 Iason Keramidis
Iason Keramidis Ergina Vourkou1,2†
Ergina Vourkou1,2† Katerina Papanikolopoulou
Katerina Papanikolopoulou Efthimios M. C. Skoulakis
Efthimios M. C. Skoulakis- 1Biomedical Sciences Research Centre “Alexander Fleming”, Institute for Fundamental Biomedical Research, Vari, Greece
- 21st Department of Neurology, Memory and Movement Disorders Clinic, Eginition Hospital, Medical School, National and Kapodistrian University of Athens, Athens, Greece
Hyperphosphorylated Tau protein is the main component of the neurofibrillary tangles, characterizing degenerating neurons in Alzheimer’s disease and other Tauopathies. Expression of human Tau protein in Drosophila CNS results in increased toxicity, premature mortality and learning and memory deficits. Herein we use novel transgenic lines to investigate the contribution of specific phosphorylation sites previously implicated in Tau toxicity. These three different sites, Ser238, Thr245, and Ser262 were tested either by blocking their phosphorylation, by Ser/Thr to Ala substitution, or pseudophosphorylation, by changing Ser/Thr to Glu. We validate the hypothesis that phosphorylation at Ser262 is necessary for Tau-dependent learning deficits and a “facilitatory gatekeeper” to Ser238 occupation, which is linked to Tau toxicity. Importantly we reveal that phosphorylation at Thr245 acts as a “suppressive gatekeeper”, preventing phosphorylation of many sites including Ser262 and consequently of Ser238. Therefore, we elucidate novel interactions among phosphosites central to Tau mediated neuronal dysfunction and toxicity, likely driven by phosphorylation-dependent conformational plasticity.
Introduction
Tau is a multifunctional neuronal microtubule-associated protein (Sotiropoulos et al., 2017) important for regulation of axonal transport and the length of the labile domains of axonal microtubules (Qiang et al., 2018). A single gene gives rise to 6 protein isoforms in the adult human brain via alternative splicing that results in isoforms that contain 1, 2 or none sequence blocks at their amino-terminus (0N, 1N, 2N), as well as inclusion of exon 10, or its exclusion from the mRNA (Andreadis et al., 1992). This leads to isoforms with 3 (3R isoforms), or 4 (4R isoforms) caboxy-terminal repeats, which mediate interactions with the microtubular cytoskeleton (Buee et al., 2000). A predominant characteristic of all 6 Tau isoforms present in the human brain is their highly elevated steady state phosphorylation load mediated by the opposing actions of several kinases and phosphatases (Johnson and Stoothoff, 2004). Tau has 84 putative phosphorylation sites and it is reported more extensively phosphorylated during development than in mature neurons (Brion et al., 1993; Yu et al., 2009). Multiple kinases have been shown to phosphorylate Tau in vitro and in cells, but whether they also target Tau in vivo and under what circumstances remains largely elusive (Hosoi et al., 1995; Hong et al., 1997; Zheng-Fischhofer et al., 1998; Gong et al., 2005).
Interestingly, studies indicate that Tau phosphorylation might be primed by occupation of a particular site before the occurrence of additional phosphorylations (Hanger et al., 2009). Tau contains multiple “intrinsically disordered regions” (IDRs), which interfere with structural stability of the protein (Uversky, 2015). Then, this “gatekeeper” phosphorylation effect suggests that it may enable or inhibit local tertiary structures that expose or occlude other, often distant, phosphorylation sites (Jeganathan et al., 2008; Sibille et al., 2012; Schwalbe et al., 2015). Hence, the effect of specific phosphorylations may be to regulate this “structural plasticity” of Tau, contribute to the subcellular localization of Tau isoforms (Sotiropoulos et al., 2017) and modulate their functional properties (Xia et al., 2015).
Irrespective of whether it is mutated, or wild-type, pathological Tau presents increased phosphorylation at sites occupied physiologically, but also on sites occupied only when pathology is present and are referred to as “disease-associated” epitopes (Morris et al., 2015; Arendt et al., 2016). Although the mechanisms that trigger hyper-phosphorylation are unclear at present, the result is neuronal deposition of hyper-phosphorylated Tau (Martin et al., 2011). If indeed phosphorylations modulate the structure and functional properties of Tau isoforms, then this hyper-phosphorylation is likely to underlie significant changes in the properties of the protein that underlie its pathobiology (Regan et al., 2017). In fact, extensive literature has led to the widely held notion that aberrant Tau phosphorylation is central to neuronal pathology (Stoothoff and Johnson, 2005) and provided evidence that soluble hyper-phosphorylated Tau contributes to neuronal dysfunction before its aggregation (Fath et al., 2002; Santacruz et al., 2005; Brandt et al., 2009; Decker et al., 2015).
Antibodies that recognize non-physiologically phosphorylated Tau at specific sites (phosphoepitopes) in patient neurons but not in age-matched healthy individuals have been developed and used as specific diagnostic markers of Tauopathies (Sergeant et al., 2005). However, the mechanistic understanding of the sequential phosphorylation events that occur on Tau and which sites are essential for maintenance and evolution of pathology are still unclear. Identification of phosphorylation sites on Tau that either trigger or are essential for pathogenesis are pivotal to our understanding of Tau-dependent neuronal malfunction and toxicity.
Drosophila models of Tauopathies contribute significantly to the concept that accumulation of prefibrillar hyper-phosphorylated forms of Tau correlate with human Tau-mediated toxicity in flies (Wittmann et al., 2001; Steinhilb et al., 2007a, b; Feuillette et al., 2010). Recently, we have identified two novel phosphorylation sites on Tau, Ser238 and Thr245, as essential for its toxic effects on mushroom body (MB) integrity (Kosmidis et al., 2010; Papanikolopoulou et al., 2010) and premature lethality (Papanikolopoulou and Skoulakis, 2015). The MBs are neuronal assemblies that constitute major insect brain centers for learning and memory (Davis, 2005). Significantly, blocking Ser238 and Thr245 phosphorylation by substituting them with alanines (STA mutant), yielded animals with structurally normal but profoundly dysfunctional MBs, as flies accumulating the mutant protein exhibited impaired associative learning (Kosmidis et al., 2010). Moreover, our results strongly suggested that Ser238 occupation is a critical mediator of Tau neurotoxicity in vivo. However, neuronal dysfunction measured as olfactory learning deficits appeared to rely on phosphorylation at Ser262, whose enhanced occupation appeared to account for the learning deficits of animals expressing the STA mutant Tau (Papanikolopoulou and Skoulakis, 2015).
Our previous analyses were performed on the double mutant STA and with Tau transgenes in different genomic locations with consequent variable transgenic protein expression, which attenuated accurate quantitative measures of toxicity and dysfunction and their dependence on relative hTau levels in the CNS. Therefore, we aimed to investigate the contribution of each of these two sites independently and investigate the potential interplay between them with respect to neuronal toxicity and dysfunction with quantifiably equally expressed transgenes. Hence, we generated new single mutants that block or mimic phosphorylation at these sites. We also focus on the role of Ser262 as a “gatekeeper” of disease-associated phospho-epitope appearance and whether as previously suggested (Papanikolopoulou and Skoulakis, 2015), its occupation is regulated by phosphorylation of Ser238 or Thr245 or vice versa.
Materials and Methods
Drosophila Culture and Strains
Drosophila crosses were set up en masse in standard wheat-flour-sugar food supplemented with soy flour and CaCl2 and cultured at 25°C and 50–70% humidity in a 12 h light/dark cycle unless noted otherwise. The Elavc155-Gal4 and Ras2-Gal4 have been described before (Gouzi et al., 2011). The Elav-Gal4 line on the second chromosome was obtained from Bloomington Drosophila Stock center (#8765). The dual Gal 4 driver strains Elavc155-Gal4;Ras2-Gal4 (henceforth Elav;Ras2) and Elavc155-Gal4;Elav/CyO (henceforth Elav;Elav), were constructed by standard crosses. The hTau0N4R (0N4R) transgenic flies were a gift from M. Feany (Harvard Medical School, Boston, MA, United States).
To generate the transgenics carrying point mutations, the hTau0N4R cDNA (kind gift from Dr. Martin Chow, University of Kentucky), was subcloned into the pUAS.attB vector (Bischof et al., 2007) as a BglII/XbaI fragment. The mutants were generated by replacing the indicated Ser and Thr residues with Ala or Glu using the QuickChange XL site-directed mutagenesis kit (Agilent) according to the manufacturer’s instructions. The complementary mutagenic oligonucleotides pairs (5’ to 3’) for each mutant are shown below. The silent restriction sites introduced for effective screening of positive clones appear underlined in italics whereas the amino acid substitution is shown in bold.
S238A (PvuII): 5’ CCACCCAAGTCGCCGTCA GCTGCCAAGAGCCGCCTGCAGACAGC.
GGTGGGTTCAGCGGCAGTCGACGGTTCTCGGCG GACGTCTGTCG 5’.
S238E (XbaI): 5’ CCAAGTCGCCGTCTGAGGCCAAG TCTAGACTGCAGACAGCCCC.
GGTTCAGCGGCAGACTCCGGTTCAGATCTGACGTCTG TCGGGG 5’.
T245A (SalI): 5’ CTTCCGCCAAGAGTCGACTGCAGGCAG CCCCCGTGCCCATG.
GAAGGCGGTTCTCAGCTGACGTCCGTCGGGG GCACGGGTAC 5’
T245E (SalI): 5’ CGTCTTCCGCCAAGAGTCGACTGCAG GAGGCCCCCGTGCCCATG.
GCAGAAGGCGGTTCTCAGCTGACGTCCTCCGGGGG CACGGGTAC 5’.
S262A (BglII): 5’ GCCAGATCTGAAGAATGTCAAGTCCAA GATCGGCGCCACTGAG.
CGGTCTAGACTTCTTACAGTTCAGGTTCTAGCCGCGG TGACTC 5’.
S262E (BglII): 5’ GCCAGATCTGAAGAATGTCAA GTCCAAGATCGGCGAGACTGAG.
CGGTCTAGACTTCTTACAGTTCAGGTTCT AGCCGCTCTGACTC 5’.
STA (PvuII): 5’ CCAAGTCGCCGTCAGCTGCCAAGAGCC GCCTGCAGGCAGCCCCCG.
GGTTCAGCGGCAGTCGAC GGTTCTCGGCGGACGTCCGTCGGGGGC 5’.
The sequence of the mutants was confirmed by sequencing (VBC-biotech). Transgenic flies were generated by phiC31-mediated transgenesis by BestGene Inc. (Chino Hills, CA, United States). DNAs were injected into genomic landing site 53B2 on the second chromosome (BDSC #9736).
To obtain flies for experiments detailed herein, virgins from the driver strains were crossed to males bearing the hTau transgenes and experimental genotypes were selected from the progeny as appropriate and as detailed below:
Female Elav X 0N4R or 0N4RII or w1118 (for controls).
Female Elav;Elav/CyO X 0N4R or 0N4RII: Select male and female non-CyO progeny.
Female Elav;Ras2 × 0N4R or 0N4RII: Select male and female non-CyO progeny.
Female Elav;Ras2 × 0N4RS238A or 0N4RS238E or 0N4RT245A or 0N4RT245E or 0N4RS262A or 0N4RS262E or 0N4RSTA: Select male and female progeny.
Female Elav;Ras2 X w1118: Select male and female progeny for controls.
Histology
Immunohistochemistry on paraffin sections of Drosophila adult heads were performed on paraffin sections as previously described (Kosmidis et al., 2010) using the anti-Leo primary antibody at 1:4000.
Western Blot and Antibodies
Total Tau levels and occupation of particular phosphosites were determined in 2–5 adult female head homogenates in 1x Laemmli buffer (50 mM Tris pH 6.8, 5% 2-mercaptoethanol, 2% SDS, 10% glycerol, and 0.01% bromophenol blue). The monoclonal antibodies T46 (recognizing hTau irrespective of phosphorylation- Invitrogen, 1:3,000), AT8 (recognizing phosphorylated Ser202/Thr205-Pierce Endogen, 1:1,000), AT100 (recognizing phosphorylated Thr212/Ser214-Pierce Endogen 1:1,000) and the polyclonal antibodies pS396 (Source Bioscience, 1:3,000) and pS262 (Source Bioscience, 1:1,000) were used. We generated a novel polyclonal antibody against phospho-Ser238 of hTau, raised in rabbits (Pocono Rabbit Farm and Laboratory) against peptide TPPKSPSpSAKSRLQTAPVPMP and affinity purified before use. In order to normalize sample loading, anti-Syntaxin (mAb 8C3, Developmental Studies Hybridoma Studies) was used at 1:3,000. Secondary antibodies were applied at 1:5,000.
Lifespan Determination
Flies accumulating hTau0N4R variants under Elavc155-Gal4;Ras2-Gal4 were raised at 18°C and subsequently 17 groups of 20 (340 flies per genotype in two biological replicates), 1–2 day-old males per genotype were maintained at 30°C transferring to fresh vials every 3 days until they expired (Papanikolopoulou and Skoulakis, 2015).
Drug Feeding
Flies accumulating hTau0N4R variants under Elavc155-Gal4; Ras2-Gal4 were raised at 25°C together with control heterozygotes. Paraquat feeding was performed as previously described (Dias-Santagata et al., 2007). Briefly fifteen groups of 20, (two biological replicates of 300 flies total) 1–2 day-old males per genotype were fed 30 mM of methyl viologen (Acros Organics) in standard food for 24 h or as indicated and the number of surviving flies per vial were counted.
Behavioral Assessment
Behavioral assays were performed under dim red light at 24–25°C and 70-75% humidity. All flies were 2–5 days old, collected under CO2 anesthesia one day prior to the experiment and kept in food vials in groups of 50–70 flies each at 30°C. Pavlovian olfactory aversive conditioning was performed using aversive odors as conditioned stimuli (CS+ and CS−) with the electric shock unconditioned stimulus (US). The odors used were benzaldehyde (BNZ) and 3-octanol (OCT) diluted in oil (5% v/v for BNZ and 50% v/v for OCT). One hour before training flies were transferred to fresh food vials. For training, a group of 50–70 flies were transferred into a tube lined with electrifiable grid and presented with air (500 mL/min). Flies were first exposed to an odor for 30 s paired with 90 V shock (consisting of six 1.25 s pulses with 4.5 s inter-pulse intervals, so 6 US/CS pairings were delivered within 30 s of odor presentation) and then 30 s of air. Subsequently, flies were exposed to the second odor for 30 s without shock and then 30 s of air. Two groups of animals of the same genotype were trained simultaneously such as, one to avoid BNZ and the other to avoid OCT, while the complimentary odorant was used as control odor. The animals were transferred to a T-maze apparatus immediately and flies were tested simultaneously for preferential avoidance of the conditioned odorant allowed to choose between the two odors for 90 s. A performance index (PI) was calculated as the fraction of flies that avoided the CS+ minus the fraction that avoided the CS− odors divided by the total number of flies in the experiment. A final PI is the average of the scores from the two groups of flies trained to complementary conditioning stimuli and ranges from 0 to 100. Because all new transgenes are inserted in the same site we used one of the cognates as heterozygous controls.
Statistical Analyses
Quantification of all Western blots was performed by densitometry and the ratio of Tau relative to that of Syntaxin (Syx) was calculated. The ratio of the control genotype was set to 1 and all experimental ratios were reported as relative to that. The means and SEMs were compared following an initial, significant differences- indicating ANOVA (positive ANOVA), using Dunnett’s tests relative to the designated control. Similarly, learning performance indices were calculated for each genotype as indicated above and following positive ANOVA, means and SEMs were compared to that of controls using Dunnett’s tests.
Survival data were examined for differences at each assessment day using Wilcoxon/Kruskal-Wallis tests. If significant differences were uncovered then the means and SEMs from each genotype for that day were compared relative to controls using the Steel with control tests. Means and SEMs of survival upon oxidative stress toxicity were compared to that that of controls or the variants as designated using Dunnett’s or planned multiple comparisons tests following an initial positive ANOVA. All statistical details are presented in Supplementary Tables 1–5.
Results
Learning Deficits Are Independent of hTau Levels in Adult Drosophila
Because the randomly inserted hTauSTA was generated in the 2N4R isoform (Kosmidis et al., 2010), to generalize the effects of that double mutation and validate the derived conclusions (Papanikolopoulou and Skoulakis, 2015) independently, we generated the STA double mutation in the 0N4R isoform. In addition, comparison of the effects of transgenics on neuronal function and neurotoxicity is expected to be facilitated if the protein levels were similar or the same, rather than the variable levels attained by randomly inserted transgenes. Therefore, we generated new 0N4R, STA and single phosphomutant strains via phiC31-mediated transgenesis (Bischof et al., 2007), with all transposons inserted in the same landing site on the second chromosome (55B2), expected to yield similar expression levels to facilitate cross variant comparisons. Importantly, all new transgenes used herein presented similar levels of expression (Supplementary Figure 1), ascertaining that potential phenotypic differences among the transgenic proteins would not be due to differences in expression, but rather reflect differential functional effects.
Interestingly, unlike for the randomly inserted hTau0N4R (henceforth 0N4R) (Wittmann et al., 2001), pan-neuronal expression of the same transgene integrated in the second chromosome landing site (0N4RII) did not result in MB ablation (Figure 1A2 vs. Figure 1A3), as previously reported (Kosmidis et al., 2010). We hypothesized that integration within the attp sites, or at the specific location on the second chromosome must have attenuated 0N4RII expression resulting in intact MBs. To address this and because all phosphosite variants we generated were integrated in the same chromosomal site, we sought to increase expression of these transgenes to match that of 0N4R. To that end we combined two copies of the pan-neuronal Elav driver, the typical one on the X chromosome with an independent insertion on the second (Elav;Elav), or the pan-neuronal Elav driver on the X with the ubiquitous adult CNS driver Ras2 (Gouzi et al., 2011) as detailed in Materials and Methods.
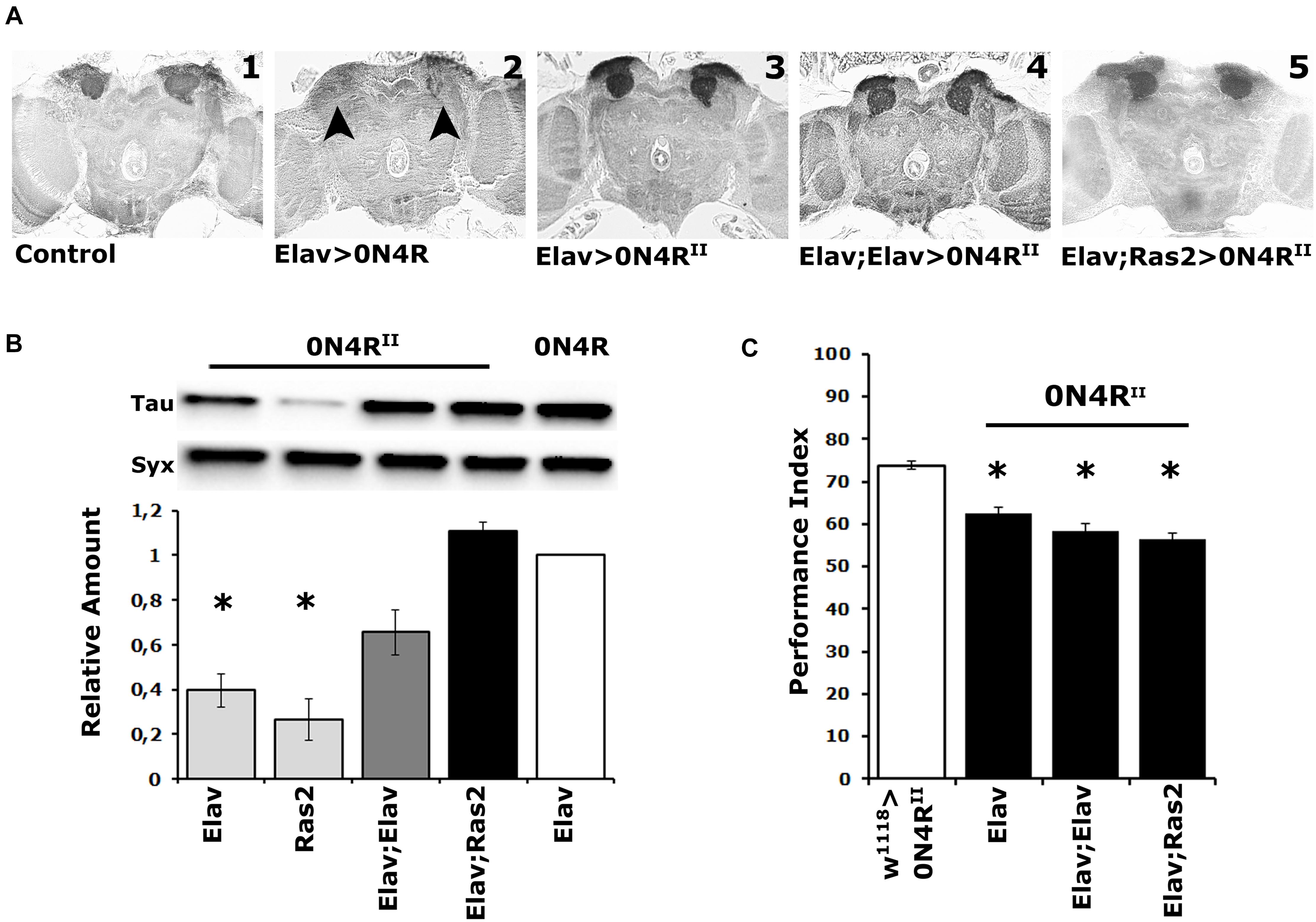
Figure 1. Elevation of hTau in the fly CNS does not precipitate MB structural deficits, and the MBs remain dysfunctional over a range of transgenic protein accumulation. (A) Carnoy’s-fixed paraffin-embedded frontal sections at the level of the MB dendrites (calyces) from control (Elav/ +) and animals expressing the different 0N4R-encoding transgenes with the indicated Gal4 driver. The morphology of the MBs was evaluated with the anti-Leonardo antibody. Arrowheads point to the missing calyces in animals expressing the randomly inserted 0N4R human transgene. (B) Representative Western blot of head lysates from animals expressing the 0N4RII and 0N4R transgenes under the indicated Gal4 driver using the T46 anti-Tau antibody. The genotype of animals used is indicated below the quantification. For the quantification, Tau levels were normalized using the Syx loading control and are shown as a ratio of their mean ± SEM values relative to respective levels in Elav > 0N4R flies, which was set to 1. The star indicates significant differences from the control genotype indicative of reduced Tau levels. Statistical details in Supplementary Table 1. (C) Learning performance of animals accumulating pan-neuronally the indicated 0N4R transgenes (black bars), compared with transgene heterozygotes (white bars). The genotypes of all animals are indicated below each bar. The means ± standard error of the mean (SEM) are shown. Stars indicate significant differences from control. Statistical details in Supplementary Table 1.
Quantification of 0N4RII levels relative to those of the original 0N4R revealed significant differences under Elav, Ras2, but not the double driver Elav;Elav, or Elav;Ras2 (Figure 1B and Supplementary Table 1). Importantly, 0N4RII accumulation under Elav;Ras2 was the closest to the levels of 0N4R under Elav (Figure 1B and Supplementary Table 1) as reported before (Kosmidis et al., 2010). Therefore, the Elav;Ras2 composite driver was used for all subsequent experiments. Interestingly, however, despite the similar levels of expression the MBs remained structurally intact in animals expressing 0N4RII under Elav;Ras2 (Figure 1A5), as they also did under Elav;Elav (Figure 1A4). In fact quantification of the dendritic areas (calyces) stained by the anti-Leo antibody in controls (0.1376 mm2) compared to those in animals expressing 0N4RII under Elav;Ras2 (0.1268 mm2) did not yield significant differences (Student’s t-test, p = 0.1716, n = 6). This indicates that the structural deficits of the MBs are highly sensitive to the amount of hTau present during their development (Kosmidis et al., 2010) and levels under Elav;Ras2 are likely just under the threshold requisite to precipitate defects. Alternatively, the presence of the attp sites flanking the new transgenes may insulate them from non-specific effects of neighboring chromatin on their levels, or temporal expression pattern. This likely leads to bypassing the sensitive period during early embryogenesis when hTau elevation results in aberrant MBs (Kosmidis et al., 2010).
Importantly, expression of the 0N4RII transgene under the single Elav, the Elav;Elav or the Elav;Ras2 drivers yielded similar deficits in associative learning (Figure 1C and Supplementary Table 1). Although the lowest performance index was consistently attained under Elav;Ras2, it was marginally (p = 0.002) different than that under Elav and not different from the performance of animals expressing 0N4RII under Elav;Elav (Supplementary Table 1). Since 0N4RII expression under all pan-neuronal drivers tested yielded similar learning deficits, these results demonstrate that the magnitude of learning impairment is not proportional to the levels of hTau and is not consequent of altered MB integrity. Therefore, we used Elav;Ras2 to drive 0N4RII for subsequent analyses because it induces expression levels similar to previously used and reported conditions (Kosmidis et al., 2010; Papanikolopoulou et al., 2010; Papanikolopoulou and Skoulakis, 2015), which would facilitate comparisons and extrapolations.
Differential Contributions of Ser238 and Thr245 to hTau-Dependent Toxicity
Initially we determined whether the Ser238 and Thr245 to Ala double mutation (STA), on the 0N4R isoform suppresses hTau toxicity measured as elevated age-dependent mortality as did when on the 2N4R isoform (Kosmidis et al., 2010). To that end, we raised animals at 18C to keep transgene expression minimal and then transferred and maintained the adults to 30C to maximize transgene expression and scored for survivors every 2 days. Importantly, expression of 0N4RII resulted in premature lethality relative to controls (Figure 2A and Supplementary Table 2), similar to the randomly inserted transgene (Papanikolopoulou and Skoulakis, 2015). In contrast, expression of 0N4RSTA did not yield significantly different survival profile from that of controls (Figure 2A and Supplementary Table 2). Significantly, on the 29th day when the population of control animals is reduced by 50% (50% attrition-dotted line on Figure 2A), the population of surviving 0N4RII animals was significantly different from controls while that of 0N4RSTA-expressing flies was not (Supplementary Table 2). Therefore, the new transgenes on chromosome II fully recapitulate and verify the survival results obtained with the randomly inserted transgenic strains (Papanikolopoulou and Skoulakis, 2015). Because the new transgenes are expressed at the same level (Supplementary Figure 1), these results confirm that toxicity is independent of hTau protein levels and the assertion that Ser238 or Thr245, or both are required for hTau dependent toxicity manifested as premature mortality.
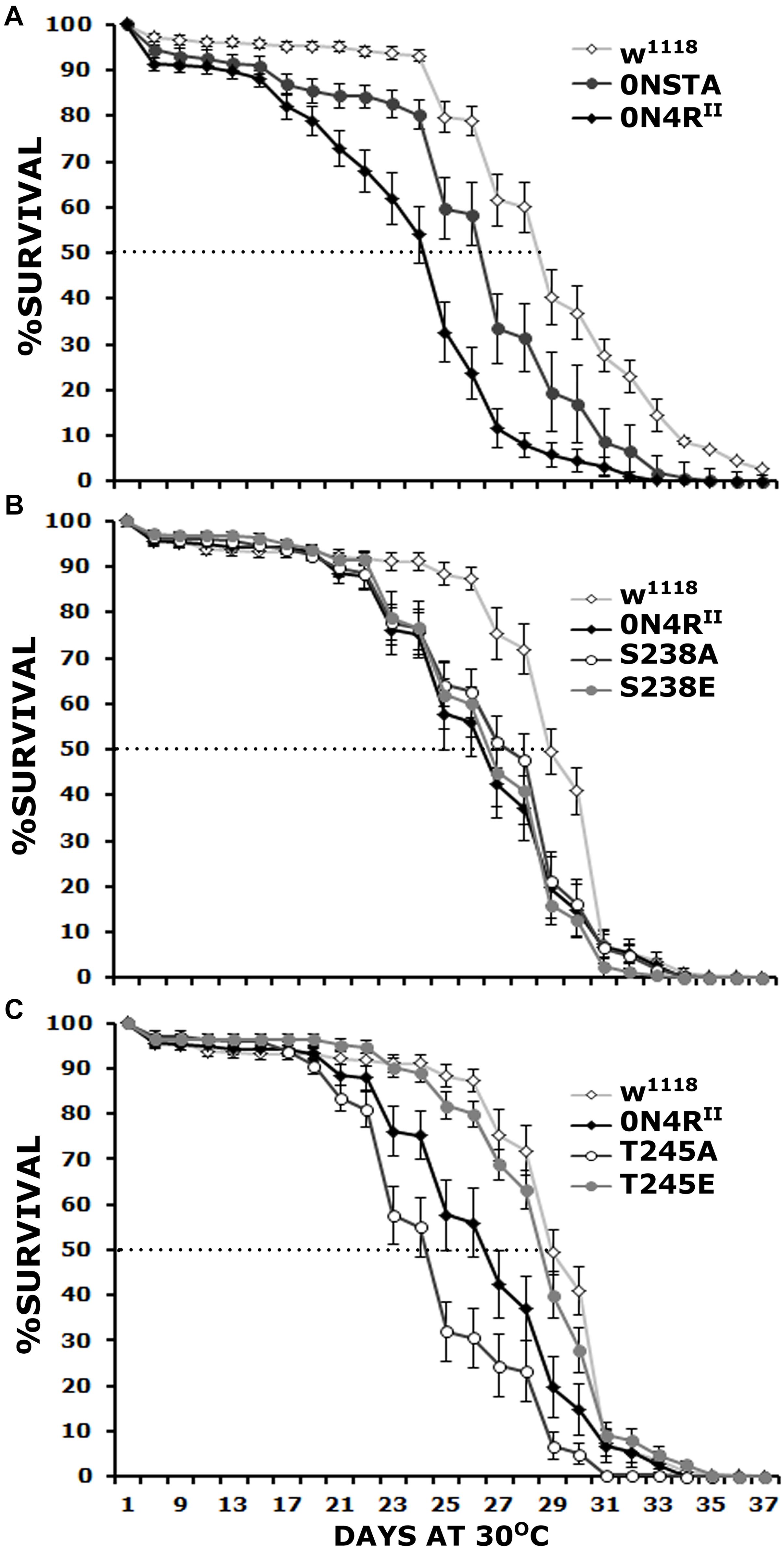
Figure 2. Differential effects of mutants on Ser238 and Thr245 on premature lethality. Survival curves for animals expressing 0N4RII, 0NSTA (A), S238A, S238E (B), T245A, and T245E variants (C) expressed in the CNS in comparison with Elav/+; Ras/ + control heterozygotes (w1118). The data represent the mean ± SEM from two independent experiments with a total of 340 flies assessed per genotype. After 29 days, the population of Elav/+; Ras/ + control heterozygotes (w1118) was reduced by 50% (50% attrition). Statistical details in Supplementary Table 2.
To determine whether both, or which of the two phosphorylation sites mutated in the STA transgene mediate hTau toxicity, we examined the effects of single substitutions of Ser238 and Thr245 either to the phosphorylation-blocking Ala, or the potential phosphomimic Glu. Ser238 has been reported phosphorylated immediately before the 50% attrition point on 0N4R (Papanikolopoulou and Skoulakis, 2015), leading to the hypothesis that its occupation predicts toxicity. Surprisingly, however, blocking phosphorylation at Ser238 did not ameliorate the toxicity of the 0N4RS238A protein (Figure 2B), which remained significantly different from that of control flies from days 23 until 30, including day 29 when the control population reached the 50% attrition day (Figure 2B and Supplementary Table 2). The pseudophosphorylated 0N4RS238E also remained as toxic as the parental 0N4RII (Figure 2B and Supplementary Table 2). The results suggest that phosphorylation at Ser238 may be necessary, but it is not sufficient to induce of hTau toxicity underlying early mortality, unlike our prior hypothesis (Papanikolopoulou and Skoulakis, 2015).
Significantly, the 0N4RT245A protein where Thr245 phosphorylation was blocked by the Ala substitution presented consistently elevated mortality from days 23 to 30 (Figure 2C and Supplementary Table 2), earlier than in animals expressing 0N4RII and presenting a 50% attrition on day 24 instead of day 29 in controls (Figure 2C). This suggests that occupation of this site is essential to suppress toxicity leading to early mortality. Surprisingly and in accord with this hypothesis, the phosphomimic 0N4RT245E mutation ameliorated toxicity behaving much like controls throughout the curve including the 50% attrition day (Figure 2C and Supplementary Table 2). These results strongly suggest that phosphorylation at Thr245 is protective or averts toxicity, whereas lack of occupation at that site is necessary for hTau toxicity linked to early mortality.
A different measure of toxicity that underlies the level of oxidative stress upon pathological hTau accumulation is resistance to paraquat. This is a redox-active bipyridine heterocyclic compound, which in tissues produces superoxide anions and therefore exacerbates already extant oxidative stress (Rzezniczak et al., 2011). Consequently, flies experiencing increased oxidative stress due to pathological hTau accumulation are expected to be more sensitive to added paraquat-mediated stress (Dias-Santagata et al., 2007). In fact, after 24 h of exposure to 30 mM paraquat-laced food, control flies presented a baseline 10% mortality, while 40% of 0N4RII animals expired in the same time frame. In contrast, the Ser238 and Thr245 to Ala double mutation 0N4RSTA-expressing animals suppressed mortality to basal levels similar to that of controls (Figure 3 and Supplementary Table 3), suggesting a role for both Ser238 and/or Thr245 in hTau-mediated oxidative stress toxicity.
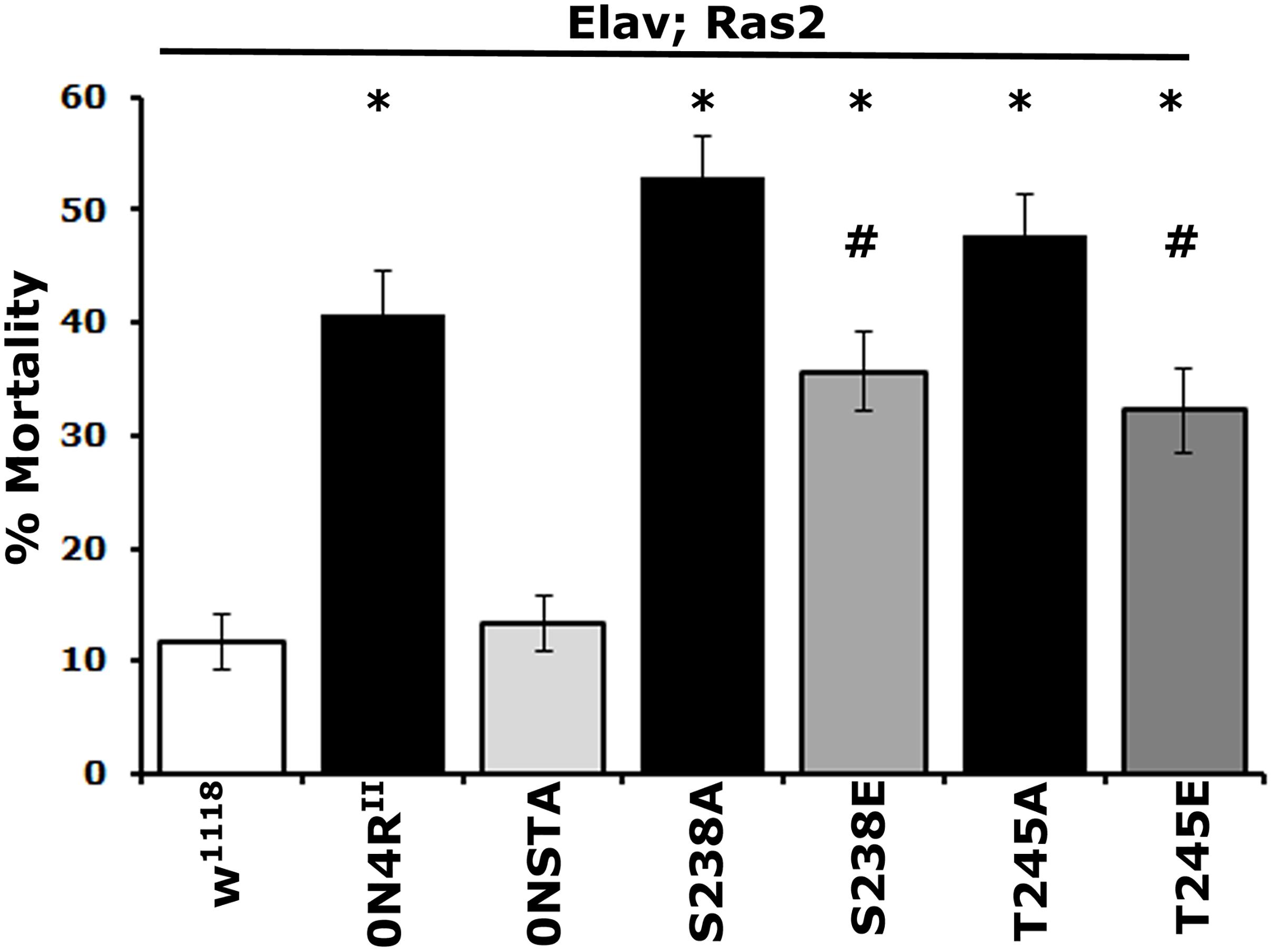
Figure 3. Differential effects of mutants on Ser238 and Thr245 on oxidative stress. One to 2-day-old flies were exposed to 30 mM paraquat, and mortality was scored after 24 h. The data represent the mean ± SEM from two independent experiments with a total of 300 flies assessed per genotype. The genotypes of all animals are indicated below each bar. Stars indicate significant difference from controls and # significant difference from the cognate mutant. Compared with the control driver heterozygote flies (Elav/+; Ras/+) administration of paraquat for 24 h resulted in significant lethality for all Tau variants except for 0NSTA expressing flies. Statistical details in Supplementary Table 3.
As for mortality, the 0N4RS238A protein with blocked phosphorylation at Ser238 and the pseudophosphorylated 0N4RS238E were as toxic as 0N4RII (Figure 3 and Supplementary Table 3). However, toxicity of 0N4RS238E was significantly milder than that of 0N4RS238A (Supplementary Table 3). Blocking Thr245 phosphorylation presented elevated hTau toxicity compared to 0N4RII-expressing animals (Figure 3 and Supplementary Table 3). Interestingly 0N4RT245A was ameliorated to 0N4RII levels by pseudophophorylation at that site, but it was not eliminated to 0N4RSTA basal levels (Figure 3 and Supplementary Table 3). Because expression of the double phosphoblock mutant 0N4RSTA presented only baseline toxicity unlike the single site mutants (Supplementary Table 3), these results suggest that both sites likely need to be occluded from phosphorylation to eliminate or suppress hTau-mediated oxidative stress toxicity. The dependence of oxidative stress-dependent lethality on occupation of Ser238 and Thr245 is distinctly different from that for premature mortality, suggesting that the two measures of toxicity may in fact reflect distinct pathological mechanisms.
Multiple Phosphorylations Depend on the State of Thr245 and Ser238 Occupation
To investigate the proposed interaction between occupation of Ser238 and Thr245 and its consequences on distant sites, we used phospho-specific antibodies against sites associated with pathology. We selected the AT8 epitope formed by phosphorylation of Ser202 and Thr205 and AT100 signifying occupation of Thr212 and Ser214 in the Proline Rich Region of hTau, amino-terminal to Ser238 and Thr245. In addition, we assayed phosphorylation at Ser396 near the carboxy-terminus of the protein. As demonstrated again on Figures 4A–C, the expression levels of the new transgenes are the same and equivalent with that of 0N4RII. In addition, we verified the specificity of the new anti-pSer238 antibody against 0N4RSTA (Supplementary Figure 2). A residual band of higher molecular mass was detectable in all lysates including 0N4RSTA (Supplementary Figure 2) and is considered non-specific.
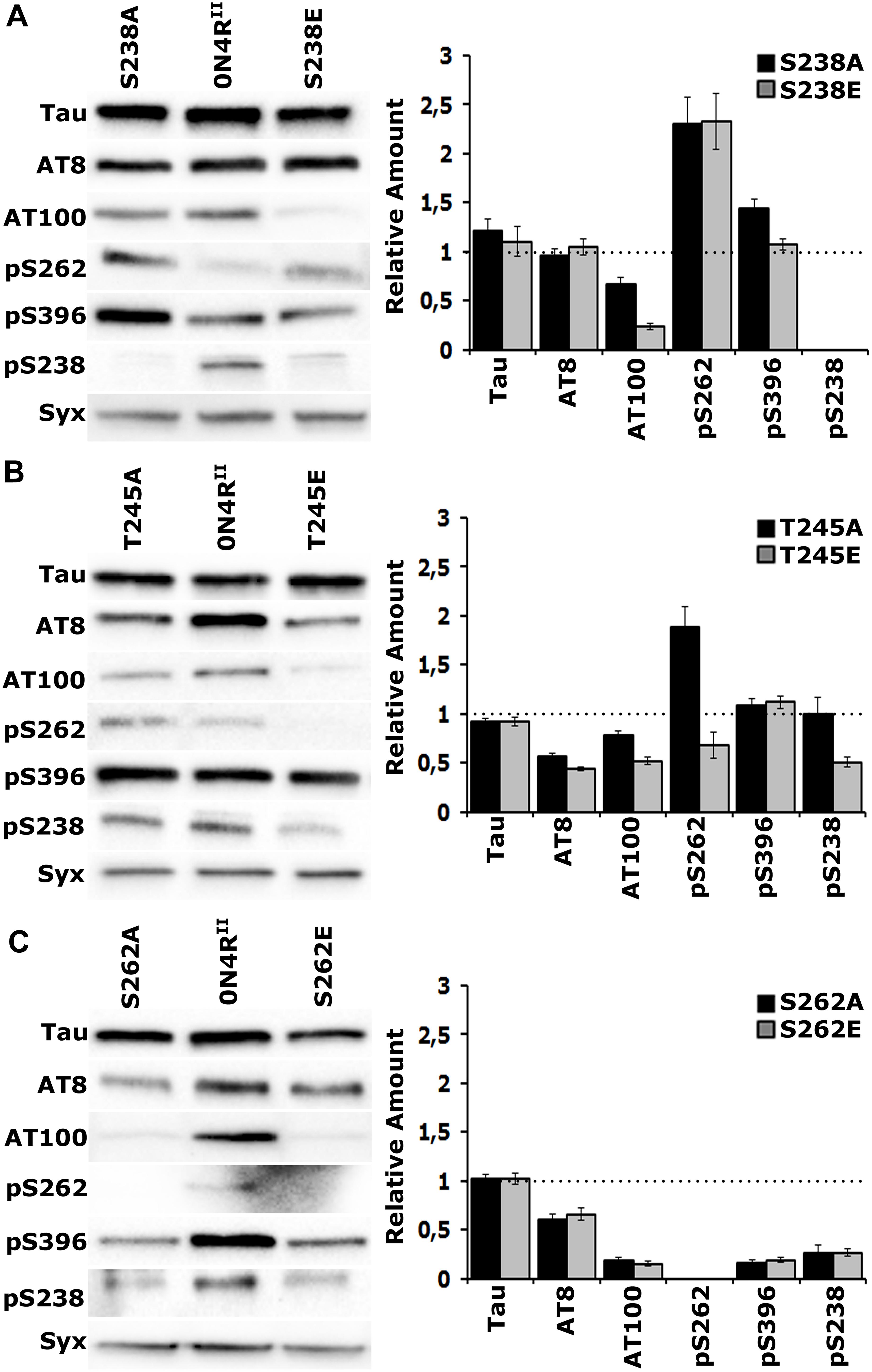
Figure 4. Gatekeeper phosphorylation interactions among Ser238, Thr245, and Ser262. Representative Western blots from head lysates of flies accumulating 0N4RII pan-neuronally compared with similar lysates from site-specific Ala and Glu mutants probed with the antibodies indicated on the left. The level of syntaxin (Syx) in the lysates was used as loading control for quantifications. The first antibody measures total Tau in the lysates, whereas all others target particular phosphorylated residues. Quantification of at least three independent biological replicates of at least six technical replicates are shown on the right. The syntaxin-normalized level of 0N4RII for each quantification was fixed to 1 and represented by the horizontal dotted line. The bars represent the mean ± SEM relative levels of mutants phosphorylated at the given sites, over that of the 0N4RII control. Bars and their respective error bars that are not in contact with the dotted line are significantly different from control. Statistical details in Supplementary Table 4. (A–C) Representative Western blots from head lysates of flies accumulating 0N4RII pan-neuronally compared with similar lysates from S238A and S238E mutants (A), T245A and T245E mutants (B) and S262A and S262E mutants (C) probed with the antibodies indicated on the left. Dunnett’s tests followed initial ANOVA to indicate significant differences from control for all phosphosites.
Blocking phosphorylation, or pseudophosphorylation of Ser238 reduced AT100 phosphorylation (Figure 4A and Supplementary Table 4) and elevated occupation of Ser262 (Figure 4A and Supplementary Table 4) relative to that on 0N4RII. This suggests that Ser262 occupation is independent of the state of Ser238 phosphorylation. Phosphorylation of the distant Ser396 was elevated upon blockade of Ser238 occupation, but remained unaffected by pseudophosphorylation of that site (Figure 4A and Supplementary Table 4). As expected, the anti-pSer238 antibody also did not recognize the 0N4RS238A, or the 0N4RS238E proteins (Figure 4A).
Importantly, although Ser238 occupation appeared unaffected in the 0N4RT245A protein, it was significantly reduced in the pseudo-phosphorylated 0N4RT245E (Figure 4B and Supplementary Table 4). This is consistent with the notion that occupation of Thr245 suppresses Ser238 phosphorylation. Given that Glu substitution provides a single charge at the site vs. the two charges carried by the phosphate group, the suppression may in fact be higher in the normal wild type hTau. Because an anti- pThr245 antibody is not currently available, we cannot currently directly test whether the reciprocal that is, Ser238 occupation suppressing Thr245 phosphorylation may be true as well. Conversely, blockade of Thr245 occupation enhanced Ser262 relative to that on 0N4RII, while pseudophosphorylation left it at control levels if not marginally lower (Figure 4B and Supplementary Table 4). This is consistent with the notion that loss or suppression of Thr245 phosphorylation leads to Ser238 and Ser262 occupation. Inasmuch as the phosphomimic substitution may not fully reflect the consequence of bona fide phosphorylation at the site, the data support the notion Thr245 occupation suppresses both Ser238 and Ser262 phosphorylation. Therefore, in effect it acts as a “gatekeeper” for the consequent pathological manifestations of Ser238 and Ser262 occupation (Papanikolopoulou and Skoulakis, 2015).
The proposed role of Thr245 as the critical “gatekeeper” instead of the Ser238, the two residues concurrently changed in the STA mutants is also reflected in the consequences of blocking, or pseudophosphorylating it on distant phosphosites. Phosphorylation at the proximal phospho epitopes recognized by the AT8 and AT100 antibodies are significantly depressed in both Thr245 mutants (Figure 4B and Supplementary Table 4), but Ser396 occupation remains unaffected. In contrast, only AT100 occupation is suppressed in Ser238 mutants, while Ser396 phosphorylation appears significantly elevated when Ser238 phosphorylation is blocked (Figure 4A and Supplementary Table 4). This suggests that Thr245 occupation is required for phosphorylation at the AT8 and AT100 epitopes, while Ser238 phosphorylation suppresses occupation of Ser396, another manifestation of potentially conformation-mediated interactions among distant phosphosites.
The essential role of Ser262 as a broad-acting “gatekeeper” of pathology (Papanikolopoulou and Skoulakis, 2015) in accord to independently-derived similar proposal (Nishimura et al., 2004) is demonstrated in Figure 4C where all phosphorylations in question are drastically suppressed on the 0N4RS262A and 0N4RS262E proteins. This includes pSer238, which drastically reduced, albeit not completely eliminated (Figure 4C and Supplementary Table 4). Although the effect of blocking Ser262 phosphorylation (0N4RS262A), supports the notion that its occupation is required for subsequent Ser238 phosphorylation, the opposite might be expected for the phospho-mimic 0N4RS262E. The fact that Ser238 is under-phosphorylated on the 0N4RS262E protein argues that the single charge- bearing Glu is not an efficient substitute for phosphorylated Ser262 in mediating conformational changes promoting phosphorylation at proximal and distant sites (Figure 4C). Rather it appears that it may in fact have a negative effect by occluding bona fide phosphorylation at that site.
Differential Contribution of Ser238 and Thr245 to hTau-Mediated Learning Deficits
In addition to its “gatekeeper” effects, phosphorylation at Ser262 was proposed to be necessary for and predict learning deficits (Papanikolopoulou and Skoulakis, 2015). The Ser262 variants were in fact generated for a second reason in addition to facilitating comparison and validation of the effects of the Ser238 and Thr245 variants. Because the expression level of the previously used randomly inserted Ser262A variant was low (Papanikolopoulou and Skoulakis, 2015), it raised the possibility that the lack of learning deficits upon expression of that transgene may have been consequent of its low expression.
Pan-neuronal expression of the new 0N4RSTA variant did not precipitate structural defects in the MBs (Figure 5A), in agreement with prior reports (Kosmidis et al., 2010; Papanikolopoulou and Skoulakis, 2015). Moreover, as for the 2N4RSTA (Kosmidis et al., 2010), expression of 0N4RSTA yielded significant deficits in associative learning (Figure 5B and Supplementary Table 5), equivalent to those presented by 0N4RII (Supplementary Table 5), demonstrating that although grossly intact structurally, the CNS of animals expressing this hTau variant is dysfunctional. Importantly, expression of the single variants 0N4RS238A and 0N4RT245A resulted in a similar decrease in learning. Given that Ser262 is hyper-phosphorylated in these variants (Figures 4A,B), the data support the proposal that elevated occupation at this site is in fact requisite for the learning deficits (Papanikolopoulou and Skoulakis, 2015).
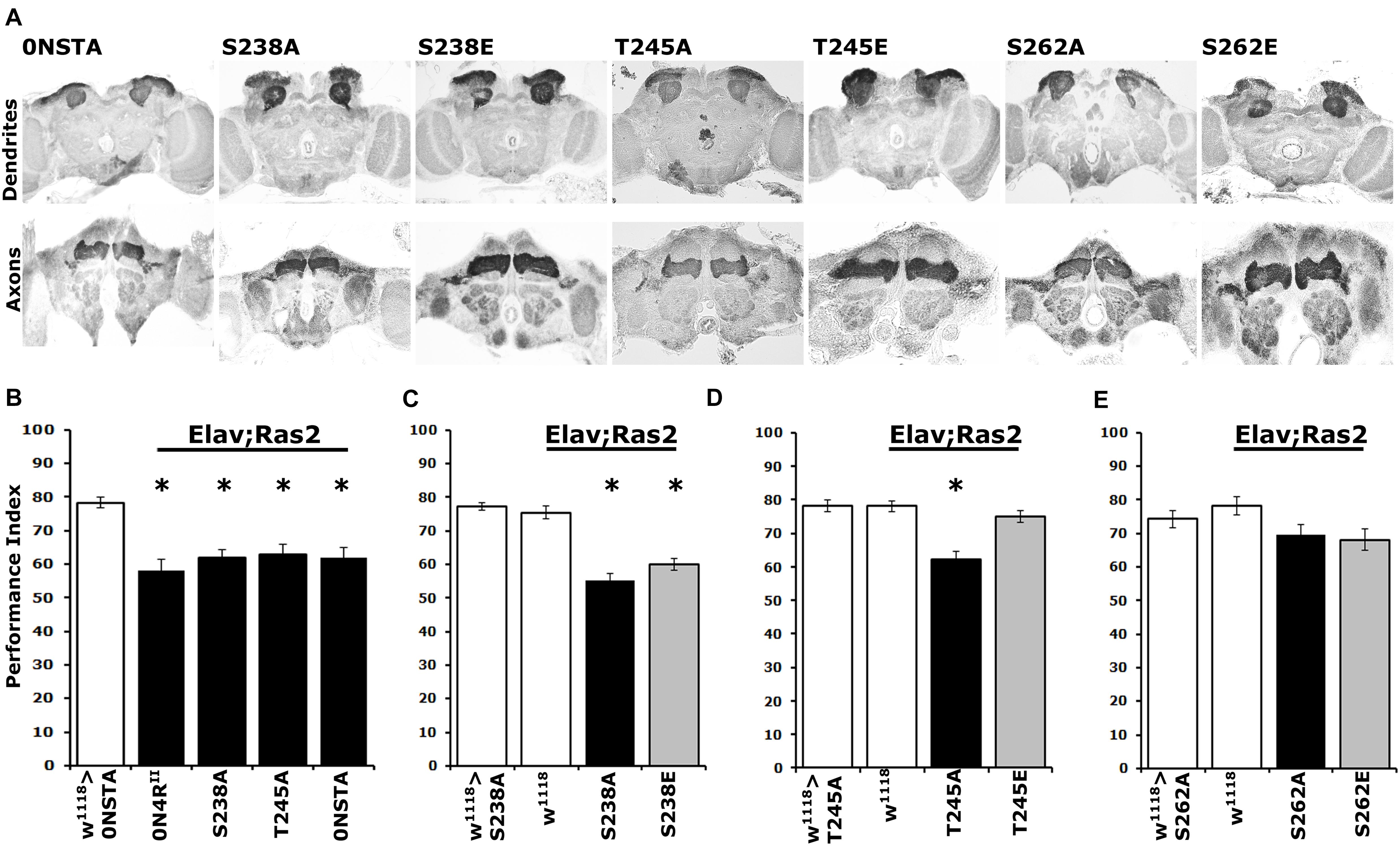
Figure 5. Differential learning defects of mutants on Ser238 and Thr245 and Ser262. (A) Carnoy’s-fixed paraffin-embedded frontal sections stained with anti-Leonardo at the level of MB dendrites in the posterior of the head and of MB axons of the γ-subtype-lobe from animals expressing the indicated Tau variants. (B–E) Learning performance of animals accumulating pan-neuronally the indicated Tau variants (black bars) compared with driver or transgene heterozygotes (white bar). The genotypes of all animals are indicated below each bar. The means ± standard error of the mean (SEM) are shown. Stars indicate significant differences from control. Statistical details in Supplementary Table 5.
Further toward this notion, we assayed associative learning in animals expressing the phosphomimic variants 0N4RS238E and 0N4RT245E alongside the respective Ala substituted proteins at these sites. Interestingly, expression of 0N4RS238E precipitated a learning deficit equivalent to that of the phospho-blocked variant 0N4RS238A (Figure 5C and Supplementary Table 5), as predicted by the hyper-phosphorylation of this protein at Ser262 (Figure 4A) and in agreement with the inefficiency of Glu substitution as a phospho-mimic at that site. In contrast, unlike the deficit presented by animals expressing 0N4RT245A, expression of the pseudo-phosphorylated variant 0N4RT245E did not affect learning (Figure 5D and Supplementary Table 5). This agrees with the phosphorylation state of Ser262 on this variant protein and in support of the proposed role of Thr245 phosphorylation as suppressive of hTau-mediated toxicity and dysfunction. This in turn suggests that phosphorylation at this epitope may be constitutive on non-pathogenic hTau.
As expected, expression of the phosphoblocked 0N4RS262A protein did not affect learning (Figure 5E and Supplementary Table 5), confirming published results (Papanikolopoulou and Skoulakis, 2015). Moreover, since 0N4RS262A is expressed at the same level as controls (Supplementary Figure 1 and Figure 4C), this result verifies that lack of deficient learning was not consequent of lower expression of the randomly inserted transgene (Papanikolopoulou and Skoulakis, 2015). Again, lack of the expected learning deficit upon expression of the pseudo-phosphorylated 0N4RS262E is a likely consequence of the inefficient Glu-mediated mimic of phosphorylation at that site (Figure 5E).
Discussion
Although hTau phosphorylation and its regulation has received waning attention lately, hyper-phosphorylation at disease linked phosphoepitopes remain strong pathology-linked biomarkers (Blennow and Zetterberg, 2018). Therefore, understanding the patterns and regulation of hTau phosphorylation is essential to monitor pathologies and their progression, but just as importantly, to understand its contribution to the IDR-mediated structural plasticity of this protein. The results herein describe a sequence of apparent “gatekeeper” phosphorylations, which affect both hTau toxicity and neuronal dysfunction in Drosophila that could also serve as disease biomarkers in patients (Papanikolopoulou and Skoulakis, 2015).
A hypothesis put forward before (Papanikolopoulou and Skoulakis, 2015), suggested that Ser262 is required for Ser238 occupation, which was verified experimentally herein (Figure 4). Importantly, generation of the single mutants on Ser238 and Thr245 revealed an important new regulatory point not evident with the STA double mutant analyzed before. Collectively, the toxicity (Figures 2, 3) and learning (Figure 5) data in the context of the phospho-profile analyses (Figure 4), strongly indicate that Thr245 phosphorylation attenuates or blocks Ser262 occupation, which in turn is required for Ser238 phosphorylation leading to toxicity. Therefore, Ser262 phosphorylation precedes and acts as a “gatekeeper” to Ser238 occupation, which promotes hTau-dependent premature mortality or decreased resistance to oxidative stress, two very likely distinct manifestations of hTau toxicity.
Currently we do not know the mechanism or trigger of Thr245 dephosphorylation, as well as the speed and mechanism of the consequent Ser262 occupation, although it is likely mediated by conformational changes. As the resultant learning deficits require time after hTau expression to be manifested (Papanikolopoulou et al., 2010; Papanikolopoulou and Skoulakis, 2015), the process is likely relatively slow. Since Ser262 occupation precedes that of Ser238, this may account for the reported significant delay in phosphorylation of the latter (Papanikolopoulou and Skoulakis, 2015), a potential link to the age dependent manifestation of degenerative Tauopathies in humans as well. This is turn suggests that monitoring Ser238 occupation may be a useful biomarker of disease progression.
Domains such as the highly conserved microtubule binding repeats and the amino terminal region of the protein appear functionally specialized (Trushina et al., 2019). Notably, the work described herein suggests IDR/conformation-dependent interactions with the 24 aminoacids that separate Ser238 just amino-terminal to the first microtubule binding repeat to Ser262 within it, with significant roles in hTau-mediated toxicity and dysfunction. Therefore, we propose that this region defines a new potential hTau domain of importance to toxicity and neuronal dysfunction. This may be reflected in phospho-profiles of Tau fragments in Cerebrospinal fluid (CSF) samples from Alzheimer’s disease (AD) patients that show significant enrichment in Ser238 and Ser262 occupation (Russell et al., 2017). Moreover, an independent study indicated that phosphorylated Ser238 and Ser262 appear specifically associated with pathological Tau in AD patients (Martin et al., 2013). We propose that this “toxicity domain” contributes to Tau structural plasticity, as the occupation state of Ser238, Thr245, and Ser262 affects the phosphorylation state of additional proximal sites such as Ser202/Thr205 (AT8), Thr212/Ser214 (AT100), but also the distant Ser396 at the far carboxy-terminus of the protein (Figure 4), which when phosphorylated is also enriched in the CSF from AD patients (Russell et al., 2017). As all the sites under consideration are in invariant hTau regions, the regulatory mechanisms we propose are pertinent to all isoforms.
Importantly, Thr245 phosphorylation appears to be inhibitory not only to Ser262 and consequently to Ser238 occupation, but also on Ser202/Thr205 and Thr212/Ser214 (Figure 4B) and perhaps it represents a broader suppressor of toxicity and neuronal dysfunction (Figure 6). Hence, occupation of this site appears to act as a “gatekeeper” against additional phosphorylations linked to pathologies. In agreement with this notion, phosphorylated Thr245 appears exclusively in physiological human brain lysates (Martin et al., 2013). To our knowledge, the only other phosphorylation reported to decrease Tau/Aβ-induced toxicity is on Thr205 (Ittner et al., 2016). This contrasts with the apparent role of Ser262 phosphorylation as an enabler of phosphorylation at many additional sites, probably a lot more than we have tested herein (Figure 4C), including the essential for toxicity Ser238. To that end ongoing efforts aim at generating an anti p-Thr245 antibody, which along with either pSer238and pSer262 may be useful biomarkers in monitoring progression of Tauopathies.
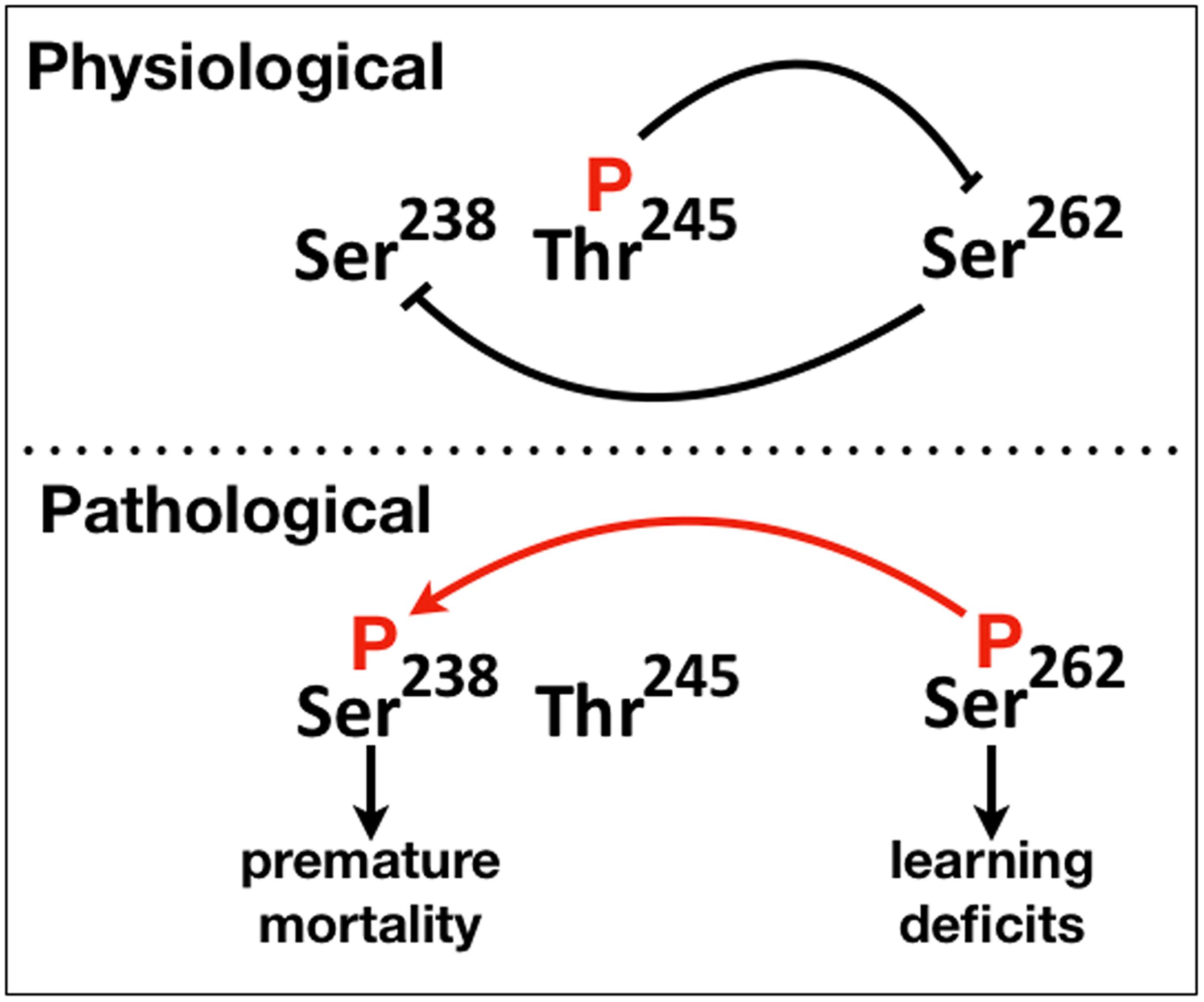
Figure 6. A schematic of the proposed phosphorylation-mediated interaction between “suppressive” and “facilitatory gatekeeper” phosphosite interactions proximal to and within the first microtubule binding repeat of hTau. Blunt lines represent “suppressive” while the red arrow “facilitatory” interactions. The direction of the arrow indicates that Ser238 is phosphorylated after occupation of Ser262.
Collectively therefore, there appear to be two types of “gatekeeper” phosphosites, those that when occupied suppress additional phosphorylations such as pThr245 and others like pSer262 that enable them. This is reflected in the schematic on Figure 6, where we propose that abrogation or attenuation of Thr245 phosphorylation removes the effect of the toxicity and dysfunction “suppressing gatekeeper,” enabling occupation of the “facilitatory gatekeeper” phospho-Ser262 (Nishimura et al., 2004; Papanikolopoulou and Skoulakis, 2015). In agreement with others (Trushina et al., 2019), we posit that these changes in hTau toxicity are manifestations of the IDR-mediated structural plasticity of the protein with local or broad conformational changes favored by the presence or absence of particular phosphorylations leading to physiological or pathogenic states.
One notable result from the work herein is that the single charge contributed by Glu replacing the relevant Serines and Threonines does not mimic the double charge of the phosphate group in the case of Ser238 (Figures 2B, 3, 4A, 5C) and Ser262 (Figures 4C, 5E). This indicates that the proposed local structural plasticity is at least in part mediated and possibly stabilized by weak charge based interactions, a proposed characteristic of intrinsically disordered proteins (Trushina et al., 2019). Interestingly, our data suggests that with respect to these sites, Glu substitution may in fact mimic blocking phosphorylation at these sites (Figures 2B, 3, 4A,C, 5C,E), possibly by occlusion and this approach should be used with caution.
Although they are obvious pharmaceutical targets, it remains a challenge to identify and interfere with the phosphatases and kinases that target specific phosphoepitopes in part because of overlapping consensus sequences and participation in many other physiological cellular processes (Martin et al., 2013). For Thr245, whose phosphorylation appears to function as “suppressive gatekeeper” to subsequent pathology-linked phosphorylations, the therapeutic challenge is to maintain its occupation. In addition, development of an anti-phospho Thr245 antibody for early detection of its dephosphorylated state may be a good disease prognostic biomarker.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
IK and EV performed research and prepared the manuscript. EV wrote the manuscript initial draft. KP performed research and guidance. ES directed the research and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the funds from the “Strategic Development of the Biomedical Research Institute “Alexander Fleming” (MIS 5002562) implemented under the “Action for the Strategic Development on the Research and Technological Sector,” funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund) and Infrafrontier GR (co-funded by Greece and the European Union, ERDF and NSRF 2014-2020).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the help of Maro Loizou for expert technical support and of Best Gene (CA, United States) for generating the transgenics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2020.569520/full#supplementary-material
References
Andreadis, A., Brown, W. M., and Kosik, K. S. (1992). Structure and novel exons of the human tau gene. Biochemistry 3110626–10633. doi: 10.1021/bi00158a027
Arendt, T., Stieler, J. T., and Holzer, M. (2016). Tau and tauopathies. Brain Res. Bull. 126(Pt 3), 238–292. doi: 10.1016/j.brainresbull.2016.08.018
Bischof, J., Maeda, R. K., Hediger, M., Karch, F., and Basler, K. (2007). An optimized transgenesis system for Drosophila using germ-line-specific φC31 integrases. Proc. Natl. Acad. Sci. U.S.A. 104, 3312–3317. doi: 10.1073/pnas.0611511104
Blennow, K., and Zetterberg, H. (2018). Biomarkers for Alzheimer’s disease: current status and prospects for the future. J. Intern. Med. 284, 643–663. doi: doi.org/10.1111/joim.12816
Brandt, R., Gergou, A., Wacker, I., Fath, T., and Hutter, H. (2009). A Caenorhabditis elegans model of tau hyperphosphorylation: induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 30, 22–33. doi: 10.1016/j.neurobiolaging.2007.05.011
Brion, J. P., Smith, C., Couck, A. M., Gallo, J. M., and Anderton, B. H. (1993). Developmental changes in tau phosphorylation: fetal tau is transiently phosphorylated in a manner similar to paired helical filament-tau characteristic of Alzheimer’s disease. J. Neurochem. 61, 2071–2080. doi: 10.1111/j.1471-4159.1993.tb07444.x
Buee, L., Bussiere, T., Buee-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130. doi: 10.1016/S0165-0173(00)00019-9
Davis, R. L. (2005). Olfactory memory formation in Drosophila: from molecular to systems neuroscience. Annu. Rev. Neurosci. 28, 275–302. doi: 10.1146/annurev.neuro.28.061604.135651
Decker, J. M., Kruger, L., Sydow, A., Zhao, S., Frotscher, M., Mandelkow, E., et al. (2015). Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca(++) dysregulation. Acta Neuropathol. Commun. 3:23. doi: 10.1186/s40478-015-0193-3
Dias-Santagata, D., Fulga, T. A., Duttaroy, A., and Feany, M. B. (2007). Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J. Clin. Invest. 117, 236–245. doi: 10.1172/JCI28769
Fath, T., Eidenmuller, J., and Brandt, R. (2002). Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease. J. Neurosci. 22, 9733–9741. doi: 10.1523/JNEUROSCI.22-22-09733.2002
Feuillette, S., Miguel, L., Frebourg, T., Campion, D., and Lecourtois, M. (2010). Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J. Neurochem. 113, 895–903. doi: 10.1111/j.1471-4159.2010.06663.x
Gong, C. X., Liu, F., Grundke-Iqbal, I., and Iqbal, K. (2005). Post-translational modifications of tau protein in Alzheimer’s disease. J. Neural. Transm. 112, 813–838. doi: 10.1007/s00702-004-0221-0
Gouzi, J. Y., Moressis, A., Walker, J. A., Apostolopoulou, A. A., Palmer, R. H., Bernards, A., et al. (2011). The receptor tyrosine kinase Alk controls neurofibromin functions in Drosophila growth and learning. PLoS Genet. 7:e1002281. doi: 10.1371/journal.pgen.1002281
Hanger, D. P., Anderton, B. H., and Noble, W. (2009). Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 15, 112–119. doi: 10.1016/j.molmed.2009.01.003
Hong, M., Chen, D. C., Klein, P. S., and Lee, V. M. (1997). Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem. 272, 25326–25332. doi: 10.1074/jbc.272.40.25326
Hosoi, T., Uchiyama, M., Okumura, E., Saito, T., Ishiguro, K., Uchida, T., et al. (1995). Evidence for cdk5 as a major activity phosphorylating tau protein in porcine brain extract. J. Biochem. 117, 741–749. doi: 10.1093/oxfordjournals.jbchem.a124771
Ittner, A., Chua, S., Bertz, J., Volkerling, A., van der Hoven, J., Gladbach, A., et al. (2016). Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 354, 904–908. doi: 10.1126/science.aah6205
Jeganathan, S., Hascher, A., Chinnathambi, S., Biernat, J., Mandelkow, E. M., and Mandelkow, E. (2008). Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 283, 32066–32076. doi: 10.1074/jbc.M805300200
Johnson, G. V., and Stoothoff, W. H. (2004). Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 117(Pt 24), 5721–5729. doi: 10.1242/jcs.01558
Kosmidis, S., Grammenoudi, S., Papanikolopoulou, K., and Skoulakis, E. M. C. (2010). Differential effects of Tau on the integrity and function of neurons essential for learning in Drosophila. J. Neurosci. 30, 464–477. doi: 10.1523/JNEUROSCI.1490-09.2010
Martin, L., Latypova, X., and Terro, F. (2011). Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem. Int. 58, 458–471. doi: 10.1016/j.neuint.2010.12.023
Martin, L., Latypova, X., Wilson, C., Magnaudeix, A., Perrin, M., Yardin, C., et al. (2013). Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res. Rev. 12, 289–309. doi: 10.1016/j.arr.2012.06.003
Morris, M., Knudsen, G. M., Maeda, S., Trinidad, J. C., Ioanoviciu, A., Burlingame, A. L., et al. (2015). Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 18, 1183–1189. doi: 10.1038/nn.4067
Nishimura, I., Yang, Y., and Lu, B. (2004). PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 116, 671–682. doi: 10.1016/S0092-8674(04)00170-9
Papanikolopoulou, K., Kosmidis, S., Grammenoudi, S., and Skoulakis, E. M. (2010). Phosphorylation differentiates tau-dependent neuronal toxicity and dysfunction. Biochem. Soc. Trans. 38, 981–987. doi: 10.1042/BST0380981
Papanikolopoulou, K., and Skoulakis, E. M. (2015). Temporally distinct phosphorylations differentiate Tau-dependent learning deficits and premature mortality in Drosophila. Hum. Mol. Genet. 24, 2065–2077. doi: 10.1093/hmg/ddu726
Qiang, L., Sun, X., Austin, T. O., Muralidharan, H., Jean, D. C., Liu, M., et al. (2018). Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr. Biol. 28, 2181–2189.e2184. doi: 10.1016/j.cub.2018.05.045
Regan, P., Whitcomb, D. J., and Cho, K. (2017). Physiological and pathophysiological implications of synaptic Tau. Neuroscientist 23, 137–151. doi: 10.1177/1073858416633439
Russell, C., Mitra, V., Hansson, K., Blennow, K., Gobom, J., Zetterberg, H., et al. (2017). Comprehensive quantitative profiling of tau and phosphorylated tau peptides in cerebrospinal fluid by mass spectrometry provides new biomarker candidates. J. Alzheimers. Dis. 55, 303–313. doi: 10.3233/JAD-160633
Rzezniczak, T., Douglas, L. A., Watterson, J. H., and Merritt, T. J. S. (2011). Paraquat administration in Drosophila for use in metabolic studies of oxidative stress. Anal. Biochem. 419, 345–347. doi: 10.1016/j.ab.2011.08.023
Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., et al. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. doi: 10.1126/science.1113694
Schwalbe, M., Kadavath, H., Biernat, J., Ozenne, V., Blackledge, M., Mandelkow, E., et al. (2015). Structural impact of Tau phosphorylation at threonine 231. Structure 23, 1448–1458. doi: 10.1016/j.str.2015.06.002
Sergeant, N., Delacourte, A., and Buee, L. (2005). Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta 1739, 179–197. doi: 10.1016/j.bbadis.2004.06.020
Sibille, N., Huvent, I., Fauquant, C., Verdegem, D., Amniai, L., Leroy, A., et al. (2012). Structural characterization by nuclear magnetic resonance of the impact of phosphorylation in the proline-rich region of the disordered Tau protein. Proteins 80, 454–462. doi: 10.1002/prot.23210
Sotiropoulos, I., Galas, M. C., Silva, J. M., Skoulakis, E., Wegmann, S., Maina, M. B., et al. (2017). Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 5:91. doi: 10.1186/s40478-017-0489-6
Steinhilb, M. L., Dias-Santagata, D., Fulga, T. A., Felch, D. L., and Feany, M. B. (2007a). Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell 18, 5060–5068. doi: 10.1091/mbc.e07-04-0327
Steinhilb, M. L., Dias-Santagata, D., Mulkearns, E. E., Shulman, J. M., Biernat, J., Mandelkow, E. M., et al. (2007b). S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J. Neurosci. Res. 85, 1271–1278. doi: 10.1002/jnr.21232
Stoothoff, W. H., and Johnson, G. V. (2005). Tau phosphorylation: physiological and pathological consequences. Biochim. Biophys. Acta 1739, 280–297. doi: 10.1016/j.bbadis.2004.06.017
Trushina, N., Bakota, L., Mulkidjanian, A., and Brandt, R. (2019). The evolution of Tau phosphorylation and interactions. Front. Aging Neurosci. 11:256. doi: 10.3389/fnagi.2015.00256
Uversky, V. N. (2015). Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 7:18. doi: 10.3389/fnagi.2015.00018
Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., Hutton, M., et al. (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714. doi: 10.1126/science.1062382
Xia, D., Li, C., and Gotz, J. (2015). Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule-associated protein Tau to dendritic spines. Biochim. Biophys. Acta 1852, 913–924. doi: 10.1016/j.bbadis.2014.12.017
Yu, Y., Run, X., Liang, Z., Li, Y., Liu, F., Liu, Y., et al. (2009). Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J. Neurochem. 108, 1480–1494. doi: 10.1111/j.1471-4159.2009.05882.x
Zheng-Fischhofer, Q., Biernat, J., Mandelkow, E. M., Illenberger, S., Godemann, R., and Mandelkow, E. (1998). Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur. J. Biochem. 252, 542–552. doi: 10.1046/j.1432-1327.1998.2520542.x
Keywords: Tau, Tau phosphorylation, toxicity, neuronal dysfunction, learning deficits, gatekeeper phosphorylation, Drosophila
Citation: Keramidis I, Vourkou E, Papanikolopoulou K and Skoulakis EMC (2020) Functional Interactions of Tau Phosphorylation Sites That Mediate Toxicity and Deficient Learning in Drosophila melanogaster. Front. Mol. Neurosci. 13:569520. doi: 10.3389/fnmol.2020.569520
Received: 04 June 2020; Accepted: 15 September 2020;
Published: 21 October 2020.
Edited by:
Isabel Lastres-Becker, Autonomous University of Madrid, SpainReviewed by:
Karim Belarbi, Université de Lille, FranceMarie-Christine Galas, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2020 Keramidis, Vourkou, Papanikolopoulou and Skoulakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Efthimios M. C. Skoulakis, c2tvdWxha2lzQGZsZW1pbmcuZ3I=
†These authors have contributed equally to this work
‡Present address: Iason Keramidis, CERVO Brain Research Center, Quebec City, QC, Canada