 Lacramioara Fabian
Lacramioara Fabian James J. Dowling
James J. Dowling- 1Program for Genetics and Genome Biology, Hospital for Sick Children, Toronto, ON, Canada
- 2Division of Neurology, Hospital for Sick Children, Toronto, ON, Canada
- 3Departments of Pediatrics and Molecular Genetics, University of Toronto, Toronto, ON, Canada
LAMA2-related congenital muscular dystrophy (CMD; LAMA2-MD), also referred to as merosin deficient CMD (MDC1A), is a severe neonatal onset muscle disease caused by recessive mutations in the LAMA2 gene. LAMA2 encodes laminin α2, a subunit of the extracellular matrix (ECM) oligomer laminin 211. There are currently no treatments for MDC1A, and there is an incomplete understanding of disease pathogenesis. Zebrafish, due to their high degree of genetic conservation with humans, large clutch sizes, rapid development, and optical clarity, have emerged as an excellent model system for studying rare Mendelian diseases. They are particularly suitable as a model for muscular dystrophy because they contain at least one orthologue to all major human MD genes, have muscle that is similar to human muscle in structure and function, and manifest obvious and easily measured MD related phenotypes. In this review article, we present the existing zebrafish models of MDC1A, and discuss their contribution to the understanding of MDC1A pathomechanisms and therapy development.
Introduction
LAMA2-related congenital muscular dystrophy (CMD; LAMA2-MD), also called merosin deficient CMD or MDC1A, is the most common subtype of CMD (Schorling et al., 2017; Sframeli et al., 2017; Mohassel et al., 2018; Mercuri et al., 2019). MDC1A is an autosomal recessive neuromuscular disorder caused by mutations in laminin α2 (LAMA2, Helbling-Leclerc et al., 1995; Holmberg and Durbeej, 2013). Complete loss of LAMA2 protein leads to an early onset clinical phenotype featuring severe, diffuse muscle weakness and wasting, demyelinating peripheral neuropathy, and pauci-clinical central nervous system abnormalities, including white matter changes and, in some cases, structural brain lesions (Quijano-Roy et al., 1993; Menezes et al., 2014; Oliveira et al., 2018). The disease is associated with significant co-morbidities, including wheelchair dependence and respiratory failure, and early mortality in some cases (Dimova and Kremensky, 2018). A less common entity is partial merosin deficiency, a disorder caused by partial loss of LAMA2 expression/function that is associated with a later onset, milder form of muscular dystrophy (Nguyen et al., 2019). Both MDC1A and partial merosin deficiency, as well as other rare clinical phenotypes associated with LAMA2 mutations, are all classified as LAMA2-MD (Oliveira et al., 2018; Verma et al., 2018; Amin et al., 2019). Currently, there are no treatments for LAMA2-MD, and there is an incomplete understanding of disease pathogenesis.
Laminins
Laminins are high molecular weight glycoproteins expressed abundantly in the basal lamina, a specialized layer of the extracellular matrix (ECM; Aumailley, 2013). Laminins are multidomain heterotrimeric proteins comprised of α, β and γ polypeptide chains (Mohassel et al., 2018), which come in five (LAMA1–5), four (LAMB1–4) and three (LAMC1–3) genetic variants, respectively (Aumailley, 2013; Yurchenco et al., 2018). The polypeptide chains fold in a similar cross-shaped pattern, with distinct structural domains performing specific functions, such as facilitation of self-assembly of most laminins into large polymers by the globular laminin N-terminal (LN) domain (Hohenester, 2019). A few of the polypeptide chains, such as a4 lack the LN domain and therefore do not self-assemble (Aumailley, 2013). Based on the chain composition, more than 15 laminins have been identified in humans (Colognato and Yurchenco, 2000; Sztal et al., 2011). In zebrafish, 12 laminin-encoding genes have been found, out of which 10 have mammalian orthologs, with evolutionarily conserved function (Sztal et al., 2011), whereas two of them, lamb1b and lamb2l, have no mammalian orthologs (Sztal et al., 2011). Two human laminin-encoding genes, LAMB3 and LAMC2, have not been found in zebrafish (Sztal et al., 2011). Laminins play essential roles in many tissues and organs during development (Yao, 2017). In zebrafish, laminins are involved in myriad developmental processes spanning multiple organ systems (Table 1).
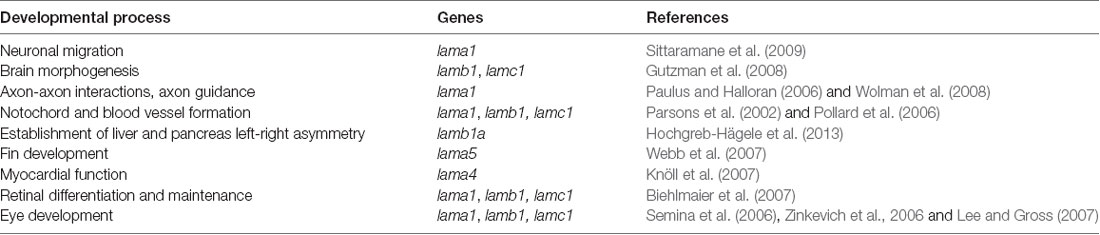
Table 1. Examples of developmental processes where laminins are involved.
Several laminin genes are expressed during skeletal muscle development in zebrafish. Some of these, like lama2, lama4, lamb2, and lamc1, are detected as early as 24 hpf (when myogenesis begins) and persist in the post-juvenile stages, whereas others (lamb1, lamb4, and lamc3) have only a brief-expression during early muscle development (Sztal et al., 2011). For example, during zebrafish early skeletal muscle development, lamb1 and lamc1 are required for fast muscle fiber elongation, orientation, and their attachment at the myotendinous junctions (MTJs), the primary site of force transmission (Snow et al., 2008; Snow and Henry, 2009). Zebrafish lamb1 and lamc1 mutants and morphants show delayed or impaired muscle fiber elongation, non-parallel orientation of fibers in the myotome, and defects in MTJ morphogenesis (Snow et al., 2008). Lama4 is essential for mechanical stability in zebrafish skeletal muscle (Postel et al., 2008). In lama4 morphants, recruitment of focal adhesion proteins integrin-linked kinase (ilk) and paxillin at the MTJs is impaired, resulting in detachment of myofibers and their surrounding sarcolemma from the MTJ complex (Postel et al., 2008). Lama2 was also shown to be important for zebrafish muscle development and relevant studies on its role in this process are discussed below.
Laminin α2
The laminin α2 (LAMA2) gene encodes the alpha2 chain and constitutes a subunit of several laminin proteins (Tunggal et al., 2000; Aumailley et al., 2005): Laminin 211 (Laminin α2β1γ1, Laminin 2 or merosin; Durbeej, 2015; Barraza-Flores et al., 2020), Laminin α2β2γ1 (Laminin 221, Laminin 4 or S-merosin; Patton et al., 1997) and Laminin 213 (Laminin α2β1γ3, Laminin 12; Koch et al., 1999; Ido et al., 2008). LAMA2 is the major laminin isoform expressed in the vertebrate muscle system (Sztal et al., 2012).
The zebrafish lama2 gene, representing the ortholog of human LAMA2, maps to chromosome 20 and is expressed in the nervous system, head, otic vesicle, adaxial cells, and skeletal muscle (Sztal et al., 2011). Mutations in zebrafish lama2 results in a type of muscular dystrophy phenotypically similar to the human MDC1A (Hall et al., 2007), which identifies zebrafish as a suitable model for understanding this disease and for development of therapies.
Zebrafish Models of Muscular Dystrophies
Studies of animal models of muscular dystrophies have proven essential for a better understanding of the pathogenesis of these disorders and for developing disease-specific therapies (Saunier et al., 2016; Widrick et al., 2019). Research using mouse models for LAMA2-MD have identified potential therapeutic strategies, which, in turn, have led to improvements in murine disease pathology and survival (Miyagoe-Suzuki et al., 2000; Meinen et al., 2007, 2011; Vishnudas and Miller, 2009; McKee et al., 2017; Reinhard et al., 2017; Willmann et al., 2017; Mohassel et al., 2018; Yurchenco et al., 2018).
Recently, zebrafish have emerged as an excellent animal model for studying human muscle diseases, mainly due to their highly similar skeletal muscle, with conserved genetic, molecular and histological features (Telfer et al., 2010; Berger and Currie, 2012; Gibbs et al., 2013). Also, external fertilization, a large number of offspring, rapid embryonic development, optical transparency of embryos and larvae, combined with the ability to easily absorb pharmacological compounds, make zebrafish an excellent tool for studying disease pathomechanisms and identifying potential therapeutic targets (Zon and Peterson, 2005; Gibbs et al., 2013; Waugh et al., 2014; MacRae and Peterson, 2015; Cassar et al., 2020; Fazio et al., 2020). Importantly, readily available and easily applied experimental approaches allow for efficient and rapid assessment of structural and functional damage of the muscular system in the numerous zebrafish dystrophy models. For example, comprehensive phenotypic analysis of muscle damage can be easily done by using birefringence assay (Figures 1A,A′; Berger et al., 2012), histochemistry or immunohistochemistry staining (Figures 1B,B′), injections with fluorescently-tagged markers (Figures 1C,C′; Lombardo et al., 2012) or vital dyes, such as Evans Blue Dye (EBD; Smith et al., 2015) followed by live imaging, swimming assay to assess motor behavior (Figures 1D,D′; Zon and Peterson, 2005; Gibbs et al., 2013; Smith et al., 2013), and other equally useful techniques (Gibbs et al., 2013; MacRae and Peterson, 2015).
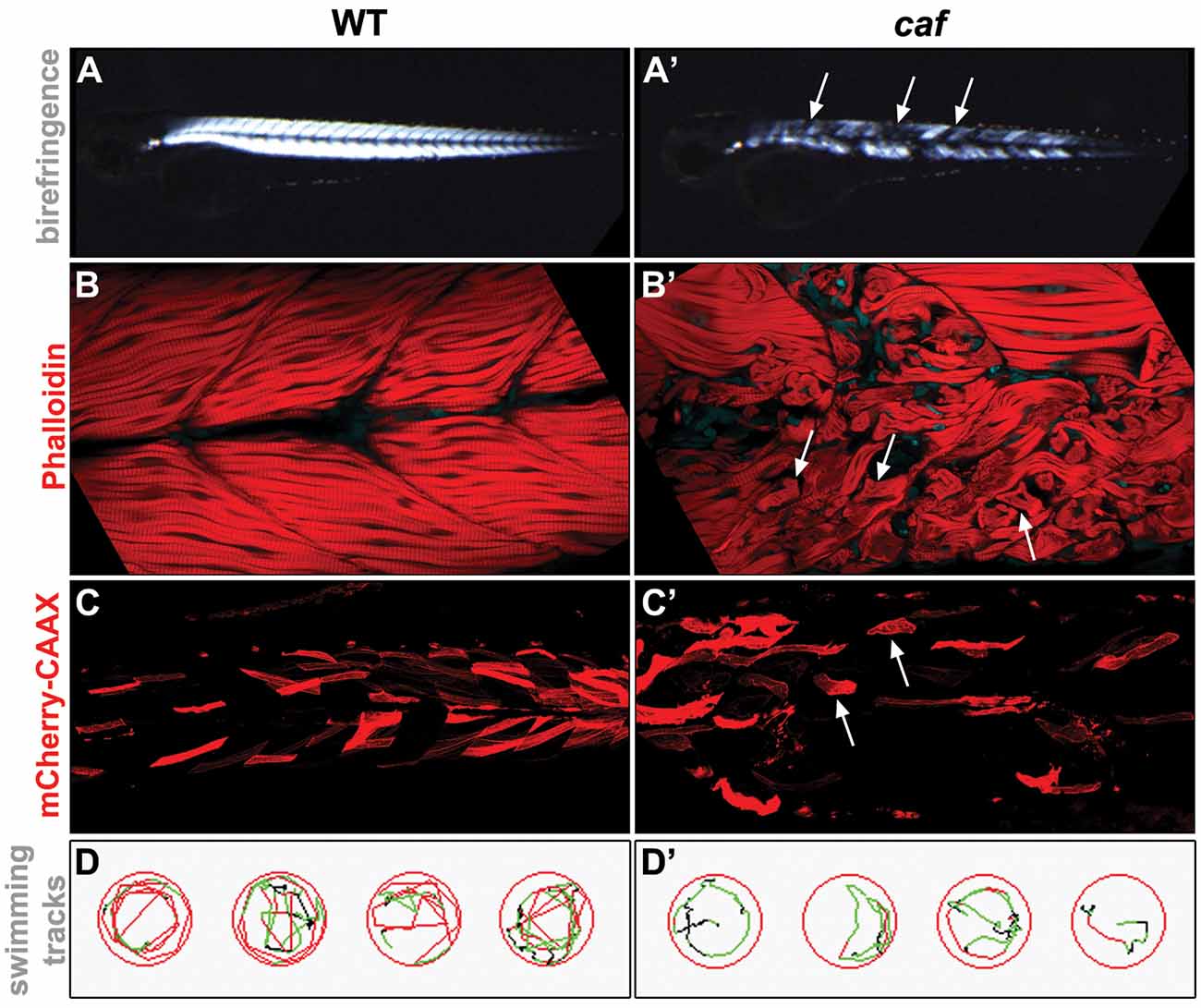
Figure 1. Examples of experimental approaches used for the phenotypical analysis of the zebrafish LAMA2-related congenital muscular dystrophy (CMD, LAMA2-MD) model. (A,A’) Birefringence assay. The organization of muscle fibers can be seen by polarized light. Detached muscle fibers in the caf mutant show up as dark regions in the muscle (arrows). (B,B’) Whole-mount staining. Phalloidin stains the actin filaments in the muscle. Muscle fibers detached from the myotendinous junctions (MTJs) in the caf mutant can be easily identified (arrows). (C,C’) Injected fluorescent marker. unc53:mCherry-CAAX-pA construct (Zhao et al., 2019), which marks muscle cells, was injected into 1-cell stage embryos and visualized by live imaging at 3 dpf. Detached fibers in the caf mutant can be easily identified (arrows). (D,D’) Swimming assay. Swim behavior can be tracked and quantified using Viewpoint Zebrabox software (Viewpoint Life Sciences Inc.). Time spent moving, distance traveled and speed of movement are useful indicators of muscle function. Fewer tracks and lower speed (indicated by green tracks) are seen in the recorded caf mutants.
Zebrafish models have been developed for many human diseases, such as glioblastoma (Gamble et al., 2018), eye diseases (Moosajee et al., 2008; Bryan et al., 2016), cardiovascular disorders (reviewed in Gut et al., 2017), and kidney diseases (reviewed in Jerman and Sun, 2017), to name a few. Various human muscle disorders, such as Duchenne muscular dystrophy (DMD; Bassett and Currie, 2003; Bassett et al., 2003; Widrick et al., 2016), Laminin α2-associated muscular dystrophy (Jacoby et al., 2009), Ullrich CMD (Telfer et al., 2010), dystroglycanopathies (reviewed in Hewitt, 2009; Lin et al., 2011; Bailey et al., 2019), facioscapulohumeral muscular dystrophy (Mitsuhashi et al., 2013; Pakula et al., 2019), X-linked myotubular myopathy (Dowling et al., 2009; Lawlor et al., 2016; Sabha et al., 2016), and nemaline myopathies (Telfer et al., 2012; Sztal et al., 2015), also have been modeled in zebrafish (reviewed in Nance et al., 2012; Gibbs et al., 2013; Lek et al., 2015; Li et al., 2017).
Zebrafish Models for Lama2-MD
To date, only a handful of zebrafish models for LAMA2-MD have been developed, even though there are more than three hundred LAMA2 gene variants associated with human disease (Oliveira et al., 2018).
The first zebrafish model of LAMA2-MD was described by Currie and colleagues (Hall et al., 2007) and was identified through complementation studies between dystrophic mutants generated through an N-ethyl-N-nitrosourea (ENU) mutagenesis screen at the University of Tubingen, Germany (Granato et al., 1996). Homozygous mutant zebrafish carrying either teg15a or tk209 recessive mutant allele, show impaired swimming behavior, severe muscle loss, and detached myofibers. Based on the specific shape of the detached fibers, which resemble cotton candy, they named this mutant candyfloss (caf). The two caf alleles, cafteg15 and caftk209, represent loss-of-function mutations in lama2 gene, and both homozygous mutants exhibit a loss of lama2 protein expression, with similar degenerative muscle phenotype, death by 16 dpf in the majority of cases, and lack of progeny for the small percent of surviving mutants. The mutations have been mapped to the globular domain of lama2, which is required for binding to dystroglycan (Hall et al., 2007), a component of the dystrophin-associated glycoprotein complex (DGC) involved in attaching the muscle fibers to the ECM (Sztal et al., 2012). caf mutations are located within amino acid residues conserved in humans and mice where known human LAMA2-MD mutations have been identified (Hall et al., 2007).
Using a birefringence assay as a screening tool, it was shown that the muscle defects present in the caf zebrafish resemble those described in human patients with LAMA2-MD, namely a stochastic pattern of myofiber detachment from the MTJs. This detachment affects both slow and fast muscle fibers (Sztal et al., 2012), is muscle cell-autonomous, and is dependent on the motor activity of the muscle (Hall et al., 2007; Thomasi et al., 2018). Notably, even though the detachment of the damaged fibers happens rapidly, they maintain the integrity of their sarcolemma, in contrast to what is happening in the muscle of the DMD zebrafish model sapje (Bassett et al., 2003; Smith et al., 2015). This is well demonstrated through the use of EBD injections, with caf zebrafish showing limited uptake into the sarcoplasm (Hall et al., 2007; Smith et al., 2015). A similar finding of limited/reduced EBD uptake is observed in the dy mouse model of LAMA2-MD (Straub et al., 1997), suggesting that impairment of membrane integrity is less prominent in LAMA2-MD vs. other MDs, and also supporting the validity of the muscle phenotype of caf zebrafish.
Of note, through elegant in vivo time-lapse experiments using various fluorescently-tagged constructs, the specific properties of the lama2-deficient myofibers were characterized in detail (Hall et al., 2019). The authors showed these fibers are long-lived, and undergo extensive cellular remodeling by extending protrusions to re-attach to the ECM. They display the formation of new pre-myofibers and undergo nuclear fusion with nearby satellite cells, all processes that indicate that repair, regeneration, and survival mechanisms are activated in the lama2-deficient myofibers. Importantly, the authors showed this is not the case in dystrophin-deficient fibers (Hall et al., 2019).
More recently, another zebrafish model for LAMA2-MD has been characterized (Gupta et al., 2012; Smith et al., 2017). The lama2cl501 mutant, also identified through an ENU mutagenesis screen (Gupta et al., 2011), carries a mutation in a highly conserved splice site located in the coiled-coil α-helical domain in the long arm of lama2, which is required for binding of LAMA2 to the other laminins in the heterotrimeric complex. This mutation results in a complete loss-of-function due to defective splicing of the lama2 mRNA. The phenotype of lama2cl501 mutants is essentially identical to that of caf zebrafish, with early-onset muscle degeneration due to detachment of fibers from the MTJs and death by 15 dpf (Gupta et al., 2012). Importantly, the detachment of the myofibers from the MTJs in lama2cl501 happens without the loss of sarcolemmal integrity, similar to caf mutants. These mutants show reduced laminin expression in the basal membrane at the MTJs complexes, smaller myotomes indicative of growth defects, disorganized sarcomere structure, and increased number of necrotic fibers. Also, lama2cl501 mutants exhibit brain and eye defects (Gupta et al., 2012). Pathogenesis of lama2cl501 is similar to that of human patients with MDC1A, making this mutant another excellent animal model for identifying potential therapies for MDC1A.
Additional research looking at genetic interactions between lama2 and other dystrophic genes has contributed to our understanding of the specific pathomechanism(s) by which the muscle damage occurs in LAMA2-MD (Sztal et al., 2012). LAMA2, as the major muscle isoform, regulates attachment of myofibers to the ECM either through the dystroglycan complex or through integrin pathways (Tunggal et al., 2000; Pozzi et al., 2017). However, proteins such as dystroglycan (Ervasti and Campbell, 1993; Lisi and Cohn, 2007), dystrophin (Bassett and Currie, 2004), integrin-α7 (Postel et al., 2008), or ilk (Postel et al., 2008) that interact directly or indirectly with LAMA2, play important roles in modifying the LAMA2-MD phenotype. Systematic epistatic experiments in this study (Sztal et al., 2012) showed that concomitant loss of ilk and dmd (dystrophin), or ilk and DAG1 (dystroglycan) result in a more severe dystrophic phenotype than the loss of lama2 or either one alone. Also, the authors show that the phenotype of lama2/ilk, lama2/dmd, or lama2/dag1 double homozygous mutants is less severe than the one exhibited by the ilk/dmd or ilk/dag1 mutants, implicating other laminins, in addition to lama2, in maintaining the attachment of myofibers to the ECM. Further, by injecting either lama4 or lama1 morpholino in lama2 mutants, Sztal et al. (2012) showed that lama1, but not lama4, also plays a significant role in this process.
A key outcome of the studies using the caf and lama2cl501 zebrafish models was the ability to discriminate between this disorder and DMD, another severe form of muscular dystrophy (Bassett et al., 2003; Bassett and Currie, 2004; Widrick et al., 2016). In muscle from the DMD zebrafish model sapje the detached fibers show significant sarcolemmal damage, followed by rapid and increased apoptosis and/or necrosis (Bassett et al., 2003), whereas muscle from the caf and lama2cl501 zebrafish fully detaches without concomitant sarcolemmal damage (Hall et al., 2007; Gupta et al., 2012). Also, detached myofibers in lama2 zebrafish show increased survival and regeneration due to the up-regulation of lama4 in detached fibers (Sztal et al., 2012).
Therapeutic Strategies for LAMA2-MD—Lessons from Zebrafish
Several studies using lama2 zebrafish models identified potential therapeutic strategies for LAMA2-MD (Sztal et al., 2012; Smith et al., 2017; Hall et al., 2019). Results from studies in other dystrophic zebrafish models can also be translated and applied to LAMA2-MD (Goody et al., 2012; Kawahara et al., 2014; Widrick et al., 2019; Wood et al., 2019).
Drug Screening and Drug Therapy
Studies by Smith et al. (2017) identified and characterized a very early coiling defect in the lama2cl501 fish, which can be used as a measurable and reliable phenotype for drug screening. The mutant fish complete significantly fewer tail coiling movements compared to their wild type siblings (Smith et al., 2017). Importantly, this phenotype manifests only in caf and lama2cl501fish, not in DMD mutants. This early phenotype is consistent with the early perinatal changes observed in LAMA2-MD mouse models (Mehuron et al., 2014), and mirrors the congenital onset phenotype of patients. Therefore, this zebrafish model may be effectively used to identify drug therapies that act at early stages in the LAMA2-MD disease process, which then could be translated into mouse models and clinical trials.
Recent studies using an integrin beta1 zebrafish (itgβ1) showed that targeting LAMA2 binding partners, such as integrin, could also provide insights into putative drug therapies for LAMA2-MD (Wood et al., 2019). itgβ1-deficient fish displayed increased amounts of LAMA2 and collagen at the ECM, indicating that inhibition of itgβ1 in lama2-deficient models might ameliorate the LAMA2-MD phenotype. Injections of the peptide RGD, an itgβ1 inhibitor, led to increased myofiber stability at the basal lamina in caf zebrafish, by increasing the levels of lama2 at the ECM (Wood et al., 2019).
Additional insights into using zebrafish models for the development of drug therapies for muscular dystrophies were provided by studies modulating nicotinamide adenine dinucleotide (NAD+) biosynthesis in dag1 and itga7 dystrophic morphants (Goody et al., 2012). NAD+ synthesis, mediated by the muscle-specific nicotinamide riboside kinase 2b (nrk2b), was shown to be essential for lamc1 polymerization at the MTJs and identified additional regulators of muscle morphogenesis in the cell adhesion signaling pathway (Goody et al., 2010). Exogenous supplementation of NAD+ or overexpression of its downstream effector, paxillin, ameliorate the dystrophic phenotype, by increasing the MTJ-basement membrane organization through laminin augmentation (Goody et al., 2012).
Gene Therapy and Protein Replacement Therapy
Recent studies in the cafteg15 zebrafish model showed that expressing lama2 or injecting lama2 rescues the LAMA2-MD dystrophic fiber phenotype (Hall et al., 2019). Generalized expression of lama2 under a heat-shock promoter during embryonic development or muscle-specific overexpression of lama2 in caf fish led to normal levels and correct distribution of laminin at the MTJs and complete rescue of the dystrophic phenotype (Hall et al., 2019). Driving the expression of lama2 later in development, after the dystrophic phenotype is fully established, resulted in a significant decrease in the number of detached fibers, increased survival, remodeling, repair and reattachment of detached fibers (Hall et al., 2019). Intramuscular delivery of Laminin111, a laminin complex similar to Laminin211 shown to functionally replace Laminin211 in an MCD1A mouse model (Van Ry et al., 2014), increased the population of muscle stem cells and resulted in significant improvement of the caf phenotype (Hall et al., 2019). This is similar to what has been described for laminin replacement therapy in DMD and alpha7 integrin-null mouse models (Rooney et al., 2009a,b; Goudenege et al., 2010) and provides additional validation of the therapy, as well as of the model as a vehicle for discovery and development of therapies.
Caveats of Using Zebrafish as the LAMA2-MD Disease Model
The above studies describing LAMA2-MD zebrafish models, together with the increasing number of studies modeling other human diseases in zebrafish (Steffen et al., 2007; Wood and Currie, 2014), prove the amenability of zebrafish as an organism for advancing our understanding of pathogenic mechanisms and therapies development. However, we should mention that a few caveats should be taken into consideration when translating the results from the LAMA2-MD zebrafish to human patients with LAMA2-MD.
LAMA2-MD pathophysiology shows slight differences between human patients and zebrafish models. For example, in humans, the LAMA2-MD dystrophic phenotype is associated, besides other features, with increased atrophy and apoptosis, defective regeneration and repair, depletion of satellite cell pools, upregulated autophagy and abnormal proteasome-dependent degradation (Durbeej, 2015). These changes have yet to be thoroughly examined in zebrafish models of LAMA2-MD. Also, evaluating non-muscle phenotypes associated with LAMA2 mutations presents challenges in the zebrafish. Importantly, myelination is distinctly different in zebrafish compared to mammals, with peripheral myelin expressing myelin basic protein and not MPZ or PMP22. Thus, studying mechanisms related to white matter disease and peripheral neuropathy may not be feasible in zebrafish, and addressing the impact of therapeutic interventions on these features of disease not possible.
Designing drug screens for LAMA2-MD in zebrafish requires taking into consideration that detachment of myofibers in caf mutants is movement- and mechanical load-dependent (Hall et al., 2007). Therefore, it is necessary to ensure the drugs tested do not affect swimming behavior. Fish immobilized due to highly toxic drugs, for example, would lead to the identification of false-positive drug hits.
Overall, there is therefore the need to balance the advantage of the zebrafish with these shortcomings. The integration of observations in caf mutants with other in vivo models of LAMA2-MD should greatly aid in their translatability. In particular, several mutant mouse models accurately phenocopy key aspects of the human disease (Gawlik and Durbeej, 2020), and provide an opportunity to test and validate findings from the caf mutants and to determine their relevance to non-muscle systems. This is particularly true concerning therapy development, where a pipeline of large scale drug screening in zebrafish combined with testing and validation in the mouse may yield candidate therapeutics with the highest potential for successful translation to patients. The establishment of a similar pipeline crossing multiple species was recently reported for congenital muscle disease due to RYR1 mutation (Volpatti et al., 2020).
Conclusions
Zebrafish models of human diseases contribute significantly to our understanding of underlying pathogenic mechanisms, characterization of signaling pathways regulating them, and development of therapeutic strategies. The main strengths of the zebrafish model are a large number of offspring, rapid embryonic development, and optical transparency of the embryos, which allow for successful screening approaches, from drug discovery to genome-scale CRISPR and genetic modifiers screening (reviewed in Gut et al., 2017).
Zebrafish is an excellent model organism to study LAMA2-MD, as they mirror the genetics and motor phenotypes of patients and carry important advantages for pathway analyses and drug discovery. Zebrafish are extremely amenable to high-throughput chemical screening to identify therapeutic drugs for LAMA2-MD (MacRae and Peterson, 2015). This approach has been successfully used in other zebrafish models of human disease (Bootorabi et al., 2017; Jardine et al., 2020; Gut et al., 2017; Matsuda et al., 2018). Furthermore, genome-editing technologies such as TALENs and CRISPR/Cas systems are easily applied to zebrafish and could be used to generate and study a large number of patient-specific mutations (Zhang et al., 2018; Giardoglou and Beis, 2019; Lek et al., 2020). Despite some limitations (Gut et al., 2017), these genome-editing approaches allow for the generation of a theoretically unlimited number of zebrafish mutants, which could ultimately enable scientists to systematically and comprehensively study full allele series for disorders such as LAMA2-MD. Lastly, performing genetic modifiers screens in caf zebrafish with methodologies including ENU mutagenesis and CRISPR gene editing (McGovern et al., 2015; Quattrocelli et al., 2017a,b; Rahit and Tarailo-Graovac, 2020; Volpatti et al., 2020) should enable the identification of genetic interactions and novel disease modifiers, data which would greatly advance our understanding of the pathomechanisms and phenotypic variability of LAMA2-MD.
Author Contributions
JD: conception and final approval. JD and LF: design and critical revisions. LF: drafting the article.
Funding
LF is supported by grants from National Institutes of Health (NIH) and Natural Sciences and Engineering Research Council of Canada (NSERC). JD is supported by grants from Canadian Institutes of Health Research (CIHR), NIH, Muscular Dystrophy Association (MDA), NSERC and Genome Canada.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Mo Zhao (Hospital for Sick Children, Toronto) for providing the unc53-mcherry-CAAX construct and Sarah Smith (Hospital for Sick Children, Toronto) for allowing us to use some of her unpublished birefringence and viewpoint images.
References
Amin, M., Bakhit, Y., Koko, M., Ibrahim, M. O. M., Salih, M. A., Ibrahim, M., et al. (2019). Rare variant in LAMA2 gene causing congenital muscular dystrophy in a Sudanese family. A case report. Acta Myol. 38, 21–24.
Aumailley, M., Bruckner-Tuderman, L., Carter, W. G., Deutzmann, R., Edgar, D., Ekblom, P., et al. (2005). A simplified laminin nomenclature. Matrix Biol. 24, 326–332. doi: 10.1016/j.matbio.2005.05.006
Bailey, E. C., Alrowaished, S. S., Kilroy, E. A., Crooks, E. S., Drinkert, D. M., Karunasiri, C. M., et al. (2019). NAD+ improves neuromuscular development in a zebrafish model of FKRP-associated dystroglycanopathy. Skelet. Muscle 9:21. doi: 10.1186/s13395-019-0206-1
Barraza-Flores, P., Bates, C. R., Oliveira-Santos, A., and Burkin, D. J. (2020). Laminin and integrin in LAMA2-related congenital muscular dystrophy: from disease to therapeutics. Front. Mol. Neurosci. 13:1. doi: 10.3389/fnmol.2020.00001
Bassett, D. I., Bryson-Richardson, R. J., Daggett, D. F., Gautier, P., Keenan, D. G., and Currie, P. D. (2003). Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 130, 5851–5860. doi: 10.1242/dev.00799
Bassett, D. I., and Currie, P. D. (2003). The zebrafish as a model for muscular dystrophy and congenital myopathy. Hum. Mol. Genet. 12, R265–R270. doi: 10.1093/hmg/ddg279
Bassett, D., and Currie, P. D. (2004). Identification of a zebrafish model of muscular dystrophy. Clin. Exp. Pharmacol. Physiol. 31, 537–540. doi: 10.1111/j.1440-1681.2004.04030.x
Berger, J., and Currie, P. D. (2012). Zebrafish models flex their muscles to shed light on muscular dystrophies. Dis. Model. Mech. 5, 726–732. doi: 10.1242/dmm.010082
Berger, J., Sztal, T., and Currie, P. D. (2012). Quantification of birefringence readily measures the level of muscle damage in zebrafish. Biochem. Biophys. Res. Commun. 423, 785–788. doi: 10.1016/j.bbrc.2012.06.040
Biehlmaier, O., Makhankov, Y., and Neuhauss, S. C. (2007). Impaired retinal differentiation and maintenance in zebrafish laminin mutants. Invest. Ophthalmol. Vis. Sci. 48, 2887–2894. doi: 10.1167/iovs.06-1212
Bootorabi, F., Manouchehri, H., Changizi, R., Barker, H., Palazzo, E., Saltari, A., et al. (2017). Zebrafish as a model organism for the development of drugs for skin cancer. Int. J. Mol. Sci. 18:1550. doi: 10.3390/ijms18071550
Bryan, C. D., Chien, C. B., and Kwan, K. M. (2016). Loss of laminin α1 results in multiple structural defects and divergent effects on adhesion during vertebrate optic cup morphogenesis. Dev. Biol. 416, 324–337. doi: 10.1016/j.ydbio.2016.06.025
Cassar, S., Adatto, I., Freeman, J. L., Gamse, J. T., Iturria, I., Lawrence, C., et al. (2020). Use of zebrafish in drug discovery toxicology. Chem. Res. Toxicol. 33, 95–118. doi: 10.1021/acs.chemrestox.9b00335
Colognato, H., and Yurchenco, P. D. (2000). Form and function: the laminin family of heterotrimers. Dev. Dyn. 218, 213–234. doi: 10.1002/(sici)1097-0177(200006)218:2<213::aid-dvdy1>3.0.co;2-r
Dimova, I., and Kremensky, I. (2018). LAMA2 congenital muscle dystrophy: a novel pathogenic mutation in bulgarian patient. Case Rep. Genet. 2018:3028145. doi: 10.1155/2018/3028145
Dowling, J. J., Vreede, A. P., Low, S. E., Gibbs, E. M., Kuwada, J. Y., Bonnemann, C. G., et al. (2009). Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 5:e1000372. doi: 10.1371/journal.pgen.1000372
Durbeej, M. (2015). Laminin-α2 chain-deficient congenital muscular dystrophy: pathophysiology and development of treatment. Curr. Top. Membr. 76, 31–60. doi: 10.1016/bs.ctm.2015.05.002
Ervasti, J. M., and Campbell, K. P. (1993). A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122, 809–823. doi: 10.1083/jcb.122.4.809
Fazio, M., Ablain, J., Chuan, Y., Langenau, D. M., and Zon, L. I. (2020). Zebrafish patient avatars in cancer biology and precision cancer therapy. Nat. Rev. Cancer. 20, 263–273. doi: 10.1038/s41568-020-0252-3
Gamble, J. T., Reed-Harris, Y., Barton, C. L., La Du, J., Tanguay, R., and Greenwood, J. A. (2018). Quantification of glioblastoma progression in zebrafish xenografts: adhesion to laminin α5 promotes glioblastoma microtumor formation and inhibits cell invasion. Biochem. Biophys. Res. Commun. 506, 833–839. doi: 10.1016/j.bbrc.2018.10.076
Gawlik, K. I., and Durbeej, M. (2020). A family of laminin α2 chain-deficient mouse mutants: advancing the research on LAMA2-CMD. Front. Mol. Neurosci. 13:59. doi: 10.3389/fnmol.2020.00059
Giardoglou, P., and Beis, D. (2019). On zebrafish disease models and matters of the heart. Biomedicines 7:15. doi: 10.3390/biomedicines7010015
Gibbs, E. M., Horstick, E. J., and Dowling, J. J. (2013). Swimming into prominence: the zebrafish as a valuable tool for studying human myopathies and muscular dystrophies. FEBS J. 280, 4187–4197. doi: 10.1111/febs.12412
Goody, M. F., Kelly, M. W., Lessard, K. N., Khalil, A., and Henry, C. A. (2010). Nrk2b-mediated NAD+ production regulates cell adhesion and is required for muscle morphogenesis in vivo: Nrk2b and NAD+ in muscle morphogenesis. Dev. Biol. 344, 809–826. doi: 10.1016/j.ydbio.2010.05.513
Goody, M. F., Kelly, M. W., Reynolds, C. J., Khalil, A., Crawford, B. D., and Henry, C. A. (2012). NAD+ biosynthesis ameliorates a zebrafish model of muscular dystrophy. PLoS Biol. 10:e1001409. doi: 10.1371/journal.pbio.1001409
Goudenege, S., Lamarre, Y., Dumont, N., Rousseau, J., Frenette, J., Skuk, D., et al. (2010). Laminin-111: a potential therapeutic agent for Duchenne muscular dystrophy. Mol. Ther. 18, 2155–2163. doi: 10.1038/mt.2010.165
Granato, M., van Eeden, F. J., Schach, U., Trowe, T., Brand, M., Furutani-Seiki, M., et al. (1996). Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 123, 399–413.
Gupta, V., Kawahara, G., Gundry, S. R., Chen, A. T., Lencer, W. I., Zhou, Y., et al. (2011). The zebrafish dag1 mutant: a novel genetic model for dystroglycanopathies. Hum. Mol. Genet. 20, 1712–1725. doi: 10.1093/hmg/ddr047
Gupta, V. A., Kawahara, G., Myers, J. A., Chen, A. T., Hall, T. E., Manzini, M. C., et al. (2012). A splice site mutation in laminin-α2 results in a severe muscular dystrophy and growth abnormalities in zebrafish. PLoS One 7:e43794. doi: 10.1371/journal.pone.0043794
Gut, P., Reischauer, S., Stainier, D. Y. R., and Arnaout, R. (2017). Little fish, big data: zebrafish as a model for cardiovascular and metabolic disease. Physiol. Rev. 97, 889–938. doi: 10.1152/physrev.00038.2016
Hall, T. E., Bryson-Richardson, R. J., Berger, S., Jacoby, A. S., Cole, N. J., Hollway, G. E., et al. (2007). The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin α2-deficient congenital muscular dystrophy. Proc. Natl. Acad. Sci. U S A 104, 7092–7097. doi: 10.1073/pnas.0700942104
Hall, T. E., Wood, A. J., Ehrlich, O., Li, M., Sonntag, C. S., Cole, N. J., et al. (2019). Cellular rescue in a zebrafish model of congenital muscular dystrophy type 1A. NPJ Regen. Med. 4:21. doi: 10.1038/s41536-019-0084-5
Helbling-Leclerc, A., Zhang, X., Topaloglu, H., Cruaud, C., Tesson, F., Weissenbach, J., et al. (1995). Mutations in the laminin α2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 11, 216–218. doi: 10.1038/ng1095-216
Hewitt, J. E. (2009). Abnormal glycosylation of dystroglycan in human genetic disease. Biochim. Biophys. Acta 1792, 853–861. doi: 10.1016/j.bbadis.2009.06.003
Hochgreb-Hägele, T., Yin, C., Koo, D. E., Bronner, M. E., and Stainier, D. Y. (2013). Laminin β1a controls distinct steps during the establishment of digestive organ laterality. Development 140, 2734–2745. doi: 10.1242/dev.097618
Hohenester, E. (2019). Laminin G-like domains: dystroglycan-specific lectins. Curr. Opin. Struct. Biol. 56, 56–63. doi: 10.1016/j.sbi.2018.11.007
Holmberg, J., and Durbeej, M. (2013). Laminin-211 in skeletal muscle function. Cell Adh. Migr. 7, 111–121. doi: 10.4161/cam.22618
Ido, H., Ito, S., Taniguchi, Y., Hayashi, M., Sato-Nishiuchi, R., Sanzen, N., et al. (2008). Laminin isoforms containing the γ3 chain are unable to bind to integrins due to the absence of the glutamic acid residue conserved in the C-terminal regions of the γ1 and γ2 chains. J. Biol. Chem. 283, 28149–28157. doi: 10.1074/jbc.m803553200
Jacoby, A. S., Busch-Nentwich, E., Bryson-Richardson, R. J., Hall, T. E., Berger, J., Berger, S., et al. (2009). The zebrafish dystrophic mutant softy maintains muscle fibre viability despite basement membrane rupture and muscle detachment. Development 136, 3367–3376. doi: 10.1242/dev.034561
Jardine, S., Anderson, S., Babcock, S., Leung, G., Pan, J., Dhingani, N., et al. (2020). Drug screen identifies leflunomide for treatment of inflammatory bowel disease caused by TTC7A deficiency. Gastroenterology 158, 1000–1015. doi: 10.1053/j.gastro.2019.11.019
Jerman, S., and Sun, Z. (2017). Using zebrafish to study kidney development and disease. Curr. Top. Dev. Biol. 124, 41–79. doi: 10.1016/bs.ctdb.2016.11.008
Kawahara, G., Gasperini, M. J., Myers, J. A., Widrick, J. J., Eran, A., Serafini, P. R., et al. (2014). Dystrophic muscle improvement in zebrafish via increased heme oxygenase signaling. Hum. Mol. Genet. 23, 1869–1878. doi: 10.1093/hmg/ddt579
Knöll, R., Postel, R., Wang, J., Krätzner, R., Hennecke, G., Vacaru, A. M., et al. (2007). Laminin-α4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation 116, 515–525. doi: 10.1161/circulationaha.107.689984
Koch, M., Olson, P. F., Albus, A., Jin, W., Hunter, D. D., Brunken, W. J., et al. (1999). Characterization and expression of the laminin γ3 chain: a novel, non-basement membrane-associated, laminin chain. J. Cell Biol. 145, 605–618. doi: 10.1083/jcb.145.3.605
Lawlor, M. W., Beggs, A. H., Buj-Bello, A., Childers, M. K., Dowling, J. J., James, E. S., et al. (2016). Skeletal muscle pathology in X-linked myotubular myopathy: review with cross-species comparisons. J. Neuropathol. Exp. Neurol. 75, 102–110. doi: 10.1093/jnen/nlv020
Lee, J., and Gross, J. M. (2007). Laminin beta1 and gamma1 containing laminins are essential for basement membrane integrity in the zebrafish eye. Invest. Ophthalmol. Vis. Sci. 48, 2483–2490. doi: 10.1167/iovs.06-1211
Lek, A., Rahimov, F., Jones, P. L., and Kunkel, L. M. (2015). Emerging preclinical animal models for FSHD. Trends Mol. Med. 21, 295–306. doi: 10.1016/j.molmed.2015.02.011
Lek, A., Zhang, Y., Woodman, K. G., Huang, S., DeSimone, A. M., Cohen, J., et al. (2020). Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci. Transl. Med. 12:eaay0271. doi: 10.1126/scitranslmed.aay0271
Li, M., Hromowyk, K. J., Amacher, S. L., and Currie, P. D. (2017). Muscular dystrophy modeling in zebrafish. Methods Cell Biol. 138, 347–380. doi: 10.1016/bs.mcb.2016.11.004
Lin, Y. Y., White, R. J., Torelli, S., Cirak, S., Muntoni, F., and Stemple, D. L. (2011). Zebrafish Fukutin family proteins link the unfolded protein response with dystroglycanopathies. Hum. Mol. Genet. 20, 1763–1775. doi: 10.1093/hmg/ddr059
Lisi, M. T., and Cohn, R. D. (2007). Congenital muscular dystrophies: new aspects of an expanding group of disorders. Biochim. Biophys. Acta 1772, 159–172. doi: 10.1016/j.bbadis.2006.09.006
Lombardo, V. A., Sporbert, A., and Abdelilah-Seyfried, S. (2012). Cell tracking using photoconvertible proteins during zebrafish development. J. Vis. Exp. 67:4350. doi: 10.3791/4350
MacRae, C. A., and Peterson, R. T. (2015). Zebrafish as tools for drug discovery. Nat. Rev. Drug Discov. 14, 721–731. doi: 10.1038/nrd4627
Matsuda, H., Mullapudi, S. T., Yang, Y. H. C., Masaki, H., Hesselson, D., and Stainier, D. Y. R. (2018). Whole-organism chemical screening identifies modulators of pancreatic β-cell function. Diabetes 67, 2268–2279. doi: 10.2337/db17-1223
McGovern, V. L., Massoni-Laporte, A., Wang, X., Le, T. T., Le, H. T., Beattie, C. E., et al. (2015). Plastin 3 expression does not modify spinal muscular atrophy severity in the 7 SMA mouse. PLoS One 10:e0132364. doi: 10.1371/journal.pone.0132364
McKee, K. K., Crosson, S. C., Meinen, S., Reinhard, J. R., Ruegg, M. A., and Yurchenco, P. D. (2017). Chimeric protein repair of laminin polymerization ameliorates muscular dystrophy phenotype. J. Clin. Invest. 127, 1075–1089. doi: 10.1172/jci90854
Mehuron, T., Kumar, A., Duarte, L., Yamauchi, J., Accorsi, A., and Girgenrath, M. (2014). Dysregulation of matricellular proteins is an early signature of pathology in laminin-deficient muscular dystrophy. Skelet. Muscle 4:14. doi: 10.1186/2044-5040-4-14
Meinen, S., Barzaghi, P., Lin, S., Lochmuller, H., and Ruegg, M. A. (2007). Linker molecules between laminins and dystroglycan ameliorate laminin-α2-deficient muscular dystrophy at all disease stages. J. Cell Biol. 176, 979–993. doi: 10.1083/jcb.200611152
Meinen, S., Lin, S., Thurnherr, R., Erb, M., Meier, T., and Ruegg, M. A. (2011). Apoptosis inhibitors and mini-agrin have additive benefits in congenital muscular dystrophy mice. EMBO Mol. Med. 3, 465–479. doi: 10.1002/emmm.201100151
Menezes, M. J., McClenahan, F. K., Leiton, C. V., Aranmolate, A., Shan, X., and Colognato, H. (2014). The extracellular matrix protein laminin α2 regulates the maturation and function of the blood-brain barrier. J. Neurosci. 34, 15260–15280. doi: 10.1523/JNEUROSCI.3678-13.2014
Mercuri, E., Bonnemann, C. G., and Muntoni, F. (2019). Muscular dystrophies. Lancet 394, 2025–2038. doi: 10.1016/S0140-6736(19)32910-1
Mitsuhashi, H., Mitsuhashi, S., Lynn-Jones, T., Kawahara, G., and Kunkel, L. M. (2013). Expression of DUX4 in zebrafish development recapitulates facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 22, 568–577. doi: 10.1093/hmg/dds467
Miyagoe-Suzuki, Y., Nakagawa, M., and Takeda, S. (2000). Merosin and congenital muscular dystrophy. Microsc. Res. Tech. 48, 181–191. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<181::AID-JEMT6>3.0.CO;2-Q
Mohassel, P., Foley, A. R., and Bönnemann, C. G. (2018). Extracellular matrix-driven congenital muscular dystrophies. Matrix Biol. 71–72, 188–204. doi: 10.1016/j.matbio.2018.06.005
Moosajee, M., Gregory-Evans, K., Ellis, C. D., Seabra, M. C., and Gregory-Evans, C. Y. (2008). Translational bypass of nonsense mutations in zebrafish rep1, pax2.1 and lamb1 highlights a viable therapeutic option for untreatable genetic eye disease. Hum. Mol. Genet. 17, 3987–4000. doi: 10.1093/hmg/ddn302
Nance, J. R., Dowling, J. J., Gibbs, E. M., and Bonnemann, C. G. (2012). Congenital myopathies: an update. Curr. Neurol. Neurosci. Rep. 12, 165–174. doi: 10.1007/s11910-012-0255-x
Nguyen, Q., Lim, K. R. Q., and Yokota, T. (2019). Current understanding and treatment of cardiac and skeletal muscle pathology in laminin-α2 chain-deficient congenital muscular dystrophy. Appl. Clin. Genet. 12, 113–130. doi: 10.2147/tacg.s187481
Oliveira, J., Gruber, A., Cardoso, M., Taipa, R., Fineza, I., Goncalves, A., et al. (2018). LAMA2 gene mutation update: toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum. Mutat. 39, 1314–1337. doi: 10.1002/humu.23599
Pakula, A., Lek, A., Widrick, J., Mitsuhashi, H., Bugda Gwilt, K. M., Gupta, V. A., et al. (2019). Transgenic zebrafish model of DUX4 misexpression reveals a developmental role in FSHD pathogenesis. Hum. Mol. Genet. 28, 320–331. doi: 10.1093/hmg/ddy348
Parsons, M. J., Pollard, S. M., Saude, L., Feldman, B., Coutinho, P., Hirst, E. M., et al. (2002). Zebrafish mutants identify an essential role for laminins in notochord formation. Development 129, 3137–3146.
Patton, B. L., Miner, J. H., Chiu, A. Y., and Sanes, J. R. (1997). Distribution and function of laminins in the neuromuscular system of developing, adult and mutant mice. J. Cell Biol. 139, 1507–1521. doi: 10.1083/jcb.139.6.1507
Paulus, J. D., and Halloran, M. C. (2006). Zebrafish bashful/laminin-alpha 1 mutants exhibit multiple axon guidance defects. Dev. Dyn. 235, 213–224. doi: 10.1002/dvdy.20604
Pollard, S. M., Parsons, M. J., Kamei, M., Kettleborough, R. N., Thomas, K. A., Pham, V. N., et al. (2006). Essential and overlapping roles for laminin alpha chains in notochord and blood vessel formation. Dev. Biol. 289, 64–76. doi: 10.1016/j.ydbio.2005.10.006
Postel, R., Vakeel, P., Topczewski, J., Knoll, R., and Bakkers, J. (2008). Zebrafish integrin-linked kinase is required in skeletal muscles for strengthening the integrin-ECM adhesion complex. Dev. Biol. 318, 92–101. doi: 10.1016/j.ydbio.2008.03.024
Pozzi, A., Yurchenco, P. D., and Iozzo, R. V. (2017). The nature and biology of basement membranes. Matrix Biol. 57–58, 1–11. doi: 10.1016/j.matbio.2016.12.009
Quattrocelli, M., Capote, J., Ohiri, J. C., Warner, J. L., Vo, A. H., Earley, J. U., et al. (2017a). Genetic modifiers of muscular dystrophy act on sarcolemmal resealing and recovery from injury. PLoS Genet. 13:e1007070. doi: 10.1371/journal.pgen.1007070
Quattrocelli, M., Spencer, M. J., and McNally, E. M. (2017b). Outside in: the matrix as a modifier of muscular dystrophy. Biochim. Biophys. Acta Mol. Cell Res. 1864, 572–579. doi: 10.1016/j.bbamcr.2016.12.020
Quijano-Roy, S., Sparks, S. E., and Rutkowski, A. (1993). “LAMA2-related muscular dystrophy,” in GeneReviews (R), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens (Seattle, WA: University of Washington). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK97333/.
Gutzman, J. H., Graeden, E. G., Lowery, L. A., Holley, H. S., and Sive, H. (2008). Formation of the zebrafish midbrain-hindbrain boundary constriction requires laminin-dependent basal constriction. Mech. Dev. 125, 974–983. doi: 10.1016/j.mod.2008.07.004
Rahit, K., and Tarailo-Graovac, M. (2020). Genetic modifiers and rare mendelian disease. Genes 11:239. doi: 10.3390/genes11030239
Reinhard, J. R., Lin, S., McKee, K. K., Meinen, S., Crosson, S. C., Sury, M., et al. (2017). Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci. Transl. Med. 9:eaal4649. doi: 10.1126/scitranslmed.aal4649
Rooney, J. E., Gurpur, P. B., and Burkin, D. J. (2009a). Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U S A 106, 7991–7996. doi: 10.1073/pnas.0811599106
Rooney, J. E., Gurpur, P. B., Yablonka-Reuveni, Z., and Burkin, D. J. (2009b). Laminin-111 restores regenerative capacity in a mouse model for α7 integrin congenital myopathy. Am. J. Pathol. 174, 256–264. doi: 10.2353/ajpath.2009.080522
Sabha, N., Volpatti, J. R., Gonorazky, H., Reifler, A., Davidson, A. E., Li, X., et al. (2016). PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J. Clin. Invest. 126, 3613–3625. doi: 10.1172/JCI86841
Saunier, M., Bönnemann, C. G., Durbeej, M., Allamand, V., and CMD Animal Model Consortium. (2016). 212th ENMC international workshop: animal models of congenital muscular dystrophies, naarden, the netherlands, 29–31 May 2015. Neuromuscul. Disord. 26, 252–259. doi: 10.1016/j.nmd.2016.02.002
Schorling, D. C., Kirschner, J., and Bonnemann, C. G. (2017). Congenital muscular dystrophies and myopathies: an overview and update. Neuropediatrics 48, 247–261. doi: 10.1055/s-0037-1604154
Semina, E. V., Bosenko, D. V., Zinkevich, N. C., Soules, K. A., Hyde, D. R., Vihtelic, T. S., et al. (2006). Mutations in laminin α1 result in complex, lens-independent ocular phenotypes in zebrafish. Dev. Biol. 299, 63–77. doi: 10.1016/j.ydbio.2006.07.005
Sframeli, M., Sarkozy, A., Bertoli, M., Astrea, G., Hudson, J., Scoto, M., et al. (2017). Congenital muscular dystrophies in the UK population: clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul. Disord. 27, 793–803. doi: 10.1016/j.nmd.2017.06.008
Sittaramane, V., Sawant, A., Wolman, M. A., Maves, L., Halloran, M. C., and Chandrasekhar, A. (2009). The cell adhesion molecule Tag1, transmembrane protein Stbm/Vangl2 and Lamininalpha1 exhibit genetic interactions during migration of facial branchiomotor neurons in zebrafish. Dev. Biol. 325, 363–373. doi: 10.1016/j.ydbio.2008.10.030
Smith, L. L., Beggs, A. H., and Gupta, V. A. (2013). Analysis of skeletal muscle defects in larval zebrafish by birefringence and touch-evoke escape response assays. J. Vis. Exp. 82:e50925. doi: 10.3791/50925
Smith, S. J., Horstick, E. J., Davidson, A. E., and Dowling, J. (2015). Analysis of zebrafish larvae skeletal muscle integrity with evans blue dye. J. Vis. Exp. 105:53183. doi: 10.3791/53183
Smith, S. J., Wang, J. C., Gupta, V. A., and Dowling, J. J. (2017). A novel early onset phenotype in a zebrafish model of merosin deficient congenital muscular dystrophy. PLoS One 12:e0172648. doi: 10.1371/journal.pone.0172648
Snow, C. J., Goody, M., Kelly, M. W., Oster, E. C., Jones, R., Khalil, A., et al. (2008). Time-lapse analysis and mathematical characterization elucidate novel mechanisms underlying muscle morphogenesis. PLoS Genet. 4:e1000219. doi: 10.1371/journal.pgen.1000219
Snow, C. J., and Henry, C. A. (2009). Dynamic formation of microenvironments at the myotendinous junction correlates with muscle fiber morphogenesis in zebrafish. Gene Expr. Patterns 9, 37–42. doi: 10.1016/j.gep.2008.08.003
Steffen, L. S., Guyon, J. R., Vogel, E. D., Beltre, R., Pusack, T. J., Zhou, Y., et al. (2007). Zebrafish orthologs of human muscular dystrophy genes. BMC Genomics 8:79. doi: 10.1186/1471-2164-8-79
Straub, V., Rafael, J. A., Chamberlain, J. S., and Campbell, K. P. (1997). Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 139, 375–385. doi: 10.1083/jcb.139.2.375
Sztal, T., Berger, S., Currie, P. D., and Hall, T. E. (2011). Characterization of the laminin gene family and evolution in zebrafish. Dev. Dyn. 240, 422–431. doi: 10.1002/dvdy.22537
Sztal, T. E., Sonntag, C., Hall, T. E., and Currie, P. D. (2012). Epistatic dissection of laminin-receptor interactions in dystrophic zebrafish muscle. Hum. Mol. Genet. 21, 4718–4731. doi: 10.1093/hmg/dds312
Sztal, T. E., Zhao, M., Williams, C., Oorschot, V., Parslow, A. C., Giousoh, A., et al. (2015). Zebrafish models for nemaline myopathy reveal a spectrum of nemaline bodies contributing to reduced muscle function. Acta Neuropathol. 130, 389–406. doi: 10.1007/s00401-015-1430-3
Telfer, W. R., Busta, A. S., Bonnemann, C. G., Feldman, E. L., and Dowling, J. J. (2010). Zebrafish models of collagen VI-related myopathies. Hum. Mol. Genet. 19, 2433–2444. doi: 10.1093/hmg/ddq126
Telfer, W. R., Nelson, D. D., Waugh, T., Brooks, S. V., and Dowling, J. J. (2012). Neb: a zebrafish model of nemaline myopathy due to nebulin mutation. Dis. Model. Mech. 5, 389–396. doi: 10.1242/dmm.008631
Thomasi, A. B. D., Sonntag, C., Pires, D. F., Zuidema, D., Benci, A., Currie, P. D., et al. (2018). A low-cost pulse generator for exacerbating muscle fiber detachment phenotypes in zebrafish. Zebrafish 15, 420–424. doi: 10.1089/zeb.2017.1512
Tunggal, P., Smyth, N., Paulsson, M., and Ott, M. C. (2000). Laminins: structure and genetic regulation. Microsc. Res. Tech. 51, 214–227. doi: 10.1002/1097-0029(20001101)51:3<214::aid-jemt2>3.0.co;2-j
Van Ry, P. M., Minogue, P., Hodges, B. L., and Burkin, D. J. (2014). Laminin-111 improves muscle repair in a mouse model of merosin-deficient congenital muscular dystrophy. Hum. Mol. Genet. 23, 383–396. doi: 10.1093/hmg/ddt428
Verma, S., Goyal, P., Guglani, L., Peinhardt, C., Pelzek, D., and Barkhaus, P. E. (2018). COL6A and LAMA2 mutation congenital muscular dystrophy: a clinical and electrophysiological study. J. Clin. Neuromuscul. Dis. 19, 108–116. doi: 10.1097/cnd.0000000000000198
Vishnudas, V. K., and Miller, J. B. (2009). Ku70 regulates Bax-mediated pathogenesis in laminin-α2-deficient human muscle cells and mouse models of congenital muscular dystrophy. Hum. Mol. Genet. 18, 4467–4477. doi: 10.1093/hmg/ddp399
Volpatti, J. R., Endo, Y., Knox, J., Groom, L., Brennan, S., Noche, R., et al. (2020). Identification of drug modifiers for RYR1 related myopathy using a multi-species discovery pipeline. Elife 9:e52946. doi: 10.7554/eLife.52946
Waugh, T. A., Horstick, E., Hur, J., Jackson, S. W., Davidson, A. E., Li, X., et al. (2014). Fluoxetine prevents dystrophic changes in a zebrafish model of Duchenne muscular dystrophy. Hum. Mol. Genet. 23, 4651–4662. doi: 10.1093/hmg/ddu185
Webb, A. E., Sanderford, J., Frank, D., Talbot, W. S., Driever, W., and Kimelman, D. (2007). Laminin alpha5 is essential for the formation of the zebrafish fins. Dev. Biol. 311, 369–382. doi: 10.1016/j.ydbio.2007.08.034
Widrick, J. J., Alexander, M. S., Sanchez, B., Gibbs, D. E., Kawahara, G., Beggs, A. H., et al. (2016). Muscle dysfunction in a zebrafish model of Duchenne muscular dystrophy. Physiol. Genomics 48, 850–860. doi: 10.1152/physiolgenomics.00088.2016
Widrick, J. J., Kawahara, G., Alexander, M. S., Beggs, A. H., and Kunkel, L. M. (2019). Discovery of novel therapeutics for muscular dystrophies using zebrafish phenotypic screens. J. Neuromuscul. Dis. 6, 271–287. doi: 10.3233/jnd-190389
Willmann, R., Gordish-Dressman, H., Meinen, S., Ruegg, M. A., Yu, Q., Nagaraju, K., et al. (2017). Improving reproducibility of phenotypic assessments in the dyw mouse model of laminin-α2 related congenital muscular dystrophy. J. Neuromuscul. Dis. 4, 115–126. doi: 10.3233/jnd-170217
Wolman, M. A., Sittaramane, V. K., Essner, J. J., Yost, H. J., Chandrasekhar, A., and Halloran, M. C. (2008). Transient axonal glycoprotein-1 (TAG-1) and laminin-alpha1 regulate dynamic growth cone behaviors and initial axon direction in vivo. Neural Dev. 3:6. doi: 10.1186/1749-8104-3-6
Wood, A. J., Cohen, N., Joshi, V., Li, M., Costin, A., Hersey, L., et al. (2019). RGD inhibition of itgb1 ameliorates laminin-α2-deficient zebrafish fibre pathology. Hum. Mol. Genet. 28, 1403–1413. doi: 10.1093/hmg/ddy426
Wood, A. J., and Currie, P. D. (2014). Analysing regenerative potential in zebrafish models of congenital muscular dystrophy. Int. J. Biochem. Cell Biol. 56, 30–37. doi: 10.1016/j.biocel.2014.10.021
Yao, Y. (2017). Laminin: loss-of-function studies. Cell. Mol. Life Sci. 74, 1095–1115. doi: 10.1007/s00018-016-2381-0
Yurchenco, P. D., McKee, K. K., Reinhard, J. R., and Ruegg, M. A. (2018). Laminin-deficient muscular dystrophy: molecular pathogenesis and structural repair strategies. Matrix Biol. 71–72, 174–187. doi: 10.1016/j.matbio.2017.11.009
Zhang, Y., Zhang, Z., and Ge, W. (2018). An efficient platform for generating somatic point mutations with germline transmission in the zebrafish by CRISPR/Cas9-mediated gene editing. J. Biol. Chem. 293, 6611–6622. doi: 10.1074/jbc.ra117.001080
Zhao, M., Smith, L., Volpatti, J., Fabian, L., and Dowling, J. J. (2019). Insights into wild-type dynamin 2 and the consequences of DNM2 mutations from transgenic zebrafish. Hum. Mol. Genet. 28, 4186–4196. doi: 10.1093/hmg/ddz260
Zinkevich, N. S., Bosenko, D. V., Link, B. A., and Semina, E. V. (2006). Laminin alpha 1 gene is essential for normal lens development in zebrafish. BMC Dev. Biol. 6:13. doi: 10.1186/1471-213X-6-13
Keywords: laminin, LAMA2 gene, muscular dystrophy, zebrafish model, MDC1A
Citation: Fabian L and Dowling JJ (2020) Zebrafish Models of LAMA2-Related Congenital Muscular Dystrophy (MDC1A). Front. Mol. Neurosci. 13:122. doi: 10.3389/fnmol.2020.00122
Received: 30 April 2020; Accepted: 11 June 2020;
Published: 09 July 2020.
Edited by:
Stefano Carlo Previtali, San Raffaele Scientific Institute (IRCCS), ItalyReviewed by:
Hanns Lochmuller, University of Ottawa, CanadaPeter Currie, Australian Regenerative Medicine Institute (ARMI), Australia
Copyright © 2020 Fabian and Dowling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James J. Dowling, amFtZXMuZG93bGluZ0BzaWNra2lkcy5jYQ==