 LiJun Zhou1,2
LiJun Zhou1,2 RenShi Xu1,2*
RenShi Xu1,2*- 1Department of Neurology, National Regional Center for Neurological Diseases, Clinical College of Nanchang Medical College, Jiangxi Provincial People's Hospital, First Affiliated Hospital of Nanchang Medical College, Xiangya Hospital of Central South University Jiangxi Hospital, Nanchang, Jiangxi, China
- 2Medical College of Nanchang University, Nanchang, China
Amyotrophic lateral sclerosis (ALS) is a common adult-onset neurodegenerative disease characterized by the progressive death of motor neurons in the cerebral cortex, brain stem, and spinal cord. The exact mechanisms underlying the pathogenesis of ALS remain unclear. The current consensus regarding the pathogenesis of ALS suggests that the interaction between genetic susceptibility and harmful environmental factors is a promising cause of ALS onset. The investigation of putative harmful environmental factors has been the subject of several ongoing studies, but the use of transgenic animal models to study ALS has provided valuable information on the onset of ALS. Here, we review the current common invertebrate genetic models used to study the pathology, pathophysiology, and pathogenesis of ALS. The considerations of the usage, advantages, disadvantages, costs, and availability of each invertebrate model will also be discussed.
1 Introduction
Amyotrophic lateral sclerosis (ALS) is a multifactorial non-cell-autonomous neurodegenerative disease. Since the 1990s, several genes and mutations have been found to be involved in the pathogenesis of ALS. Based on this, several genetic animal models have been developed to study the pathology and pathogenesis of ALS, including invertebrates and vertebrates, such as yeast, flies, worms, zebrafish, mice, rats, guinea pigs, dogs, and non-human primates. Although each model displays different phenotypes, these genetic models are complementary to the pathological mechanisms of motor neuron (MNs) degeneration and the progression of ALS and are thus beneficial for research on the potential common pathogenesis of ALS through different genetic animal models, furthering the search for novel medicine treatments (Bonifacino et al., 2021).
Commonly used invertebrate genetic models mainly consist of yeast, Drosophila, and Caenorhabditis. These invertebrate experimental models were originally used to study genetics and development. These were also sometimes used to study the mechanisms associated with molecular and cell biology and are now presently propelled into the study of functional genomics and proteomics. The highly manipulable genomes of these models allow us to rapidly reproduce the transgenic lines, providing the ideal models for studying gene functions and gene and protein network interaction (Surguchov, 2021).
The full sequence of the invertebrate model genome is also easy to access for comparison with higher vertebrates and mammals in facilitating the evolution of genomic studies to rapidly produce transgenic animals by DNA transformation. Invertebrate organisms have more significant experimental advantages than their mammalian counterparts, such as a short generation time, small size, easy maintenance, and low cost. Another advantage of the invertebrate model is that it is amenable to genetic studies forward from phenotype to gene and reverse from gene to phenotype. Classic forward genetics studies of invertebrate models allow us to identify new molecules or pathways involved in special cell processes, which is a key advantage of invertebrate models. Forward genetics studies that apply a chemical mutagen to invertebrate models can generate target gene mutants to elucidate target gene functions. Reverse genetic studies using invertebrate models can rapidly identify possible molecular and biological pathways of certain target genes. The knockdown of target gene mutants using RNA interference (RNAi) technologies in invertebrate models can dramatically reduce target gene products by introducing double-stranded RNA (dsRNA) in models and provide related data about the roles of some target genes in biological processes.
In addition to being amenable to forward and reverse genetic approaches, the advantages of invertebrate models include the following: yeast models possess a controlled environment, with a single system, an individual cell or tissue mechanism, easy gene manipulation, and easy culture. A special advantage of Drosophila is that it possesses large chromosomes, while a unique advantage of Caenorhabditis is its transparent body. Brenner (1974), Horvitz and Sulston (1980), and Sulston and Horvitz (1981) were awarded the Nobel Prize in Physiology or Medicine in 2002 for their seminal contributions concerning the genetic regulation of organ development and programmed cell death using the Caenorhabditis elegans (C. elegans) model as an investigative tool. The findings from these models established by them revealed some key genes involved in cell division, differentiation, and apoptosis and confirmed that both the structures and functions of these genes were highly conserved in higher animals, including humans. Based on their findings, they provided a framework in which simple animals can be used to identify potential molecular pathways and related biological processes. In addition, the advantages of Drosophila and Caenorhabditis also include a short life cycle, high fertility, a less complex nervous system, fully sequenced genome, which is homologous by more than 65%-75% to human genes, molecular pathways similar to humans, availability of mutagenic and transgenic techniques, and low cost (Gois et al., 2020). An important advantage of these models is that their data and information are often directly applied to research mechanisms of human diseases. Another advantage of establishing a model that imitates human disease using small invertebrate organisms is the high degree of conservation in mammalian organisms, which is useful for identifying the molecular components involved in pathogenesis. However, a major disadvantage of invertebrate models is that they are evolutionarily distant from mammals. In addition, compared to higher animals, the organs of invertebrate models are extremely undeveloped and simple, and alterations in many physiological functions and pathological and pathophysiological processes during the disease course in higher animals and mammals cannot be observed in invertebrate models. Another major disadvantage is the limited cellular diversity, which is not beneficial for studying pathological alterations at the organ and cellular levels (Gois et al., 2020) (Figure 1).
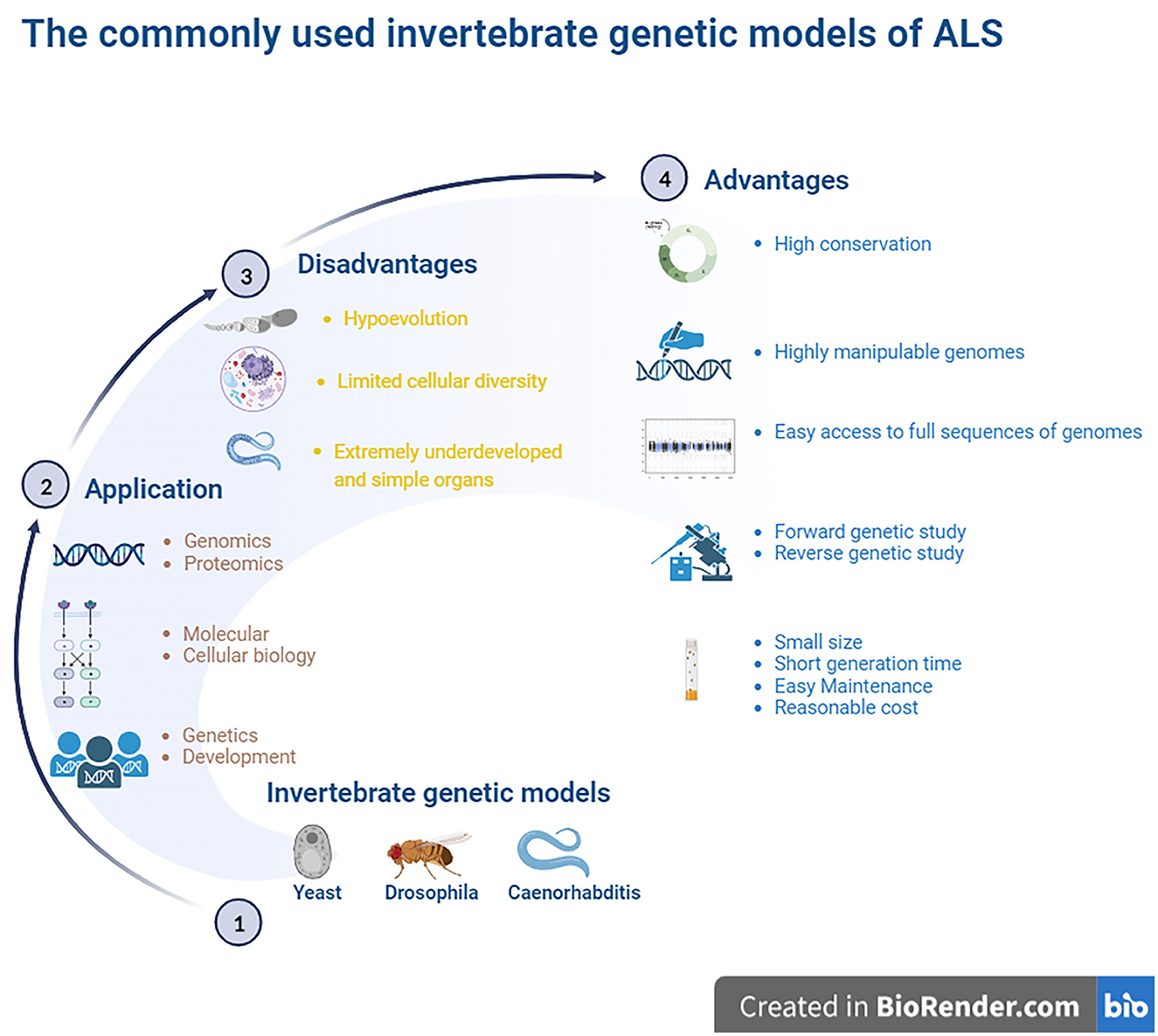
Figure 1. The commonly used invertebrate genetic models of ALS. The highly manipulative genomes of these models allow us to rapidly reproduce the transgenic lines for providing the ideal models for studying the gene functions and protein network interactions. The finds from the established models identified some key genes to involve in cell division, differentiation and apoptosis, which structures and functions were the high conservation with higher animals including humans, and it is easy access to the full sequence of model genomes. The model organisms have more significant experimental advantages, such as the short generate time, small sizes, easy maintenance as well as less cost. Besides, it is amenable to the genetic approaches of both forward and reverse. However, these invertebrate animal models are evolutionarily far from higher animal and mammalians, therefore, lots of physiologic functions can't be observed. The organs are extremely undeveloped as well as the limited cellular diversity, so it isn't beneficial to study the pathological alteration of organs and cellular levels. ALS, amyotrophic lateral sclerosis.
2 Genes involved in ALS
The majority of ALS cases are sporadic, and ~5%–10% of ALS cases are familial, with accurate Mendelian hereditary features and obvious penetrance (Gros-Louis et al., 2006). Since several missense mutations in the Cu2+/Zn2+ superoxide dismutase 1 (SOD1) gene were discovered in the subsets of familial ALS patients in 1993 (Rosen et al., 1993), various ALS-related gene mutations have been reported in the pathogenesis of ALS. Based on current genetic evidence, genetic mutations are thought to be the key cause of ALS pathogenesis. Mutations in some RNA metabolism-related genes and their related potential pathogenic mechanisms, such as disorders of cell transportation, axon outgrowth, protein metabolism, glutamatergic signaling, angiogenesis, neurotransmission, and antioxidant functions, have been confirmed to participate in the pathogenesis of ALS (Rosen et al., 1993; Neumann et al., 2006; Kwiatkowski et al., 2009; Renton et al., 2011; Morgan and Orrell, 2016). Mutations in SOD1 (Rosen et al., 1993), TAR DNA-binding protein-43 (TDP-43), fused in sarcoma/translocated in liposarcoma (FUS/TLS, FUS) (Zou et al., 2017), and open reading frame 72 on chromosome 9 (C9ORF72) genes (DeJesus-Hernandez et al., 2011) are common in major cases of ALS. In addition, several genes with less frequent mutations, such as VABP, OPTN, VCP, UBQLN2, MATR3, TBK1, NEK1, C21ORF2, ANXA11, and KIF5A are also found to participate in the pathogenesis of partial ALS patients (Leblond et al., 2014; Chia et al., 2018; Brenner and Freischmidt, 2022). Mutations in ~30 genes have already been identified in patients with ALS to date; among them, major pathological mechanisms of the known ALS genes have been identified, including SOD1, TDP43, FUS and C9orf72. Recently discovered ALS-related candidates and risk genes include SPTLC1, KANK1, CAV1, HTT, and WDR7 (Brenner and Freischmidt, 2022). To investigate the possible genetic mechanisms underlying ALS pathogenesis, various models have been established, including invertebrate models. In this review, we briefly discuss the usage, advantages, disadvantages, costs, and availability of current common invertebrate genetic models.
3 Nomenclature yeast models
Yeast models of ALS have been widely adopted to model human neurodegenerative diseases, such as ALS, since these have the advantages of easy manipulation and recapitulation compared to more complex eukaryotic cells. In particular, yeast models offer special insights into the potential relationships between the gain-of-function (GOF) toxicity of some protein aggregations. Although yeast models cannot reproduce the numerous pathogenic mechanisms involved in the degeneration of neuronal networks, they have proven to be ideal for discovering ALS-related pathogenic proteins. Among them, the yeast models of prion-like neurodegenerative disease-related proteins provide clues and valuable information for future research using a higher model system and ultimately developing therapeutics (Monahan et al., 2018).
Approximately 6,000 human genes are homologous to those of yeast; among them, ~10% can be complemented with yeast (Cherry et al., 2012). Moreover, ~500 genes involved in human diseases are orthologs in yeast (Kryndushkin and Shewmaker, 2011). The most straightforward method for searching candidate pathogenic genes of human diseases among yeast is in situations where the disease genes are complemented by the yeast ortholog genes. The absence of human protein quality control factors in yeast can be an advantage in yeast models. Because of the deficiency of chaperones that regulate human pathogenic proteins among yeast, they can be useful for evaluating the chaperone-mediated changing mechanisms of aggregation and toxicity of human pathogenic proteins (De Graeve et al., 2013; Kumar et al., 2018; Park et al., 2018) (Figure 2).
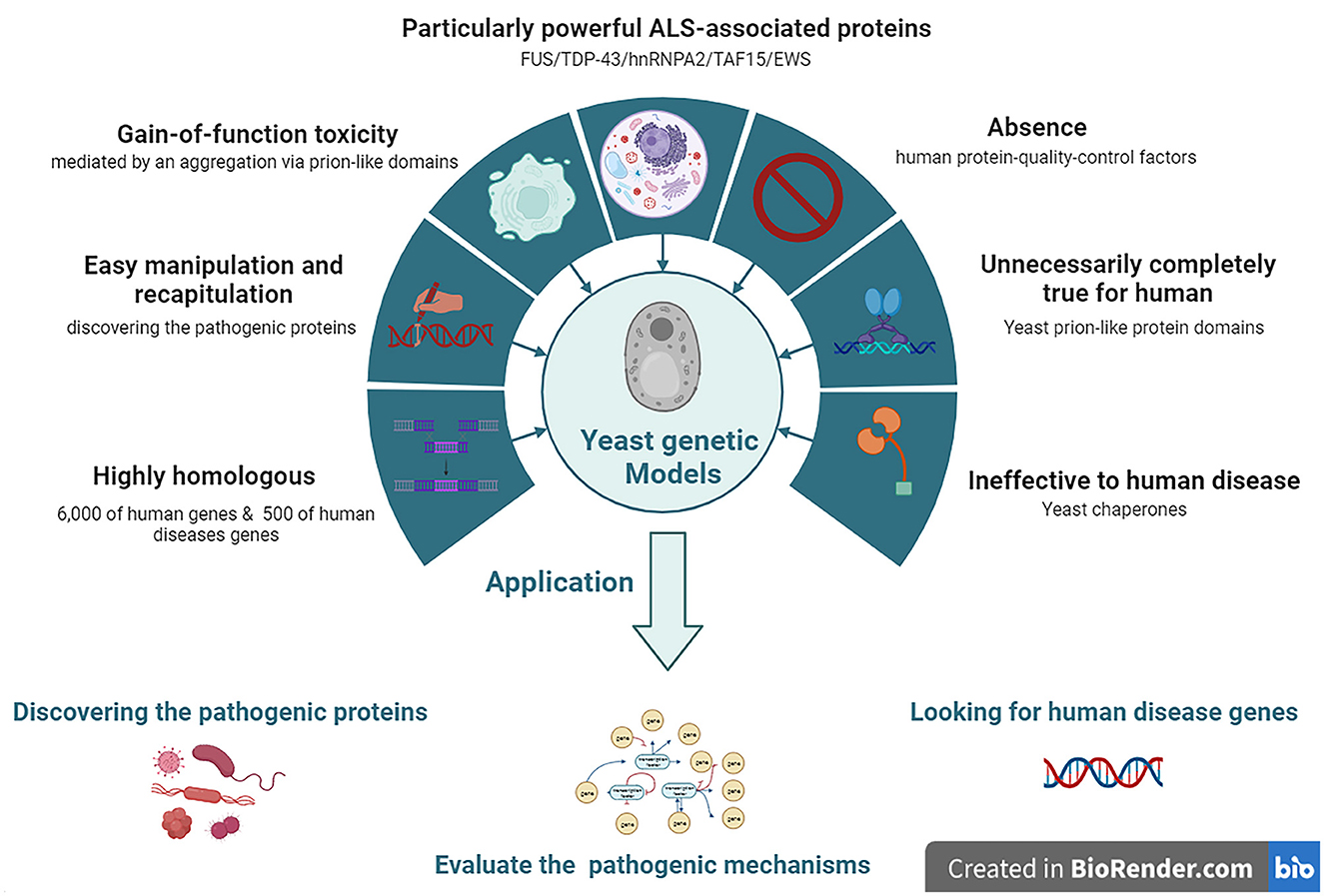
Figure 2. The features and applications of Yeasts genetic models in ALS. ALS, amyotrophic lateral sclerosis; EWS, Ewing sarcoma breakpoint region 1; FUS, fused in sarcoma; hnRNPA2, heterogeneous nuclear ribonucleoprotein A2; TAF15, TATA-box binding protein associated factor 15; TDP-43, TAR DNA-binding protein 43.
3.1 Yeasts carrying SOD1 and OPTN mutations
Many neurodegenerative disease-related genes/proteins such as SOD1 and OPTN in ALS (Rabizadeh et al., 1995; Kryndushkin et al., 2012) have been established as models among yeast (Braun, 2015). To develop SOD1 yeast models, various ALS-related SOD1 mutations such as A4V, G37R, H48Q, G93A, and S134N have been introduced into the yeast SOD1 gene. The SOD1 yeast model demonstrated that the mutant human SOD1 (hmSOD1) protein is unstable and reduces cell viability but does not form insoluble SOD1 protein aggregates. Moreover, the hmSOD1 toxic effects seem not to depend on mitochondrial dysfunction or oxidative stress, but rather on the inability to control central metabolic processes, which is most probably due to the severe disruption of vacuolar compartments (Bonifacino et al., 2021).
3.2 Yeasts carrying FUS and TDP-43 mutations
ALS-related proteins with prion-like domains have proven to be particularly powerful in yeast models because they are similar to naturally existing yeast prion-like proteins, such as FUS, TDP-43, heterogeneous nuclear ribonucleoprotein A2 (hnRNPA2), TATA-box binding protein associated factor 15 (TAF15), and Ewing sarcoma breakpoint region 1 (EWS) proteins, which have similar protein architectures. Moreover, these proteins were found in the neuronal cytoplasm of post-mortem ALS patients. In yeast models, ALS-related proteins and their mutant isoforms typically induce GOF toxicity, which is partially mediated by aggregation via prion-like domains. Yeast did not possess TDP-43 orthologous genes. Yeast models overexpressing human wild-type (WT) TDP-43 showed that TDP-43 accumulated and formed subcellular aggregates in the cytoplasm, which inhibited cell growth, disrupted cell morphology, and generated cytotoxicity. Yeast models of ALS-related TDP-43 mutations, such as K, M337V, Q343R, N345K, R361S, and N390D, accelerate protein aggregation, increase the number of cytosolic aggregates, and lead to growth arrest and cell death. Yeasts do not express FUS orthologs; thus, yeasts FUS models have been established by ectopically transforming human WT or mutant FUS genes into yeast. FUS yeast models are tightly associated with the translocation of FUS protein from the nucleus to the cytoplasm, forming aggregates co-localized with P bodies and stress granules in yeast cytoplasm and inhibiting the ubiquitin proteasome systems (Bonifacino et al., 2021).
Yeast models also have some limitations and disadvantages. Yeast protein domains are not the same as those in humans; thus, they must be carefully considered in studies using yeast models. The formation of prion-like proteins in yeast is its (True et al., 2004; Halfmann et al., 2012); however, this is not the case for human ALS proteins with prion-like domains. Almost all mammalian all mammalian prion-like proteins are harmful to humans (An and Harrison, 2016) since the properties of yeast prion-like protein domains are not completely conserved, but produce distinct alterations of constructs and functions that lead to contrary functions between yeast and humans during the human evolution process. Another limitation of yeast models is yeast cells do not have resident interacting proteins or chaperones that regulate human pathogenic proteins compared to mammalian cells. Chaperones are especially important for quality control and are integral to the propagation of endogenous prion proteins. Owing to the deficiency of resident interacting proteins and chaperones in yeast, the interaction of ectopically expressing human prion-like proteins in yeast is naturally different from that in humans; thus, the conclusions derived from studying protein interactions in yeast models may not be suitable for human diseases (Masison and Reidy, 2015).
4 Drosophila melanogaster models
Drosophila melanogaster is a good genetic model for studying neurodegenerative diseases, including ALS. D. melanogaster is a relatively complicated animal with a well-developed brain and other neural systems and display various behaviors such as learning, motor, and vision. D. melanogaster models are the earliest multiple cellular eukaryotes used as genetic models and have been applied in studying various basic biological principles, such as genetic phenotypes and genotypes, and behavioral and developed processes. The gene was first described as a functional genetic unit in D. melanogaster models. These models can be used to easily observe the neurodegenerative phenotype of rough eye generation under a light microscope. In addition, it can also be used to screen for genetic modifiers of gene enhancers and suppressors and can be used to study genetic inheritance and behavioral and developmental processes associated with human neurodegenerative diseases, including ALS. However, one main disadvantage of D. melanogaster models is that forward genetic screening by RNAi is relatively complex compared to the C. elegans model. Gene knockdown by RNAi cannot be conducted by feeding with dsRNA as simply as in C. elegans model; therefore, D. melanogaster models are made by injecting dsRNA into their embryos (Figure 3).
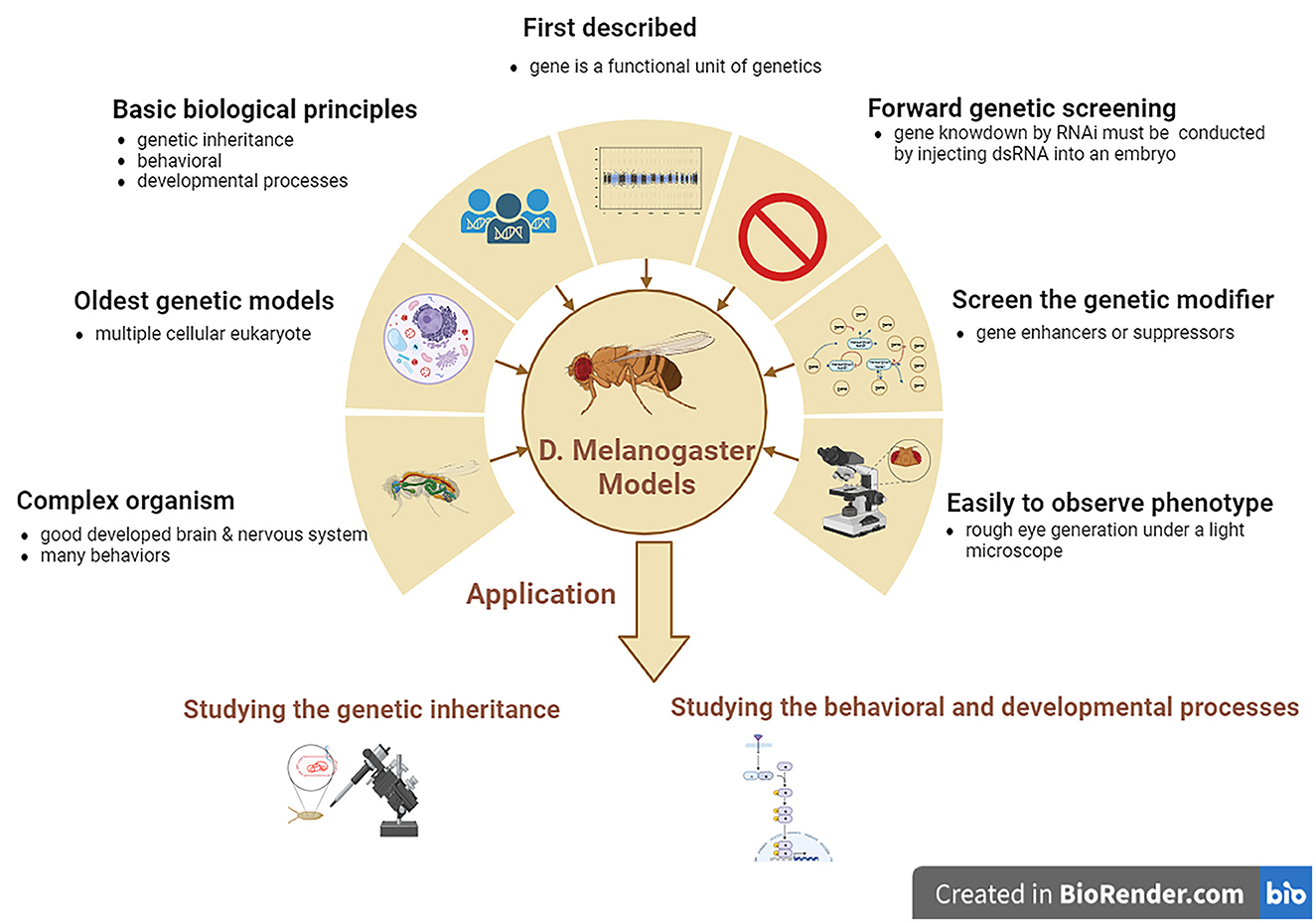
Figure 3. The features and applications of D. melanogaster genetic models in ALS. ALS, amyotrophic lateral sclerosis; D. melanogaster, Drosophila melanogaster.
4.1 D. melanogaster carrying SOD1 mutations
The hSOD1 D. melanogaster models were produced by amplifying the hSOD1 gene through missense mutation technology and replacing the nitrogenous base among DNA correspondence for hSOD1 reading site with a directive mutation. The hmSOD1 D. melanogaster models exhibit movement dysfunction, local accumulation of hmSOD1 in MNs, enlargement of glial cells, neuronal degeneration, muscle contraction, decreased survival rates, and mitochondrial dysfunction (Gois et al., 2020). It reduces lifespan and fecundity and increases susceptibility to oxidative stress, movement defects, and necrotic cell death in hmSOD1 D. melanogaster models with SOD1 mutations (Phillips et al., 1989). The hmSOD1 D. melanogaster model expresses four hSOD1 mutants – G85R, H71Y, H48R and G37R. Among the endogenous D. melanogaster, SOD1 (dSOD1) produces a large amount of dSOD1 protein in neural cells (Layalle et al., 2021). Models expressing G85R, H71Y, and H48R mutations show decreased survival, developmental defects, and larval and adult dyskinesia due to muscle axon contraction (Braems et al., 2021). In addition, some antioxidant compounds have neuroprotective effects that improve exercise performance, prolong lifespan, and reduce hSOD1 cytoplasmic inclusion bodies in the hSOD1 D. melanogaster model (Liguori et al., 2021).
The transgenic D. melanogaster model expressing only hSOD1WT in MNs has been demonstrated to extend its longevity, but does not affect the movement or survival of MNs (Parkes et al., 1998). Moreover, the hSOD1WTtransgenic D. melanogaster model extended the longevity of the SOD1 deletion mutant and normal D. melanogaster but did not prevent age-dependent movement disorders. The D. melanogaster model's widespread downregulation of SOD1 expression accelerates age-dependent movement disorders and shortens the lifespan of D. melanogaster (Braems et al., 2021). Meanwhile, the D. melanogaster model selectively expressing WT or causative-related hSOD1 mutations (A4V and G85R) in MNs induces progressive movement dysfunction with electrophysiological defect, and the SOD1 aberrant accumulates stress responses around the glial cells (Watson et al., 2008). Moreover, the transgenic D. melanogaster model specifically expressing hmSOD1 in MNs showed a progressive motor protective effect, prevented the accumulation of hmSOD1 aggregation, and increased the glial cell stress response in the ventral nervous cord, accompanied by electrophysiological defects in neural circuits. The hmSOD1 transgenic D. melanogaster overexpressing mutated SOD1 can model both cellular and non-cellular autonomic damage (Walters et al., 2019). The transgenic D. melanogaster model expressing WT or mutant (A4V and G85R) hSOD1 among D. melanogaster MNs would lead to D. melanogaster climbing defects that progress over time and defects among neural circuits accompanied by the stress responses of glial cells and local accumulation of mutant SOD1 proteins among MNs (Braems et al., 2021). Meanwhile, the hSOD1 A4V transgenic D. melanogaster model showed synaptic transmission defects and focal mutation SOD1 accumulation in MNs, HSP upregulation in glial cells, cellular autonomic lesions in MNs, and abnormal alterations in glial cells. This D. melanogaster model reveals that ALS is not confined to damaging MNs, and toxic SOD1 transfers from neurons to glial cells (Clement et al., 2003; Boillée et al., 2006). The hSOD1 transgenic D. melanogaster models are usually established by hSOD1 gene expression among the MNs of D. melanogaster SOD1 null background applying GAL4/UAS yeast systems. In hSOD1 transgenic D. melanogaster models, very low levels of hSOD1WT expression are sufficient to reverse the lifespan reduction, oxidative stress increase, and physiological dysfunctions related to the SOD1 null D. melanogaster model (Mockett et al., 2003). In patients with ALS, it is very difficult to identify the complex genetics involved in its pathogenesis and to functionally investigate new candidate disease genes. D. melanogaster models can allow for an in-depth study of ALS progression from the earliest signs to terminal stages. They can rapidly perform a large range of simple assays from lifespan and motor assays to anatomical screens and can provide important functional information on the potential machinery required for proper motor neuron function and how this machinery may be dysregulated during ALS, which would not be possible in human neural systems. The D. melanogaster model is useful for addressing complex genetic diseases such as ALS. The UAS/Gal4/Gal80 system can upregulate and knock down D. melanogaster genes, including SOD1, and the ectopic expression of human SOD1 genes or mutations in a tissue-specific manner, exhibiting the typical pathologies of hmSOD1 ALS, which is beneficial for further investigation to identify disease-modifying genes, mutations, and disease pathways (Walters et al., 2019).
4.2 D. melanogaster carrying TDP-43 mutations
TDP-43 is a highly conserved and ubiquitously expressed nuclear protein that participates in various cellular processes, including mRNA splicing, transcription, stability, and transportation (Langellotti et al., 2018; Lembke et al., 2019). TDP-43 is crucial for promoting the formation and growth of neuromuscular junctions (NMJ) (Strah et al., 2020). A number of D. melanogaster models generated by the TDP-43 toxicity of endogenous D. melanogaster TDP-43 (dTDP) and hTDP-43 transgenic expression revealed that the TDP-43 protein showed toxicity, and the major phenotypes were largely similar in these D. melanogaster models. The D. melanogaster model lacking dTDP appears externally normal, but presents a deficiency in locomotion behaviors, a reduction in lifespan, anatomical defects of NMJ, and a decrease in dendrite branches (Feiguin et al., 2009; Lu et al., 2009; Lin et al., 2011). Among the above-mentioned TDP-43 D. melanogaster models, these phenotypes can be recovered and repaired by expressing the normal hTDP-43 protein in neurons, such as MNs (Feiguin et al., 2009). The phenotypes of TDP-43 D. melanogaster models revealed that the dysfunction of TDP-43 might result in the pathogenesis of ALS. In addition, overexpression of either dTDP or hTDP-43 in D. melanogaster also displayed the main pathological characteristics of ALS, premature lethality, neuronal loss, and defects in NMJ architecture and movement. TDP-43 D. melanogaster models revealed that the mechanisms of toxic GOF are related to TDP-43 proteinopathy (Lu et al., 2009; Elden et al., 2010; Hanson et al., 2010; Li et al., 2010; Ritson et al., 2010; Voigt et al., 2010; Estes et al., 2011; Miguel et al., 2011).
To study and identify the possible roles and potential pathogenic mechanisms of the TDP-43 protein in the pathogenesis of ALS, several TDP-43 transgenic D. melanogaster models have been developed to further explore whether gene modification of TDP-43 can increase or decrease the generation of TDP-43 toxicity (Elden et al., 2010; Hanson et al., 2010; Ritson et al., 2010). In these TDP-43 transgenic D. melanogaster models, upregulation of the human ATXN2 gene ortholog Pab1-binding protein 1 enhanced TDP-43 toxicity, resulting in more severe TDP-43-induced phenotypes (Elden et al., 2010). Moreover, the TDP-43 interacting partner ubiquilin 1 overexpression also produces similar TDP-43-induced phenotypes (Kim et al., 2009), reduced steady expression of TDP-43, and enhanced TDP-43 phenotypes (Hanson et al., 2010). The TDP-43-induced phenotypes among TDP-43 transgenic D. melanogaster models can also be modulated by co-expressing an ATPase member, valosin-containing protein (VCP), which is related to the protein family members of cellular activities that regulate many cell processes (Ritson et al., 2010). The above-described TDP-43 transgenic D. melanogaster models may enable the development of novel therapeutic targets that regulate TDP-43 expression in TDP-43-associated ALS patients, which is expected to prevent or treat TDP-43-associated ALS patients.
Bis-(2-ethylhexyl)-2,3,4,5-tetrabromophthalate (TBPH), a D. melanogaster TDP-43 homolog, promotes target gene transcription by combining with sequences rich in uridine guanine, thus stabilizing the binding of chromatin regulators to DNA (Zhang et al., 2018a). Overexpression of mutant or WT hTDP-43 and TBPH in transgenic D. melanogaster models affects lifespan, mobility, axonal transport, and pupal shell sealing, whereas TBPH depletion leads to movement disorders and a shortened lifespan (Walters et al., 2019; Liguori et al., 2021). Among them, hTDP-43WT overexpression in neurons results in an increase in NMJ buttons and branches associated with hTDP-43WT protein aggregation (Kankel et al., 2020). However, the phenotypes induced by the mutant hTDP-43 were not distinguishable from the hTDP-43WT induced phenotypes (Feldman et al., 2022).
Increased TDP-43 expression in TDP-43 transgenic D. melanogaster models induces mitochondrial dysfunction, including ridge abnormality and loss, resulting in a decrease in mitochondrial membrane potential and an increase in reactive oxygen species (ROS) production, usually accompanied by the loss of striated muscle tissue, and affects the survival and function of neurons due to excessive ROS production. Moreover, the TDP-43 expression increase also inhibited mitochondrial complex I activation and reduced mitochondrial ATP synthesis. Compared with the D. melanogaster control model, the volume of mitochondria in the eyes of D. melanogaster expressing TDP-43WT or TDP−43A315T was significantly reduced. TDP-43WT and TDP-43A315TGOF induced mitochondrial unfolded protein responses, including Lon and LonP1. LonP1 is a major mitochondrial matrix protease that interacts with TDP-43 to reduce mitochondrial TDP-43 protein levels, which can prevent mitochondrial damage and neurodegeneration in the D. melanogaster model induced by TDP-43 (Khalil et al., 2017; Wang et al., 2019; Layalle et al., 2021). Highly fragmented mitochondria have also been found among the axons of MNs in a D. melanogaster model expressing TDP-43 (Layalle et al., 2021). Sigma-1 receptor enhances the resistance of D. melanogaster to oxidative stress and exerts positive effects on motor activity and ATP levels (Couly et al., 2020).
In TDP-43 D. melanogaster models, significant changes were observed in glucose metabolism, including an increase in pyruvic acid levels. Among the MNs of TDP-43 D. melanogaster models, the mRNA level of phosphofructose kinase (an enzyme that controls the rate of fermentation) significantly increased (Manzo et al., 2019; Layalle et al., 2021). A high sugar diet can improve the exercise and life defect induced by TDP-43 protein lesions among MNs or/and glial cells, but it does not improve the muscles lesions in the TDP-43 D. melanogaster models, indicating that the metabolic disorders participate in the neural systems damage (Manzo et al., 2019). In addition, it has been revealed that some changes in the lipid metabolism, such as the reduction of both carnitine shuttle and lipid β oxidation, occur among TDP-43 D. melanogaster models (Layalle et al., 2021).
Among TDP-43 transgenic D. melanogaster models, other factors were found to have an impact on TDP-43. For example, the inhibition of protein tyrosine kinase 2 (PTK2) significantly reduces ubiquitin aggregation and cytotoxicity of TDP-43. Non-phosphorylated sequencesome 1 (SQSTM1) inhibits the accumulation and neurotoxicity of insoluble polyubiquitin proteins induced by TDP-43 overexpression in neuronal cells. TANK-binding kinase 1 (TBK1) participates in PTK2-mediated SQSTM1 phosphorylation. Therefore, the PTK2-TBK1-SQSTM1 axis plays a key role in the pathogenesis of TDP-43 by modulating neurotoxicity caused by damage to the ubiquitin-proteasome system in the TDP-43 transgenic D. melanogaster model (Lee et al., 2020). Recently, it was found that the generation of reversible droplet-like nuclear bodies (Wang et al., 2020), the overexpression of long non-coding RNA nuclear enriched abundant transcript 1–1 (Matsukawa et al., 2021), Mucuna pruriens and Withania somnifera (Maccioni et al., 2018; Paul et al., 2021; Zahra et al., 2022), and the production of CG5445 (a previously uncharacterized D. melanogaster gene) (Uechi et al., 2018, p. 3) alleviate the cytotoxicity of TDP-43 in neurons and improve movement or eye symptoms in the TDP-43 transgenic D. melanogaster model.
Gemin3 (DDX20 or DP103) is a dead-box RNA helicase that participates in a variety of cellular processes. The combination of Gemin3 and TDP-43 or FUS destruction aggravates vitality defects, motor dysfunction, and muscle atrophy while inhibiting the overgrowth of NMJ in the TDP-43 transgenic D. melanogaster model (Cacciottolo et al., 2019). A 3-fold knockdown of the lethal gene inhibitor (elongation factor of RNA polymerase II) significantly inhibited the morphological defects of the compound eye and medial retinae induced by TDP-43 in TDP-43 transgenic D. melanogaster models. D. melanogaster has five histone deacetylases (HDAC), among which Rpd3 knockdown significantly inhibits the rough eye phenotype caused by hTDP-43 in the TDP-43 transgenic D. melanogaster model (Yamaguchi et al., 2021). TBPH deficiency leads to larval death and reduced HDAC6 levels (Braems et al., 2021). Among the many single or double hydrophilic tag conjugating peptides D1-D8, D4 has the strongest ability to degrade TDP-43 protein and can penetrate the cell wall in short periods, inducing TDP-43 protein degradation in a dose- and time-dependent manner (Gao et al., 2019).
Reverse transposable element (RTE) expression was strongly upregulated in the head of TBPH-null D. melanogaster. TBPH regulates the silencing mechanism of small interfering RNA inhibiting RTE, and TBPH modulates Dicer 2 expression levels through direct protein mRNA interaction in TDP-43 transgenic D. melanogaster models (Romano et al., 2020). TBPH also interacted with VCP to inhibit VCP-induced degeneration in a TDP-43 transgenic D. melanogaster model (Braems et al., 2021).
4.3 D. melanogaster carrying FUS mutations
Although FUS mutations obviously affect MNs as well as other neurons found in FUS transgenic D. melanogaster models, its pathogenic mechanism of proteinopathy remains largely unknown. To study the alterations of FUS-associated functions in neurodegenerative diseases, including ALS, we reproduced the FUS transgenic D. melanogaster model expressing mutant human FUS (hmFUS), such as R518K, R521C, and R521H. These models cause a severe neurodegeneration in D. melanogaster eyes, whereas WT human FUS (hFUSWT) expression only led to mild neurodegeneration (Lanson et al., 2011).
Both movement defects and premature death have been observed in mutant FUS transgenic D. melanogaster. Moreover, mutant FUS overexpression increased the accumulation of the FUS protein. Causative roles on ALS-related hFUS mutants including both R524S and P525L in the FUS transgenic D. melanogaster model have also been described (Chen et al., 2011). Among the D. melanogaster models overexpressing WT and ALS-related FUS mutations among various neuron subsets, such as photoreceptors, mushroom bodies, and MNs, progressive age-related neuronal degeneration, such as axon loss, morphological alterations, and functional impairment of MNs, has been observed. The model of Cabeza (hFUS D. melanogaster homolog)-deficient D. melanogaster is used to study FUS functions, and the results showed both reduced lifespan and locomotor defects compared to controls. It was also found that transferring hFUSWT gene into this model could fully recover these phenotypes, but in this model co-expressing the ALS-related mutant FUS proteins, the phenotypes of reduced lifespan and locomotor defects were not recovered. This suggests that ALS-linked FUS mutations are toxic in FUS transgenic D. melanogaster models (Walker et al., 2011). In a transgenic D. melanogaster model coupled FUS with TDP-43 gene in neurons, it was demonstrated that FUS acts on ALS pathogen-associated effects together with TDP-43 via a common genetic pathway. Moreover, FUS and TDP-43 exert synergistic effects on RNA-dependent complexes. These models show that FUS and TDP-43 exert partial damage during the pathogenesis of ALS (Layalle et al., 2021).
D. melanogaster FUS homolog Cabeza is extensively expressed in the majority of tissues, including the neural system. Most Cabeza-D. melanogaster models with Cabeza functional loss are fatal, and only a few D. melanogaster are able to develop into adulthood, but exhibit severely shortened lifespans and motor disorders. Importantly, co-expression of wild-type Cabeza in the neurons of Cabeza-mutant D. melanogaster models can repair climbing and flight defects. Compared with Cabeza-mutant D. melanogaster models, larvae overexpressing Cabeza/FUSWT exhibited opposite NMJ electrophysiological phenotypes, characterized by reduced excitatory junction potentials and miniature excitatory junction potential amplitudes. In addition, it was found that the D. Melanogaster model may have a self-inhibitory mechanism for FUS/Cabeza, as the overexpression of FUS reduces the intracellular Cabeza content (Zhang et al., 2018a). D. melanogaster overexpressing hFUSWT exhibits abnormal eye morphology, shortened lifespan, eclosion defects, climbing defects, a reduced number of synaptic buttons, a reduced active region of the NMJ, and axonal degeneration (Azuma et al., 2018). hFUS is overexpressed in photoreceptor neurons in the eyes of D. melanogaster, resulting in mild retinal degeneration, rough eye surfaces, and a decrease in red pigment. The changes in the different FUS regions have different effects, such as in HEK293 cells, where the C-terminal truncated FUS chelates the FUSWT protein from the nucleus to the cytoplasm, thereby exacerbating FUS-induced retinal degeneration. The FUS-P525L mutation exacerbates FUS-induced retinal degeneration by increasing the FUS cytoplasmic distribution (Matsumoto et al., 2018; Layalle et al., 2021).
The arginine residue in the low-complexity domain of the FUS C-terminus is necessary for the maturation of FUS, while the N-terminal glutamine-glycine-serine-tyrosine (QGSY)-rich region (amino acids 1–164) and the C-terminal RGG2 domain are necessary for the toxicity of FUS, including the N-terminal QGSY, prion-like domains, and C-terminal RGG2 domains. It can distinguish between FUS and Cabeza toxicity based on its QGSY domain (Bogaert et al., 2018). The expression of different FUS mutations (R521C, 521H, 518K, R524S, or P525L) resulted in more severe rough-eye phenotypes in FUS transgenic D. Melanogaster models. FUS expression triggers Hippo activation and c-Jun N-terminal kinase signaling, leading to neuronal degeneration in FUS transgenic D. Melanogaster models. In addition, the Hippo signaling pathway is a modifier of Cabeza knockdown phenotypes (Layalle et al., 2021).
In FUS transgenic D. melanogaster models, it has been shown that inhibiting nuclear output can reduce hFUS-induced toxicity, thereby confirming the involvement of nuclear-cytoplasmic transport in the pathogenesis of ALS. It is worth noting that, recently, a series of complex enhancers and inhibitors have been found for ALS modifying factors screened in two different transgenic D. melanogaster models carrying human mutant FUS genes (Liguori et al., 2021). In the Cabeza knockdown D. melanogaster models, there are 14 inhibitory mutations knockdown models that effectively inhibit rough eye phenotypes caused by Cabeza knockdown. Mutations in Chameau and N-alpha-acetyltransferase 60 inhibit locomotion and cause morphological defects in NMJ synapses caused by Cabeza knockdown (Yamaguchi et al., 2021). FUS is similar to TDP-43 gene and regulates synaptic transmission among D. melanogaster NMJ; synaptic transmission defects precede the degeneration and loss of MNs in FUS transgenic D. melanogaster models. The nuclear-cytoplasmic transport proteins exportin-1 and nucleoporin-154 are modifiers of FUS toxicity, which prevent FUS-induced toxicity and reduce apoptosis of ventral nerve cord neurons (Braems et al., 2021).
In third-instar larvae of a Cabeza mutant D. melanogaster model, it was shown that the Cabeza mutant leads to an increase in induced and spontaneous neurotransmitter release in the NMJ (Zhang et al., 2018a). In addition, overexpression of hFUS led to a decrease in presynaptic activity areas and impaired synaptic transmission in hFUS transgenic D. melanogaster model. The loss and acquisition of FUS function in FUS and Cabeza transgenic D. melanogaster models may be achieved through dominantly negative mechanisms or the downregulation of endogenous Cabeza, which are mediated by interference with vesicular and mitochondrial transportation. This indicates that the Cabeza mutation leads to developmental and motor defects, which become more severe with age (Walters et al., 2019). Mutant FUS or D. melanogaster homolog Cabeza expressing type IV dendritic neurons leads to FUS or Cabeza cytoplasm mislocalization and axon transportation toward the pre-synapse end in FUS or Cabeza transgenic D. melanogaster models. Moreover, FUS or Cabeza overexpression leads to the progressive loss of neuronal projections and the reduction of synaptic mitochondria, while a large number of calcium transients appear in synapses by controlling the expression of calcium channels. In addition, mutant FUS overexpression results in a decrease in presynaptic synaptic-binding proteins, specifically disrupting axonal transport and inducing excessive excitability. Therefore, the mutation FUS/Cabeza damages the axon transportation of synapse vesicular proteins, thereby reducing the levels of synaptic-binding proteins, which are the main components of presynaptic release mechanisms. The overexpression of FUS can disrupt mitochondrial transport in D. melanogaster MNs, and an increase in FUS transport during neural biological processes alters the focal levels of synapse transcription or induces the formation of stress particles, thereby disrupting local translation and causing synaptic hyperexcitability (Machamer et al., 2018).
Karyopherin beta-2, also known as Kap-β-2 or transportin-1, participates in proline/tyrosine nuclear localization signal (PY-NLS), which inhibits and reverses the FUS fibrosis. In addition, it is worth noting that Kap-β2 can prevent RNA binding protein (RBP) with PY-NLS from accumulating among stress granules, restore the nucleus RBP localization and function, and rescue the neurodegeneration induced by ALS-related FUS in the FUS transgenic D. melanogaster models. The increase of Kap-β2 saves neurodegeneration and lifespan related to FUS in the FUS transgenic D. melanogaster models and reduces the accumulation of FUS in stress granules (Guo et al., 2018).
Knockdown of heat shock response gense in the D. melanogaster model expressing hFUSWT ω-LncRNA can reduce the level of hFUSWT mRNA and induce the generation of insoluble inclusion bodies comprising non-toxic hFUSWT proteins and lysosomal-associated membrane protein 1 in cytoplasm (Yamaguchi et al., 2021). Nuclear chromatin-binding protein (Xrp1), the main modifier of Cabeza mutative phenotypes, is strongly upregulated in the Cabeza-mutant D. melanogaster model, effectively reducing motor and life defects. Moreover, the Cabeza-mutant D. melanogaster model exhibits an imbalance in gene expression, and the Xrp1 heterozygosity can alleviate this (Mallik et al., 2018). Several genes, including TDP-43, Transportin-1, TER94/VCP, arginine methyltransferase, and the mitochondrial companion Hsp60 have been shown to interact with FUS/Cabeza genetically in the FUS/Cabeza transgenic D. melanogaster model (Zhang et al., 2018a).
4.4 D. melanogaster carrying C9orf72 mutations
C9orf72 is a common pathogenic gene involved in ALS. The present study showed that different cellular processes, including transcription, translation, nuclear-cytoplasmic transport, and protein degradation, are involved in the pathogenesis of C9orf72 ALS (Liguori et al., 2021). The D. melanogaster genome does not contain a direct homolog of human C9orf72, thus, an ALS-linked D. melanogaster model related to C9orf72 was developed by expressing the GGGGCC (G4C2) repeat (Yamaguchi et al., 2021). Although C9orf72 in ALS patients usually comprises over hundreds of G4C2 repeat sequences, the overexpression of 30–58 G4C2 repeat sequences in D. melanogaster eyes or MNs is sufficient to result in neuronal degeneration. Based on the expression level and time, the G4C2 repeat or arginine-rich dipeptide repeat (DPR) expressed among MNs leads to serious NMJ defects in the third-instar larvae of the C9orf72 G4C2 repeat transgenic D. melanogaster model (Zhang et al., 2018a). The amplification of the G4C2 hexanucleotide repeat expansion (HRE) of C9orf72 leads to the loss of C9orf72 function by inhibiting transcriptional extension and splicing of the first intron (Zhang et al., 2018a). The characteristic pathological hallmark observed among the different tissues of C9orf72 ALS, including MNs, is RNA lesions. RNA lesions are the result of HRE transcription, leading to the accumulation of repetitive RNA aggregates, usually located in the nucleus or cytosol (Layalle et al., 2021). This suggests that RNA-carrying HRE do not cause toxicity, but that the arginine-rich DPR sequence encoded by the HRE sequence mediates neurotoxicity (Moens et al., 2018). Consistent with this, the abnormal translation of HRE leads to the accumulation of DPR protein in the brains of ALS patients (Azoulay-Ginsburg et al., 2021). When comparing the effects of RNA expression among the HRE transgenic D. melanogaster model, only DPR proteins containing arginine have neurotoxicity on the G4C2 motifs encoding different dipeptide combinations (Bolus et al., 2020).
Poly (glycine-arginine) (GR) is a common genetic cause of ALS and FTD, occupying forty percent of fALS cases (Cunningham et al., 2020). Partial functional loss of an essential DNA repair protein Ku80 inhibits retinal degeneration induced by poly (GR) in poly (GR) transgenic D. melanogaster models. In the neurons of D. melanogaster model expressing poly (GR) and in patients with C9orf72, the Ku80 expression is significantly increased. Elevated Ku80 expression contributes to GR aggregation and induces neurodegeneration in a poly (GR) transgenic D. melanogaster model (Lopez-Gonzalez et al., 2019).
Arginine-rich DPR is a highly toxic product derived from the C9orf72 repeat amplification mutation and is a common cause of fALS. However, their role in synaptic regulation and excitatory toxicity remains unclear. Applying C9orf72 DPRs with different toxic intensities revealed that arginine-rich DPRs induced selective degeneration of glutamate neurons in the C9orf72-DPR transgenic D. melanogaster model. Among them, the C9orf72-DPR can stimulate synaptic overgrowth and promote glutamate release. Furthermore, an increase in glutamate release from excitatory neurons expressing DPR not only causes excitotoxicity in postsynaptic neurons, including MNs, but also produces cell-autonomous excitotoxicity in presynaptic neurons (Xu and Xu, 2018).
Up-frameshift 1 (UPF1) is an RBP with helicase activity that reduces DPR-mediated toxicity. Overexpression of UPF1 significantly reduces the severity of known neurodegenerative phenotypes and poly (GR) content without changing the amount of repeat RNA, indicating that UPF1 has a neuroprotective effect on C9orf72-ALS (Zaepfel et al., 2021). The present study shows that UPF1 expression regulates the DPR- or HRE-induced neurodegenerative phenotypes of C9orf72-ALS transgenic D. melanogaster models, including eye symptoms (Xu et al., 2019; Ortega et al., 2020; Sun et al., 2020). In addition, DPR can directly bind to nucleoporins, and knockdown of karyopherin alpha3 in the C9orf72-ALS transgenic D. melanogaster model can enhance the toxicity of DPR (Walters et al., 2019; Braems et al., 2021). Furthermore, the loss of C9orf72 function disrupts autophagy and lysosomal functions in many types of cells, including MNs (Ji et al., 2017; Shi et al., 2018; Zhu et al., 2020).
The key regulatory factor of autophagy, which is refractory to Sigma P/sequestosome 1 [Ref(2)P/p62], is an effective inhibitor of ALS induced by G4C2 HRE in C9orf72 (Cunningham et al., 2020). The microphthalmia/transcription factor E family of transcription factors is a key regulator of autophagy-lysosome function. Transcription factor EB (TFEB) is a candidate therapeutic target for ALS (Cortes and La Spada, 2019). It was found that C9orf72-HRE impaired the microphthalmia-associated transcription factor/transcription factor EB nuclear input, disrupted autophagy, and exacerbated the protein balance defect in the C9orf72 ALS transgenic D. melanogaster model. Increased TFEB activity may prevent neuronal death and promote recovery of neuronal function through different mechanisms in the C9orf72-ALS transgenic D. melanogaster model (Torra et al., 2018).
The G4C2 amplification repeat sequence and tau protein are traditionally considered to be related to partial clinical manifestations in several neurodegenerative diseases, such as ALS. Co-expression of the G4C2 repeat sequence and Tau can lead to the synergistic deterioration of rough eyes, movement functions, longevity, and abnormal NMJ morphology in the G4C2 repeat sequence and Tau co-expression transgenic D. melanogaster model. In addition, 30 G4C2 repeats increase tau phosphorylation levels (He et al., 2019). Moreover, the downregulation of D. melanogaster tau homologous protein can reduce neurodegeneration and motor lesions and prolong the lifespan of C9ORF72 G4C2 repeatedly expressing D. melanogaster model (Braems et al., 2021).
5 C. elegans models
The C. elegans genome was the first multicellular organism to be completely sequenced in 1998 (C. elegans Sequencing Consortium, 1998). C. elegans contains ~20,000 genes that are highly similar to the 25,000 human genes. Therefore, it is considered as an ideal model for analyzing the genetic and molecular mechanisms underlying neuronal development, function, and disease. Moreover, more than 42% of human disease-linked genes have an organology with C. elegans, suggesting that almost all biochemical pathways have been conserved throughout the evolution of C. elegans and humans (Culetto and Sattelle, 2000). C. elegans was the first model used to study the biochemical pathways of human diseases.
C. elegans is genetically tractable, which indicates that its genome can be easily manipulated. This characteristic is particularly advantageous in ALS research, in which the identification of genetic factors contributing to the disease is essential. Researchers can introduce specific mutations associated with ALS into the C. elegans genome to observe the resulting phenotypic changes. This approach helps elucidate the genetic basis of ALS and identify potential therapeutic targets (Clarke et al., 2018). Twenty-six neurons in five different neuronal types are gamma-aminobutyric acid (GABA)-ergic neurons in C. elegans (McIntire et al., 1993). Among them, 19 GABAergic neurons, called type D neurons, exert regular locomotion by providing dorsal-ventral cross inhibition to the body wall muscle (White et al., 1986; McIntire et al., 1993). In addition, C. elegans contains more than 50 cholinergic MNs that control locomotion (Liu et al., 2020). Therefore, C. elegans transgenic models are useful for studying the pathological alterations related to locomotor dysfunction. C. elegans is a hermaphrodite, ~1 mm in length, with almost 3 days of short generation cycles, and ~300 progenies in a large brood. It has a short lifespan of ~3 weeks and undergoes rapid generation cycles, allowing for quick observation of the effects of genetic manipulations across multiple generations. In ALS studies, this characteristic enables researchers to assess the progression of neurodegeneration and the inheritance patterns of genetic mutations associated with the disease in a relatively short timeframe (von Mikecz, 2022). C. elegans has a transparent anatomical body that can be used to visualize all cell types at developmental stages (Brenner, 1974). The transparent body of C. elegans is another advantage, especially for the visualization of cellular and subcellular events. Researchers can observe the development and degeneration of neurons in real-time, providing valuable insights into the mechanisms of ALS pathogenesis. This transparency aids in monitoring changes, such as protein aggregation, cellular death, and alterations in neuronal morphology. The experimental techniques available for C. elegans make it easy to observe and quantify cellular events such as neuronal cell death and protein inclusion. This is critical for ALS research, in which an understanding of the dynamics of these events is essential. The C. elegans transparent body, combined with advanced imaging techniques, facilitates detailed analysis of cellular changes associated with ALS. Another advantage of the C. elegans model is the ease of observation and quantification of neuronal cell death and protein inclusion using experimental techniques (Ma et al., 2022).
The whole embryo cellular lineages of C. elegans are completely clear, which makes it possible to track organogenesis from the earliest stage of embryo formation to the terminal stage of organ differentiation and morphogenesis (Sulston et al., 1983). In addition, the nervous system of C. elegans is very simple; it possess 302 neurons among 959 cells in an adult C. elegans body. Each neuron is located in a unique anatomical position (Sulston et al., 1983). Twenty neurons are located in the larynx, while the 282 remaining neurons are located in various ganglia, from head to tail, along the main longitudinal axon tract in the ventral spinal cord. Among the neurons that develop during embryogenesis, 80 MNs develop at the post-embryonic stage. C. elegans has a relatively simple nervous system with 302 neurons, making it an excellent model for studying neural circuits and functions (Takeishi et al., 2020). Understanding the molecular and cellular events that underlie neurodegeneration is crucial for ALS research. The simplicity of the C. elegans nervous system allows researchers to map and monitor individual neurons, thereby facilitating the identification of key genes and pathways involved in ALS pathology (Gois et al., 2020).
The structures of the C. elegans nervous system were highly reproducible in each generation, as described previously, and high-resolution images were obtained by electron microscopic reconstruction. Approximately 5,000 chemical synapses, 2,000 neuromuscular junctions, and 500 gap junctions have been described, and all connections of the entire neuronal circuit have been mapped in C. elegans models (White et al., 1986). In C. elegans models, it is relatively easy to achieve target gene knockdown or overexpression by RNAi by injecting dsRNA into the unique genes of interest, simply soaking C. elegans in the dsRNA medium or feeding C. elegans the bacterium carrying the targeted dsRNA (Fire et al., 1998; Maeda et al., 2001). In addition, primary culture resources for neurons and muscle cells can also be obtained by dissecting C. elegans, which can further optimize and stabilize the growth of embryonic cells (Christensen et al., 2002).
Although many disease-relevant genes have been identified in neurodegenerative disorders, including ALS, the mechanisms underlying the dysfunction and death of selective MNs remain poorly understood. Based on this, molecular conservation in the neuronal signaling pathway between invertebrates and vertebrates is high, and C. elegans contains almost all known signaling and neurotransmitter systems found in the nervous system of mammals (Bargmann, 1998). Therefore, C. elegans models have generally been used by researchers to investigate the mechanisms underlying neurodegenerative diseases, including ALS. Previous studies on C. elegans have provided valuable data and evidence for elucidating the molecular pathways involved in many human diseases, including ALS. Because of these advantages, C. elegans has attracted many researchers to use it as an in vivo model for researching pathological mechanisms in neurodegenerative diseases such as ALS, providing some perspective evidence for identifying potential pathogenic targets and developing new therapeutic measures against human diseases such as ALS (Figure 4).
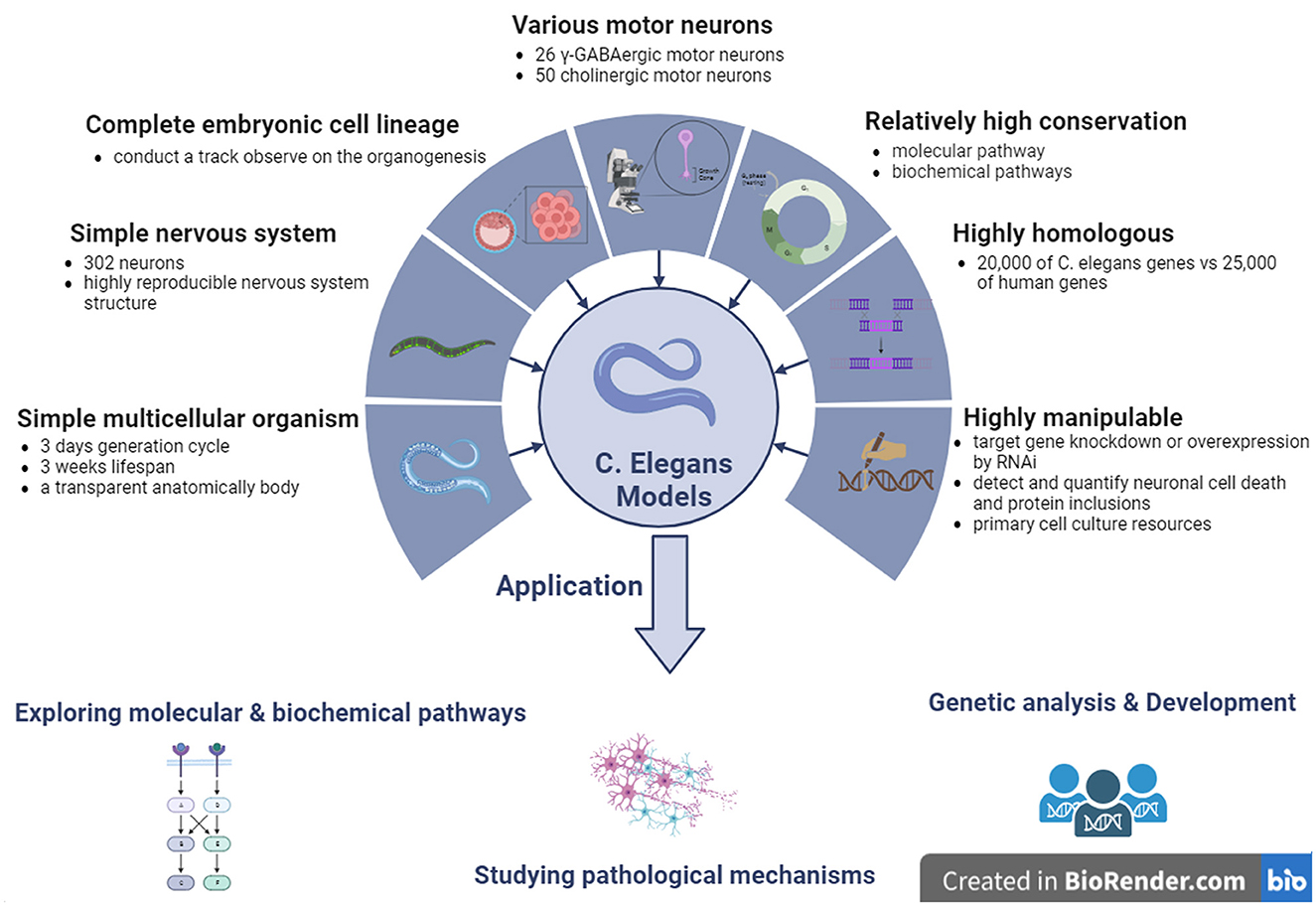
Figure 4. The features and applications of C. elegans genetic models in ALS. ALS, amyotrophic lateral sclerosis; C. elegans, Caenorhabditis elegans; γ-GABA, gamma-aminobutyric acid.
5.1 C. elegans carrying SOD1 mutations
Since C. elegans ALS models were first established in 2001 (Oeda et al., 2001), a series of C. elegans models have been developed, and many potential mechanisms associated with ALS pathogenesis have been identified. Among them, the hmSOD1 C. elegans model was produced by transferring wild-type human SOD1 (WThSOD1) and familial ALS (fALS) SOD1 mutations, such as A4V, G37R, and G93A, by controlling several promoters of the heat shock protein (HSP)-16.2 and myosin heavy chain 3 (myo-3) genes. The hmSOD1 transgenic C. elegans model controlled by the inducible HSP-16.2 promoter expresses hmSOD1 in nearly all tissues, including neurons, and the hmSOD1 C. elegan models controlled by the muscle-specific promoter of myo-3 largely express hmSOD1 among all muscles besides the pharynx.
The universal expression of hmSOD1 in C. elegans blocks natural biological responses to oxidative stress and induces the aggregation of toxic proteins, while the expression of hmSOD1 in all C. elegans neural systems leads to motor defects and neuron transmission damage in C. elegans models. The establishment of a SOD1 single copy (A4V, H71Y, L84V, G93A, and G85R) mutation knockout C. elegans model can analyze the unique effect of the special mutation on the degeneration of cholinergic and glutamic MNs in C. elegans models. These findings indicate that the pathogenesis of ALS is a neuronal subtype-specific gain of toxic functions, as well as a loss of physiological functions (Liguori et al., 2021). Moreover, loss of SOD1 function is the main cause of glutamate neuron degeneration after oxidative stress in C. elegans models, and the toxic functions gaining of SOD1 protein may cause cholinergic MNs degeneration in C. elegans models (Baskoylu et al., 2018). In the ALS-like hmSOD1 C. elegans model containing the ALS G85R mutation, a relatively high level of hmSOD1 was expressed in the neurons of C. elegans, resulting in movement defects of C. elegans expressing SOD1 G85R (Baskoylu et al., 2018).
Although morphological abnormalities of neurons and discernible survival or behavioral changes were not observed in the C. elegans model of hmSOD1 expression, the hmSOD1 C. elegans model demonstrated decreased resistance to paraquat-induced oxidative stress, significantly reducing the degradation ability of hmSOD1 proteins, leading to the abnormal accumulation of hmSOD1 proteins among muscular cells. Moreover, the final pathological phenotypes of hmSOD1 proteins in the C. elegans models were similar to the pathologically altered features observed in the post-mortem tissues of ALS patients.
The transgenic C. elegans model of entire neuronal expression of hSOD1 G85R using the synaptobrevin (snb-1) gene promoter coupled with yellow fluorescent protein (YFP) exhibits serious movement defects and paralysis (Wang et al., 2019). The observed phenotypes in this C. elegans model correlated with abnormal intra-neuronal aggregation of the mutated SOD1 protein. Other hmSOD1 genetic C. elegans models expressing various SOD1 mutants, such as G85R, G93A, and 127X, can be used to directly compare the differences in the aggregation, toxicity, and cellular interactions of hmSOD1 proteins induced by different SOD1 mutants. The hmSOD1-YFP C. elegans models expressing the SOD1 G85R, G93A, and 127X mutants in muscular cells using the muscle-specific promoter of unc-54 gene did not exhibit significant cell dysfunction (Gidalevitz et al., 2009). When these SOD1 mutations were transferred into the genetic background of temperature-sensitive mutations in C. elegans, the toxins produced by the mutated SOD1 protein were significantly enhanced, resulting in a variety of toxic phenotypes. Therefore, it was suggested that the hmSOD1 produced toxic phenotypes that might not only be the aggregate toxic products of hmSOD1 proteins but are also closely associated with genetic interactions in the genetic background.
The C. elegans body locomotion is controlled by excitatory (acetylcholine, glutamate) and inhibitory (GABA) neurons. Stable overexpression of hmSOD1 G93A mutation causes the degeneration of GABAergic MNs, whereas single-copy expression of hmSOD1 G93A mutation only causes oxidative stress-induced cholinergic and glutamic neurodegeneration (Braems et al., 2021). Endogenous SOD1 mutations, such as hmSOD1 G93A, trigger glutamate neuronal death in C. elegans (Baskoylu et al., 2018).
Metformin increased daf-1 and lgg-1 gene expression in hmSOD1 C. elegans model. The effects of Metformin on the lifespan of hmSOD1 C. elegans models were modulated by daf-16 and lgg-1 genes. In lgg-1 deficient C. elegans, metformin did not restore the movement, amount, or morphology of MNs (Senchuk et al., 2018). Metformin significantly prolonged the lifespan, improved exercise performance, and enhanced the antioxidant activity of hmSOD1 C. elegans, further indicating that metformin enhanced the expression of lgg-1, daf-16, skn-1, and other genes that regulate autophagy, longevity, and oxidative stress in hmSOD1 C. elegans models. Therefore, lgg-1 or daf-16 overexpression alleviates aging and pathological abnormalities in hmSOD1 C. elegans models, whereas genetic deletion of lgg-1 or daf-16 eliminates the beneficial effect of metformin in hmSOD1 C. elegans models. The median survival period of lgg-1 overexpression of hmSOD1G93A C. elegans increased relative to hmSOD1G93A C. elegans, indicating that activation of the lgg-1 gene reduces the neurotoxicity induced by hmSOD1 protein aggregation by inducing hmSOD1 protein degradation, thereby promoting the lifespan of hmSOD1 C. elegans. Although both lgg-1 and daf-16 extend the hmSOD1 C. elegans, no additive effects are observed when these two genes are simultaneously overexpressed (Alberti et al., 2010). A regulatory factor, ubiquitin-specific processing protease 7 (USP7), affects the elimination of misfolded proteins such as hmSOD1 and TDP-43. The USP7 effect is modulated by both the transforming growth factor β and the small mother against decapentaplegic (SMAD) pathways, is mediated by USP7-NEDD4L-SMAD2 axis, and its activation promotes the protein quality control. Furthermore, Math-33 deletion inhibited protein toxicity induced by hmSOD1 G85R mutation in the hmSOD1 G85R C. elegans model (Zhang et al., 2020).
5.2 C. elegans carrying TDP-43 mutations
The TDP-43 transgenic C. elegans models were reproduced by expressing wild-type human TDP-43 (hTDP-43WT) protein in neurons to study TDP-43 functional alterations and neurotoxicity in the pathogenesis of ALS (Ash et al., 2010). TDP-43 transgenic C. elegans models were generated using snb-1 gene promoters to drive human TDP-43 (hTDP-43) cDNA expression in whole neurons. This TDP-43 transgenic C. elegans model exhibited distinctive uncoordinated phenotypes, such as slow movement and inappropriate responses to stimuli. The C. elegans model first exhibits these phenotypes at the larval stage and constantly retains these phenotypes throughout adulthood. This C. elegans model indicated that these uncoordinated phenotypes correlated with the synaptic lesions of abnormal MNs. The mechanism by which abnormal nuclear TDP-43 activity leads to abnormal synapses is not yet known, but it indicates that excessive activation of the nuclear TDP-43 protein might change some components of RNA metabolism, such as alternative RNA splicing, resulting in the dysfunction of some special proteins necessary for normal synapse functions.
TDP-43 transgenic C. elegans models show that TDP-43 dysfunction results in insufficient chromatin processing and dsRNA accumulation, which may activate natural immunological systems and promote neuroinflammation (Milstead et al., 2023). It is currently understood that the neurodegenerative disease-related proteins, especially TDP-43 (ALS), amyloid beta and tau (Alzheimer's disease), α-synuclein (Parkinson's disease, PD), and FUS (frontotemporal dementia, FTD), partially show some characteristics of misfolding prion proteins. In addition, the TDP-43 C. elegans model also showed that the autophagy-lysosome pathway is the prion-like transmission pathway of TDP-43 protein (Sandhof et al., 2020). Meanwhile, it was discovered that the regulation of autophagy or exocytosis/endocytosis can affect the transfer of these misfolded proteins (Tyson et al., 2017). The TDP-43 protein is an intracellular protein but can promote prion-like transmission of TDP-43 by entering the lysosomal system in the TDP-43 C. elegans model (Peng et al., 2020). This suggests that the autophagy-lysosome system and endocytosis-dependent endolysosomal pathway are related to the elimination of TDP-43 protein aggregates (Liu et al., 2017).
The currently developed TDP-43 transgenic C. elegans model also showed some similar phenotypes in that normal hTDP-43WT overexpression in all neurons caused different scales of movement defects (Liachko et al., 2010). Moreover, the overexpression of many human ALS-related TDP-43 mutations, such as G290A, A315T, and M337V, causes a series of movement dysfunction phenotypes similar to those in ALS patients. It also indicates that these phenotypes of motor dysfunction worsen over time. The above-described similar phenotypes reproduced by the mutant TDP-43 transgenic C. elegans models reproduce major features seen in ALS patients, such as progressive muscle weakness and atrophy, shortened life span, and MNs degeneration accompanied by both hyperphosphorylation, truncation, and ubiquitination of TDP-43 proteins and the accumulation of detergent-insoluble TDP-43 proteins. The TDP-43 C. elegans model provides an ideal in vivo model for exploring the cellular and molecular mechanisms underlying ALS. These TDP-43 C. elegans models may be used to search for potential alterations in TDP-43 function in the pathogenesis of ALS, possibly revealing the neurotoxic mechanisms of TDP-43 associated with ALS, and ultimately finding possible targets for developing novel therapeutic measures.
Twenty-four human homologous genes that inhibit TDP-43 motor dysfunction after a reduction in RNAi-targeted gene expression have been identified. These genes involve multiple pathways, including the energy production and metabolism genes cox-10, F23F12.3, F55G1.5, paqr-1, and cox-6A, the extracellular matrix and cytoskeleton genes col-89, glie-8, hs-5, sax-2, vab-9, and zig3, the ion transport genes cnnm-3, C13B4.1, and unc-77, the nucleic acid function genes dna-2, tbx-11, and umps-1, protein balance genes C47E12.3, gpx7, and pcp-5 and the signal transduction genes F31E3.2, F40B5.2, and Y44E3A4 (Liachko et al., 2019). Human TDP-43 and C. elegans TDP-1 are functional homologs with similar RNA-binding activity, conserved N-terminus, nuclear localization signal (NLS), and RNA recognition motifs (Mitra et al., 2019). The C. elegans models of human TDP-43 or C. elegans TDP-1 expression in entire neurons produce the uncoordinated, slow, and GABAergic MNs constriction. In addition, the C. elegans model of targeted mutant TDP-43 expression in C. elegans GABAergic MNs can lead to age-dependent progressive paralysis, GABAergic neuronal degeneration, and synaptic lesions. Mutated exogenous TDP-43 induces oxidative stress and aberrant expression of endogenous TDP-1 decreases neuronal function and lifespan in C. elegans models. Excessive phosphorylation of TDP-43 can interfere with protein homeostasis, leading to neurotoxicity in C. elegans model. Among the kinases involved, cyclin-dependent kinase 7 is directly involved in TDP-43 phosphorylation and decreases TDP-43 phosphorylation by inhibiting the produce of this enzyme in C. elegans model (Liguori et al., 2021). TDP-1 deficiency or deletion leads to dsRNA accumulation and enhances exogenous RNAi by increasing nuclear RNAi in C. elegans. C. elegans nuclear RNAi involves chromatin alterations that are regulated by heterochromatin protein like-2 (HPL-2) and the homolog of heterochromatin protein 1 (HP1). TDP-1 interacts directly with HPL-2, and the TDP-1 deletion significantly changed the chromatin association of HPL-2. These molecular changes, replicated in C. elegans models, indicate that the nuclear depletion of hTDP-43 might result in alterations in disease-related RNA metabolism during the pathogenesis of ALS (Saldi et al., 2018).
5.3 C. elegans carrying FUS mutations
FUS is involved in various functions associated with DNA and RNA processing (Birsa et al., 2020). Prominent features of the mutant FUS C. elegans model include cytoplasmic mislocalization, generation of FUS aggregates, age-dependent motor defects, paralysis, and impaired GABAergic neurotransmission. In addition, the transmission of MNs to muscle tissues is reduced through ultrastructural and electrophysiological methods, indicating a role for FUS in synaptic vesicle tissue and nerve transmission (Braems et al., 2021). To establish the ALS-FUS C. elegans model, the FUS R524S and P525L allele mutations are inserted into the c-terminus NLS of endogenous fust-1 gene in chromosome 2 to generate an ALS-FUS animal model. Overexpression of FUS leads to severe motor defects and damages neurons and muscle autophagy without damaging the neuronal ubiquitin-proteasome pathway in FUS C. elegans models (Baskoylu et al., 2022). FUS C. elegans models have also shown that autophagy dysfunction may contribute to the development of ALS. However, the mode and degree of autophagy dysfunction in ALS pathogenesis are not fully understood (Chua et al., 2022). Excessive expression of FUS in C. elegans models affects the self-regulation of FUS, thereby affecting RNA processing, stability, and protein homeostasis. The irreversible hydrogel formed by the mutant FUS gene impairs the function of heterogeneous ribonucleoprotein particles, reducing the speed of new protein synthesis in mutant FUS C. elegans models (Markert et al., 2020). Disruption of these functions occurs by negatively affecting autophagy. The steady-state level of FUS is crucial for normal neuronal function, as increased FUS levels accelerate diseases such as ALS (Zhang et al., 2018b).
5.4 C. elegans carrying C9ORF72 mutations
The mutative C9ORF72 gene results in the loss of its functions, gain of toxic functions, or both; however, whether these C9ORF72 dysfunctions are involved in the pathogenesis of ALS is not yet fully understood. It has been revealed that the C9ORF72 pathogenic GGGGCC (G4P2) repeat mutation significantly decreases C9ORF72 expression. The C. elegans model harboring the 3rd and 4th exon deletion in C. elegans ALS-associated gene homolog alfa-1 led to a drastic reduction in C9ORF72 expression, exhibiting a movement defect resulting in age-dependent paralysis and the degeneration of GABA-ergic MNs. Moreover, the mutant alfa-1 C. elegans model showed osmotic stress sensitivity to provoke MNs degeneration.
The causative G4P2 repeat among the C9ORF72 gene is the key cause of familial and sporadic ALS found up to date. It is not known whether the C9ORF72 functions are the same as those of other ALS-related genes such as TDP-43 and FUS. It is important to note that C9ORF72 expression is also reduced in many ALS cases with the negative pathogenic G4P2 repeat, suggesting that C9ORF72 may exert synergistic effects with other ALS-related genes. In order to further understand the C9ORF72 potential synergistic effects with other ALS-related genes, the C. elegans model carrying mutative TDP-43 A315T or FUSΔUS5 combined with the null alfa-1 mutation were established, which showed that the null alfa-1 expression decreases and the paralytic phenotypes exacerbates but does not change the toxic of mutative FUS protein, which suggests that the mutative C9ORF72 gene exerts a synergistic effect with the mutative TDP-43 gene in the same MNs toxic pathway but not the mutative FUS gene. Reduced alfa-1/C9ORF72 expression enhanced the toxicity of the mutated TDP-43 protein but did not affect the FUS protein in the C. elegans model. This suggests that the genetic network is composed of many genes involved in the pathogenesis of ALS, where ALS-related genes interact each other based on the study in this C. elegans model. The model development of genetic networks in C. elegans is helpful for understanding the synergistic toxic mechanisms of ALS-related genes in neuronal loss in ALS (Therrien and Parker, 2014a).
C. elegans models studying the mechanism associated with the gain or loss of C9ORF72 gene functions revealed that the deletion of C9ORF72 gene leads to early serious paralysis, nuclear transport impairment, lysosomal homeostasis dysregulation, neurodegeneration, and neuronal death. The C9ORF72 C. elegans model can be used to investigate endocytosis, which reveals that alfa-1 deletion causes a defect in lysosome homeostasis, such as the dysfunction of lysosome reformate and the degradation of endocytosal elements; thus, the C9ORF72 C. elegans model is the first chosen model that is used to identify whether or not C9ORF72 is involved in the endocytosis pathway (Therrien and Parker, 2014b).
Partial neurodegenerative diseases have common or overlapping pathogenic mechanisms at the cellular level, such as protein misfolding, excitotoxicity, and altered RNA homeostasis. Moreover, genetic factors contributing to common, cross, or overlapping pathogenesis imply the existence of a possible genetic network of neurodegeneration in the pathogenesis of ALS. The recently discovered overlapping pathogenic mutation C9ORF72 in both ALS and FTD is perhaps the best illustration. It was thought that the different neurodegenerative diseases were distinct entities and possessed a single genetic spectrum; however, the current study findings are changing this opinion, considering that some neurodegenerative diseases have a similar genetic spectrum. Following the progression of the related genetic discovery, there is an urgent need to establish a novel genetic model to not only investigate the causative mechanisms associated with the pathogenic mutations of neurodegeneration but also to study the possible genetic interactions among the different mutated genes, which may reveal novel pathogenic targets. Based on the evolutionary conservation of various pathogenic genes with C. elegans, the C. elegans genetic models may be the first candidates to study the genetic interactions among these pathogenic genes; therefore, C. elegans genetic models, such as the mutative C9ORF72 gene, may be used to model both ALS and FTD to study the potential genetic interactions and networks in the pathogenesis of ALS and FTD (Therrien and Parker, 2014b).
6 Conclusions and future perspectives
Establishing invertebrate models provides new approaches and tools for a better understanding of ALS (Tables 1–3). Various transgenic invertebrate models have been widely used to explore ALS, especially fALS. Transgenic invertebrate models are not ideal for studying sporadic ALS or searching for drugs to cure and/or prevent ALS. Novel evidence from the experimental results of small animal models, such as invertebrate animals, to human clinical trials is very limited and challenging in the investigation of ALS because of the significant differences in the genomes, molecules, and anatomy between invertebrate animals and humans. For example, some invertebrate genetic models display mild ALS phenotypes without neurodegeneration. The shorter lifetime of most invertebrates is not long enough to induce the typical neurodegeneration that occurs in humans. In addition, only a small portion of alternative splicing transcription exons is conserved between humans and invertebrates. These differences between invertebrates and humans might result in substantial differences between the results from invertebrates and humans for ALS, such as pathogenesis, pathology, and medical treatment (Zhu et al., 2023). Of course, the large animal models such as vertebrates, monkeys, and orangutans have smaller species differences in neuropathology. These are the optimal models for studying the pathogenesis, pathology, and treatment of human diseases. However, there are also many disadvantages such as high cost, gene targeting inefficiency, the limitations of resources and ethics, and longer time, which are not beneficial for the extensive use of large animal models (Zhu et al., 2023). The combining application of invertebrate and vertebrate animal models can better mimic ALS and is beneficial for detecting potential causative pathways and effective measures to cure and/or prevent ALS. Generally, a better understanding of the advantages and limitations of different animal models will enable better use of different animal models to mimic ALS. In the future, the development of new invertebrate models will focus on establishing invertebrate models with different mechanisms, including genetics, which will hopefully reveal novel evidence in the pathogenesis and etiology of ALS and reveal for novel strategies to cure or prevent ALS (Myszczynska and Ferraiuolo, 2016).
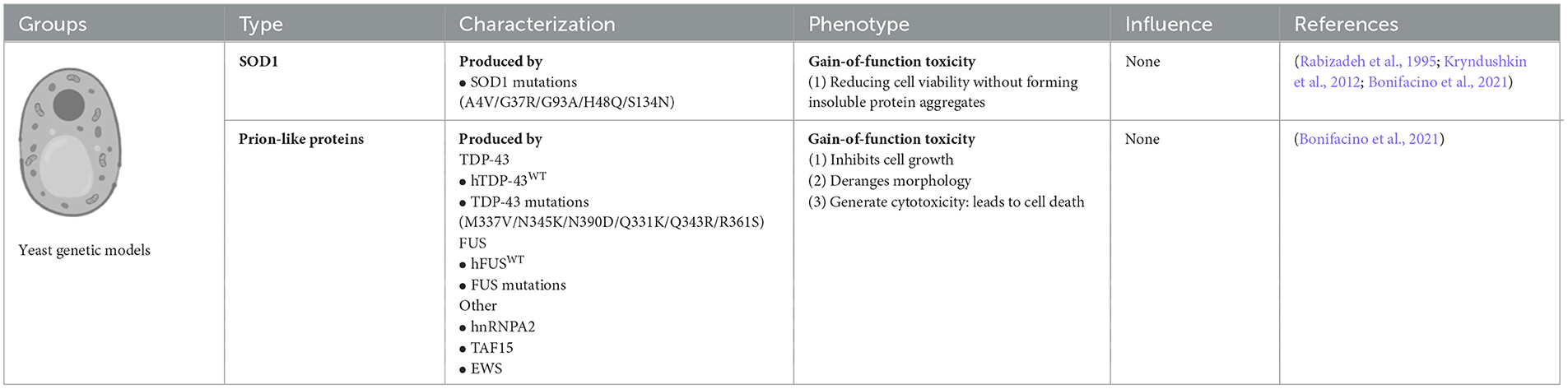
Table 1. Comparison of invertebrate genetic models of ALS: yeast.

Table 2. Comparison of invertebrate genetic models of ALS: Drosophila melanogaster.
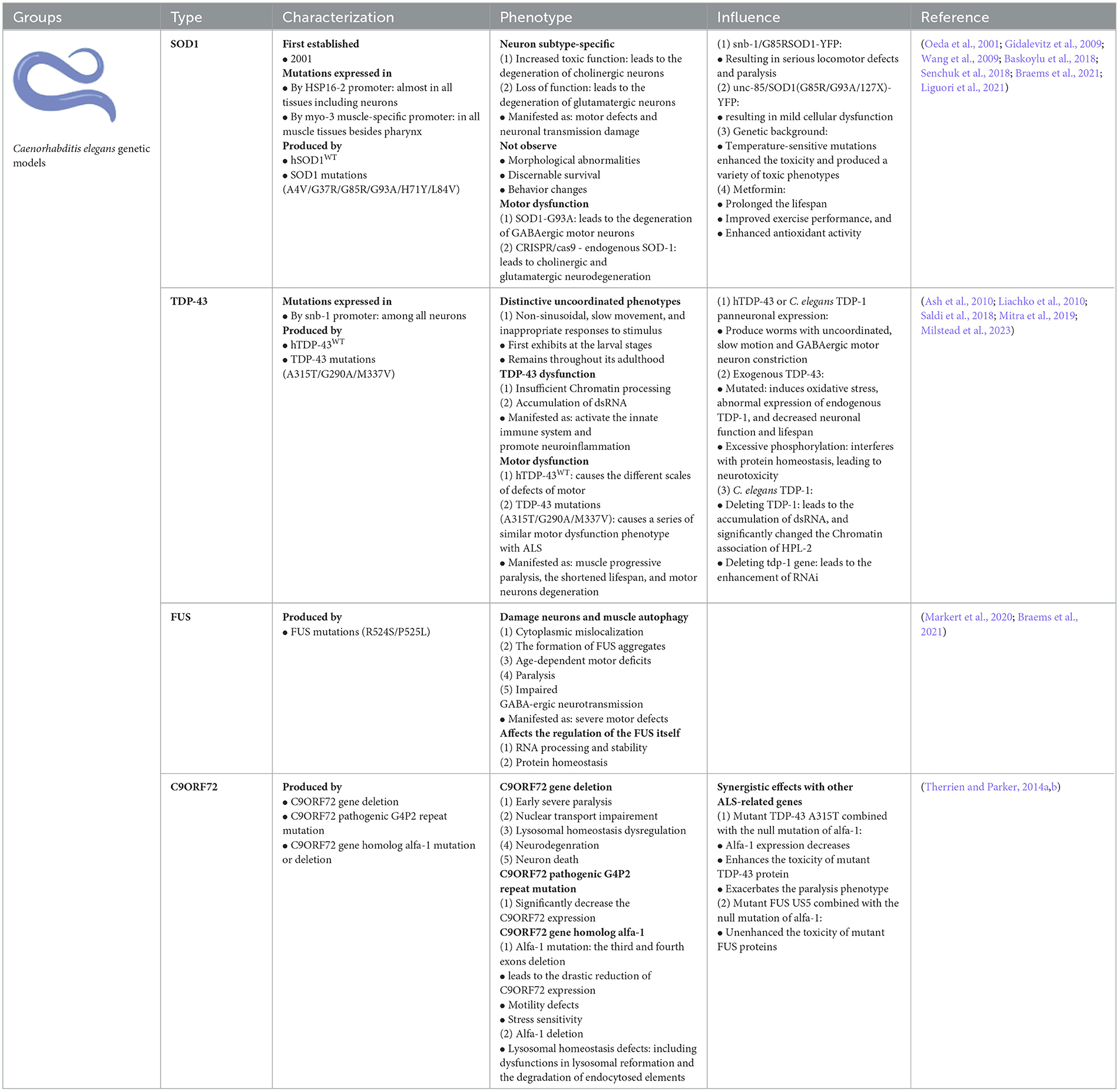
Table 3. Comparison of invertebrate genetic models of ALS: Caenorhabditis elegans.
There are advantages of using invertebrate models over zebrafish, mice, and induced pluripotent stem cell (iPSC) models in ALS studies ALS is an aggressive and fatal degenerative disease that damages the nervous system. To study the pathogenesis of ALS, a series of in vivo and in vitro models, including yeast, flies, worms, zebrafish, mice, and human iPSCs from patients with ALS, have been established one after another. Although mouse models and invertebrate model zebrafish are the major small-animal models of ALS, the discovery of iPSCs provides a novel opportunity to study the molecular phenotypes of ALS within human cells. It is important that iPSC technology can model both familial and sporadic ALS in relevant human genetic backgrounds as well as research drug development and the potential pathogenesis of ALS through personalized or targeted iPSC-intervening mice and zebrafish models. Further identification of iPSCs using mouse and zebrafish models might still be necessary to determine which phenotypes, responsible genes, and putative target compounds most likely reflect upstream disease driving, as opposed to epiphenomenon-related or even compensatory mechanisms (Hawrot et al., 2020). In general, invertebrate zebrafish, vertebrate mice, and iPSCs models are complementary to ALS studies.
There are also advantages of using in vivo over in vitro studies. Using in vivo models such as animal models or human trials allows for a better simulation of the complexity of diseases, including their etiology, progression, and response to treatment. In contrast, in vitro studies typically provide limited information and struggle to capture the intricate changes that occur throughout the disease process. In vitro studies are often conducted under simplified conditions, making it challenging to replicate diverse interactions within living organisms. This may lead to inaccuracies in the results when compared with the in vivo situation (Myszczynska and Ferraiuolo, 2016; Gois et al., 2020).
Maintaining physiological relevance can be a challenge in in vitro studies, which require adjustments to align with the physiological conditions found in living organisms. However, complete replication may not be possible. In contrast, in vivo research maintains higher biological complexity throughout the organism. The in vivo environment encompasses cells, tissues, organs, and their interactions, providing a realistic representation of biological phenomena (Myszczynska and Ferraiuolo, 2016; Gois et al., 2020).
A rich network of interactions between different cells and organs within living organisms is crucial for studying the comprehensive impact of diseases. In vivo models capture the dynamics of these networks. In vitro studies may struggle to accurately replicate the complex interactions between cells and organs, limiting our understanding of the overall biological system. Although in vitro studies offer insights into cellular responses, their limitations are evident when assessing the effects of drugs on an entire living organism. In this context, in vivo research provides a more direct assessment of the effects of drugs on the overall biological system, thereby enhancing the accuracy of predicting potential therapeutic efficacy and side effects (Myszczynska and Ferraiuolo, 2016; Gois et al., 2020; Bonifacino et al., 2021).
Optogenetic strategies are promising candidates for exploring the use of invertebrate models to develop novel genetic models to investigate the pathological mechanisms of ALS. Optogenetics is a novel advanced technology used to identify potential therapeutic strategies that are achieved by regulating ion flow and electrical signals in neuronal cells and neural circuits damaged by disease. At present, optogenetics has not been extensively applied to animal models of neurodegenerative diseases such as ALS and PD. Recently, human and D. melanogaster receptors were activated by light and rearranged during transfection, creating one-component optogenetic tools called Opto-hRET and Opto-dRET. These rearranged receptors strongly induced the MAPK/ERK proliferative signaling pathway in cells cultured under blue light stimulation. Light activation of Opto-dRET suppressed mitochondrial defects, tissue degeneration, and behavioral deficits in PINK1B9 D. melanogaster of a familial PD-associated kinase, PTEN-induced putative kinase 1 (PINK1) loss. Mitochondrial fragmentation was rescued by applying Opto-dRET through the PI3K/NF-κB pathway in human cells with a loss of PINK1 function. This optogenetic evidence demonstrated that a light-activated receptor can ameliorate disease hallmarks in a genetic model of PD. Similarly, optogenetic methods may be used in ALS models, including in vivo and in vitro models, to study the pathogenesis, pathology, and potential treatments for ALS. The use of invertebrate models to develop novel genetic and optogenetic strategies to investigate the pathological mechanisms of ALS is a promising research tool (Ingles-Prieto et al., 2021).
Author contributions
LZ: Writing—original draft, Writing—review & editing. RX: Conceptualization, Funding acquisition, Writing—original draft, Writing—review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Open access funding was provided by the Committee of the National Natural Science Foundation of China (82160255), Jiangxi Provincial Department of Science and Technology (20192BAB205043), and the Health and Family Planning Commission of Jiangxi Province (202210002 and 202310119).
Acknowledgments
This review is partially adapted from the P81–84, 85–92, 109–124 in chapter 4 of the book of in vivo and in vitro Models to Study Amyotrophic Lateral Sclerosis edited by François Berthod and François Gros-Louis in Center LOEX de l'Université Laval, Center de recherche FRSQ du Center hospitalier affilié universitaire de Québec, Département de Chirurgie, Faculté de Médecine, Université Laval, Québec, Canada. We are honestly and sincerely grateful to them for their valuable work, and some information cited in their book is in our manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alberti, A., Michelet, X., Djeddi, A., and Legouis, R. (2010). The autophagosomal protein LGG-2 acts synergistically with LGG-1 in dauer formation and longevity in C. elegans. Autophagy 6, 622–633. doi: 10.4161/auto.6.5.12252
An, L., and Harrison, P. M. (2016). The evolutionary scope and neurological disease linkage of yeast-prion-like proteins in humans. Biol. Direct 11, 32. doi: 10.1186/s13062-016-0134-5
Ash, P. E. A., Zhang, Y.-J., Roberts, C. M., Saldi, T., Hutter, H., Buratti, E., et al. (2010). Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 19, 3206–3218. doi: 10.1093/hmg/ddq230
Azoulay-Ginsburg, S., Di Salvio, M., Weitman, M., Afri, M., Ribeiro, S., Ebbinghaus, S., et al. (2021). Chemical chaperones targeted to the endoplasmic reticulum (ER) and lysosome prevented neurodegeneration in a C9orf72 repeat expansion Drosophila amyotrophic lateral sclerosis (ALS) model. Pharmacol. Rep. 73, 536–550. doi: 10.1007/s43440-021-00226-2
Azuma, Y., Mizuta, I., Tokuda, T., and Mizuno, T. (2018). Amyotrophic lateral sclerosis model. Adv. Exp. Med. Biol. 1076, 79–95. doi: 10.1007/978-981-13-0529-0_6
Bargmann, C. I. (1998). Neurobiology of the Caenorhabditis elegans genome. Science. 282, 2028–2033. doi: 10.1126/science.282.5396.2028
Baskoylu, S. N., Chapkis, N., Unsal, B., Lins, J., Schuch, K., Simon, J., et al. (2022). Disrupted autophagy and neuronal dysfunction in C. elegans knockin models of FUS amyotrophic lateral sclerosis. Cell Rep. 38, 110195. doi: 10.1016/j.celrep.2021.110195
Baskoylu, S. N., Yersak, J., O'Hern, P., Grosser, S., Simon, J., Kim, S., et al. (2018). Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 14, e1007682. doi: 10.1371/journal.pgen.1007682
Birsa, N., Bentham, M. P., and Fratta, P. (2020). Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. Cell Dev. Biol. 99, 193–201. doi: 10.1016/j.semcdb.2019.05.023
Bogaert, E., Boeynaems, S., Kato, M., Guo, L., Caulfield, T. R., Steyaert, J., et al. (2018). Molecular dissection of FUS points at synergistic effect of low-complexity domains in toxicity. Cell Rep. 24, 529–537. doi: 10.1016/j.celrep.2018.06.070
Boillée, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 312, 1389–1392. doi: 10.1126/science.1123511
Bolus, H., Crocker, K., Boekhoff-Falk, G., and Chtarbanova, S. (2020). Modeling neurodegenerative disorders in Drosophila melanogaster. Int. J. Mol. Sci. 21, 3055. doi: 10.3390/ijms21093055
Bonifacino, T., Zerbo, R. A., Balbi, M., Torazza, C., Frumento, G., Fedele, E., et al. (2021). Nearly 30 years of animal models to study amyotrophic lateral sclerosis: a historical overview and future perspectives. IJMS 22, 12236. doi: 10.3390/ijms222212236
Braems, E., Tziortzouda, P., and Van Den Bosch, L. (2021). Exploring the alternative: fish, flies and worms as preclinical models for ALS. Neurosci. Lett. 759, 136041. doi: 10.1016/j.neulet.2021.136041
Braun, R. J. (2015). Ubiquitin-dependent proteolysis in yeast cells expressing neurotoxic proteins. Front. Mol. Neurosci. 8, 8. doi: 10.3389/fnmol.2015.00008
Brenner, D., and Freischmidt, A. (2022). Update on genetics of amyotrophic lateral sclerosis. Curr. Opin. Neurol. 35, 672–677. doi: 10.1097/WCO.0000000000001093
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. doi: 10.1093/genetics/77.1.71
Cacciottolo, R., Ciantar, J., Lanfranco, M., Borg, R. M., Vassallo, N., Bordonn,é, R., et al. (2019). SMN complex member Gemin3 self-interacts and has a functional relationship with ALS-linked proteins TDP-43, FUS and Sod1. Sci. Rep. 9, 18666. doi: 10.1038/s41598-019-53508-4
C. elegans Sequencing Consortium (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018. doi: 10.1126/science.282.5396.2012
Chen, Y., Yang, M., Deng, J., Chen, X., Ye, Y., Zhu, L., et al. (2011). Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell 2, 477–486. doi: 10.1007/s13238-011-1065-7
Cherry, J. M., Hong, E. L., Amundsen, C., Balakrishnan, R., Binkley, G., Chan, E. T., et al. (2012). Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res. 40, D700–705. doi: 10.1093/nar/gkr1029
Chia, R., Chi,ò, A., and Traynor, B. J. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. The Lancet Neurol. 17, 94–102. doi: 10.1016/S1474-4422(17)30401-5
Christensen, M., Estevez, A., Yin, X., Fox, R., Morrison, R., McDonnell, M., et al. (2002). A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron 33, 503–514. doi: 10.1016/S0896-6273(02)00591-3
Chua, J. P., De Calbiac, H., Kabashi, E., and Barmada, S. J. (2022). Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy 18, 254–282. doi: 10.1080/15548627.2021.1926656
Clarke, E. K., Rivera, G., omez, K. A., Mustachi, Z., Murph, M. C., and Schvarzstein, M. (2018). Manipulation of ploidy in Caenorhabditis elegans. J. Vis. Exp. 57296. doi: 10.3791/57296
Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillée, S., Rule, M., et al. (2003). Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302, 113–117. doi: 10.1126/science.1086071
Cortes, C. J., and La Spada, A. R. (2019). TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: Molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol. Dis. 122, 83–93. doi: 10.1016/j.nbd.2018.05.012
Couly, S., Khalil, B., Viguier, V., Roussel, J., Maurice, T., and Liévens, J.-C. (2020). Sigma-1 receptor is a key genetic modulator in amyotrophic lateral sclerosis. Hum. Mol. Genet. 29, 529–540. doi: 10.1093/hmg/ddz267
Culetto, E., and Sattelle, D. B. (2000). A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum. Mol. Genet. 9, 869–877. doi: 10.1093/hmg/9.6.869
Cunningham, K. M., Maulding, K., Ruan, K., Senturk, M., Grima, J. C., Sung, H., et al. (2020). TFEB/Mitf links impaired nuclear import to autophagolysosomal dysfunction in C9-ALS. Elife 9, e59419. doi: 10.7554/eLife.59419.sa2
De Graeve, S., Marinelli, S., Stolz, F., Hendrix, J., Vandamme, J., Engelborghs, Y., et al. (2013). Mammalian ribosomal and chaperone protein RPS3A counteracts α-synuclein aggregation and toxicity in a yeast model system. Biochem. J. 455, 295–306. doi: 10.1042/BJ20130417
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Elden, A. C., Kim, H.-J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
Estes, P. S., Boehringer, A., Zwick, R., Tang, J. E., Grigsby, B., and Zarnescu, D. C. (2011). Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 20, 2308–2321. doi: 10.1093/hmg/ddr124
Feiguin, F., Godena, V. K., Romano, G., D'Ambrogio, A., Klima, R., and Baralle, F. E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592. doi: 10.1016/j.febslet.2009.04.019
Feldman, E. L., Goutman, S. A., Petri, S., Mazzini, L., Savelieff, M. G., Shaw, P. J., et al. (2022). Amyotrophic lateral sclerosis. Lancet 400, 1363–1380. doi: 10.1016/S0140-6736(22)01272-7
Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., and Mello, C. C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. doi: 10.1038/35888
Gao, N., Huang, Y.-P., Chu, T.-T., Li, Q.-Q., Zhou, B., Chen, Y.-X., et al. (2019). TDP-43 specific reduction induced by Di-hydrophobic tags conjugated peptides. Bioorg. Chem. 84, 254–259. doi: 10.1016/j.bioorg.2018.11.042
Gidalevitz, T., Krupinski, T., Garcia, S., and Morimoto, R. I. (2009). Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet 5:e1000399. doi: 10.1371/journal.pgen.1000399
Gois, A. M., Mendonça, D. M. F., Freire, M. A. M., and Santos, J. R. (2020). In Vitro and in vivo models of amyotrophic lateral sclerosis: an updated overview. Brain Res. Bull. 159, 32–43. doi: 10.1016/j.brainresbull.2020.03.012
Gros-Louis, F., Gaspar, C., and Rouleau, G. A. (2006). Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 956–972. doi: 10.1016/j.bbadis.2006.01.004
Guo, L., Kim, H. J., Wang, H., Monaghan, J., Freyermuth, F., Sung, J. C., et al. (2018). Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173, 677–692. doi: 10.1016/j.cell.2018.03.002
Halfmann, R., Wright, J. R., Alberti, S., Lindquist, S., and Rexach, M. (2012). Prion formation by a yeast GLFG nucleoporin. Prion 6, 391–399. doi: 10.4161/pri.20199
Hanson, K. A., Kim, S. H., Wassarman, D. A., and Tibbetts, R. S. (2010). Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS). J. Biol. Chem. 285, 11068–11072. doi: 10.1074/jbc.C109.078527
Hawrot, J., Imhof, S., and Wainger, B. J. (2020). Modeling cell-autonomous motor neuron phenotypes in ALS using iPSCs. Neurobiol. Dis. 134, 104680. doi: 10.1016/j.nbd.2019.104680
He, H., Huang, W., Wang, R., Lin, Y., Guo, Y., Deng, J., et al. (2019). Amyotrophic Lateral Sclerosis-associated GGGGCC repeat expansion promotes Tau phosphorylation and toxicity. Neurobiol. Dis. 130, 104493. doi: 10.1016/j.nbd.2019.104493
Horvitz, H. R., and Sulston, J. E. (1980). Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics 96, 435–454. doi: 10.1093/genetics/96.2.435
Ingles-Prieto, A., Furthmann, N., Crossman, S. H., Tichy, A. M., Hoyer, N., Petersen, M., et al. (2021). Optogenetic delivery of trophic signals in a genetic model of Parkinson's disease. PLoS Genet. 17, e1009479. doi: 10.1371/journal.pgen.1009479
Ji, Y. J., Ugolino, J., Brady, N. R., Hamacher-Brady, A., and Wang, J. (2017). Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy 13, 1254–1255. doi: 10.1080/15548627.2017.1299312
Kankel, M. W., Sen, A., Lu, L., Theodorou, M., Dimlich, D. N., McCampbell, A., et al. (2020). Amyotrophic lateral sclerosis modifiers in Drosophila reveal the phospholipase D pathway as a potential therapeutic target. Genetics 215, 747–766. doi: 10.1534/genetics.119.302985
Khalil, B., Cabirol-Pol, M.-J., Miguel, L., Whitworth, A. J., Lecourtois, M., and Liévens, J.-C. (2017). Enhancing mitofusin/marf ameliorates neuromuscular dysfunction in Drosophila models of TDP-43 proteinopathies. Neurobiol. Aging 54, 71–83. doi: 10.1016/j.neurobiolaging.2017.02.016
Kim, S. H., Shi, Y., Hanson, K. A., Williams, L. M., Sakasai, R., Bowler, M. J., et al. (2009). Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1. J. Biol. Chem. 284, 8083–8092. doi: 10.1074/jbc.M808064200
Kryndushkin, D., Ihrke, G., Piermartiri, T. C., and Shewmaker, F. (2012). A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity: ALS-linked optineurin forms toxic aggregates in yeast cells. Mol. Microbiol. 86, 1531–1547. doi: 10.1111/mmi.12075
Kryndushkin, D., and Shewmaker, F. (2011). Modeling ALS and FTLD proteinopathies in yeast: an efficient approach for studying protein aggregation and toxicity. Prion 5, 250–257. doi: 10.4161/pri.17229
Kumar, J., Kline, N. L., and Masison, D. C. (2018). Human DNAJB6 antiamyloid chaperone protects yeast from polyglutamine toxicity separately from spatial segregation of aggregates. Mol. Cell. Biol. 38, e00437–e00418. doi: 10.1128/MCB.00437-18
Kwiatkowski, T. J., Bosco, D. A., LeClerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Langellotti, S., Romano, G., Feiguin, F., Baralle, F. E., and Romano, M. (2018). RhoGAPp190: a potential player in tbph-mediated neurodegeneration in Drosophila. PLoS ONE 13, e0195845. doi: 10.1371/journal.pone.0195845
Lanson, N. A., Maltare, A., King, H., Smith, R., Kim, J. H., Taylor, J. P., et al. (2011). A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet. 20, 2510–2523. doi: 10.1093/hmg/ddr150
Layalle, S., They, L., Ourghani, S., Raoul, C., and Soustelle, L. (2021). Amyotrophic lateral sclerosis genes in Drosophila melanogaster. Int. J. Mol. Sci. 22, 904. doi: 10.3390/ijms22020904
Leblond, C. S., Kaneb, H. M., Dion, P. A., and Rouleau, G. A. (2014). Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp. Neurol. 262, 91–101. doi: 10.1016/j.expneurol.2014.04.013
Lee, S., Jeon, Y.-M., Cha, S. J., Kim, S., Kwon, Y., Jo, M., et al. (2020). PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy 16, 1396–1412. doi: 10.1080/15548627.2019.1686729
Lembke, K. M., Law, A. D., Ahrar, J., and Morton, D. B. (2019). Deletion of a specific exon in the voltage-gated calcium channel gene cacophony disrupts locomotion in Drosophila larvae. J. Exp. Biol. 222, jeb191106. doi: 10.1242/jeb.191106
Li, Y., Ray, P., Rao, E. J., Shi, C., Guo, W., Chen, X., et al. (2010). A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U. S. A. 107, 3169–3174. doi: 10.1073/pnas.0913602107
Liachko, N. F., Guthrie, C. R., and Kraemer, B. C. (2010). Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 30, 16208–16219. doi: 10.1523/JNEUROSCI.2911-10.2010
Liachko, N. F., Saxton, A. D., McMillan, P. J., Strovas, T. J., Keene, C. D., Bird, T. D., et al. (2019). Genome wide analysis reveals heparan sulfate epimerase modulates TDP-43 proteinopathy. PLoS Genet. 15, e1008526. doi: 10.1371/journal.pgen.1008526
Liguori, F., Amadio, S., and Volonté, C. (2021). Where and why modeling amyotrophic lateral sclerosis. Int. J. Mol. Sci. 22, 3977. doi: 10.3390/ijms22083977
Lin, M.-J., Cheng, C.-W., and Shen, C.-K. J. (2011). Neuronal function and dysfunction of Drosophila dTDP. PLoS ONE 6, e20371. doi: 10.1371/journal.pone.0020371
Liu, G., Coyne, A. N., Pei, F., Vaughan, S., Chaung, M., Zarnescu, D. C., et al. (2017). Endocytosis regulates TDP-43 toxicity and turnover. Nat. Commun. 8, 2092. doi: 10.1038/s41467-017-02017-x
Liu, P., Chen, B., and Wang, Z.-W. (2020). GABAergic motor neurons bias locomotor decision-making in C. elegans. Nat. Commun. 11, 5076. doi: 10.1038/s41467-020-18893-9
Lopez-Gonzalez, R., Yang, D., Pribadi, M., Kim, T. S., Krishnan, G., Choi, S. Y., et al. (2019). Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD. Proc. Natl. Acad. Sci. USA. 116, 9628–9633. doi: 10.1073/pnas.1901313116
Lu, Y., Ferris, J., and Gao, F.-B. (2009). Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol. Brain 2, 30. doi: 10.1186/1756-6606-2-30
Ma, L., Li, X., Liu, C., Yan, W., Ma, J., Petersen, R. B., Peng, A., and Huang, K. (2022). Modelling Parkinson's disease in C. elegans: strengths and limitations. Curr. Pharm. Des. 28, 3033–3048. doi: 10.2174/1381612828666220915103502
Maccioni, R., Setzu, M. D., Talani, G., Solari, P., Kasture, A., Sucic, S., et al. (2018). Standardized phytotherapic extracts rescue anomalous locomotion and electrophysiological responses of TDP-43 Drosophila melanogaster model of ALS. Sci. Rep. 8, 16002. doi: 10.1038/s41598-018-34452-1
Machamer, J. B., Woolums, B. M., Fuller, G. G., and Lloyd, T. E. (2018). FUS causes synaptic hyperexcitability in Drosophila dendritic arborization neurons. Brain Res. 1693, 55–66. doi: 10.1016/j.brainres.2018.03.037
Maeda, I., Kohara, Y., Yamamoto, M., and Sugimoto, A. (2001). Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr. Biol. 11, 171–176. doi: 10.1016/S0960-9822(01)00052-5
Mallik, M., Catinozzi, M., Hug, C. B., Zhang, L., Wagner, M., Bussmann, J., et al. (2018). Xrp1 genetically interacts with the ALS-associated FUS orthologue caz and mediates its toxicity. J. Cell Biol. 217, 3947–3964. doi: 10.1083/jcb.201802151
Manzo, E., Lorenzini, I., Barrameda, D., O'Conner, A. G., Barrows, J. M., Starr, A., et al. (2019). Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. Elife 8, e45114. doi: 10.7554/eLife.45114.037
Markert, S. M., Skoruppa, M., Yu, B., Mulcahy, B., Zhen, M., Gao, S., et al. (2020). Overexpression of an ALS-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol. Open 9, bio055129. doi: 10.1242/bio.055129
Masison, D. C., and Reidy, M. (2015). Yeast prions are useful for studying protein chaperones and protein quality control. Prion 9, 174–183. doi: 10.1080/19336896.2015.1027856
Matsukawa, K., Kukharsky, M. S., Park, S.-K., Park, S., Watanabe, N., Iwatsubo, T., et al. (2021). Long non-coding RNA NEAT1_1 ameliorates TDP-43 toxicity in in vivo models of TDP-43 proteinopathy. RNA Biol. 18, 1546–1554. doi: 10.1080/15476286.2020.1860580
Matsumoto, T., Matsukawa, K., Watanabe, N., Kishino, Y., Kunugi, H., Ihara, R., et al. (2018). Self-assembly of FUS through its low-complexity domain contributes to neurodegeneration. Hum. Mol. Genet. 27, 1353–1365. doi: 10.1093/hmg/ddy046
McIntire, S. L., Jorgensen, E., and Horvitz, H. R. (1993). Genes required for GABA function in Caenorhabditis elegans. Nature 364, 334–337. doi: 10.1038/364334a0
Miguel, L., Frébourg, T., Campion, D., and Lecourtois, M. (2011). Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol. Dis. 41, 398–406. doi: 10.1016/j.nbd.2010.10.007
Milstead, R. A., Link, C. D., Xu, Z., and Hoeffer, C. A. (2023). TDP-43 knockdown in mouse model of ALS leads to dsRNA deposition, gliosis, and neurodegeneration in the spinal cord. Cereb. Cortex 33, 5808–5816. doi: 10.1093/cercor/bhac461
Mitra, J., Guerrero, E. N., Hegde, P. M., Liachko, N. F., Wang, H., Vasquez, V., et al. (2019). Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. U. S. A. 116, 4696–4705. doi: 10.1073/pnas.1818415116
Mockett, R. J., Radyuk, S. N., Benes, J. J., Orr, W. C., and Sohal, R. S. (2003). Phenotypic effects of familial amyotrophic lateral sclerosis mutant Sod alleles in transgenic Drosophila. Proc. Natl. Acad. Sci. U. S. A. 100, 301–306. doi: 10.1073/pnas.0136976100
Moens, T. G., Mizielinska, S., Niccoli, T., Mitchell, J. S., Thoeng, A., Ridler, C. E., et al. (2018). Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathol. 135, 445–457. doi: 10.1007/s00401-017-1798-3
Monahan, Z. T., Rhoads, S. N., Yee, D. S., and Shewmaker, F. P. (2018). Yeast models of prion-like proteins that cause amyotrophic lateral sclerosis reveal pathogenic mechanisms. Front. Mol. Neurosci. 11:453. doi: 10.3389/fnmol.2018.00453
Morgan, S., and Orrell, R. W. (2016). Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 119, 87–98. doi: 10.1093/bmb/ldw026
Myszczynska, M., and Ferraiuolo, L. (2016). New in vitro models to study amyotrophic lateral sclerosis. Brain Pathol. 26, 258–265. doi: 10.1111/bpa.12353
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 314, 130–133. doi: 10.1126/science.1134108
Oeda, T., Shimohama, S., Kitagawa, N., Kohno, R., Imura, T., Shibasaki, H., et al. (2001). Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum. Mol. Genet. 10, 2013–2023. doi: 10.1093/hmg/10.19.2013
Ortega, J. A., Daley, E. L., Kour, S., Samani, M., Tellez, L., Smith, H. S., et al. (2020). Nucleocytoplasmic proteomic analysis uncovers eRF1 and nonsense-mediated decay as modifiers of ALS/FTD C9orf72 toxicity. Neuron 106, 90–107. doi: 10.1016/j.neuron.2020.01.020
Park, S.-K., Arslan, F., Kanneganti, V., Barmada, S. J., Purushothaman, P., Verma, S. C., et al. (2018). Overexpression of a conserved HSP40 chaperone reduces toxicity of several neurodegenerative disease proteins. Prion 12, 16–22. doi: 10.1080/19336896.2017.1423185
Parkes, T. L., Elia, A. J., Dickinson, D., Hilliker, A. J., Phillips, J. P., and Boulianne, G. L. (1998). Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat. Genet. 19, 171–174. doi: 10.1038/534
Paul, S., Chakraborty, S., Anand, U., Dey, S., Nandy, S., Ghorai, M., et al. (2021). Withania somnifera (L.) Dunal (Ashwagandha): a comprehensive review on ethnopharmacology, pharmacotherapeutics, biomedicinal and toxicological aspects. Biomed. Pharmacother. 143, 112175. doi: 10.1016/j.biopha.2021.112175
Peng, C., Trojanowski, J. Q., and Lee, V. M.-Y. (2020). Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 16, 199–212. doi: 10.1038/s41582-020-0333-7
Phillips, J. P., Campbell, S. D., Michaud, D., Charbonneau, M., and Hilliker, A. J. (1989). Null mutation of copper/zinc superoxide dismutase in Drosophila confers hypersensitivity to paraquat and reduced longevity. Proc. Natl. Acad. Sci. U.S.A. 86, 2761–2765. doi: 10.1073/pnas.86.8.2761
Rabizadeh, S., Gralla, E. B., Borchelt, D. R., Gwinn, R., Valentine, J. S., Sisodia, S., et al. (1995). Mutations associated with amyotrophic lateral sclerosis convert superoxide dismutase from an antiapoptotic gene to a proapoptotic gene: studies in yeast and neural cells. Proc. Natl. Acad. Sci. U.S.A. 92, 3024–3028. doi: 10.1073/pnas.92.7.3024
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Ritson, G. P., Custer, S. K., Freibaum, B. D., Guinto, J. B., Geffel, D., Moore, J., et al. (2010). TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 30, 7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010
Romano, G., Klima, R., and Feiguin, F. (2020). TDP-43 prevents retrotransposon activation in the Drosophila motor system through regulation of Dicer-2 activity. BMC Biol. 18, 82. doi: 10.1186/s12915-020-00816-1
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Saldi, T. K., Gonzales, P., Garrido-Lecca, A., Dostal, V., Roberts, C. M., Petrucelli, L., et al. (2018). The Caenorhabditis elegans ortholog of TDP-43 regulates the chromatin localization of the heterochromatin protein 1 homolog HPL-2. Mol. Cell. Biol. 38, e00668–e00617. doi: 10.1128/MCB.00668-17
Sandhof, C. A., Hoppe, S. O., Tittelmeier, J., and Nussbaum-Krammer, C. (2020). C. elegans models to study the propagation of prions and prion-like proteins. Biomolecules 10, 1188. doi: 10.3390/biom10081188
Senchuk, M. M., Dues, D. J., Schaar, C. E., Johnson, B. K., Madaj, Z. B., Bowman, M. J., et al. (2018). Activation of DAF-16/FOXO by reactive oxygen species contributes to longevity in long-lived mitochondrial mutants in Caenorhabditis elegans. PLoS Genet. 14, e1007268. doi: 10.1371/journal.pgen.1007268
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W.-H., Hung, S.-T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Strah, N., Romano, G., Introna, C., Klima, R., Marzullo, M., Ciapponi, L., et al. (2020). TDP-43 promotes the formation of neuromuscular synapses through the regulation of Disc-large expression in Drosophila skeletal muscles. BMC Biol. 18, 34. doi: 10.1186/s12915-020-00767-7
Sulston, J. E., and Horvitz, H. R. (1981). Abnormal cell lineages in mutants of the nematode Caenorhabditis elegans. Dev. Biol. 82, 41–55. doi: 10.1016/0012-1606(81)90427-9
Sulston, J. E., Schierenberg, E., White, J. G., and Thomson, J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119. doi: 10.1016/0012-1606(83)90201-4
Sun, Y., Eshov, A., Zhou, J., Isiktas, A. U., and Guo, J. U. (2020). C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat. Commun. 11, 3354. doi: 10.1038/s41467-020-17129-0
Surguchov, A. (2021). Invertebrate models untangle the mechanism of neurodegeneration in Parkinson's disease. Cells 10, 407. doi: 10.3390/cells10020407
Takeishi, A., Takagaki, N., and Kuhara, A. (2020). Temperature signaling underlying thermotaxis and cold tolerance in Caenorhabditis elegans. J. Neurogenet. 34, 351–362. doi: 10.1080/01677063.2020.1734001
Therrien, M., and Parker, J. A. (2014a). Deciphering genetic interactions between ALS genes using C. elegans. Worm 3, e29047. doi: 10.4161/worm.29047
Therrien, M., and Parker, J. A. (2014b). Worming forward: amyotrophic lateral sclerosis toxicity mechanisms and genetic interactions in Caenorhabditis elegans. Front. Genet. 5, 85. doi: 10.3389/fgene.2014.00085
Torra, A., Parent, A., Cuadros, T., Rodríguez-Galván, B., Ruiz-Bronchal, E., Ballabio, A., et al. (2018). Overexpression of TFEB drives a pleiotropic neurotrophic effect and prevents parkinson's disease-related neurodegeneration. Mol. Ther. 26, 1552–1567. doi: 10.1016/j.ymthe.2018.02.022
True, H. L., Berlin, I., and Lindquist, S. L. (2004). Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 431, 184–187. doi: 10.1038/nature02885
Tyson, T., Senchuk, M., Cooper, J. F., George, S., Van Raamsdonk, J. M., and Brundin, P. (2017). Novel animal model defines genetic contributions for neuron-to-neuron transfer of α-synuclein. Sci. Rep. 7, 7506. doi: 10.1038/s41598-017-07383-6
Uechi, H., Kuranaga, E., Iriki, T., Takano, K., Hirayama, S., Miura, M., et al. (2018). Ubiquitin-binding protein CG5445 suppresses aggregation and cytotoxicity of amyotrophic lateral sclerosis-linked TDP-43 in Drosophila. Mol. Cell. Biol. 38, e00195–e00117. doi: 10.1128/MCB.00195-17
Voigt, A., Herholz, D., Fiesel, F. C., Kaur, K., Müller, D., Karsten, P., et al. (2010). TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE 5, e12247. doi: 10.1371/journal.pone.0012247
von Mikecz, A. (2022). Exposome, molecular pathways and one health: the invertebrate Caenorhabditis elegans. Int. J. Mol. Sci. 23, 9084. doi: 10.3390/ijms23169084
Walker, L. M., Huber, M., Doores, K. J., Falkowska, E., Pejchal, R., Julien, J.-P., et al. (2011). Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470. doi: 10.1038/nature10373
Walters, R., Manion, J., and Neely, G. G. (2019). Dissecting Motor neuron disease with Drosophila melanogaster. Front. Neurosci. 13, 331. doi: 10.3389/fnins.2019.00331
Wang, C., Duan, Y., Duan, G., Wang, Q., Zhang, K., Deng, X., et al. (2020). Stress induces dynamic, cytotoxicity-antagonizing TDP-43 nuclear bodies via paraspeckle LncRNA NEAT1-mediated liquid-liquid phase separation. Mol. Cell 79, 443–4587. doi: 10.1016/j.molcel.2020.06.019
Wang, J., Farr, G. W., Hall, D. H., Li, F., Furtak, K., Dreier, L., et al. (2009). An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 5:e1000350. doi: 10.1371/journal.pgen.1000350
Wang, P., Deng, J., Dong, J., Liu, J., Bigio, E. H., Mesulam, M., et al. (2019). TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 15, e1007947. doi: 10.1371/journal.pgen.1007947
Watson, M. R., Lagow, R. D., Xu, K., Zhang, B., and Bonini, N. M. (2008). A Drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J. Biol. Chem. 283, 24972–24981. doi: 10.1074/jbc.M804817200
White, J. G., Southgate, E., Thomson, J. N., and Brenner, S. (1986). The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 314, 1–340. doi: 10.1098/rstb.1986.0056
Xu, W., Bao, P., Jiang, X., Wang, H., Qin, M., Wang, R., et al. (2019). Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. Brain 142, 1349–1364. doi: 10.1093/brain/awz070
Xu, W., and Xu, J. (2018). C9orf72 dipeptide repeats cause selective neurodegeneration and cell-autonomous excitotoxicity in Drosophila glutamatergic neurons. J. Neurosci. 38, 7741–7752. doi: 10.1523/JNEUROSCI.0908-18.2018
Yamaguchi, M., Omori, K., Asada, S., and Yoshida, H. (2021). Epigenetic regulation of ALS and CMT: a lesson from Drosophila models. Int. J. Mol. Sci. 22, 491. doi: 10.3390/ijms22020491
Zaepfel, B. L., Zhang, Z., Maulding, K., Coyne, A. N., Cheng, W., Hayes, L. R., et al. (2021). UPF1 reduces C9orf72 HRE-induced neurotoxicity in the absence of nonsense-mediated decay dysfunction. Cell Rep. 34, 108925. doi: 10.1016/j.celrep.2021.108925
Zahra, W., Birla, H., Singh, S. S., Rathore, A. S., Dilnashin, H., Singh, R., et al. (2022). Neuroprotection by Mucuna pruriens in neurodegenerative diseases. Neurochem. Res. 47, 1816–1829. doi: 10.1007/s11064-022-03591-3
Zhang, K., Coyne, A. N., and Lloyd, T. E. (2018a). Drosophila models of amyotrophic lateral sclerosis with defects in RNA metabolism. Brain Res. 1693, 109–120. doi: 10.1016/j.brainres.2018.04.043
Zhang, T., Periz, G., Lu, Y.-N., and Wang, J. (2020). USP7 regulates ALS-associated proteotoxicity and quality control through the NEDD4L-SMAD pathway. Proc. Natl. Acad. Sci. USA. 117, 28114–28125. doi: 10.1073/pnas.2014349117
Zhang, T., Wu, Y.-C., Mullane, P., Ji, Y. J., Liu, H., He, L., et al. (2018b). FUS regulates activity of MicroRNA-mediated gene silencing. Mol. Cell 69, 787–801. doi: 10.1016/j.molcel.2018.02.001
Zhu, L., Li, S., Li, X. J., and Yin, P. (2023). Pathological insights from amyotrophic lateral sclerosis animal models: comparisons, limitations, and challenges. Transl. Neurodegener. (2023) 12, 46. doi: 10.1186/s40035-023-00377-7
Zhu, Q., Jiang, J., Gendron, T. F., McAlonis-Downes, M., Jiang, L., Taylor, A., et al. (2020). Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 23, 615–624. doi: 10.1038/s41593-020-0619-5
Keywords: amyotrophic lateral sclerosis, invertebrate models, yeast, Drosophila melanogaster, Caenorhabditis elegans, SOD1, TDP-43, FUS
Citation: Zhou L and Xu R (2024) Invertebrate genetic models of amyotrophic lateral sclerosis. Front. Mol. Neurosci. 17:1328578. doi: 10.3389/fnmol.2024.1328578
Received: 27 October 2023; Accepted: 24 January 2024;
Published: 04 March 2024.
Edited by:
Andrei Surguchov, University of Kansas Medical Center, United StatesReviewed by:
Irina G. Sourgoutcheva, University of Kansas Medical Center, United StatesYan Liu, Lanzhou University, China
Copyright © 2024 Zhou and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: RenShi Xu, eHVyZW5zaGlAbmN1LmVkdS5jbg==