 Mitsuto Sato1,2
Mitsuto Sato1,2 Hotake Takizawa1
Hotake Takizawa1 Akinori Nakamura3
Akinori Nakamura3 Bradley J. Turner4
Bradley J. Turner4 Fazel Shabanpoor4
Fazel Shabanpoor4 Yoshitsugu Aoki1*
Yoshitsugu Aoki1*- 1Department of Molecular Therapy, National Institute of Neuroscience, National Center of Neurology and Psychiatry, Kodaira, Japan
- 2Department of Medicine (Neurology and Rheumatology), Shinshu University School of Medicine, Matsumoto, Japan
- 3Department of Clinical Research, National Hospital Organization Matsumoto Medical Center, Matsumoto, Japan
- 4The Florey Institute of Neuroscience and Mental Health, University of Melbourne, Parkville, VIC, Australia
Neuromuscular and neurodegenerative diseases are mostly modeled using genetically modified animals such as mice. However, animal models do not recapitulate all the phenotypes that are specific to human disease. This is mainly due to the genetic, anatomical and physiological difference in the neuromuscular systems of animals and humans. The emergence of direct and indirect human somatic cell reprogramming technologies may overcome this limitation because they enable the use of disease and patient-specific cellular models as enhanced platforms for drug discovery and autologous cell-based therapy. Induced pluripotent stem cells (iPSCs) and urine-derived stem cells (USCs) are increasingly employed to recapitulate the pathophysiology of various human diseases. Recent cell-based modeling approaches utilize highly complex differentiation systems that faithfully mimic human tissue- and organ-level dysfunctions. In this review, we discuss promising cellular models, such as USC- and iPSC-based approaches, that are currently being used to model human neuromuscular and neurodegenerative diseases.
Introduction
Understanding the mechanisms underlying the pathology of human disease is essential for drug development. Studies on fundamental principles of human disease and testing of therapeutic modalities are commonly conducted in mouse models (Partridge, 2013). The use of animal models to mimic human diseases presents challenges arising from genetic and physiological differences between humans and animals, in pathologic mechanisms and therapeutic effect. However, due to the lack of biologically-relevant human disease models, animal models represent the only available approach, which could recapitulate, a physiological and anatomical condition in vivo. In contrast, cellular models of human disease, which recapitulate the pathophysiology of various neuromuscular and neurodegenerative diseases, bring us closer to achieving personalized therapy for the individual patient.
It is still being debated as to which cells can be used for cell-based studies. For example, primary myoblasts are usually employed for the study of Duchenne muscular dystrophy (DMD). DMD is a devastating muscle disorder caused by frameshift mutations in the DMD gene. The responsible gene (DMD) encodes the subsarcolemmal protein, dystrophin. In DMD, various frameshift mutations in DMD prevent the full translation of dystrophin. Primary myoblasts express enough levels of dystrophin mRNA but do not express dystrophin protein. This renders them a suitable surrogate for DMD, but collecting myoblasts requires invasive muscle biopsy. Dystrophin mRNA has also been detected in lymphocytes and fibroblasts, as shown by nested real-time polymerase chain reaction (RT-PCR). However, dystrophin protein is not detected in these cells because of illegitimate dystrophin transcripts present at a very low level. Fibroblasts can be converted to myotubes by virally-mediated MyoD1 transduction. Although the transduced cells express dystrophin mRNA and protein, achieving enough expression at the protein level remains challenging. In our previous study, we overcame this issue by designing an in vitro assay based on MyoD1-converted fibroblasts isolated using fluorescence-activated cell sorting (FACS) to determine patient eligibility before clinical trials (Saito et al., 2010).
In the neuromuscular diseases, it is challenging to generate a disease model that faithfully represent the patient’s pathology, and there are situations in which enough efficacy and safety cannot be confirmed in clinical trials. This is in contradiction to the results in animal models. Human-induced pluripotent stem cells (iPSCs) are an attractive platform for overcoming these limitations. Therefore, patient-specific iPSCs can provide unlimited disease-relevant cells in a personalized manner. This serves as an essential resource for cell types previously considered rare or inaccessible, including skeletal and cardiac myocytes, neurons, and glia. However, there are some limitations about genome instability and epigenetic memory associated with the reprogramming of iPSC, integrity of iPSC derivatives, inherent biological and technical variability between iPSC lines and differentiated cells, and modeling of diseases that are epigenetically influenced by environmental factors or largely sporadic in etiology. Disease modeling using somatic stem cells have also been conducted as a way to solve epigenetic and environmental factors.
In this review, we discuss the current status of cellular modeling of neuromuscular and neurodegenerative diseases, and how such models can contribute towards developing precision therapies for patients with these diseases. In addition, we review a new approach to disease modeling based on urine-derived stem cells (USCs) that is used as a model for neuromuscular disease.
Stem Cells Used for Modeling Disease
Stem cells are a valuable research tool for basic, pre-clinical, and clinical studies. Stem cells are defined by two essential characteristics; one is the ability to divide indefinitely and self-replicate, and the other is the ability to differentiate into mature cells under appropriate conditions and specific signals (Malaver-Ortega et al., 2012). According to differentiation potential, stem cells are classified as follows; totipotent, pluripotent, multipotent, oligopotent, and unipotent (Malaver-Ortega et al., 2012). Totipotent and pluripotent cells, which can differentiate into three embryonic lineages and change to any cell type, correspond to embryonic stem cells. Pluripotent stem cells can be obtained from adult somatic cells by incorporating pluripotent transcription factors into the cell’s genome. These cells are called iPSCs that have been fully reprogrammed to achieve an induced pluripotent state. Somatic stem cells, multipotent cells that can differentiate into a limited number of mature cell types, can be found among the tissues such as the brain, skeletal muscle, skin, bone marrow, blood, adipose tissue, and liver. These cells have the role of repairing damaged tissue and obtaining tissue homeostasis when tissue damage occurs. One type of somatic stem cell is a mesenchymal stem cell (MSC) that can differentiate into various mesodermal cells such as osteoblasts, chondrocytes, muscle cells, and adipocytes (Nombela-Arrieta et al., 2011). MSCs are an example of multipotent stem cells that are characterized by adherence to plastic surfaces with a wide range of proliferative potentials in vitro and in vivo. Lymphoid and myeloid cells are called oligopotent stem cells, and skeletal muscle satellite cells are examples of unipotent cells involved in muscle regeneration. These stem cells are used in cellular modeling of neuromuscular diseases, especially iPSCs are extensively applied. The generation of disease model cells with somatic cells has technical difficulties, and the number of reports on this approach is limited (Grath and Dai, 2019).
Modeling of Muscle and Neuronal Diseases Using iPSCs
Human iPSCs are an attractive platform for overcoming the limitations of animal models in disease modeling and drug discovery. In 2006, a study from Japan showed that murine adult fibroblasts could be successfully reprogrammed by the introduction of four transcription genes including Oct3/4, Sox2, Klf4, and c-Myc via retroviral vectors (Takahashi and Yamanaka, 2006). In 2007, differentiated human somatic cells were reprogrammed to enter a pluripotent state allowing for the creation of patient and disease-specific stem cells (Takahashi et al., 2007). iPSCs have the capacity for self-renewal and differentiation and can also be directly generated from skin fibroblasts and blood cells of the patients as well as from other somatic cell sources. However, recently developed approaches employ lymphocytes, squamous cells, and urine-derived cells, which can be obtained in a less invasive manner. iPSCs can differentiate into almost any cell type including skeletal and cardiac myocytes, neurons, and glias. Because these cell lines are patient-specific, they are expected to recapitulate disease-specific phenotypes and elucidate the molecular mechanisms that drive neuromuscular and neurodegenerative diseases.
Obtaining tissues from patients with muscle and neuronal diseases is difficult because muscle biopsy is invasive, and brain biopsy is almost impractical, and involve risks such as pain, bleeding, infection, anesthesia-related complications, and seizures. Therefore, numerous studies are developing methods to derive myogenic and neuronal cells from iPSCs.
There are two different approaches that are commonly used to differentiate iPSCs into myogenic precursor cells (Kodaka et al., 2017). One approach involves overexpressing myogenic transcription factors, MyoD1 and Pax7, in iPSCs using integrated vectors such as lentiviruses (Maffioletti et al., 2015). This approach is highly efficient, but vector integration can lead to genotoxicity. Another method involves mimicking key signaling events such as dual modulation of the Wnt and bone morphogenetic protein signaling pathways to induce myogenesis in iPSCs (Chal et al., 2016). This direct reprogramming approach requires a month to generate robust myogenesis but avoids the need for genetic modification or cell sorting, thereby enabling abundant production of myogenic promoters for therapeutic applications.
Although optimized methods of iPSC differentiation induction have been developed (Revilla et al., 2016), the protocol for neuronal differentiation induction varies greatly depending on the desired cell type. Neuronal differentiation, development, and maturation are modeled on the progression of chemical signaling that occurs in vivo. The appropriate composition, concentration, and timing of the growth factor signals induce the differentiation of the target neuron. Furthermore, in order to convert to fully differentiated cells with sufficient differentiation efficiency, it is necessary to consider the environment suitable for the development and maturation of the target neuron (Engel et al., 2016). Evaluation using a disease-specific marker has been reported to have a differentiation efficiency of around 90% in cholinergic neurons (Crompton et al., 2013) and astrocytes (Krencik et al., 2011; Serio et al., 2013).
Using current protocols, iPSCs can be differentiated into cells with phenotypes resembling those of dopaminergic, glutamatergic, GABAergic, and motor neurons, those of medium spiny neurons of the striatum, and those of glial progenitors (Ross and Akimov, 2014). iPSCs are used to study various neuromuscular and neurodegenerative diseases such as spinal muscular atrophy (SMA; Sareen et al., 2012), amyotrophic lateral sclerosis (ALS; Egawa et al., 2012), Huntington’s (Kaye and Finkbeiner, 2013), Parkinson’s disease (PD; Devine et al., 2011), and Alzheimer’s disease (AD; Ooi et al., 2013).
In the study of the effects of a single genetic abnormality, it is necessary to consider the genetic background and disease-related mutations that are thoroughly permeated in the iPSCs used. Since disease-specific iPSCs exhibit a disease phenotype, their genetic background might be considered to be permissible in the phenotype. But in the genome-edited wild-type iPSCs, if a genetic background is not taken into account, the disease state is not accurately reflected (Musunuru et al., 2018). Genomic editing techniques such as CRISPR/Cas9 are currently used to minimize these variations due to genetic background. That is, use genome editing techniques to correct genetic abnormalities in patient-derived cells or introduce putative genetic abnormalities into cells derived from healthy individuals. By creating two patterns of isogenic cell pairs and comparing their phenotypes, it is thought that the underlying pathological mechanisms can be understood in more detail (Bassett, 2017).
Although iPSCs are excellent for cellular modeling in neuromuscular and neurodegenerative diseases, some limitations still remain and hinder the use of iPSC-based assays. First, collecting somatic cells for iPSC preparation may require invasive procedures. For example, the harvesting of patient-derived fibroblasts requires a skin biopsy, which is not ideal for young patients. Inducing iPSC differentiation into specific cells can be time-consuming, and may require special techniques and equipments. Differentiation efficiency of iPSCs derived from a single patient can vary among clones. Additionally, these iPSCs are heterogeneous which complicates reproducibility of directed differentiation and analyses such as high-throughput screening. Future studies will increase iPSC homogeneity which will improve differentiation efficiency. Methods used to differentiate iPSC into neuronal cells often require long-term culture during which the cells develop mature functional properties. iPSCs may require 1–2 months to differentiate into dopamine neurons, often achieving only 10–20% on the tyrosine hydroxylase index, which is used as an indicator of dopaminergic neurons (Playne and Connor, 2017). Cerebellar Purkinje cells are challenging to culture in vitro because of their large size, complex morphology, unique firing properties, and extended period of maturation (over 150 days; Watson et al., 2015). Patient-derived iPSCs used to model neurologic diseases need to be developed with greater efficiency than one provided by currently available methods. Additionally, several studies have shown that iPSC-derived neurons recapitulate only early-, but not late-onset, phenotypes of neurologic diseases (Nguyen et al., 2011; Patterson et al., 2012). This is primarily due to the fetal nature and immature phenotype of iPSC-derived neurons (Ho et al., 2016). The process of iPSC reprogramming, which involves an embryo-like pluripotent state, results in the loss of specific age-related characteristics (Lapasset et al., 2011). Environmental and aging-related factors also present a significant risk in late-onset neuromuscular and neurodegenerative diseases. Therefore, it is essential to establish cellular models corresponding to the fundamental features of these diseases.
Direct Conversion of Somatic Cells Into Neurons and Muscle Cells
Previous studies have examined whether one differentiated cell type can be directly converted into another desired cell type by genetic manipulation without passing through an intermediate or pluripotent state. Such direct differentiation of mature somatic cells into other cell types was first reported by Lassar and co-workers who showed that introducing the MyoD1 transcription factor can convert fibroblasts into skeletal muscle cells (Davis et al., 1987; Tapscott et al., 1988). Thereafter, Xie et al. (2004) differentiated mature B lymphocytes into macrophages, Zhou et al. (2008) transdifferentiated exocrine cells into pancreatic endocrine cells, Ieda et al. (2010) converted fibroblasts into functional cardiomyocytes. Currently, direct cell conversion is not limited to cell types originating from the same germ layer. Human fibroblasts and hepatocytes can also be converted into functional neurons using transcription factors and/or microRNAs. Such direct cell conversion generates targeted cell types more rapidly than do iPSC-based techniques. However, methods to verify cellular identity, uniform phenotype, functionality, and safety throughout the process of transformation have not yet been established.
Several studies have shown that brain neurons and cardiomyocytes converted directly from somatic cells preserve the cellular ageing markers and possibly even the state of maturation (Qian et al., 2012; Mertens et al., 2015; Huh et al., 2016). Specifically, Tang et al. (2017) have shown that motor neurons, generated by direct reprogramming from fibroblasts can maintain the characteristics of aging donors including extensive DNA damage, loss of heterochromatin and nuclear tissue, and increased SA-β-Gal activity. Preservation of these characteristics has not been observed in iPSC-based models. Furthermore, Liu et al. (2016) demonstrated that direct reprogramming of motor neurons from fibroblasts maintain the biological age of ALS patients, showing degenerative morphology, hypoactivity, and reduced survival. These studies indicate that somatic cells converted directly into neurons without undergoing the intermediate pluripotent state may retain the age-related biochemical phenotype of the donor. As mentioned above, in neurodegenerative diseases, ageing and environmental factors influence the onset and progression of the disease state. Therefore, maintenance of the age-related phenotype is essential in the modeling of these diseases. Overall, these findings suggest that neurons obtained directly from converted somatic cells may be more appropriate than iPSCs for modeling of neurological diseases.
Using Urine-Derived Stem Cells to Model Neuromuscular and Neurodegenerative Diseases
Cells used in in vitro disease research should be obtained from patients of all ages, genders, and genetic origin by procedures that are non-invasive, low-cost, and straightforward to implement (Zhang et al., 2008). Obtaining cells from urine presents a non-invasive approach, and urine is a readily available and nearly unlimited source of biological samples. In recent years, cells with stem-like characteristics have been identified in urine samples and have been recognized as useful materials in disease modeling (Falzarano and Ferlini, 2019).
Characterization of Urine-Derived Stem Cells
USCs are progenitor cells that can self-renew and differentiate (Zhang et al., 2008). USCs can be induced to differentiate into several cell types, including endothelial cells, uroepithelial cells, smooth muscle cells, neural stem cells, and beta cells (Bharadwaj et al., 2011). USCs are thought to originate specifically from kidney glomerular parietal epithelial cells (PECs). USCs isolated from the upper urinary tract and voided USCs are similar in morphology, cell growth pattern, and differentiation potential (Zhang et al., 2014). Furthermore, USCs from a woman who received a kidney transplanted from a male donor contained a Y chromosome and exhibited normal kidney cell markers (PAX2 and PAX8; Bharadwaj et al., 2013). These findings indicate that USCs originate from the kidney and/or the upper urinary tract. USCs express specific genes, protein markers (synaptopodin and podocin) and a high percentage of CD146+/CD31− that expressed in glomerular wall cells and podocytes (Bharadwaj et al., 2013). These markers are not detected in other cells of the urinary system, such as urinary tract epithelium and smooth muscle cells of the bladder and ureter (Bharadwaj et al., 2013). PECs have been reported to self-renew and regenerate podocytes and proximal tubule cells (Sagrinati et al., 2006; Poulsom and Little, 2009; Miesen et al., 2017). These facts strongly support that USCs are derived from PECs. USCs show high expandability that is comparable to that of other widely used stem cells such as bone marrow stem cells, blood progenitor cells, keratinocyte progenitor cells, umbilical cord stem cells, and adipose-derived stem cells (Terstegge et al., 2007; Zhang et al., 2014; Abdelalim and Emara, 2015; Guan et al., 2015). USCs express high levels of mesenchymal stem-cell markers, such as CD44, CD73, CD29, CD105, CD166, CD90, and CD13 (He et al., 2016), and pluripotent stem cell markers including POU5F1 or Oct 3/4, c-Myc, SSEA-1/4, and Klf-4 (Bharadwaj et al., 2011). USCs are multipotent and can differentiate into cells of mesodermal, endodermal and ectodermal lineage (Bharadwaj et al., 2013). USCs also show a high proliferative ability that is comparable to that of other commonly used stem cells such as bone marrow stem cells (Zhang et al., 2014), umbilical cord stem cells (Liu et al., 2018), and adipose-derived stem cells (Kang et al., 2015).
Isolation of USCs is straightforward and reproducible. The method for USC isolation has been described previously (Zhou et al., 2012). In our previous study, we used an existing protocol with some modifications (Takizawa et al., 2019); briefly, our methods are described in Figure 1. Urine collection is a straightforward, repeatable procedure that is non-invasive to the patient. Additionally, reducing the costs associated with cell culture presents considerable advantages. The isolation of USCs requires simple centrifugation and standard culture plates without particular substrates, which decreases isolating cost to less than US$70 per sample (Pavathuparambil Abdul Manaph et al., 2018). Conversely, iPSC reprogramming requires expertise and various types of equipment, raising the cost to more than US$120–200 per sample (Beers et al., 2015).
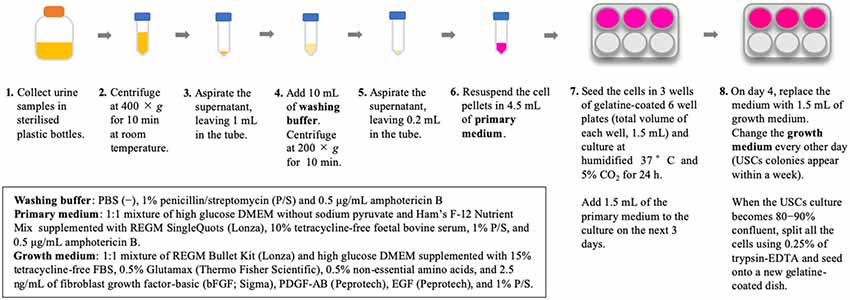
Figure 1. Method for isolation of urine-derived stem cells (USCs).
Application of Urine-Derived Stem Cells in Neuromuscular Disease
Few studies have evaluated the usefulness of USCs in disease modeling and drug screening. The following summarizes the reports on the generation of cellular models for muscle and neurological diseases using USCs. One is to induce USCc into iPSC and create a cellular model (urine-derived induced pluripotent stem cells, UiPSCs), and another is to induce USC into the target cell by direct reprogramming Supplementary Table S1.
Urine-Derived Induced Pluripotent Stem Cells
Presently, human iPSCs can be generated from various donor sources. Urine may represent an ideal source of cells for generating iPSCs. After Zhou et al. (2012) generated iPSCs from USCs, numerous studies revealed that iPSCs generated from USCs might have several advantages over iPSCs generated from other somatic cells. Induction of UiPSCs requires less time than do iPSCs derived from fibroblasts, lymphocytes, and keratinocytes (Zhou et al., 2012). Moreover, UiPSCs show a high reprogramming efficiency of 0.1%–4% (Benda et al., 2013) and generating efficiency of approximately 1.5% which is a hundredfold higher than that of fibroblasts (0.01%; Ousterout et al., 2015). It has been demonstrated that mesenchymal-to-epithelial transition (MET) is an essential early step in reprogramming fibroblasts to iPSCs, and a critical rate-limiting step during conversion (Li et al., 2010; Samavarchi-Tehrani et al., 2010). The USCs, which mainly originated from epithelial cells, do not require MET. This fact may affect their differentiation potential. Furthermore, iPSCs derived from fibroblasts may possess epigenetic memory, which is easily differentiated into a lineage related to the donor cell type, the mesoderm lineage (Kim et al., 2010, 2011). However, this tendency has not been observed in UiPSC (Shi et al., 2016).
UiPSCs have already been used in numerous studies on disease modeling (Ji et al., 2017) such as those on Type 2 long QT syndrome (Jouni et al., 2015), dilated cardiomyopathy (Lin et al., 2016), multiple endocrine neoplasia type 1 syndrome (Guo et al., 2017), hemophilia A (Jia et al., 2014), systemic lupus erythematosus (Chen et al., 2013), Down syndrome (Lee et al., 2017), SMA (Zhou et al., 2018), spinal cord injury (Liu et al., 2017), and muscular dystrophy (Afzal and Strande, 2015).
Direct Reprogramming of Urine-Derived Stem Cells Into Myogenic Lineage
Several studies on skeletal muscle diseases indicate that USC can be induced into myogenic lineage by direct reprogramming via muscle transcription factor MyoD1. Falzarano et al. (2016) demonstrated that USC derived from patients with DMD retain the patient-specific DMD mutation and that USCs converted via MyoD1 show no dystrophin expression. Falzarano et al. (2016) additionally showed that truncated dystrophin is restored by in-framing with antisense oligonucleotides against exon 44 of DMD. Kim et al. (2016) demonstrated that myogenic reprogramming of urine cells derived from patients with DMD and limb-girdle muscular dystrophy (LGMD) type 2 could recapitulate the disease phenotype. They additionally showed that USC genomes could be edited using CRISPR/Cas9.
Recently, we developed a novel MyoD1-converted, urine-derived cell to in vitro model of the pathological processes of muscle cells affected by DMD (Takizawa et al., 2019). In that study, we showed that 3-deazaneplanocin A hydrochloride (DZNep) promotes the differentiation of USCs into myotubes. DZNep-treated USCs, differentiated via MyoD1, are excellent in vitro models of muscle cells affected by DMD. Moreover, this system, which is based on urine-derived cells obtained from patients with DMD, can be successfully used to evaluate exon skipping therapy using antisense oligonucleotide for DMD. This newly-established in vitro assay will be used in a wide range of studies regardless of age, sex, and muscular disease type of patients. Direct reprogramming of USCs could potentially be used to study the pathophysiology of various diseases, and to diagnose and develop novel therapies for patients with these conditions.
Direct Reprogramming of Urine-Derived Stem Cells Into Neuronal Lineage
Urine-derived cells can be differentiated into neural-lineage cells by culture in neural induction medium supplemented with basic fibroblast growth factor (Bharadwaj et al., 2013; Guan et al., 2014; Zhang et al., 2016). However, few studies have examined the direct differentiation of neuronal cells from urine-derived cells. Wang et al. (2013) developed integration-free and expandable human neural progenitor cells which can self-renew and differentiate into multi-functional neuronal subtypes and glial cells in vitro. Several groups reported that approximately 40% of the induced cells express several neural markers and show neurogenic extensions and processes (Bharadwaj et al., 2013; Guan et al., 2014). USCs treated with growth factors and cultured on laminin-treated plates readily convert into immature neuronal cells (Guan et al., 2014; Kim et al., 2018). Human urinary cells can be converted into neural stem cells by a non-integration-free method using small molecules, and it takes less time than through iPSCs (Cheng et al., 2014). The induced neural progenitor cells can then be converted into astrocytes, oligodendrocytes, and neurons, and may play an essential role in identifying and developing safe and effective therapies for patients with neurodegenerative conditions.
Future Aspects of Using Urine-Derived Stem Cells as Cellular Models of Human Disease
As discussed previously, USCs also present an advantageous in vitro model to study disease mechanisms, identify new biomarkers, evaluate therapeutic approaches, and screen drugs (Figure 2). USCs can be obtained reliably and non-invasively in a short period of time and cultured at low cost. This newly-established in vitro assay can, therefore, be adapted for various studies and platforms. Direct reprogramming is an efficient and economical approach because it can be used to generate patient-specific cell lineages without the presence of iPSC intermediates. USCs could show superior differentiation, and can, therefore, be used to delineate the mechanisms of common and rare genetic diseases and to screen drugs for the treatment of these conditions. However, the directed differentiation efficiency of USCs used to produce the mature target-cell types, needs to be optimized. Models recapitulating neurological diseases in vitro need to possess not only the gene and protein expression of neuron-surface markers but also characteristics of functional maturation such as synapses and cellular homeostasis. Neurons obtained directly from reprogrammed USCs must be evaluated via genetic, biological, and electrophysiological assessment. Future differentiation of neural cells from USCs will be inspired by the previous studies on iPSC reprogramming and other somatic cells direct reprogramming. USCs may play a complementary role in developing methods for rapid and efficient creation of iPSCs, and can be used to directly generate relevant cells for in vitro disease models.
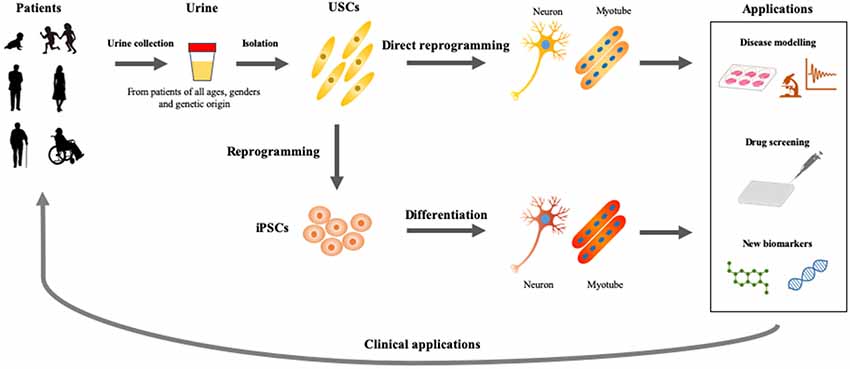
Figure 2. Applications of USCs as cellular models of human diseases. Urine represents an ideal material, which could be obtained from patients of all ages and genders by non-invasive and straightforward procedures. USCs- and induced pluripotent stem cells (iPSCs)-based disease modelings would be useful for basic and applied research, which accelerate the development of personalized medicine.
There is some essential and critical issue about USCs that there is limited understanding about the biological characteristics and the ability to differentiate into other cell lines of USC as mentioned above. The neurons differentiated from USCs are not enough to determine neuronal model cells by functional evaluation, and cannot clarify the pathological mechanism. However, as mentioned above, there is a lot of potentials for USC to become a new platform, making it possible to supplement the issues of iPSC technology and to approach pathophysiology from a new aspect.
The USCs platform is still in the infancy stage and expected that this technology will be further developed and adapted by a wider research community. We believe that USCs will play an essential role in the study of diseases and drug screening. Future studies should develop more advanced USC-based differentiation systems, which will faithfully recapitulate human tissue-level and organ-level dysfunction (Rowe and Daley, 2019).
Conclusions
The difficulties present in obtaining brain and muscular tissue, and lack of adequate preclinical models with high predictive and translational power, pose limitations in the study of neuromuscular diseases and also in developing effective drugs for patients with these disorders. Currently, advances in human iPSC-based technologies are clearly helping to overcome the limitations. Additionally, USC-based modeling will provide valuable information for establishing a diagnosis and providing effective treatment options, although USCs have not been extensively investigated in disease modeling. It should be a critical question of whether the iPSCs and USCs will be able to mimic the complexity of the human neuromuscular system or not.
Application of iPSCs and USCs will be useful for predicting drug response and assessing environmental disease triggers in neuromuscular and neurodegenerative diseases. The development of USCs- and iPSCs-based technology provides a new platform in the field of disease modeling and works in complementary ways, it is expected to benefit research and clinical applications in personalized medicine.
Author Contributions
MS and YA wrote the initial draft of the manuscript. HT assisted in the preparation and wrote the manuscript. BT, FS, and AN have contributed critically and reviewed the manuscript.
Funding
This work was supported by a Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (C) (Grant No. 18K07544 to YA), Grants-in-Aid for Research on Nervous and Mental Disorders (Grant No. 28-6 to YA), and the Japan Agency for Medical Research and Development (Grant No. 18ek0109239h0002, 18lm0203066h0001, and 18lm0203069h0001 to YA) and an Australian NHMRC-ARC Dementia Research Leadership Fellowship (APP1137024 to BT).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2019.00297/full#supplementary-material.
References
Abdelalim, E. M., and Emara, M. M. (2015). Advances and challenges in the differentiation of pluripotent stem cells into pancreatic β cells. World J. Stem Cells 7, 174–181. doi: 10.4252/wjsc.v7.i1.174
Afzal, M. Z., and Strande, J. L. (2015). Generation of induced pluripotent stem cells from muscular dystrophy patients: efficient integration-free reprogramming of urine derived cells. J. Vis. Exp. 95:52032. doi: 10.3791/52032
Bassett, A. R. (2017). Editing the genome of hiPSC with CRISPR/Cas9: disease models. Mamm. Genome 28, 348–364. doi: 10.1007/s00335-017-9684-9
Beers, J., Linask, K. L., Chen, J. A., Siniscalchi, L. I., Lin, Y., Zheng, W., et al. (2015). A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Sci. Rep. 5:11319. doi: 10.1038/srep11319
Benda, C., Zhou, T., Wang, X., Tian, W., Grillari, J., Tse, H. F., et al. (2013). Urine as a source of stem cells. Adv. Biochem. Eng. Biotechnol. 129, 19–32. doi: 10.1007/10_2012_157
Bharadwaj, S., Liu, G., Shi, Y., Markert, C., Andersson, K.-E., Atala, A., et al. (2011). Characterization of urine-derived stem cells obtained from upper urinary tract for use in cell-based urological tissue engineering. Tissue Eng. Part A 17, 2123–2132. doi: 10.1089/ten.TEA.2010.0637
Bharadwaj, S., Liu, G., Shi, Y., Wu, R., Yang, B., He, T., et al. (2013). Multipotential differentiation of human urine-derived stem cells: potential for therapeutic applications in urology. Stem Cells 31, 1840–1856. doi: 10.1002/stem.1424
Chal, J., Al Tanoury, Z., Hestin, M., Gobert, B., Aivio, S., Hick, A., et al. (2016). Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc. 10, 1833–1850. doi: 10.1038/nprot.2016.110
Chen, Y., Luo, R., Xu, Y., Cai, X., Li, W., Tan, K., et al. (2013). Generation of systemic lupus erythematosus-specific induced pluripotent stem cells from urine. Rheumatol. Int. 33, 2127–2134. doi: 10.1007/s00296-013-2704-5
Chen, W., Xie, M., Yang, B., Bharadwaj, S., Song, L., Liu, G., et al. (2017). Skeletal myogenic differentiation of human urine-derived cells as a potential source for skeletal muscle regeneration. J. Tissue Eng. Regen. Med. 11, 334–341. doi: 10.1002/term.1914
Cheng, L., Hu, W., Qiu, B., Zhao, J., Yu, Y., Guan, W., et al. (2014). Generation of neural progenitor cells by chemical cocktails and hypoxia. Cell Res. 24, 665–679. doi: 10.1038/cr.2014.32
Crompton, L. A., Byrne, M. L., Taylor, H., Kerrigan, T. L., Bru-Mercier, G., Badger, J. L., et al. (2013). Stepwise, non-adherent differentiation of human pluripotent stem cells to generate basal forebrain cholinergic neurons via hedgehog signaling. Stem Cell Res. 11, 1206–1221. doi: 10.1016/j.scr.2013.08.002
Davis, R. L., Weintraub, H., and Lassar, A. B. (1987). Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000. doi: 10.1016/0092-8674(87)90585-x
Devine, M. J., Ryten, M., Vodicka, P., Thomson, A. J., Burdon, T., Houlden, H., et al. (2011). Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2:440. doi: 10.1038/ncomms1453
Egawa, N., Kitaoka, S., Tsukita, K., Naitoh, M., Takahashi, K., Yamamoto, T., et al. (2012). Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4:145ra104. doi: 10.1126/scitranslmed.3004052
Engel, M., Do-Ha, D., Muñoz, S. S., and Ooi, L. (2016). Common pitfalls of stem cell differentiation: a guide to improving protocols for neurodegenerative disease models and research. Cell. Mol. Life Sci. 73, 3693–36709. doi: 10.1007/s00018-016-2265-3
Falzarano, M. S., D’Amario, D., Siracusano, A., Massetti, M., Amodeo, A., La Neve, F., et al. (2016). Duchenne muscular dystrophy myogenic cells from urine-derived stem cells recapitulate the dystrophin genotype and phenotype. Hum. Gene Ther. 27, 772–783. doi: 10.1089/hum.2016.079
Falzarano, M. S., and Ferlini, A. (2019). Urinary stem cells as tools to study genetic disease: overview of the literature. J. Clin. Med. 8:627. doi: 10.3390/jcm8050627
Gaignerie, A., Lefort, N., Rousselle, M., Forest-Choquet, V., Flippe, L., Francois-Campion, V., et al. (2018). Urine-derived cells provide a readily accessible cell type for feeder-free mRNA reprogramming. Sci. Rep. 8:14363. doi: 10.1038/s41598-018-32645-2
Grath, A., and Dai, G. (2019). Direct cell reprogramming for tissue engineering and regenerative medicine. J. Biol. Eng. 13:14. doi: 10.1186/s13036-019-0144-9
Guan, J.-J., Niu, X., Gong, F.-X., Hu, B., Guo, S.-C., Lou, Y.-L., et al. (2014). Biological characteristics of human-urine-derived stem cells: potential for cell-based therapy in neurology. Tissue Eng. Part A 20, 1794–1806. doi: 10.1089/ten.TEA.2013.0584
Guan, J., Zhang, J., Li, H., Zhu, Z., Guo, S., Niu, X., et al. (2015). Human urine derived stem cells in combination with β-TCP can be applied for bone regeneration. PLoS One 10:e0125253. doi: 10.1371/journal.pone.0125253
Guo, D., Wu, F., Liu, H., Gao, G., Kou, S., Yang, F., et al. (2017). Generation of non-integrated induced pluripotent stem cells from a 59-year-old female with multiple endocrine neoplasia type 1 syndrome. Stem Cell Res. 18, 64–66. doi: 10.1016/j.scr.2016.12.009
He, W., Zhu, W., Cao, Q., Shen, Y., Zhou, Q., Yu, P., et al. (2016). Generation of mesenchymal-like stem cells from urine in pediatric patients. Transplant. Proc. 48, 2181–2185. doi: 10.1016/j.transproceed.2016.02.078
Ho, R., Sances, S., Gowing, G., Amoroso, M. W., O’Rourke, J. G., Sahabian, A., et al. (2016). ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat. Neurosci. 19, 1256–1267. doi: 10.1038/nn.4345
Huh, C. J., Zhang, B., Victor, M. B., Dahiya, S., Batista, L. F., Horvath, S., et al. (2016). Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. Elife 20:e18648. doi: 10.7554/eLife.18648
Ieda, M., Fu, J. D., Delgado-Olguin, P., Vedantham, V., Hayashi, Y., Bruneau, B. G., et al. (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. doi: 10.1016/j.cell.2010.07.002
Ji, X., Wang, M., Chen, F., and Zhou, J. (2017). Urine-derived stem cells: the present and the future. Stem Cells Int. 2017:4378947. doi: 10.1155/2017/4378947
Jia, B., Chen, S., Zhao, Z., Liu, P., Cai, J., Qin, D., et al. (2014). Modeling of hemophilia A using patient-specific induced pluripotent stem cells derived from urine cells. Life Sci. 108, 22–29. doi: 10.1016/j.lfs.2014.05.004
Jouni, M., Si-Tayeb, K., Es-Salah-Lamoureux, Z., Latypova, X., Champon, B., Caillaud, A., et al. (2015). Toward personalized medicine: using cardiomyocytes differentiated from urine-derived pluripotent stem cells to recapitulate electrophysiological characteristics of type 2 long QT syndrome. J. Am. Heart Assoc. 4:e002159. doi: 10.1161/JAHA.115.002159
Kang, H. S., Choi, S. H., Kim, B. S., Choi, J. Y., Park, G. B., Kwon, T. G., et al. (2015). Advanced properties of urine derived stem cells compared to adipose tissue derived stem cells in terms of cell proliferation, immune modulation and multi differentiation. J. Korean Med. Sci. 30, 1764–1776. doi: 10.3346/jkms.2015.30.12.1764
Kaye, J. A., and Finkbeiner, S. (2013). Modeling Huntington’s disease with induced pluripotent stem cells. Mol. Cell. Neurosci. 56, 50–64. doi: 10.1016/j.mcn.2013.02.005
Kim, E. Y., Barefield, D. Y., Vo, A. H., Gacita, A. M., Schuster, E. J., Wyatt, E. J., et al. (2019). Distinct pathological signatures in human cellular models of myotonic dystrophy subtypes. JCI Insight 4:122686. doi: 10.1172/jci.insight.122686
Kim, J. Y., Chun, S. Y., Park, J. S., Chung, J. W., Ha, Y. S., Lee, J. N., et al. (2018). Laminin and platelet-derived growth factor-BB promote neuronal differentiation of human urine-derived stem cells. Tissue Eng. Regen. Med. 15, 195–209. doi: 10.1007/s13770-017-0102-x
Kim, K., Doi, A., Wen, B., Ng, K., Zhao, R., Cahan, P., et al. (2010). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290. doi: 10.21236/ada544181
Kim, E. Y., Page, P., Dellefave-Castillo, L. M., McNally, E. M., and Wyatt, E. J. (2016). Direct reprogramming of urine-derived cells with inducible MyoD for modeling human muscle disease. Skelet. Muscle 6:32. doi: 10.1186/s13395-016-0103-9
Kim, K., Zhao, R., Doi, A., Ng, K., Unternaehrer, J., Cahan, P., et al. (2011). Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 29, 1117–1119. doi: 10.1038/nbt.2052
Kodaka, Y., Rabu, G., and Asakura, A. (2017). Skeletal muscle cell induction from pluripotent stem cells. Stem Cells Int. 2017:1376151. doi: 10.1155/2017/1376151
Krencik, R., Weick, J. P., Liu, Y., Zhang, Z. J., and Zhang, S. C. (2011). Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat. Biotechnol. 29, 528–534. doi: 10.1038/nbt.1877
Lapasset, L., Milhavet, O., Prieur, A., Besnard, E., Babled, A., Ät-Hamou, N., et al. (2011). Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 25, 2248–2253. doi: 10.1101/gad.173922.111
Lee, Y. M., Zampieri, B. L., Scott-McKean, J. J., Johnson, M. W., and Costa, A. C. S. (2017). Generation of integration-free induced pluripotent stem cells from urine-derived cells isolated from individuals with down syndrome. Stem Cells Transl. Med. 6, 1465–1476. doi: 10.1002/sctm.16-0128
Li, R., Liang, J., Ni, S., Zhou, T., Qing, X., Li, H., et al. (2010). A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63. doi: 10.1016/j.stem.2010.04.014
Lin, Y. H., Chen, X. M., Zhang, J. W., He, X. Q., Dai, W. J., and Chen, M. S. (2016). Preclinical study on induction of pluripotent stem cells from urine of dilated cardiomyopathy patients. Eur. Rev. Med. Pharmacol. Sci. 20, 1450–1457.
Liu, G., Pareta, R. A., Wu, R., Shi, Y., Zhou, X., Liu, H., et al. (2013). Skeletal myogenic differentiation of urine-derived stem cells and angiogenesis using microbeads loaded with growth factors. Biomaterials 34, 1311–1326. doi: 10.1016/j.biomaterials.2012.10.038
Liu, G., Wu, R., Yang, B., Deng, C., Lu, X., Walker, S. J., et al. (2018). Human urine-derived stem cell differentiation to endothelial cells with barrier function and nitric oxide production. Stem Cells Transl. Med. 7, 686–698. doi: 10.1002/sctm.18-0040
Liu, M.-L., Zang, T., and Zhang, C.-L. (2016). Direct lineage reprogramming reveals disease-specific phenotypes of motor neurons from human ALS patients. Cell Rep. 14, 115–128. doi: 10.1016/j.celrep.2015.12.018
Liu, Y., Zheng, Y., Li, S., Xue, H., Schmitt, K., Hergenroeder, G. W., et al. (2017). Human neural progenitors derived from integration-free iPSCs for SCI therapy. Stem Cell Res. 19, 55–64. doi: 10.1016/j.scr.2017.01.004
Maffioletti, S. M., Gerli, M. F. M., Ragazzi, M., Dastidar, S., Benedetti, S., Loperfido, M., et al. (2015). Efficient derivation and inducible differentiation of expandable skeletal myogenic cells from human ES and patient-specific iPS cells. Nat. Protoc. 10, 941–958. doi: 10.1038/nprot.2015.057
Malaver-Ortega, L. F., Sumer, H., Liu, J., and Verma, P. J. (2012). The state of the art for pluripotent stem cells derivation in domestic ungulates. Theriogenology 78, 1749–1762. doi: 10.1016/j.theriogenology.2012.03.031
Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Böhnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718. doi: 10.1016/j.stem.2015.09.001
Miesen, L., Steenbergen, E., and Smeets, B. (2017). Parietal cells—new perspectives in glomerular disease. Cell Tissue Res. 369, 237–244. doi: 10.1007/s00441-017-2600-5
Musunuru, K., Sheikh, F., Gupta, R. M., Houser, S. R., Maher, K. O., Milan, D. J., et al. (2018). Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: a scientific statement from the American Heart Association. Circ. Genomic Precis. Med. 11:e000043. doi: 10.1161/hcg.0000000000000043
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P., et al. (2011). LRRK2 mutant iPSC-derived da neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Nombela-Arrieta, C., Ritz, J., and Silberstein, L. E. (2011). The elusive nature and function of mesenchymal stem cells. Nat. Rev. Mol. Cell Biol. 12, 126–131. doi: 10.1038/nrm3049
Ooi, L., Sidhu, K., Poljak, A., Sutherland, G., O’Connor, M. D., Sachdev, P., et al. (2013). Induced pluripotent stem cells as tools for disease modelling and drug discovery in Alzheimer’s disease. J. Neural Transm. 120, 103–111. doi: 10.1007/s00702-012-0839-2
Ousterout, D. G., Kabadi, A. M., Thakore, P. I., Majoros, W. H., Reddy, E., and Gersbach, C. A. (2015). Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 6:6244. doi: 10.1038/ncomms7244
Partridge, T. A. (2013). The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 280, 4177–4186. doi: 10.1111/febs.12267
Patterson, M., Chan, D. N., Ha, I., Case, D., Cui, Y., Van Handel, B., et al. (2012). Defining the nature of human pluripotent stem cell progeny. Cell Res. 22, 178–193. doi: 10.1038/cr.2011.133
Pavathuparambil Abdul Manaph, N., Al-Hawaas, M., Bobrovskaya, L., Coates, P. T., and Zhou, X. F. (2018). Urine-derived cells for human cell therapy. Stem Cell Res. Ther. 9:189. doi: 10.1186/s13287-018-0932-z
Playne, R., and Connor, B. (2017). Understanding Parkinson’s disease through the use of cell reprogramming. Stem Cell Rev. Rep. 13, 151–169. doi: 10.1007/s12015-017-9717-5
Poulsom, R., and Little, M. H. (2009). Parietal epithelial cells regenerate podocytes. J. Am. Soc. Nephrol. 20, 231–233. doi: 10.1681/asn.2008121279
Qian, L., Huang, Y., Spencer, C. I., Foley, A., Vedantham, V., Liu, L., et al. (2012). in vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593–598. doi: 10.1038/nature11044
Revilla, A., González, C., Iriondo, A., Fernández, B., Prieto, C., Marín, C., et al. (2016). Current advances in the generation of human iPS cells: implications in cell-based regenerative medicine. J. Tissue Eng. Regen. Med. 10, 893–907. doi: 10.1002/term.2021
Ross, C. A., and Akimov, S. S. (2014). Human-induced pluripotent stem cells: potential for neurodegenerative diseases. Hum. Mol. Genet. 23, R17–R26. doi: 10.1093/hmg/ddu204
Rowe, R. G., and Daley, G. Q. (2019). Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 20, 377–388. doi: 10.1038/s41576-019-0100-z
Sagrinati, C., Netti, G. S., Mazzinghi, B., Lazzeri, E., Liotta, F., Frosali, F., et al. (2006). Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J. Am. Soc. Nephrol. 17, 2443–2456. doi: 10.1681/ASN.2006010089
Saito, T., Nakamura, A., Aoki, Y., Yokota, T., and Okada, T. (2010). Antisense PMO found in dystrophic dog model was effective in cells from exon 7-deleted DMD patient. PLoS One 5:e12239. doi: 10.1371/journal.pone.0012239
Samavarchi-Tehrani, P., Golipour, A., David, L., Sung, H. K., Beyer, T. A., Datti, A., et al. (2010). Functional genomics reveals a BMP-Driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7, 64–77. doi: 10.1016/j.stem.2010.04.015
Sareen, D., Ebert, A. D., Heins, B. M., McGivern, J. V., Ornelas, L., and Svendsen, C. N. (2012). Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS One 7:e39113. doi: 10.1371/journal.pone.0039113
Serio, A., Bilican, B., Barmada, S. J., Ando, D. M., Zhao, C., Siller, R., et al. (2013). Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U S A 110, 4697–4702. doi: 10.1073/pnas.1300398110
Shi, L., Cui, Y., Luan, J., Zhou, X., and Han, J. (2016). Urine-derived induced pluripotent stem cells as a modeling tool to study rare human diseases. Intractable Rare Dis. Res. 5, 192–201. doi: 10.5582/irdr.2016.01062
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Takizawa, H., Hara, Y., Mizobe, Y., Ohno, T., Suzuki, S., Inoue, K., et al. (2019). Modelling duchenne muscular dystrophy in MYOD1-converted urine-derived cells treated with 3-deazaneplanocin A hydrochloride. Sci. Rep. 9:3807. doi: 10.1038/s41598-019-40421-z
Tang, Y., Liu, M.-L., Zang, T., and Zhang, C.-L. (2017). Direct reprogramming rather than iPSC-based reprogramming maintains aging hallmarks in human motor neurons. Front. Mol. Neurosci. 10:359. doi: 10.3389/fnmol.2017.00359
Tapscott, S. J., Davis, R. L., Thayer, M. J., Cheng, P. F., Weintraub, H., and Lassar, A. B. (1988). MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science 242, 405–411. doi: 10.1126/science.3175662
Terstegge, S., Laufenberg, I., Pochert, J., Schenk, S., Itskovitz-Eldor, J., Endl, E., et al. (2007). Automated maintenance of embryonic stem cell cultures. Biotechnol. Bioeng. 96, 195–201. doi: 10.1002/bit.21061
Wang, L., Wang, L., Huang, W., Su, H., Xue, Y., Su, Z., et al. (2013). Generation of integration-free neural progenitor cells from cells in human urine. Nat. Methods 10, 84–89. doi: 10.1038/nmeth.2283
Watson, L. M., Wong, M. M. K., and Becker, E. B. E. (2015). Induced pluripotent stem cell technology for modelling and therapy of cerebellar ataxia. Open Biol. 5:150056. doi: 10.1098/rsob.150056
Xie, H., Ye, M., Feng, R., and Graf, T. (2004). Stepwise reprogramming of B cells into macrophages. Cell 117, 663–676. doi: 10.1016/s0092-8674(04)00419-2
Yi, H., Xie, B., Liu, B., Wang, X., Xu, L., Liu, J., et al. (2018). Derivation and identification of motor neurons from human urine-derived induced pluripotent stem cells. Stem Cells Int. 2018:3628578. doi: 10.1155/2018/3628578
Zhang, S. Z., Ma, L. X., Qian, W. J., Li, H. F., Wang, Z. F., Wang, H. X., et al. (2016). Modeling neurological disease by rapid conversion of human urine cells into functional neurons. Stem Cells Int. 2016:2452985. doi: 10.1155/2016/2452985
Zhang, Y., McNeill, E., Tian, H., Soker, S., Andersson, K. E., Yoo, J. J., et al. (2008). Urine derived cells are a potential source for urological tissue reconstruction. J. Urol. 180, 2226–2233. doi: 10.1016/j.juro.2008.07.023
Zhang, D., Wei, G., Li, P., Zhou, X., and Zhang, Y. (2014). Urine-derived stem cells: a novel and versatile progenitor source for cell-based therapy and regenerative medicine. Genes Dis. 1, 8–17. doi: 10.1016/j.gendis.2014.07.001
Zhou, T., Benda, C., Dunzinger, S., Huang, Y., Ho, J. C., Yang, J., et al. (2012). Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 7, 2080–2089. doi: 10.1038/nprot.2012.115
Zhou, Q., Brown, J., Kanarek, A., Rajagopal, J., and Melton, D. A. (2008). in vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature 455, 627–632. doi: 10.1038/nature07314
Zhou, M., Hu, Z., Qiu, L., Zhou, T., Feng, M., Hu, Q., et al. (2018). Seamless genetic conversion of SMN2 to SMN1 via CRISPR/Cpf1 and single-stranded oligodeoxynucleotides in spinal muscular atrophy patient-specific induced pluripotent stem cells. Hum. Gene Ther. 29, 1252–1263. doi: 10.1089/hum.2017.255
Keywords: urine-derived stem cells (USCs), induced pluripotent stem cells (iPSCs), disease modeling, direct-reprogramming, precision medicine
Citation: Sato M, Takizawa H, Nakamura A, Turner BJ, Shabanpoor F and Aoki Y (2019) Application of Urine-Derived Stem Cells to Cellular Modeling in Neuromuscular and Neurodegenerative Diseases. Front. Mol. Neurosci. 12:297. doi: 10.3389/fnmol.2019.00297
Received: 19 July 2019; Accepted: 20 November 2019;
Published: 05 December 2019.
Edited by:
Robert J. Harvey, University of the Sunshine Coast, AustraliaReviewed by:
Ludo M. Van Den Bosch, KU Leuven, BelgiumPeter Claus, Hannover Medical School, Germany
Copyright © 2019 Sato, Takizawa, Nakamura, Turner, Shabanpoor and Aoki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshitsugu Aoki, dHN1Z3U1NkBuY25wLmdvLmpw