 Yuan Zhang
Yuan Zhang Lili Qian1
Lili Qian1 Ying Liu
Ying Liu- 1Institute for Translational Medicine, The Affiliated Hospital of Qingdao University, Qingdao University, Qingdao, China
- 2Institute of Translational Medicine, The Affiliated Hospital of Hangzhou Normal University, Hangzhou, China
- 3School of Basic Medical Sciences, Qingdao University, Qingdao, China
- 4Institute of Biomedical Research, School for Life Sciences, Shandong University of Technology, Zibo, China
Background: Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disease. The characteristic pathologies include extracellular senile plaques formed by β-amyloid protein deposition, neurofibrillary tangles formed by hyperphosphorylation of tau protein, and neuronal loss with glial cell hyperplasia. Circular RNAs (circRNAs) are rich in miRNA-binding sites (miRNA response elements, MREs), which serve as miRNA sponges or competitive endogenous RNAs (ceRNAs). Although several research groups have identified dysregulated circRNAs in the cerebral cortex of SAMP8 mice or APP/PS1 mice using deep RNA-seq analysis, we need to further explore circRNA expression patterns, targets, functions and the signaling pathways involved in the pathogenesis of AD and in particular the hippocampal circRNA expression profiles in AD.
Methods: We used deep RNA sequencing to investigate circRNA-ceRNA network patterns in the hippocampus of APP/PS1 mice.
Results: In our study, 70 dysregulated circRNAs, 39 dysregulated miRNAs and 121 dysregulated mRNAs were identified between the APP/PS1 group and the wild-type group at 8 months in the hippocampus of the mice. Through correlation analysis, we identified 11 dysregulated circRNAs, 7 dysregulated miRNAs and 8 dysregulated mRNAs forming 16 relationships in the circRNA-miRNA-mRNA regulatory network. Gene ontology (GO) analysis indicated that the dysregulated circRNAs were most enriched in biological metabolic processes. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed that the dysregulation of circRNAs was enriched in the cGMP-PKG signaling pathway, cAMP signaling pathway, Hippo signaling pathway, platelet activation, long-term potentiation and axon guidance. In addition, our findings preliminarily verified that the novel_circ_0003012/mmu-miR-298-3p/Smoc2 signaling axis may regulate the pathophysiology of AD by affecting the cGMP-PKG signaling pathway.
Conclusions: These newly identified circRNAs in networks and signaling pathways reveal potential diagnostic or therapeutic targets for AD.
Introduction
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disease and is the most common type of senile dementia (Tiwari et al., 2019). The main characteristics are memory impairment, cognitive decline, personality change and language impairment, which seriously affect people’s daily lives. However, the pathogenesis of AD has not been fully elucidated. The characteristic pathologies include extracellular senile plaques formed by β-amyloid protein deposition, neurofibrillary tangles formed by hyperphosphorylation of the tau protein, and neuronal loss with glial cell hyperplasia (Wang et al., 2014; Gouras et al., 2015; Wilkins and Swerdlow, 2017).
Circular RNA (circRNA) is a non-coding RNA with a unique covalent closed loop structure. CircRNAs are rich in miRNA-binding sites (miRNA response elements, MREs), which serve as miRNA sponges or competitive endogenous RNAs (ceRNAs) (Lei et al., 2018; Kristensen et al., 2019). Currently, several studies have shown that circRNAs play an important role in the regulation of neurodegenerative diseases via their interaction with disease-associated miRNAs (D’Ambra et al., 2019).
In previous studies, several research groups have identified dysregulated circRNAs in the cerebral cortex of SAMP8 mice or APP/PS1 mice using deep RNA-seq analysis (Zhang et al., 2017; Ma et al., 2019). Other groups have also identified dysregulated circRNAs in the hippocampal tissues of an AD mouse model by circRNA microarray (Huang et al., 2018; Wang et al., 2018). Currently, it is believed that the nerve loss caused by the development of AD is mainly in the cortex and hippocampus. The hippocampus is very important for learning and memory. Changes in the function and structure of the hippocampus are critical for learning and memory, such as long-term potentiation (LTP) and synaptic remodeling (Matsuzaki et al., 2004; Mu and Gage, 2011). Several key molecules influence the generation of new hippocampal neurons in AD, and significant changes in neurogenesis occur earlier than the onset of hallmark lesions or neuronal impairment (Lazarov and Marr, 2010).
Despite these findings, we need to further explore the expression patterns, targets, and functions of circRNAs and the signaling pathways involved in the pathogenesis of AD. Therefore, further research is of great importance. Here, we detected dysregulation of the circRNA-ceRNA profile in the hippocampus of APP/PS1 mice using deep RNA-seq analysis. We performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses to predict the biological roles and potential signaling pathways of these differentially expressed circRNAs. Furthermore, we conducted circRNA-ceRNA network pattern analysis to further explore the potential roles of dysregulated circRNAs in AD pathogenesis. Taken together, our findings may promote a better understanding of the role of circRNAs in the neuropathogenesis of AD.
Materials and Methods
Animals
Eight-month-old APP/PS1 mice and their age-matched wild-type mice were purchased from Model Animal Research Institute of Myhalic (Wuhan, China). The mice were housed two per cage under the standard conditions (12 h light/dark cycle at 25°C and 50 ± 10% relative humidity). We randomly selected nine animals from each group, three animals for RNA-seq, and six animals for Real-time qPCR. Animals administered general anesthesia and then collected hippocampal tissue. Animal care and experimental procedures were implemented according to the document “Guidance Suggestions for Caring for Laboratory Animals” produced by the Ministry of Science and Technology of China in 2006.
RNA Extraction and Qualification
Total RNA was extracted from each hippocampal tissue sample by RNAprep Pure Tissue Kit (TIANGEN BIOTECH, Beijing, China) in accordance with the manufacturer’s instructions.
Using 1% agarose gels to monitor RNA degradation and contamination. Then, using the NanoPhotometer® spectrophotometer to check the RNA purity (IMPLEN, CA, United States) and using Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer to measure the RNA concentration (Life Technologies, CA, United States). Finally, using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system to assess the RNA integrity (Agilent Technologies, CA, United States).
RNA-Seq
Details of the mRNA-seq, miRNA-seq, and circRNA-seq methods are described in Supplementary Materials.
Real-Time qPCR
To validate the RNA-Seq data, we randomly selected 3 of circRNA, miRNA and mRNA for qRT-PCR analysis, respectively. Total RNA was extracted from each hippocampal tissue sample, and then reverse-transcribed into cDNA using PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara, Dalian, China) according to the manufacturer’s instruction.
Real-time quantitative PCR (RT-qPCR) was performed using the SYBR® Premix Ex TaqTM II (Tli RNase H Plus) Kit with a Bio-Rad CFX Manager 3.1 real-time PCR system (CFX96TM Real-Time PCR, Bio-Rad, United States). The relative circRNA and mRNA expression levels were calculated using the 2–ΔΔCt method and were normalized to GAPDH as an endogenous reference transcript. miRNA expression levels were normalized to that of U6. The specific primers for each gene are listed in Supplementary Table 1. Data shown represent the means of three experiments.
GO Annotations and KEGG Pathway Analyses
Gene Ontology (GO) enrichment analysis of differentially expressed genes was conducted by clusterProfiler, an R package for functional classification and enrichment of gene clusters using hypergeometric distribution. KEGG is a database resource for understanding high-level functions and utilities of the biological system1. We used clusterProfiler R package to test the statistical enrichment of aberrantly expressed circRNAs in KEGG pathways. GO and KEGG terms with corrected P-value < 0.05 were considered significantly enrichment of aberrantly expressed circRNAs.
Annotation for CircRNA-miRNA-mRNA Interaction
We have selected the differentially expressed circRNA, miRNA and mRNA that have been identified. CircRNA-miRNA interactions and miRNA-mRNA interactions were predicted with Arraystar’s home-made miRNA target prediction software based on TargetScan2 and miRanda3. The circRNA-miRNA-mRNA network covered two cases: upregulated circRNA-downregulated miRNA-upregulated mRNA, and downregulated circRNA-upregulated miRNA-upregulated mRNA. Then, we constructed circRNA-miRNA-mRNA network using the Cytoscape software V2.7.0 (San Diego, CA, United States).
Statistical Analysis
Statistical analyses were performed using SPSS v16.0 software (SPSS, Inc., Chicago, IL, United States). All data were expressed as the mean ± SEM. p < 0.05 was statistically significant.
Results
Overview of CircRNA-Seq
A total of 514,529,568 raw reads were generated, 255,871,400 for wild-type (WT) mice, and 258,658,168 for APP/PS1 mice. Removed poly(N)-containing, low-quality, and adaptor-containing reads from the raw data, then remained 506,341,272 clean reads including 251,602,810 for wild-type and 254,738,462 for APP/PS1 mice. The high-quality clean data were mapped to the mouse reference sequence by Hisat24 and the unmapped reads were subsequently selected (Pertea et al., 2016). circRNAs were detected and identified using find_circ and CIRI, 5,683 circRNAs were detected (Memczak et al., 2013; Gao et al., 2015). These circRNAs were used for subsequent analyses.
Overview of miRNA-Seq
A total of 90,306,346 raw reads were generated, 42,973,163 for WT mice, and 47,333,183 for APP/PS1 mice. After removal of low-quality and adaptor sequences, 41,501,993 clean reads for WT group and 46,256,666 clean reads for APP/PS1 group were remained. The reads we selected are mostly based on the length of 21–22 nt in both groups. These reads were annotated and classified based on previous studies (Ma et al., 2019). Finally, 1,351 matured miRNAs (1,275 known and 76 novel) were detected. These miRNAs were used for the subsequent analysis.
Overview of mRNA-Seq
A total of 267,484,154 raw reads were generated: 135,704,866 for APP/PS1 mice, and 131,779,288 for wild-type mice. After discarding the reads with adapters, poly-N > 10%, and discarding the low-quality reads. 211,535,266 UMI reads were obtained: 107,820,630 for APP/PS1 mice, and 103,714,636 for wild-type mice. The clean reads were mapped to the mouse reference genome, and the Dedup2MappedUMI rates were approximately 80.50% for APP/PS1 mice and 85.08% for wild-type mice. The cufflink results indicated that 57,077 protein-coding transcripts were identified. These mRNAs were used for the subsequent analysis.
Differential Expression Analysis Between APP/PS1 and Wild-Type Mice
We identified 70 significantly aberrantly expressed circRNAs between APP/PS1 mice and wild-type (WT) mice at 8 months in the hippocampus (p < 0.05), of which 44 circRNAs were upregulated and 26 were downregulated (Table 1). We performed cluster analysis on the differential circRNA expression and generated a heatmap to visualize the results of the cluster analysis (Figures 1A,B).
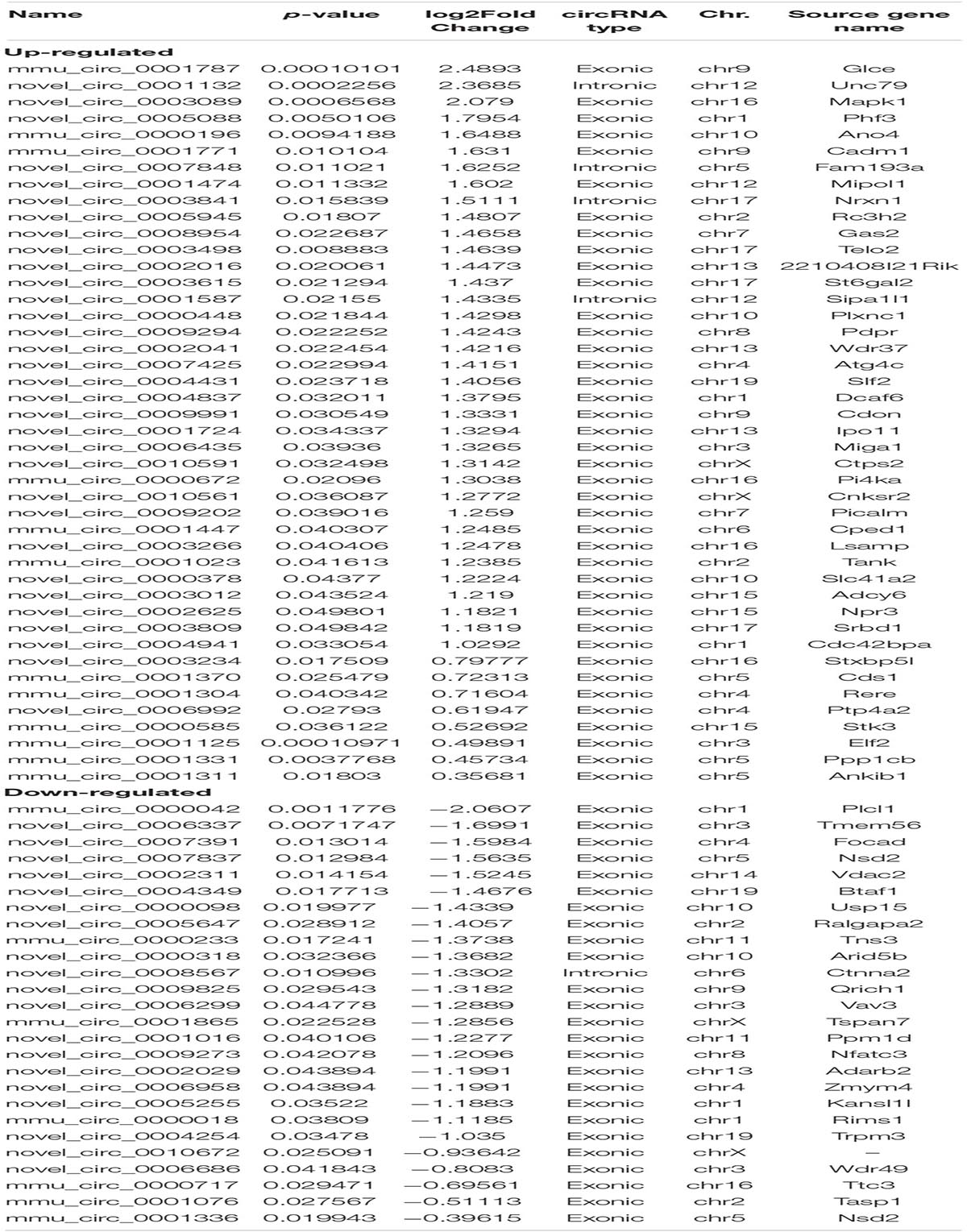
Table 1. Differently expressed circRNAs between APP/PS1 and WT mice.
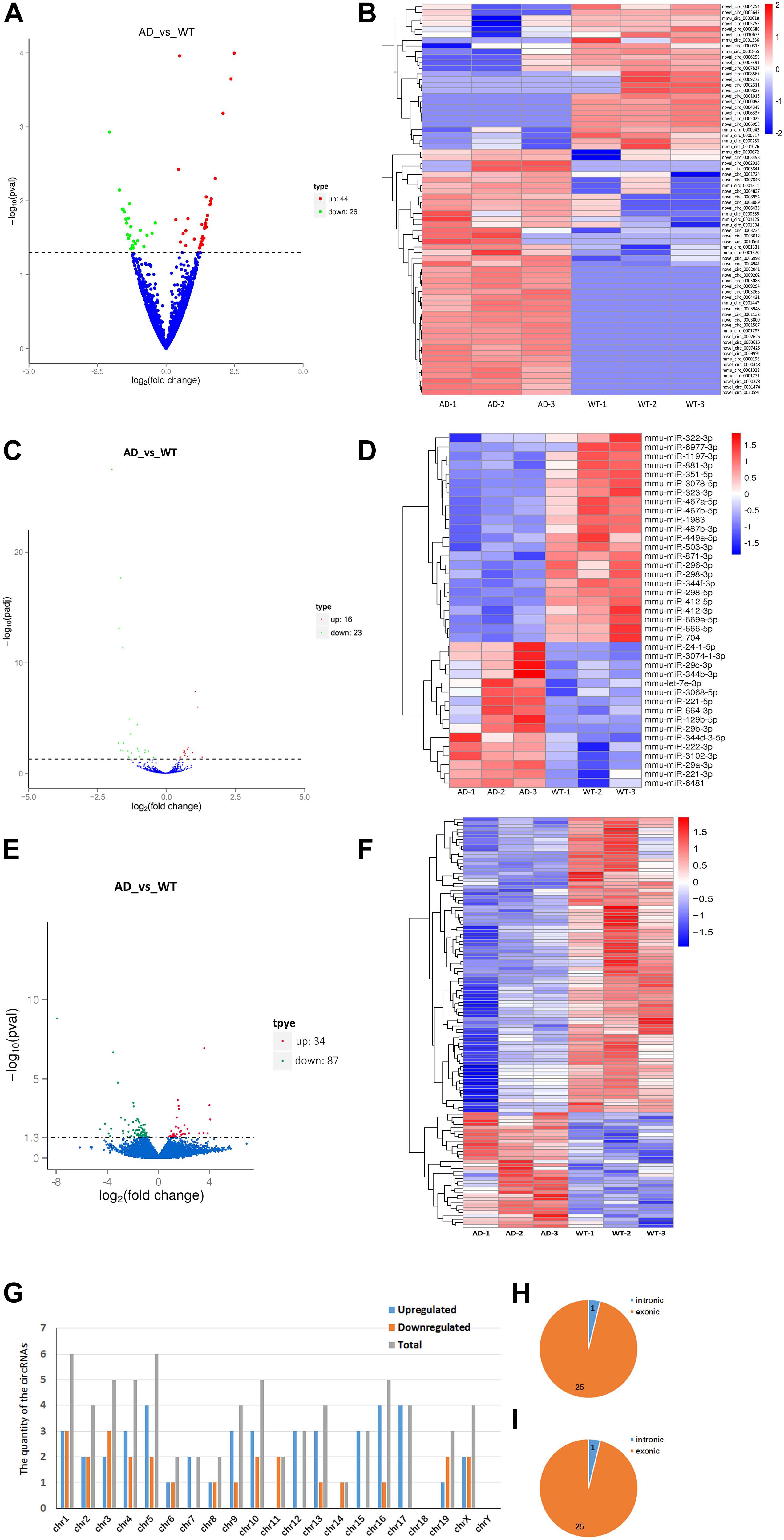
Figure 1. Expression profiles of distinct RNAs. (A,B) Expression profiles of circRNAs. (A) In the volcano plots, green, red, and blue points represent circRNAs that were downregulated, upregulated, and not significantly different in 8-month APP/PS1 mice relative to 8-month WT mice. (B) Cluster analysis of expression of circRNAs. Red and blue: increased and decreased expression, respectively. (C,D) Expression profiles of miRNAs. (C) In the volcano plots, green, red, and blue points represent miRNAs that were downregulated, upregulated, and not significantly different in 8-month APP/PS1 mice relative to 8-month WT mice. (D) Cluster analysis of expression of miRNAs. Red and blue: increased and decreased expression, respectively. (E,F) Expression profiles of mRNAs. (E) In the volcano plots, green, red, and blue points represent mRNAs that were downregulated, upregulated, and not significantly different in 8-month APP/PS1 mice relative to 8-month WT mice. (F) Cluster analysis of expression of mRNAs. Red and blue: increased and decreased expression, respectively. (G–I) Chromosomal distribution of differentially expressed circRNAs in hippocampal of APP/PS1 mice. (G) Chromosomal localization of differentially expressed circRNAs in hippocampal of APP/PS1 mice. (H) Gene localization of upregulated circRNAs in hippocampal of APP/PS1 mice. (I) Gene localization of downregulated circRNAs in hippocampal of APP/PS1 mice.
Next, we identified 39 significantly aberrantly expressed miRNAs between APP/PS1 mice and WT mice (p < 0.05), of which 16 miRNAs were upregulated and 23 were downregulated (Table 2). Cluster analysis and heatmapping were performed to show the results of the differential miRNA expression (Figures 1C,D).
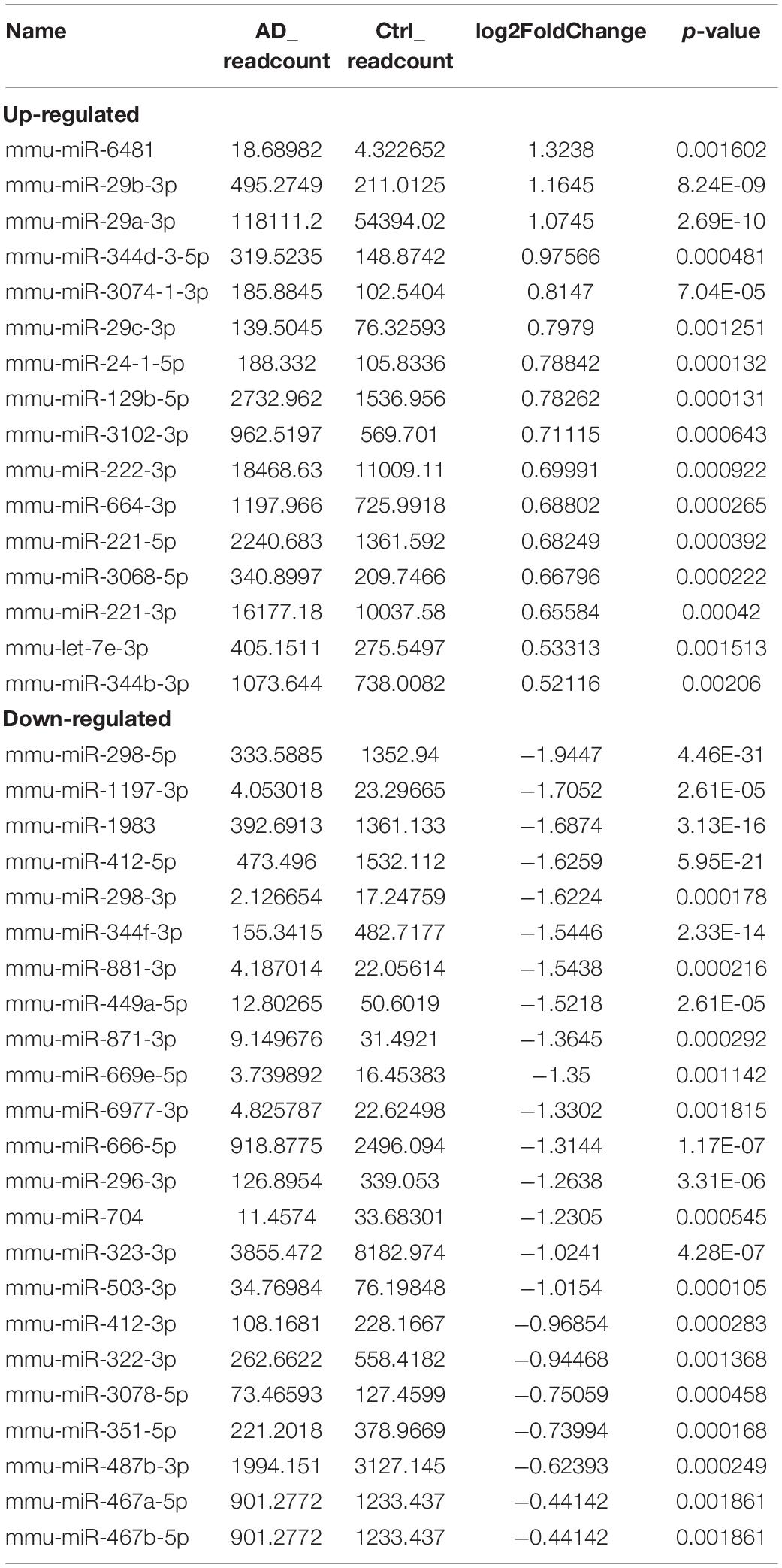
Table 2. Differently expressed miRNAs between APP/PS1 and WT mice.
Finally, we estimated the expression levels of the mRNA transcripts. A total of 121 mRNAs were significantly aberrantly expressed between the APP/PS1 mice and the WT mice (p < 0.05), with 34 upregulated mRNAs and 87 downregulated mRNAs (Table 3). Cluster analysis and heatmapping were performed to show the results of the differential mRNA expression (Figures 1E,F).
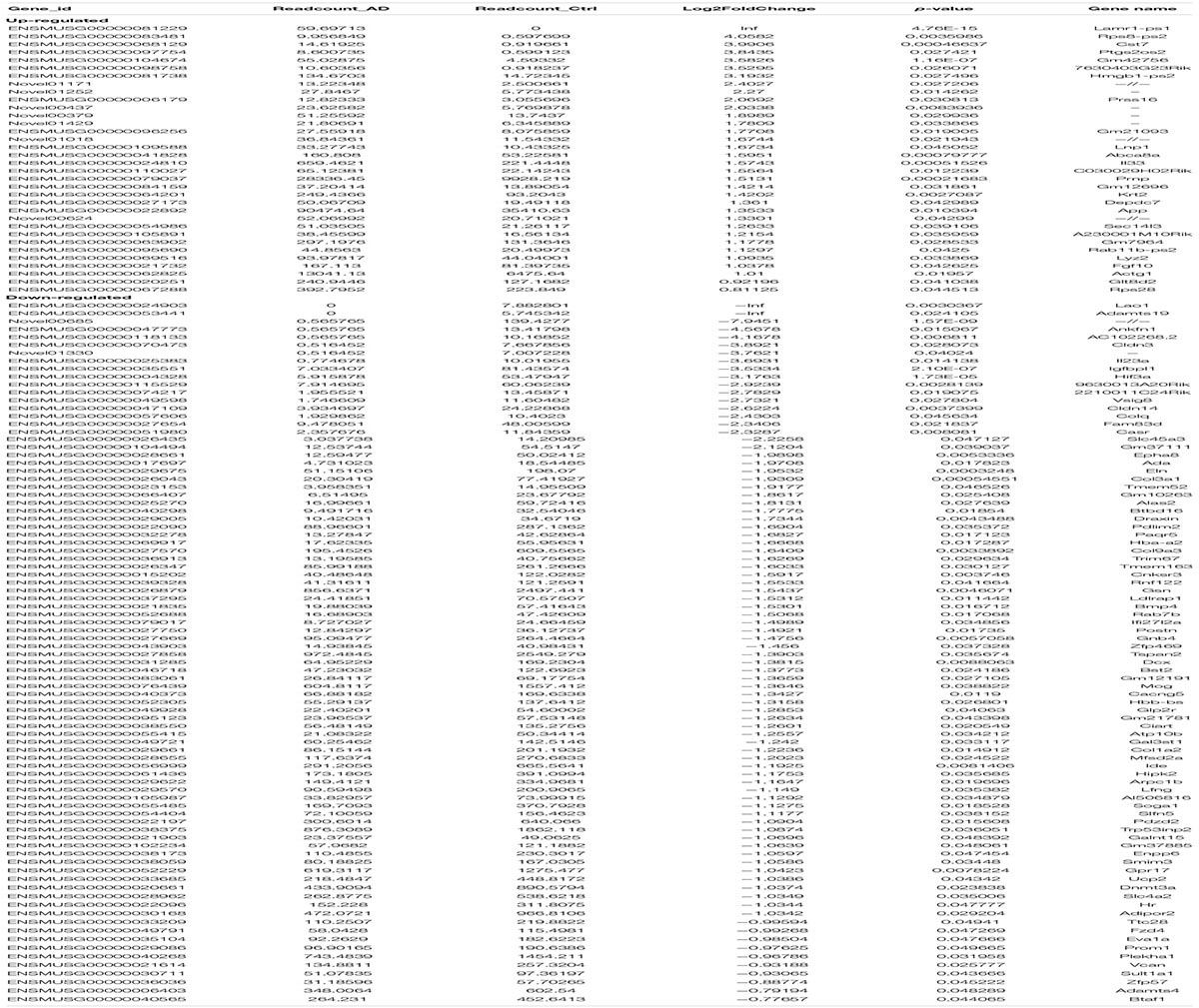
Table 3. Differently expressed mRNAs between APP/PS1 and WT mice.
The data showed that the significantly aberrantly expressed circRNAs were scattered across different chromosomes: the 44 upregulated circRNAs were located on 17 chromosomes, and the 26 downregulated circRNAs were located on 15 chromosomes. The top three chromosomes for the upregulated circRNAs were chromosome (chr.) 5 (4/44), chr. 16 (4/44), and chr. 17 (4/44), while the top two chromosomes for the downregulated circRNAs were chr. 1 (3/26) and chr. 3 (3/26). As for localization of the dysregulated circRNAs, there were 40 exonic and 4 intronic in the upregulated circRNAs and 25 exonic and 1 intronic in the downregulated circRNAs (Figures 1G–I and Table 1).
qPCR Confirmation
We used RT-qPCR to confirm the differentially expressed RNAs in our RNA-seq experiments. We randomly selected three circRNAs, three miRNAs and three mRNAs to perform RT-qPCR. As shown in Figure 2A, all selected transcripts were detected in the hippocampus of the APP/PS1 mice and WT mice and nearly exhibited significant differential expression. In summary, near consistency was observed between the qPCR results and the RNA-seq data.
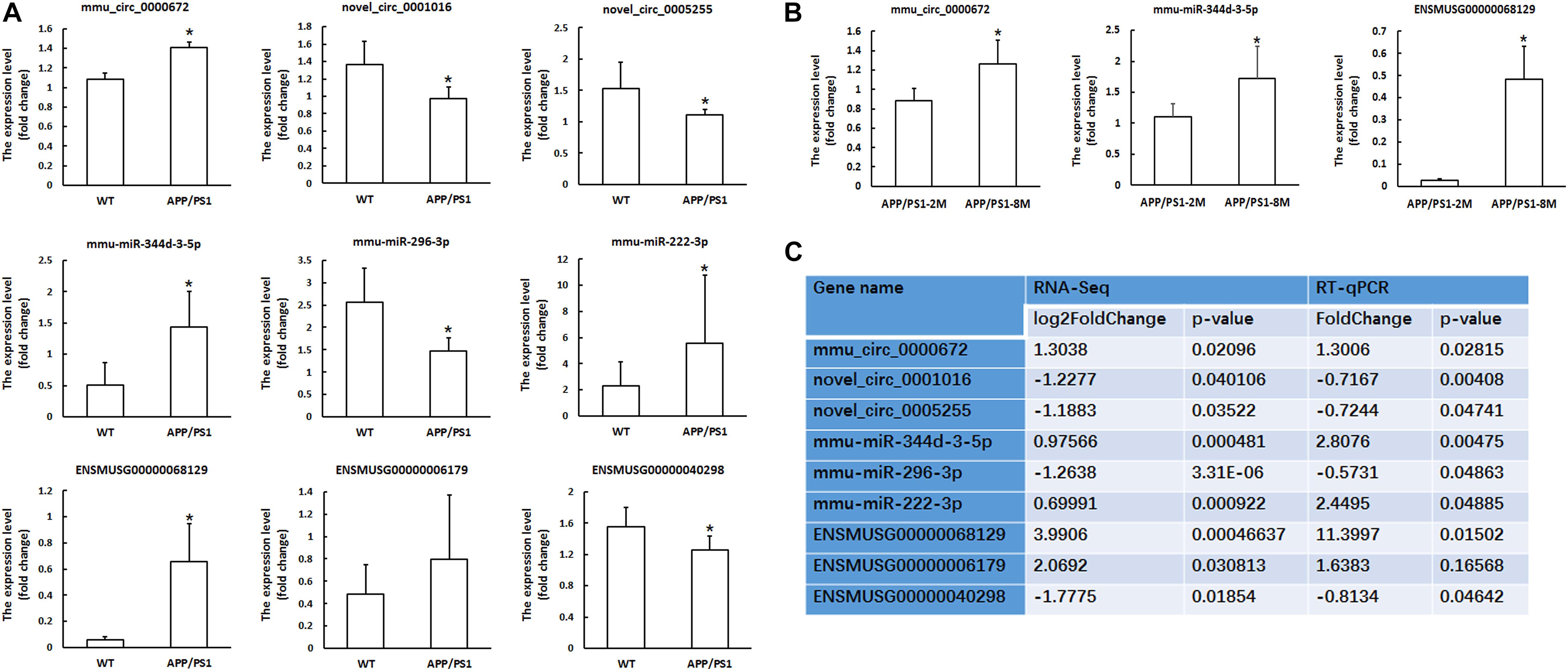
Figure 2. Validation of circRNA, miRNA and mRNAs expression by qPCR. (A) The expression levels of candidate circRNAs, miRNAs and mRNAs for validation by qPCR in hippocampal tissues of 8-month-old APP/PS1 mice and WT mice. (B) The expression levels of candidate circRNAs, miRNAs and mRNAs for validation by qPCR in hippocampal tissues of 8-month-old APP/PS1 mice and 2-month-old APP/PS1 mice. CircRNA and mRNA expression was quantified relative to Gapdh expression level by using the comparative cycle threshold (ΔCT) method. MiRNA expression was quantified relative to U6 expression level by using the comparative cycle threshold (ΔCT) method. Data are presented as means ± SD (n = 6; *p < 0.05). (C) The comparison information of the RNA-Seq and qRT-PCR data of 8-month-old APP/PS1 mice vs. WT mice.
Furthermore, we confirmed the differential expression of circRNAs, miRNAs and mRNAs in 8-month-old APP/PS1 mice relative to 2-month-old APP/PS1 mice. The results showed that mmu_circ_0000672, mmu-miR-344d-3-5p and ENSMUSG00000068129 (Cst7) were significantly different between the hippocampal tissues of 8-month-old APP/PS1 mice and 2-month-old APP/PS1 mice (P < 0.05) (Figure 2B). This result indicated that these genes changed significantly with the age of the APP/PS1 mice.
GO and KEGG Analyses
Gene Ontology (GO) analyses were performed on the circRNAs, and the top highly significantly enriched GO terms of the dysregulated circRNAs on biological process (BP) and molecular function (MF) are shown in Figure 3A. The 5 top terms were phosphorus metabolic process (GO: 0006793), phosphate-containing compound metabolic process (GO: 0006796),
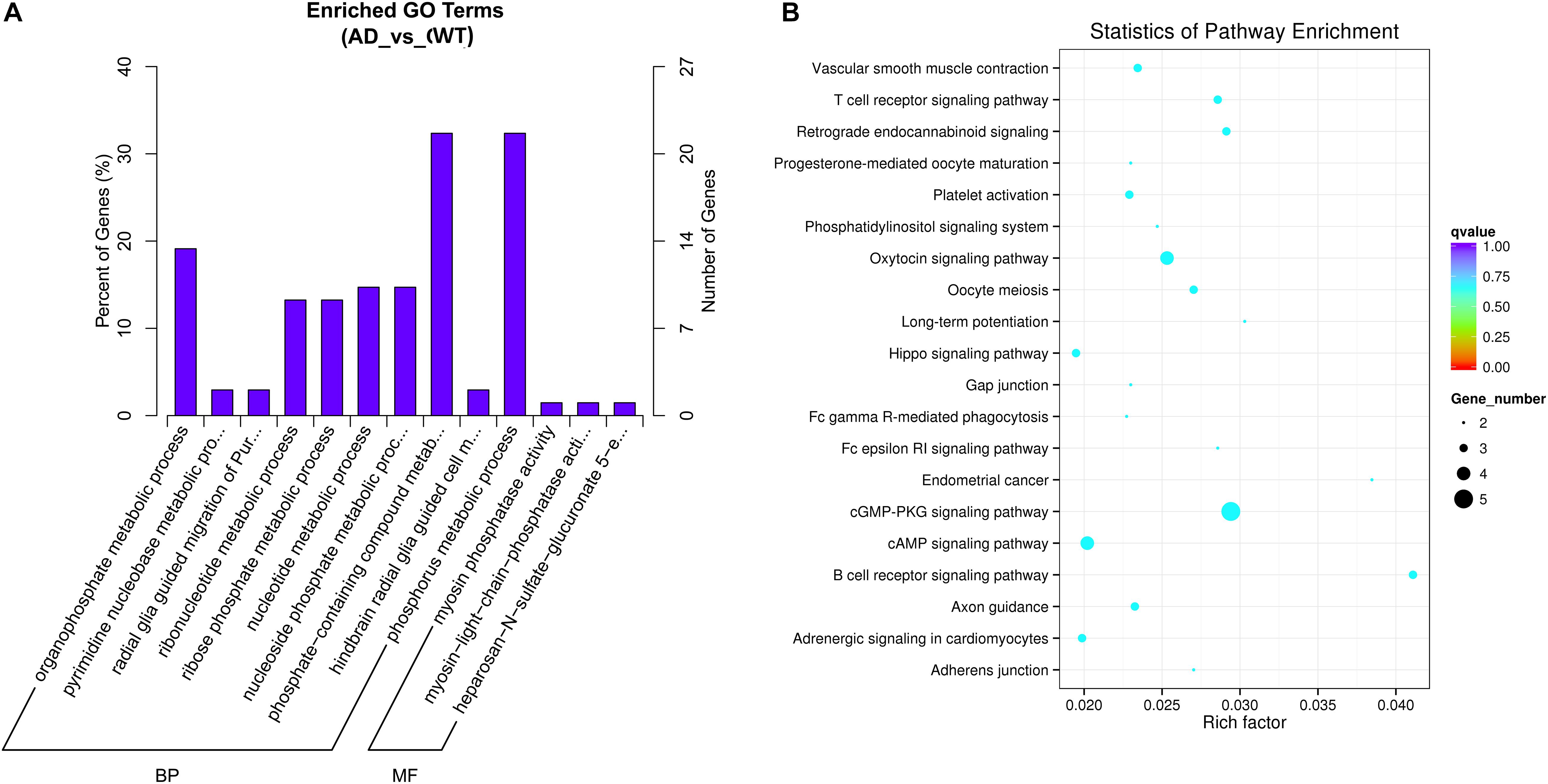
Figure 3. (A) Gene Ontology (GO) Enrichment Annotations of pathological progression of AD: Biological Process (BP) and Molecular Function (MF). (B) Significantly Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG). The aberrantly expressed circRNAs in distinct aspects of AD pathology.
organophosphate metabolic process (GO: 0019637), nucleotide metabolic process (GO: 0009117) and nucleoside phosphate metabolic process (GO: 0006753). Several metabolic pathway-related terms were also observed, such as pyrimidine nucleobase metabolic process (GO: 0006206), ribonucleotide metabolic process (GO: 0009259) and ribose phosphate metabolic process (GO: 0019693). In summary, the pathological progression of AD may be associated with several metabolic pathways regulated by circRNAs.
In addition, we also performed GO analysis of miRNAs and mRNAs. Through GO analysis of miRNAs, we found that the 20 top terms enriched in BP, cellular component (CC) and MF were almost all associated with cellular metabolic process, intracellular organelle/part and binding functions (Supplementary Figure 1A): cellular metabolic process (GO: 0044237), metabolic process (GO: 0008152), cellular macromolecule metabolic process (GO: 0044260), intracellular part (GO: 0044424), intracellular organelle (GO: 0043229), membrane-bounded organelle (GO: 0043227), intracellular membrane-bounded organelle (GO:0043231), protein binding (GO:0005515), and binding (GO: 0005488). Moreover, GO analysis indicated that the most enriched mRNAs correlated with single-organism developmental process (GO: 0044767), developmental process (GO: 0032502), multicellular organismal development (GO: 0007275), anatomical structure development (GO: 0048856), system development (GO: 0048731), gliogenesis (GO:0042063), extracellular region (GO: 0005576), extracellular region part (GO: 0044421), extracellular space (GO: 0005615) and structural molecule activity (GO: 0005198) (Supplementary Figure 2A). This result indicated that the dysregulated mRNAs were mostly enriched in the cellular/organism development process or cell differentiation.
Kyoto Encyclopedia of Genes and Genomes pathway analysis was performed to determine the signaling pathways related to the dysregulated circRNAs. By using the Q-value scale from 0 to 1, the top 20 significantly enriched pathways were identified, as shown in Figure 3B. Specifically, the cGMP-PKG signaling pathway, cAMP signaling pathway, axon guidance, platelet activation, LTP, Hippo signaling pathway and phosphatidylinositol signaling system were demonstrated to be closely related to the onset and development of AD.
Kyoto Encyclopedia of Genes and Genomes pathways were associated with dysregulated miRNAs involved in the MAPK signaling pathway, Ras signaling pathway, endocytosis, focal adhesion, axon guidance, neurotrophin signaling pathway and glycerophospholipid metabolism (Supplementary Figure 1B). KEGG pathway analysis of the dysregulated mRNAs identified enrichment in metabolic pathways, protein digestion and absorption, ribosomes, regulation of actin cytoskeleton, PI3K-Akt signaling pathway, platelet activation, spliceosome, tight junction, and the Hippo signaling pathway (Supplementary Figure 2B).
Construction of the CircRNA-ceRNA Regulatory Networks
CircRNAs have MREs, which can be used as miRNA sponges to competitively bind miRNAs, thereby inhibiting miRNA targets to mRNA and indirectly regulating mRNA expression. Based on ceRNA theory, to search for circRNA-miRNA-gene pairs with the same MREs, circRNA-miRNA-gene pairs were constructed with the circRNA as a decoy, the miRNA as the core, and the mRNA as the target to construct a ceRNA regulatory network. The circRNA-ceRNA network pattern can show the regulation of the circRNA on the related mRNA-encoding genes.
Based on the RNA-seq data, we selected 11 dysregulated circRNAs, 7 dysregulated miRNAs and 8 dysregulated mRNAs, and there were 16 relationships contained in the constructed circRNA-miRNA-mRNA regulatory network (Figure 4 and Table 4). The ceRNA network covered two cases: one was circRNA (7 circRNAs upregulated in APP/PS1 mice)-miRNA (3 miRNAs downregulated in APP/PS1 mice)-mRNA (3 mRNAs upregulated in APP/PS1 mice), and the other was circRNA (4 circRNAs downregulated)-miRNA (4 miRNAs upregulated)-mRNA (5 mRNAs upregulated). These circRNA-miRNA-mRNA interactions may supply a novel perspective for the pathogenesis of AD. We observed that one circRNA could interact with different miRNAs and that one miRNA could be regulated by multiple circRNAs; for example, mmu_circ_0000717 could interact with mmu-miR-222-3p, mmu-miR-221-5p, mmu-miR-3102-3p and mmu-miR-344d-3-5p, and mmu-miR-298-3 could co-associate with mmu_circ_0001370, novel_circ_0007425, and novel_circ_0003012.
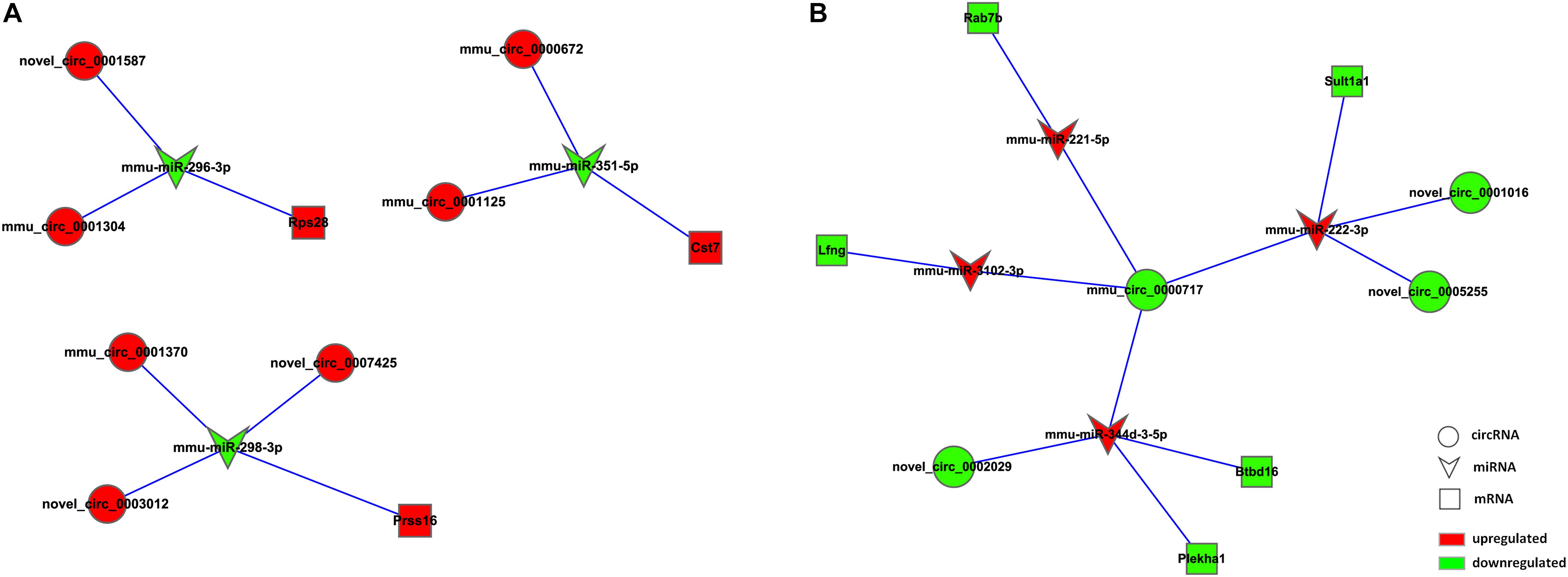
Figure 4. CircRNA-ceRNA network analysis in hippocampal tissue of APP/PS1 Mice. The ceRNA networks were based on circRNA-miRNA and miRNA-mRNA interactions. Circle nodes represent circRNAs, triangle nodes represent miRNAs, and rectangle nodes represent mRNAs. Red represent upregulated, green represent downregulated. (A) circRNA (down in APP/PS1 mice)-miRNA (up in APP/PS1 mice)-mRNA (down in APP/PS1 mice). (B) circRNA (up in APP/PS1 mice)-miRNA (down in APP/PS1 mice)-mRNA (up in APP/PS1 mice).

Table 4. CircRNA-ceRNA networks in AD.
Verification of the Potential Regulatory Mechanism of circRNAs in the Key Signaling Pathways
Through KEGG analysis, we obtained key regulatory signaling pathways, including the cGMP-PKG signaling pathway, cAMP signaling pathway, and Hippo signaling pathway. These pathways have been reported to participate in key regulatory roles in neurodegenerative diseases.
We further explored the regulatory effects of the differential expression of circRNAs on the cGMP-PKG signaling pathway, cAMP signaling pathway, and Hippo signaling pathway. We searched for the differentially expressed circRNAs enriched in the 3 signaling pathways and obtained five circRNAs that might be involved in the cGMP-PKG signaling pathway: novel_circ_0002311, novel_circ_0009273, novel_circ_0003012, novel_circ_0003089, and novel_circ_0001331. Four circRNAs might be involved in the cAMP signaling pathway: novel_circ_0006299, novel_circ_0003012, novel_circ_0003089, and novel_circ_0001331. Two circRNAs might be involved in the Hippo signaling pathway: novel_circ_0008567 and mmu_circ_0000585.
We predicted differentially expressed miRNAs that interact with those circRNAs and found that novel_circ_0009273, novel_circ_0003012, novel_circ_0006299, novel_circ_0008567, and mmu_circ_0000585 could target mmu-miR-3074-1-3p, mmu-miR-298-3p, mmu-miR-296-3p, mmu-miR-298-5p, mmu-miR-3074-1-3p, and mmu-miR-669e-5p, respectively.
Through the predictive analysis of miRNA-mRNA interactions, we identified the downstream target mRNAs that might be regulated and constructed a circRNA-ceRNA network related to the three signaling pathways (Figure 5A and Table 5). Based on the regulatory mechanism of circRNA-ceRNA, we ultimately screened the novel_circ_0003012/mmu-miR-298-3p/Smoc2 signaling axis, which might affect the cGMP-PKG signaling pathway (Figure 5B). We used qPCR to verify the differential expression of these genes, and used WB to verify the level of Smoc2, the results are shown in Figures 5C,D.
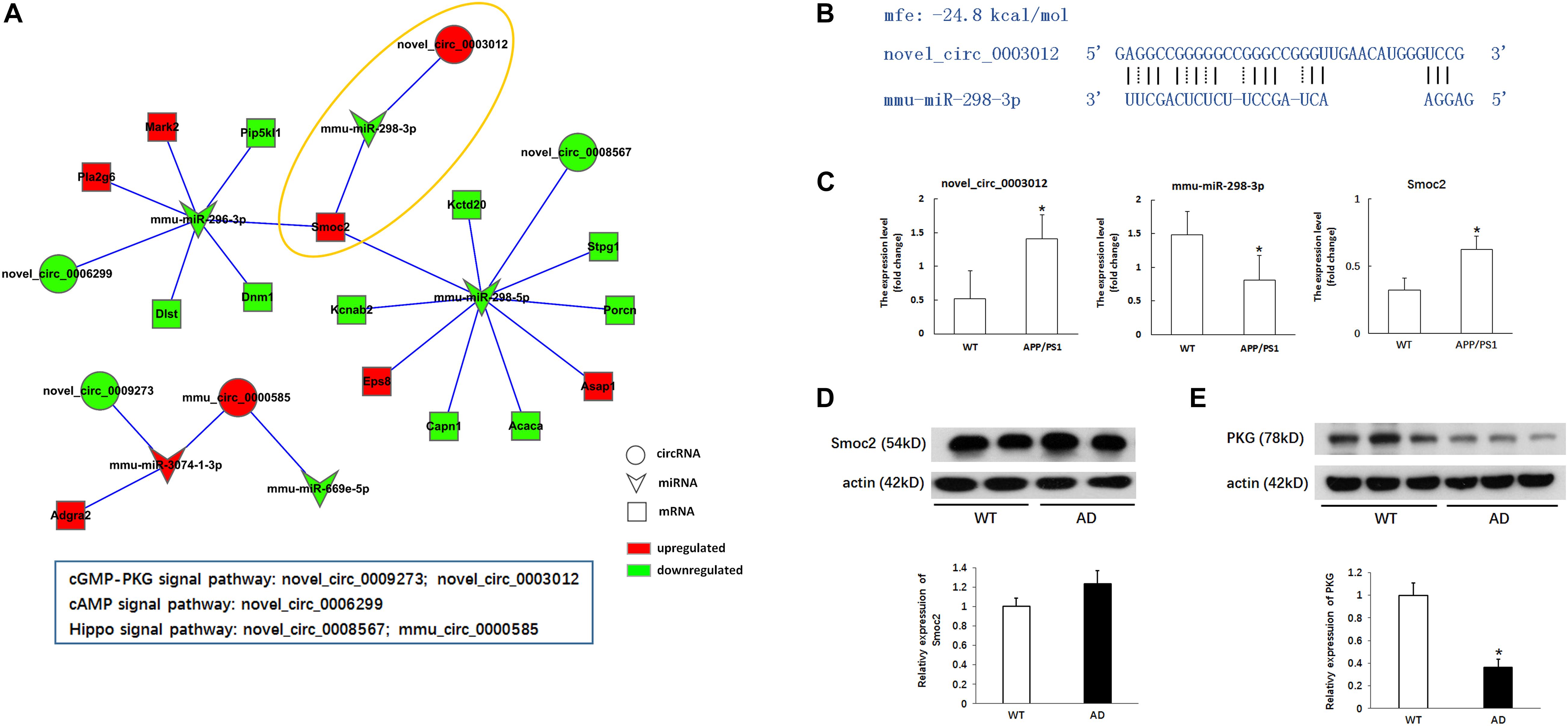
Figure 5. Verification of the potential regulation mechanism of circRNAs in the key signaling pathways. (A) CircRNA-ceRNA network of circRNAs in the key signaling pathways. Circle nodes represent circRNAs, triangle nodes represent miRNAs, and rectangle nodes represent mRNAs. Red represent upregulated, green represent downregulated. (B) The interaction of novel_circ_0003012/mmu-miR-298-3p. (C) The expression levels of novel_circ_0003012, mmu-miR-298-3p and SMOC2 by qPCR in hippocampal tissues of 8-month-old APP/PS1 mice and WT mice. CircRNA and mRNA expression was quantified relative to Gapdh expression level by using the comparative cycle threshold (ΔCT) method. MiRNA expression was quantified relative to U6 expression level by using the comparative cycle threshold (ΔCT) method. Data are presented as means ± SD (n = 6; *p < 0.05). (D) The expression of Smoc2 by Western blot in hippocampal tissues of 8-month-old APP/PS1 mice and WT mice. Data are presented as means ± SD (n = 2). (E) The expression of PKG by Western blot in hippocampal tissues of 8-month-old APP/PS1 mice and WT mice. Data are presented as means ± SD (n = 3; *p < 0.05).
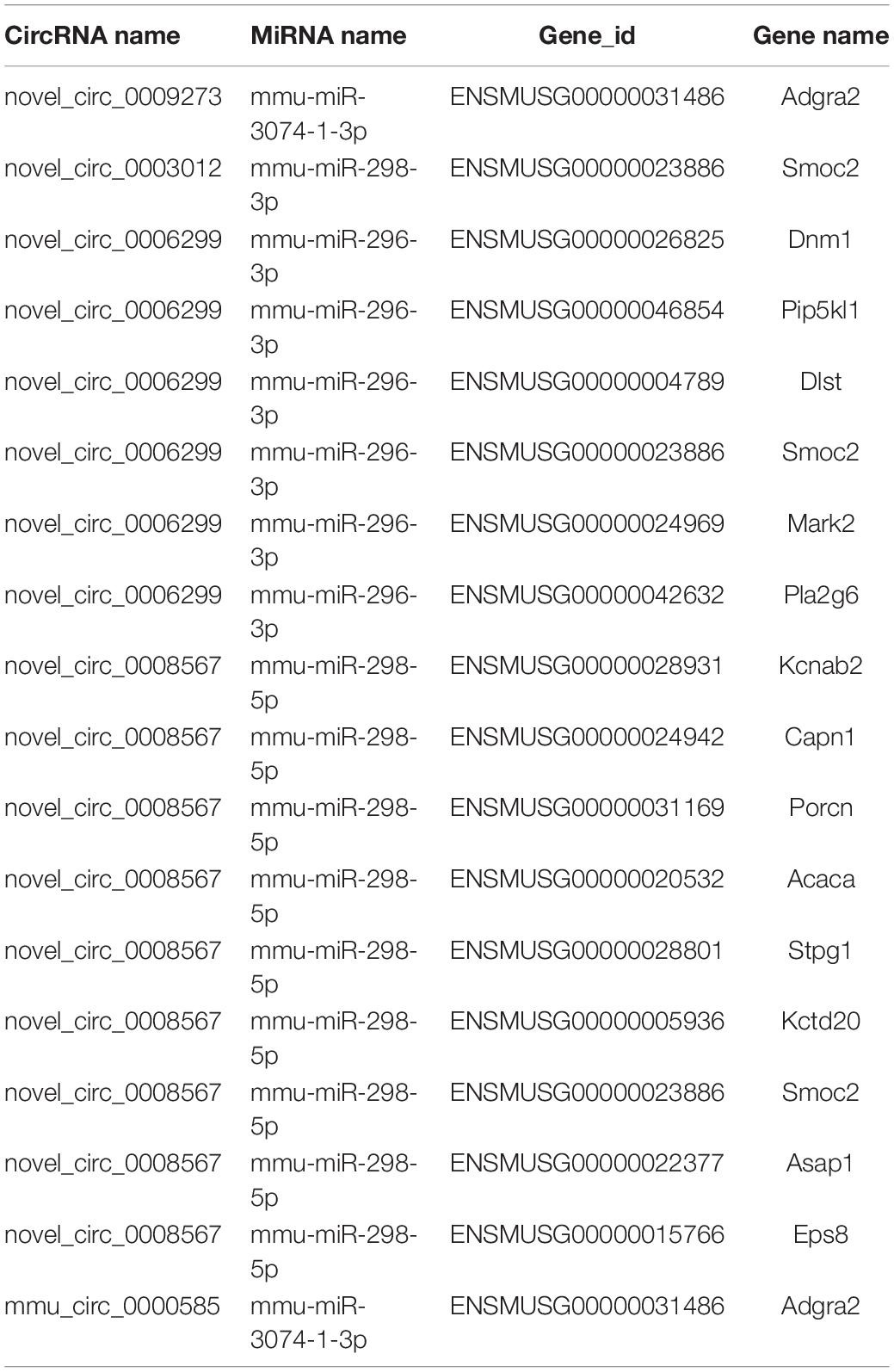
Table 5. CircRNA-ceRNA networks in cGMP-PKG, cAMP, and Hippo signaling pathway.
Furthermore, we also verified whether the circRNA-ceRNA network affects the cGMP-PKG signaling pathway. As shown in Figure 5E, we used Western blotting to detect the expression of PKG, a key factor in the cGMP-PKG signaling pathway, and the results showed that the expression of PKG in the hippocampus of APP/PS1 mice was significantly reduced compared with that in the WT group (p < 0.05).
Preliminary verification of the regulatory role of the novel_circ_0003012/mmu-miR-298-3p/Smoc2 signaling axis in the pathology of AD showed that it involved the downregulation of PKG.
Discussion
Analyzing the expression profiles of circRNA-ceRNA may provide new insights into our understanding of the pathophysiology of AD. In our study, we found 70 dysregulated circRNAs, 39 dysregulated miRNAs and 121 dysregulated mRNA between the APP/PS1 group and wild-type group at 8 months in the hippocampus of the mice; 44 circRNAs, 16 miRNAs and 34 mRNAs were upregulated, and 26 circRNAs, 23 miRNAs, and 87 mRNAs were downregulated in APP/PS1 mice relative to their levels in wild-type mice. Through correlation analysis, we obtained 11 dysregulated circRNAs (7 upregulated circRNAs and 4 downregulated), 7 dysregulated miRNAs (4 upregulated miRNAs and 3 downregulated) and 8 dysregulated mRNAs (3 upregulated mRNAs and 5 downregulated), forming 16 relationships in the circRNA-miRNA-mRNA regulatory network. Our results showed that the aberrantly expressed circRNAs had miRNA-binding sites and were thus predicted to play a regulatory role via the ceRNA mechanism (Zhang et al., 2020). These circRNA-miRNA-mRNA regulatory networks may play an important role in the onset and development of AD. For instance, mmu_circ_0001125 and mmu_circ_0000672 were found to be ceRNAs of mmu-miR-351-5p, which targets Cst7 (ENSMUSG00000068129). Cst7 (cystatin F) encodes an endosomal/lysosomal cathepsin inhibitor that regulates cathepsin activity in the lysosomal pathway (Magister and Kos, 2013). The expression of Cst7 is important in microglia for reducing the phagocytic capacity of activated microglia. Reducing the expression of Cst7 might promote the clearance of Aβ species through microglia and macrophages (Ofengeim et al., 2017).
The GO analysis was performed to further annotate the biological functions related to the aberrantly expressed circRNAs. The top GO terms of the differentially expressed circRNAs were most enriched in biological metabolic processes, such as phosphorus metabolic process, organophosphate metabolic process, nucleotide metabolic process, nucleoside phosphate metabolic process, pyrimidine nucleobase metabolic process, ribonucleotide metabolic process and ribose phosphate metabolic process. This result indicated that the pathological progression of AD may be associated with several metabolic pathways regulated by circRNAs. In addition, we also performed GO analysis of miRNAs and mRNAs. The top terms of miRNAs were almost all associated with cellular metabolic process, intracellular organelle/part and binding functions. The dysregulated mRNAs were mostly enriched in cellular/organism development processes or cell differentiation.
As the main components of nucleic acids, nucleobases, nucleosides, nucleotides and related phosphorylated metabolites have many important roles as intermediates in biosynthetic pathways in biological systems (Lane and Fan, 2015; Swerdlow, 2018; Muguruma et al., 2020). There is growing evidence that nucleotide metabolism is involved in pathological mechanisms in many different neurodegenerative diseases, such as Alzheimer’s disease. As Gonzalez-Dominguez et al. (2015) revealed, numerous metabolites, including purine and pyrimidine metabolites, show significant differences between AD and WT mice in all brain tissues, especially in hippocampal and cortical regions. This result indicated that alterations in the metabolism of nucleotides play an important role in the pathological process of AD. For instance, purinergic signaling plays a critical role in the development of AD. Studies have demonstrated that adenosine receptors in the frontal cortex of the affected brain are upregulated and that these receptors are redistributed. Furthermore, these receptors have higher activity in neurons affected by Aβ deposition (Albasanz et al., 2008; Cieslak and Wojtczak, 2018).
In addition, abnormal synthesis or metabolism of pyrimidine nucleotides is also considered to be an important factor in the pathological process of AD, and its disorder can cause dysfunction of the oxidative phosphorylation (OXPHOS) system (Pesini et al., 2019). The OXPHOS system plays an important role in the mechanism of synaptic failure and neurodegeneration triggered by Aβ (Pesini et al., 2014). After Aβ deposition, OXPHOS dysfunction appears to be a frequent finding in many AD patients (Swerdlow et al., 2014). OXPHOS participates in many cellular processes, and defects in this system affect many biochemical pathways. One of these biochemical pathways is de novo pyrimidine biosynthesis. A decrease in the de novo synthesis of pyrimidine nucleotides leads to dysfunction of the OXPHOS system and to the pathogenesis of late-onset AD (Pesini et al., 2019). Disorders of pyrimidine metabolism, with decreased uridine monophosphate and increased uracil, ultimately lead to synaptic plasticity and neuronal impairment (Czech et al., 2012). Several studies have also indicated that oxidative stress is closely related to the abnormal metabolism of purines and pyrimidines in AD (Lyras et al., 1997; Gonzalez-Dominguez et al., 2015). All these results indicate that abnormal nucleotide metabolism is also an important factor in the onset and development of AD. These circRNAs in the hippocampus of AD mice may play a critical role in the pathological progression of AD by regulating nucleotide metabolism.
Kyoto Encyclopedia of Genes and Genomes analysis showed that the dysregulation of circRNAs was enriched in many signaling pathways, which are closely related to the pathogenesis of AD, including the cGMP-PKG signaling pathway, cAMP signaling pathway, Hippo signaling pathway, platelet activation, LTP and axon guidance. KEGG pathway analysis of dysregulated miRNAs and mRNAs also identified enrichment in focal adhesion, axon guidance, platelet activation and the Hippo signaling pathway. In particular, the signaling pathways of miRNA and mRNA enrichment, such as platelet activation, axon guidance and the Hippo signaling pathway, were consistent with the KEGG analysis of circRNAs.
Other studies have also reported that several dysregulated circRNAs in the cerebral cortex of AD mice are enriched in the PI3K-Akt signaling pathway, tight junctions, Hippo signaling pathway, LTP and axon guidance (Zhang et al., 2017; Ma et al., 2019). These results are consistent with our study that the dysregulated circRNAs in the hippocampus of AD mice were also enriched in the Hippo signaling pathway, LTP and axon guidance. The Hippo signaling pathway, also known as the Salvador/Warts/Hippo (SWH) pathway, is named after the protein kinase Hippo (Hpo) in Drosophila and is a key regulator in the pathway (Zheng and Pan, 2019). The Hippo pathway is composed of a series of conserved kinases (Huang et al., 2005). Numerous studies have confirmed that the Hippo signaling pathway plays an important role in cell functions. Hippo signaling activates induced cell death, whereas inactivation of Hippo signaling triggers cell proliferation (Grusche et al., 2011; Sun and Irvine, 2011). A recent study indicated that the Hippo pathway plays an important role in the pathogenesis of AD. The Hippo pathway affects Aβ42-mediated neurodegeneration due to the excessive activation of Hippo signaling, leading to enhanced Aβ42 toxicity; however, downregulation of the Hippo signaling pathway can rescue Aβ42-mediated neurodegeneration (Irwin et al., 2020).
Interestingly, we also found that the signaling pathways of the cGMP-PKG signaling pathway, cAMP signaling pathway and platelet activation in the hippocampus of AD mice were associated with dysregulated circRNAs in the pathogenesis of AD (Kelly, 2018; Ricciarelli and Fedele, 2018). Cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) are well-established second messengers required for LTP and memory formation and consolidation (Ricciarelli and Fedele, 2018). Recent evidence indicates that excessive Aβ deposition inhibits both the cAMP and cGMP pathways and impairs LTP signal transduction. Changes in cAMP signals in specific brain regions may be related to the pathology of dementia. Reduced cAMP signaling is an important factor in AD pathology. Increasing cAMP signaling in specific regions of the brain can resist age-related declines in brain function. Studies have shown that cAMP levels in the hippocampus can be reduced by the overexpression of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) or the infusion of Aβ1–42 (Chen et al., 2012; Zhang et al., 2014). Furthermore, cAMP-elevating agents can reverse or prevent Aβ-induced hippocampal deficits.
Cyclic guanosine monophosphate-dependent protein kinase (PKG) and the cGMP controller phosphodiesterase are critical participants in the neuroinflammatory process, which may lead to neurological dysfunction, cell death and further neurodegeneration (Ricciarelli and Fedele, 2018). The increase in cGMP levels decreases the Aβ load in transgenic models of AD and in models of physiological aging (Sierksma et al., 2013). In addition, cGMP-dependent Akt activation and GSK3β inactivation can reduce tau hyperphosphorylation. PKG, as the key downstream target of cGMP, has been reported to be significantly decreased in both the cortex and hippocampus after treatment with Aβ (Wang et al., 2017). The cGMP-PKG pathway plays a crucial role in preventing apoptosis and promoting neural cell survival. It has been shown that the activation of PKG in hippocampal neurons is involved in the LTP induced by NO and carbon monoxide (Fiscus, 2002). Inhibition of PKG activity in hippocampal neurons can partially block the prosurvival effects of APPS, suggesting that cGMP, via activation of PKG, mediates the neuroprotective effect of APPS (Barger et al., 1995; Fiscus, 2002). Our results are consistent with the above research, confirming that the expression of PKG in the hippocampus was obviously decreased in AD mice and that the cGMP-PKG pathway might play an essential role in the pathophysiology of AD.
From the results of the circRNA-ceRNA network constructed on the basis of the differentially expressed circRNAs, miRNAs and mRNAs obtained from the sequencing analysis results and the series of circRNAs predicted by KEGG analysis that are closely related to the cGPM-PKG signaling pathway, we found that the novel_circ_0003012/mmu-miR-298-3p/Smoc2 signaling axis may be closely related to the pathological mechanism of AD.
Through preliminary verification, we found that the differential expression of novel_circ_0003012 and mmu-miR-298-3p may regulate the pathological mechanism of AD by affecting the cGPM-PKG signaling pathway.
Conclusion
In summary, we elucidated the circRNA-ceRNA network patterns in the hippocampus of APP/PS1 and WT mice by using deep RNA-seq analysis. Our findings further expand the current knowledge regarding the biology of circRNA-ceRNA, their involved signaling pathways, such as the dysregulated circRNAs in nucleotide metabolism, cGMP-PKG signaling pathway, cAMP signaling pathway, platelet activation and Hippo signaling pathway, and their regulatory roles in AD pathogenesis. In addition, our findings preliminarily verified that the novel_circ_0003012/mmu-miR-298-3p/Smoc2 signaling axis may regulate the pathophysiology of AD by affecting the cGMP-PKG signaling pathway. These newly identified circRNAs in networks and signaling pathways reveal potential diagnostic or therapeutic targets for AD.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA712946.
Ethics Statement
The animal study was reviewed and approved by Medical Ethics Committee of Qingdao University.
Author Contributions
YZ conceived and wrote the manuscript. YZ and LQ reviewed and edited the manuscript. YLy, YLg, WY, and YZf participatedin literature search, data collection, and figures design. All authors read and approved the manuscript.
Funding
This work was supported by funding from the National Natural Science Foundation of China (No. 81901085), the China Postdoctoral Science Foundation (No. 2019M662302), and the Qingdao Applied Basic Research Project (No. 19-6-2-50-cg).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2021.665788/full#supplementary-material
Abbreviations
AD, Alzheimer’s disease; BACE1, β-site amyloid precursor protein-cleaving enzyme 1; BP, biological process; cAMP, cyclic adenosine monophosphate; CC, cellular component; ceRNAs, competitive endogenous RNAs; circRNAs, circular RNAs; cGMP, cyclic guanosine monophosphate; Cst7, cystatin F; GO, Gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; LTP, long-term potentiation; MF, molecular function; MREs, miRNA response elements; OXPHOS, oxidative phosphorylation; PKG, cGMP-dependent protein kinase; WT, wild-type.
Footnotes
- ^ http://www.genome.jp/kegg/
- ^ www.targetscan.org
- ^ www.microrna.org/microrna/home.do
- ^ http://ccb.jhu.edu/software/hisat2
References
Albasanz, J. L., Perez, S., Barrachina, M., Ferrer, I., and Martin, M. (2008). Up-regulation of adenosine receptors in the frontal cortex in Alzheimer’s disease. Brain Pathol. 18, 211–219. doi: 10.1111/j.1750-3639.2007.00112.x
Barger, S. W., Fiscus, R. R., Ruth, P., Hofmann, F., and Mattson, M. P. (1995). Role of cyclic GMP in the regulation of neuronal calcium and survival by secreted forms of beta-amyloid precursor. J. Neurochem. 64, 2087–2096. doi: 10.1046/j.1471-4159.1995.64052087.x
Chen, Y., Huang, X., Zhang, Y. W., Rockenstein, E., Bu, G., Golde, T. E., et al. (2012). Alzheimer’s beta-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of beta-amyloid. J. Neurosci. 32, 11390–11395. doi: 10.1523/jneurosci.0757-12.2012
Cieslak, M., and Wojtczak, A. (2018). Role of purinergic receptors in the Alzheimer’s disease. Purinergic Signal. 14, 331–344. doi: 10.1007/s11302-018-9629-0
Czech, C., Berndt, P., Busch, K., Schmitz, O., Wiemer, J., Most, V., et al. (2012). Metabolite profiling of Alzheimer’s disease cerebrospinal fluid. PLoS One 7:e31501. doi: 10.1371/journal.pone.0031501
D’Ambra, E., Capauto, D., and Morlando, M. (2019). Exploring the regulatory role of circular RNAs in Neurodegenerative Disorders. Int. J. Mol. Sci. 20:5477. doi: 10.3390/ijms20215477
Fiscus, R. R. (2002). Involvement of cyclic GMP and protein kinase G in the regulation of apoptosis and survival in neural cells. Neuro Signals 11, 175–190. doi: 10.1159/000065431
Gao, Y., Wang, J., and Zhao, F. (2015). CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 16:4.
Gonzalez-Dominguez, R., Garcia-Barrera, T., Vitorica, J., and Gomez-Ariza, J. L. (2015). Metabolomic screening of regional brain alterations in the APP/PS1 transgenic model of Alzheimer’s disease by direct infusion mass spectrometry. J. Pharm. Biomed. Anal. 102, 425–435. doi: 10.1016/j.jpba.2014.10.009
Gouras, G. K., Olsson, T. T., and Hansson, O. (2015). beta-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 12, 3–11.
Grusche, F. A., Degoutin, J. L., Richardson, H. E., and Harvey, K. F. (2011). The Salvador/Warts/Hippo pathway controls regenerative tissue growth in Drosophila melanogaster. Dev. Biol. 350, 255–266. doi: 10.1016/j.ydbio.2010.11.020
Huang, J., Wu, S., Barrera, J., Matthews, K., and Pan, D. (2005). The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122, 421–434. doi: 10.1016/j.cell.2005.06.007
Huang, J. L., Qin, M. C., Zhou, Y., Xu, Z. H., Yang, S. M., Zhang, F., et al. (2018). Comprehensive analysis of differentially expressed profiles of Alzheimer’s disease associated circular RNAs in an Alzheimer’s disease mouse model. Aging 10, 253–265. doi: 10.18632/aging.101387
Irwin, M., Tare, M., Singh, A., Puli, O. R., Gogia, N., Riccetti, M., et al. (2020). A positive feedback loop of Hippo- and c-Jun-Amino-terminal kinase signaling pathways regulates amyloid-beta-mediated Neurodegeneration. Front. Cell Dev. Biol. 8:117. doi: 10.3389/fcell.2020.00117
Kelly, M. P. (2018). Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal. 42, 281–291. doi: 10.1016/j.cellsig.2017.11.004
Kristensen, L. S., Andersen, M. S., Stagsted, L. V. W., Ebbesen, K. K., Hansen, T. B., and Kjems, J. (2019). The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 20, 675–691.
Lane, A. N., and Fan, T. W. (2015). Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 43, 2466–2485. doi: 10.1093/nar/gkv047
Lazarov, O., and Marr, R. A. (2010). Neurogenesis and Alzheimer’s disease: at the crossroads. Exp. Neurol. 223, 267–281. doi: 10.1016/j.expneurol.2009.08.009
Lei, K., Bai, H., Wei, Z., Xie, C., Wang, J., Li, J., et al. (2018). The mechanism and function of circular RNAs in human diseases. Exp. Cell Res. 368, 147–158. doi: 10.1016/j.yexcr.2018.05.002
Lyras, L., Cairns, N. J., Jenner, A., Jenner, P., and Halliwell, B. (1997). An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J, Neurochem. 68, 2061–2069. doi: 10.1046/j.1471-4159.1997.68052061.x
Ma, N., Pan, J., Ye, X., Yu, B., Zhang, W., and Wan, J. (2019). Whole-transcriptome analysis of APP/PS1 mouse brain and identification of circRNA-miRNA-mRNA networks to investigate AD pathogenesis. Mol. Ther. Nucleic Acids 18, 1049–1062. doi: 10.1016/j.omtn.2019.10.030
Magister, S., and Kos, J. (2013). Cystatins in immune system. J. Cancer 4, 45–56. doi: 10.7150/jca.5044
Matsuzaki, M., Honkura, N., Ellis-Davies, G. C., and Kasai, H. (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. doi: 10.1038/nature02617
Memczak, S., Jens, M., Elefsinioti, A., Torti, F., Krueger, J., Rybak, A., et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. doi: 10.1038/nature11928
Mu, Y., and Gage, F. H. (2011). Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 6:85. doi: 10.1186/1750-1326-6-85
Muguruma, Y., Tsutsui, H., Akatsu, H., and Inoue, K. (2020). Comprehensive quantification of purine and pyrimidine metabolism in Alzheimer’s disease postmortem cerebrospinal fluid by LC-MS/MS with metal-free column. Biomed. Chromatogra. 34:e4722. doi: 10.1002/bmc.4722
Ofengeim, D., Mazzitelli, S., Ito, Y., DeWitt, J. P., Mifflin, L., Zou, C., et al. (2017). RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, E8788–E8797.
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT. StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Pesini, A., Iglesias, E., Bayona-Bafaluy, M. P., Garrido-Perez, N., Meade, P., Gaudo, P., et al. (2019). Brain pyrimidine nucleotide synthesis and Alzheimer disease. Aging 11, 8433–8462. doi: 10.18632/aging.102328
Pesini, A., Iglesias, E., Garrido, N., Bayona-Bafaluy, M. P., Montoya, J., and Ruiz-Pesini, E. (2014). OXPHOS, pyrimidine nucleotides, and Alzheimer’s disease: a pharmacogenomics approach. J. Alzheimer’s Dis 42, 87–96. doi: 10.3233/jad-140384
Ricciarelli, R., and Fedele, E. (2018). cAMP, cGMP and amyloid beta: three ideal partners for memory formation. Trends Neurosci. 41, 255–266. doi: 10.1016/j.tins.2018.02.001
Sierksma, A. S., Rutten, K., Sydlik, S., Rostamian, S., Steinbusch, H. W., van den Hove, D. L., et al. (2013). Chronic phosphodiesterase type 2 inhibition improves memory in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neuropharmacology 64, 124–136. doi: 10.1016/j.neuropharm.2012.06.048
Sun, G., and Irvine, K. D. (2011). Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev. Biol. 350, 139–151. doi: 10.1016/j.ydbio.2010.11.036
Swerdlow, R. H. (2018). Mitochondria and mitochondrial cascades in Alzheimer’s Disease. J. Alzheimer’s Dis 62, 1403–1416. doi: 10.3233/jad-170585
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2014). The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231. doi: 10.1016/j.bbadis.2013.09.010
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A., and Nair, M. (2019). Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 14, 5541–5554.
Wang, J. Z., Wang, Z. H., and Tian, Q. (2014). Tau hyperphosphorylation induces apoptotic escape and triggers neurodegeneration in Alzheimer’s disease. Neurosci. Bull. 30, 359–366. doi: 10.1007/s12264-013-1415-y
Wang, L., Xiaokaiti, Y., Wang, G., Xu, X., Chen, L., Huang, X., et al. (2017). Inhibition of PDE2 reverses beta amyloid induced memory impairment through regulation of PKA/PKG-dependent neuro-inflammatory and apoptotic pathways. Sci. Rep. 7:12044.
Wang, Z., Xu, P., Chen, B., Zhang, Z., Zhang, C., Zhan, Q., et al. (2018). Identifying circRNA-associated-ceRNA networks in the hippocampus of Abeta1-42-induced Alzheimer’s disease-like rats using microarray analysis. Aging 10, 775–788. doi: 10.18632/aging.101427
Wilkins, H. M., and Swerdlow, R. H. (2017). Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 133, 71–79. doi: 10.1016/j.brainresbull.2016.08.009
Zhang, C., Cheng, Y., Wang, H., Wang, C., Wilson, S. P., Xu, J., et al. (2014). RNA interference-mediated knockdown of long-form phosphodiesterase-4D (PDE4D) enzyme reverses amyloid-beta42-induced memory deficits in mice. J. Alzheimer’s Dis 38, 269–280. doi: 10.3233/jad-122236
Zhang, L., Ding, H., Zhang, Y., Wang, Y., Zhu, W., and Li, P. (2020). Circulating MicroRNAs: biogenesis and clinical significance in acute myocardial infarction. Front. Physiol. 11:1088. doi: 10.3389/fphys.2020.01088
Zhang, S., Zhu, D., Li, H., Li, H., Feng, C., and Zhang, W. (2017). Characterization of circRNA-Associated-ceRNA networks in a senescence-accelerated mouse prone 8 brain. Mol. Ther. 25, 2053–2061. doi: 10.1016/j.ymthe.2017.06.009
Keywords: Alzheimer’s disease, circRNA, hippocampus, expression profiles, ceRNA
Citation: Zhang Y, Qian L, Liu Y, Liu Y, Yu W and Zhao Y (2021) CircRNA-ceRNA Network Revealing the Potential Regulatory Roles of CircRNA in Alzheimer’s Disease Involved the cGMP-PKG Signal Pathway. Front. Mol. Neurosci. 14:665788. doi: 10.3389/fnmol.2021.665788
Received: 08 February 2021; Accepted: 26 April 2021;
Published: 21 May 2021.
Edited by:
Javier Egea, Princess University Hospital, SpainReviewed by:
Ursula Susan Sandau, Oregon Health & Science University, United StatesLili Cui, Guangdong Medical University, China
Copyright © 2021 Zhang, Qian, Liu, Liu, Yu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Zhang, eXVhbl96aGFuZzg0QDEyNi5jb20=