibley et al., 1982; Leff et al., 1985), serotonergic (Battaglia et al., 1984; Gozlan et al., 1995; Watson et al., 2000) muscarinic (Ehlert, 1985), cannabinoid (Gullapalli et al., 2010), and opioid receptors (Law et al., 1985). The pharmacology of high-affinity receptors features specific phenomena: negative GTP cycle feedback loop, oligo-heterodimerization, internalization. While many in vitro studies revealed dysregulation of the balance between coupled and uncoupled states in these receptors, the implications for neurological disorders remains to be demonstrated, and new tools are needed. At present, few GPCRs can actually be studied using both antagonist and agonist radiotracers. Thus the different functional states of the receptors in vivo cannot be distinguished, making it impossible to specifically study the receptors in the high-affinity state which must represent the true (synapse) responsivity of the system to endogenous neurotransmission. Given that agonists bind preferentially to coupled receptors, PET imaging could disclose the active state of a receptor population.
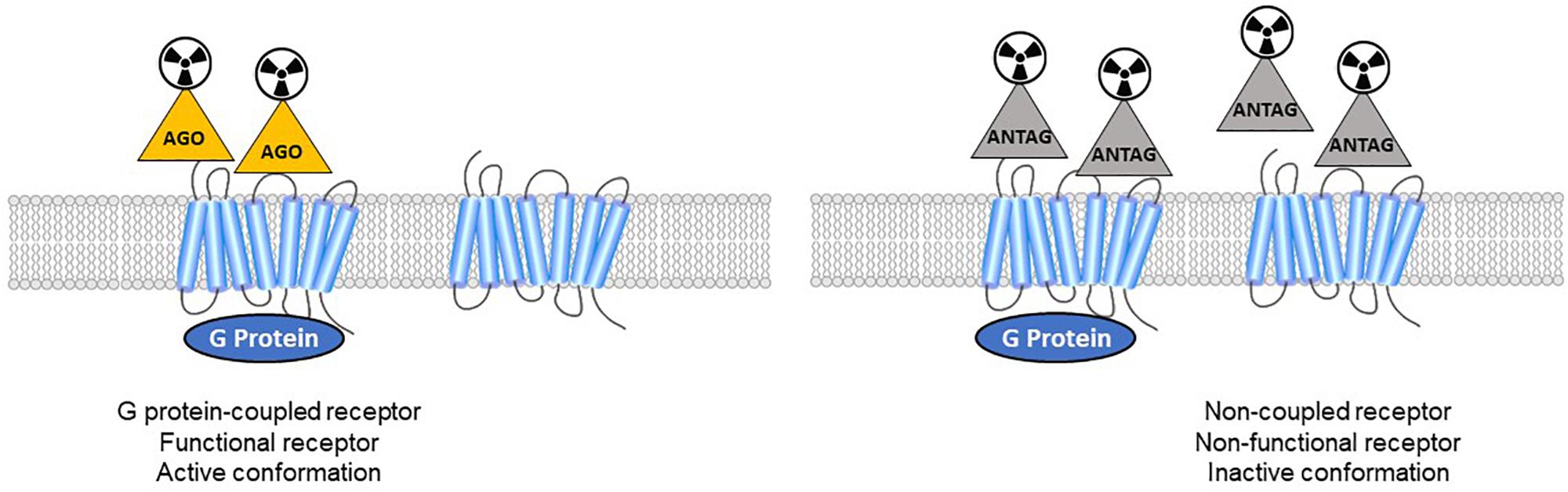
Figure 1. Antagonist radioligands bind both coupled and non-coupled GPCRs with the same affinity. Agonist radioligands discriminate high-affinity (active GPCRs) versus low-affinity sites (inactive receptors).
Antagonists Are Often Used in PET Neuroimaging
The primary reason was that radiochemists and radiopharmacologists had access to a larger choice of antagonist molecules, initially developed as neuropharmacological tools; but there are also other reasons. Firstly, in terms of pharmacology, it is simpler to use an antagonist, which has only one affinity for a given receptor, as it does not discriminate between different subpopulations but rather provides an image of the global density of receptors. In contrast, using an agonist complicates the analysis due to its dual affinity for both high- and low-affinity receptors, providing an image with lower signal-to-noise ratio due to the lower density of the targeted receptors. Moreover, agonists are more likely to induce undesirable side effects for the patient if the quantity injected is too great (i.e., higher than a tracer dose): the specific activity needs to be high enough for a microdose of agonist to be injected, at subpharmacological level. Another problem is that the rate of conversion from high- to low-affinity state can cause the agonist to dissociate from its receptor too quickly: after stimulation by the agonist, the receptor is likely to switch to the low-affinity state within seconds (Mathis et al., 1997). Although dissociation is slower than the conversion from high- to low-affinity state, this can cast doubt on the functional state of the receptors actually targeted by the agonist (Seeman, 2012). All these reasons explain why PET neuroimaging using agonist radiotracers is considered challenging, as it raises a number of difficulties relating to radiopharmacy (e.g., the potential pharmacological effects of the radioligand if the specific activity is too low), or to pharmacological concepts that are not at present fully elucidated, such as the conformational model of GPCR signaling itself. In fact, GPCRs can be considered as either pre-coupled to their respective G proteins or not (De Lean et al., 1980; Kent et al., 1980; Mongeau et al., 1992), or as initially non-coupled and interacting with G proteins after agonist stimulation (Laruelle, 2000; Skinbjerg et al., 2010).
Agonist Radiotracers to Explore the Coupling of GPCRs
On the other hand, the fact that neuroimaging mainly uses antagonist radiotracers incurs a number of limitations, in terms of both neurophysiology and pathophysiology. For neurophysiological exploration, the lack of sensitivity to endogenous neurotransmitter level or exogenous agonists at pharmacological dose, as will be discussed below, calls for the use of agonist radioligands. The physiological impact of GPCR functional state plays a major role in maintaining cellular homeostasis and effective neurotransmission. Compared to ion channels, which produce relatively simple and quick responses after stimulation by a ligand, GPCR signaling involves complex signaling cascades with production of numerous secondary messengers, interaction with various channels (Reboreda et al., 2018) and phosphorylation of diverse proteins, which may have varying long-term effects in the cell. Therefore, GPCRs are of critical functional importance in the CNS and are one of the most important drug targets in pharmacology, and especially neuropharmacology (Albizu et al., 2010; Björk and Svenningsson, 2011; Jastrzebska et al., 2011; Hauser et al., 2017). The same GPCR can interact with multiple signaling pathways that may vary across the brain [e.g., 5-HT1A receptors (la Cour, 2006)]. For example, according to the recent concept of biased agonism, each ligand may preferentially stimulate a few of the numerous pathways that can interact with a GPCR. This may explain the diverse pharmacological effects observed for a given class of GPCR ligands (Albizu et al., 2010). The complexity of GPCRs needs to be studied more extensively in vivo, and requires the development of suitable tools such as agonist radiotracers and, ultimately, biased agonists. Pathophysiologically, G protein signaling is strongly involved in many neurodegenerative or neuropsychiatric disorders (Schreiber and Avissar, 2000; Avissar and Schreiber, 2006; Thathiah and De Strooper, 2011; Heese, 2013). Numerous in vitro studies showed that GPCR coupling state is modified in pathological conditions (Schreiber et al., 2009; Becker et al., 2014; Vidal et al., 2016). The exploration of coupled and non-coupled populations of receptors in vivo could be a key point in developing new therapies, monitoring treatment effects and identifying treatment responders. At present, it is difficult to explore G protein coupling in the brain in vivo due to a lack of non-invasive tools; it is measured either on brain slices in vitro (Pejchal et al., 2002; Shen et al., 2002) or in peripheral cells such as leukocytes (Golan et al., 2011).
According to in vitro pharmacological findings, agonist radiotracers may provide new tools, challenging the standard ligand receptor model in pharmacology (Laruelle, 2000). They seem to be involved in GPCR activity and reflect the responsiveness of the synapse signaling system (Shalgunov, 2017). Therefore, agonist radioligands could provide precise in vivo pharmacology by imaging only active neuroreceptors (Zimmer, 2016). PET would thus seem to be the means to shed light on GPCR properties in vivo.
State of the Art of Existing PET Agonist Radiotracers for Neuroimaging
The following section comprises an exhaustive review of PET radiotracers with a translation to first in human are presented (Figure 2 and Table 1). See Supplementary Material for exhaustive review.
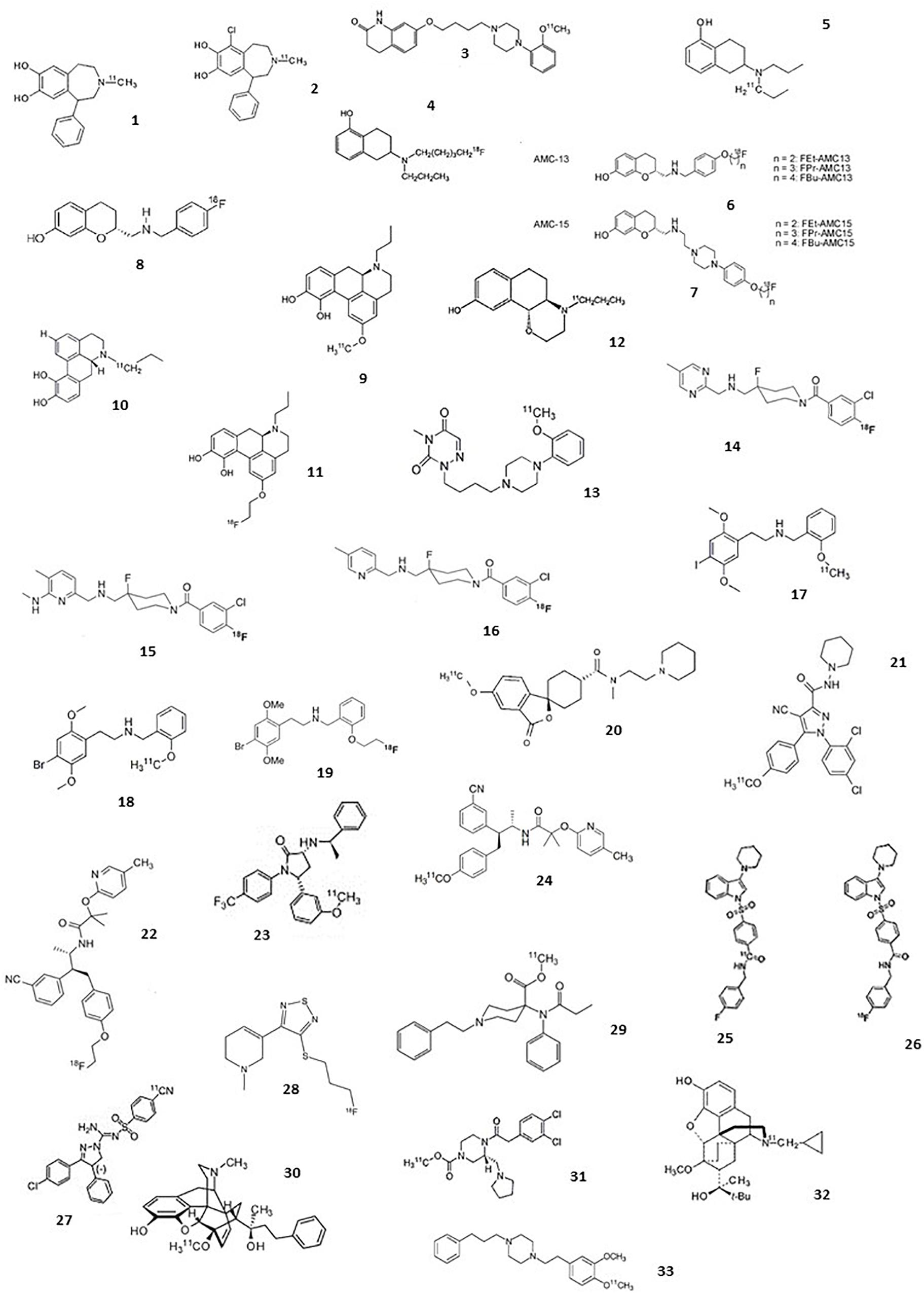
Figure 2. Chemical structures of current agonist radiotracers of GPCRs. Dopamine receptors: (1) [11C]SKF 82957, (2) [11C]-SKF75670, (3) [11C]SV-III-130, (4) [11C]5-OH-DPAT, (5) [18F]5-OH-FPPAT, (6) [18F]FBu-AMC13 and derivatives, (7) [18F]FEt-AMC15 and derivatives, (8) [18F]AMC20, (9) [11C]NPA, (10) [11C]MNPA, (11) [18F]MCL-524, (12) [11C]PHNO; serotonin receptors: (13) [11C]CUMI-101, (14) [18F]F15599, (15) [18F]F13714, (16) [18F]F13640, (17) [11C]Cimbi-5, (18) [11C]Cimbi-36, (19) [18F]FECimbi-36; histamine receptors: (20) [11C]MK-8278; cannabinoid receptors: (21) [11C]OMAR or [11C]JHU75528; (22) [18F]MK-9470, (23) [11C]MePPEP, (24) [11C]CB-119, (25) [11C]PipISB, (26) [18F]PipISB, (27) [11C]SD5024; acetylcholine receptors: (28) [18F]FP-TZTP; Opioïd receptors: (29) [11C]carfentanil, (30) [11C]PEO, (31) [11C]GR103545, (32) [11C]buprenorphine; Sigma 1 receptors: (33) [11C]SA4503.
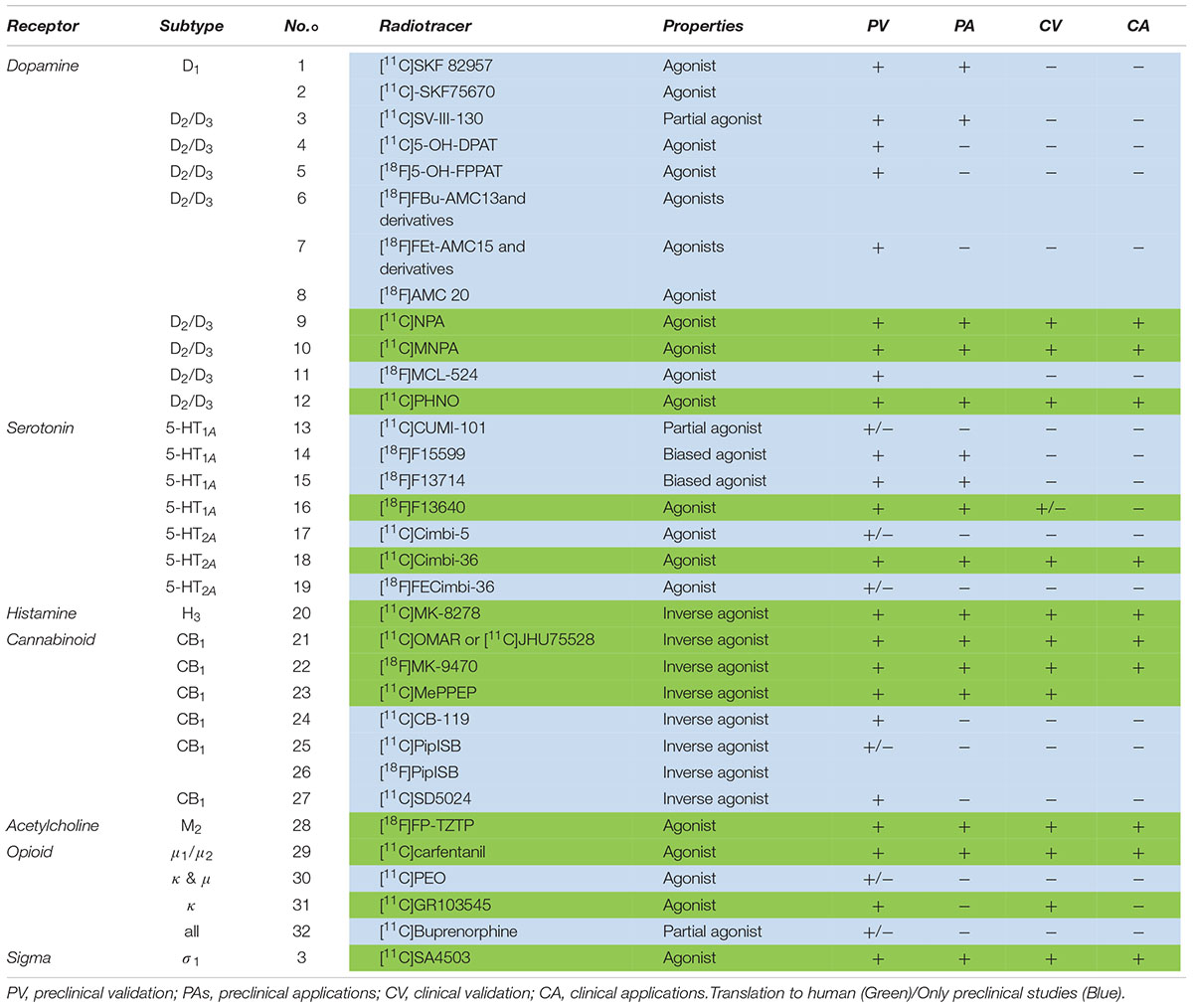
Table 1. Current agonist radiotracers of GPCRs with at least preclinical validation.
Dopaminergic Receptors
The dopaminergic system has five formally described subtypes of receptors (D1, D2, D3, D4, D5). The dopaminergic system has benefited from the development of a large number of PET radiotracers. The majority of these radiotracers are derived from the many pharmacological tools and drug candidates that have been developed for psychiatry and then neurology. Among these, the use in humans of three agonist radiotracers of D2/D3R, allowed to investigate sensitivity to neurotransmitter release and estimate the proportion of coupling in comparison with the reference antagonist radiotracer, [11C]raclopride.
D2/D3 Receptors
[11C]NPA
Ernst (1967) reported that apomorphine interacted with dopamine receptors. This observation launched intensive research about apomorphine’s structure/activity relationship, to define precisely the pharmacophore enabling dopamine receptor binding/agonism/stimulation. [11C]apomorphine itself was synthesized and evaluated in rat brain as a radioligand of D1-like and D2-like receptors (Zijlstra et al., 1993; Finnema et al., 2010). Brain uptake and specific binding ratios were too low for satisfactory application in PET imaging. Hwang et al. (2000) proposed [11C] radiolabeling of NPA, first synthesized in Neumeyer et al. (1973). NPA is a D2 agonist with higher affinity for D2 receptors at high affinity than for D2 receptors at low affinity (Sibley et al., 1982). Previous studies demonstrated that its tritiated analog crossed the BBB, with high uptake in striatum (van der Werf et al., 1983). Hwang and colleagues described the radiosynthesis of [11C]NPA, and biodistribution studies in rodents confirmed high uptake in striatum and a high striatum/cerebellum ratio (3.4 at 30 min post-injection). Haloperidol pretreatment decreased the striatum/cerebellum ratio to 1.3 at 30 min in rat brain. PET imaging studies on a single baboon also revealed a high striatum/cerebellum ratio of 2.8 at 45 min post-injection. A blockade study with haloperidol strongly decreased the striatum/cerebellum ratio, confirming specific binding to D2/D3 receptors in vivo. Cumming et al. (2002) demonstrated that [3H]NPA was more sensitive to endogenous dopamine in striatum than the antagonist [11C]raclopride in a study on living mice: both dopamine depletion by reserpine and dopamine increase by amphetamine had greater effects on the binding potential of tritiated NPA. A study in anesthetized Göttingen miniature pigs, using compartmental analyses, showed fast tracer metabolism, compensated by high brain uptake during the first minutes; the striatal binding potential was comparable with [11C]raclopride, and both MPTP pretreatment and deep brain stimulation of the subthalamus failed to produce obvious effects on [11C]NPA binding in vivo (Cumming et al., 2003). Another quantitative study in baboon validated the use of model-based approaches to quantify [11C]NPA binding in vivo (Hwang et al., 2004). Other PET studies in baboons confirmed its higher sensitivity to dopamine release in striatum compared to [11C]raclopride (Narendran et al., 2004; Hwang et al., 2005). The proportion of D2 receptors at high affinity was estimated to be 79% in the striatum using a bolus plus constant infusion (Narendran, 2005). A study in 1 male baboon did not show significant difference between time of recovery after amphetamine-induced dopamine release as measured by [11C]raclopride and [11C]NPA (Narendran et al., 2006). Subsequent studies focused on translation to humans. An initial study concluded that administration of a common dose of radiotracer (370 Mbq) yielded an acceptable dosimetric range in all organs (Laymon et al., 2009). Reproducibility studies and kinetic modeling in a second human study confirmed that [11C]NPA was a reliable tool to image D2/D3 receptors in high-affinity state in striatum (Narendran et al., 2009). Narendran and colleagues performed a comparative evaluation of [11C]NPA and [11C]raclopride to measure amphetamine-induced dopamine release in the human striatum after oral amphetamine pretreatment: decreases in BPND level were slightly greater for the agonist radiotracer, whereas no significant difference was found for BPP (Narendran et al., 2010). McCormick et al. (2010), in ex vivo studies, demonstrated that isoflurane increased the binding and amphetamine sensitivity of [11C]NPA and other agonists in comparison with [11C]raclopride, which may have been a confounding factor in several preclinical studies that reported higher sensitivity of agonist radiotracers to endogenous dopamine.
An initial clinical study in cocaine abusers versus controls did not find any differences in D2/D3 binding in striatum, contrary to several studies that previously reported lower binding of [11C]raclopride in cocaine abusers. The authors concluded that D2/D3 receptors in high-affinity state may be unaltered in cocaine dependence (Narendran et al., 2011). Other preclinical studies were performed using [11C]NPA: one ex vivo study demonstrated that it was more effective than [11C]raclopride in detecting an increase in receptor availability following unilateral injections of 6-OH-DA in rat, a classical model of Parkinson’s disease (Palner et al., 2011); another study in rat suggested differential distribution of tritiated NPA and raclopride in the striatum, with comparable Bmax in the dorsal striatum and lower Bmax for [3H]NPA in the ventral striatum (Minuzzi and Cumming, 2010).
[11C]MNPA
MNPA, a methoxy-NPA derivative, was initially described in Gao et al. (1990), and was characterized as a D2R agonist (Ki = 0.17 nM). A preliminary PET study in cynomolgus monkey in Halldin et al. (1992) demonstrated high binding of [11C]MNPA in the striatum. Finnema et al. (2005) described an optimized radiosynthesis and further in vivo PET experiments in cynomolgus monkey. The study confirmed previous findings about D2 specificity: pretreatment with unlabeled raclopride considerably decreased the signal in regions known to contain D2 receptors. Two advantages were mentioned in comparison with [11C]NPA: a fivefold higher affinity, which might enable quantitative analysis in extrastriatal regions such as thalamus, and easier radiosynthesis of [11C]MNPA, which only needs [11C]methylation. Metabolite analysis did not show any lipophilic metabolites which could interfere with the parent-compound signal in the brain. The sensitivity of the radiotracer to synaptic dopamine levels was then compared versus [11C]raclopride in cynomolgus monkeys; [11C]MNPA showed higher sensitivity. Using the same methodology of high-affinity state quantification proposed by Narendran with [11C]NPA, the authors suggested that about 60% of D2 receptors were in the high-affinity state (Seneca et al., 2006). In preparation for future human studies, a kinetic brain analysis and whole-body imaging study was performed in monkeys (Seneca et al., 2008a). Brain distribution volumes were identified using a 2-tissue compartment model and in accordance with the known distribution of D2/D3 receptors, and the estimated dosimetry in human, extrapolated from the preclinical results, was moderate to low. However, the authors reported that BPND values of [11C]MNPA were lower than [11C]raclopride or other agonist radiotracers such as [11C]PHNO and [11C]NPA, probably due to higher uptake in the cerebellum. Seneca et al. (2008b), the same team conducted a PET study in rat using a bolus/infusion paradigm following dopamine depletion pretreatment with reserpine plus alpha-methyl-para-tyrosine. Dopamine occupancy at baseline was estimated at 53% in rat brain, in agreement with previous results with other agonist radiotracers. In the same study, binding in striatum was displaceable with raclopride but not by BP897, a selective D3 ligand, suggesting that [11C]MNPA binds specifically to D2 and not D3 receptors. Skinbjerg et al. (2009) conducted a precise pharmacological characterization of MNPA with in vitro studies on recombinant cells and membrane preparations. They concluded that MNPA was a full agonist of D2 and also D3 receptors (in contrast to previous in vivo findings). Also, two high- and low-affinity binding states were observed only for membrane preparations and not for cells. Other preclinical studies were conducted the same year: Finnema et al. (2009) determined the occupancy of the agonist apomorphine at increasing doses in cynomolgus monkey using [11C]raclopride and [11C]MNPA. Contrary to the hypothesis that agonist radioligands are more sensitive than antagonists to competition with pharmacological doses of agonists, there was no difference between Ki and ID50 determined with both radiotracers, suggesting that all D2/3 receptors are in high-affinity state or that there is only a single receptor state. Tokunaga et al. (2009) reported an example of a drug discovery approach using in vivo imaging with [11C]MNPA to detect dopamine neurotransmission system modulation by MPEP, an antagonist of group mGlu1 receptors. Steiger et al. (2009), an optimization of radiosynthesis was also described, with a time of about 40 min after radionuclide production.
The first clinical trial, on 10 subjects, in Otsuka et al. (2009), used a classical protocol with arterial blood sampling and metabolite analysis (and PET procedure). Results showed a distribution pattern in concordance with the D2 receptor distribution. The SRTM and transient equilibrium methods were validated to estimate binding potentials. Another clinical study investigated binding of the antipsychotic risperidone on high- or low-affinity D2 receptors with [11C]raclopride and [11C]MNPA and found that risperidone bound indifferently to both states of D2 receptors, with comparable occupancies and ED50 values for both tracers (Kodaka et al., 2010). In 2013, a study investigated the reproducibility of the binding potential ratio of [11C]MNPA to [11C]raclopride, reflecting the proportion of receptors in high-affinity state compared to overall receptor density; reproducibility was satisfactory in the caudate and putamen (Kodaka et al., 2012). More recently, the same team studied the different D2 receptor affinity states in 11 antipsychotic-free schizophrenic patients, using [11C]raclopride and [11C]MNPA; the binding potential ratio (agonist/antagonist) was significantly higher in the putamen in patients than control subjects, despite unchanged levels of total D2 receptors (Kubota et al., 2017).
Other preclinical studies using [11C]MNPA were conducted over the years. Skinbjerg et al. (2010) showed that in vivo striatal binding of the tracer was unchanged in dopamine beta-hydroxylase-deficient mice, concluding that their increased sensitivity to psychostimulants was not due to a higher proportion of receptors in the high-affinity state. The same year, the D2 receptor occupancies of quinpirole, aripiprazole, and haloperidol were estimated in conscious rats, using tritiated MNPA, PHNO, and raclopride (Peng et al., 2010). All compounds showed similar occupancy values with the different radioligands, presumably due to a high proportion of receptors in the high-affinity state in vivo. Another study, focusing on stress in conscious monkeys, showed that stress level correlated negatively with [11C]raclopride binding, and positively with [11C]MNPA binding (Tsukada et al., 2011).
[11C]PHNO
The first radiolabeling and preclinical evaluation of the napthoxazine derivative D2-agonist (+)-4-Propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b]-[1,4]-oxazin-9-ol, or (+)-PHNO, was reported in Wilson et al. (2005). [11C]-(+)-PHNO binding in rat brain was highly selective and specific to D2 receptors. The tracer also showed sensitivity to increases and decreases in endogenous dopamine levels. The full D2 agonistic properties of (+)-PHNO were previously documented (Jones et al., 1984). The first-in-man study was performed 1 year later (Willeit et al., 2006). [11C]-(+)-PHNO displayed good brain uptake and favorable kinetics; test–retest data suggested BP estimates to be reliable, and pre-treatment with haloperidol reduced specific binding without detectable changes in cerebellum, validating its utility as a D2-receptor agonist radioligand for PET. Binding in the globus pallidus was greater than with the D2 antagonist [11C]raclopride, suggesting that [11C]-(+)-PHNO also binds significantly to D3 receptors in humans. Tracer distribution, displaceability by endogenous dopamine, specificity and modeling properties were further explored in cat and compared versus [11C]raclopride (Ginovart et al., 2006a). Although Scatchard analysis showed comparable Bmax values with both [11C]-(+)-PHNO and [11C]-raclopride, the agonist was more sensitive than the antagonist to dopamine release (BP inhibition up to 83 versus 56%, respectively). The signal-to-noise ratio in the striatum was also 2.5-fold higher than that of [11C]NPA. Unusually high binding in the globus pallidus was reported in baboons and explained by higher affinity of [11C]-(+)-PHNO for D3 receptors than other D2/D3 radioligands, possibly contributing to its greater vulnerability to endogenous dopamine (dopamine affinity being higher for D3 than D2 receptors) compared to other radioligands (Narendran et al., 2006).
Kinetic modeling of the tracer was then described in humans (Ginovart et al., 2006b), to enable [11C]-(+)-PHNO binding to be quantified in clinical studies. Willeit et al. (2008), the D2–D3 agonist radioligand was reported to be sensitive to competition with endogenous dopamine in humans. Several studies were then performed with [11C]-(+)-PHNO, to study the high-affinity state of D2 receptors, D3 receptors and endogenous dopamine release in schizophrenia, addiction or according to social status (Graff-Guerrero et al., 2008; Mizrahi et al., 2011, 2012; Le Foll et al., 2013; Matuskey et al., 2015; Caravaggio et al., 2016). This radiotracer was often compared to [11C]raclopride, but it is unclear if the differences between the two radiotracers are due to the agonistic properties of [11C]-(+)-PHNO or to its higher affinity for D3 receptors. For instance, [11C]-(+)-PHNO shows preferential uptake in the ventral striatum and globus pallidus, due to the high density of D3 receptors in these areas, whereas [11C]raclopride shows preferential uptake in the dorsal striatum; [11C]-(+)-PHNO wash-out is also slower in the globus pallidus compared to the other regions (Graff-Guerrero et al., 2008). Similarly, a PET study in Parkinson’s disease showed a significant decrease in [11C]-(+)-PHNO levels in the globus pallidus, in contrast to [11C]raclopride, and an agonist/antagonist ratio that decreased proportionally to motor deficit and lowered mood, interpreted as the consequence of D3 receptor density modifications (Boileau et al., 2009). Searle et al. (2010) suggested that [11C]-(+)-PHNO uptake is due to high-affinity D2 receptors in the dorsal striatum, to high-affinity D2 receptors and D3 receptors in the ventral striatum, globus pallidus and thalamus, and only to D3 receptors in the substantia nigra). Numerous preclinical studies were also performed using [11C]-(+)-PHNO. Seeman et al. (2007) reported a 2-to-3-fold increase in high-affinity D2 receptors in rat following repeated injection of amphetamine, explaining why the animals were more sensitive to dopaminergic agonists. One year later, the sensitivity was compared between [11C]-(+)-PHNO and [3H]raclopride after various pharmacological challenges in conscious rats (McCormick et al., 2008). Similar degrees of inhibition were shown for both radiotracers following the pre-injection of amphetamine, cold NPA (a full agonist), aripiprazole (a partial agonist), haloperidol and clozapine (D2 antagonists). However, these results were contradicted by another study that showed greater ex vivo inhibition of [3H]PHNO binding by NPA than by [3H]raclopride, and greater displacement of the agonist radiotracer in amphetamine-sensitized rats (Kumar et al., 2006; Seeman, 2009). Contrasting results were published the same year by McCormick et al. (2009), underlining significant discrepancies in the field which require further study.
D4 Receptors
Although no convincing radiotracer specific to D4 receptors is presently available for PET neuroimaging in the human brain, the discovery of an inverse agonist was reported in Prante et al. (2008). The compound was derived from FAUC 113 and FAUC 213 and was more selective for D4 receptors than for D2 and D3 receptors. The [18F]-labeled molecule showed specific binding in rat brain in vitro, but no further investigations have yet been reported.
Serotonergic Receptors
Serotonin neurotransmission is characterized by a large number of subfamilies of receptors (14 are currently described). After the dopaminergic system, it is the second neurotransmission system to have benefited from the development of many PET radiotracers. While a significant number of them can be used in humans as radiopharmaceuticals, most are antagonists and currently very few agonists are available. However, the therapeutic potential of these many sub-families of receptors in psychiatry and neurology justifies further research in this area.
5-HT1A Receptors
From [18F]F15599 to [18F]F13640
Much effort has been made to develop a radiotracer agonist of 5-HT1A receptors, with initially limited success. These attempts included exploration of derivatives of various structures (analogs of 8-OH-DPAT and apomorphine, arylpiperazine, or thiochromine based ligands) which showed promising properties in vitro but were generally not suitable for in vivo imaging because of lack of evidence of specific binding (Thorell, 1995; Mathis et al., 1997; Suehiro et al., 1998; Hwang et al., 2001; Fujio et al., 2002; Zimmer et al., 2003; Vandecapelle et al., 2004). Lemoine et al. (2010), the well-characterized agonist F15599, initially seen as a drug candidate, was radiolabeled with [18F] and explored by PET imaging in rats and cats. It showed high affinity (Ki = 2.2 nM) and excellent specificity for 5-HT1A receptors, acting as a full agonist both in vitro and in pharmacological tests in rats, with preferential activity at post-synaptic receptors (Newman-Tancredi, 2011). Despite encouraging in vitro results, the signal-to-noise ratio was insufficient for PET imaging. Its structural analog, [18F]F13714, displaying higher affinity for 5-HT1A receptors (Ki = 0.1 nM), was also evaluated (Lemoine et al., 2012). Despite a better SNR and evidence of binding to 5-HT1A receptors in vivo, [18F]F13714 binding was irreversible in rat, cat and rhesus monkey, which suggests it would be difficult to quantify binding parameters in humans. Interestingly, [18F]F13714 was also compared with the antagonist [18F]MPPF in conscious and anesthetized marmosets; it displayed a markedly different distribution pattern from [18F]MPPF, with highest uptake in raphe and cortical areas, as opposed to hippocampus and amygdala for the antagonist radiotracer. It also showed region-specific uptake changes in isoflurane-anesthetized animals, contrary to [18F]MPPF for which global increase throughout the brain was observed (Yokoyama et al., 2016). Top design a radiotracer that would be easier to quantify, the structural analog [18F]F13640 was recently evaluated (Vidal et al., 2018). F13640 also exhibits high selectivity for 5-HT1A, but intermediate affinity (Ki = 1 nM) compared to the previous two attempts. [18F]F13640 showed specific binding to 5-HT1A receptors and agonistic properties in vitro and in vivo in rats, cats and rhesus monkeys, despite a distribution pattern contrasting with antagonist radiotracers (and similar to that of [18F]F13714). Moreover, ex vivo autoradiography experiments using pharmacological challenge with d-fenfluramine in rats suggested that [18F]F13640 is far more sensitive to competition with endogenous serotonin than the antagonist [18F]MPPF. Despite a slow washout, tracer binding is reversible, with increased washout after administration of fenfluramine, a serotonin releaser, during scanning in rats and cats (unpublished data). An autoradiographic study performed on postmortem hippocampus slices from AD patients at different Braak stages also demonstrated a decrease in [18F]F13640 binding in the CA1 area, occurring earlier than the decrease in [18F]MPPF binding in AD subjects. Further studies are ongoing to characterize the properties of [18F]F13640, and a first-in-man study is currently underway (clinicaltrials.gov number: NCT03347331). The first images in human showed a binding pattern different from that seen with the conventional antagonist 5-HT1A radiopharmaceutical [18F]-MPPF (Colom et al., 2019).
5-HT2A Receptors
From [11C]Cimbi5 to [11C]Cimbi36
Ettrup et al. (2010) reported [11C]-labeling and evaluation of the N-benzyl phenylethylamine derivative Cimbi-5, previously described as a selective and very potent agonist for 5-HT2A receptors (Braden et al., 2006). [11C]Cimbi-5 distribution was consistent with the known 5-HT2A distribution, and it was blocked by the antagonist ketanserin in pig brain (Ettrup et al., 2010). In an attempt to optimize the target-to-background binding ratio, the same team evaluated 9 other phenylethylamine analogs of [11C]Cimbi-5 in pig (Ettrup et al., 2011). The analog [11C]Cimbi-36 was identified as the most promising candidate for PET imaging of 5-HT2A receptors as it showed the highest target-to-background binding ratio and was displaceable by ketanserin. In vitro studies confirmed that Cimbi-36 was a potent and selective 5-HT2A agonist (Ki = 1 nM, ED50 = 0.5 nM). Its properties were then characterized in non-human primate and compared with the antagonist [11C]MDL-100907 (Finnema et al., 2014). [11C]Cimbi-36 distribution was again consistent with the known 5-HT2A distribution and blocked by ketanserin in all brain regions except the cerebellum, which was found to be a suitable reference region. Binding potential was approximately half that of [11C]MDL-100907 across cortical regions but higher in other brain regions such as the choroid plexus, which was found to be related to 5-HT2C receptor binding as it was blocked by the 5-HT2C ligand SB 242084. The authors concluded that [11C]Cimbi-36 was an agonist radioligand suitable for examination of 5-HT2A receptors in cortical regions and of 5-HT2C receptors in the choroid plexus of the primate brain. The first-in-man study was performed in 29 healthy volunteers, with arterial input measurement and pretreatment with ketanserin in 5 subjects (Ettrup et al., 2014). The authors concluded that [11C]Cimbi-36 was a suitable agonist radioligand for PET imaging of high-affinity 5-HT2A receptors in the cortex, and that cerebellum was an appropriate reference tissue for quantification without blood sampling. Recently, test–retest reproducibility was investigated and the distribution was compared to the antagonist [18F]altanserin in humans (Ettrup et al., 2016). The results showed excellent test–retest reproducibility and a high correlation between the two radiotracers, Cimbi-36 and altanserin, except in regions with high 5-HT2C receptor density (choroid plexus and hippocampus), where [11C]Cimbi-36 binding is probably to both 5-HT2A and 5-HT2C receptors. Sensitivity in detecting changes in endogenous 5-HT levels was also explored in pig brain using various pharmacological challenges, by simultaneous measurement of extracellular 5-HT concentration with microdialysis and PET imaging (Jorgensen et al., 2017). There was a significant correlation between 5-HT levels and 5-HT2A occupancy, indicating that [11C]Cimbi-36 is sensitive to competition with serotonin, although only at sufficiently high release. Another study in rhesus monkey demonstrated significant decrease in [11C]Cimbi-36 binding in most brain regions following administration of fenfluramine at 5 mg/kg (Yang et al., 2017). The tracer was found to be more sensitive to 5-HT release than the antagonist [11C]MDL 100907, and with sensitivity comparable to [11C]AZ10419369, a 5-HT1B partial agonist currently considered to be one of the most sensitive radioligands.
Finally, a study in humans recently compared two positions of [11C]-labeling for the tracer and concluded that the position initially chosen in the previous studies produced a higher signal-to-noise ratio (Johansen et al., 2018).
Cannabinoid Receptors
The endocannabinoid system is more recent in discovery compared to previous monoaminergic systems. This neurotransmission system seems to play key modulatory roles in the brain and much effort has been made to try to understand precisely its pathophysiological role in various behavioral and neurological diseases. While there are currently two known subtypes of cannabinoid receptors, termed CB1 and CB2, only the first have benefited from the development of agonist radiotracers.
CB1 Receptors
[11C]JHU75528 or [11C]OMAR
Fan et al. (2006) reported the synthesis of [11C]JHU75528, to image CB1 receptors. The tracer showed higher in vitro affinity and lower lipophilicity than the two prototypical CB1 agonists, rimonaban and AM281). Autoradiography studies in mice and PET studies in baboon showed high cerebral uptake, good SNR and specific binding displaced by the cold ligand or rimonaban pretreatment. Metabolite analysis demonstrated that a few fractions cross the BBB (Horti et al., 2006). The first clinical assays on humans confirmed a good CB1 receptor quantification (Horti, 2007; Wong et al., 2008). In terms of quantification, plasma reference graph analysis (Logan et al., 1990) was found to be more reliable than the two-compartmental model for estimating Vt. It was not possible to use pons or white matter as reference regions, due to small size and lack of favorable kinetics, respectively (Wong et al., 2010). Comparison between healthy volunteers and schizophrenic patients found elevated values in patients, especially in the pons region. This preliminary study showed the potential of Vt values to prove CB1 involvement in schizophrenia and to predict the type and severity of clinical symptoms. Gao et al. (2012) proposed a new synthesis route for [11C]OMAR and analogs, and Normandin explored more precisely the quantification of the tracer in humans (Normandin et al., 2015). They found that multilinear analysis was the most robust method. Test–retest reproducibility was satisfactory. There were significant sex differences in tracer properties, and especially in metabolism and brain uptake. These findings suggest that [11C]OMAR is a reliable radiotracer and that, in this case, gender differences must be considered in PET analysis.
[18F]MK-9470
Continuing previous efforts to develop a CB1 receptor radiotracer, Burns et al. (2007) developed [18F]MK-9470, a specific inverse agonist, in a context of drug development. The in vitro affinity of MK-9470 was 0.7 nM with a 60-fold selectivity for CB1 receptors in comparison with CB2 receptors. Autoradiography studies on rhesus brain slices showed a signal consistent with CB1 receptor distribution. PET studies in monkeys showed rapid uptake with displaceable binding by the specific inverse agonist MK-0364. In vivo PET study in humans showed slow kinetics with high uptake in striatum, frontal cortex and posterior cingulate. Metabolite analysis and test–retest reproducibility were satisfactory enough to envisage [18F]MK-9470 as a suitable radiotracer to explore CB1 receptor density. These findings were applied to determine the occupancy of the inverse agonist MK-0364. One year later, a biodistribution and radiation dosimetry study demonstrated acceptable dose exposure and the feasibility of multiple scans (Van Laere et al., 2008b). The tracer was also used to assess gender differences in CB1 receptor distribution and changes in receptor expression with healthy aging (Van Laere et al., 2008a). Several clinical studies were then performed, including a drug occupancy study of the CB1 receptor inverse agonist taranabant (Addy et al., 2008), and studies exploring the relationship between CB1 receptors and personality traits (Van Laere et al., 2009), temporal lobe epilepsy (Goffin et al., 2011), Parkinson’s disease (Van Laere et al., 2012), eating disorder (Gérard et al., 2011; Ceccarini et al., 2016), migraine (Van der Schueren et al., 2012), schizophrenia (Ceccarini et al., 2013), AD (Ahmad et al., 2014), chronic cannabis use (Ceccarini et al., 2015), alcohol abuse (Ceccarini et al., 2014), and Huntington’s disease (Ceccarini et al., 2019). The tracer was also used in numerous preclinical studies (Goffin et al., 2008; Casteels et al., 2010a,b,c,d, 2011, 2014; Gérard et al., 2010; Ooms et al., 2014; Cleeren et al., 2018). Several studies were also performed to optimize quantification in humans (Sanabria-Bohórquez et al., 2010) and rats (Casteels et al., 2012; Miederer et al., 2018) and radiosynthesis (Thomae et al., 2014).
Muscarinic Receptors
Muscarinic acetylcholine neurotransmission have been described for several decades and its interest has resurfaced more recently because of their implications in neurodegenerative diseases, justifying research work in PET neuroimaging. Five subtypes of muscarinic receptors have been determined, named M1–M5. Among them, only the M2 family has benefited from the development of agonist PET radiotracers used in humans.
M2 Receptors
[18F]FP-TZTP
In this context, Sauerberg et al. (1992) developed a series of muscarinic agonists containing a thiadiazolyl moiety. Based on these data, three fluorinated derivatives of TZTP were evaluated in vitro for their affinity toward the various muscarinic receptors (Kiesewetter et al., 1995). The derivative FP-TZTP displayed higher affinity for M2 than M1 receptors (Ki = 2.2 vs. 7.4 nM) and was radiolabeled with [18F] and further evaluated in rat. [18F]FP-TZTP displayed specific binding, being dose-dependently blocked by the analog P-TZTP, also a M2-preferring agonist, but only partially blocked by antagonists of M1 or M2 receptors., [18F]FP-TZTP was then evaluated in preclinical in vitro and in vivo studies in rats and monkeys: [18F]FP-TZTP showed specific binding to M2 receptors, significantly inhibited by cold compound and L-687,306, a muscarinic agonist. Uptake was significant in cortical and subcortical regions, with low uptake in cerebellum. Metabolism study in rats showed no significant presence of radiometabolites in the brain (Kiesewetter et al., 1999). The 1-compartmental model was the most reliable for determining distribution volume in rhesus monkey (Carson et al., 1998). Finally, [18F]FP-TZTP was sensitive to variations in ACh levels induced by physostigmine, an AChEI. Kiesewetter et al. (2003) reported 1-step automated radiosynthesis of [18F]FP-TZTP, and Ma et al. (2003) described a method using liquid–liquid PE and solid phase extraction to rapidly measure concentrations of tracer and metabolites thanks to the previous identification of the metabolite structures by LC-MS-MS (Ma et al., 2002).
The first-in-man study was performed in Podruchny et al. (2003) on healthy young and older volunteers. The binding pattern of [18F]FP-TZTP was consistent with the known M2 receptor distribution. Older subjects had significantly greater distribution volumes, which was explained by lower synaptic acetylcholine concentrations. Jagoda et al. (2003) used different models of KO mice to confirm the M2 selectivity of [18F]FP-TZTP, demonstrating a significant decrease in binding (from 51 to 61%) only in M2R KO mice, almost none in M1R KO mice (about 20% in amygdala and hippocampus), and none in M3R and M4R KO mice. Considering the fact that P-TZTP and the cold agonist FP-TZTP used in competition studies could produce changes in cerebral blood flow, decreasing the PET signal by reduced tracer delivery rather than by competition for receptors, Shimoji et al. (2003) showed that inhibition of [18F]FP-TZTP by these compounds was not due to agonist-induced reduction in CBF: the degree of tracer uptake inhibition was unchanged when a peripheral muscarinic antagonist was combined with muscarinic agonists to prevent the CBF changes induced by agonists alone. In a new clinical study, [18F]FP-TZTP was used to compare two populations of healthy subjects with and without apolipoprotein E-epsilon 4 allele, which is associated with increased susceptibility to AD and age-related memory problems (Cohen et al., 2003). APOE-epsilon4+ subjects had greater distribution volumes in gray matter than APOE-epsilon4- subjects, which was again interpreted in terms of synaptic acetylcholine concentration differences. [18F]FP-TZTP was then used to understand the cholinergic contribution to the emotional and sensory effects of procaine. Procaine dose-dependently decreased [18F]FP-TZTP specific binding (Benson et al., 2004). Following the 2003 clinical study, a new study was performed to evaluate the influence of age and APOE-epsilon4 genotype on the increase in acetylcholine concentration induced by physostigmine infusion and the distribution volumes of [18F]FP-TZTP (Cohen et al., 2006). It was also demonstrated that physostigmine induced a decrease in [18F]FP-TZTP uptake, and that both age and APOE-e4 genotype influenced the modulation of PET signal by physostigmine infusion. Furthermore, [18F]FP-TZTP was used to demonstrate the involvement of M2 receptors in mood disorders: there was decreased binding in patients suffering from bipolar disorder, which could be due to a reduction in M2 receptor density or affinity, or to an increase in endogenous acetylcholine levels (Cannon et al., 2006). van Oosten et al. (2009) reported an optimized radiosynthesis using a new precursor. Cannon et al. (2011), another clinical study was performed combining [18F]FP-TZTP-PET and genetic analyses: it was shown that single nucleotide polymorphisms for the M2R gene were associated with changes in [18F]FP-TZTP binding in bipolar disorder patients. Finally, Ravasi et al. (2012) found that constant infusion of [18F]FP-TZTP was better than bolus injection for performing microPET in rodents: blood clearance and metabolism were too rapid to measure a reproducible input function after bolus injection.
Histaminergic Receptors
There are four known histamine receptors, H1, H2, H3, and H4. The first imaging works focused on the H1 receptor but without the development of PET agonists. More recently, H3 receptor, a target with emerging pathophysiological implications, has led to the development of agonist radiotracers.
H3 Receptors
The H3 receptor has a presynaptic location and is involved in the regulation of histamine neurotransmission and modulation of release of other neurotransmitters (Van Laere et al., 2013). Thus, it has been demonstrated that, instead of only interfering with the negative feedback loop of histamine like an antagonist, H3 inverse agonists potentialize histaminergic neurotransmission by decreasing constitutive H3 signaling. These pharmacological properties suggest new treatments for various psychiatric or neurodegenerative diseases. According to the two-state model of agonist action, inverse agonists may have higher affinity for the inactive state of the receptor (Leff et al., 1985; Berg and Clarke, 2018). Concomitant development of H3 receptor inverse agonist radiotracers therefore seems important for the development of new H3 inverse agonists as therapeutic agents, as such radiotracers may better reflect the population of receptors actually targeted by these new ligands.
Spiro-Isobenzofuranone Derivative: [11C]MK-8278
In this context, in Hamill et al. (2009) reported the radiosynthesis and evaluation of two promising inverse agonists, as shown by the inhibition of basal [35S]GTPgammaS binding to membrane homogenates expressing recombinant H3 receptor derived from a family of spiro-isobenzofuranone-based compounds (Jitsuoka et al., 2008). The study described a radiosynthesis with high specific activity and revealing appropriate in vitro autoradiographic distribution in rhesus monkey and human brain, and specific binding in PET experiments in rhesus monkey for both compounds, with greater brain uptake for [11C]MK-8278. Using a bolus plus infusion method and in vivo PET imaging with [11C]MK-8278 in rhesus monkeys, the authors also determined the occupancy of diverse H3 receptor inverse agonists in relation to their plasma concentration. Van Laere et al. (2014) confirmed the utility of [11C]MK-8278 as a specific radioligand to evaluate in vivo occupancy of new H3 inverse agonists in human brain.
They first described whole-body biodistribution and dosimetry in humans, and found that the effective dose was in the typical range of other [11C]-labeled radiopharmaceuticals. The binding parameters of [11C]MK-8278 were quantified using a metabolite-corrected arterial input function. 1TCM and SRTM methods, considering pons as a reference region, showed reproducible estimates of Vt and BPnd values, respectively. Finally, the authors determined the human pharmacological profile of two inverse agonists, MK-024 and MK-3134, taken orally at various doses 6 h prior the PET scan; they thus obtained the receptor occupancy of both compounds as a function of oral dose or plasma concentration, demonstrating the key role of [11C]MK-8278 for characterizing target engagement of H3 inverse agonists (by calculating RO as a function of plasma concentration).
Opioid Receptors
There are four major subtypes of opioid receptors named delta (δ), kappa (κ), mu (μ), and nociceptin receptors. Agonist radiotracers of opioid receptors development was mainly derived by radiolabeling of existing drugs and displayed extensive use to understand physiopathological mechanisms in various diseases.
μ Opioid Receptors
[11C]Carfentanil
Radiosynthesis of the very potent μ-opioid agonist [11C]carfentanil was reported in Dannals et al. (1985), quickly followed by a first PET study in humans and baboons (Frost et al., 1985). High radioactivity levels were found in the striatum and thalamus and low levels in the cerebellum and occipital cortex, consistent with the known regional density of μ receptors. [11C]carfentanil binding was also strongly reduced by pretreatment with the antagonist naloxone, confirming its high specificity and suitability as an opioid receptor agonist radiotracer. It was then used in a clinical PET study to demonstrate elevated μ receptor concentration in temporal lobe epilepsy (Frost et al., 1988). A multicompartmental analysis was performed to quantify the binding parameters of [11C]carfentanil in human brain (Frost et al., 1989), and an in vitro binding study with the tritiated molecule further demonstrated its selectivity for the μ receptor subtype in human and rat brain (Titeler et al., 1989).
Frost et al. (1990), the binding patterns of [11C]carfentanil and the antagonist [11C]diprenorphine were compared in humans, showing different regional distributions that were explained by the non-selectivity of diprenorphine for the different subtypes of opioid receptors. In addition, a study focusing on temporal epilepsy demonstrated significant changes in opioid receptors with [11C]carfentanil but not [11C]diprenorphine (Mayberg et al., 1991). Zubieta et al. (1996), a study demonstrated the involvement of the opioid system in addiction by showing that [11C]carfentanil binding was increased in cocaine-dependent subjects compared to healthy controls, and correlated positively with cocaine craving. Since then, a huge number of clinical PET studies of μ receptors have been performed with [11C]carfentanil, focusing on epilepsy (Madar et al., 1997), the menstrual cycle (Smith et al., 1998), gender and age differences (Zubieta et al., 1999), addiction (Zubieta et al., 2000; Bencherif et al., 2004; Gorelick et al., 2005, 2008; Heinz et al., 2005; Greenwald et al., 2007; Scott et al., 2007a; Weerts et al., 2008, 2011, 2014; Ghitza et al., 2010; Ray et al., 2011; Falcone et al., 2012; Minkowski et al., 2012; Mitchell et al., 2012; Wand et al., 2012; Kuwabara et al., 2014; Domino et al., 2015; Mick et al., 2016; Nuechterlein et al., 2016; Hermann et al., 2017; Majuri et al., 2017), eating disorders (Bencherif et al., 2005; Karlsson et al., 2015, 2016; Tuominen et al., 2015; Joutsa et al., 2018), PTSD (Liberzon et al., 2007), major depression (Kennedy et al., 2006; Prossin et al., 2011; Hsu et al., 2015; Peciña et al., 2015a; Light et al., 2017) pain (Bencherif et al., 2002; Scott et al., 2007b, 2008; Wager et al., 2007; Harris et al., 2009; DosSantos et al., 2012; Hagelberg et al., 2012; Campbell et al., 2013; Martikainen et al., 2013; DaSilva et al., 2014a, b; Peciña et al., 2015b; Karjalainen et al., 2017) and behavior or emotions (Hsu et al., 2013; Mitchell et al., 2013; Nummenmaa et al., 2015, 2018; Karjalainen et al., 2016; Manninen et al., 2017; Tuulari et al., 2017; Saanijoki et al., 2018). [11C]carfentanil was used to measure the receptor occupancy of buprenorphin in heroin-dependent subjects (Zubieta et al., 2000; Greenwald et al., 2007), and of nalmefene in healthy subjects (Ingman et al., 2005). A multimodal study also evaluated μ receptor occupancy by the opioid receptor antagonist naltrexone and the inverse agonist GSK1521498 in relation with the modulation of the fMRI response to a food stimulus (Rabiner et al., 2011). [11C]carfentanil appeared to be sensitive to endogenous opioid fluctuations in studies showing decreased binding potential during somatic pain (Bencherif et al., 2002; Scott et al., 2007b), after placebo administration (Zubieta et al., 2005; Scott et al., 2008) and after pharmacological challenge associated with release of opioid peptides (Colasanti et al., 2012).
[18F]-labeled derivatives of carfentanil, [18F]fluoro-pentyl carfentanil and the analog sufentanil, [18F]fluoro-propyl-sufentanil were developed by Henriksen et al. (2005a). Both compounds had nanomolar affinity for μ-opioid human receptors, and their distribution in rat brain slices was consistent with μ-opioid receptor expression. The derivative of sufentanil produced almost no radioactive metabolites in mouse brain (Henriksen et al., 2005b). However, no further results have yet been reported.
κ Opioid Receptors
[11C]GR89696 and [11C]GR103545
[11C]GR89696, a racemate that is an antagonist of κ1 receptors and agonist of κ2 receptors, was synthesized and evaluated in mice in Ravert et al. (1999). Uptake was in good agreement with known kappa opioid receptor distribution and was inhibited by kappa opioid-selective drugs. The R and S enantiomers of [11C]GR89696 were later characterized separately, showing that only the R enantiomer [11C]GR103545 exhibited selective and saturable binding to kappa receptors (Ravert et al., 2002). [11C]GR103545 regional binding patterns in baboon brain were also consistent with the established distribution of kappa receptors, and binding was blocked by naloxone pretreatment (Talbot et al., 2005). Another study showed that [11C]GR103545 also had high affinity for kappa receptors in humans in vitro (Ki = 0.02 nM) and in awake rhesus monkeys (Schoultz et al., 2010). Kd and Bmax were estimated using a Scatchard plot in a bolus/infusion protocol, in the same species (Tomasi et al., 2013).
The first-in-man study was performed in Naganawa et al. (2014) and showed the suitability of the tracer for imaging and quantifying kappa receptors in humans, although quantification of kinetic parameters can be difficult due to lack of a reference region and to slow kinetics. Recently, a pilot study of kappa opioid receptor binding in major depression was conducted, using [11C]GR103545 to compare distribution volumes between healthy volunteers and patients suffering from major depressive disorder; no significant differences were detected (Miller et al., 2018). The tracer was also used to investigate the effect of various ligands on the kappa opioid receptor in rodents (Placzek et al., 2015). First, the authors validated the use of [11C]GR103545 to measure drug occupancy at kappa receptors by showing that specific binding was blocked by pre-injection of GR89696 and the antagonists naloxone and LY2795050. Then, they showed that injections of the kappa receptor agonist salvinorin A 1 min before the PET scan induced a dose-dependent decrease in [11C]GR103545 binding potential. At sufficiently high dose, this decrease persisted up to 2.5h after administration, although the half-life of salvinorin A is only few minutes, suggesting an agonist-induced adaptive response by kappa receptors. The same authors demonstrated that, although the agonist [11C]GR103545 and the antagonist [11C]LY2459989 have similar distribution patterns in rat brain, they differed in sensitivity to competition with various kappa receptor ligands (Placzek et al., 2018): the binding potential of both tracers was reduced to a similar extent by pre-injection of the opioid receptor antagonists naloxone and naltrexone, and the selective kappa receptor antagonist LY2795050, whereas other kappa antagonists blocked [11C]GR103545 binding more effectively (Bruchas et al., 2007). Finally, the kappa agonists butorphan and GR89696 showed comparable impact on the binding potentials of [11C]GR103545 and [11C]LY2459989, whereas the other agonists, salvinorin A and U-50488, significantly decreased [11C]GR103545 uptake and had no effect on [11C]LY2459989 (Placzek et al., 2018). The authors explained these findings by a likely different conformation recognized by LY2459989, as the mutation of the residue D138 dramatically decreased the affinity of LY2459989 and not GR103545 for kappa opioid receptors.
Sigma Receptors
Initially considered as part of opioid receptors, pharmacological properties of sigma receptors identified them as a specific family of receptors. Two subfamilies of sigma receptors are currently identified, σ1 and σ2 receptors. If the role of σ1 receptors is not well-defined, potential therapeutic applications emerge in experimental neurology, justifying the research of PET agonist radiotracers.
σ1 Receptors
[11C]SA4503
Kawamura et al. (1999) reported [11C]-radiolabeling and evaluation of SA6298, a selective σ1 receptor agonist. The compound showed high brain uptake in vivo in rats and 1 cat, but the signal was mostly non-specific. The same team evaluated the analog [11C]SA4503, which has slightly lower affinity but better specificity for σ1 receptors, with more encouraging results (Kawamura et al., 2000): there was high specific uptake in rat brain in vivo, as shown by blocking studies which decreased the signal proportionally to the σ1 affinity of the various ligands. Moreover, no radiolabeled metabolites were found in the brain. [11C]SA4503 binding in mouse and cat brain was also highly specific (Kawamura et al., 2000). Further experiments in conscious monkeys confirmed it as a promising radiotracer (Ishiwata et al., 2001). Although uptake increased continuously during control scans, tracer binding was displaced by haloperidol, which has high affinity for σ1 receptors. [11C]SA4503 was then used to investigate the time-course occupancy of σ1 receptors by haloperidol in mice (Ishiwata et al., 2003) and humans (Ishiwata et al., 2006), and its tritiated analog was used to measure age-related changes in σ1 receptor expression in rat brain in vitro (Kawamura et al., 2003a). The density of σ1 receptors significantly increased with age, a finding that was confirmed in a PET study in monkeys (Kawamura et al., 2003b), but not in rat brain in vivo by Ramakrishnan et al. (2015), who showed a decrease in BP in several brain regions in aged rats.
Mishina et al. (2005), a clinical study compared [11C]SA4503 binding in healthy volunteers and Parkinson’s disease patients, and found no difference between controls and patients but a significant reduction in BP in the more injured side of the anterior putamen in patients, as assessed by [11C]CFT binding. Quantitative analysis of σ1 receptors in the human brain using [11C]SA4503 was reported in Sakata et al. (2007). Another study was performed in AD patients: compared to elderly volunteers, AD patients had lower BP in the cortex and cerebellum (Mishina et al., 2008). The high occupancy of σ1 receptors by the SSRI fluvoxamine and not by paroxetine (Ishikawa et al., 2007) and high occupancy by the AChEI donepezil (Ishikawa et al., 2009) were demonstrated in living human brain at therapeutic doses. Several fluorinated analogs of [11C]SA4503 were synthesized and evaluated, including [18F]FE-SA4503 (Elsinga et al., 2002, 2004), which is non-selective for the different subtypes of sigma receptor, [18F]FE-SA5845, less favorable in terms of kinetics, and [18F]FM-SA4503, which showed high specific binding and is more selective of σ1 receptors (Kawamura et al., 2007).
What Is Different With Antagonist Radiotracers?
Here, we propose to discuss the in vivo differences between agonist and antagonist radiotracers. The initial concept supporting the use of agonists as radiotracers is based on their preference for the high-affinity state of GPCR receptors as opposed to the total population of receptors, as observed in vitro. This concept should be associated to obvious differences between agonists and antagonists, such as differential sensitivity to competition with various ligands, to pharmacological alterations of G-protein/receptor coupling and to pathological alterations. Although the issue is likely to be much more complex in vivo, and it has proved difficult to demonstrate the existence of different coupling states of GPCR receptors in living organisms, a number of studies did highlight the above-mentioned differences. In addition, some data even suggest distinct brain distribution patterns between agonist and antagonist radiotracers for certain GPCR.
In vivo Binding of Agonists Versus Antagonists
According to in vitro data, agonist radiotracers are expected to display lower specific binding and available receptor density than reference antagonist radiotracers. Some studies directly compared the BP (Bmax/Kd) of an agonist and an antagonist radiotracer specific for the same target in the same subjects. For instance, Kodaka et al. (2012) compared the binding potentials of the D2/D3 receptor radiotracers [11C]MNPA and [11C]raclopride in two humans, showing that the agonist’s binding potential was about four times lower than the antagonist’s. The BP ratio between the two radiotracers was highly reproducible on test–retest, and was suggested as a possible estimate of the proportion of receptors in high-affinity state as compared to overall D2/D3R density. A similar approach was used for 5-HT1A receptors, using the partial agonist [11C]CUMI-101 and the antagonist [11C]WAY-100635 in non-human primates (Kumar et al., 2012). The authors reported an average 45% lower binding potential for [11C]CUMI-101, with some regional variations (highest proportion of coupled receptors in the parahippocampal gyrus, and lowest in the amygdala and putamen). Another study in marmosets, comparing the full 5-HT1A agonist [18F]F13714 and the antagonist [18F]MPPF, showed that antagonist binding potential was approximatively threefold higher in 5-HT1AR-rich regions (such as hippocampus and amygdala), whereas in striatum and thalamus BPND levels were similar between the two tracers (Yokoyama et al., 2016). These regional variations in the proportion of 5-HT1AR in high-affinity state were even greater in conscious animals. Taken together, these studies advocate differential targeting of GPCR receptors by agonists, which display lower binding potential likely because they bind preferentially to high-affinity receptor states. Therefore, if both tracers are available for a given GPCR, the proportion of highly effective receptors can be determined as compared to overall receptor density, in physiological or pathological conditions. However, interpretation of the above results is limited by a number of factors.
Firstly, comparison of binding potentials reflects the differences in Bmax/Kd ratio rather than Bmax directly; although the affinity of radiotracers is classically known from in vitro binding studies, the actual in vivo affinity can differ significantly. Unfortunately, very few studies directly compared the in vivo density of receptors targeted by an agonist versus an antagonist radiotracer. Using Scatchard analyses of PET data in 2 cats, Ginovart et al. (2006a) estimated the Bmax of the agonist [11C]PHNO to be similar to that of [11C]raclopride, casting doubt on differential binding of agonist/antagonist radiotracers in vivo. On the other hand, the Bmax of [11C]NPA was shown to be about 79% of that of [11C]raclopride in baboon (Narendran, 2005).
Another problem in comparing agonists and antagonists is the selectivity of the compounds: it is rather common for them not to be fully selective for a given GPCR, complicating the interpretation of results. This is precisely the case concerning [11C]PHNO, which has higher affinity for the D3R receptor subtype than other dopaminergic radiotracers (Narendran et al., 2006). Consequently, most clinical findings using this radiotracer were interpreted in terms of D3R alterations rather than D2/D3R coupling state. Another example is the 5-HT2AR agonist radiotracer [11C]Cimbi-36, which displayed lower binding than the antagonist [11C]MDL-100907 in cortical regions in rhesus monkey, but not in the hippocampus or choroid plexus, due to significant binding to 5-HT2C receptors (Finnema et al., 2014). In human brain, [11C]Cimbi-36 provided BPs that were comparable to (in cortical regions) or higher than (in 5-HT2CR-rich regions) the antagonist [18F]altanserin (Ettrup et al., 2016). Estimated Bavail values (knowing the plasma protein binding of each tracer and the in vitro affinities of each ligand) were also similar. Finally, the partial μ opioid receptor agonist [11C]carfentanil and the antagonist [11C]diprenorphine were also compared in baboons (Shiue et al., 1991) and humans (Frost et al., 1990): the greater uptake of [11C]diprenorphine in the striatum or cingulate cortex was explained by its significant affinity for other opioid receptor subtypes or different kinetic properties compared to [11C]carfentanil. In this regard, it is obvious that direct comparison of agonist and antagonist radiotracers can also be hindered by large differences in the kinetic parameters K1, k2, k3 and k4, especially as different modeling approaches may be needed to quantify BP.
Considering the existence of two affinity sites for the agonist, the kinetics of displacement by endogenous neurotransmitters or exogenous drugs differs between agonist and antagonist radiotracers. This is the case for [11C]raclopride and [11C]NPA, where quantitative autoradiography showed biphasic displacement for the agonist and monophasic displacement for the antagonist with increasing concentration of LSD (Minuzzi and Cumming, 2010). This phenomenon introduces another degree of complexity in comparing antagonist and agonist displacement in pharmacological challenge. Furthermore, it was demonstrated that activated 5-HT1A receptors induced a specific dynamics on the cell surface in vivo, which can modify in vivo receptor distribution (Pucadyil and Chattopadhyay, 2007). This could explain the difference between agonist and antagonist radiotracers and also the frequent discrepancies between in vitro and in vivo data.
As the occupancy of a GPCR by its specific endogenous neurotransmitter is expected to be greater in the high- than in the low-affinity state, the estimated Bavail value for an agonist may be closer to the Bavail value for an antagonist than the theoretical Bmax values, which adds another level of complexity in comparing agonists versus antagonists. Therefore, considering the number of parameters that influence radiotracer binding quantification in vivo, it seems reasonable to conclude that it will generally be difficult to calculate directly the ratio of coupled receptors to total receptors density reliably enough to provide meaningful pathophysiological information. Likewise, simply comparing binding potentials or even Bavail between agonist and antagonist radiotracers in physiological conditions is unlikely to answer the question of the actual existence of a high-affinity GPCR state in vivo.
Greater Sensitivity to Neurotransmitter Release?
The dopamine system is the system most widely explored in terms of neurotransmitter release monitoring using PET (Finnema et al., 2015). Several PET radioligands of D2/D3 receptors are sensitive to dopamine release, such as the benzamide derivative [11C]raclopride, an antagonist that has been extensively used to evaluate changes in dopamine release in the striatum, providing new insights into the role of dopamine in pathological and physiological conditions. Other antagonists with higher D2 affinity, such as [11C]FLB-457 and [18F]fallypride, have been used to monitor extracellular dopamine fluctuations in extrastriatal regions where the density of D2 receptors is lower. In theory, the sensitivity of these radioligands is limited by the fact that endogenous dopamine preferentially targets the high-affinity state of D2/D3 receptors, which is only a part of total receptor density as measured by antagonist radiotracers (Laruelle, 2000). It was demonstrated that agonist radiotracers of D2/D3 receptors such as [11C]MNPA, [11C]NPA and [11C]PHNO were even more sensitive to DA release, both in animals (Ginovart et al., 2006a; Gallezot et al., 2014 for [11C]PHNO; Narendran et al., 2004 for [11C]NPA; Seneca et al., 2006; Skinbjerg et al., 2010 for [11C]MNPA and humans Narendran et al., 2010; Shotbolt et al., 2012; Caravaggio et al., 2014). These experiments determined the proportion of high- and low-affinity states of D2/D3 receptors by means of amphetamine challenge or Scatchard analysis (Table 1).
Of the numerous attempts to develop a radiotracer sensitive to serotonin release, only a few experiments with [11C]CUMI-101 (Milak et al., 2011) ([18F]F13640 (Vidal et al., 2018) and [11C]Cimbi36 (Jorgensen et al., 2017; Yang et al., 2017) showed sensitivity. These difficulties suggest notable differences between dopamine and serotonin competition systems: degree of receptor availability, proportion of high-affinity state receptors, and size of the accessible receptor pool (Paterson et al., 2010). However, agonist radiotracers seem to be more appropriate than antagonist radiotracers to evaluate neurotransmitter release. For example, the literature does not report significant sensitivity for the 5-HT1A receptor antagonist [18F]MPPF, but only then in the case of a huge release of serotonin (Zimmer et al., 2002; Rbah et al., 2003), suggesting a small proportion of coupled receptors in basal state (Udo de Haes et al., 2006). Higher sensitivity to neurotransmitter release than for antagonist radiotracers was also suggested in vivo for [11C]GR103545 (for complete details, see Table 1). However, in pharmacological challenge paradigms, many agonist radiotracers lack direct comparison with antagonist radiotracers.
Finally, the effect of anesthesia has to be taken into account, particularly in preclinical studies. As observed by several teams, anesthesia is also responsible for changes in cerebral blood flow, receptor affinity and, finally, neurotransmission (Tsukada et al., 2002; Hassoun et al., 2003; Yokoyama et al., 2016). More precisely, it is also known to affect GPCR coupling (Seeman and Kapur, 2003). In vivo experiments on conscious subjects are consequently recommended, but assessment of such a protocol is not always possible in animals. Quick translation to human experiments is therefore desirable. Further investigation must be envisaged to explore the in vivo behavior of agonist and antagonist radioligands. This will certainly affect the current ligand-receptor paradigm.
The Concept of Internalization
Internalization is a phenomenon that is induced by agonist stimulation. Briefly, variations in the neurotransmitter, especially increasing levels in the synapse, can influence receptor crossing from cell surface to intracellular compartment. This has been demonstrated for dopamine, serotonin (Riad et al., 2001), muscarinic (Keith et al., 1998) mu opioid receptors (Quelch et al., 2014), and α2 receptors (Olli-Lähdesmäki et al., 1999). This adaptive process can interfere with the binding of agonist radiotracers, especially in pharmacological challenge, which induces a massive release of neurotransmitter into the synapse. Thus, the observed decrease in binding following pharmacological challenge could be due to internalization more than to direct competition (Zimmer et al., 2004; Ginovart, 2005). Consequently, the level of lipophilicity could explain differences in ligand binding: lipophilic radioligands bind both free and sequestered receptors, whereas hydrophilic ligands bind receptors only at the cell surface (Aznavour et al., 2006).
For example, it has be proven that, after amphetamine challenge, the acute effect of neurotransmitter release is responsible for a large decrease in the levels of both types of radiotracer (Narendran et al., 2004; Ginovart et al., 2006a; Seneca et al., 2006). In the case of [11C]MNPA and [18F]fallypride, an antagonist, amplitude was greater for the agonist. On a short time-scale, the phenomenon of displacement was dominant; then, on a longer scale, internalization caused incomplete recovery of both radiotracers (Skinbjerg et al., 2010). However, [18F]fallypride is known to bind internalized receptors with affinity twofold lower than free receptors. Consequently, [18F]fallypride was also affected by internalization. The process of internalization remains unclear: Narendran et al. (2004) found no difference between NPA and raclopride in recovery time after amphetamine challenge.
In the case of 5-HT2A receptors, Ettrup et al. (2010) found no differences in binding between the agonist [11C]Cimbi36 and the antagonist [18F]altanserin. They found a correlation between BPnd for both radiotracers. Bavail was almost the same (164 for the agonist and 173 for the antagonist). But these results were not in agreement with in vitro data suggesting internalization of 80% of receptors.
The Case of Biased Agonism
G protein-coupled receptors display two different states, an active state (coupled receptor) and an inactive state (non-coupled receptor), and it was demonstrated in vitro that a given receptor may be coupled to different subtypes of G protein (Offermanns et al., 1994; Laugwitz et al., 1996) depending on its location in the brain (Jin et al., 2001). In this context, studies demonstrated that an agonist can selectively trigger a single transduction pathway among the numerous transduction pathways of GPCR (Berg and Clarke, 2006; Kenakin, 2015; Luttrell et al., 2015). Consequently, agonist ligands for a single target provide their own functional signature by selecting a specific transduction pathway. This biased agonism is related to allosteric modification of the receptor defined by multiple conformations, each depending on ligand interaction with signaling proteins (Berg and Clarke, 2006; Kenakin and Christopoulos, 2012). In this context, Yokoyama et al. (2016) compared agonist and antagonist radiotracers on 5-HT1A receptors in non-human primates. The binding of the biased agonist [18F]F13714 was not only lower than [18F]MPPF but very different. Although images revealed binding all consistent with 5-HT1A receptor distribution (cortical regions, amygdala, hypothalamus, and raphe nucleus), there were notable differences in intensity : e.g., lower in hippocampus and amygdala and higher in the cingulate and insular cortices for the agonist radiotracer. [18F]MPPF showed twofold higher binding in the hippocampus and amygdala. The authors attributed these differences to the biased agonism of F13714, interacting with specific G protein subtypes and targeting a specific brain region composed of presynaptic receptors: raphe striatum and thalamus. The notion of biased agonism was also recently explored by PET/MR imaging, and contributes to defining the existence of biased agonism on 5-HT1A receptors (Vidal et al., 2018). In the kappa-opioid receptor, differences in the dynamics of receptor structure induced by the agonist [11C]GR103545 versus the antagonists [11C]LY2795050 and [11C]LY2459989 were used to explain the in vivo discrepancies observed on PET imaging (Placzek et al., 2018). Biased agonism requires exploring different transduction pathways composed of a single receptor and is a key point in the hypothetical in vivo differences between agonist and antagonist radiotracers. In vitro data clearly show the existence of two different affinity states for GPCR, but there are still difficulties in demonstrating this on PET imaging, suggesting that there may be multiple receptor conformations rather than just two affinity sites. Going further, it was also demonstrated that some 5-HT2A antagonists are able to trigger arrestin pathways and induce paradoxical desensitization of GPCR (Gray and Roth, 2001).
What Is the Role of PET Agonists in Neuroimaging?
Improving the Measure of Endogenous Neurotransmitter Release
It is assumed from the in vitro data that antagonists are less sensitive to neurotransmitter release than agonists. Agonists bind only to high-affinity state receptors, whereas antagonists bind to both high- and low-affinity state receptors equally. Therefore, when initiating competition, the antagonist is not effectively involved (Paterson et al., 2010; Finnema et al., 2015). On the other hand, agonist radiotracers provide direct estimation of the target affinity of endogenous neurotransmitters (Narendran et al., 2004). Considering pharmacological findings suggesting that agonists are more sensitive to neurotransmitter release, it is of great interest to test this hypothesis in vivo. However, as seen before, it is difficult to demonstrate high sensitivity; experimental conditions are a determining factor. Measuring neurotransmitter release involves knowing the exact neurotransmitter level, by microdialysis. The endogenous levels are too low to be estimated at baseline with a radiotracer (Finnema et al., 2015), and it is necessary to perform pharmacological challenge to obtain huge neurotransmitter release. The development of modeling of neurotransmitter release contributes to understanding these mechanisms (Normandin et al., 2012).
Precision Pharmacology to Evaluate Neurologic Disorders and New Therapeutics
Agonist radiotracers are useful tools to develop new agonist drug candidates. In this context, it seems to be more appropriate to choose the same type of ligand when performing drug-occupancy studies. Also, in activation studies it is possible to visualize the impact of drug or task on the active population of receptors. Therefore, there is great interest in testing this clinically. Although this has not yet been formally demonstrated, it can be proposed that pathological conditions lead to a specific decoupling of GCPRs, leading to a functional deficit of neurotransmission. PET imaging by radiopharmaceutical agonists may enable precise definition of which GPCR pathways are damaged in CNS disorders and which pathways are still functional.
Firstly, it is possible to evaluate the difference in basal binding between control and pathologic conditions. Secondly, basal binding can contribute to therapeutic optimization of a drug and of early drug development by using the drug occupancy paradigm. However, for example, a study on cynomolgus monkeys found no differences in drug occupancy of apomorphine measured with [11C]raclopride or [11C]MNPA (Finnema et al., 2009). On the other hand, an ex vivo study showed that the D2 agonist NPA was more effective in detecting an increase in receptor availability in the early stages of Parkinson’s disease. Likewise, concerning 5-HT1A receptors in a postmortem study in Alzheimer patients, the radiolabeled agonist F13640 was more effective in detecting transient over-expression of 5-HT1A receptors, followed by functional decoupling of these same receptors, before a decrease in their total density at a later Braak stage (Vidal et al., 2016).
In therapeutics, it could be possible to stimulate only specific pathways with biased agonists, to offset GPCR damage in brain. Thus, it appears that agonist radiotracers could be useful for developing precision pharmacology (Schaffhausen, 2017), with numerous applications in pharmacotherapeutics. In any case, it is now recommended to confirm these hypotheses with in vivo PET studies.
Future Challenges and Conclusion
Agonist radiotracers provide many opportunities to decipher the ligand-GPCR paradigm in neuropharmacology. It is possible to pursue in vitro findings with an in vivo design. The concomitant use of antagonist and agonist radiotracers sheds light on complex phenomena such as reversible conversion of high- to low-affinity receptors and internalization. However, the existence of two receptor affinity states has not yet been clearly demonstrated in vivo (Finnema et al., 2010; Skinbjerg et al., 2012). There are discrepancies between in vitro and in vivo findings, certainly because the in vivo environment of the neuron is complex. Study conditions are also a determining factor. Anesthesia or changes in blood flow modify radiotracer binding. There is also great variability in the methods used to quantify high-affinity receptors in vitro and in vitro (Richfield et al., 1986; De Haes, 2005; Shalgunov, 2017). The notion of biased agonism introduces more complexity in molecular effects of agonists. A combination of PET and MRI may shed light on agonist functional activities (Vidal et al., 2018). Finally, there is a need to develop new tools to demonstrate in vivo coupling of GPCR. Indeed, the binding of an agonist radioligand involves intracellular molecular remodeling, which is probably not the case with the binding of a “silent” antagonist. These differences in docking justify that the brain images of agonists, and in particular their distribution patterns, are not directly comparable to those currently obtained by PET imaging of receptors using mainly antagonists. It is therefore necessary to develop new models for interpreting this molecular and functional imaging of receptors. The interpretation of the models will be based in particular on the implementation of PET imaging studies that will compare the binding patterns of an antagonistic radiotracer and an agonist radiotracer directed toward the same receptor in the same subject. Studies on animal models will also provide valuable information, in particular by pharmacologically modulating the G-protein coupling state of receptors during PET acquisitions. Finally, molecular modeling, whose bioinformatics tools are constantly evolving, will make it possible to simulate the docking specificity of agonist molecules in receptor molecular niches.
All these new tools will lead to new paradigms for neuroimaging which, in turn, will contribute to new advances in neurology and psychiatry.
Author Contributions
LZ initiated the research topic of the manuscript, proposed its initial plan and made its revision. MC and BV wrote the manuscript.
Funding
This work was performed within the framework of the LABEX PRIMES (ANR-11-LABX-0063) of Université de Lyon, within the program “Investissements d’Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2019.00255/full#supplementary-material
Abbreviations
μ Opioid Receptors, mu opioid receptors; mGlu1, metabotropic glutamate receptor 1; 11C, carbon-11; 18F, Fluor-18; 1TCM, one-tissue compartmental model; 2TCM, two-tissue compartmental model; 5-HT, serotonin; 5-HT1A, subtype 1A of serotonin receptors; 5-HT1B, subtype 1B of serotonin receptors; 5-HT2A, subtype 2A of serotonin receptors; 5-HT2C, subtype 2C of serotonin receptors; 5-HT4, subtype 4 of serotonin receptors; 5-HT7, subtype 7 of serotonin receptors; 6-OH-DA, 6 hydroxy-dopamine; A1 Receptor, subtype 1 of adenosine receptors; A2A Receptor, subtype 2A of adenosine receptors; Ach, acetylcholine; AChEI, acetylcholinesterase inhibitors; AD, Alzheimer’s disease; APOE-e4, apoenzyme 4; Bavail, available density of receptors; BBB, blood–brain barrier; Bmax, maximal density of receptors; BP, binding potential; BPND, non-displaceable binding potential; BPP, plasma binding potential; CB1, subtype 1 of cannabinoid receptors; CNS, central nervous system; D2, type 2 dopaminergic receptors; D3, type 3 dopaminergic receptors; Emax, maximal effect; GABAB, subtype B of GABA receptors; GPCRs, G-protein -coupled-receptors; GTP, guanosine triphosphate; H3 receptors, subtype 3 of histamine receptors; IC50, half-maximal inhibitory concentration; Kd/Ki, affinity of receptors; KO, knock-out; LSD, diethylamide lysergic acid; M1R, type 1 of muscarinic receptors; M2R, type 2 of muscarinic receptors; M3R, type 3 of muscarinic receptors; M4R, type 4 of muscarinic receptors; nor-BNI, norbinaltorphimine; PET/fMRI, Positron emission tomography/functional magnetic resonance imaging; PET, Positron emission tomography; PgP, P glycoprotein; SRTM, simplified tissue reference model; κ Opioid Receptors, kappa opioid receptors; σ1, sigma 1 receptors
References
Addy, C., Wright, H., Van Laere, K., Gantz, I., Erondu, N., Musser, B. J., et al. (2008). The Acyclic CB1R inverse agonist taranabant mediates weight loss by increasing energy expenditure and decreasing caloric intake. Cell Metab. 7, 68–78. doi: 10.1016/j.cmet.2007.11.012
Ahmad, R., Goffin, K., Van den Stock, J., De Winter, F.-L., Cleeren, E., Bormans, G., et al. (2014). In vivo type 1 cannabinoid receptor availability in Alzheimer’s disease. Eur. Neuropsychopharmacol. 24, 242–250. doi: 10.1016/j.euroneuro.2013.10.002
Albizu, L., Moreno, J. L., Gonzalez-Maeso, J., and Sealfon, S. C. (2010). Heteromerization of G protein-coupled receptors: relevance to neurological disorders and neurotherapeutics. CNS Neurol. Disord. Drug Targets 9, 636–650. doi: 10.2174/187152710793361586
Avissar, S., and Schreiber, G. (2006). The involvement of G proteins and regulators of receptor–G protein coupling in the pathophysiology, diagnosis and treatment of mood disorders. Clin. Chim. Acta 366, 37–47. doi: 10.1016/j.cca.2005.11.003
Aznavour, N., Rbah, L., Riad, M., Reilhac, A., Costes, N., Descarries, L., et al. (2006). A PET imaging study of 5-HT1A receptors in cat brain after acute and chronic fluoxetine treatment. Neuroimage 33, 834–842. doi: 10.1016/j.neuroimage.2006.08.012
Battaglia, G., Shannon, M., and Titeler, M. (1984). Guanyl nucleotide and divalent cation regulation of cortical S 2 serotonin receptors. J. Neurochem. 43, 1213–1219. doi: 10.1111/j.1471-4159.1984.tb05375.x
Becker, G., Streichenberger, N., Billard, T., Newman-Tancredi, A., and Zimmer, L. (2014). A postmortem study to compare agonist and antagonist 5-HT1A receptor-binding sites in Alzheimer’s disease. CNS Neurosci. Ther. 20, 930–934. doi: 10.1111/cns.12306
Bencherif, B., Fuchs, P., Sheth, R., Dannals, R., Campbell, J., and Frost, J. (2002). Pain activation of human supraspinal opioid pathways as demonstrated by [11C]carfentanil and positron emission tomography (PET). Pain 99, 589–598. doi: 10.1016/s0304-3959(02)00266-x
Bencherif, B., Guarda, A. S., Colantuoni, C., Ravert, H. T., Dannals, R. F., and Frost, J. J. (2005). Regional mu-opioid receptor binding in insular cortex is decreased in bulimia nervosa and correlates inversely with fasting behavior. J. Nucl. Med. 46, 1349–1351.
Bencherif, B., Wand, G. S., McCaul, M. E., Kim, Y. K., Ilgin, N., Dannals, R. F., et al. (2004). Mu-opioid receptor binding measured by [11C]carfentanil positron emission tomography is related to craving and mood in alcohol dependence. Biol. Psychiatry 55, 255–262. doi: 10.1016/j.biopsych.2003.07.007
Benson, B., Carson, R., Kiesewetter, D., Herscovitch, P., Eckelman, W., Post, R., et al. (2004). A potential cholinergic mechanism of procaine’s limbic activation. Neuropsychopharmacology 29, 1239–1250. doi: 10.1038/sj.npp.1300404
Berg, K. A., and Clarke, W. P. (2006). Development of functionally selective agonists as novel therapeutic agents. Drug Discov. Today Ther. Strateg. 3, 421–428. doi: 10.1016/j.ddstr.2006.10.017
Berg, K. A., and Clarke, W. P. (2018). Making sense of pharmacology: inverse agonism and functional selectivity. Int. J. Neuropsychopharmacol. 21, 962–977. doi: 10.1093/ijnp/pyy071
Björk, K., and Svenningsson, P. (2011). Modulation of monoamine receptors by adaptor proteins and lipid rafts: role in some effects of centrally acting drugs and therapeutic agents. Annu. Rev. Pharmacol. Toxicol. 51, 211–242. doi: 10.1146/annurev-pharmtox-010510-100520
Boileau, I., Guttman, M., Rusjan, P., Adams, J. R., Houle, S., Tong, J., et al. (2009). Decreased binding of the D3 dopamine receptor-preferring ligand [11C]-(+)-PHNO in drug-naive Parkinson’s disease. Brain 132, 1366–1375. doi: 10.1093/brain/awn337
Braden, M. R., Parrish, J. C., Naylor, J. C., and Nichols, D. E. (2006). Molecular interaction of serotonin 5-HT2A receptor residues Phe339(6.51) and Phe340(6.52) with superpotent N-Benzyl phenethylamine agonists. Mol. Pharmacol. 70, 1956–1964. doi: 10.1124/mol.106.028720
Bruchas, M. R., Yang, T., Schreiber, S., Defino, M., Kwan, S. C., Li, S., et al. (2007). Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J. Biol. Chem. 282, 29803–29811. doi: 10.1074/jbc.M705540200
Burns, H. D., Van Laere, K., Sanabria-Bohorquez, S., Hamill, T. G., Bormans, G., Eng, W.-S., et al. (2007). [18F]MK-9470, a positron emission tomography (PET) tracer for in vivo human PET brain imaging of the cannabinoid-1 receptor. Proc. Natl. Acad. Sci. U.S.A. 104, 9800–9805. doi: 10.1073/pnas.0703472104
Campbell, C. M., Bounds, S. C., Kuwabara, H., Edwards, R. R., Campbell, J. N., Haythornthwaite, J. A., et al. (2013). individual variation in sleep quality and duration is related to cerebral mu opioid receptor binding potential during tonic laboratory pain in healthy subjects. Pain Med. Malden Mass 14, 1882–1892. doi: 10.1111/pme.12231
Cannon, D., Carson, R., Nugent, A., Eckelman, W., Kiesewetter, D., Williams, J., et al. (2006). Reduced muscarinic type 2 receptor binding in subjects with bipolar disorder. Arch. Gen. Psychiatry 63:741. doi: 10.1001/archpsyc.63.7.741
Cannon, D. M., Klaver, J. K., Gandhi, S. K., Solorio, G., Peck, S. A., Erickson, K., et al. (2011). Genetic variation in cholinergic muscarinic-2 receptor gene modulates M2 receptor binding in vivo and accounts for reduced binding in bipolar disorder. Mol. Psychiatry 16, 407–418. doi: 10.1038/mp.2010.24
Caravaggio, F., Chung, J., Gerretsen, P., Fervaha, G., Nakajima, S., Plitman, E., et al. (2016). Exploring the relationship between social attachment and dopamine D 2/3 receptor availability in the brains of healthy humans using. Soc. Neurosci. 12, 163–173. doi: 10.1080/17470919.2016.1152997
Caravaggio, F., Nakajima, S., Borlido, C., Remington, G., Gerretsen, P., Wilson, A., et al. (2014). Estimating endogenous dopamine levels at D2 and D3 receptors in humans using the agonist radiotracer [(11)C]-(+)-PHNO. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 39, 2769–2776. doi: 10.1038/npp.2014.125
Carson, R. E., Kiesewetter, D. O., Jagoda, E., Der, M. G., Herscovitch, P., and Eckelman, W. C. (1998). Muscarinic cholinergic receptor measurements with [18F]FP-TZTP: control and competition studies. J. Cereb. Blood Flow Metab. 18, 1130–1142. doi: 10.1097/00004647-199810000-00010
Casteels, C., Bormans, G., and Van Laere, K. (2010a). The effect of anaesthesia on [(18)F]MK-9470 binding to the type 1 cannabinoid receptor in the rat brain. Eur. J. Nucl. Med. Mol. Imaging 37, 1164–1173. doi: 10.1007/s00259-010-1383-7
Casteels, C., Lauwers, E., Baitar, A., Bormans, G., Baekelandt, V., and Van Laere, K. (2010b). In vivo type 1 cannabinoid receptor mapping in the 6-hydroxydopamine lesion rat model of Parkinson’s disease. Brain Res. 1316, 153–162. doi: 10.1016/j.brainres.2009.12.026
Casteels, C., Martinez, E., Bormans, G., Camon, L., de Vera, N., Baekelandt, V., et al. (2010c). Type 1 cannabinoid receptor mapping with [18F]MK-9470 PET in the rat brain after quinolinic acid lesion: a comparison to dopamine receptors and glucose metabolism. Eur. J. Nucl. Med. Mol. Imaging 37, 2354–2363. doi: 10.1007/s00259-010-1574-2
Casteels, C., Vanbilloen, B., Vercammen, D., Bosier, B., Lambert, D. M., Bormans, G., et al. (2010d). Influence of chronic bromocriptine and levodopa administration on cerebral type 1 cannabinoid receptor binding. Synapse 64, 617–623. doi: 10.1002/syn.20769
Casteels, C., Gérard, N., van Kuyck, K., Pottel, L., Nuttin, B., Bormans, G., et al. (2014). Small animal PET imaging of the type 1 cannabinoid receptor in a rodent model for anorexia nervosa. Eur. J. Nucl. Med. Mol. Imaging 41, 308–321. doi: 10.1007/s00259-013-2522-8
Casteels, C., Koole, M., Celen, S., Bormans, G., and Van Laere, K. (2012). Preclinical evaluation and quantification of [18F]MK-9470 as a radioligand for PET imaging of the type 1 cannabinoid receptor in rat brain. Eur. J. Nucl. Med. Mol. Imaging 39, 1467–1477. doi: 10.1007/s00259-012-2163-3
Casteels, C., Vandeputte, C., Rangarajan, J. R., Dresselaers, T., Riess, O., Bormans, G., et al. (2011). Metabolic and type 1 cannabinoid receptor imaging of a transgenic rat model in the early phase of Huntington disease. Exp. Neurol. 229, 440–449. doi: 10.1016/j.expneurol.2011.03.014
Ceccarini, J., Ahmad, R., Van de Vliet, L., Casteels, C., Vandenbulcke, M., Vandenberghe, W., et al. (2019). Behavioral symptoms in premanifest huntington disease correlate with reduced frontal CB 1 R levels. J. Nucl. Med. 60, 115–121. doi: 10.2967/jnumed.118.210393
Ceccarini, J., Hompes, T., Verhaeghen, A., Casteels, C., Peuskens, H., Bormans, G., et al. (2014). Changes in cerebral CB1 receptor availability after acute and chronic alcohol abuse and monitored abstinence. J. Neurosci. Off. J. Soc. Neurosci. 34, 2822–2831. doi: 10.1523/JNEUROSCI.0849-13.2014
Ceccarini, J., Kuepper, R., Kemels, D., van Os, J., Henquet, C., and Van Laere, K. (2013). MK-9470 PET measurement of cannabinoid CB1receptor availability in chronic cannabis users. Addict. Biol. 20, 357–367. doi: 10.1111/adb.12116
Ceccarini, J., Kuepper, R., Kemels, D., van Os, J., Henquet, C., and Van Laere, K. (2015). [18 F]MK-9470 PET measurement of cannabinoid CB 1 receptor availability in chronic cannabis users: CB 1 receptor in cannabis users. Addict. Biol. 20, 357–367. doi: 10.1111/adb.12116
Ceccarini, J., Weltens, N., Ly, H. G., Tack, J., Van Oudenhove, L., and Van Laere, K. (2016). Association between cerebral cannabinoid 1 receptor availability and body mass index in patients with food intake disorders and healthy subjects: a [18F]MK-9470 PET study. Transl. Psychiatry 6:e853. doi: 10.1038/tp.2016.118
Cleeren, E., Casteels, C., Goffin, K., Koole, M., Van Laere, K., Janssen, P., et al. (2018). Positron emission tomography imaging of cerebral glucose metabolism and type 1 cannabinoid receptor availability during temporal lobe epileptogenesis in the amygdala kindling model in rhesus monkeys. Epilepsia 59, 959–970. doi: 10.1111/epi.14059
Cohen, R., Carson, R., Filbey, F., Szczepanik, J., and Sunderland, T. (2006). Age and APOE-ε4 genotype influence the effect of physostigmine infusion on the in-vivo distribution volume of the muscarinic-2-receptor dependent tracer [18F] FP-TZTP. Synapse 60, 86–92. doi: 10.1002/syn.20276
Cohen, R. M., Podruchny, T. A., Bokde, A. L. W., Carson, R. E., Herscovitch, P., Kiesewetter, D. O., et al. (2003). Higher in vivo muscarinic-2 receptor distribution volumes in aging subjects with an apolipoprotein E-?4 allele. Synapse 49, 150–156. doi: 10.1002/syn.10225
Colasanti, A., Searle, G., Long, C., Hill, S., Reiley, R., Quelch, D., et al. (2012). Endogenous opioid release in the human brain reward system induced by acute amphetamine administration. Biol. Psychiatry 72, 371–377. doi: 10.1016/j.biopsych.2012.01.027
Colom, M., Costes, N., Redouté, J., Dailler, F., Gobert, F., Le Bars, D., et al. (2019). 18F-F13640 PET imaging of functional receptors in humans. Eur. J. Nucl. Med. Mol. Imaging doi: 10.1007/s00259-019-04473-7 [Epub ahead of print].
Cumming, P., Gillings, N., Jensen, S., Bjarkam, C., and Gjedde, A. (2003). Kinetics of the uptake and distribution of the dopamine D2,3 agonist (R)-N-[1-11C]n-propylnorapomorphine in brain of healthy and MPTP-treated Göttingen miniature pigs. Nucl. Med. Biol. 30, 547–553. doi: 10.1016/s0969-8051(02)00448-1
Cumming, P., Wong, D., Gillings, N., Hilton, J., Scheffel, U., and Gjedde, A. (2002). Specific binding of [11C]Raclopride and N-[3H]Propyl-norapomorphine to dopamine receptors in living mouse striatum: occupancy by endogenous dopamine and guanosine triphosphate-free G protein. J. Cereb. Blood Flow Metab. 22, 596–604. doi: 10.1097/00004647-200205000-00011
Dannals, R., Ravert, H., James Frost, J., Wilson, A., Donald Burns, H., and Wagner, H. (1985). Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int. J. Appl. Radiat. Isot. 36, 303–306. doi: 10.1016/0020-708x(85)90089-4
DaSilva, A. F., Nascimento, T. D., DosSantos, M. F., Lucas, S., van HolsbeecK, H., DeBoer, M., et al. (2014a). Association of μ-Opioid activation in the prefrontal cortex with spontaneous migraine attacks - brief report I. Ann. Clin. Transl. Neurol. 1, 439–444. doi: 10.1002/acn3.65
DaSilva, A. F., Nascimento, T. D., DosSantos, M. F., and Zubieta, J.-K. (2014b). Migraine and the Mu-opioidergic system-Can we directly modulate it? Evidence from neuroimaging studies. Curr. Pain Headache Rep. 18:429. doi: 10.1007/s11916-014-0429-0
De Haes, U. (2005). In Vivo Imaging of Dopamine and Serotonin Release: Response to Psychopharmacological Challenges. Master’s thesis, University of Groningen, Groningen.
De Lean, A., Stadel, J. M., and Lefkowitz, R. J. (1980). A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 255, 7108–7117.
Domino, E. F., Hirasawa-Fujita, M., Ni, L., Guthrie, S. K., and Zubieta, J. K. (2015). Regional brain [(11)C]carfentanil binding following tobacco smoking. Prog. Neuropsychopharmacol. Biol. Psychiatry 59, 100–104. doi: 10.1016/j.pnpbp.2015.01.007
DosSantos, M., Martikainen, I., Nascimento, T., Love, T., Deboer, M., Maslowski, E., et al. (2012). Reduced basal ganglia μ-opioid receptor availability in trigeminal neuropathic pain: a pilot study. Mol. Pain 8:74. doi: 10.1186/1744-8069-8-74
Ehlert, F. J. (1985). The relationship between muscarinic receptor occupancy and adenylate cyclase inhibition in the rabbit myocardium. Mol. Pharmacol. 28, 410–421.
Elsinga, P., Kawamura, K., Kobayashi, T., Tsukada, H., Senda, M., Vaalburg, W., et al. (2002). Synthesis and evaluation of [18F] fluoroethyl SA4503 as a PET ligand for the sigma receptor. Synapse 43, 259–267. doi: 10.1002/syn.10045
Elsinga, P., Tsukada, H., Harada, N., Kakiuchi, T., Kawamura, K., Vaalburg, W., et al. (2004). Evaluation of [18F] fluorinated sigma receptor ligands in the conscious monkey brain. Synapse 52, 29–37. doi: 10.1002/syn.20001
Ernst, A. (1967). Mode of action of apomorphine and dexamphetamine on gnawing compulsion in rats. Psychopharmacologia 10, 316–323. doi: 10.1007/bf00403900
Ettrup, A., da Cunha-Bang, S., McMahon, B., Lehel, S., Dyssegaard, A., Skibsted, A. W., et al. (2014). Serotonin 2A receptor agonist binding in the human brain with [11 C]Cimbi-36. J. Cereb. Blood Flow Metab. 34, 1188–1196. doi: 10.1038/jcbfm.2014.68
Ettrup, A., Hansen, M., Santini, M. A., Paine, J., Gillings, N., Palner, M., et al. (2011). Radiosynthesis and in vivo evaluation of a series of substituted 11C-phenethylamines as 5-HT2A agonist PET tracers. Eur. J. Nucl. Med. Mol. Imaging 38, 681–693. doi: 10.1007/s00259-010-1686-8
Ettrup, A., Palner, M., Gillings, N., Santini, M. A., Hansen, M., Kornum, B. R., et al. (2010). Radiosynthesis and evaluation of 11C-CIMBI-5 as a 5-HT2A receptor agonist radioligand for PET. J. Nucl. Med. 51, 1763–1770. doi: 10.2967/jnumed.109.074021
Ettrup, A., Svarer, C., McMahon, B., da Cunha-Bang, S., Lehel, S., Møller, K., et al. (2016). Serotonin 2A receptor agonist binding in the human brain with [11 C]Cimbi-36: test–retest reproducibility and head-to-head comparison with the antagonist [18 F]altanserin. Neuroimage 130, 167–174. doi: 10.1016/j.neuroimage.2016.02.001
Falcone, M., Gold, A. B., Wileyto, E. P., Ray, R., Ruparel, K., Newberg, A., et al. (2012). μ-Opioid receptor availability in the amygdala is associated with smoking for negative affect relief. Psychopharmacology 222, 701–708. doi: 10.1007/s00213-012-2673-5
Fan, H., Ravert, H., Holt, D., Dannals, R., and Horti, A. (2006). Synthesis of 1-(2,4-dichlorophenyl)-4-cyano-5-(4-[11C]methoxyphenyl)-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide ([11C]JHU75528) and 1-(2-bromophenyl)-4-cyano-5-(4-[11C]methoxyphenyl)-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide ([11C]JHU75575) as potential radioligands for PET imaging of cerebral cannabinoid receptor. J. Label. Compd. Radiopharm. 49, 1021–1036. doi: 10.1002/jlcr.1125
Finnema, J. S., Bang-Andersen, B., Wikstrom, H. V., and Halldin, C. (2010). Current state of agonist radioligands for imaging of brain dopamine D2/D3 receptors in vivo with positron emission tomography. Curr. Top. Med. Chem. 10, 1477–1498. doi: 10.2174/156802610793176837
Finnema, S., Halldin, C., Bang-Andersen, B., Gulyás, B., Bundgaard, C., Wikström, H., et al. (2009). Dopamine D(2/3) receptor occupancy of apomorphine in the nonhuman primate brain–a comparative PET study with [11C]raclopride and [11C]MNPA. Synapse 63, 378–389. doi: 10.1002/syn.20615
Finnema, S. J., Scheinin, M., Shahid, M., Lehto, J., Borroni, E., Bang-Andersen, B., et al. (2015). Application of cross-species PET imaging to assess neurotransmitter release in brain. Psychopharmacology 232, 4129–4157. doi: 10.1007/s00213-015-3938-6
Finnema, S. J., Seneca, N., Farde, L., Shchukin, E., Sóvágó, J., Gulyás, B., et al. (2005). A preliminary PET evaluation of the new dopamine D2 receptor agonist [11C]MNPA in cynomolgus monkey. Nucl. Med. Biol. 32, 353–360. doi: 10.1016/j.nucmedbio.2005.01.007
Finnema, S. J., Stepanov, V., Ettrup, A., Nakao, R., Amini, N., Svedberg, M., et al. (2014). Characterization of [11C]Cimbi-36 as an agonist PET radioligand for the 5-HT2A and 5-HT2C receptors in the nonhuman primate brain. Neuroimage 84, 342–353. doi: 10.1016/j.neuroimage.2013.08.035
Frost, J., Douglass, K., Mayberg, H., Dannals, R., Links, J., Wilson, A., et al. (1989). Multicompartmental analysis of [11C]Carfentanil binding to opiate receptors in humans measured by positron emission tomography. J. Cereb. Blood Flow Metab. 9, 398–409. doi: 10.1038/jcbfm.1989.59
Frost, J., Mayberg, H., Fisher, R., Douglass, K., Dannals, R., Links, J., et al. (1988). Mu-opiate receptors measured by positron emission tomography are increased in temporal lobe epilepsy. Ann. Neurol. 23, 231–237. doi: 10.1002/ana.410230304
Frost, J., Mayberg, H., Sadzot, B., Dannals, R., Lever, J., Ravert, H., et al. (1990). Comparison of [11C]Diprenorphine and [11C]Carfentanil binding to opiate receptors in humans by positron emission tomography. J. Cereb. Blood Flow Metab. 10, 484–492. doi: 10.1038/jcbfm.1990.90
Frost, J., Wagner, H., Dannals, R., Ravert, H., Links, J., Wilson, A., et al. (1985). Imaging opiate receptors in the human brain by positron tomography. J. Comput. Assist. Tomogr. 9, 231–236. doi: 10.1097/00004728-198503000-00001
Fujio, M., Nagata, S., Kawamura, K., Sugiyama, N., Tanaka, H., Uno, K., et al. (2002). Synthesis and evaluation of 11C-labeled (S)-N-[1-(2-phenylethyl) pyrrolidin-2-yl]methyl-3-methylthiobenzamide as a PET 5-HT1A receptor ligand. Nucl. Med. Biol. 29, 657–663. doi: 10.1016/s0969-8051(02)00305-0
Gallezot, J., Esterlis, I., Bois, F., Zheng, M., Lin, S., Kloczynski, T., et al. (2014). Evaluation of the sensitivity of the novel α4β2∗ nicotinic acetylcholine receptor PET radioligand 18 F-(-)-NCFHEB to increases in synaptic acetylcholine levels in rhesus monkeys. Synapse 68, 556–564. doi: 10.1002/syn.21767
Gao, M., Wang, M., and Zheng, Q. (2012). A new high-yield synthetic route to PET CB1 radioligands [11C]OMAR and its analogs. Bioorg. Med. Chem. Lett. 22, 3704–3709. doi: 10.1016/j.bmcl.2012.04.030
Gao, Y., Baldessarini, R., Kula, N., and Neumeyer, J. (1990). Synthesis and dopamine receptor affinities of enantiomers of 2-substituted apomorphines and their N-n-propyl analogs. J. Med. Chem. 33, 1800–1805. doi: 10.1021/jm00168a040
Gérard, N., Ceccarini, J., Bormans, G., Vanbilloen, B., Casteels, C., Goffin, K., et al. (2010). Influence of chronic nicotine administration on cerebral type 1 cannabinoid receptor binding: an vivo micro-PET study rat using 18F 42, MK-9470. J. Mol. Neurosci. 42, 162–167. doi: 10.1007/s12031-010-9340-2
Gérard, N., Pieters, G., Goffin, K., Bormans, G., and Van Laere, K. (2011). Brain type 1 cannabinoid receptor availability in patients with anorexia and bulimia nervosa. Biol. Psychiatry 70, 777–784. doi: 10.1016/j.biopsych.2011.05.010
Ghitza, U. E., Preston, K. L., Epstein, D. H., Kuwabara, H., Endres, C. J., Bencherif, B., et al. (2010). Brain mu-opioid receptor binding predicts treatment outcome in cocaine-abusing outpatients. Biol. Psychiatry 68, 697–703. doi: 10.1016/j.biopsych.2010.05.003
Ginovart, N. (2005). Imaging the dopamine system with in vivo [11C]raclopride displacement studies: understanding the true mechanism. Mol. Imaging Biol. Off. Publ. Acad. Mol. Imaging 7, 45–52. doi: 10.1007/s11307-005-0932-0
Ginovart, N., Galineau, L., Willeit, M., Mizrahi, R., Bloomfield, P., Seeman, P., et al. (2006a). Binding characteristics and sensitivity to endogenous dopamine of [11C](+)-PHNO, a new agonist radiotracer for imaging the high-affinity state of D2 receptors in vivo using positron emission tomography. J. Neurochem. 97, 1089–1103. doi: 10.1111/j.1471-4159.2006.03840.x
Ginovart, N., Willeit, M., Rusjan, P., Graff, A., Bloomfield, P., Houle, S., et al. (2006b). Positron emission tomography quantification of [11C](+)-PHNO binding in the human brain. J. Cereb. Blood Flow Metab. 27, 857–871. doi: 10.1038/sj.jcbfm.9600411
Goffin, K., Bormans, G., Casteels, C., Bosier, B., Lambert, D., Grachev, I., et al. (2008). An in vivo [18F] MK-9470 microPET study of type 1 cannabinoid receptor binding in Wistar rats after chronic administration of valproate and levetiracetam. Neuropharmacology 54, 1103–1106. doi: 10.1016/j.neuropharm.2008.02.018
Goffin, K., Van Paesschen, W., and Van Laere, K. (2011). In vivo activation of endocannabinoid system in temporal lobe epilepsy with hippocampal sclerosis. Brain 134, 1033–1040. doi: 10.1093/brain/awq385
Golan, M., Schreiber, G., and Avissar, S. (2011). Antidepressants elevate GDNF expression and release from C6 glioma cells in a β-arrestin1-dependent, CREB interactive pathway. Int. J. Neuropsychopharmacol. 14, 1289–1300. doi: 10.1017/S1461145710001550
Gorelick, D. A., Kim, Y. K., Bencherif, B., Boyd, S. J., Nelson, R., Copersino, M., et al. (2005). Imaging brain mu-opioid receptors in abstinent cocaine users: time course and relation to cocaine craving. Biol. Psychiatry 57, 1573–1582. doi: 10.1016/j.biopsych.2005.02.026
Gorelick, D. A., Kim, Y. K., Bencherif, B., Boyd, S. J., Nelson, R., Copersino, M. L., et al. (2008). Brain mu-opioid receptor binding: relationship to relapse to cocaine use after monitored abstinence. Psychopharmacology 200, 475–486. doi: 10.1007/s00213-008-1225-5
Gozlan, H., Thibault, S., Laporte, A.-M., Lima, L., and Hamon, M. (1995). The selective 5-HT1A antagonist radioligand [3H]WAY 100635 labels both G-protein-coupled and free 5-HT1A receptors in rat brain membranes. Eur. J. Pharmacol. Mol. Pharmacol. 288, 173–186. doi: 10.1016/0922-4106(95)90192-2
Graff-Guerrero, A., Mizrahi, R., Agid, O., Marcon, H., Barsoum, P., Rusjan, P., et al. (2008). The dopamine D2 receptors in high-affinity state and d3 receptors in schizophrenia: a clinical [11C](+)-PHNO PET Study. Neuropsychopharmacology 34, 1078–1086. doi: 10.1038/npp.2008.199
Gray, J. A., and Roth, B. L. (2001). Paradoxical trafficking and regulation of 5-HT(2A) receptors by agonists and antagonists. Brain Res. Bull. 56, 441–451. doi: 10.1016/s0361-9230(01)00623-2
Greenwald, M., Johanson, C.-E., Bueller, J., Chang, Y., Moody, D. E., Kilbourn, M., et al. (2007). Buprenorphine duration of action: mu-opioid receptor availability and pharmacokinetic and behavioral indices. Biol. Psychiatry 61, 101–110. doi: 10.1016/j.biopsych.2006.04.043
Gullapalli, S., Amrutkar, D., Gupta, S., Kandadi, M. R., Kumar, H., Gandhi, M., et al. (2010). Characterization of active and inactive states of CB1 receptor and the differential binding state modulation by cannabinoid agonists, antagonists and inverse agonists. Neuropharmacology 58, 1215–1219. doi: 10.1016/j.neuropharm.2010.03.001
Hagelberg, N., Aalto, S., Tuominen, L., Pesonen, U., Någren, K., Hietala, J., et al. (2012). Striatal μ-opioid receptor availability predicts cold pressor pain threshold in healthy human subjects. Neurosci. Lett. 521, 11–14. doi: 10.1016/j.neulet.2012.05.042
Halldin, C., Farde, L., Litton, J., Hall, H., and Sedvall, G. (1992). [11C]Ro 15-4513, a ligand for visualization of benzodiazepine receptor binding. Psychopharmacology 108, 16–22. doi: 10.1007/bf02245279
Halldin, C., Gulyas, B., and Farde, L. (2001). PET studies with carbon-11 radioligands in neuropsychopharmacological drug development. Curr. Pharm. Des. 7, 1907–1929. doi: 10.2174/1381612013396871
Hamill, T., Sato, N., Jitsuoka, M., Tokita, S., Sanabria, S., Eng, W., et al. (2009). Inverse agonist histamine H3 receptor PET tracers labelled with carbon-11 or fluorine-18. Synapse 63, 1122–1132. doi: 10.1002/syn.20689
Harris, R. E., Zubieta, J.-K., Scott, D. J., Napadow, V., Gracely, R. H., and Clauw, D. J. (2009). Traditional Chinese acupuncture and placebo (sham) acupuncture are differentiated by their effects on mu-opioid receptors (MORs). Neuroimage 47, 1077–1085. doi: 10.1016/j.neuroimage.2009.05.083
Hassoun, W., Le Cavorsin, M., Ginovart, N., Zimmer, L., Gualda, V., Bonnefoi, F., et al. (2003). PET study of the [11C]raclopride binding in the striatum of the awake cat: effects of anaesthetics and role of cerebral blood flow. Eur. J. Nuclear Med. Mol. Imaging 30, 141–148. doi: 10.1007/s00259-002-0904-4
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schiöth, H. B., and Gloriam, D. E. (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842. doi: 10.1038/nrd.2017.178
Heese, K. (2013). G Proteins, p60TRP, and neurodegenerative diseases. Mol. Neurobiol. 47, 1103–1111. doi: 10.1007/s12035-013-8410-1
Heinz, A., Reimold, M., Wrase, J., Hermann, D., Croissant, B., Mundle, G., et al. (2005). Correlation of stable elevations in striatal mu-opioid receptor availability in detoxified alcoholic patients with alcohol craving: a positron emission tomography study using carbon 11-labeled carfentanil. Arch. Gen. Psychiatry 62, 57–64. doi: 10.1001/archpsyc.62.1.57
Henriksen, G., Platzer, S., Hauser, A., Willoch, F., Berthele, A., Schwaiger, M., et al. (2005a). 18F-labeled sufentanil for PET-imaging of mu-opioid receptors. Bioorg. Med. Chem. Lett. 15, 1773–1777. doi: 10.1016/j.bmcl.2005.02.049
Henriksen, G., Platzer, S., Marton, J., Hauser, A., Berthele, A., Schwaiger, M., et al. (2005b). Syntheses, biological evaluation, and molecular modeling of 18F-labeled 4-anilidopiperidines as mu-opioid receptor imaging agents. J. Med. Chem. 48, 7720–7732. doi: 10.1021/jm0507274
Hermann, D., Hirth, N., Reimold, M., Batra, A., Smolka, M. N., Hoffmann, S., et al. (2017). Low μ-Opioid receptor status in alcohol dependence identified by combined positron emission tomography and post-mortem brain analysis. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 42, 606–614. doi: 10.1038/npp.2016.145
Hoffman, B. B., and Lefkowitz, R. J. (1980). Radioligand binding studies of adrenergic receptors: new insights into molecular and physiological regulation. Annu. Rev. Pharmacol. Toxicol. 20, 581–608. doi: 10.1146/annurev.pa.20.040180.003053
Honer, M., Gobbi, L., Martarello, L., and Comley, R. A. (2014). Radioligand development for molecular imaging of the central nervous system with positron emission tomography. Drug Discov. Today 19, 1936–1944. doi: 10.1016/j.drudis.2014.08.012
Horti, A. (2007). “[11C]JHU75528, a PET radioligand for imaging of cerebral cannabinoid CB1 receptors,” in Proceedings of the 39-th Meeting of European Brain and Behaviour Society, Trieste.
Horti, A. G., Fan, H., Kuwabara, H., Hilton, J., Ravert, H. T., Holt, D. P., et al. (2006). 11C-JHU75528: a radiotracer for PET imaging of CB1 cannabinoid receptors. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 47, 1689–1696.
Hsu, D. T., Sanford, B. J., Meyers, K. K., Love, T. M., Hazlett, K. E., Walker, S. J., et al. (2015). It still hurts: altered endogenous opioid activity in the brain during social rejection and acceptance in major depressive disorder. Mol. Psychiatry 20, 193–200. doi: 10.1038/mp.2014.185
Hsu, D. T., Sanford, B. J., Meyers, K. K., Love, T. M., Hazlett, K. E., Wang, H., et al. (2013). Response of the μ-opioid system to social rejection and acceptance. Mol. Psychiatry 18, 1211–1217. doi: 10.1038/mp.2013.96
Hwang, D., Huang, Y., Ngo, K., Savenkova, L., Guo, N., Zhu, Z., et al. (2001). Preparation and evaluation of [11C]labeled 5-HT1A agonist: (R)-10-Methyl-11-hydroxyaporphine. J. Label. Compd. Radiopharm. 44, 176–178. doi: 10.1002/jlcr.2580440161
Hwang, D., Narendran, R., Hwang, Y., Slifstein, M., Talbot, P., Sudo, Y., et al. (2004). Quantitative analysis of ()-N-11C-Propyl-Norapomorphine in vivo binding in nonhumanprimates. J. Nucl. Med. 45, 338–346.
Hwang, D., Narendran, R., and Laruelle, M. (2005). Positron-labeled dopamine agonists for probing the high affinity states of dopamine subtype 2 receptors. Bioconjugate Chem. 16, 27–31. doi: 10.1021/bc049834n
Hwang, D. R., Kegeles, L. S., and Laruelle, M. (2000). (-)-N-[(11)C]propyl-norapomorphine: a positron-labeled dopamine agonist for PET imaging of D(2) receptors. Nucl. Med. Biol. 27, 533–539. doi: 10.1016/s0969-8051(00)00144-x
Ingman, K., Hagelberg, N., Aalto, S., Någren, K., Juhakoski, A., Karhuvaara, S., et al. (2005). Prolonged central mu-opioid receptor occupancy after single and repeated nalmefene dosing. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 30, 2245–2253. doi: 10.1038/sj.npp.1300790
Ishikawa, M., Ishiwata, K., Ishii, K., Kimura, Y., Sakata, M., Naganawa, M., et al. (2007). High occupancy of sigma-1 receptors in the human brain after single oral administration of fluvoxamine: a positron emission tomography study using [11C]SA4503. Biol. Psychiatry 62, 878–883. doi: 10.1016/j.biopsych.2007.04.001
Ishikawa, M., Sakata, M., Ishii, K., Kimura, Y., Oda, K., Toyohara, J., et al. (2009). High occupancy of σ1 receptors in the human brain after single oral administration of donepezil: a positron emission tomography study using [11C]SA4503. Int. J. Neuropsychopharmacol. 12, 1127–1131. doi: 10.1017/s1461145709990204
Ishiwata, K., Kobayashi, T., Kawamura, K., and Matsuno, K. (2003). Age-related changes of the binding of [3H]SA4503 to sigma1 receptors in the rat brain. Ann. Nucl. Med. 17, 73–77. doi: 10.1007/bf02988264
Ishiwata, K., Oda, K., Sakata, M., Kimura, Y., Kawamura, K., Oda, K., et al. (2006). A feasibility study of [11C]SA4503-PET for evaluating sigma1 receptor occupancy by neuroleptics: the binding of haloperidol to sigma1 and dopamine D2-like receptors. Ann. Nucl. Med. 20, 569–573. doi: 10.1007/bf03026824
Ishiwata, K., Tsukada, H., Kawamura, K., Kimura, Y., Nishiyama, S., Kobayashi, T., et al. (2001). Mapping of CNS sigma1 receptors in the conscious monkey: preliminary PET study with [11C]SA4503. Synapse 40, 235–237. doi: 10.1002/syn.1046
Jagoda, E., Kiesewetter, D., Shimoji, K., Ravasi, L., Yamada, M., Gomeza, J., et al. (2003). Regional brain uptake of the muscarinic ligand, [18F] FP-TZTP, is greatly decreased in M2 receptor knockout mice but not in M1, M3 and M4 receptor knockout mice. Neuropharmacology 44, 653–661. doi: 10.1016/s0028-3908(03)00050-9
Jastrzebska, B., Debinski, A., Filipek, S., and Palczewski, K. (2011). Role of membrane integrity on G protein-coupled receptors: rhodopsin stability and function. Prog. Lipid Res. 50, 267–277. doi: 10.1016/j.plipres.2011.03.002
Jin, L. Q., Wang, H. Y., and Friedman, E. (2001). Stimulated D(1) dopamine receptors couple to multiple Galpha proteins in different brain regions. J. Neurochem. 78, 981–990. doi: 10.1046/j.1471-4159.2001.00470.x
Jitsuoka, M., Tsukahara, D., Ito, S., Tanaka, T., Takenaga, N., Tokita, S., et al. (2008). Synthesis and evaluation of a spiro-isobenzofuranone class of histamine H3 receptor inverse agonists. Bioorg. Med. Chem. Lett. 18, 5101–5106. doi: 10.1016/j.bmcl.2008.07.125
Johansen, A., Hansen, H. D., Svarer, C., Lehel, S., Leth-Petersen, S., Kristensen, J. L., et al. (2018). The importance of small polar radiometabolites in molecular neuroimaging: a PET study with [11C]Cimbi-36 labeled in two positions. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 38, 659–668. doi: 10.1177/0271678X17746179
Jones, J. H., Anderson, P. S., Baldwin, J. J., Clineschmidt, B. V., McClure, D. E., Lundell, G. F., et al. (1984). Synthesis of 4-substituted 2H-naphth[1,2-b]-1,4-oxazines, a new class of dopamine agonists. J. Med. Chem. 27, 1607–1613. doi: 10.1021/jm00378a014
Jorgensen, L. M., Weikop, P., Villadsen, J., Visnapuu, T., Ettrup, A., Hansen, H. D., et al. (2017). Cerebral 5-HT release correlates with [11 C]Cimbi36 PET measures of 5-HT2A receptor occupancy in the pig brain. J. Cereb. Blood Flow Metab. 37, 425–434. doi: 10.1177/0271678X16629483
Joutsa, J., Karlsson, H. K., Majuri, J., Nuutila, P., Helin, S., Kaasinen, V., et al. (2018). Binge eating disorder and morbid obesity are associated with lowered mu-opioid receptor availability in the brain. Psychiatry Res. Neuroimaging 276, 41–45. doi: 10.1016/j.pscychresns.2018.03.006
Karjalainen, T., Karlsson, H. K., Lahnakoski, J. M., Glerean, E., Nuutila, P., Jääskeläinen, I. P., et al. (2017). Dissociable roles of cerebral μ-Opioid and type 2 dopamine receptors in vicarious pain: a combined PET-fMRI study. Cereb. Cortex 27, 4257–4266. doi: 10.1093/cercor/bhx129
Karjalainen, T., Tuominen, L., Manninen, S., Kalliokoski, K. K., Nuutila, P., Jääskeläinen, I. P., et al. (2016). Behavioural activation system sensitivity is associated with cerebral μ-opioid receptor availability. Soc. Cogn. Affect. Neurosci. 11, 1310–1316. doi: 10.1093/scan/nsw044
Karlsson, H. K., Tuominen, L., Tuulari, J. J., Hirvonen, J., Parkkola, R., Helin, S., et al. (2015). Obesity is associated with decreased μ-opioid but unaltered dopamine D2 receptor availability in the brain. J. Neurosci. Off. J. Soc. Neurosci. 35, 3959–3965. doi: 10.1523/JNEUROSCI.4744-14.2015
Karlsson, H. K., Tuulari, J. J., Tuominen, L., Hirvonen, J., Honka, H., Parkkola, R., et al. (2016). Bariatric surgery normalizes brain opioid receptors. Mol. Psychiatry 21:989. doi: 10.1038/mp.2016.116
Kawamura, K., Elsinga, P., Kobayashi, T., Ishii, S., Wang, W., Matsuno, K., et al. (2003a). Synthesis and evaluation of 11C- and 18F-labeled 1-[2-(4-alkoxy-3-methoxyphenyl)ethyl]4-(3-phenylpropyl)piperazines as sigma receptor ligands for positron emission tomography studies. Nucl. Med. Biol. 30, 273–284. doi: 10.1016/s0969-8051(02)00439-0
Kawamura, K., Kimura, Y., Tsukada, H., Kobayashi, T., Nishiyama, S., Kakiuchi, T., et al. (2003b). An increase of sigma1 receptors in the aged monkey brain. Neurobiol. Aging 24, 745–752. doi: 10.1016/s0197-4580(02)00152-5
Kawamura, K., Ishiwata, K., Tajima, H., Ishii, S., Matsuno, K., Homma, Y., et al. (2000). In vivo evaluation of [11C]SA4503 as a PET ligand for mapping CNS sigma1 receptors. Nucl. Med. Biol. 27, 255–261. doi: 10.1016/s0969-8051(00)00081-0
Kawamura, K., Ishiwata, K., Tajima, H., Ishii, S., Shimada, Y., Matsuno, K., et al. (1999). Synthesis and in vivo evaluation of [11C]SA6298 as a PET sigma1 receptor ligand. Nucl. Med. Biol. 26, 915–922. doi: 10.1016/s0969-8051(99)00069-4
Kawamura, K., Tsukada, H., Shiba, K., Tsuji, C., Harada, N., Kimura, Y., et al. (2007). Synthesis and evaluation of fluorine-18-labeled SA4503 as a selective sigma1 receptor ligand for positron emission tomography. Nuclear Med. Biol. 34, 571–577. doi: 10.1016/j.nucmedbio.2007.03.009
Keith, D. E., Anton, B., Murray, S. R., Zaki, P. A., Chu, P. C., Lissin, D. V., et al. (1998). mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol. Pharmacol. 53, 377–384. doi: 10.1124/mol.53.3.377
Kenakin, T. (2015). New lives for seven transmembrane receptors as drug targets. Trends Pharmacol. Sci. 36, 705–706. doi: 10.1016/j.tips.2015.09.004
Kenakin, T., and Christopoulos, A. (2012). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 12, 205–216. doi: 10.1038/nrd3954
Kennedy, S. E., Koeppe, R. A., Young, E. A., and Zubieta, J.-K. (2006). Dysregulation of endogenous opioid emotion regulation circuitry in major depression in women. Arch. Gen. Psychiatry 63, 1199–1208. doi: 10.1001/archpsyc.63.11.1199
Kent, R. S., De Lean, A., and Lefkowitz, R. J. (1980). A quantitative analysis of beta-adrenergic receptor interactions: resolution of high and low affinity states of the receptor by computer modeling of ligand binding data. Mol. Pharmacol. 17, 14–23.
Kiesewetter, D., Carson, R., Jagoda, E., Herscovitch, P., and Eckelman, W. (1999). In vivo muscarinic binding of 3-(alkylthio)-3-thiadiazolyl tetrahydropyridines. Synapse 31, 29–40.
Kiesewetter, D., Lee, J., Lang, L., Park, S., Paik, C., and Eckelman, W. (1995). Preparation of 18F-Labeled muscarinic agonist with M2 selectivity. J. Med. Chem. 38, 5–8. doi: 10.1021/jm00001a002
Kiesewetter, D., Vuong, B., and Channing, M. (2003). The automated radiosynthesis of [18F] FP-TZTP. Nucl. Med. Biol. 30, 73–77. doi: 10.1016/s0969-8051(02)00354-2
Kodaka, F., Ito, H., Kimura, Y., Fujie, S., Takano, H., Fujiwara, H., et al. (2012). Test-retest reproducibility of dopamine D2/3 receptor binding in human brain measured by PET with [11C]MNPA and [11C]raclopride. Eur. J. Nucl. Med. Mol. Imaging 40, 574–579. doi: 10.1007/s00259-012-2312-8
Kodaka, F., Ito, H., Takano, H., Takahashi, H., Arakawa, R., Miyoshi, M., et al. (2010). Effect of risperidone on high-affinity state of dopamine D2 receptors: a PET study with agonist ligand [11C](R)-2-CH3O-N-n-propylnorapomorphine. Int. J. Neuropsychopharmacol. 14, 83–89. doi: 10.1017/s1461145710001148
Kubota, M., Nagashima, T., Takano, H., Kodaka, F., Fujiwara, H., Takahata, K., et al. (2017). Affinity states of striatal dopamine D2 receptors in antipsychotic-free patients with schizophrenia. Int. J. Neuropsychopharmacol. 20, 928–935. doi: 10.1093/ijnp/pyx063
Kumar, J. S. D., Majo, V. J., Hsiung, S.-C., Millak, M. S., Liu, K.-P., Tamir, H., et al. (2006). Synthesis and in Vivo Validation of [O -Methyl- 11 C]2-{4-[4-(7-methoxynaphthalen-1-yl)piperazin- 1-yl]butyl}-4-methyl-2 H -[1,2,4]triazine-3,5-dione: A Novel 5-HT 1A receptor agonist positron emission tomography ligand. J. Med. Chem. 49, 125–134. doi: 10.1021/jm050725j
Kumar, J. S. D., Milak, M. S., Majo, V. J., Prabhakaran, J., Mali, P., Savenkova, L., et al. (2012). Comparison of high and low affinity serotonin 1A receptors by PET in vivo in nonhuman primates. J. Pharmacol. Sci. 120, 254–257. doi: 10.1254/jphs.12100SC
Kuwabara, H., Heishman, S. J., Brasic, J. R., Contoreggi, C., Cascella, N., Mackowick, K. M., et al. (2014). Mu opioid receptor binding correlates with nicotine dependence and reward in smokers. PLoS One 9:e113694. doi: 10.1371/journal.pone.0113694
la Cour, C. M. (2006). Regional differences in the coupling of 5-Hydroxytryptamine-1A receptors to G proteins in the rat brain. Mol. Pharmacol. 70, 1013–1021. doi: 10.1124/mol.106.022756
Laruelle, M. (2000). Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J. Cereb. Blood Flow Metab. 20, 423–451. doi: 10.1097/00004647-200003000-00001
Laugwitz, K. L., Allgeier, A., Offermanns, S., Spicher, K., Van Sande, J., Dumont, J. E., et al. (1996). The human thyrotropin receptor: a heptahelical receptor capable of stimulating members of all four G protein families. Proc. Natl. Acad. Sci. U.S.A. 93, 116–120. doi: 10.1073/pnas.93.1.116
Law, P. Y., Hom, D. S., and Loh, H. H. (1985). Multiple affinity states of opiate receptor in neuroblastoma x glioma NG108-15 hybrid cells. Opiate agonist association rate is a function of receptor occupancy. J. Biol. Chem. 260, 3561–3569.
Laymon, C., Mason, N., Frankle, W., Carney, J., Lopresti, B., Litschge, M., et al. (2009). Human biodistribution and dosimetry of the D2/3 agonist 11C-N-Propylnorapomorphine (11C-NPA) determined from PET. J. Nucl. Med. 50, 814–817. doi: 10.2967/jnumed.108.058131
Le Foll, B., Guranda, M., Wilson, A., Houle, S., Rusjan, P., Wing, V., et al. (2013). Elevation of dopamine induced by cigarette smoking: novel insights from a [11C](+)-PHNO PET study in humans. Neuropsychopharmacology 39, 415–424. doi: 10.1038/npp.2013.209
Leff, S. E., Hamblin, M. W., and Creese, I. (1985). Interactions of dopamine agonists with brain D1 receptors labeled by 3H-antagonists. Evidence for the presence of high and low affinity agonist-binding states. Mol. Pharmacol. 27, 171–183.
Lemoine, L., Becker, G., Vacher, B., Billard, T., Lancelot, S., Newman-Tancredi, A., et al. (2012). Radiosynthesis and preclinical evaluation of 18F-F13714 as a fluorinated 5-HT1A receptor agonist radioligand for PET neuroimaging. J. Nucl. Med. 53, 969–976. doi: 10.2967/jnumed.111.101212
Lemoine, L., Verdurand, M., Vacher, B., Blanc, E., Le Bars, D., Newman-Tancredi, A., et al. (2010). [18F]F15599, a novel 5-HT1A receptor agonist, as a radioligand for PET neuroimaging. Eur. J. Nucl. Med. Mol. Imaging 37, 594–605. doi: 10.1007/s00259-009-1274-y
Liberzon, I., Taylor, S. F., Phan, K. L., Britton, J. C., Fig, L. M., Bueller, J. A., et al. (2007). Altered central micro-opioid receptor binding after psychological trauma. Biol. Psychiatry 61, 1030–1038. doi: 10.1016/j.biopsych.2006.06.021
Light, S. N., Bieliauskas, L. A., and Zubieta, J.-K. (2017). Top-Down” mu-opioid system function in humans: mu-opioid receptors in ventrolateral prefrontal cortex mediate the relationship between hedonic tone and executive function in major depressive disorder. J. Neuropsychiatry Clin. Neurosci. 29, 357–364. doi: 10.1176/appi.neuropsych.16090171
Logan, J., Fowler, J., Volkow, N., Wolf, A., Dewey, S., Schlyer, D., et al. (1990). Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl](-)-Cocaine PET studies in human subjects. J. Cereb. Blood Flow Metab. 10, 740–747. doi: 10.1038/jcbfm.1990.127
Luttrell, L. M., Maudsley, S., and Bohn, L. M. (2015). Fulfilling the promise of “Biased” G protein-coupled receptor agonism. Mol. Pharmacol. 88, 579–588. doi: 10.1124/mol.115.099630
Ma, Y., Kiesewetter, D., Lang, L., and Eckelman, W. C. (2003). Application of LC-MS to the analysis of new radiopharmaceuticals. Mol. Imaging Biol. Off. Publ. Acad. Mol. Imaging 5, 397–403. doi: 10.1016/j.mibio.2003.09.013
Ma, Y., Kiesewetter, D. O., Jagoda, E. M., Huang, B. X., and Eckelman, W. C. (2002). Identification of metabolites of fluorine-18-labeled M2 muscarinic receptor agonist, 3-(3-[(3-fluoropropyl)thio]-1,2,5-thiadiazol-4-yl)-1,2,5,6-tetrahydro-1-methylpyridine, produced by human and rat hepatocytes. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 766, 319–329. doi: 10.1016/s0378-4347(01)00517-5
Madar, I., Lesser, R. P., Krauss, G., Zubieta, J. K., Lever, J. R., Kinter, C. M., et al. (1997). Imaging of?- and?-opioid receptors in temporal lobe epilepsy by positron emission tomography. Ann. Neurol. 41, 358–367. doi: 10.1002/ana.410410311
Majuri, J., Joutsa, J., Johansson, J., Voon, V., Alakurtti, K., Parkkola, R., et al. (2017). Dopamine and opioid neurotransmission in behavioral addictions: a comparative PET study in pathological gambling and binge eating. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 42, 1169–1177. doi: 10.1038/npp.2016.265
Manninen, S., Tuominen, L., Dunbar, R. I., Karjalainen, T., Hirvonen, J., Arponen, E., et al. (2017). Social laughter triggers endogenous opioid release in humans. J. Neurosci. Off. J. Soc. Neurosci. 37, 6125–6131. doi: 10.1523/JNEUROSCI.0688-16.2017
Martikainen, I. K., Peciña, M., Love, T. M., Nuechterlein, E. B., Cummiford, C. M., Green, C. R., et al. (2013). Alterations in endogenous opioid functional measures in chronic back pain. J. Neurosci. Off. J. Soc. Neurosci. 33, 14729–14737. doi: 10.1523/JNEUROSCI.1400-13.2013
Mathis, C. A., Huang, Y., and Simpson, N. R. (1997). Synthesis and evaluation of 5-HT1A agonists a radioligands: failure of G protein-coupled receptor agonists as in vivo imaging agents. J. Label Compd. Radiopharm. 40, 563–564.
Matuskey, D., Gaiser, E., Gallezot, J., Angarita, G., Pittman, B., Nabulsi, N., et al. (2015). A preliminary study of dopamine D2/3 receptor availability and social status in healthy and cocaine dependent humans imaged with [11C](+)PHNO. Drug Alcohol Depend. 154, 167–173. doi: 10.1016/j.drugalcdep.2015.06.039
Mayberg, H., Sadzot, B., Meltzer, C., Fisher, R., Lesser, R., Dannals, R., et al. (1991). Quantification of mu and non-mu opiate receptors in temporal lobe epilepsy using positron emission tomography. Ann. Neurol. 30, 3–11. doi: 10.1002/ana.410300103
McCormick, P., Ginovart, N., and Wilson, A. (2010). Isoflurane anaesthesia differentially affects the amphetamine sensitivity of agonist and antagonist D2/D3 positron emission tomography radiotracers: implications for in vivo imaging of dopamine release. Mol. Imaging Biol. 13, 737–746. doi: 10.1007/s11307-010-0380-3
McCormick, P. N., Kapur, S., Reckless, G., and Wilson, A. A. (2009). Ex vivo [11 C]-(+)-PHNO binding is unchanged in animal models displaying increased high-affinity states of the D 2 receptor in vitro. Synapse 63, 998–1009. doi: 10.1002/syn.20671
McCormick, P. N., Kapur, S., Seeman, P., and Wilson, A. A. (2008). Dopamine D2 receptor radiotracers [11C](+)-PHNO and [3H]raclopride are indistinguishably inhibited by D2 agonists and antagonists ex vivo. Nucl. Med. Biol. 35, 11–17. doi: 10.1016/j.nucmedbio.2007.08.005
Mick, I., Myers, J., Ramos, A. C., Stokes, P. R. A., Erritzoe, D., Colasanti, A., et al. (2016). Blunted endogenous opioid release following an oral amphetamine challenge in pathological gamblers. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 41, 1742–1750. doi: 10.1038/npp.2015.340
Miederer, I., Buchholz, H.-G., Kronfeld, A., Maus, S., Weyer-Elberich, V., Mildenberger, P., et al. (2018). Pharmacokinetics of the cannabinoid receptor ligand [18 F]MK-9470 in the rat brain - Evaluation of models using microPET. Med. Phys. 45, 725–734. doi: 10.1002/mp.12732
Milak, M. S., Severance, A. J., Prabhakaran, J., Kumar, J. D., Majo, V. J., Ogden, R. T., et al. (2011). In vivo serotonin-sensitive binding of [11 C]CUMI-101: a serotonin 1A receptor agonist positron emission tomography radiotracer. J. Cereb. Blood Flow Metab. 31, 243–249. doi: 10.1038/jcbfm.2010.83
Miller, J. M., Zanderigo, F., Purushothaman, P. D., DeLorenzo, C., Rubin-Falcone, H., Ogden, R. T., et al. (2018). Kappa opioid receptor binding in major depression: a pilot study. Synapse 72:e22042. doi: 10.1002/syn.22042
Minkowski, C. P., Epstein, D., Frost, J. J., and Gorelick, D. A. (2012). Differential response to IV carfentanil in chronic cocaine users and healthy controls. Addict. Biol. 17, 149–155. doi: 10.1111/j.1369-1600.2010.00256.x
Minuzzi, L., and Cumming, P. (2010). Agonist binding fraction of dopamine D2/3 receptors in rat brain: a quantitative autoradiographic study. Neurochem. Int. 56, 747–752. doi: 10.1016/j.neuint.2010.01.010
Mishina, M., Ishiwata, K., Ishii, K., Kitamura, S., Kimura, Y., Kawamura, K., et al. (2005). Function of sigma1 receptors in Parkinson’s disease. Acta Neurol. Scand. 112, 103–107. doi: 10.1111/j.1600-0404.2005.00432.x
Mishina, M., Ohyama, M., Ishii, K., Kitamura, S., Kimura, Y., Oda, K., et al. (2008). Low density of sigma1 receptors in early Alzheimer’s disease. Ann. Nucl. Med. 22, 151–156. doi: 10.1007/s12149-007-0094-z
Mitchell, J. M., O’Neil, J. P., Jagust, W. J., and Fields, H. L. (2013). Catechol-O-methyltransferase genotype modulates opioid release in decision circuitry. Clin. Transl. Sci. 6, 400–403. doi: 10.1111/cts.12075
Mitchell, J. M., O’Neil, J. P., Janabi, M., Marks, S. M., Jagust, W. J., and Fields, H. L. (2012). Alcohol consumption induces endogenous opioid release in the human orbitofrontal cortex and nucleus accumbens. Sci. Transl. Med. 4:116ra6. doi: 10.1126/scitranslmed.3002902
Mizrahi, R., Agid, O., Borlido, C., Suridjan, I., Rusjan, P., Houle, S., et al. (2011). Effects of antipsychotics on D3 receptors: a clinical PET study in first episode antipsychotic naive patients with schizophrenia using [11C]-(+)-PHNO. Schizophr. Res. 131, 63–68. doi: 10.1016/j.schres.2011.05.005
Mizrahi, R., Suridjan, I., Kenk, M., George, T., Wilson, A., Houle, S., et al. (2012). Dopamine response to psychosocial stress in chronic cannabis users: a PET study with 11C-PHNO. Neuropsychopharmacology 38, 673–682. doi: 10.1038/npp.2012.232
Mongeau, R., Welner, S. A., Quirion, R., and Suranyi-Cadotte, B. E. (1992). Further evidence for differential affinity states of the serotonin1A receptor in rat hippocampus. Brain Res. 590, 229–238. doi: 10.1016/0006-8993(92)91100-s
Naganawa, M., Jacobsen, L. K., Zheng, M.-Q., Lin, S.-F., Banerjee, A., Byon, W., et al. (2014). Evaluation of the agonist PET radioligand [11C]GR103545 to image kappa opioid receptor in humans: kinetic model selection, test–retest reproducibility and receptor occupancy by the antagonist PF-04455242. Neuroimage 99, 69–79. doi: 10.1016/j.neuroimage.2014.05.033
Narendran, R. (2005). Measurement of the proportion of D2 receptors configured in state of high affinity for agonists in vivo: a positron emission tomography study using [11C]N-Propyl-norapomorphine and [11C]Raclopride in Baboons. J. Pharmacol. Exp. Ther. 315, 80–90. doi: 10.1124/jpet.105.090068
Narendran, R., Frankle, W., Mason, N., Laymon, C., Lopresti, B., Price, J., et al. (2009). Positron emission tomography imaging of D 2/3 agonist binding in healthy human subjects with the radiotracer. Synapse 63, 574–584. doi: 10.1002/syn.20633
Narendran, R., Hwang, D., Slifstein, M., Talbot, P., Erritzoe, D., Huang, Y., et al. (2004). In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (-)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist radiotracer [11C]raclopride. Synapse 52, 188–208. doi: 10.1002/syn.20013
Narendran, R., Martinez, D., Mason, N. S., Lopresti, B. J., Himes, M. L., Chen, C.-M., et al. (2011). Imaging of dopamine D2/3 agonist binding in cocaine dependence: a [11C]NPA positron emission tomography study. Synapse 65, 1344–1349. doi: 10.1002/syn.20970
Narendran, R., Mason, N., Laymon, C., Lopresti, B., Velasquez, N., May, M., et al. (2010). A comparative evaluation of the dopamine D2/3 agonist radiotracer [11C](-)-N-Propyl-norapomorphine and Antagonist [11C]Raclopride to measure amphetamine-induced dopamine release in the human striatum. J. Pharmacol. Exp. Ther. 333, 533–539. doi: 10.1124/jpet.109.163501
Narendran, R., Slifstein, M., Hwang, D., Hwang, Y., Scher, E., Reeder, S., et al. (2006). Amphetamine-induced dopamine release: duration of action as assessed with the D2/3 receptor agonist radiotracer (—-)-N-[11C]propyl-norapomorphine ([11C]NPA) in an anesthetized nonhuman primate. Synapse 61, 106–109. doi: 10.1002/syn.20346
Neumeyer, J., Neustadt, B., Oh, K., Weinhardt, K., Boyce, C., Rosenberg, F., et al. (1973). Aporphines. 8. total synthesis and pharmacological evaluation of (±)-Apomorphine, (±)-Apocodeine, (±)-N-n-Propylnorapomorphine, and (±)-N-n–Propylnorapocodeine. J. Med. Chem. 16, 1223–1228. doi: 10.1021/jm00269a601
Newman-Tancredi, A. (2011). Biased agonism at serotonin 5-HT 1A receptors: preferential postsynaptic activity for improved therapy of CNS disorders. Neuropsychiatry 1, 149–164. doi: 10.2217/npy.11.12
Normandin, M., Zheng, M., Lin, K., Mason, N., Lin, S., Ropchan, J., et al. (2015). Imaging the cannabinoid CB1 receptor in humans with. J. Cereb. Blood Flow Metab. 35, 1313–1322. doi: 10.1038/jcbfm.2015.46
Normandin, M. D., Schiffer, W. K., and Morris, E. D. (2012). A linear model for estimation of neurotransmitter response profiles from dynamic PET data. Neuroimage 59, 2689–2699. doi: 10.1016/j.neuroimage.2011.07.002
Nuechterlein, E. B., Ni, L., Domino, E. F., and Zubieta, J.-K. (2016). Nicotine-specific and non-specific effects of cigarette smoking on endogenous opioid mechanisms. Prog. Neuropsychopharmacol. Biol. Psychiatry 69, 69–77. doi: 10.1016/j.pnpbp.2016.04.006
Nummenmaa, L., Manninen, S., Tuominen, L., Hirvonen, J., Kalliokoski, K. K., Nuutila, P., et al. (2015). Adult attachment style is associated with cerebral μ-opioid receptor availability in humans. Hum. Brain Mapp. 36, 3621–3628. doi: 10.1002/hbm.22866
Nummenmaa, L., Saanijoki, T., Tuominen, L., Hirvonen, J., Tuulari, J. J., Nuutila, P., et al. (2018). μ-opioid receptor system mediates reward processing in humans. Nat. Commun. 9:1500. doi: 10.1038/s41467-018-03848-y
Offermanns, S., Wieland, T., Homann, D., Sandmann, J., Bombien, E., Spicher, K., et al. (1994). Transfected muscarinic acetylcholine receptors selectively couple to Gi-type G proteins and Gq/11. Mol. Pharmacol. 45, 890–898.
Olli-Lähdesmäki, T., Kallio, J., and Scheinin, M. (1999). Receptor subtype-induced targeting and subtype-specific internalization of human α 2 -Adrenoceptors in PC12 cells. J. Neurosci. 19, 9281–9288. doi: 10.1523/JNEUROSCI.19-21-09281.1999
Ooms, M., Rietjens, R., Rangarajan, J. R., Vunckx, K., Valdeolivas, S., Maes, F., et al. (2014). Early decrease of type 1 cannabinoid receptor binding and phosphodiesterase 10A activity in vivo in R6/2 Huntington mice. Neurobiol. Aging 35, 2858–2869. doi: 10.1016/j.neurobiolaging.2014.06.010
Otsuka, T., Ito, H., Halldin, C., Takahashi, H., Takano, H., Arakawa, R., et al. (2009). Quantitative PET analysis of the dopamine D2 receptor agonist radioligand 11C-(R)-2-CH3O-N-n-Propylnorapomorphine in the human brain. J. Nucl. Med. 50, 703–710. doi: 10.2967/jnumed.108.058503
Palner, M., Kjaerby, C., Knudsen, G., and Cumming, P. (2011). Effects of unilateral 6-OHDA lesions on [3H]N-propylnorapomorphine binding in striatum ex vivo and vulnerability to amphetamine-evoked dopamine release in rat. Neurochem. Int. 58, 243–247. doi: 10.1016/j.neuint.2010.12.007
Paterson, L. M., Tyacke, R. J., Nutt, D. J., and Knudsen, G. M. (2010). Measuring endogenous 5-HT release by emission tomography: promises and pitfalls. J. Cereb. Blood Flow Metab. 30, 1682–1706. doi: 10.1038/jcbfm.2010.104
Peciña, M., Bohnert, A. S. B., Sikora, M., Avery, E. T., Langenecker, S. A., Mickey, B. J., et al. (2015a). Association between placebo-activated neural systems and antidepressant responses: neurochemistry of placebo effects in major depression. JAMA Psychiatry 72:1087. doi: 10.1001/jamapsychiatry.2015.1335
Peciña, M., Love, T., Stohler, C. S., Goldman, D., and Zubieta, J.-K. (2015b). Effects of the Mu opioid receptor polymorphism (OPRM1 A118G) on pain regulation, placebo effects and associated personality trait measures. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 40, 957–965. doi: 10.1038/npp.2014.272
Pejchal, T., Foley, M. A., Kosofsky, B. E., and Waeber, C. (2002). Chronic fluoxetine treatment selectively uncouples raphe 5-HT 1A receptors as measured by [35 S]-GTP γ S autoradiography. Br. J. Pharmacol. 135, 1115–1122. doi: 10.1038/sj.bjp.0704555
Peng, T., Zysk, J., Dorff, P., Elmore, C. S., Ström, P., Malmquist, J., et al. (2010). D2 receptor occupancy in conscious rat brain is not significantly distinguished with [3H]-MNPA, [3H]-(+)-PHNO, and [3H]-raclopride. Synapse 64, 624–633. doi: 10.1002/syn.20771
Pike, V. W. (2009). PET radiotracers: crossing the blood–brain barrier and surviving metabolism. Trends Pharmacol. Sci. 30, 431–440. doi: 10.1016/j.tips.2009.05.005
Placzek, M. S., Schroeder, F. A., Che, T., Wey, H.-Y., Neelamegam, R., Wang, C., et al. (2018). Discrepancies in kappa opioid agonist binding revealed through PET imaging. ACS Chem. Neurosci. 10, 384–395. doi: 10.1021/acschemneuro.8b00293
Placzek, M. S., Van de Bittner, G. C., Wey, H.-Y., Lukas, S. E., and Hooker, J. M. (2015). Immediate and persistent effects of salvinorin A on the kappa opioid receptor in rodents, monitored in vivo with PET. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 40, 2865–2872. doi: 10.1038/npp.2015.159
Podruchny, T. A., Connolly, C., Bokde, A., Herscovitch, P., Eckelman, W. C., Kiesewetter, D. O., et al. (2003). In vivo muscarinic 2 receptor imaging in cognitively normal young and older volunteers. Synapse 48, 39–44. doi: 10.1002/syn.10165
Prante, O., Tietze, R., Hocke, C., Löber, S., Hübner, H., Kuwert, T., et al. (2008). Synthesis, radiofluorination, and in vitro evaluation of pyrazolo[1,5-a]pyridine-based dopamine D4 receptor ligands: discovery of an inverse agonist radioligand for PET. J. Med. Chem. 51, 1800–1810. doi: 10.1021/jm701375u
Prossin, A. R., Koch, A. E., Campbell, P. L., McInnis, M. G., Zalcman, S. S., and Zubieta, J.-K. (2011). Association of plasma interleukin-18 levels with emotion regulation and μ-opioid neurotransmitter function in major depression and healthy volunteers. Biol. Psychiatry 69, 808–812. doi: 10.1016/j.biopsych.2010.10.014
Pucadyil, T. J., and Chattopadhyay, A. (2007). The human serotonin1A receptor exhibits G-protein-dependent cell surface dynamics. Glycoconj. J. 24, 25–31. doi: 10.1007/s10719-006-9008-x
Quelch, D., De Santis, V., Strege, A., Myers, J., Wells, L., Nutt, D., et al. (2014). Influence of agonist induced internalization on [3H]Ro15-4513 binding-an application to imaging fluctuations in endogenous GABA with positron emission tomography. Synapse 69, 60–65. doi: 10.1002/syn.21780
Rabiner, E. A., Beaver, J., Makwana, A., Searle, G., Long, C., Nathan, P. J., et al. (2011). Pharmacological differentiation of opioid receptor antagonists by molecular and functional imaging of target occupancy and food reward-related brain activation in humans. Mol. Psychiatry 16:785. doi: 10.1038/mp.2011.29
Ramakrishnan, N., Visser, A., Rybczynska, A., Nyakas, C., Luiten, P., Kwizera, C., et al. (2015). Sigma-1 agonist binding in the aging rat brain: a MicroPET study with [11C]SA4503. Mol. Imaging Biol. 18, 588–597. doi: 10.1007/s11307-015-0917-6
Ravasi, L., Tokugawa, J., Nakayama, T., Seidel, J., Sokoloff, L., Eckelman, W., et al. (2012). Imaging of the muscarinic acetylcholine neuroreceptor in rats with the M2 selective agonist [18F] FP-TZTP. Nucl. Med. Biol. 39, 45–55. doi: 10.1016/j.nucmedbio.2011.06.003
Ravert, H., Mathews, W., Musachio, J., Scheffel, U., Finley, P., and Dannals, R. (1999). [11C]methyl 4-[(3,4-dichlorophenyl)acetyl]3-[(1-pyrrolidinyl)methyl]1-piperazinecarboxylate. Nucl. Med. Biol. 26, 737–741. doi: 10.1016/s0969-8051(99)00043-8
Ravert, H., Scheffel, U., Mathews, W., Musachio, J., and Dannals, R. (2002). [11C]GR89696, a potent kappa opiate receptor radioligand; in vivo binding of the R and S enantiomers. Nucl. Med. Biol. 29, 47–53. doi: 10.1016/s0969-8051(01)00285-2
Ray, R., Ruparel, K., Newberg, A., Wileyto, E., Loughead, J., Divgi, C., et al. (2011). Human Mu Opioid Receptor (OPRM1 A118G) polymorphism is associated with brain mu-opioid receptor binding potential in smokers. Proc. Natl. Acad. Sci. U.S.A. 108, 9268–9273. doi: 10.1073/pnas.1018699108
Rbah, L., Leviel, V., and Zimmer, L. (2003). Displacement of the PET ligand 18F-MPPF by the electrically evoked serotonin release in the rat hippocampus. Synapse 49, 239–245. doi: 10.1002/syn.10235
Reboreda, A., Theissen, F. M., Valero-Aracama, M. J., Arboit, A., Corbu, M. A., and Yoshida, M. (2018). Do TRPC channels support working memory? Comparing modulations of TRPC channels and working memory through G-protein coupled receptors and neuromodulators. Behav. Brain Res. 354, 64–83. doi: 10.1016/j.bbr.2018.02.042
Riad, M., Watkins, K. C., Doucet, E., Hamon, M., and Descarries, L. (2001). Agonist-induced internalization of serotonin-1a receptors in the dorsal raphe nucleus (autoreceptors) but not hippocampus (heteroreceptors). J. Neurosci. 21, 8378–8386. doi: 10.1523/JNEUROSCI.21-21-08378.2001
Richfield, E. K., Young, A. B., and Penney, J. B. (1986). Properties of D2 dopamine receptor autoradiography: high percentage of high-affinity agonist sites and increased nucleotide sensitivity in tissue sections. Brain Res. 383, 121–128. doi: 10.1016/0006-8993(86)90013-2
Saanijoki, T., Nummenmaa, L., Tuulari, J. J., Tuominen, L., Arponen, E., Kalliokoski, K. K., et al. (2018). Aerobic exercise modulates anticipatory reward processing via the μ-opioid receptor system. Hum. Brain Mapp. 39, 3972–3983. doi: 10.1002/hbm.24224
Sakata, M., Kimura, Y., Naganawa, M., Oda, K., Ishii, K., Chihara, K., et al. (2007). Mapping of human cerebral sigma1 receptors using positron emission tomography and [11C]SA4503. Neuroimage 35, 1–8. doi: 10.1016/j.neuroimage.2006.11.055
Sanabria-Bohórquez, S. M., Hamill, T. G., Goffin, K., De Lepeleire, I., Bormans, G., Burns, H. D., et al. (2010). Kinetic analysis of the cannabinoid-1 receptor PET tracer [18F]MK-9470 in human brain. Eur. J. Nucl. Med. Mol. Imaging 37, 920–933. doi: 10.1007/s00259-009-1340-5
Sauerberg, P., Olesen, P., Nielsen, S., Treppendahl, S., Sheardown, M., Honore, T., et al. (1992). Novel functional M1 selective muscarinic agonists. Synthesis and structure-activity relationships of 3-(1,2,5-thiadiazolyl)-1,2,5,6-tetrahydro-1-methylpyridines. J. Med. Chem. 35, 2274–2283. doi: 10.1021/jm00090a019
Schaffhausen, J. (2017). What precisely is precision medicine? Trends Pharmacol. Sci. 38, 1–2. doi: 10.1016/j.tips.2016.11.004
Schoultz, B., Hjornevik, T., Willoch, F., Marton, J., Noda, A., Murakami, Y., et al. (2010). Evaluation of the kappa-opioid receptor-selective tracer [11C]GR103545 in awake rhesus macaques. Eur. J. Nucl. Med. Mol. Imaging 37, 1174–1180. doi: 10.1007/s00259-010-1384-6
Schreiber, G., and Avissar, S. (2000). G proteins as a biochemical tool for diagnosis and monitoring treatments of mental disorders. Isr. Med. Assoc. J. 2(Suppl.) 86–91.
Schreiber, G., Golan, M., and Avissar, S. (2009). Beta-arrestin signaling complex as a target for antidepressants and as a depression marker. Drug News Perspect. 22, 467–480.
Scott, D. J., Domino, E. F., Heitzeg, M. M., Koeppe, R. A., Ni, L., Guthrie, S., et al. (2007a). Smoking modulation of μ-Opioid and dopamine D2 receptor-mediated neurotransmission in humans. Neuropsychopharmacology 32, 450–457. doi: 10.1038/sj.npp.1301238
Scott, D. J., Stohler, C. S., Koeppe, R. A., and Zubieta, J.-K. (2007b). Time-course of change in [11C]carfentanil and [11C]raclopride binding potential after a nonpharmacological challenge. Synapse 61, 707–714. doi: 10.1002/syn.20404
Scott, D. J., Stohler, C. S., Egnatuk, C. M., Wang, H., Koeppe, R. A., and Zubieta, J.-K. (2008). Placebo and nocebo effects are defined by opposite opioid and dopaminergic responses. Arch. Gen. Psychiatry 65, 220–231. doi: 10.1001/archgenpsychiatry.2007.34
Searle, G., Beaver, J. D., Comley, R. A., Bani, M., Tziortzi, A., Slifstein, M., et al. (2010). Imaging dopamine D3 receptors in the human brain with positron emission tomography, [11C]PHNO, and a selective D3 receptor antagonist. Biol. Psychiatry 68, 392–399. doi: 10.1016/j.biopsych.2010.04.038
Seeman, P. (2009). Dopamine D2 High receptors measured ex vivo are elevated in amphetamine-sensitized animals. Synapse 63, 186–192. doi: 10.1002/syn.20595
Seeman, P. (2012). Dopamine agonist radioligand binds to both D2High and D2Low receptors, explaining why alterations in D2High are not detected in human brain scans. Synapse 66, 88–93. doi: 10.1002/syn.20987
Seeman, P., Hall, F. S., and Uhl, G. (2007). Increased dopamine D2High receptors in knockouts of the dopamine transporter and the vesicular monoamine transporter may contribute to spontaneous hyperactivity and dopamine supersensitivity. Synapse 61, 573–576. doi: 10.1002/syn.20402
Seeman, P., and Kapur, S. (2003). Anesthetics inhibit high-affinity states of dopamine D2 and other G-linked receptors. Synapse 50, 35–40. doi: 10.1002/syn.10221
Seneca, N., Finnema, S., Farde, L., Gulyás, B., Wikström, H., Halldin, C., et al. (2006). Effect of amphetamine on dopamine D2 receptor binding in nonhuman primate brain: a comparison of the agonist radioligand [11C]MNPA and antagonist [11C]raclopride. Synapse 59, 260–269. doi: 10.1002/syn.20238
Seneca, N., Skinbjerg, M., Zoghbi, S. S., Liow, J., Gladding, R. L., Hong, J., et al. (2008a). Kinetic brain analysis and whole-body imaging in monkey of [11 C]MNPA: a dopamine agonist radioligand. Synapse 62, 700–709. doi: 10.1002/syn.20544
Seneca, N., Zoghbi, S., Skinbjerg, M., Liow, J., Hong, J., Sibley, D., et al. (2008b). Occupancy of dopamine D 2/3 receptors in rat brain by endogenous dopamine measured with the agonist positron. Emiss. Tomogr. Radioligand 62, 756–763. doi: 10.1002/syn.20549
Shalgunov, D. (2017). Development of 18F-Labeled Agonist Radioligands for PET Imaging of the High-Affinity State of Cerebral Dopamine D2/3 Receptors. Master’s thesis, Groningen: University of Groningen.
Shalgunov, V., van Waarde, A., Booij, J., Michel, M. C., Dierckx, R. A. J. O., and Elsinga, P. H. (2019). Hunting for the high-affinity state of G-protein-coupled receptors with agonist tracers: theoretical and practical considerations for positron emission tomography imaging. Med. Res. Rev. 39, 1014–1052. doi: 10.1002/med.21552
Shen, C., Li, H., and Meller, E. (2002). Repeated treatment with antidepressants differentially alters 5-HT1A agonist-stimulated [35S]GTPγS binding in rat brain regions. Neuropharmacology 42, 1031–1038. doi: 10.1016/S0028-3908(02)00064-3
Shimoji, K., Esaki, T., Itoh, Y., Ravasi, L., Cook, M., Jehle, J., et al. (2003). Inhibition of [18F] FP-TZTP binding by loading doses of muscarinic agonists P-TZTP or FP-TZTP in vivo is not due to agonist-induced reduction in cerebral blood flow. Synapse 50, 151–163. doi: 10.1002/syn.10257
Shiue, C.-Y., Bai, L.-Q., Teng, R.-R., Arnett, C. D., Dewey, S. L., Wolf, A. P., et al. (1991). A comparison of the brain uptake of N-(cyclopropyl[11C]methyl) norbuprenorphine ([11C]buprenorphine) and N-(cyclopropyl[11C]methyl) nordiprenorphme ([11C]diprenorphine) in baboon using PET. Int. J. Radiat. Appl. Instrum. Part B Nuclear Med. Biol. 18, 281–288. doi: 10.1016/0883-2897(91)90123-3
Shotbolt, P., Tziortzi, A. C., Searle, G. E., Colasanti, A., van der Aart, J., Abanades, S., et al. (2012). Within-subject comparison of [11C]-(+)-PHNO and [11C]raclopride sensitivity to acute amphetamine challenge in healthy humans. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 32, 127–136. doi: 10.1038/jcbfm.2011.115
Sibley, D. R., De Lean, A., and Creese, I. (1982). Anterior pituitary dopamine receptors. Demonstration of interconvertible high and low affinity states of the D-2 dopamine receptor. J. Biol. Chem. 257, 6351–6361.
Skinbjerg, M., Liow, J.-S., Seneca, N., Hong, J., Lu, S., Thorsell, A., et al. (2010). D2 dopamine receptor internalization prolongs the decrease of radioligand binding after amphetamine: a PET study in a receptor internalization-deficient mouse model. Neuroimage 50, 1402–1407. doi: 10.1016/j.neuroimage.2010.01.055
Skinbjerg, M., Namkung, Y., Halldin, C., Innis, R. B., and Sibley, D. R. (2009). Pharmacological characterization of 2-methoxy- N -propylnorapomorphine’s interactions with D 2 and D 3 dopamine receptors. Synapse 63, 462–475. doi: 10.1002/syn.20626
Skinbjerg, M., Sibley, D. R., Javitch, J. A., and Abi-Dargham, A. (2012). Imaging the high-affinity state of the dopamine D2 receptor in vivo: fact or fiction? Biochem. Pharmacol. 83, 193–198. doi: 10.1016/j.bcp.2011.09.008
Smith, Y. R., Zubieta, J.-K., del Carmen, M. G., Dannals, R. F., Ravert, H. T., Zacur, H. A., et al. (1998). Brain opioid receptor measurements by positron emission tomography in normal cycling women: relationship to luteinizing hormone pulsatility and gonadal steroid Hormones1. J. Clin. Endocrinol. Metab. 83, 4498–4505. doi: 10.1210/jcem.83.12.5351
Steiger, C., Finnema, S., Raus, L., Schou, M., Nakao, R., Suzuki, K., et al. (2009). A two-step one-pot radiosynthesis of the potent dopamine D 2/D 3 agonist. J. Label Compd. Radiopharm. 52, 158–165. doi: 10.1002/jlcr.1583
Suehiro, M., Underwood, M., Arango, V., Wang, T., Kassir, S., Bakalian, M., et al. (1998). In vivo biodistribution of a radiotracer for imaging serotonin-1a receptor sites with pet: [11C]Ly274601. Life Sci. 63, 1533–1542. doi: 10.1016/s0024-3205(98)00420-2
Talbot, P. S., Narendran, R., Butelman, E. R., Huang, Y., Ngo, K., Slifstein, M., et al. (2005). 11C-GR103545, a radiotracer for imaging kappa-opioid receptors in vivo with PET: synthesis and evaluation in baboons. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 46, 484–494.
Thathiah, A., and De Strooper, B. (2011). The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat. Rev. Neurosci. 12, 73–87. doi: 10.1038/nrn2977
Thomae, D., Morley, T., Hamill, T., Carroll, V., Papin, C., Twardy, N., et al. (2014). Automated one-step radiosynthesis of the CB1receptor imaging agent [18F] MK-9470. J. Label. Compd. Radiopharm. 57, 611–614. doi: 10.1002/jlcr.3219
Thorell, J. O. (1995). (R)-[N-11C-methyl]11-hydroxy-10-methylaporphine as a ligand for 5-HT1A receptors: synthesis and evaluation of its biodistribution in monkey with PET. J. Label Compd. Radiopharm. 44:179.
Titeler, M., Lyon, R., Kuhar, M., Frost, J., Dannals, R., Leonhardt, S., et al. (1989). μ Opiate receptors are selectively labelled by [3H]carfentanil in human and rat brain. Eur. J. Pharmacol. 167, 221–228. doi: 10.1016/0014-2999(89)90582-7
Tokunaga, M., Seneca, N., Shin, R., Maeda, J., Obayashi, S., Okauchi, T., et al. (2009). Neuroimaging and physiological evidence for involvement of glutamatergic transmission in regulation of the striatal dopaminergic system. J. Neurosci. 29, 1887–1896. doi: 10.1523/jneurosci.2559-08.2009
Tomasi, G., Nabulsi, N., Zheng, M., Weinzimmer, D., Ropchan, J., Blumberg, L., et al. (2013). Determination of in vivo Bmax and Kd for 11C-GR103545, an agonist PET Tracer for κ-Opioid receptors: a study in nonhuman primates. J. Nucl. Med. 54, 600–608. doi: 10.2967/jnumed.112.112672
Tsukada, H., Miyasato, K., Kakiuchi, T., Nishiyama, S., Harada, N., and Domino, E. F. (2002). Comparative effects of methamphetamine and nicotine on the striatal [11C]raclopride binding in unanesthetized monkeys. Synapse 45, 207–212. doi: 10.1002/syn.10102
Tsukada, H., Ohba, H., Nishiyama, S., and Kakiuchi, T. (2011). Differential effects of stress on [11C]raclopride and [11C]MNPA binding to striatal D2/D3 dopamine receptors: a PET study in conscious monkeys. Synapse 65, 84–89. doi: 10.1002/syn.20845
Tuominen, L., Tuulari, J., Karlsson, H., Hirvonen, J., Helin, S., Salminen, P., et al. (2015). Aberrant mesolimbic dopamine-opiate interaction in obesity. Neuroimage 122, 80–86. doi: 10.1016/j.neuroimage.2015.08.001
Tuulari, J. J., Tuominen, L., de Boer, F. E., Hirvonen, J., Helin, S., Nuutila, P., et al. (2017). Feeding releases endogenous opioids in humans. J. Neurosci. Off. J. Soc. Neurosci. 37, 8284–8291. doi: 10.1523/JNEUROSCI.0976-17.2017
Udo de Haes, J. I., Harada, N., Elsinga, P. H., Maguire, R. P., and Tsukada, H. (2006). Effect of fenfluramine-induced increases in serotonin release on [18F]MPPF binding: a continuous infusion PET study in conscious monkeys. Synapse 59, 18–26. doi: 10.1002/syn.20209
Van de Bittner, G. C., Ricq, E. L., and Hooker, J. M. (2014). A Philosophy for CNS radiotracer design. Acc. Chem. Res. 47, 3127–3134. doi: 10.1021/ar500233s
Van der Schueren, B. J., Van Laere, K., Gérard, N., Bormans, G., and De Hoon, J. N. (2012). Interictal type 1 cannabinoid receptor binding is increased in female migraine patients. Headache J. Head Face Pain 52, 433–440. doi: 10.1111/j.1526-4610.2011.02030.x
van der Werf, J. F., Sebens, J. B., Vaalburg, W., and Korf, J. (1983). In vivo binding of N-n-propylnorapomorphine in the rat brain: regional localization, quantification in striatum and lack of correlation with dopamine metabolism. Eur. J. Pharmacol. 87, 259–270. doi: 10.1016/0014-2999(83)90336-9
Van Laere, K., Casteels, C., Lunskens, S., Goffin, K., Grachev, I. D., Bormans, G., et al. (2012). Regional changes in type 1 cannabinoid receptor availability in Parkinson’s disease in vivo. Neurobiol. Aging 33:620.e1-8. doi: 10.1016/j.neurobiolaging.2011.02.009
Van Laere, K., Goffin, K., Bormans, G., Casteels, C., Mortelmans, L., de Hoon, J., et al. (2009). Relationship of Type 1 cannabinoid receptor availability in the human brain to novelty-seeking temperament. Arch. Gen. Psychiatry 66:196. doi: 10.1001/archgenpsychiatry.2008.530
Van Laere, K., Goffin, K., Casteels, C., Dupont, P., Mortelmans, L., de Hoon, J., et al. (2008a). Gender-dependent increases with healthy aging of the human cerebral cannabinoid-type 1 receptor binding using [18F]MK-9470 PET. Neuroimage 39, 1533–1541. doi: 10.1016/j.neuroimage.2007.10.053
Van Laere, K., Koole, M., Sanabria Bohorquez, S., Goffin, K., Guenther, I., Belanger, M., et al. (2008b). Whole-body biodistribution and radiation dosimetry of the human cannabinoid type-1 receptor ligand 18F-MK-9470 in healthy subjects. J. Nucl. Med. 49, 439–445. doi: 10.2967/jnumed.107.047290
Van Laere, K., Sanabria-Bohorquez, S., Mozley, D., Burns, D., Hamill, T., Van Hecken, A., et al. (2013). 11C-MK-8278 PET as a tool for pharmacodynamic brain occupancy of histamine 3 receptor inverse agonists. J. Nucl. Med. 55, 65–72. doi: 10.2967/jnumed.113.122515
Van Laere, K. J., Sanabria-Bohorquez, S. M., Mozley, D. P., Burns, D. H., Hamill, T. G., Van Hecken, A., et al. (2014). 11C-MK-8278 PET as a tool for pharmacodynamic brain occupancy of histamine 3 receptor inverse agonists. J. Nucl. Med. 55, 65–72. doi: 10.2967/jnumed.113.122515
van Oosten, E. M., Wilson, A. A., Stephenson, K. A., Mamo, D. C., Pollock, B. G., Mulsant, B. H., et al. (2009). An improved radiosynthesis of the muscarinic M2 radiopharmaceutical, [18F]FP-TZTP. Appl. Radiat. Isot. 67, 611–616. doi: 10.1016/j.apradiso.2008.12.015
Vandecapelle, M., Dumont, F., De Vos, F., Strijckmans, K., Leysen, D., Audenaert, K., et al. (2004). Synthesis and preliminaryin vivo evaluation of 4-[18F] fluoro-N-2-[4-(6-trifluoromethylpyridin-2-yl)piperazin-1-yl]ethylbenzamide, a potential PET radioligand for the 5-HT1A receptor. J. Label. Compd. Radiopharm. 47, 531–542. doi: 10.1002/jlcr.837
Vidal, B., Fieux, S., Redouté, J., Villien, M., Bonnefoi, F., Le Bars, D., et al. (2018). In vivo biased agonism at 5-HT1A receptors: characterisation by simultaneous PET/MR imaging. Neuropsychopharmacology 43, 2310–2319. doi: 10.1038/s41386-018-0145-2
Vidal, B., Sebti, J., Verdurand, M., Fieux, S., Billard, T., Streichenberger, N., et al. (2016). Agonist and antagonist bind differently to 5-HT 1A receptors during Alzheimer’s disease: a post-mortem study with PET radiopharmaceuticals. Neuropharmacology 109, 88–95. doi: 10.1016/j.neuropharm.2016.05.009
Wager, T. D., Scott, D. J., and Zubieta, J.-K. (2007). Placebo effects on human mu-opioid activity during pain. Proc. Natl. Acad. Sci. U.S.A. 104, 11056–11061. doi: 10.1073/pnas.0702413104
Wand, G. S., Weerts, E. M., Kuwabara, H., Wong, D. F., Xu, X., and McCaul, M. E. (2012). The relationship between naloxone-induced cortisol and mu opioid receptor availability in mesolimbic structures is disrupted in alcohol dependent subjects. Alcohol Fayettev. N 46, 511–517. doi: 10.1016/j.alcohol.2012.04.006
Watson, J., Collin, L., Ho, M., Riley, G., Scott, C., Selkirk, J. V., et al. (2000). 5-HT1A receptor agonist-antagonist binding affinity difference as a measure of intrinsic activity in recombinant and native tissue systems. Br. J. Pharmacol. 130, 1108–1114. doi: 10.1038/sj.bjp.0703394
Weerts, E. M., Kim, Y. K., Wand, G. S., Dannals, R. F., Lee, J. S., Frost, J. J., et al. (2008). Differences in delta- and mu-opioid receptor blockade measured by positron emission tomography in naltrexone-treated recently abstinent alcohol-dependent subjects. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 33, 653–665. doi: 10.1038/sj.npp.1301440
Weerts, E. M., Wand, G. S., Kuwabara, H., Munro, C. A., Dannals, R. F., Hilton, J., et al. (2011). Positron emission tomography imaging of mu- and delta-opioid receptor binding in alcohol-dependent and healthy control subjects. Alcohol. Clin. Exp. Res. 35, 2162–2173. doi: 10.1111/j.1530-0277.2011.01565.x
Weerts, E. M., Wand, G. S., Kuwabara, H., Xu, X., Frost, J. J., Wong, D. F., et al. (2014). Association of smoking with μ-opioid receptor availability before and during naltrexone blockade in alcohol-dependent subjects. Addict. Biol. 19, 733–742. doi: 10.1111/adb.12022
Willeit, M., Ginovart, N., Graff, A., Rusjan, P., Vitcu, I., Houle, S., et al. (2008). First human evidence of d-amphetamine induced displacement of a D2/3 agonist radioligand: a [11C]-(+)-PHNO positron emission tomography study. Neuropsychopharmacology 33, 279–289. doi: 10.1038/sj.npp.1301400
Willeit, M., Ginovart, N., Kapur, S., Houle, S., Hussey, D., Seeman, P., et al. (2006). High-Affinity states of human brain dopamine D2/3 receptors imaged by the agonist [11C](+)-PHNO. Biol. Psychiatry 59, 389–394. doi: 10.1016/j.biopsych.2005.09.017
Wilson, A. A., McCormick, P., Kapur, S., Willeit, M., Garcia, A., Hussey, D., et al. (2005). Radiosynthesis and evaluation of [11 C]-(+)-4-Propyl-3,4,4a,5,6,10b-hexahydro-2 H -naphtho[1,2- b][1,4]oxazin-9-ol as a potential radiotracer for in vivo imaging of the dopamine D2 high-affinity state with positron emission tomography. J. Med. Chem. 48, 4153–4160. doi: 10.1021/jm050155n
Wong, D., Kuwabara, H., Horti, A., Raymont, V., Brasic, J., Guevara, M., et al. (2010). Quantification of cerebral cannabinoid receptors subtype 1 (CB1) in healthy subjects and schizophrenia by the novel PET radioligand [11C]OMAR. Neuroimage 52, 1505–1513. doi: 10.1016/j.neuroimage.2010.04.034
Wong, D. F., Kuwabara, H., Horti, A. G., Kumar, A., Brasic, J., Ye, W., et al. (2008). “Imaging of Human Cannaboid CB1 Type Human Receptors with [11C]OMAR,” in Proceedings of the 55th Annual Meeting of the Society of Nuclear Medicine, New Orleans, LA.
Yang, K.-C., Stepanov, V., Martinsson, S., Ettrup, A., Takano, A., Knudsen, G. M., et al. (2017). Fenfluramine reduces [11C]Cimbi-36 binding to the 5-HT2A receptor in the nonhuman primate brain. Int. J. Neuropsychopharmacol. 20, 683–691. doi: 10.1093/ijnp/pyx051
Yokoyama, C., Mawatari, A., Kawasaki, A., Takeda, C., Onoe, K., Doi, H., et al. (2016). Marmoset serotonin 5-HT 1A receptor mapping with a biased agonist PET probe 18 F-F13714: comparison with an antagonist tracer 18 F-MPPF in awake and anesthetized states. Int. J. Neuropsychopharmacol. 19:yw079. doi: 10.1093/ijnp/pyw079
Zijlstra, S., van der Worp, H., Wiegman, T., Visser, G. M., Korf, J., and Vaalburg, W. (1993). Synthesis and in vivo distribution in the rat of a dopamine agonist: N-([11C]methyl)norapomorphine. Nucl. Med. Biol. 20, 7–12. doi: 10.1016/0969-8051(93)90131-d
Zimmer, L. (2016). Pharmacological agonists for more-targeted CNS radio-pharmaceuticals. Oncotarget 7, 80111–80112. doi: 10.18632/oncotarget.13418
Zimmer, L., Fournet, G., Benoît, J., Guillaumet, G., and Le Bars, D. (2003). Carbon-11 labelling of 8[[3-[4-(2-[(11)C]methoxyphenyl)piperazin-1-yl]-2-hydroxypropyl]oxy]thiochroman, a presynaptic 5-HT(1A) receptor agonist, and its in vivo evaluation in anaesthetised rat and in awake cat. Nucl. Med. Biol. 30, 541–546. doi: 10.1016/s0969-8051(03)00027-1
Zimmer, L., Mauger, G., Le Bars, D., Bonmarchand, G., Luxen, A., and Pujol, J.-F. (2002). Effect of endogenous serotonin on the binding of the 5-HT1A PET ligand 18F-MPPF in the rat hippocampus: kinetic beta measurements combined with microdialysis. J. Neurochem. 80, 278–286. doi: 10.1046/j.0022-3042.2001.00696.x
Zimmer, L., Riad, M., Rbah, L., Belkacem-Kahlouli, A., Le Bars, D., Renaud, B., et al. (2004). Toward brain imaging of serotonin 5-HT1A autoreceptor internalization. Neuroimage 22, 1421–1426. doi: 10.1016/j.neuroimage.2004.03.020
Zubieta, J., Dannals, R., and Frost, J. (1999). Gender and age influences on human brain mu-opioid receptor binding measured by PET. Am. J. Psychiatry 156, 842–848. doi: 10.1176/ajp.156.6.842
Zubieta, J., Gorelick, D., Stauffer, R., Ravert, H., Dannals, R., and Frost, J. (1996). Increased mu opioid receptor binding detected by PET in cocaine–dependent men is associated with cocaine craving. Nat. Med. 2, 1225–1229. doi: 10.1038/nm1196-1225
Zubieta, J., Greenwald, M. K., Lombardi, U., Woods, J. H., Kilbourn, M. R., Jewett, D. M., et al. (2000). Buprenorphine-induced changes in mu-opioid receptor availability in male heroin-dependent volunteers: a preliminary study. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 23, 326–334. doi: 10.1016/S0893-133X(00)00110-X
Keywords: radiopharmaceutical, receptor, agonist, Positron emission tomography (PET), neuroimaging
Citation: Colom M, Vidal B and Zimmer L (2019) Is There a Role for GPCR Agonist Radiotracers in PET Neuroimaging? Front. Mol. Neurosci. 12:255. doi: 10.3389/fnmol.2019.00255
Received: 25 July 2019; Accepted: 02 October 2019;
Published: 18 October 2019.
Edited by:
Philippe De Deurwaerdere, Université de Bordeaux, FranceReviewed by:
Diana Martinez, Columbia University, United StatesHartmut Lüddens, Johannes Gutenberg University Mainz, Germany
Copyright © 2019 Colom, Vidal and Zimmer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luc Zimmer, bHVjLnppbW1lckB1bml2LWx5b24xLmZy