 Kunie Ando1*
Kunie Ando1* Sarah Houben1
Sarah Houben1 Mégane Homa1Marie-Ange de Fisenne1
Mégane Homa1Marie-Ange de Fisenne1 Marie-Claude Potier2
Marie-Claude Potier2 Christophe Erneux3†
Christophe Erneux3† Jean-Pierre Brion1†
Jean-Pierre Brion1† Karelle Leroy1†
Karelle Leroy1†- 1Laboratory of Histology, Neuroanatomy and Neuropathology, Faculty of Medicine, Université Libre de Bruxelles, ULB Neuroscience Institute, Brussels, Belgium
- 2ICM Institut du Cerveau et de la Moelle épinière, CNRS UMR7225, INSERM U1127, UPMC, Hôpital de la Pitié-Salpêtrière, Paris, France
- 3Institut de Recherche Interdisciplinaire en Biologie Humaine et moléculaire (IRIBHM), Université Libre de Bruxelles, Brussels, Belgium
Introduction
Alzheimer's disease (AD) is the most common form of dementia. Its prevalence will significantly increase in the coming decades, whereas no efficient treatment is currently available. AD has two main neuropathological lesions: amyloid plaques and neurofibrillary tangles (NFTs). Amyloid plaques are composed of amyloid ß (Aß) peptides cleaved from the Amyloid Precursor Protein (APP) (Glenner and Wong, 1984). NFTs, present in the brain of AD and related neurodegenerative diseases, are constituted of tau proteins (Brion et al., 1985) in hyperphosphorylated and aggregated form (Wang and Mandelkow, 2016).
Endolysosomal Abnormalities in AD
Evidence from both genetic and biochemical studies supports the involvement of endolysosomal abnormalities in the development of Alzheimer brain lesions (Van Acker et al., 2019). Enlargement of endosomes in neurons and peripheral cells is observed at early stage of AD and of Down syndrome (DS) individuals who are at high risk for AD (Cataldo et al., 1996; Botte and Potier, 2020). The endocytic machinery may thus be a highly vulnerable cascade that undergoes alterations at the early stages of AD. Importantly, endocytosis is closely linked to the development of both Aß and tau pathologies. On one hand, it is believed that modifications of the endolysosomal compartment in AD and DS are mostly linked to increased Aβ production as the amyloidogenic processing of APP occurs in the endosomal pathway (Koo and Squazzo, 1994; Botte et al., 2020). On the other hand, uptake of abnormal tau by endocytosis is an important step to sustain tau spreading from cell to cell (Wu et al., 2013; Evans et al., 2018; Puangmalai et al., 2020; Rauch et al., 2020). Tau pathology is strongly correlated to functional deficits in AD brains (Nelson et al., 2012). The brain propagation of tau pathology in AD follows neuroanatomical pathways and can reflect transmission of abnormal tau proteins from cell to cell in a “prion-like” manner. This transcellular transfer of abnormal tau is thought to induce recruitment and seeding of the normal soluble tau proteins into pathological aggregated tau and would need cellular internalization of abnormal tau through endocytic mechanisms (Mudher et al., 2017). In this review, we mainly focus on endocytic abnormalities in AD brains, the underlying potential mechanisms, and the relationship with tau pathology.
Genetic risk Factors for Late-Onset AD (LOAD) and LOAD-Related Endolysosomal Proteins
Familial AD (FAD) accounts for <5% of AD cases and is well characterized by mutations in three genes (APP, PSEN1, and PSEN2). Although genetic factors are estimated to represent 60% of the risk to develop LOAD, they remained largely unknown for a long time except for APOE (Gatz et al., 2006). Genome-wide association studies (GWAS) and whole genome sequencing studies (WGS) have identified more than 45 genes/loci increasing or decreasing the susceptibility to develop LOAD (Lambert et al., 2013; Dourlen et al., 2019). Some GWAS hit genes encode key proteins involved in endocytosis and membrane dynamics such as INPP5D (SHIP1), Bridging Integrator 1 (BIN1), Phosphatidylinositol Binding Clathrin Assembly Protein (PICALM), Ras and Rab Interactor 3 (RIN3), CD2 Associated Protein (CD2AP), Sortilin Related Receptor 1 (SORL1), Cas scaffolding protein family member 4 (CASS4) (Lambert et al., 2013). In addition, several independent studies have reported the involvement of genes encoding endolysosomal proteins in AD such as INPPL1 (SHIP2) (Mostafavi et al., 2018), Synaptojanin-1 (SYNJ1) (Miranda et al., 2018) and phospholipase D3 (PLD3) (Cruchaga et al., 2014). The pathophysiological mechanisms by which these genes may modulate the risk for LOAD are still not fully understood.
Dynamin-Dependent Endocytosis and LOAD-Related Endolysosomal Proteins
The endolysosomal proteins listed above play critical roles at various steps of dynamin-dependent endocytosis and further steps (Figure 1A). Endocytosis starts with the recruitment of endocytic proteins to the plasma membrane subdomain enriched with phosphatidylinositol (PI) 4,5-bisphosphate [PI(4,5)P2] to form a clathrin-coated pit (CCP) (Ferguson and De Camilli, 2012; Wang et al., 2019). SHIP2 negatively regulates the dynamics of CCP formation (Nakatsu et al., 2010) by engaging a change in PI(3,4)P2 (Ghosh et al., 2018) and PI(4,5)P2 (Elong Edimo et al., 2016). Membrane invagination starts from this pit by assembling clathrin and AP-2 with the adaptor protein PICALM (Tebar et al., 1999). BIN1 is involved in membrane curvature and remodeling but BIN1 is also detected in the early endosomes of axons in neurons and is also implicated in recycling BACE1, a ß-site APP cleaving enzyme present in early endosomes (Ubelmann et al., 2017). Deep invagination of the bud and narrow neck formation are assisted by actin polymerization where SYNJ1 and dynamin interact with their protein partners possessing BAR domains (Chang-Ileto et al., 2011). After scission of newly formed vesicles by dynamin, SYNJ1 plays a critical role in clathrin-coated vesicle uncoating (Cremona et al., 1999). Then, RIN3 joins in the transport pathway from plasma membrane to early endosomes (Kajiho et al., 2003). Similarly, CD2AP is detected in the early endosomes of the dendrites in cultured neurons and is involved in actin remodeling, membrane trafficking (Lehtonen et al., 2002) and in APP sorting (Ubelmann et al., 2017). SORL1, directly interacting with APP, is localized primarily to early endosomes and the trans-Golgi network, shuttling between these two membrane compartments (Willnow et al., 2008). Lastly, PLD3 is implicated in endolysosomal system (Fazzari et al., 2017). CASS4 is rather implicated in focal adhesion integrity and tau toxicity (Dourlen et al., 2019). Most of these proteins are directly or indirectly involved in interactions with actin networks as described for dynamin (Zhang et al., 2020), SHIP1 (Lesourne et al., 2005), SHIP2 (Ghosh et al., 2018), BIN1 (Butler et al., 1997; Drager et al., 2017), SYNJ1 (Sakisaka et al., 1997), CD2AP (Lehtonen et al., 2002; Lynch et al., 2003; Tang and Brieher, 2013) and CASS4 (Deneka et al., 2015). Tau itself is also involved in organizing actin networks (Elie et al., 2015). Given that many of these endocytic proteins are also implicated in synaptic vesicle endocytosis and focal adhesion formation at the synaptic cleft, they are assumed to be involved in synaptic dysfunctions observed in AD (Dourlen et al., 2019; Perdigao et al., 2020).
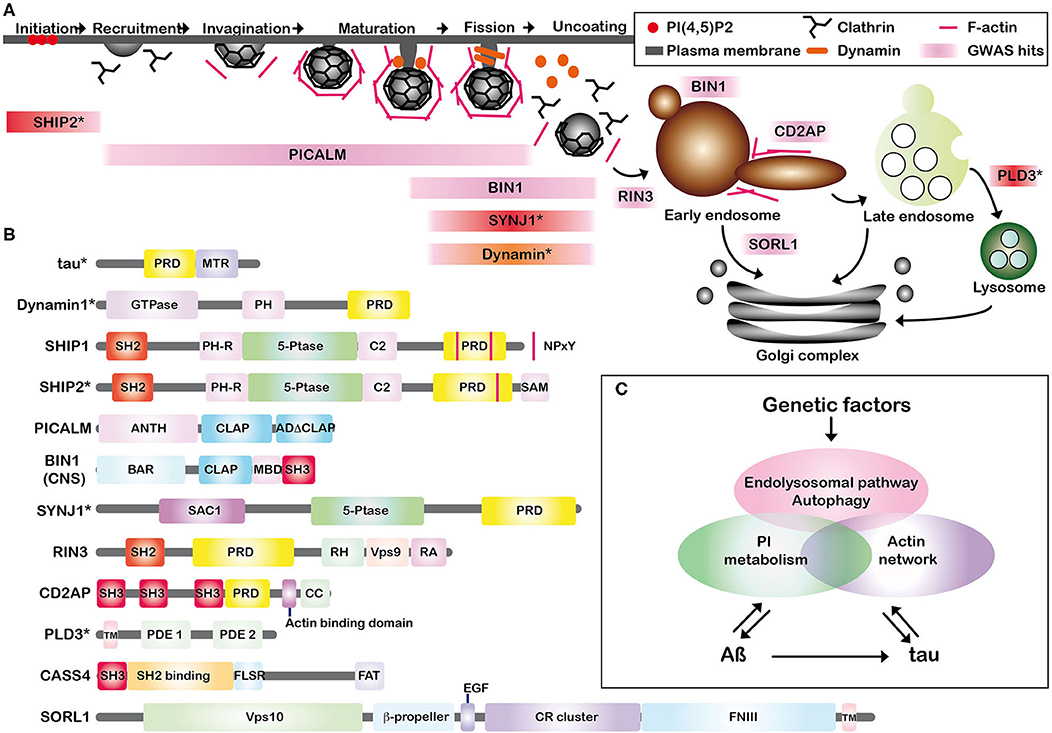
Figure 1. (A) Schematic representation of the associations of Alzheimer key proteins in CCP formation and dynamin-dependent endocytosis. Some of the recently identified AD susceptibility factors are involved in the early and late stages of dynamin-dependent endocytosis. GWAS-hits are shown in pink. SHIP1 is rather implicated in phagosome formation in macrophages and CASS4 in focal adhesion. SHIP1 and CASS4 are not included in (A). *SHIP2, SYNJ1, and PLD3 are endolysosomal proteins involved in AD pathogenesis but not categorized as GWAS hits and shown in red. Dynamin is shown in orange. (B) The schematic illustration of the endolysosomal proteins. Some of them possess SH2, SH3 domains, or PRD. PRD, proline-rich domain; MTR, microtubule binding repeats; PH, pleckstrin-homology; SH2, Src homology 2; 5-Ptase, phosphoinositide 5-phosphatase domain; SAM, sterile alpha motif; NPxY, a conserved tyrosine phosphorylation motif (Asn-Pro-x-Tyr); ANTH, AP180 N-terminal homology (ANTH) domain; CLAP, clathrin-associated protein-binding; ADΔCLAP, A second sub-domain without clearly crucial motifs for clathrin binding. CNS, central nervous system; BAR, BIN/Amphiphysin/Rvs; SH3, Src homology 3; SAC1, suppressor of actin 1; RH, RIN-homology; Vps, vacuolar protein sorting; RA, Ras-association (RA); CC, coiled coil; TM, transmembrane; PDE, phosphodiesterase; SH2 Binding, tyrosine phosphosite-enriched domain containing binding sites for partners with SH2 domains; FLSR, central serine rich region; FAT, focal adhesion targeting domain; EGF, epidermal growth factor; CR cluster, complement-type repeat domains; FNIII, fibronectin type III repeats. *It has to be noted that tau, dynamin, SHIP2, SYNJ1, and PLD3 are not GWAS hits. (C) Schematic summary of our hypothesis. Genetic factors are linked to both APP processing (Aß production) and tau pathology via actin network, PI metabolism and endolysosomal pathway/autophagy, leading to neuronal dysfunctions. PI metabolism is modulated by both Aß (Berman et al., 2008) and tau (Hwang et al., 1996). Aß is linked to tau by accelerating brain tau-seeded pathology (He et al., 2018; Vergara et al., 2019).
Involvement of Endocytic Proteins With Proline-Rich Domain (PRD) and Src-Homology3 (SH3)-Domain in Endocytic Alterations in AD
Dynamin-dependent endocytosis is regulated by the interplay of the interactions between PRD of dynamin and SH3 domain-containing proteins (Ferguson and De Camilli, 2012). Some of the endocytic proteins discussed above possess SH2 or SH3 domains and/or PRD (Figure 1B). These endocytic proteins form an interconnected protein network by direct or indirect protein-protein interactions. For example, BIN1 directly interacts with PICALM, SYNJ1, dynamin, RIN3 and tau (Chapuis et al., 2013; Shen et al., 2020). We hypothesize that dynamin-dependent endocytosis could be highly vulnerable and sensitive. Firstly, cyclin-dependent kinase 5 (CDK5), a kinase activated in AD brains (Patrick et al., 1999), phosphorylates PRD of both dynamin1 and SYNJ1 to block the interaction with SH3 domains of their protein partners (Ferguson and De Camilli, 2012). Secondly, the expression level of each of these endocytic proteins has a profound effect on endosomal structures. For example, depletion of SHIP2 accelerates the maturation of CCPs (Nakatsu et al., 2010) and depletion of PICALM leads to enlargement of clathrin-coated vesicle sizes (Miller et al., 2015). Likewise, depletion of BIN1, SYNJ1, CD2AP, and SORL1 results in an enlargement of early endosomes in cultured cells (Calafate et al., 2016; Ubelmann et al., 2017; Fasano et al., 2018; Knupp et al., 2020). In view of the fact that most of the LOAD-related SNPs reside in noncoding regions of the genome, they are supposed to play a role in regulating gene expression. Long-term up- or down-regulation of even one of these endocytic proteins encoded by LOAD-susceptibility genes might thus provoke endosomal abnormalities. In other words, endocytic alterations might begin much earlier than the appearance of AD lesions in the individuals bearing risk alleles of these GWAS-hit genes.
Interactions Between Tau, Endocytosis Regulation, and Phosphoinositides
Given that endocytic proteins such as SHIP1, SHIP2, and SYNJ1 are PI-5-phosphatases involved in PI metabolism (Ramos et al., 2019), we hypothesize that upstream dysregulation of PI metabolism in AD brains may as well accelerate AD pathology. PIs act as signaling molecules in several biological functions, including membrane dynamics, cell adhesion, autophagy, and endocytosis. The homeostasis of PIs, tightly regulated by PI kinases and phosphatases in healthy cells (Di Paolo and De Camilli, 2006), is dysregulated in AD brains (Stokes and Hawthorne, 1987; Jope et al., 1994; Morel et al., 2013). While Aß modulates PI(4,5)P2 metabolism (Berman et al., 2008; He et al., 2019), PI(4,5)P2 may as well be involved in the formation of tau pathology. PI(4,5)P2 directly interacts with tau (Surridge and Burns, 1994) and can induce fibrillization of recombinant tau in vitro (Talaga et al., 2018). Indeed, PI(4,5)P2 is abnormally concentrated with lipid raft markers in NFTs and in granulovacuolar degeneration bodies in post-mortem brain tissues of AD and other tauopathies (Nishikawa et al., 2014) and is associated with several tau kinases in the raft structures (Nishikawa et al., 2016). Furthermore, tau possesses a PRD composed of seven Pro-X-X-Pro (PXXP) motifs, in its central domain (Figure 1B). Tau interacts with various SH3-containing proteins including phospholipase C (PLC) γ1 (Reynolds et al., 2008). PLC hydrolyses PI(4,5)P2 to generate diacylglycerol (DAG) and inositol 1,4,5-trisphosphate [Ins(1,4,5)P3], an important second messenger to mobilize calcium from internal stores. Tau modulates cellular signaling by interacting with PLC γ1 thereby enhancing the cleavage of PI(4,5)P2 (Hwang et al., 1996). In vitro phosphorylation of tau by GSK3ß, a kinase abnormally activated in AD brains (Leroy et al., 2007), significantly decreases interaction with some of its SH3-containing partners such as PLC γ1 (Reynolds et al., 2008). This implies that interactions between tau and its SH3-containing partners including PLC γ1 are likely to be disrupted in AD brains. Tau hyperphosphorylation may thus trigger dysregulation of PI metabolism and the upstream cascade of endocytosis (Wallroth and Haucke, 2018). It is also speculated that some of the proteins possessing SH2 and SH3 domains and/or PRD may be influenced by the release of “free” SH3 domains of tau partners due to tau detachment. On the other hand, the somatodendritic tau concentration is ~8-fold higher in AD compared to age-matched controls (Khatoon et al., 1992). Tau is associated with some of these endocytic proteins such as BIN1 (Calafate et al., 2016; Sartori et al., 2019), PICALM (Ando et al., 2013, 2016, 2020a) and SYNJ1 (Ando et al., 2020b). By direct interaction with tau, these endocytic proteins may (i) play roles in internalization of pathological tau during endocytosis by directly binding to tau, (ii) undergo a significant alteration in their subcellular localizations due to sequestration by tau. Some endocytic proteins interacting with phosphorylated tau are significantly decreased from the soluble fraction of AD brain lysates as observed for PICALM (Ando et al., 2016) and SYNJ1 (Ando et al., 2020b). While PICALM and SYNJ1 play critical roles in endocytosis, they also modulate autophagy (Moreau et al., 2014; Vanhauwaert et al., 2017). It is presumed that tau sequestration of such endocytic proteins could also lead to defects in both endolysosomal and autophagy pathways, central network to clearance of cellular macromolecules including Aß and tau.
Discussion
Many of the endocytic machinery proteins implicated in AD risk possess SH2, SH3 domains, and/or PRD and are involved in actin dynamics as well as in regulation of PIs. Because the endocytic machinery needs fine-tuned regulation of PIs and endocytic protein-protein interactions, the endocytic pathway must be highly vulnerable. Dysregulation of even one of these endocytic proteins could lead to significant endocytic abnormalities. Hyperphosphorylation of tau may further accelerate endocytic dysregulation. Genetic risk factors and tau pathology might well have profound impacts on synaptic functions, endolysosomal/autophagic pathways, and APP processing via dysfunction of endocytosis, actin network, and PI metabolism (Figure 1C). Endolysosomal/autophagic abnormalities are also linked to both Aß and tau pathologies. Aß and tau are also tightly linked: Aß inhibits proteasome pathway (Almeida et al., 2006) and accelerates tau pathology progression (He et al., 2018; Vergara et al., 2019). Several genetic risk factors for LOAD may have pathological effects by inducing endocytic abnormalities leading to Aß production, tau pathology progression, synaptic failure, and deficits in membrane dynamics, all events observed in the progression of AD.
Author Contributions
KA constructed the main concept of the manuscript by exchanging opinions with the other authors. All authors participated in writing the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
This study was supported by grants from the Belgian Fonds de la Recherche Scientifique Médicale (T.0023.15 to J-PB and J.0078.18 to CE), the Fund Aline (King Baudouin Foundation) to J-PB, the Foundation for Alzheimer Research (FRA/SAO) to KL and the Génicot Fund of ULB to J-PB and KL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize that many interesting studies had to be omitted due to the word limitations.
References
Almeida, C. G., Takahashi, R. H., and Gouras, G. K. (2006). Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 26, 4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006
Ando, K., Brion, J. P., Stygelbout, V., Suain, V., Authelet, M., Dedecker, R., et al. (2013). Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer's brains. Acta Neuropathol. 125, 861–878. doi: 10.1007/s00401-013-1111-z
Ando, K., De Decker, R., Vergara, C., Yilmaz, Z., Mansour, S., Suain, V., et al. (2020a). Picalm reduction exacerbates tau pathology in a murine tauopathy model. Acta Neuropathol. 139, 773–789. doi: 10.1007/s00401-020-02125-x
Ando, K., Ndjim, M., Turbant, S., Fontaine, G., Pregoni, G., Dauphinot, L., et al. (2020b). The lipid phosphatase Synaptojanin 1 undergoes a significant alteration in expression and solubility and is associated with brain lesions in Alzheimer's disease. Acta Neuropathol. Commun. 8, 79. doi: 10.1186/s40478-020-00954-1
Ando, K., Tomimura, K., Sazdovitch, V., Suain, V., Yilmaz, Z., Authelet, M., et al. (2016). Level of PICALM, a key component of clathrin-mediated endocytosis, is correlated with levels of phosphotau and autophagy-related proteins and is associated with tau inclusions in AD, PSP and Pick disease. Neurobiol. Dis. 94, 32–43. doi: 10.1016/j.nbd.2016.05.017
Berman, D. E., Dall'armi, C., Voronov, S. V., Mcintire, L. B., Zhang, H., Moore, A. Z., et al. (2008). Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat. Neurosci 11, 547–554. doi: 10.1038/nn.2100
Botte, A., Laine, J., Xicota, L., Heiligenstein, X., Fontaine, G., Kasri, A., et al. (2020). Ultrastructural and dynamic studies of the endosomal compartment in Down syndrome. Acta Neuropathol. Commun. 8, 89. doi: 10.1186/s40478-020-00956-z
Botte, A., and Potier, M. C. (2020). Focusing on cellular biomarkers: the endo-lysosomal pathway in Down syndrome. Prog. Brain Res. 251, 209–243. doi: 10.1016/bs.pbr.2019.10.002
Brion, J. P., Couck, A. M., Passareiro, E., and Flament-Durand, J. (1985). Neurofibrillary tangles of Alzheimer's disease: an immunohistochemical study. J. Submicrosc. Cytol. 17, 89–96.
Butler, M. H., David, C., Ochoa, G. C., Freyberg, Z., Daniell, L., Grabs, D., et al. (1997). Amphiphysin II (SH3P9; BIN1), a member of the amphiphysin/Rvs family, is concentrated in the cortical cytomatrix of axon initial segments and nodes of ranvier in brain and around T tubules in skeletal muscle. J. Cell Biol. 137, 1355–1367. doi: 10.1083/jcb.137.6.1355
Calafate, S., Flavin, W., Verstreken, P., and Moechars, D. (2016). Loss of Bin1 promotes the propagation of tau pathology. Cell Rep. 17, 931–940. doi: 10.1016/j.celrep.2016.09.063
Cataldo, A. M., Hamilton, D. J., Barnett, J. L., Paskevich, P. A., and Nixon, R. A. (1996). Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J. Neurosci. 16, 186–199. doi: 10.1523/JNEUROSCI.16-01-00186.1996
Chang-Ileto, B., Frere, S. G., Chan, R. B., Voronov, S. V., Roux, A., and Di Paolo, G. (2011). Synaptojanin 1-mediated PI(4,5)P2 hydrolysis is modulated by membrane curvature and facilitates membrane fission. Dev. Cell 20, 206–218. doi: 10.1016/j.devcel.2010.12.008
Chapuis, J., Hansmannel, F., Gistelinck, M., Mounier, A., Van Cauwenberghe, C., Kolen, K. V., et al. (2013). Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234. doi: 10.1038/mp.2013.1
Cremona, O., Di Paolo, G., Wenk, M. R., Luthi, A., Kim, W. T., Takei, K., et al. (1999). Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179–188. doi: 10.1016/S0092-8674(00)81649-9
Cruchaga, C., Karch, C. M., Jin, S. C., Benitez, B. A., Cai, Y., Guerreiro, R., et al. (2014). Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature 505, 550–554. doi: 10.1038/nature12825
Deneka, A., Korobeynikov, V., and Golemis, E. A. (2015). Embryonal Fyn-associated substrate (EFS) and CASS4: the lesser-known CAS protein family members. Gene 570, 25–35. doi: 10.1016/j.gene.2015.06.062
Di Paolo, G., and De Camilli, P. (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. doi: 10.1038/nature05185
Dourlen, P., Kilinc, D., Malmanche, N., Chapuis, J., and Lambert, J. C. (2019). The new genetic landscape of Alzheimer's disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 138, 221–236. doi: 10.1007/s00401-019-02004-0
Drager, N. M., Nachman, E., Winterhoff, M., Bruhmann, S., Shah, P., Katsinelos, T., et al. (2017). Bin1 directly remodels actin dynamics through its BAR domain. EMBO Rep. 18, 2051–2066. doi: 10.15252/embr.201744137
Elie, A., Prezel, E., Guerin, C., Denarier, E., Ramirez-Rios, S., Serre, L., et al. (2015). Tau co-organizes dynamic microtubule and actin networks. Sci. Rep. 5:9964. doi: 10.1038/srep09964
Elong Edimo, W., Ghosh, S., Derua, R., Janssens, V., Waelkens, E., Vanderwinden, J. M., et al. (2016). SHIP2 controls plasma membrane PI(4,5)P2 thereby participating in the control of cell migration in 1321 N1 glioblastoma cells. J. Cell Sci. 129, 1101–1114. doi: 10.1242/jcs.179663
Evans, L. D., Wassmer, T., Fraser, G., Smith, J., Perkinton, M., Billinton, A., et al. (2018). Extracellular monomeric and aggregated tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep. 22, 3612–3624. doi: 10.1016/j.celrep.2018.03.021
Fasano, D., Parisi, S., Pierantoni, G. M., De Rosa, A., Picillo, M., Amodio, G., et al. (2018). Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 9, 385. doi: 10.1038/s41419-018-0410-7
Fazzari, P., Horre, K., Arranz, A. M., Frigerio, C. S., Saito, T., Saido, T. C., et al. (2017). PLD3 gene and processing of APP. Nature 541, E1–E2. doi: 10.1038/nature21030
Ferguson, S. M., and De Camilli, P. (2012). Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13, 75–88. doi: 10.1038/nrm3266
Gatz, M., Reynolds, C. A., Fratiglioni, L., Johansson, B., Mortimer, J. A., Berg, S., et al. (2006). Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174. doi: 10.1001/archpsyc.63.2.168
Ghosh, S., Scozzaro, S., Ramos, A. R., Delcambre, S., Chevalier, C., Krejci, P., et al. (2018). Inhibition of SHIP2 activity inhibits cell migration and could prevent metastasis in breast cancer cells. J. Cell Sci. 131:jcs216408. doi: 10.1242/jcs.216408
Glenner, G. G., and Wong, C. W. (1984). Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/S0006-291X(84)80190-4
He, Y., Wei, M., Wu, Y., Qin, H., Li, W., Ma, X., et al. (2019). Amyloid beta oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nat. Commun. 10, 1193. doi: 10.1038/s41467-019-09114-z
He, Z., Guo, J. L., Mcbride, J. D., Narasimhan, S., Kim, H., Changolkar, L., et al. (2018). Amyloid-beta plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 24, 29–38. doi: 10.1038/nm.4443
Hwang, S. C., Jhon, D. Y., Bae, Y. S., Kim, J. H., and Rhee, S. G. (1996). Activation of phospholipase C-gamma by the concerted action of tau proteins and arachidonic acid. J. Biol. Chem. 271, 18342–18349. doi: 10.1074/jbc.271.31.18342
Jope, R. S., Song, L., Li, X., and Powers, R. (1994). Impaired phosphoinositide hydrolysis in Alzheimer's disease brain. Neurobiol. Aging 15, 221–226. doi: 10.1016/0197-4580(94)90116-3
Kajiho, H., Saito, K., Tsujita, K., Kontani, K., Araki, Y., Kurosu, H., et al. (2003). RIN3: a novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J. Cell Sci. 116, 4159–4168. doi: 10.1242/jcs.00718
Khatoon, S., Grundke-Iqbal, I., and Iqbal, K. (1992). Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein. J. Neurochem. 59, 750–753. doi: 10.1111/j.1471-4159.1992.tb09432.x
Knupp, A., Mishra, S., Martinez, R., Braggin, J. E., Szabo, M., Kinoshita, C., et al. (2020). Depletion of the AD risk gene SORL1 selectively impairs neuronal endosomal traffic independent of amyloidogenic APP processing. Cell Rep. 31, 107719. doi: 10.1016/j.celrep.2020.107719
Koo, E. H., and Squazzo, S. L. (1994). Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389.
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lehtonen, S., Zhao, F., and Lehtonen, E. (2002). CD2-associated protein directly interacts with the actin cytoskeleton. Am. J. Physiol. Renal Physiol. 283, F734–743. doi: 10.1152/ajprenal.00312.2001
Leroy, K., Yilmaz, Z., and Brion, J. P. (2007). Increased level of active GSK-3beta in Alzheimer's disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 33, 43–55. doi: 10.1111/j.1365-2990.2006.00795.x
Lesourne, R., Fridman, W. H., and Daeron, M. (2005). Dynamic interactions of Fc gamma receptor IIB with filamin-bound SHIP1 amplify filamentous actin-dependent negative regulation of Fc epsilon receptor I signaling. J. Immunol. 174, 1365–1373. doi: 10.4049/jimmunol.174.3.1365
Lynch, D. K., Winata, S. C., Lyons, R. J., Hughes, W. E., Lehrbach, G. M., Wasinger, V., et al. (2003). A Cortactin-CD2-associated protein (CD2AP) complex provides a novel link between epidermal growth factor receptor endocytosis and the actin cytoskeleton. J. Biol. Chem. 278, 21805–21813. doi: 10.1074/jbc.M211407200
Miller, S. E., Mathiasen, S., Bright, N. A., Pierre, F., Kelly, B. T., Kladt, N., et al. (2015). CALM regulates clathrin-coated vesicle size and maturation by directly sensing and driving membrane curvature. Dev. Cell 33, 163–175. doi: 10.1016/j.devcel.2015.03.002
Miranda, A. M., Herman, M., Cheng, R., Nahmani, E., Barrett, G., Micevska, E., et al. (2018). Excess synaptojanin 1 contributes to place cell dysfunction and memory deficits in the aging hippocampus in three types of Alzheimer's disease. Cell Rep. 23, 2967–2975. doi: 10.1016/j.celrep.2018.05.011
Moreau, K., Fleming, A., Imarisio, S., Lopez Ramirez, A., Mercer, J. L., Jimenez-Sanchez, M., et al. (2014). PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 5:4998. doi: 10.1038/ncomms5998
Morel, E., Chamoun, Z., Lasiecka, Z. M., Chan, R. B., Williamson, R. L., Vetanovetz, C., et al. (2013). Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 4:2250. doi: 10.1038/ncomms3250
Mostafavi, S., Gaiteri, C., Sullivan, S. E., White, C. C., Tasaki, S., Xu, J., et al. (2018). A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat. Neurosci. 21, 811–819. doi: 10.1038/s41593-018-0154-9
Mudher, A., Colin, M., Dujardin, S., Medina, M., Dewachter, I., Alavi Naini, S. M., et al. (2017). What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 5, 99. doi: 10.1186/s40478-017-0488-7
Nakatsu, F., Perera, R. M., Lucast, L., Zoncu, R., Domin, J., Gertler, F. B., et al. (2010). The inositol 5-phosphatase SHIP2 regulates endocytic clathrin-coated pit dynamics. J. Cell Biol. 190, 307–315. doi: 10.1083/jcb.201005018
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Nishikawa, T., Takahashi, T., Nakamori, M., Hosomi, N., Maruyama, H., Miyazaki, Y., et al. (2016). The identification of raft-derived tau-associated vesicles that are incorporated into immature tangles and paired helical filaments. Neuropathol. Appl. Neurobiol. 42, 639–653. doi: 10.1111/nan.12288
Nishikawa, T., Takahashi, T., Nakamori, M., Yamazaki, Y., Kurashige, T., Nagano, Y., et al. (2014). Phosphatidylinositol-4,5-bisphosphate is enriched in granulovacuolar degeneration bodies and neurofibrillary tangles. Neuropathol. Appl. Neurobiol. 40, 489–501. doi: 10.1111/nan.12056
Patrick, G. N., Zukerberg, L., Nikolic, M., De La Monte, S., Dikkes, P., and Tsai, L. H. (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622. doi: 10.1038/45159
Perdigao, C., Barata, M. A., Araujo, M. N., Mirfakhar, F. S., Castanheira, J., and Guimas Almeida, C. (2020). Intracellular trafficking mechanisms of synaptic dysfunction in Alzheimer's disease. Front. Cell. Neurosci. 14:72. doi: 10.3389/fncel.2020.00072
Puangmalai, N., Bhatt, N., Montalbano, M., Sengupta, U., Gaikwad, S., Ventura, F., et al. (2020). Internalization mechanisms of brain-derived tau oligomers from patients with Alzheimer's disease, progressive supranuclear palsy and dementia with Lewy bodies. Cell Death Dis. 11, 314. doi: 10.1038/s41419-020-2503-3
Ramos, A. R., Ghosh, S., and Erneux, C. (2019). The impact of phosphoinositide 5-phosphatases on phosphoinositides in cell function and human disease. J. Lipid Res. 60, 276–286. doi: 10.1194/jlr.R087908
Rauch, J. N., Luna, G., Guzman, E., Audouard, M., Challis, C., Sibih, Y. E., et al. (2020). LRP1 is a master regulator of tau uptake and spread. Nature 580, 381–385. doi: 10.1038/s41586-020-2156-5
Reynolds, C. H., Garwood, C. J., Wray, S., Price, C., Kellie, S., Perera, T., et al. (2008). Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J. Biol. Chem. 283, 18177–18186. doi: 10.1074/jbc.M709715200
Sakisaka, T., Itoh, T., Miura, K., and Takenawa, T. (1997). Phosphatidylinositol 4,5-bisphosphate phosphatase regulates the rearrangement of actin filaments. Mol. Cell. Biol. 17, 3841–3849. doi: 10.1128/MCB.17.7.3841
Sartori, M., Mendes, T., Desai, S., Lasorsa, A., Herledan, A., Malmanche, N., et al. (2019). BIN1 recovers tauopathy-induced long-term memory deficits in mice and interacts with Tau through Thr(348) phosphorylation. Acta Neuropathol. 138, 631–652. doi: 10.1007/s00401-019-02017-9
Shen, R., Zhao, X., He, L., Ding, Y., Xu, W., Lin, S., et al. (2020). Upregulation of RIN3 induces endosomal dysfunction in Alzheimer's disease. Transl. Neurodegener. 9, 26. doi: 10.1186/s40035-020-00206-1
Stokes, C. E., and Hawthorne, J. N. (1987). Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. J. Neurochem. 48, 1018–1021. doi: 10.1111/j.1471-4159.1987.tb05619.x
Surridge, C. D., and Burns, R. G. (1994). The difference in the binding of phosphatidylinositol distinguishes MAP2 from MAP2C and Tau. Biochemistry 33, 8051–8057. doi: 10.1021/bi00192a009
Talaga, D., Smeralda, W., Lescos, L., Hunel, J., Lepejova-Caudy, N., Cullin, C., et al. (2018). PIP2 phospholipid-induced aggregation of tau filaments probed by tip-enhanced Raman spectroscopy. Angew. Chem. Int. Ed. Engl. 57, 15738–15742. doi: 10.1002/anie.201809636
Tang, V. W., and Brieher, W. M. (2013). FSGS3/CD2AP is a barbed-end capping protein that stabilizes actin and strengthens adherens junctions. J. Cell Biol. 203, 815–833. doi: 10.1083/jcb.201304143
Tebar, F., Bohlander, S. K., and Sorkin, A. (1999). Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol. Biol. Cell 10, 2687–2702. doi: 10.1091/mbc.10.8.2687
Ubelmann, F., Burrinha, T., Salavessa, L., Gomes, R., Ferreira, C., Moreno, N., et al. (2017). Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 18, 102–122. doi: 10.15252/embr.201642738
Van Acker, Z. P., Bretou, M., and Annaert, W. (2019). Endo-lysosomal dysregulations and late-onset Alzheimer's disease: impact of genetic risk factors. Mol. Neurodegener. 14, 20. doi: 10.1186/s13024-019-0323-7
Vanhauwaert, R., Kuenen, S., Masius, R., Bademosi, A., Manetsberger, J., Schoovaerts, N., et al. (2017). The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 36, 1392–1411. doi: 10.15252/embj.201695773
Vergara, C., Houben, S., Suain, V., Yilmaz, Z., De Decker, R., Vanden Dries, V., et al. (2019). Amyloid-beta pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 137, 397–412. doi: 10.1007/s00401-018-1953-5
Wallroth, A., and Haucke, V. (2018). Phosphoinositide conversion in endocytosis and the endolysosomal system. J. Biol. Chem. 293, 1526–1535. doi: 10.1074/jbc.R117.000629
Wang, H., Lo, W. T., and Haucke, V. (2019). Phosphoinositide switches in endocytosis and in the endolysosomal system. Curr. Opin. Cell Biol. 59, 50–57. doi: 10.1016/j.ceb.2019.03.011
Wang, Y., and Mandelkow, E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 22–35. doi: 10.1038/nrn.2015.1
Willnow, T. E., Petersen, C. M., and Nykjaer, A. (2008). VPS10P-domain receptors - regulators of neuronal viability and function. Nat. Rev. Neurosci. 9, 899–909. doi: 10.1038/nrn2516
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.M112.394528
Keywords: Alzheimer's disease, endocytosis, genome-wide association study, phosphoinositides, tau, amyloid ß
Citation: Ando K, Houben S, Homa M, de Fisenne M-A, Potier M-C, Erneux C, Brion J-P and Leroy K (2021) Alzheimer's Disease: Tau Pathology and Dysfunction of Endocytosis. Front. Mol. Neurosci. 13:583755. doi: 10.3389/fnmol.2020.583755
Received: 15 July 2020; Accepted: 22 December 2020;
Published: 22 January 2021.
Edited by:
Isabel Lastres-Becker, Autonomous University of Madrid, SpainReviewed by:
Cláudia Guimas Almeida, New University of Lisbon, PortugalFrancisca C. Bronfman, Andres Bello University, Chile
Maria Bullido, Autonomous University of Madrid, Spain
Copyright © 2021 Ando, Houben, Homa, de Fisenne, Potier, Erneux, Brion and Leroy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kunie Ando, S3VuaWUuQW5kb0B1bGIuYWMuYmU=
†These authors have contributed equally to this work