 Katarin Gorski1,2
Katarin Gorski1,2 Albert Spoljaric3
Albert Spoljaric3 Tuula A. Nyman4
Tuula A. Nyman4 Kai Kaila3
Kai Kaila3 Brendan J. Battersby5
Brendan J. Battersby5 Anna-Elina Lehesjoki1,2*
Anna-Elina Lehesjoki1,2*- 1Folkhälsan Research Center, Helsinki, Finland
- 2Department of Medical and Clinical Genetics, Medicum, University of Helsinki, Helsinki, Finland
- 3Molecular and Integrative Biosciences, and Neuroscience Center (HiLIFE), Faculty of Biological and Environmental Sciences, University of Helsinki, Helsinki, Finland
- 4Institute of Clinical Medicine, University of Oslo and Oslo University Hospital, Oslo, Norway
- 5Institute of Biotechnology, University of Helsinki, Helsinki, Finland
Progressive myoclonus epilepsy of Unverricht-Lundborg type (EPM1) is a neurodegenerative disorder caused by loss-of-function mutations in the cystatin B (CSTB) gene. Progression of the clinical symptoms in EPM1 patients, including stimulus-sensitive myoclonus, tonic-clonic seizures, and ataxia, are well described. However, the cellular dysfunction during the presymptomatic phase that precedes the disease onset is not understood. CSTB deficiency leads to alterations in GABAergic signaling, and causes early neuroinflammation followed by progressive neurodegeneration in brains of a mouse model, manifesting as progressive myoclonus and ataxia. Here, we report the first proteome atlas from cerebellar synaptosomes of presymptomatic Cstb-deficient mice, and propose that early mitochondrial dysfunction is important to the pathogenesis of altered synaptic function in EPM1. A decreased sodium- and chloride dependent GABA transporter 1 (GAT-1) abundance was noted in synaptosomes with CSTB deficiency, but no functional difference was seen between the two genotypes in electrophysiological experiments with pharmacological block of GAT-1. Collectively, our findings provide novel insights into the early onset and pathogenesis of CSTB deficiency, and reveal greater complexity to the molecular pathogenesis of EPM1.
Introduction
Progressive myoclonus epilepsy, EPM1 (Unverricht-Lundborg disease; OMIM 254800) is an autosomal recessive neurodegenerative disorder characterized by disease onset between 6 and 16 years, severe stimulus-sensitive, treatment-resistant and physically disabling myoclonus, tonic-clonic seizures and ataxia, with cognition essentially preserved (Koskiniemi et al., 1974; Kälviäinen et al., 2008). Magnetic resonance imaging (MRI) studies of EPM1 patient brains have shown loss of gray matter volume in the thalamus and motor cortex (Koskenkorva et al., 2009), thinning of the sensorimotor, visual and auditory cortices (Koskenkorva et al., 2012), and widespread degenerative white matter changes (Manninen et al., 2013). The histopathology have revealed widespread non-specific degenerative changes in both the cerebellum and cerebrum (Haltia et al., 1969; Koskiniemi et al., 1974; Eldridge et al., 1983; Cohen et al., 2011). MRI-navigated transcranial magnetic stimulation studies have shown significant neurophysiological changes in cortical responses, including increased prevailed inhibition in the primary motor cortex (Danner et al., 2009) and impaired intracortical interactions and coherence (Julkunen et al., 2013).
EPM1 is caused by loss-of-function mutations in cystatin B (CSTB; OMIM 601145) (Pennacchio et al., 1996; Joensuu et al., 2008). The majority of patients are homozygous for a repeat expansion mutation in the promoter region of CSTB, reducing CSTB protein expression to 5–10% of controls (Joensuu et al., 2007). When patients are compound heterozygous for the repeat expansion and a null mutation, a more severe clinical presentation of EPM1 manifests (Koskenkorva et al., 2011; Canafoglia et al., 2012). Patients homozygous for predicted null mutations in CSTB manifest a severe neonatal-onset progressive encephalopathy (Mancini et al., 2016; O’Brien et al., 2017), suggesting an important role for CSTB in nervous system development. CSTB is a ubiquitously expressed intracellular inhibitor of cysteine proteases, including cathepsins B, H, S, K, and L (Turk et al., 2008). In addition, it is reported to protect cells against apoptosis (Pennacchio et al., 1998) and oxidative stress (Lehtinen et al., 2009), and play a role in cell cycle regulation (Ceru et al., 2010). Nonetheless, the precise molecular function of CSTB at the cellular level is far from resolved.
A genetically modified mouse model with a complete loss of CSTB expression (Cstb–/–) manifests myoclonus from one month and a progressive ataxia from 6 months of age (Pennacchio et al., 1998). The onset of the clinical symptoms are preceded by early glial activation and inflammation, followed by progressive volume loss in the brain and increased oxidative damage (Lehtinen et al., 2009; Tegelberg et al., 2012; Okuneva et al., 2015). Pathological changes are detected throughout the brain, but are most striking in the cerebellum (Pennacchio et al., 1998; Tegelberg et al., 2012), where progressive atrophy reduces the cerebellar volume by almost 50% at 6 months of age (Tegelberg et al., 2012).
The characteristic symptoms in EPM1 imply alterations in the inhibitory GABAergic circuits of the brain. In older Cstb–/– mice, this presents as decreased GABAergic inhibition in the cortex (Buzzi et al., 2012), higher susceptibility to kainate-induced seizures (Franceschetti et al., 2007), and a progressive loss of inhibitory interneurons (Buzzi et al., 2012). However, altered GABAergic signaling was first noted in cerebella of presymptomatic postnatal day (P) 7 aged Cstb–/– mice during GABAergic synapse development (Joensuu et al., 2014). This presented as increased expression of the genes encoding for GABAA receptor (GABAAR) subunits α6 and δ in cerebellar tissue lysates, and as an imbalance between excitatory and inhibitory postsynaptic currents (EPSCs; IPSCs), and a complete absence of synchronous IPSC bursts in cerebellar Purkinje cells. At this early age, the number of interneurons in the cerebellum was not altered. By the early symptomatic stage (P30), the number of GABAergic presynaptic terminals and ligand binding to GABAARs were reduced. Together, the data indicate a disruption of GABAergic synapse formation in Cstb–/– mice.
To investigate the mechanisms behind the altered GABAergic synapse formation, we used a quantitative mass-spectrometry-based proteomics approach to analyze cerebellar synaptosomes of presymptomatic Cstb–/– mice. The data show a robust alteration to the mitochondrial proteome and factors important for intracellular transport and cytosolic ribosomal biogenesis.
Materials and Methods
Ethics Statement
The Animal Ethics Committee of the State Provincial Office of Southern Finland approved all animal research protocols (decisions ESAVI/10765/2015 and ESAVI/471/2019).
Mice
Cstb–/– mice were obtained from The Jackson Laboratory (Bar Harbor, ME; 129-Cstbtm1Rm/SvJ; stock #003486) (Pennacchio et al., 1998). Age-matched wild-type (wt) mice of the 129S2/SvHsd background were used as controls. The Cstb–/– mouse line was maintained by backcrossing heterozygous Cstb–/– males with inbred wt females and expanding the colony from heterozygous littermates.
Proteomics
Synaptosome Isolation
Synaptosomes were isolated from the cerebella of P14-aged Cstb–/– and wt mice (n = 5/genotype, males and females) according to the protocol described by Nolt et al. (2011), with slight modifications. Briefly, mice were sacrificed by decapitation and the cerebella dissected into ice-cold PBS. Cerebella were homogenized in 3 ml ice-cold buffer [0.32 M sucrose, 4 mM Hepes pH 7.4, protease- and phosphatase inhibitors (Pierce Protease and Phosphatase Inhibitor Mini Tablets, Thermo Fisher Scientific, Waltham, MA, United States)] and centrifuged twice at 1000 × g for 10 min at +8°C. The resulting supernatant was centrifuged at 10 000 × g for 15 min at +8°C. The resulting pellet was resuspended in 1.5 ml homogenization buffer and re-centrifuged to yield a washed crude synaptosomal fraction. This fraction was resuspended in 2 ml homogenization buffer and layered on top of 10 ml ice-cold 1.2 M sucrose and centrifuged at 230 000 × g (Sw40Ti swinging bucket rotor, Optima L-80 XP ultracentrifuge, Beckman Coulter, CA, United States) for 15 min at +4°C. The gradient interphase was diluted in homogenization buffer, layered on top of 9 ml ice-cold 0.8 M sucrose, and centrifuged at 230 000 × g for 15 min at +4°C. Synaptosomal purity and protein enrichment were analyzed by Western blot (Supplementary Figure S1).
Sample Preparation and LC-ESI-MS/MS Analysis
Lipids were removed from synaptosome samples by incubating them overnight at −20°C in five volumes ice-cold (−20°C) acetone. Samples were centrifuged twice at 1000 × g for 10 min at +8°C with an additional acetone wash in between. The pellet was air dried for 5 min and resuspended in freshly prepared 6.0 M urea/25 mM ammonium bicarbonate. Protein concentrations (μg/μl) were determined spectrophotometrically using the BCA protein assay kit (Pierce, Thermo Fisher Scientific) according to the manufacturer’s instructions. For analysis of the proteome, 25 μg of each sample was digested with trypsin (1:30 w/w, enzyme:protein; V5111 Sequencing Grade Modified Trypsin, Promega Corporation, WI, United States), and desalted by C18 cartridges. An additional phospholipid removal step was applied to samples due to impurities that interfered with ionization. Phospholipids were removed by hydrophilic interaction liquid chromatography (HILIC) using HILIC HyperSep Tips (60109-214; Thermo Fisher Scientific). Tips were conditioned by aspirating/expelling 50 μl of binding solution [15 mM ammonium acetate, pH 3.5 in 85% acetonitrile (ACN)] for five times prior to sample binding, and peptides were bound to the HILIC tips by aspirating/expelling the samples (10 μl, diluted in 85% ACN) for 20 times. Samples were washed by aspirating/expelling 20 μl binding solution for ten times, discarding the expelled solution each time. Peptide samples were released by aspirating/expelling 10 μl elution buffer (15 mM ammonium acetate, pH 3.5 in 10% ACN) for ten times, collecting the expelled solution in a 1.5 ml Eppendorf Maximum Recovery Tube (Axygen, Corning, NY, United States). Purified peptide samples were concentrated (SpeedVac, Thermo Fisher Scientific) for 10–15 min and dissolved in 1% formic acid. For liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS), 400 ng peptides/sample were analyzed in random order with several washes and blank runs in between. The analyses were performed on a nanoflow HPLC system (Easy-nLC1000, Thermo Fisher Scientific, Bremen, Germany) coupled to a Q Exactive mass spectrometer (Thermo Fisher Scientific) equipped with a nano-electrospray ionization source. Peptides were loaded on a trapping column and separated inline on a 15 cm C18 column (75 um × 15 cm, ReproSil-Pur 5 um 200 Å C18-AQ, Dr. Maisch HPLC GmbH, Ammerbuch-Entringen, Germany), with the mobile phase consisting of water with 0.1% formic acid (solvent A) and ACN/water [80:20 (v/v)] with 0.1% formic acid (solvent B). Peptides were eluted with a two-step linear gradient at a flow rate of 300 nl/min: from 2 to 20% of solvent B in 85 min, and to 40% of solvent B in 35 min, followed by a 15 min wash with 100% solvent B. MS analysis was performed using the Thermo Xcalibur 3.0 software (Thermo Fisher Scientific). The analyses were performed in a data-dependent acquisition (DDA) mode for 10 most intense peptide ions, consisting of an Orbitrap MS survey scan of mass range 300–2000 m/z, with a resolution of 140 000, mass window for precursor ion selection 2.0 m/z, and intensity threshold for triggering MS2 240, followed by fragmentation of selected peptide ions (MS2) by higher-energy collisional dissociation (HCD), with a mass resolution of 17 500 for MS/MS. Ions with unassigned charges and singly charged ions were excluded for precursor selection. The selected peptide ions were fragmented with a normalized collision energy of 27 in the collision cell. Dynamic exclusion duration was 10 s.
MS Quantification and Protein Identification
Peptides were quantified using the Progenesis LC-MS software (Non-linear Dynamics Limited, Tyne, United Kingdom), and proteins were identified using the Mascot 2.4.1 search engine (Matrix Science, MA, United States) through the Proteome Discoverer 1.4 software (Thermo Fisher Scientific). The Swiss-Prot Mus musculus database, uploaded in January 2016, was used as reference. Error tolerances on peptide and fragment ions were maximum 5 ppm and 0.02 Da, respectively. Database searches were limited to fully tryptic peptides with maximum one missed cleavage, and cysteine carbamidomethylation (+57.021464 Da) and methionine oxidation (+15.994915 Da) were set as fixed and variable modifications, respectively. Peptide-level false discovery rate (FDR) was set to 1%, and minimal peptide length to 7 amino acids. Only proteins with minimum two unique peptides were used for further analyses. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium1 via the PRIDE partner repository (Vizcaíno et al., 2013) with the dataset identifier PXD019370.
Data Processing and Analysis
The proteomics data was normalized, followed by missing value imputation of proteins with null-intensity in either sample group. Sample relations were examined by Spearman’s correlation analysis, hierarchical clustering, and principal component analysis (PCA) using R language and environment for statistical computing [version 3.2.2 (2015-08-14)] (R Core Team, 2016), and the Bioconductor module (version 3.2) (Gentleman et al., 2004). Statistical testing of abundance between sample groups (Cstb–/– vs. wt) was performed using the R package ROTS (Reproducibility-Optimized Test Statistic) (Elo et al., 2008). An abundance change with a false discover rate (FDR) -corrected p-value (q-value) ≤ 0.05 was considered as significant change. Downstream analyses of the results were conducted to proteins with a q-value ≤ 0.05 using the following softwares and statistical tests: PANTHER Overrepresentation Test (Released 20200407) using the Gene Ontology (GO) database (Mi et al., 2013) (Released 2020-02-21), using Fisher’s exact test followed by Benjamini-Hochberg correction (FDR) for multiple testing, considering FDR < 0.05 statistically significant; the web-tool ClustVis (Metsalu and Vilo, 2015), performed using singular value decomposition; the STRING database (Szklarczyk et al., 2019), using high confidence interaction score (>0.700) as cutoff, and experiments, databases, and co-expression as active interaction sources. The softwares Cytoscape (Shannon et al., 2003) and Inkscape2 were used for visualization of the results. Function and localization of mitochondrial proteins for Figure 1C were manually curated.
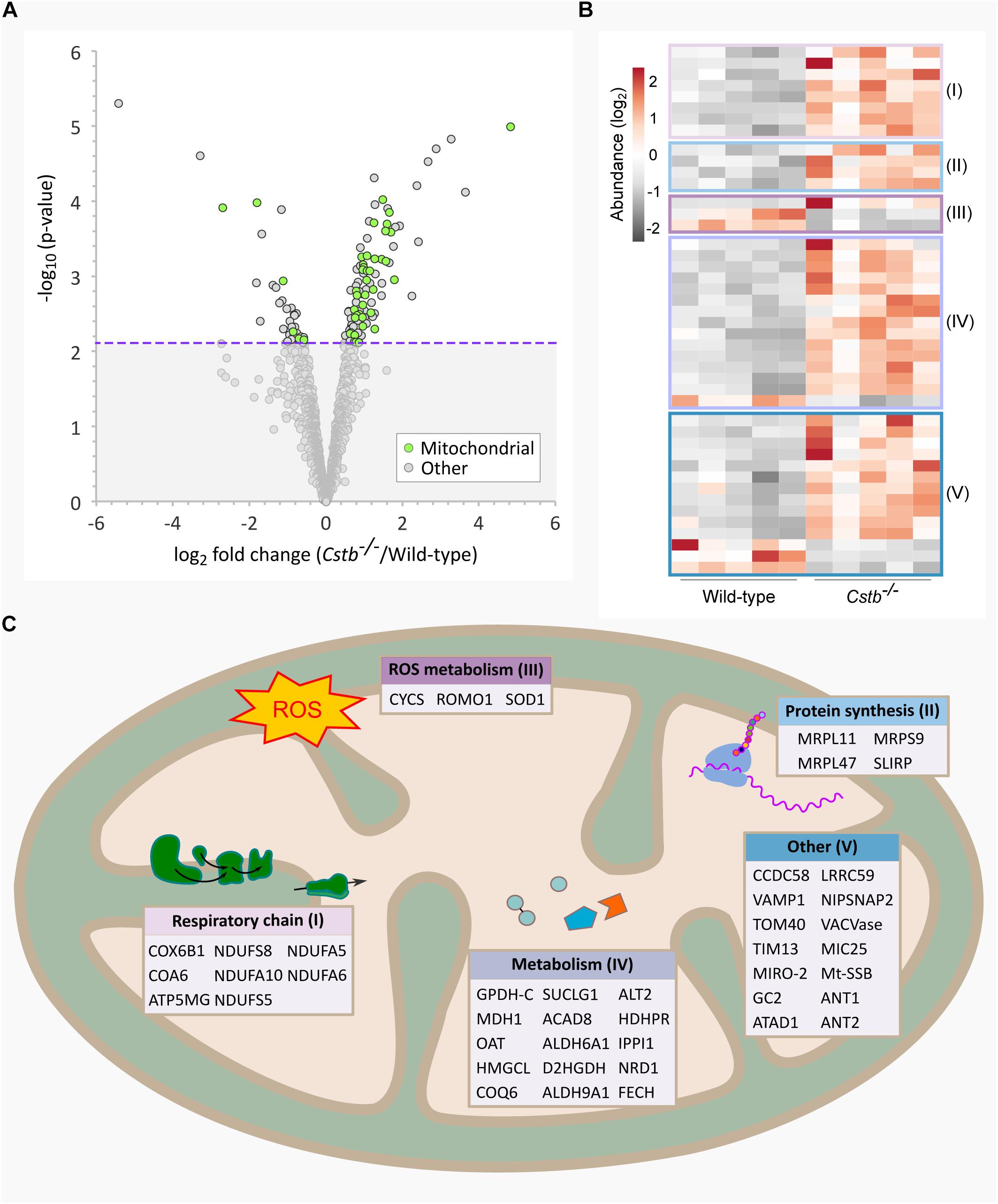
Figure 1. Differentially abundant mitochondrial proteins in Cstb–/– synaptosomes. (A) Mitochondrial proteins (green) are plotted as −log10-transformed p-value vs. log2-transformed fold change (Cstb–/–/wild-type) protein abundance. The 128 DAPs significantly differing in abundance (q-value ≤ 0.05) are plotted above the threshold limit of −log10 (p-value) 2.1 (purple dotted line). The remaining identified proteins (1973) with a non-significant q-value are plotted below the threshold limit. (B) Heatmap of 44 differentially abundant mitochondrial proteins with 36 and 6 proteins having increased and decreased fold changes, respectively. Proteins are grouped by key processes as outlined in C. Each column represents an independent biological sample. See Supplementary Table S3 for complete list of proteins. (C) Differentially abundant mitochondrial proteins detected for key processes compartmentalized within the organelle.
Electrophysiological Recordings
Cerebellar Slice Preparation
Mice (P14, n = 5 (Cstb–/–) and 10 (wt); P30, n = 5 (Cstb–/–) and 7 (wt); males and females) were deeply anesthetized with halothane, followed by cervical dislocation. Brains were quickly dissected and placed into ice-cold (+4°C) cutting solution with (in mM): 87 NaCl, 2.5 KCl, 0.5 CaCl2, 25 NaHCO3, 1.25 NaH2PO4, 7 MgCl2, 50 sucrose, and 25 D-glucose. Sagittal cerebellar slices (225 μm) were cut using a vibrating microtome (7000 SMZ-2; Campden Instruments). Slices were incubated for 1 h at +34°C in standard solution containing (in mM): 124 NaCl, 3 KCl, 2 CaCl2, 25 NaHCO3, 1.1 NaH2PO4, 2 MgSO4, and 10 D-glucose. The slices were maintained at room temperature until use in electrophysiological recordings. All solutions were equilibrated with carbogen (95% O2, 5% CO2, pH 7.4).
Electrophysiological Recordings of Tonic GABAAR Currents and Effects of Blocking GAT-1
Whole-cell voltage clamp recordings were performed in a genotype-blinded manner from cerebellar granule cells, 1-2 cells per animal. Granule cells were identified based on their morphology and electrophysiological signatures (Silver et al., 1992). Currents were amplified (EPC10, HEKA Elektronik GmbH, Lambrecht, Germany) at a sampling rate of 20–50 kHz. Data were collected using Patchmaster software (HEKA Elektronik GmbH). Recordings were corrected for a liquid junction potential of 3.9 mV. Patch pipettes from borosilicate glass had open-tip resistances of 4–7 MΩ, and they were filled with a solution containing (in mM): 140 CsCl, 10 HEPES, 5 EGTA, 4 NaCl, 1 CaCl2, 2 Mg-ATP, 5 QX-314 bromide, pH 7.3 at +20°C with NaOH; osmolarity 284 mOsm. Recordings were done in a submerged recording chamber at +32 ± 0.5°C, and slices were constantly perfused with standard solution (3.5 ml/min). GABAergic transmission was pharmacologically isolated by blocking ionotropic AMPA and NMDA receptors using CNQX (10 μM) and D-AP5 (20 μM), respectively. GABAAR antagonist picrotoxin (100 μM) was used at the end of all experiments to fully block all GABAAR -mediated currents, including the extrasynaptic tonic current. Picrotoxin and the GAT-1 inhibitor NNC-711 (10 μM) were directly applied to the perfusate.
Cells were voltage clamped at a holding potential of −70 mV. At the beginning of each experiment, current amplitudes were measured for 3 min to obtain a stable baseline.
Analysis of Data
Analysis of the electrophysiological recordings was done using the WinEDR software (Dr. J. Dempster, University of Strathclyde, Glasgow, United Kingdom). The holding currents were analyzed by taking all-point histograms derived from 30-second-windows of the recording periods (baseline; 60 s after NNC-711 application; 100 s after picrotoxin application). A Gaussian curve was fitted on the all-point histograms, and the peak value was used as the holding current value for analysis (Sipilä et al., 2005). The effect of GAT-1 activity and the total tonic GABAAR -mediated currents were defined as the net change in holding current following application of NNC-711 and picrotoxin, respectively. The change in tonic current in the presence of NNC-711 was read from the baseline of the recording during time intervals in which sIPSCs were absent. This eliminates any influence of synaptic currents on the quantification of the peak change in tonic current (see Figure 4A). In order to normalize the amount of the extrasynaptic tonic current with regard to cell size the data is presented as pA/pF.
Statistical analysis was done using GraphPad Prism version 8 for Windows (GraphPad Software, La Jolla, CA, United States)3. Testing for statistical outliers was done using the ROUT method (Q = 1%), and significance was evaluated using the two-tailed unpaired t-test and the non-parametric Mann-Whitney U test. Statistical significance was defined as p < 0.05.
Results
Cstb–/– Synaptosomes Are Quantitatively and Qualitatively Distinct
To investigate the basis for altered GABAergic signaling in Cstb–/– mice, we analyzed the proteome of cerebellar synaptosomes from presymptomatic P14 Cstb–/– and wt mice by LC-ESI-MS/MS. Altogether, a total of 2101 proteins were identified and quantified (Supplementary Table S1), with Spearman’s rank order correlation coefficients varying between 0.973 and 0.986 for Cstb–/– and 0.986 and 0.991 for wt samples, indicating strong positive relationships between the biological replicates (n = 5 / genotype) within sample groups. Downstream analyses were carried out for proteins with a q-value ≤ 0.05, narrowing the sample size to 128 differentially abundant proteins (DAPs) (Supplementary Table S2). Among these was CSTB, identified solely in synaptosomes of wt preparations, thus providing additional evidence of its location and function in the synapse.
Principal component analysis (PCA, Supplementary Figure S2A) revealed the ten replicates falling into two clusters and accounting for 75.5% (PC1) and 7.1% (PC2) of the variance, thus corresponding to the variation between genotypes. Individuals within sample groups did not cluster according to sex (females and males, n = 2 and 3 for Cstb–/–, and 3 and 2 for wt, respectively). Protein abundance ratios were consistent across biological replicates (Supplementary Figure S2B).
Cstb–/– Synaptosomes Are Distinct in Mitochondrial, Ribosomal and Intracellular Transport Proteins
To predict the biological relationships between the 128 DAPs in Cstb–/– synaptosomes, we performed Gene Ontology (GO) classification and PANTHER enrichment analyses assessing the current biological knowledge of these proteins (Supplementary Table S3).
When examining the predicted cellular components of the DAPs, we observed a striking number of proteins that localize to mitochondria (n = 44) (Figure 1A). Most of these proteins were increased in abundance (n = 36) (Figure 1B) and the fold changes were among the highest in the dataset. The differentially abundant mitochondrial proteins affect various sub-compartments of the organelle (Figure 1C), with the highest function-related fold enrichments associated to the respiratory chain (Figure 2A and Supplementary Table S3).
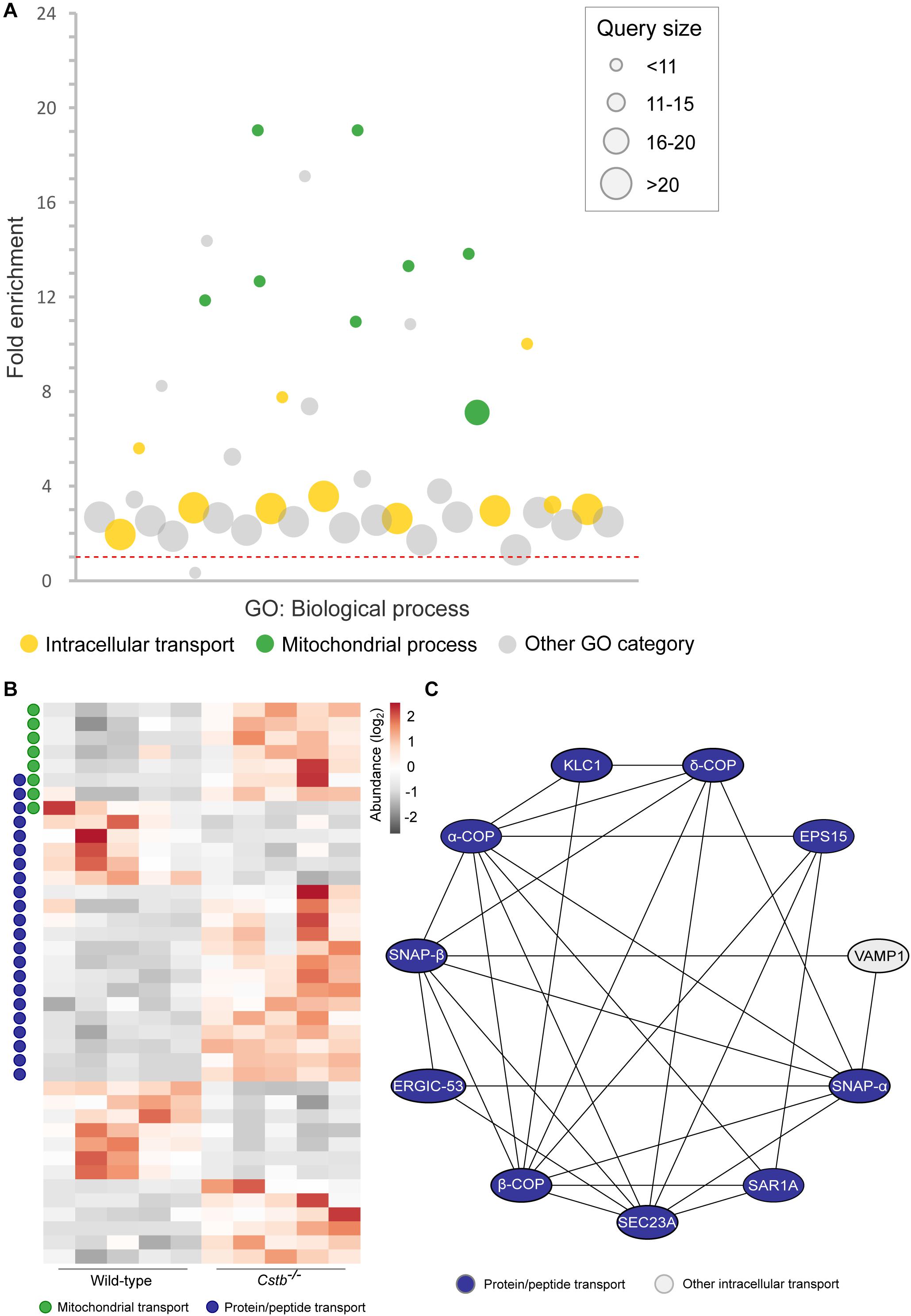
Figure 2. Differentially abundant intracellular transport proteins in Cstb–/– synaptosomes. (A) Manhattan plot of enriched (FDR < 0.05) GO terms show a high number of intracellular transport (yellow circles; GO:0006810; GO:0071702; GO:0071705; GO:0046907; GO:0015031; GO:0015833; GO:0042886; GO:0006886; GO:0006839; GO:0048193; GO:0006888) and mitochondrial process (green circles; GO:0007005; GO:0007006; GO:0022904; GO:0022900; GO:0032981; GO:0010257; GO:0006119; GO:0033108) -related biological processes in the DAPs dataset. Each circle represents a GO term, circle size corresponding to the annotated number of proteins in the DAPs dataset (query size). GO term fold enrichment is plotted on the y-axis, and the threshold limit of 1 (no fold change) is plotted as a red dotted line. (B) Heatmap of 40 differentially abundant transport proteins in Cstb–/– synaptosomes, with 13 and 27 proteins having decreased and increased fold changes, respectively. Proteins involved in mitochondrial or protein/peptide transport are marked with green or blue circles, respectively. Each column represents an independent sample, and each row represents a protein. See Supplementary Table S3 for complete list of proteins. (C) Predicted protein-protein interaction network, with a majority of interactors grouping to the GO term protein/peptide transport (blue). Networks with less than three members, or interactions below the combined interaction score threshold of 0.7, are not shown.
Next, we investigated the predicted biological processes of the DAPs. In this category, we observed an enrichment of proteins associated with intracellular transport of organelles, proteins and other cellular substances (n = 40), which were grouped to several overlapping transport-related GO terms (Figure 2A and Supplementary Table S3). The majority (n = 27) of these proteins were increased in abundance in Cstb–/– synaptosomes (Figure 2B), and most group to the GO terms protein and peptide, and mitochondrial transport. We also investigated the predicted relations between the intracellular transport proteins by protein-protein interaction network –analysis using the software STRING (Supplementary Table S4). Most of these interactions belong to the same network, consisting of the GO terms protein and peptide transport (Figure 2C).
Since axonal transport of mRNA transcripts is partly mediated by the same transport mechanisms as organelles (Kennedy and Ehlers, 2006), we asked whether the abundance of proteins involved in synaptic protein synthesis was altered. A substantial number (n = 16) of the DAPs were related to mRNA translation or to the ribonucleoprotein complex, as predicted by GO terms for molecular function and cellular component (Supplementary Table S3). Of these, 12 proteins were cytoplasmic and four mitochondrial, however, these two processes are functionally distinct. Nine of the cytosolic proteins were part of the same interaction network (Figure 3A and Supplementary Table S4) with the majority (n = 7) being structural components of the ribosome and increased in abundance in Cstb–/– synaptosomes (Figure 3B).
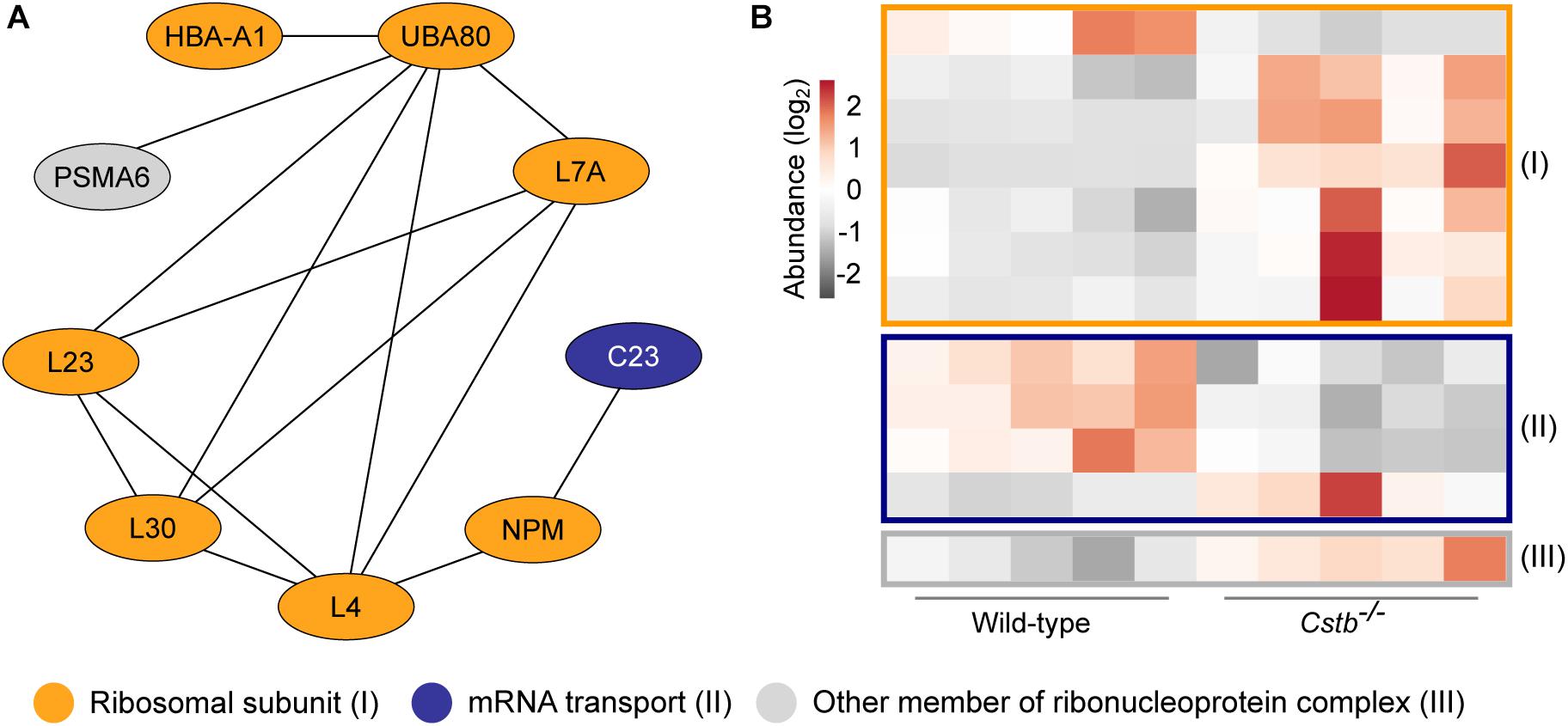
Figure 3. Differentially abundant proteins of the ribonucleoprotein complex in Cstb–/– synaptosomes. (A) Protein-protein interaction network shows the predicted relation between ribosomal subunits (orange) and proteins involved in mRNA transport (blue). Other proteins of the ribonucleoprotein complex are plotted in gray. Protein classification was performed based on GO terms. (B) Heatmap of 12 differentially abundant ribonucleoprotein complex members in Cstb–/– synaptosomes, with 4 and 8 proteins having decreased and increased fold changes, respectively. Proteins are grouped by key function. Each column represents an independent sample, and each row represents a protein. See Supplementary Table S3 for complete list of proteins.
Several Differentially Abundant Synaptic Proteins in Cstb–/– Synaptosomes
A synapse-specific location of the DAPs could provide an explanation for the previously reported alterations in GABAergic signaling. To investigate this hypothesis, we compared the DAPs dataset to the SynSysNet –online database for synaptic proteins (von Eichborn et al., 2013), and identified 31 of 128 proteins with reported synaptic location (Supplementary Table S5). Ten of these were associated with GABA- or glutamate-mediated neurotransmission (Table 1), and we studied these further by identifying their disease associations and mouse model phenotypes. GAT-1 was the only protein in the dataset exclusively associated with inhibitory GABAergic signaling, and its abundance was decreased by 0.9 fold (log2) in Cstb–/– synaptosomes. Defects in GAT-1 function have previously been associated with myoclonus, epilepsy (Johannesen et al., 2018) and altered GABAergic signaling (Jensen et al., 2003), unlike any of the other synaptic proteins we identified in our analyses.
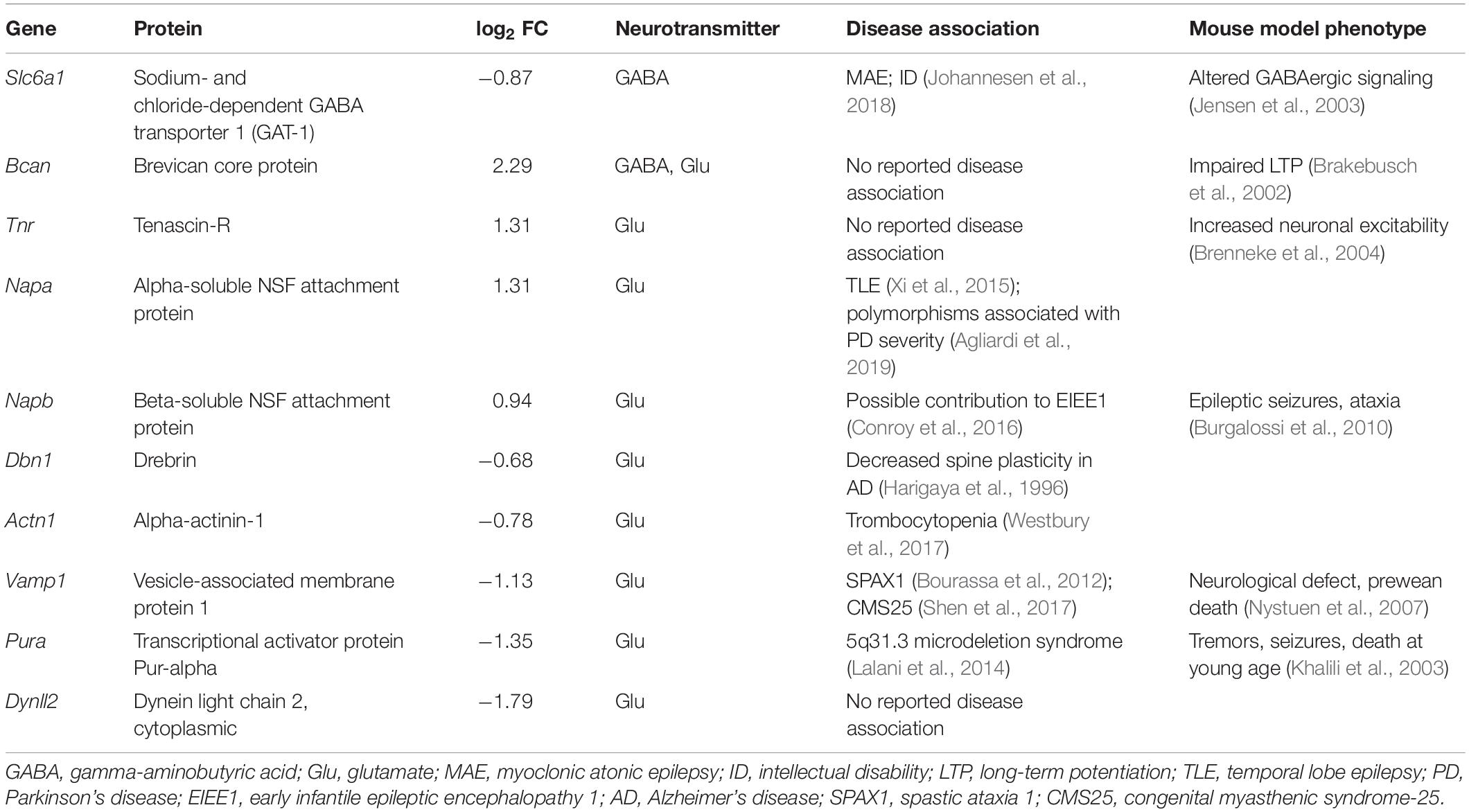
Table 1. Differentially abundant GABA and glutamate -associated proteins in Cstb–/– synaptosomes.
Unaltered GAT-1 Activity in Cstb–/– Cerebellar Granule Neurons
To investigate GAT-1 activity in cerebellar granule neurons we turned to electrophysiology. Cstb–/– mice are born healthy but develop progressive clinical symptoms at one month of age. Thus, we decided to test GAT-1 activity at two time points in development: P14 (presymptomatic) and P30 (early symptomatic). Tonic GABAAR -mediated currents are known to increase in response to GAT-1 inhibition and with increased extracellular GABA (Wall and Usowicz, 1997; Rossi et al., 2003). We inhibited GAT-1 with the antagonist NNC-711 (Suzdak et al., 1992) in voltage-clamp experiments, using the drug-induced change in tonic GABAAR -mediated current as a read-out of GAT-1 function.
Whole-cell voltage clamp recordings were obtained from a total of 39 cerebellar granule neurons (n = 6 and 13, and 8 and 12 for P14 and P30 Cstb–/– and wt, respectively) exhibiting similar morphological and electrophysiological features. The median membrane capacitance (pF), reflecting cell size, was 3.75 and 3.70 at P14 and 3.65 and 3.90 at P30 for Cstb–/– and wt, respectively (Supplementary Table S6). This is consistent with previous reports from cerebellar granule neurons (Kaneda et al., 1995; Brickley et al., 1996; Carta et al., 2004).
GAT-1 inhibition caused an increase in tonic GABAAR -mediated currents in both genotypes (Figure 4A). However, the maximum effect of blocking GAT-1 with 10 μM NNC-711 showed large variability in both age groups (Figure 4B). A high cell-to-cell variation was also observed in the total amplitude of the tonic current as seen following its complete block by 100 μM picrotoxin (Figure 4C). The median effect of blocking GAT-1 was not different between Cstb–/– and wt mice at either age point (Figure 4B and Supplementary Table S6). To normalize the quantifications of the GAT-1 mediated currents for each individual neuron, the ratio between GAT-1 induced maximum current (Figure 4B) and total available tonic current given by the picrotoxin-induced shift (Figure 4C) was calculated. The median values for these ratios did not indicate for differences between the two genotypes at either age point (Supplementary Table S6).
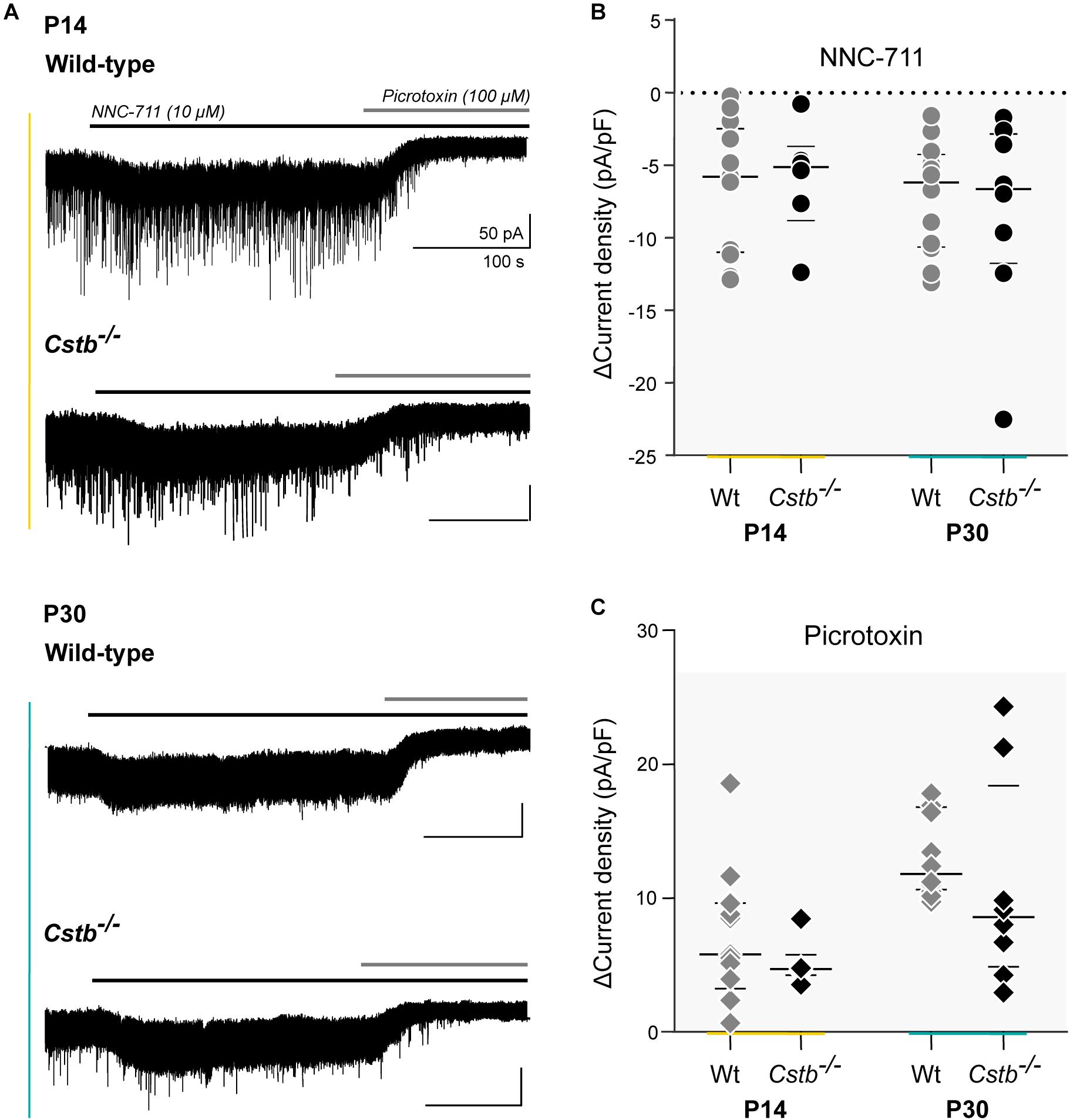
Figure 4. GAT-1 activity and total tonic GABAAR –mediated currents in cerebellar granule cells of Cstb–/– and wild-type mice. (A) Experimental traces of whole-cell voltage clamped cerebellar granule neurons of P14 and P30 Cstb–/– and wt mice. NNC-711 (10 μM) blocks GAT-1 activity. Picrotoxin (100 μM) blocks GABAAR -mediated tonic conductance. Recordings were obtained in the presence of CNQX (10 μM) and D-AP5 (20 μM). (B,C) Summary of NNC-711 and picrotoxin application. Horizontal bars represent median and interquartile range values of the change (Δ) in current density (pA/pF), induced by NNC-711 and picrotoxin application. See Supplementary Table S6 for more details.
Discussion
We undertook a proteomics approach to gain insight into the molecular basis behind altered synaptic functions in the cerebella of presymptomatic Cstb–/– mice. Our analysis reveals that mitochondrial dysfunction might be a critical factor in the early pathogenesis of CSTB deficiency, establishing for the first time an early role for mitochondria in the molecular pathogenesis of EPM1. However, in contrast to our initial hypothesis [see (Joensuu et al., 2014], we did not detect significant changes related to GABAergic signaling.
We found one third of the differentially abundant proteins identified in the cerebellar synaptosomes of presymptomatic Cstb–/– mice were part of the mitochondrial proteome. The fold changes of these proteins were among the highest in the dataset. Disruption of the mitochondrial proteome could affect ATP production and reactive oxygen species (ROS) scavenging, among other functions, shifting redox-balance to generate progressive mitochondrial dysfunction. In other neurological disorders, such as Alzheimer’s and Parkinson’s disease, early changes in synaptic mitochondrial density, energy metabolism, dynamics, and function have been reported to precede the onset of neurological symptoms and brain pathology (Arnold et al., 2011; Van Laar et al., 2018; Tang et al., 2019).
Considering the central role that mitochondria play in oxidative stress, it was perhaps not surprising to identify disruptions in the mitochondrial proteome because redox homeostasis was previously implicated in the disease mechanisms of CSTB deficiency (Lehtinen et al., 2009). Loss of CSTB function appears to sensitize cerebellar granule neurons to oxidative stress induced cell death with increased lipid peroxidation and depletion of antioxidants in the cerebellum of Cstb–/– mice. Further, loss of mitochondrial membrane integrity has been reported in lipopolysaccharide stimulated primary bone marrow -derived macrophages from Cstb–/– mice (Maher et al., 2014). A consequence of mitochondrial dysfunction is the production of ROS, which affects intracellular signal transduction pathways and generates oxidative damage (Fatokun et al., 2008). Indeed, our proteomic data show changes to the abundance of ROS-associated proteins. Notably, SOD1, an enzyme converting ROS to hydrogen peroxide limiting its potential toxicity (Wang et al., 2018) was reduced, in line with the decreased activity of SOD in cerebella of Cstb–/– mice (Lehtinen et al., 2009). We also observed alterations to the nuclear-encoded mitochondrial protein ROMO1, and the hemoglobin subunits HBA and HBB. The latter are linked to superoxide production, increased oxidative stress, and neural pathogenesis (Nishi et al., 2008; Biagioli et al., 2009; Norton et al., 2014; Lee et al., 2018). Mitochondrial ROS has been reported to modulate GABAergic signaling in cell-type-specific mechanisms (Beltrán González et al., 2019). In cerebellar granule cells, GABAAR subunit α6 -mediated inhibitory currents are strengthened in response to mitochondrial ROS, although the mechanism is currently unknown (Accardi et al., 2015). Previously, we reported increased expression of both Gabra6 and Gabrd, encoding the α6 and δ subunits of the GABAAR, in the cerebella of Cstb–/– mice (Joensuu et al., 2014).
Our proteomic data also revealed altered abundance of key transport proteins, suggesting the potential for altered transport from the soma to the synapse. These may reflect dysfunctions in global transport mechanisms or in targeted trafficking of a subset of cargo, collectively affecting synaptic plasticity. Such mechanisms have previously been described for several neurodegenerative disorders and mitochondrial dysfunction (Liu et al., 2012). Intracellular trafficking is tied to local protein synthesis and is critical for synaptic plasticity, enabling spatio-temporal regulation of translation (Steward and Schuman, 2001; Liu et al., 2012). Consistent with this interpretation, our data revealed differential abundance of several ribonucleoproteins and ribosomal subunits in the synaptosomes of Cstb–/– mice. Future studies will clarify the importance of these functions to the molecular pathogenesis of EPM1.
How does mitochondrial dysfunction and intracellular transport link to CSTB deficiency? Since 99% of the mitochondrial proteome is encoded in the nucleus, transport of the organelle back to the soma is required to maintain its function. This is paramount when the proteome has a differential turnover rate even among subunits of the same oxidative phosphorylation complex (Karunadharma et al., 2015). Thus, mitochondrial dysfunction could arise as a secondary effect of the cellular pathology at synapses.
The CSTB protein was robustly detected in our synaptosome preparations from wt mice, and has also been observed in synaptosomes from rat cortex and human cerebral organoids (Penna et al., 2019). The specific role of CSTB in synapses is not clear, but its function appears to be protective and crucial for synaptic physiology, as it is locally translated in synaptosomes of rat cerebral cortex (Penna et al., 2019). Furthermore, loss of CSTB function triggers a cascade of early pathological events, including mitochondrial dysfunction, compromised axonal transport, and altered local translation, leading to progressive neurodegeneration.
We did not identify factors in any major pathway that are known to specifically affect the GABAergic system. Our data, however, revealed a decreased abundance of GAT-1 in the synaptosomes of Cstb–/– mice. GAT-1 is a crucial member of the GABA recycling system, clearing GABA from the synaptic cleft of mature GABAergic neurons, and terminating its inhibitory actions by preventing excessive activation of extrasynaptic GABAARs that mediate tonic inhibition (Guastella et al., 1990). Moreover, mutations in SLC6A1, the gene encoding GAT-1, have previously been linked with neurological human phenotypes, including myoclonus and epilepsy (Johannesen et al., 2018). Gat-1 deficiency in mice increased the tonic postsynaptic GABAAR -mediated conductance in hippocampus (Jensen et al., 2003). No difference in GAT-1 activity in cerebellar granule cells was detected between Cstb–/– and wt mice, which may reflect heterogeneity in the cells at P14, as the cerebellum is still developing and maturing (Altman, 1972). Indeed, GAT-1 expression in the cerebellum is dependent on the maturation of neurons. Localization of GAT-1 to the GABAergic axons does not begin until the second and third postnatal weeks, paralleling synapse formation and following the expression of vesicular GABA transporter (VGAT) (Takayama and Inoue, 2005). However, our electrophysiological analysis of GAT-1 activity at P30, when the cerebellum is further matured and Cstb–/– mice enter the early symptomatic stage, also failed to detect any differences between the genotypes. These results are consistent with our previous studies, which showed less VGAT immunopositive puncta in the cerebellar molecular layer of P14 Cstb–/– mice, reaching control levels by P20 (Joensuu et al., 2014). This may indicate delayed maturation of GABAergic synapses in Cstb–/– mice. Our proteomics and electrophysiology data demonstrate that despite decreased abundance of total GAT-1 in the synaptosomes, its activity as GABA transporter in the synapse is sufficient to retain the electrophysiological properties of the cell. Its impact on other functions in the developing synapse, however, cannot be distinguished from these data, and is to be investigated in future studies.
In conclusion, our study confirms the role for CSTB in synaptic physiology and reveals a role for mitochondrial dysfunction in the early molecular pathogenesis of CSTB deficiency. The proposed hypothesis on early mitochondrial dysfunction could be the mechanism linking ROS signaling and GABAergic inhibition (Joensuu et al., 2014). Detailed understanding on how CSTB deficiency leads to mitochondrial dysfunction and the mechanisms underlying synaptic dysfunction need to be explored in future detailed studies.
Data Availability Statement
All datasets presented in this study are included in the article/supplementary material. The mass spectrometry proteomics data have been deposited to the PRIDE database with the dataset identifier PXD019370.
Ethics Statement
The animal study was reviewed and approved by the Animal Ethics Committee of the State Provincial Office of Southern Finland.
Author Contributions
KG, AS, KK, BB, and A-EL contributed the study design. KG and AS performed the experiments. KG, AS, TN, and BB analyzed the data. KG, BB, and A-EL wrote the manuscript. All authors discussed and commented on the manuscript.
Funding
We wish to acknowledge the following funding sources: Folkhälsan Research Foundation, the Sigrid Jusélius Foundation, the Finnish Epilepsy Association, Medicinska Understödsföreningen Liv och Hälsa r.f., Finnish Epilepsy Research Foundation (KG) and the Academy of Finland (KK; grant numbers 294375 and 319237). A-EL and KK are HiLIFE Fellows at the University of Helsinki.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Paula Hakala and Veronika Rezov for their skilled technical assistance, animal caretakers at the Laboratory Animal Center (University of Helsinki) for flexible cooperation and professional care of animals, Anne Rokka and Saara Tegelberg for technical advice, Tarja Joensuu, Saara Tegelberg, and Christopher Jackson for scientific advice. Mass spectrometry analysis and mass spectrometry data analysis were performed at the Turku Proteomics Facility, and at the Bioinformatics Unit at the Turku Centre for Biotechnology, University of Turku and Åbo Akademi University. The facilities are supported by Biocenter Finland.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2020.570640/full#supplementary-material
Footnotes
References
Accardi, M. V., Brown, Patricia, M. G. E., Miraucourt, L. S., Orser, B. A., and Bowie, D. (2015). α6-Containing GABA(A) receptors are the principal mediators of inhibitory synapse strengthening by insulin in cerebellar granule cells. J. Neurosci. 35, 9676–9688. doi: 10.1523/JNEUROSCI.0513-15.2015
Agliardi, C., Guerini, F. R., Zanzottera, M., Riboldazzi, G., Zangaglia, R., Sturchio, A., et al. (2019). SNAP25 gene polymorphisms protect against Parkinson’s disease and modulate disease severity in patients. Mol. Neurobiol. 56, 4455–4463. doi: 10.1007/s12035-018-1386-0
Altman, J. (1972). Postnatal development of the cerebellar cortex in the rat. II. Phases in the maturation of Purkinje cells and of the molecular layer. J. Comp. Neurol. 145, 399–463. doi: 10.1002/cne.901450402
Arnold, B., Cassady, S. J., Van Laar, V. S., and Berman, S. B. (2011). Integrating multiple aspects of mitochondrial dynamics in neurons: age-related differences and dynamic changes in a chronic rotenone model. Neurobiol. Dis. 41, 189–200. doi: 10.1016/j.nbd.2010.09.006
Beltrán González, A. N., López Pazos, M. I., and Calvo, D. J. (2019). Reactive oxygen species in the regulation of the GABA mediated inhibitory neurotransmission. Neuroscience 4522, 30395–30401. doi: 10.1016/j.neuroscience.2019.05.064
Biagioli, M., Pinto, M., Cesselli, D., Zaninello, M., Lazarevic, D., Roncaglia, P., et al. (2009). Unexpected expression of alpha- and beta-globin in mesencephalic dopaminergic neurons and glial cells. Proc. Natl. Acad. Sci. U.S.A. 106, 15454–15459. doi: 10.1073/pnas.0813216106
Bourassa, C. V., Meijer, I. A., Merner, N. D., Grewal, K. K., Stefanelli, M. G., Hodgkinson, K., et al. (2012). VAMP1 mutation causes dominant hereditary spastic ataxia in Newfoundland families. Am. J. Hum. Genet. 91, 548–552. doi: 10.1016/j.ajhg.2012.07.018
Brakebusch, C., Seidenbecher, C. I., Asztely, F., Rauch, U., Matthies, H., Meyer, H., et al. (2002). Brevican-deficient mice display impaired hippocampal CA1 long-term potentiation but show no obvious deficits in learning and memory. Mol. Cell. Biol. 22, 7417–7427. doi: 10.1128/mcb.22.21.7417-7427.2002
Brenneke, F., Bukalo, O., Dityatev, A., and Lie, A. A. (2004). Mice deficient for the extracellular matrix glycoprotein tenascin-r show physiological and structural hallmarks of increased hippocampal excitability, but no increased susceptibility to seizures in the pilocarpine model of epilepsy. Neuroscience 124, 841–855. doi: 10.1016/j.neuroscience.2003.11.037
Brickley, S. G., Cull-Candy, S. G., and Farrant, M. (1996). Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J. Physiol. 497(Pt 3), 753–759. doi: 10.1113/jphysiol.1996.sp021806
Burgalossi, A., Jung, S., Meyer, G., Jockusch, W. J., Jahn, O., Taschenberger, H., et al. (2010). SNARE protein recycling by αSNAP and βSNAP supports synaptic vesicle priming. Neuron 68, 473–487. doi: 10.1016/j.neuron.2010.09.019
Buzzi, A., Chikhladze, M., Falcicchia, C., Paradiso, B., Lanza, G., Soukupova, M., et al. (2012). Loss of cortical GABA terminals in Unverricht-Lundborg disease. Neurobiol. Dis. 47, 216–224. doi: 10.1016/j.nbd.2012.04.005
Canafoglia, L., Gennaro, E., Capovilla, G., Gobbi, G., Boni, A., Beccaria, F., et al. (2012). Electroclinical presentation and genotype-phenotype relationships in patients with Unverricht-Lundborg disease carrying compound heterozygous CSTB point and indel mutations. Epilepsia 53, 2120–2127. doi: 10.1111/j.1528-1167.2012.03718.x
Carta, M., Mameli, M., and Valenzuela, C. F. (2004). Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J. Neurosci. 24, 3746–3751. doi: 10.1523/JNEUROSCI.0067-04.2004
Ceru, S., Konjar, S., Maher, K., Repnik, U., Krizaj, I., Bencina, M., et al. (2010). Stefin B interacts with histones and cathepsin L in the nucleus. J. Biol. Chem. 285, 10078–10086. doi: 10.1074/jbc.M109.034793
Cohen, N. R., Hammans, S. R., Macpherson, J., and Nicoll, J. A. R. (2011). New neuropathological findings in Unverricht-Lundborg disease: neuronal intranuclear and cytoplasmic inclusions. Acta Neuropathol. 121, 421–427. doi: 10.1007/s00401-010-0738-2
Conroy, J., Allen, N. M., Gorman, K. M., Shahwan, A., Ennis, S., Lynch, S. A., et al. (2016). NAPB - a novel SNARE-associated protein for early-onset epileptic encephalopathy. Clin. Genet. 89, E1–E3. doi: 10.1111/cge.12648
Danner, N., Julkunen, P., Khyuppenen, J., Hukkanen, T., Könönen, M., Säisänen, L., et al. (2009). Altered cortical inhibition in Unverricht-Lundborg type progressive myoclonus epilepsy (EPM1). Epilepsy Res. 85, 81–88. doi: 10.1016/j.eplepsyres.2009.02.015
Eldridge, R., Iivanainen, M., Stern, R., Koerber, T., and Wilder, B. J. (1983). “Baltic” myoclonus epilepsy: hereditary disorder of childhood made worse by phenytoin. Lancet 2, 838–842. doi: 10.1016/s0140-6736(83)90749-3
Elo, L. L., Filén, S., Lahesmaa, R., and Aittokallio, T. (2008). Reproducibility-optimized test statistic for ranking genes in microarray studies. IEEE/ACM Trans. Comput. Biol. Bioinform. 5, 423–431. doi: 10.1109/tcbb.2007.1078
Fatokun, A. A., Stone, T. W., and Smith, R. A. (2008). Oxidative stress in neurodegeneration and available means of protection. Front. Biosci. 13:3288–3311. doi: 10.2741/2926
Franceschetti, S., Sancini, G., Buzzi, A., Zucchini, S., Paradiso, B., Magnaghi, G., et al. (2007). A pathogenetic hypothesis of Unverricht-Lundborg disease onset and progression. Neurobiol. Dis. 25, 675–685. doi: 10.1016/j.nbd.2006.11.006
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. doi: 10.1186/gb-2004-5-10-r80
Guastella, J., Nelson, N., Nelson, H., Czyzyk, L., Keynan, S., Miedel, M. C., et al. (1990). Cloning and expression of a rat brain GABA transporter. Science 249, 1303–1306. doi: 10.1126/science.1975955
Haltia, M., Kristensson, K., and Sourander, P. (1969). Neuropathological studies in three Scandinavian cases of progressive myoclonus epilepsy. Acta Neurol. Scand. 45, 63–77. doi: 10.1111/j.1600-0404.1969.tb01220.x
Harigaya, Y., Shoji, M., Shirao, T., and Hirai, S. (1996). Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J. Neurosci. Res. 43, 87–92. doi: 10.1002/jnr.490430111
Jensen, K., Chiu, C., Sokolova, I., Lester, H. A., and Mody, I. (2003). GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J. Neurophysiol. 90, 2690–2701. doi: 10.1152/jn.00240.2003
Joensuu, T., Kuronen, M., Alakurtti, K., Tegelberg, S., Hakala, P., Aalto, A., et al. (2007). Cystatin B: mutation detection, alternative splicing and expression in progressive myclonus epilepsy of Unverricht-Lundborg type (EPM1) patients. Eur. J. Hum. Genet. 15, 185–193. doi: 10.1038/sj.ejhg.5201723
Joensuu, T., Lehesjoki, A. E., and Kopra, O. (2008). Molecular background of EPM1-Unverricht-Lundborg disease. Epilepsia 49, 557–563. doi: 10.1111/j.1528-1167.2007.01422.x
Joensuu, T., Tegelberg, S., Reinmaa, E., Segerstrale, M., Hakala, P., Pehkonen, H., et al. (2014). Gene expression alterations in the cerebellum and granule neurons of Cstb(-/-) mouse are associated with early synaptic changes and inflammation. PLoS One 9:e89321. doi: 10.1371/journal.pone.0089321
Johannesen, K. M., Gardella, E., Linnankivi, T., Courage, C., de Saint Martin, A., Lehesjoki, A. E., et al. (2018). Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia 59, 389–402. doi: 10.1111/epi.13986
Julkunen, P., Säisänen, L., Könönen, M., Vanninen, R., Kälviäinen, R., and Mervaala, E. (2013). TMS-EEG reveals impaired intracortical interactions and coherence in Unverricht-Lundborg type progressive myoclonus epilepsy (EPM1). Epilepsy Res. 106, 103–112. doi: 10.1016/j.eplepsyres.2013.04.001
Kälviäinen, R., Khyuppenen, J., Koskenkorva, P., Eriksson, K., Vanninen, R., and Mervaala, E. (2008). Clinical picture of EPM1-Unverricht-Lundborg disease. Epilepsia 49, 549–556. doi: 10.1111/j.1528-1167.2008.01546.x
Kaneda, M., Farrant, M., and Cull-Candy, S. G. (1995). Whole-cell and single-channel currents activated by GABA and glycine in granule cells of the rat cerebellum. J. Physiol. 485(Pt 2), 419–435. doi: 10.1113/jphysiol.1995.sp020739
Karunadharma, P. P., Basisty, N., Chiao, Y. A., Dai, D., Drake, R., Levy, N., et al. (2015). Respiratory chain protein turnover rates in mice are highly heterogeneous but strikingly conserved across tissues, ages, and treatments. FASEB J. 29, 3582–3592. doi: 10.1096/fj.15-272666
Kennedy, M. J., and Ehlers, M. D. (2006). Organelles and trafficking machinery for postsynaptic plasticity. Annu. Rev. Neurosci. 29, 325–362. doi: 10.1146/annurev.neuro.29.051605.112808
Khalili, K., Del Valle, L., Muralidharan, V., Gault, W. J., Darbinian, N., Otte, J., et al. (2003). Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol. Cell. Biol. 23, 6857–6875. doi: 10.1128/mcb.23.19.6857-6875.2003
Koskenkorva, P., Hyppönen, J., Aikiä, M., Mervaala, E., Kiviranta, T., Eriksson, K., et al. (2011). Severer phenotype in Unverricht-Lundborg disease (EPM1) patients compound heterozygous for the dodecamer repeat expansion and the c.202C>T mutation in the CSTB gene. Neurodegener. Dis. 8, 515–522. doi: 10.1159/000323470
Koskenkorva, P., Khyuppenen, J., Niskanen, E., Könönen, M., Bendel, P., Mervaala, E., et al. (2009). Motor cortex and thalamic atrophy in Unverricht-Lundborg disease: voxel-based morphometric study. Neurology 73, 606–611. doi: 10.1212/WNL.0b013e3181b3888b
Koskenkorva, P., Niskanen, E., Hyppönen, J., Könönen, M., Mervaala, E., Soininen, H., et al. (2012). Sensorimotor, visual, and auditory cortical atrophy in Unverricht-Lundborg disease mapped with cortical thickness analysis. AJNR Am. J. Neuroradiol. 33, 878–883. doi: 10.3174/ajnr.A2882
Koskiniemi, M., Donner, M., Majuri, H., Haltia, M., and Norio, R. (1974). Progressive myoclonus epilepsy. A clinical and histopathological study. Acta Neurol. Scand. 50, 307–332.
Lalani, S. R., Zhang, J., Schaaf, C. P., Brown, C. W., Magoulas, P., Tsai, A. C., et al. (2014). Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am. J. Hum. Genet. 95, 579–583. doi: 10.1016/j.ajhg.2014.09.014
Lee, G. Y., You, D., Lee, H., Hwang, S. W., Lee, C. J., and Yoo, Y. D. (2018). Romo1 is a mitochondrial nonselective cation channel with viroporin-like characteristics. J. Cell Biol. 217, 2059–2071. doi: 10.1083/jcb.201709001
Lehtinen, M. K., Tegelberg, S., Schipper, H., Su, H., Zukor, H., Manninen, O., et al. (2009). Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J. Neurosci. 29, 5910–5915. doi: 10.1523/JNEUROSCI.0682-09.2009
Liu, X., Rizzo, V., and Puthanveettil, S. (2012). Pathologies of axonal transport in neurodegenerative diseases. Translat. Neurosci. 3, 355–372. doi: 10.2478/s13380-012-0044-7
Maher, K., Jerič Kokelj, B., Butinar, M., Mikhaylov, G., Manček-Keber, M., Stoka, V., et al. (2014). A role for stefin B (cystatin B) in inflammation and endotoxemia. J. Biol. Chem. 289, 31736–31750. doi: 10.1074/jbc.M114.609396
Mancini, G. M. S., Schot, R., de Wit, Marie Claire, Y., de Coo, R. F., Oostenbrink, R., et al. (2016). CSTB null mutation associated with microcephaly, early developmental delay, and severe dyskinesia. Neurology 86, 877–878. doi: 10.1212/WNL.0000000000002422
Manninen, O., Koskenkorva, P., Lehtimäki, K. K., Hyppönen, J., Könönen, M., Laitinen, T., et al. (2013). White matter degeneration with Unverricht-Lundborg progressive myoclonus epilepsy: a translational diffusion-tensor imaging study in patients and cystatin B-deficient mice. Radiology 269, 232–239. doi: 10.1148/radiol.13122458
Metsalu, T., and Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 43, W566–W570. doi: 10.1093/nar/gkv468
Mi, H., Muruganujan, A., Casagrande, J. T., and Thomas, P. D. (2013). Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566. doi: 10.1038/nprot.2013.092
Nishi, H., Inagi, R., Kato, H., Tanemoto, M., Kojima, I., Son, D., et al. (2008). Hemoglobin is expressed by mesangial cells and reduces oxidant stress. J. Am. Soc. Nephrol. 19, 1500–1508. doi: 10.1681/ASN.2007101085
Nolt, M. J., Lin, Y., Hruska, M., Murphy, J., Sheffler-Colins, S. I., Kayser, M. S., et al. (2011). EphB controls NMDA receptor function and synaptic targeting in a subunit-specific manner. J. Neurosci. 31, 5353–5364. doi: 10.1523/JNEUROSCI.0282-11.2011
Norton, M., Ng, A. C., Baird, S., Dumoulin, A., Shutt, T., Mah, N., et al. (2014). ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci. Signal. 7:ra10. doi: 10.1126/scisignal.2004374
Nystuen, A. M., Schwendinger, J. K., Sachs, A. J., Yang, A. W., and Haider, N. B. (2007). A null mutation in VAMP1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics 8, 1–10. doi: 10.1007/s10048-006-0068-7
O’Brien, A., Marshall, C. R., Blaser, S., Ray, P. N., and Yoon, G. (2017). Severe neurodegeneration, progressive cerebral volume loss and diffuse hypomyelination associated with a homozygous frameshift mutation in CSTB. Eur. J. Hum. Genet. 25, 775–778. doi: 10.1038/ejhg.2017.39
Okuneva, O., Korber, I., Li, Z., Tian, L., Joensuu, T., Kopra, O., et al. (2015). Abnormal microglial activation in the Cstb(-/-) mouse, a model for progressive myoclonus epilepsy, EPM1. Glia 63, 400–411. doi: 10.1002/glia.22760
Penna, E., Cerciello, A., Chambery, A., Russo, R., Cernilogar, F. M., Pedone, E. M., et al. (2019). Cystatin B involvement in synapse physiology of rodent brains and human cerebral organoids. Front. Mol. Neurosci. 12:195. doi: 10.3389/fnmol.2019.00195
Pennacchio, L. A., Bouley, D. M., Higgins, K. M., Scott, M. P., Noebels, J. L., and Myers, R. M. (1998). Progressive ataxia, myoclonic epilepsy and cerebellar apoptosis in cystatin B-deficient mice. Nat. Genet. 20, 251–258. doi: 10.1038/3059
Pennacchio, L. A., Lehesjoki, A. E., Stone, N. E., Willour, V. L., Virtaneva, K., Miao, J., et al. (1996). Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science 271, 1731–1734. doi: 10.1126/science.271.5256.1731
R Core Team (2016). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rossi, D. J., Hamann, M., and Attwell, D. (2003). Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J. Physiol. 548(Pt 1), 97–110. doi: 10.1113/jphysiol.2002.036459
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shen, X., Scola, R. H., Lorenzoni, P. J., Kay, C. S. K., Werneck, L. C., Brengman, J., et al. (2017). Novel synaptobrevin-1 mutation causes fatal congenital myasthenic syndrome. Ann. Clin. Transl. Neurol. 4, 130–138. doi: 10.1002/acn3.387
Silver, R. A., Traynelis, S. F., and Cull-Candy, S. G. (1992). Rapid-time-course miniature and evoked excitatory currents at cerebellar synapses in situ. Nature 355, 163–166. doi: 10.1038/355163a0
Sipilä, S. T., Huttu, K., Soltesz, I., Voipio, J., and Kaila, K. (2005). Depolarizing GABA Acts on intrinsically bursting pyramidal neurons to drive giant depolarizing potentials in the immature hippocampus. J. Neurosci. 25, 5280–5289. doi: 10.1523/JNEUROSCI.0378-05.2005
Steward, O., and Schuman, E. M. (2001). Protein synthesis at synaptic sites on dendrites. Annu. Rev. Neurosci. 24, 299–325. doi: 10.1146/annurev.neuro.24.1.299
Suzdak, P. D., Frederiksen, K., Andersen, K. E., Sørensen, P. O., Knutsen, L. J., and Nielsen, E. B. (1992). NNC-711, a novel potent and selective gamma-aminobutyric acid uptake inhibitor: pharmacological characterization. Eur. J. Pharmacol. 224, 189–198. doi: 10.1016/0014-2999(92)90804-d
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Takayama, C., and Inoue, Y. (2005). Developmental expression of GABA transporter-1 and 3 during formation of the GABAergic synapses in the mouse cerebellar cortex. Brain Res. Dev. Brain Res. 158, 41–49. doi: 10.1016/j.devbrainres.2005.05.007
Tang, J., Oliveros, A., and Jang, M. (2019). Dysfunctional mitochondrial bioenergetics and synaptic degeneration in Alzheimer disease. Int. Neurourol. J. 23(Suppl. 1), S5–S10. doi: 10.5213/inj.1938036.018
Tegelberg, S., Kopra, O., Joensuu, T., Cooper, J. D., and Lehesjoki, A. E. (2012). Early microglial activation precedes neuronal loss in the brain of the Cstb-/- mouse model of progressive myoclonus epilepsy, EPM1. J. Neuropathol. Exp. Neurol. 71, 40–53. doi: 10.1097/NEN.0b013e31823e68e1
Turk, V., Stoka, V., and Turk, D. (2008). Cystatins: biochemical and structural properties, and medical relevance. Front. Biosci. 13:5406–5420. doi: 10.2741/3089
Van Laar, V. S., Arnold, B., Howlett, E. H., Calderon, M. J., St. Croix, C. M., Greenamyre, J. T., et al. (2018). Evidence for compartmentalized axonal mitochondrial biogenesis: mitochondrial DNA replication increases in distal axons as an early response to parkinson’s disease-relevant stress. J. Neurosci. 38, 7505–7515. doi: 10.1523/JNEUROSCI.0541-18.2018
Vizcaíno, J. A., Côté, R. G., Csordas, A., Dianes, J. A., Fabregat, A., Foster, J. M., et al. (2013). The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069. doi: 10.1093/nar/gks1262
von Eichborn, J., Dunkel, M., Gohlke, B. O., Preissner, S. C., Hoffmann, M. F., Bauer, J. M. J., et al. (2013). SynSysNet: integration of experimental data on synaptic protein-protein interactions with drug-target relations. Nucleic Acids Res. 41, D834–D840. doi: 10.1093/nar/gks1040
Wall, M. J., and Usowicz, M. M. (1997). Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur. J. Neurosci. 9, 533–548. doi: 10.1111/j.1460-9568.1997.tb01630.x
Wang, Y., Branicky, R., Noë, A., and Hekimi, S. (2018). Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 21, 1915–1928. doi: 10.1083/jcb.201708007
Westbury, S. K., Shoemark, D. K., and Mumford, A. D. (2017). ACTN1 variants associated with thrombocytopenia. Platelets 28, 625–627. doi: 10.1080/09537104.2017.1356455
Keywords: myoclonus epilepsy, synaptosome, neurodegeneration, cerebella, electrophysiology, mitochondria
Citation: Gorski K, Spoljaric A, Nyman TA, Kaila K, Battersby BJ and Lehesjoki A-E (2020) Quantitative Changes in the Mitochondrial Proteome of Cerebellar Synaptosomes From Preclinical Cystatin B-Deficient Mice. Front. Mol. Neurosci. 13:570640. doi: 10.3389/fnmol.2020.570640
Received: 08 June 2020; Accepted: 21 October 2020;
Published: 13 November 2020.
Edited by:
Tija Carey Jacob, University of Pittsburgh, United StatesReviewed by:
Joern R. Steinert, University of Nottingham, United KingdomTiziana Bonifacino, University of Genoa, Italy
Susan T. Weintraub, University of Texas Health Science Center at San Antonio, United States
Copyright © 2020 Gorski, Spoljaric, Nyman, Kaila, Battersby and Lehesjoki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna-Elina Lehesjoki, YW5uYS1lbGluYS5sZWhlc2pva2lAaGVsc2lua2kuZmk=