 Fatma Saaoud1†
Fatma Saaoud1† Mohammed Ben Issa1†
Mohammed Ben Issa1† Lu Liu2
Lu Liu2 Keman Xu1
Keman Xu1 Yifan Lu1
Yifan Lu1 Ying Shao1Baosheng Han1Xiaohua Jiang1,2
Ying Shao1Baosheng Han1Xiaohua Jiang1,2 Xiaolei Liu1
Xiaolei Liu1 Avrum Gillespie3Jin Jun Luo4
Avrum Gillespie3Jin Jun Luo4 Laisel Martinez5
Laisel Martinez5 Roberto Vazquez-Padron5
Roberto Vazquez-Padron5 Sadia Mohsin6
Sadia Mohsin6 Beata Kosmider7
Beata Kosmider7 Hong Wang2
Hong Wang2 Silvia Fossati8
Silvia Fossati8 Xiaofeng Yang1,2*
Xiaofeng Yang1,2*- 1Department of Cardiovascular Sciences, Lewis Katz School of Medicine, Lemole Center for Integrated Lymphatics and Vascular Research, Temple University, Philadelphia, PA, United States
- 2Department of Cardiovascular Sciences, Lewis Katz School of Medicine, Center for Metabolic Disease Research and Thrombosis Research, Temple University, Philadelphia, PA, United States
- 3Section of Nephrology, Hypertension, and Kidney Transplantation, Department of Medicine, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
- 4Department of Neurology, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
- 5DeWitt Daughtry Family Department of Surgery, Leonard M. Miller School of Medicine, University of Miami, Miami, FL, United States
- 6Lewis Katz School of Medicine, Aging+Cardiovascular Discovery Center, Temple University, Philadelphia, PA, United States
- 7Lewis Katz School of Medicine, Center for Inflammation and Lung Research, Alzheimer’s Center, Temple University, Philadelphia, PA, United States
- 8Lewis Katz School of Medicine, Alzheimer’s Center, Temple University, Philadelphia, PA, United States
Introduction: Endothelial-to-mesenchymal transition (EndoMT), cell death, and fibrosis are increasingly recognized as contributing factors to Alzheimer’s disease (AD) pathology, but the underlying transcriptomic mechanisms remain poorly defined. This study aims to elucidate transcriptomic changes associated with EndoMT, diverse cell death pathways, and fibrosis in AD using the 3xTg-AD mouse model.
Methods: Using RNA-seq data and knowledge-based transcriptomic analysis on brain tissues from the 3xTg-AD mouse model of AD. This included pathway-level analysis of gene expression changes across multiple brain cell types. Mechanistic insights were further validated using single-cell RNA sequencing (scRNA-Seq) dataset from human AD brain.
Results: Our analysis showed that in the 3xTg-AD model: (i) multiple brain cell type genes are altered, promoting EndoMT through upregulation of RGCC and VCAN; (ii) genes related to various types of cell death, including apoptosis, ferroptosis, necrosis, anoikis, mitochondrial outer membrane permeability programmed cell death, mitochondrial permeability transition-driven necrosis, NETotic, and mitotic cell death, are upregulated in the several brain cell types; (iii) fibrosis-related genes are upregulated across multiple brain cell types. Further mechanistic analysis revealed: (1) mitochondrial stress through upregulation of mitochondrial genes in the brain cells; (2) upregulation of cellular, oxidative, and endoplasmic reticulum (ER) stress genes; (3) nuclear stress via upregulation of nuclear genes, transcription factors (TFs), and differentiation TFs FOSB and MEOX1; (4) metabolic reprogramming/stress through the upregulation of genes related to lipid and lipoprotein metabolism, fatty acid oxidation (FAO), glucose metabolism, and oxidative phosphorylation (OXPHOS); (5) catabolic stress via upregulation of catabolic genes. Single-cell RNA-Seq data indicated that many of these were also increased in AD patients’ brain cells. These changes were reversed by knockdown of the ER stress kinase PERK (EIF2AK3) and deficiencies in FOSB and MEOX1.
Discussion: This study uncovers previously unrecognized molecular signatures of organelle stress and bioenergetic reprogramming that drive EndoMT, cell death, and fibrosis in AD. The reversal of these changes via PERK, FOSB, and MEOX1 inhibition highlights potential therapeutic targets for mitigating neurodegenerative processes in AD.
1 Introduction
Alzheimer disease (AD) is a progressive neurodegenerative disorder primarily characterized by memory impairment, cognition decline, and behavioral changes. It is marked by the accumulation of amyloid-beta (Aβ) plaques and tau tangles in the brain, leading to neuronal death and synaptic dysfunction (Knopman et al., 2021; Ricciarelli and Fedele, 2017; Selkoe, 2001; Zhao et al., 2020). As the leading cause of senile dementia, the number of individuals aged 65 and older living with AD in the United States is projected to increase from 5.8 million in 2020 to 13.8 million by 2050 (Zhao et al., 2020). Globally, an estimated 55 million people are affected by AD and other forms of dementia (Alzheimer’s Association, 2024)1. Although the exact cause of AD remains unclear, it is believed to result from a combination of genetic, environmental, and lifestyle factors. Mutations in the amyloid precursor protein (APP) and presenilin 1 (PSEN1) genes are linked to familial forms of the disease, while the apolipoprotein E (APOE) gene (Maezawa et al., 2004) is a major genetic risk factor for late-onset AD. Neuroinflammation, vascular dysfunction, and blood-brain barrier (BBB) disruption are also integral components of AD pathology. Despite significant advances in research, effective treatments for AD remain limited, with current therapies primarily focused on alleviating symptoms rather than addressing the underlying causes. Further advancements in understanding AD progression are essential for developing new and effective therapeutics strategies.
In our recent study (Saaoud et al., 2025), RNA-sequencing (RNA-seq) analysis of brain tissue from 3xTg-AD mice, including a volcano plot and heat map, identified 316 upregulated and 412 downregulated genes. These included genes associated with the BBB, cerebrospinal fluid, and proinflammatory markers. The upregulation of genes linked to cell migration, differentiation, and trans-differentiation suggests that inflammation and cellular plasticity may play significant roles in AD pathogenesis. Additionally, genes involved in inflammasome pathways, immunometabolism, and trained immunity (innate immune memory for inflammation enhancement) were upregulated, indicating their potential contribution to AD progression. This study highlighted how trained immunity and inflammasome activation might influence AD development and revealed mechanistic insights that point to potential therapeutic targets for neuroinflammation and cellular reprogramming in AD. However, our previous study did not investigate whether organelle stress and bioenergetic mechanisms underlie cerebrovascular dysfunction and chronic inflammation in the AD brain.
We recently proposed a new concept that pathological trans-differentiation is a novel therapeutic target for cardiovascular diseases and chronic inflammation (Yang et al., 2024). Endothelial-to-mesenchymal transition (EndoMT) is a process where endothelial cells (ECs) lose their properties and differentiate into multipotent mesenchymal cells (Song et al., 2020), which occurs during both development and various pathologies, such as cancer progression, inflammation, and organ/tissue fibrosis (Yoshimatsu and Watabe, 2022). Several signaling pathways known to induce EndoMT (Xiong et al., 2018) are implicated in central nervous system pathologies associated with BBB dysfunction (Derada Troletti et al., 2016), yet transcriptomic reprogramming of the EndoMT pathway in AD remains poorly understood. Additionally, regulated cell death plays a significant role in AD (Thal et al., 2024). In 2018, the International Nomenclature Committee classified 14 types of cell death, including intrinsic and extrinsic apoptosis, mitochondrial permeability transition-driven necrosis, necroptosis (Richard and Mousa, 2022), ferroptosis, pyroptosis (Wójcik et al., 2024), parthanatos, entotic cell death, neutrophil extracellular trap (NET)otic cell death, lysosome-dependent cell death, autophagy-dependent cell death, immunogenic cell death, cellular senescence, and mitotic catastrophe. However, no comprehensive transcriptomic analysis of these cell death regulators in AD has been conducted (Brokaw et al., 2020; Galluzzi et al., 2018; Goel et al., 2022; Wang et al., 2019). Furthermore, cell death often triggers fibrosis, where fibroblasts replace dead cells and produce extracellular matrix proteins, contributing to fibrous plaque formation in the AD brain (D’Ambrosi and Apolloni, 2020; Yasuma and Gabazza, 2024). Vascular pathology, exacerbated by aging, AD, and vascular dementia, is strongly linked to chronic vascular inflammation and blood vessel dysregulation (Fang et al., 2023), but a comprehensive transcriptomic analysis of fibrosis regulators in AD remains unexplored.
Endoplasmic reticulum (ER) stress, characterized by the accumulation of unfolded or misfolded proteins within the ER, disrupts cellular homeostasis and is implicated in AD progression (Ajoolabady et al., 2022). Our research on ER-related transcriptomic changes demonstrated that the ER acts as an immune organelle (Saaoud et al., 2024), sensing various danger associated molecular patterns (DAMPs) and initiating ER stress that triggers angiotensin II (AngII)-accelerated trained immunity and differential susceptibilities of thoracic and abdominal aortas to aortic aneurysms (Lu et al., 2023). Our study also indicated that the gut microbiota-generated uremic toxin (Sun et al., 2018; Sun et al., 2024) trimethylamine-N-oxide (TMAO) (Chan et al., 2019), induces innate immune memory (also termed trained immunity)(Drummer et al., 2021a; Zhong et al., 2020)) via triggering ER stress, mitochondrial stress, and metabolic reprogramming (Lu et al., 2019; Saaoud et al., 2023). Our recent study proposed that mitochondria act as central immune organelles (Xu et al., 2024), and identified that organelle crosstalk regulators play significant roles in inflammatory diseases and cancer pathogenesis (Liu et al., 2021). We further reported that DNA checkpoint and repair factors act as nuclear sensors for intracellular organelle stresses under both physiological and pathological conditions (Zeng et al., 2018). Moreover, nuclear membrane, nucleolar, and nucleoplasm genes are upregulated in apolipoprotein E deficient (ApoE–/–) atherosclerotic aortas and in AngII-induced abdominal aortic aneurysm (AAA) in ApoE–/– mice (Yang et al., 2023). Additionally, procaspase-1 has been observed to translocate to the nucleus in proatherogenic lipid lysophosphatidylcholine (LPC)-activated human aortic endothelial cells (HAECs), where nuclear caspase-1 senses nuclear DAMPs, induces reactive oxygen species (ROS) promoter CYP1B1, and regulates numerous genes involved in HAEC activation and inflammation (Lu et al., 2021). These findings suggest that metabolic reprogramming, which plays crucial role in EndoMT and trained immunity, could be an important mechanism underlying AD pathology (Chen and Holtzman, 2022). Notably, metabolic enhancement through recombinant interferon-γ treatment has been shown to improve glycolytic metabolism and inflammatory responses in microglia, thus mitigating AD pathology in animal models (Baik et al., 2019). Despite these advances, the transcriptomic reprogramming related to organelle stress and bioenergetic metabolism in AD brain remains poorly characterized.
Despite substantial progress, several critical questions remain, highlighting gaps in our knowledge and avenues for further investigation. These questions include whether the transcriptome of the EndoMT pathway is reprogramed in AD, whether the expression of all 14 cell death type regulators remains unchanged in AD, and whether the expression of fibrosis and fibroblast regulators is altered in AD. Using our novel knowledge-based transcriptomic analysis approach, single-cell RNA-seq data from the Single Cell Portal database, and our unpublished data, we have made significant findings. Our research reveals that the 3xTg-AD induces EndoMT, triggers multiple forms of cell death, and promotes fibrosis and fibroblast proliferation in the brain. We have also identified five potential mechanisms—mitochondrial stress, ER stress, nuclear stress, metabolic reprogramming, and catabolic stress—that contribute to EndoMT, cell death, and fibrosis. These mechanistic insights provide new understanding of how AD-related processes are driven by organelle stress and bioenergetic alterations.
2 Materials and methods
2.1 Animals
Female 3xTg-AD mice (stock no. 34830-JAX) and age-matched C57BL/6J (wild-type, WT) controls were obtained from the Jackson Laboratory (Bar Harbor, ME). To control for variability due to sex and age, only female mice of the same age were used across both genotypes. Each experimental group consisted of three biological replicates (n = 3 per group). Mice were housed in a temperature-controlled environment under standard laboratory conditions and maintained on a standard chow diet with ad libitum access to food and water. At 11 months of age, mice were euthanized, and brain tissue samples—including the cortex plus hippocampus— were harvested and immediately processed for RNA-sequencing (RNA-seq) analysis.
2.2 RNA-seq and statistical analysis of RNA-seq data
Brain cortex and hippocampus tissues were pooled, homogenized, and lysed in TRIzol reagent (ThermoFisher, GE17-0891-01). RNAs were extracted according to the manufacturer’s protocol then quantified using a Nanodrop (ThermoFisher). The isolated RNA samples were then sent to Genewiz (South Plainfield, NJ) for RNA-Seq analysis. Total RNA libraries were prepared using the Pico Input SMARTer Stranded Total RNA-Seq Kit (Takara). Briefly, 10 ng of total RNA from each sample was reversely transcribed using random priming and reverse transcriptase. Full-length cDNA was synthesized using SMART (Switching Mechanism At the 5’end of RNA Template) technology, which preserves the strand orientation of the RNA. Ribosomal cDNA was hybridized to mammalian-specific R-Probes and cleaved by ZapR. Libraries containing Illumina adapters with TruSeq HT indexes were pooled and loaded onto the Hiseq 2500 platform. Single-end reads of 75 base-pair (bp), with 30 million reads per sample, were generated for subsequent bioinformatic analysis. FASTQ files were mapped to the mouse mm10 genome using the STAR Aligner. Data analysis was performed using the statistical computing environment R, along with the Bioconductor suite of packages and RStudio. Raw data underwent background subtraction, variance stabilization, and normalization via robust spline normalization. Differentially expressed genes were identified using linear modeling and Bayesian statistics with the Limma package. For comparisons between two groups, a two-tailed Student’s t-test was used to assess statistical significance.
2.3 Transcriptomic data collection
Transcriptomic datasets were obtained from the publicly accessible NIH-NCBI Gene Expression Omnibus (GEO) database.2 Relevant datasets were systematically curated and organized for further analysis. Differential gene expression analysis was performed using the GEO2R tool, an interactive web application within the GEO database, which allow comparison between experimental groups and controls using pre-processed data.
2.4 Metascape pathway analysis
Pathway enrichment analysis was performed using Metascape3, an integrated online tool designed for the functional annotation and interpretation of large-scale omics datasets. Differentially expressed genes (DEGs) identified from RNA-seq data were compiled and uploaded to the Metascape platform to investigate their associated biological pathways, molecular functions, cellular processes, and potential clinical relevance. The analysis incorporated gene ontology (GO) terms, KEGG pathways, Reactome pathways, and other functional categories to provide comprehensive insights into the biological significance of the DEGs.
3 Results
3.1 The 3xTg-AD model modulates the expression of brain EC vasculature genes, resulting in the upregulation of four specific genes
To investigate whether the 3xTg-AD model induces transcriptomic alteration in brain EC vasculature within the cortex and hippocampus, we analyzed bulk RNA-seq data from brain tissues of 3xTg-AD mice and wild-type (WT) controls, as we recently reported (Saaoud et al., 2025). Differentially expression analysis revealed that 316 genes were significantly upregulated and 412 genes were downregulated in the 3xTg-AD group compared to WT controls, using a cutoff fold change > 1.5 and p-value < 0.05, as we previously reported (Saaoud et al., 2025). Principal component analysis (PCA) was conducted to evaluate sample variance and clustering, highlighting distinct transcriptional profiles between the two groups (Figure 1A). A curated list of 221 brain vasculature genes was obtained from the Human Protein Atlas (HPA) database4 for targeted analysis. As shown in Figure 1B, 3xTg-AD modulated the expression of brain EC vasculature genes, with four genes significantly upregulated. These include GIMAP4 (GTPase, IMAP family member 4), which promotes immune cell migration (Chen et al., 2022); TINAGL1 (tubulointerstitial nephritis antigen like 1), which promotes EC tube formation (Sato et al., 2022); FAM167B (family with sequence similarity 167 member B), an EC-specific gene (Feng et al., 2019); and CAV1 (caveolin 1), a gene involved in proatherogenic process, leukocyte influx, and CD4+ regulatory T cell (Treg) suppression (Engel et al., 2011). Conversely, several genes were significantly downregulated in the 3xTg-AD brain, including PTPRB (which regulates vascular permeability and homeostasis in inflammation) (Vestweber, 2021), CD248 (which induces a maladaptive unfolded protein response in diabetic kidney disease) (Krishnan et al., 2023), TBX2 (which defines a multipotent mesenchymal progenitor pool) (Wojahn et al., 2019), SLC38A5 (which promotes retinal vascular development and pathological neovascularization in a retinopathy model (Wang et al., 2022), TMEM102 [whose overexpression may promote chemoresistance by inhibiting the mitochondria-associated apoptotic pathway (Tai et al., 2022)], Hapln3 (a novel link protein that co-localizes with versican and is upregulated by platelet-derived growth factor in arterial smooth muscle cells) (Ogawa et al., 2004), GBP4 (which serves as a central orchestrator of immune responses to infection, inflammation, and cancer) (Tretina et al., 2019), and A2M (which neutralize cytokines and modifies hemostasis) (Lagrange et al., 2022). These findings suggest that AD brain promotes brain EC activation and inflammatory cell recruitment while inhibiting vascular development and pathological neovascularization.

Figure 1. 3xTg-AD modulates the expression of brain vasculature genes, fibroblast genes, fibrosis-related genes, and endothelial-to-mesenchymal transition (EndoMT) genes. (A) Principal component analysis (PCA) of RNA-seq data demonstrating sample clustering based on genotype. The PCA plot shows clear separation between control (wild-type, WT; pink dots, n = 3) and 3xTg-AD (green dots, n = 3) samples. Principal component (PC1) and 2 (PC2) explain 23.8% and 10.9% of the variance, respectively. (B) Differential gene expression analysis (fold change > 1.5, P < 0.05) in 3xTg-AD brains reveals modulation of gene sets associated with brain vasculature (12 genes; four upregulated, eight downregulated), fibroblast (19 genes; eight upregulated, 11 downregulated), fibrosis (41 genes; 17 upregulated, 24 downregulated), and EndoMT (six genes; two upregulated—RGCC and VCAN—and 4 downregulated). (C,D) Metascape pathway enrichment analysis of the fibroblast and fibrosis-related gene sets altered in 3xTg-AD highlights key biological pathways implicated in disease pathology (log10P < –2). (E–L) Single-cell RNA sequencing data depicting expression levels of selected genes in brain cells from AD models: (E) FHIT, (F) MPZ, (G) CCR1, (H) TCAP, (I) PTGS2, (J) HSPA1B, (K) RGCC (Regulator of Cell Cycle Protein), and (L) VCAN (Versican). Data were obtained from the Single Cell Portal of the Broad Institute (https://singlecell.broadinstitute.org/single_cell/study/SCP1375/integrative-in~situ-mapping-of-single-cell-transcriptional-states-and-tissue-histopathology-in-an-alzheimer-disease-model#study-visualize). Gene sets comprising 221 brain vasculature genes and 401 fibroblast genes were collected from the Human Protein Atlas (HPA) database. The fibrosis-related gene set (777 genes) were compiled from a previous study (PMID: 33519923). EndoMT genes (111 genes) were collected from the HPA database and literature sources (PMIDs: 30864875, 29039786, 30654892).
3.2 The 3xTg-AD model modulates the expression of fibroblast- and fibrosis-associated genes, resulting in the upregulation of eight fibroblast-related genes and 17 fibrosis-related genes in brain tissue, as well as six fibrosis-related genes across multiple brain cell types
To determine whether 3xTg-AD induces transcriptomic reprogramming of fibroblasts and fibrosis, we examined the expression changes of 401 fibroblast genes from the HPA database and 777 fibrosis-related genes (Gu et al., 2020) in the 3xTg-AD brain. As shown in Figure 1B, eight out of 401 fibroblast genes and 17 out of 777 fibrosis-related genes were significantly upregulated in the 3xTg-AD brain. Notably, 3xTg-AD also downregulated the expression of eleven fibroblast genes and 24 fibrosis-associated genes. Metascape pathway analysis of the 24 upregulated fibroblast and fibrosis-related genes identified the following key pathways: negative regulation of catalytic activity, regulation of hemopoiesis, blood circulation, negative regulation of cell differentiation, olefinic compound metabolic process, positive regulation of cell migration, lipid homeostasis, viral protein interaction with cytokine and cytokine receptor, and cardiocyte (cardiomyocyte) differentiation. Conversely, the key enriched pathways for the 3xTg-AD downregulated fibroblast and fibrosis-related genes included macrophage activation, macrophage differentiation, positive regulation of ROS metabolic process, phagosome, positive regulation of secretion by cell, response to angiotensin, regulation of membrane depolarization. Among the 17 upregulated fibrosis-related genes, FHIT (fragile histidine triad diadenosine triphosphatase), MPZ (myelin protein zero), CCR1 (C-C motif chemokine receptor 1), TCAP (titin-cap), PTGS2 (prostaglandin-endoperoxide synthase 2), and HSPA1B (heat shock protein family A (Hsp70) member 1B) were found to be expressed in multiple brain cell types, based on data from the Single Cell Portal at the MIT Broad Institute5 (Figures 1E–J). Thus, 3xTg-AD model also upregulated 6 fibrosis-related genes in multiple brain cell types in AD.
3.3 The 3xTg-AD model modulated the expression of endothelial-to-mesenchymal transition (EndoMT) genes, notably upregulating regulator of cell cycle (RGCC) and versican (VCAN)
To determine whether 3xTg-AD model induces transcriptomic reprogramming in EndoMT, we analyzed the expression of 111 known EndoMT genes (Kovacic et al., 2019; Pardali et al., 2017; Piera-Velazquez and Jimenez, 2019) from the HPA database in our 3xTg-AD RNA-seq dataset. As shown in Figure 1B, 3xTg-AD modulated the expression of six EndoMT genes, with two upregulated genes—RGCC and VCAN—and four downregulated genes. RGCC (response gene to complement 32 protein, RGC-32) is known to regulate cell cycle progression and can be induced by p53 in response to DNA damage or by sublytic levels of complement system proteins, influencing cell cycle activity.6 RGCC is also stimulated by growth factors and cytokines, including transforming growth factor β (TGF-β), playing a role in the modulation of processes such as angiogenesis, fibrosis, cell migration, and differentiation (An et al., 2009). Notably, RGCC is essential for promoting the differentiation of T helper 17 (Th17) cells, which are implicated in multiple sclerosis, and in regulating significant transcriptomic changes in astrocytes, favoring the synthesis and secretion of extracellular matrix components, growth factors, axonal growth molecules, and pro-astrogliogenic factors (Tatomir et al., 2022). VCAN (Versican), an extracellular matrix (ECM) proteoglycan, is upregulated alongside other ECM-binding molecules, such as hyaluronan, tumor necrosis factor-α (TNF-α) induced protein 6 (TNFAIP6, TSG-6), and inter-alpha-trypsin inhibitor heavy chain 1 (ITIH1, IαI), during inflammation in various diseases, including cardiovascular, pulmonary, autoimmune diseases, and certain cancers. These interactions form stable scaffolds that act as “landing strips” for inflammatory cells as they migrate from the circulation into tissues (Wight et al., 2020). To validate the roles of RGCC and VCAN in mediating EndoMT in the brain, we examined their expression in multiple brain cell types in AD using single-cell RNA-seq data from the Single Cell Portal database at MIT-Broad Institute7, as shown in Figures 1K, L. Our data demonstrated that RGCC is expressed across multiple brain cell types, including ECs and vascular smooth muscle cells (VSMCs). These findings align with recent studies reporting that 14 out of 45 AD risk genes are upregulated within brain vasculature, particularly in brain ECs and VSMCs (Yang et al., 2022). Collectively, our data suggest that 3xTg-AD potentially promotes EndoMT through the upregulation of cytosol/nuclear-localized RGCC and ECM-localized VCAN.
3.4 The 3xTg-AD model upregulates several types of cell death—apoptosis, ferroptosis, necroptosis, pyroptosis, programmed necrotic death, and anoikis—in the AD brain
To assess whether the 3xTg-AD model induces transcriptomic reprogramming of cell death pathways, we analyzed the expression changes of 2021 apoptotic cell death genes, based on data from the Mouse Genome Informatics (MGI) database.8 As shown in Table 1, the 3xTg-AD model upregulated 43 apoptosis-related genes and downregulated 36. Metascape pathway analysis revealed key pathways associated with the significantly upregulated genes, including the positive regulation of apoptotic process, regulation of apoptotic signaling pathway, regulation of EC proliferation, positive regulation of protein kinase A signaling, regulation of tumor necrosis factor production, regulation of extracellular signal-regulated kinase (ERK)1 and ERK2 cascade, cell activation involved in immune response, regulation of cellular response to stress, regulation of blood vessel EC migration. Conversely, the key pathways associated with downregulated genes included the positive regulation of apoptotic process, glial cell differentiation, positive regulation of cell migration, apoptotic signaling pathway, epithelial cell proliferation, cell chemotaxis, regulation of apoptotic signaling pathway, cellular response to lipid, regulation of cell-cell adhesion, and cellular response to angiotensin.
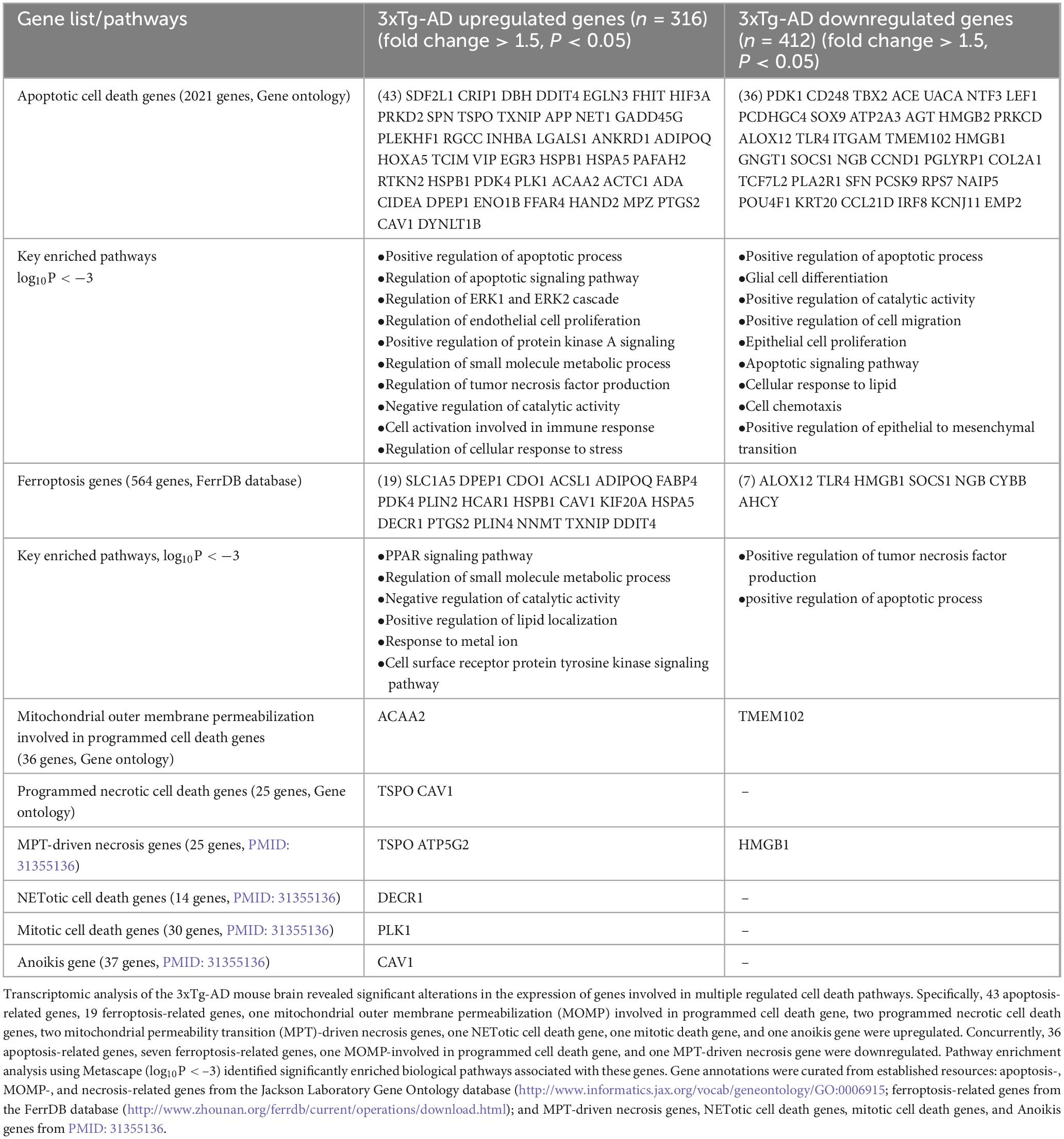
Table 1. Differential expression of cell death-related genes in the 3xTg-AD mouse brain.
Ferroptosis, a newly identified form of programmed cell death, is triggered by imbalances in iron metabolism, iron-dependent lipid peroxidation, and ROS accumulation (Zhao et al., 2023). Glutathione (GSH) depletion and Glutathione peroxidase 4 (GPX4) inactivation are known to induce ferroptosis (Jiang et al., 2021). To determine whether the 3xTg-AD model affects ferroptosis, we analyzed the expression changes of 264 ferroptosis-related genes, collected from the FerrDB database9, among the 3xTg-AD modulated genes. As shown in Table 1, 19 ferroptosis-related genes were upregulated, including solute carrier family 1 member 5 (SLC1A5) (Su et al., 2023), DPEP1, a dexamethasone-sensitized ferroptosis gene (von Mässenhausen et al., 2022), cysteine dioxygenase type 1 (CDO1), which absorb cysteine competitively, thus restricting the process of GSH synthesis and promoting ferroptosis (Li et al., 2020), and acyl-CoA synthetase long chain family member 1 (ACSL1), which mediate ferroptosis triggered by conjugated linolenic acids (Beatty et al., 2021), and other 15 ferroptosis genes, while downregulated 7 ferroptosis-related genes, including ALOX12 [the inactivation of arachidonate 12-lipoxygenase, 12S type (ALOX12) reduces p53-driven ferroptosis triggered by ROS stress] (Chu et al., 2019), Toll-like receptor 4 (TLR4), high mobility group box 1 (HMGB1) [released by anti-inflammatory M2 macrophages, binds to TLR4 on M1 macrophages, leading to the activation of signal transducer and activator of transcription 3 (STAT3) signaling in proinflammatory M1 macrophages and promoting the inflammatory response] (Feng et al., 2024), suppressor of cytokine signaling 1 (SOCS1), neuroglobin (NGB), cytochrome B-245 beta chain (CYBB), and adenosylhomocysteinase (AHCY). The key pathways of upregulated ferroptosis genes included peroxisome proliferator activated receptor (PPAR) signaling pathway, regulation of small molecule metabolic process, positive regulation of lipid localization, response to metal ion, organic acid catabolic process, cell surface receptor protein tyrosine kinase signaling pathway, however, the downregulated pathways included positive regulation of tumor necrosis factor production and positive regulation of apoptotic process.
Moreover, we identified 299 cell death regulators across 13 different types of cell death pathways, collectively termed the cell death regulatome (Galluzzi et al., 2018; Wang et al., 2019). As shown in Table 1, eight genes were upregulated in six other cell death pathways, including acetyl-CoA acyltransferase 2 (ACAA2) in the mitochondrial outer membrane permeability programmed cell death group, translocator protein (TSPO) and caveolin 1 (CAV1) in the programmed necrotic cell death group, TSPO and ATP5G2 in the mitochondrial permeability transition (MPT)-driven necrosis group, 2,4-dienoyl-CoA reductase 1 (DECR1) in the neutrophil extracellular traps (NETotic) cell death group, polo like kinase 1 (PLK1) in the mitotic cell death group, and CAV1 in the anoikis group. Notably, CAV1 was associated with three cell death categories (apoptosis, programmed necrotic cell death, and anoikis), while TSPO was linked to two categories (programmed necrotic cell death and MPT-driven necrosis). Additionally, two genes downregulated in two other cell death pathways, including transmembrane protein 102 (TMEM102) in the mitochondrial outer membrane permeability programmed cell death group, and HMGB1 in the mitochondrial permeability transition (MPT)-driven necrosis group.
The 3xTg-AD brain also downregulated GBP4 (guanylate binding protein 4) (Figure 1B), which has been shown by us and others to regulate cytosolic recruitment and activation of pro-inflammatory caspase-4 (Drummer et al., 2021b; Wandel et al., 2020). Caspase-4 (human)/caspase-11 (mouse) plays a crucial role in the non-canonical inflammasome activation, leading to inflammatory cell death (pyroptosis) as previously reported (Drummer et al., 2023; Hagar and Miao, 2014; Sun et al., 2024). Our findings suggest that caspase-4/caspase-11 activation in brain cells including ECs may be involved in AD, which was further supported by Wyss-Coray’s team in their 2022 Nature publication (Yang et al., 2022), demonstrating the involvement of microglia and brain ECs in this process. These results indicate that the 3xTg-AD model induces caspase-4/11 activation and pyroptosis in both the AD brain and brain ECs.
Taken together, our findings reveal that the 3xTg-AD brain upregulates 43 apoptosis-related genes, 19 ferroptosis-related genes, one mitochondrial outer membrane permeabilization cell death gene, two programmed necrotic cell death genes, two MPT-driven necrosis genes, one NETotic cell death gene, one mitotic cell death gene, and one anoikis gene. This suggest that the 3xTg-AD model promotes apoptosis, ferroptosis, pyroptosis, and six other types of cell death in the AD brain (Galluzzi et al., 2018; Wang et al., 2019). Additionally, genes such as RGCC (an EndoMT and apoptotic gene), cysteine rich protein 1 (CRIP1, an apoptotic gene), dopamine beta-hydroxylase (DBH, an apoptotic gene), DNA damage-inducible transcript 4 (DDIT4, associated with both apoptosis and ferroptosis), prostaglandin-endoperoxide synthase 2 (PTGS2, involved in both apoptosis and ferroptosis), perilipin 4 (PLIN4, a ferroptotic gene), caspase-4/caspase-11 (casp4, a pyroptotic gene) (Sun et al., 2024), interleukin-1β (IL1β pyroptotic cytokine) (Sun et al., 2024), and TNF-α (TNF-α involved in apoptosis, necroptosis, and trained immunity) (Saaoud et al., 2023; Wang et al., 2019) are expressed in multiple brain cells in AD (Figures 1K, I, and Supplementary Figures 1A–G). The 3xTg-AD model thus demonstrates upregulation of several types of cell death in brain cells, including apoptosis, ferroptosis, necroptosis, pyroptosis, programmed necrotic death, and anoikis.
3.5 The 3xTg-AD model induces mitochondrial stress, as evidenced by the upregulation of 32 mitochondrial genes, with a specific induction of mitochondrial stress across multiple cell types in AD brain via the upregulation of six mitochondrial genes
To explore the potential mitochondrial mechanisms underlying EndoMT, cell death, and fibrosis in the 3xTg-AD brain, we analyzed transcriptomic reprogramming within the nuclear genome-encoded mitochondrial transcriptome. As shown in Figure 2A, the 3xTg-AD brain upregulated 21 mitochondria-localized protein encoding genes, collected from the HPA database, 26 mitoCarta genes (an inventory of mammalian mitochondrial proteins and pathways, MIT-Broad Institute), and 9 genes associated with mitochondrial genetic diseases and bioenergistic metabolism (Frazier et al., 2019). These upregulated genes overlapped as shown in Figure 2B. Metascape pathway analysis of these upregulated mitochondrial genes identified the top enriched pathways (Figure 2C), including fatty acid (FA) beta oxidation (FAO), FAO using acyl-CoA dehydrogenase, mitochondrial FAO of unsaturated FAs, FA biosynthesis, PPAR signaling pathway, response to cold, lipid transport, regulation of small molecule metabolic process, and muscle structure development. Notably, three mitochondrial localized genes— solute carrier family 6 member 2 (SLC6A2), MAF BZIP transcription factor F (MAFF), troponin C1, slow skeletal and cardiac type (TNNC1)—and three mitoCarta genes— haloacid dehalogenase like hydrolase domain containing 3 (HDHD3), fragile histidine triad diadenosine triphosphatase (FHIT), interferon alpha inducible protein 27 (IFI27)—were upregulated across multiple cell types in AD brain10 (Figure 1E and Supplementary Figures 2A–E). In summary, the 3xTg-AD brain induces mitochondrial stress by upregulating 32 mitochondrial genes in brain.
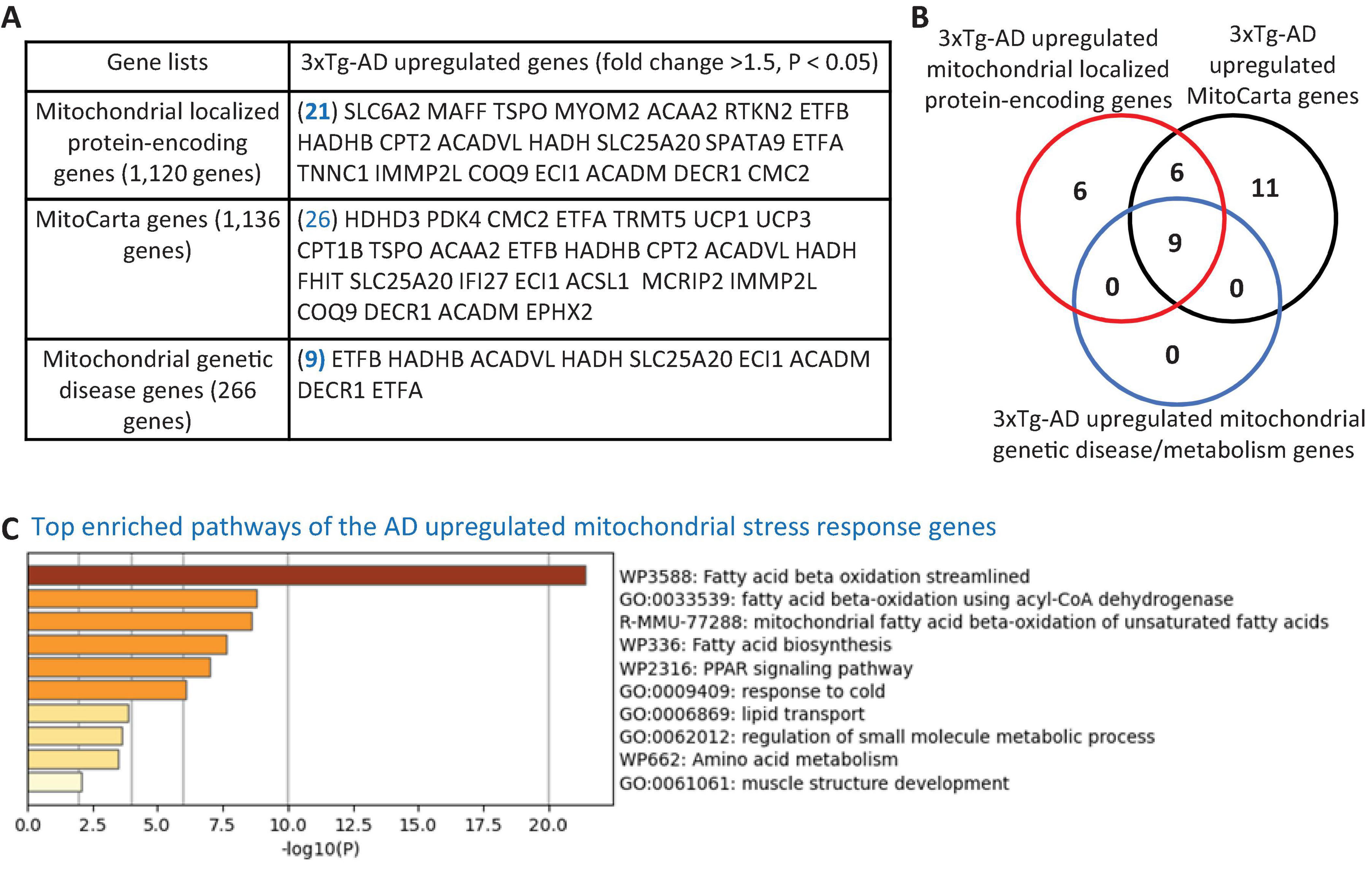
Figure 2. Transcriptional evidence of mitochondrial stress response activation in the 3xTg-AD mouse brain. (A) The 3xTg-AD mouse brain exhibit upregulation of 21 mitochondrial-localized protein-encoding genes, 26 nuclear-encoded mitochondrial genes (MitoCarta, Broad Institute), and 9 genes linked to mitochondrial genetic disorders, indicating a potential induction of mitochondrial stress in the AD brain. (B) Venn diagram illustrating the overlap among upregulated mitochondrial localized genes, MitoCarta genes, and mitochondrial genetic disease/metabolism genes in the 3xTg-AD brain. (C) Metascape pathway analysis reveals significant enrichment of mitochondrial fatty acid beta-oxidation pathways in the 3xTg-AD brain. The 1,120 mitochondrial-localized genes were sourced from the HPA, Mitocarta genes (1,136 nuclear-encoded mitochondrial genes) were retrieved from the Broad Institute (https://www.broadinstitute.org/mitocarta/mitocarta30-inventory-mammalian-mitochondrial-proteins-and-pathways), and a curated list of 266 genes linked to mitochondrial genetic disorder and metabolism were compiled from PMID: 29233888.
3.6 The 3xTg-AD model upregulated EndoMT and cell death pathways in the brain by increasing the expression of nuclear genes, DNA damage response genes, TFs, and differentiation TFs such as FOSB and MEOX1
To investigate the potential nuclear stress mechanisms underlying EndoMT, cell death, and fibrosis in the 3xTg-AD brain, we analyzed transcriptomic reprogramming in the nuclear gene transcriptome induced by 3xTg-AD. As shown in Figure 3A, the 3xTg-AD brain upregulated one nuclear membrane protein gene, 13 nucleolar genes, and 70 nucleoplasm genes. Additionally, the 3xTg-AD brain upregulated eight DNA damage response genes, four TFs [early growth response 3 (EGR3), FosB proto-oncogene, AP-1 transcription factor subunit (FOSB), MAFF, and recombination signal binding protein for immunoglobulin kappa J region like (RBPJL)], and two differentiation TFs (Ng et al., 2021), FOSB and mesenchyme homeobox 1 (MEOX1). Metascape pathway analysis of these upregulated nuclear genes revealed the key enriched pathways, including sulfur amino acid metabolic process, muscle tissue development, regulation of EC proliferation, fatty acid beta oxidation, response to glucocorticoids, regulation of cyclin-dependent protein serine/threonine kinase activity, positive regulation of DNA-binding TFs, lipid homeostasis, DNA-templated transcription, and others (Figure 3B).
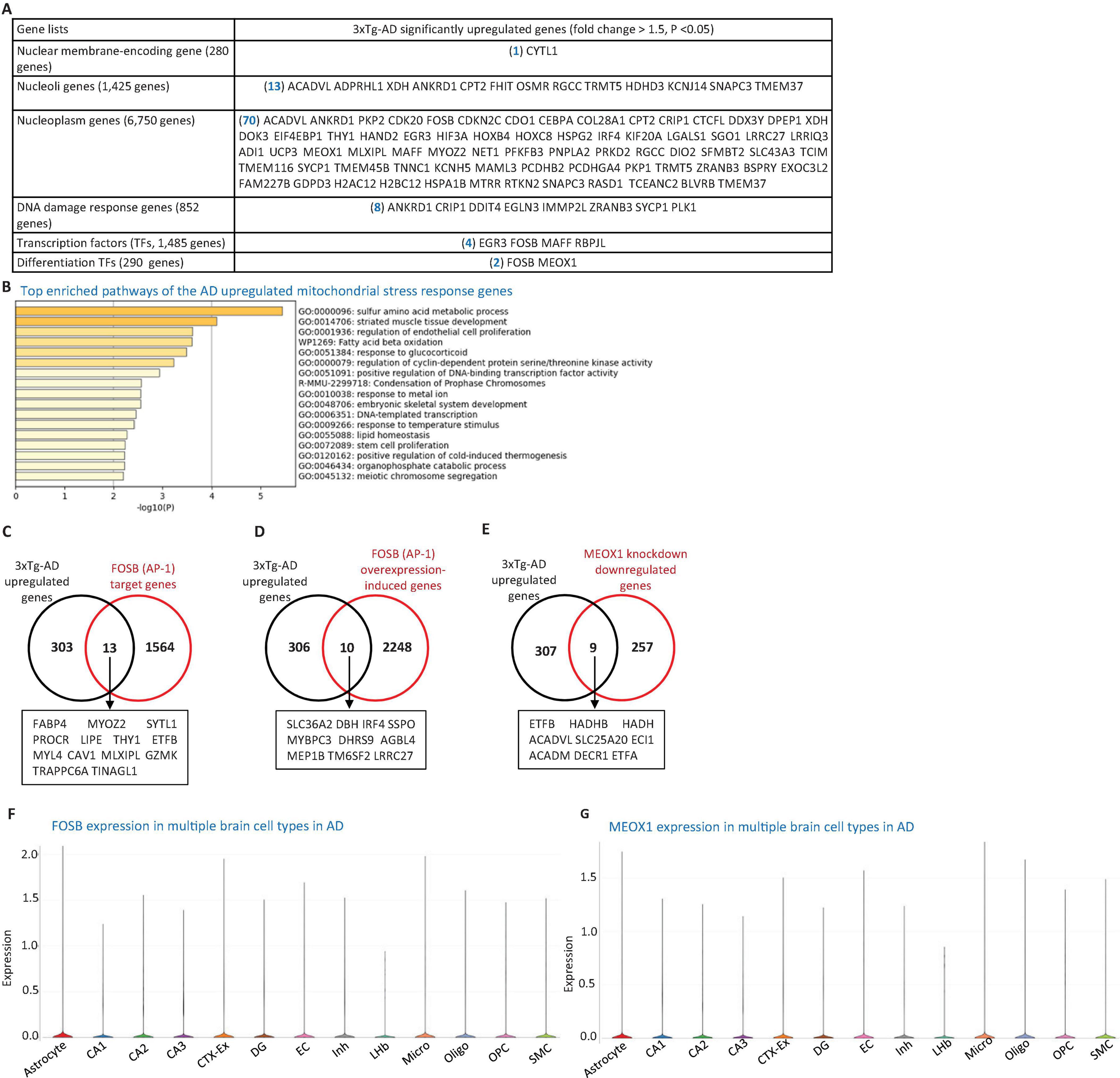
Figure 3. 3xTg-AD model induces nuclear stress responses. (A) Transcriptomic profiling of the 3xTg-AD brain reveals upregulation of genes associated with nuclear stress, including one nuclear membrane protein gene, 13 nucleolar protein genes, 70 nucleoplasmic protein genes, eight DNA damage response genes, four transcription factors (TFs), and 2 differentiation-associated TFs, suggesting activation of nuclear stress pathways and genomic instability. (B) Metascape pathway enrichment analysis of these nuclear-associated genes, including regulation of EC proliferation and fatty acid β-oxidation pathways. (C,D) 3xTg-AD-upregulated genes overlap with 13 FOSB target genes and 10 FOSB overexpression-induced genes. (E) 3xTg-AD-upregulated genes also intersect with nine genes downregulated by MEOX1 knockdown. (F,G) Single-cell RNA-seq data show the expression of FOSB and MEOX1 across multiple brain cell types in AD. Data were collected from the single-cell RNA seq deposited in the Single Cell Portal of the Broad Institute (https://singlecell.broadinstitute.org/single_cell/study/SCP1375/integrative-in~situ-mapping-of-single-cell-transcriptional-states-and-tissue-histopathology-in-an-alzheimer-disease-model#study-visualize). The 280 nuclear membrane-encoding genes, 1,425 nucleoli protein-encoding genes, 6,750 nucleoplasm protein-encoding genes, and 1485 TFs were downloaded from the HPA database. The 852 DNA damage response genes were collected from the MGI Gene Ontology database (https://www.informatics.jax.org/vocab/gene_ontology), and the 290 differentiation TFs were compiled from a previous publication (PMID: 33257861). FOSB target genes were obtained from the MotifMap dataset (https://maayanlab.cloud/Harmonizome/gene_set/AP-1/MotifMap+Predicted+Transcription+Factor+Targets), FOSB overexpression-induced genes from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111827), and MEOX1 knockdown-downregulated genes from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE133927).
To determine whether the upregulation of FOSB and MEOX1 contributed to the expression of 316 AD-upregulated genes, we collected FOSB target genes from the MotifMap dataset11, FOSB overexpression-induced genes (the dataset ID: GSE111827), and MEOX1 knockdown-downregulated genes (GSE133927) from the NIH-NCBI GEO database2. As shown in Figures 3C–E, 13 FOSB target genes, 10 FOSB overexpression-induced genes, and nine MEOX1 knockdown-downregulated genes overlapped with the 3xTg-AD upregulated genes in the brain. To explore the role of FOSB and MEOX1 in 3xTg-AD brain cells, we observed that both TFs were upregulated in multiple cell types in AD brain(see text footnote 10) Figures 3F, G. Notably, two additional TFs—EGR3 (also associated with apoptosis) and MAFF (also linked to mitochondrial-localization)—along with four nucleolar genes, including FHIT (also a mitoCarta gene), Oncostatin M Receptor (OSMR) (also a cellular response to stimulus gene), RGCC (an EndoMT and apoptotic gene), and HDHD3 (a mitoCarta gene), as well as three DNA damage response genes, cysteine rich protein 1 (CRIP1), DNA damage inducible transcript 4 (DDIT4), and Egl-9 family hypoxia inducible factor 3 (EGLN3) (all associated with apoptosis), were also upregulated in AD brain cells, as indicated by the same AD single-cell RNA-seq dataset (Figures 1E, K and Supplementary Figures 1A, C, 2B, D, 3A–C). In summary, these findings demonstrate that the 3xTg-AD brain upregulates EndoMT and cell death pathways in brain cells by enhancing the expression of nuclear genes, DNA damage response genes, TFs, and differentiation TFs FOSB and MEOX1.
3.7 The 3xTg-AD model upregulated EndoMT, cell death, and fibrosis in brain cells by increasing the expression of genes associated with cellular stress, oxidative stress, and ER stress
To investigate the potential cellular stress mechanisms driving EndoMT, cell death, and fibrosis in the 3xTg-AD brain, we examined the transcriptomic reprogramming induced by 3xTg-AD related to cellular and oxidative stress as well as ER stress. As shown in Table 2A, 3xTg-AD mice displayed upregulation of 95 genes related to cellular response to stimulus, 32 genes related to cellular response to stress, six genes associated with oxidative stress, and seven ROS regulator genes. Metascape pathway analysis of these upregulated genes (log10P < −3) revealed the key enriched pathways, including brown fat cell differentiation, fatty acid metabolic process, PPAR signaling pathway, regulation of small molecule metabolic process, response to hypoxia, regulation of extracellular signal-regulated kinase 1 (ERK1) and ERK2 cascade, positive regulation of lipid localization, lipid homeostasis, cellular response to hormone stimulus, and response to TNF.
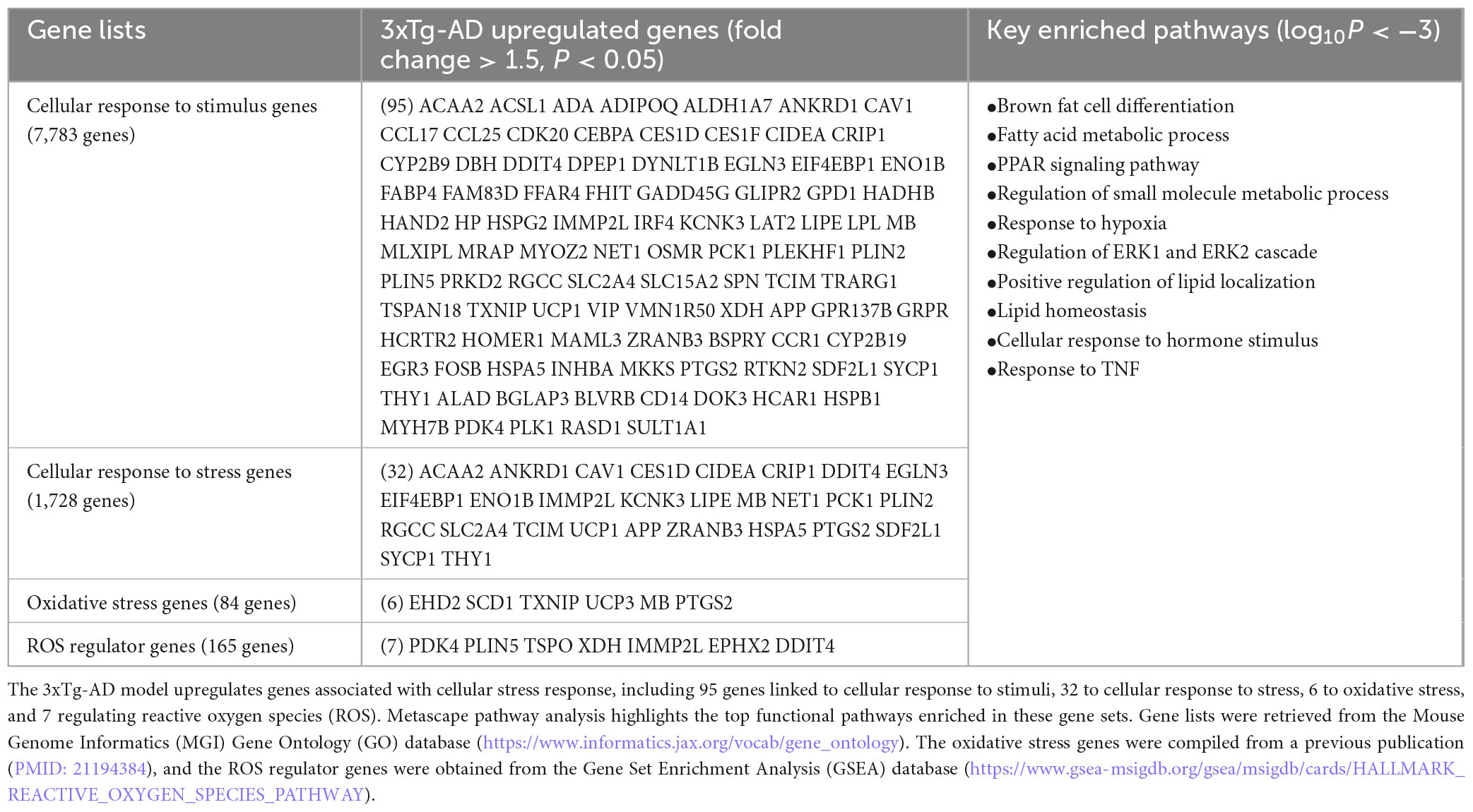
Table 2A. The 3xTg-AD model induces cellular and oxidative stress.
To explore potential mechanisms by which ER stress contributes to EndoMT, cell death, and fibrosis in the 3xTg-AD brain, we further analyzed transcriptomic reprogramming induced by 3xTg-AD across nine categories of ER stress-related genes. As shown in Table 2B, this analysis revealed: (i) upregulation of five ER-localized protein genes, (ii) upregulation of three ER processing protein genes, (iii) upregulation of four genes involved in the response to ER stress, (iv) upregulation of two AD-promoting genes (Bell et al., 2016) associated with protein kinase R-like ER kinase (EIF2AK3, also known as PERK, gut microbiota generated uremic toxin trimethylamine N-oxide, TMAO (Chan et al., 2019; Saaoud et al., 2023; Sun et al., 2018) receptor (Chen et al., 2019) interaction proteins, (v) upregulation of five genes associated with AD-suppressing (Zhang et al., 2022) activating transcription factor 6 (ATF6) interaction proteins, (vi) upregulation of two genes associated with AD- exacerbating (Duran-Aniotz et al., 2017) endoplasmic reticulum to nucleus signaling 1 (IRE1) interaction proteins, (vii) upregulation of four genes in the ER associated protein degradation (ERAD) pathway (Krshnan et al., 2022), (viii) upregulation of 22 genes induced by the ER stressor thapsigargin (a plant-derived inhibitor of calcium pumps), and (ix) upregulation of 14 genes induced by the ER stressor tunicamycin (an antibiotic produced by the bacterium streptomyces lysosuperificus). Metascape pathway analysis of these upregulated ER stress genes identified the key enriched pathways (log10P < −3), including positive regulation of cold-induced thermogenesis, regulation of small molecule metabolic process, PPAR signaling pathway, brown fat cell differentiation, cellular response to hormone stimulus, chaperone cofactor-dependent protein refolding, regulation of carbohydrate metabolic process, metabolism of vitamins and cofactors, triglyceride metabolic process, and regulation of cysteine-type endopeptidase activity involved in apoptotic process.
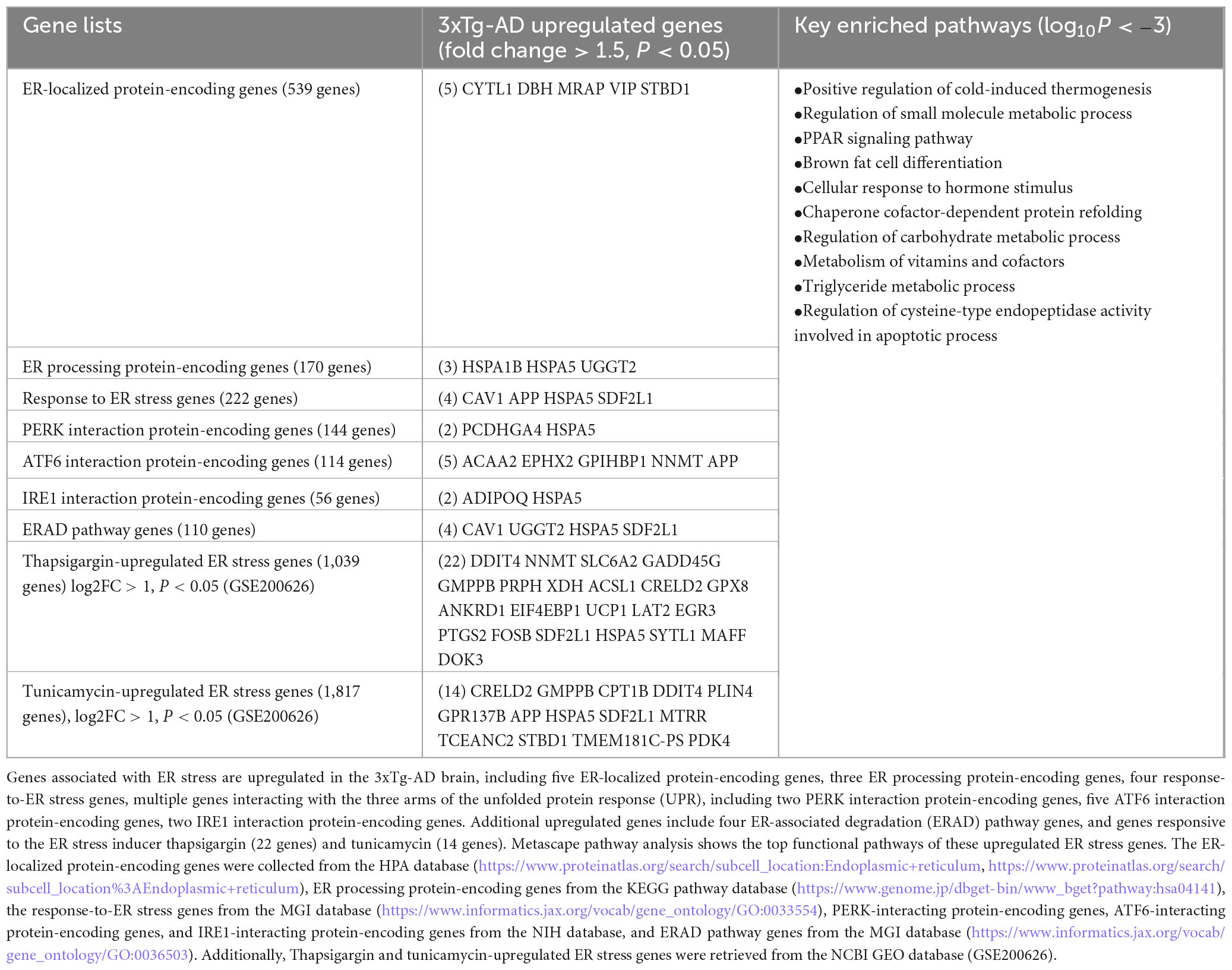
Table 2B. The 3xTg-AD model induces ER stress.
To further elucidate the role of PERK—one of the three key ER stress sensors—in the pathophysiology of AD in the brain of 3xTg mice, we analyzed two transcriptomic datasets from PERK knockout (KO) models, collected from the NCBI GEO database. As shown in Table 2C, the AGBL4 gene was significantly downregulated in the PERK KO group compared to the wild-type (WT) controls. Additionally, 22 genes were downregulated in the PERK KO group following tunicamycin treatment, relative to tunicamycin-treated WT samples. Notably, oxidative stress gene PTGS2 (prostaglandin-endoperoxide synthase 2), DDIT4 (ROS regulator, also upregulated by thapsigargin, tunicamycin, and PERK KO with tunicamycin treatment), and several other ER stress genes upregulated by PERK KO and tunicamycin treatment—such as VCAN (EndoMT gene), eukaryotic translation initiation factor 4E binding protein 1 (EIF4EBP1, cellular stress gene), perilipin 4 (PLIN4), growth arrest and DNA damage inducible gamma (GADD45G), neuronal pentraxin 2 (NPTX2), EGR3—were also upregulated across multiple brain cell types in AD (Figures 1I, L and Supplementary Figures 1C, D, 3A, 4A–C).10 Taken together, our findings demonstrate that the 3xTg-AD brain promotes EndoMT, cell death, and fibrosis in the brain and brain cells by upregulating genes related to cellular stress, oxidative stress, and ER stress.
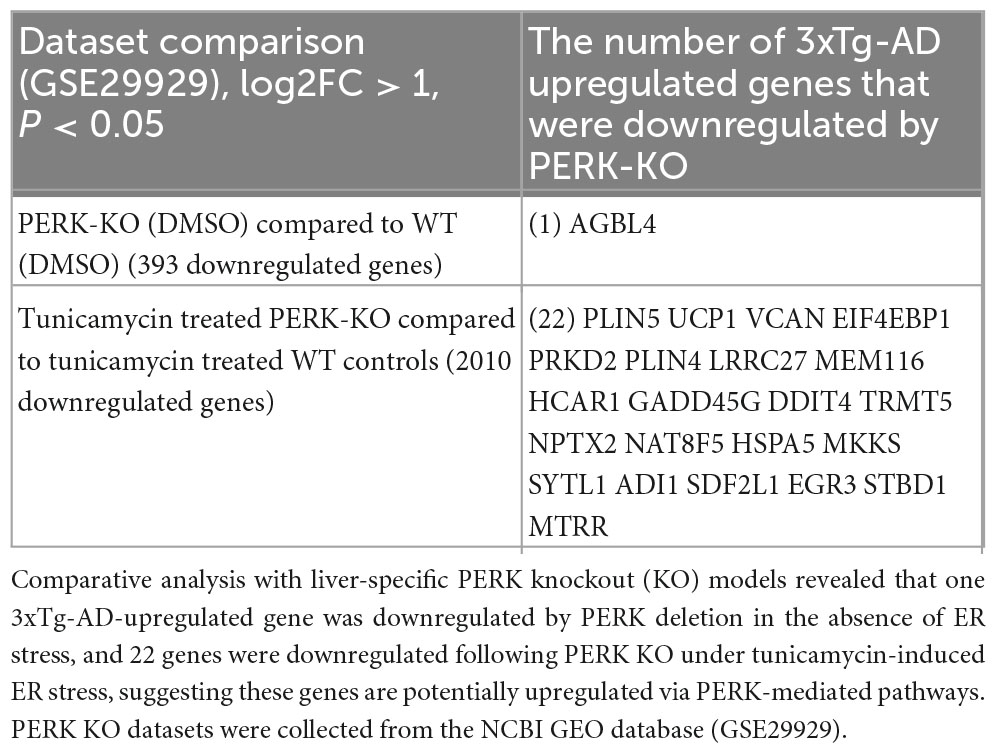
Table 2C. Transcriptomic evidence for ER stress activation in the 3xTg-AD mouse brain mediated by PERK signaling.
3.8 The 3xTg-AD model drives EndoMT, cell death, and fibrosis in both brain and brain cells through the upregulation of genes involved in lipid metabolism, glucose metabolism, and oxidative phosphorylation (OXPHOS) genes
Mitochondrial abnormalities have been reported in patients with AD, and the roles of full-length APP, and the roles of soluble APPa on mitochondrial bioenergetic metabolism have also been reported (Lopez Sanchez et al., 2019). To investigate the potential metabolic reprogramming mechanisms (via metabolites-induced post-translational modifications of histones) as we reported (Drummer et al., 2021a; Saaoud et al., 2023; Saaoud et al., 2024; Xu et al., 2024) underlying EndoMT, cell death, and fibrosis in the 3xTg-AD brain and brain cells, we examined the transcriptomic changes in metabolic pathways induced by the 3xTg-AD model. As shown in Table 3, we identified the upregulation of 25 lipid and lipoprotein metabolism genes, 8 fatty acid oxidation (FAO) genes, 7 glucose metabolism genes, and 8 OXPHOS genes in the 3xTg-AD brain. Additionally, we observed downregulation of eight lipid and lipoprotein metabolism genes, six glucose metabolism genes, and four OXPHOS genes in the 3xTg-AD brain, which aligns with previous reports of age-dependent declines in mitochondrial complex II activity in a familial AD mouse model (Emmerzaal et al., 2018), as well as reduced cytochrome c oxidase activity in AD (Morais et al., 2021). Metascape pathway analysis of AD-upregulated lipid and lipoprotein metabolism genes identified the key enriched pathways (log10P < −3), including fatty acid beta oxidation, metabolism of lipids, PPAR signaling pathway, triacylglyceride synthesis, and lipid localization. The key enriched pathways associated with AD-upregulated FAO genes included metabolism of lipids, negative regulation of catalytic activity, and membrane organization. For AD-upregulated glucose metabolism genes, the top enriched pathways included glucose metabolic process, regulation of small molecule metabolic process, small molecule catabolic process, and regulation of purine nucleotide metabolic process. Notably, several genes—including LPL (lipoprotein lipase, a lipid and lipoprotein metabolism gene), PTGS2 (lipid and lipoprotein metabolism and OXPHOS gene), and APP (amyloid beta precursor protein, OXPHOS gene)—were also upregulated among multiple cell types in AD brain (Figure 1I and Supplementary Figures 5A, B).10 Taken together, our findings demonstrate that the 3xTg-AD model promotes EndoMT, cell death, and fibrosis pathways in the brain and brain cells by upregulating genes involved in lipid and lipoprotein metabolism, glucose metabolism, and OXPHOS.
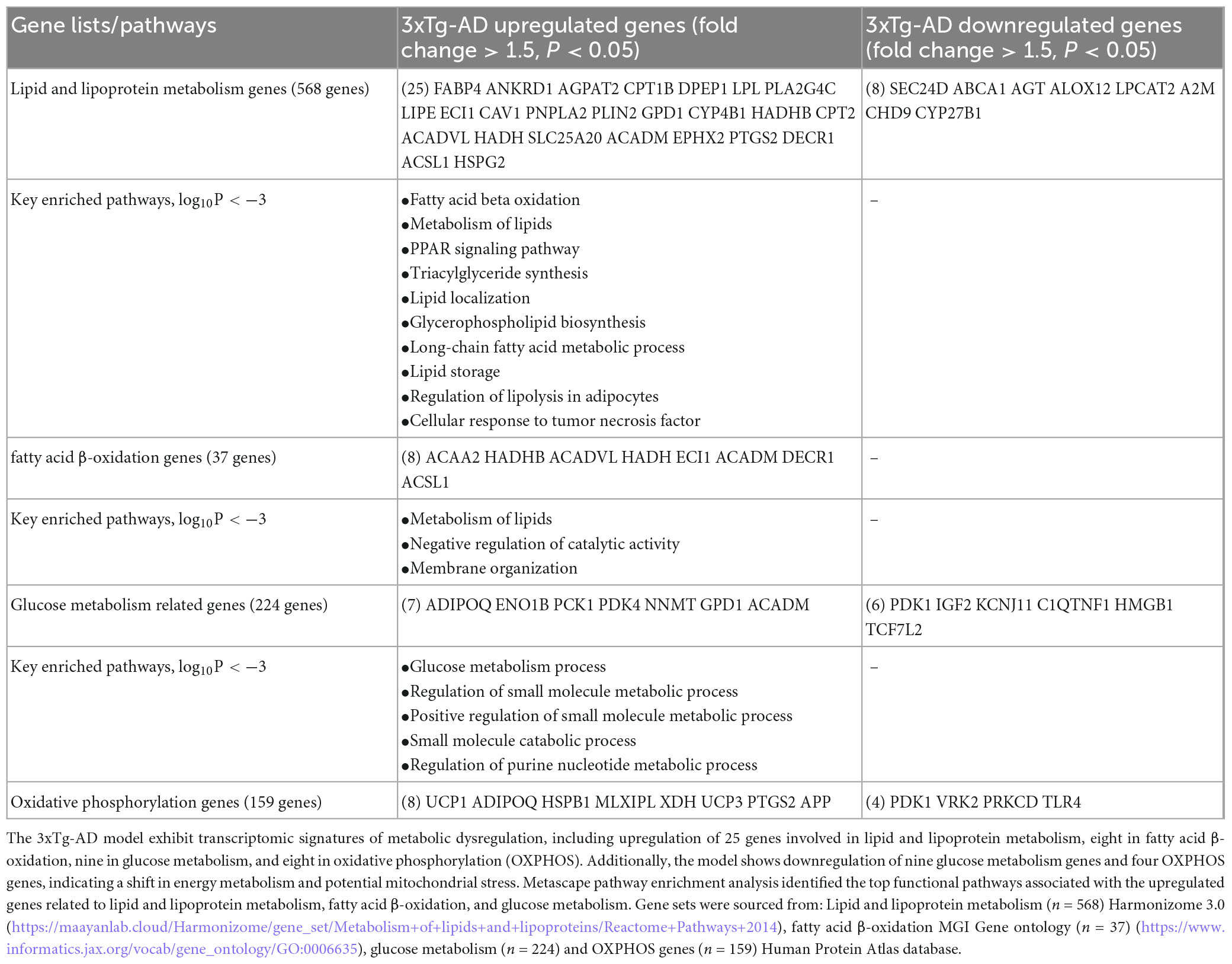
Table 3. The 3xTg-AD model induces metabolic reprogramming and stress response.
3.9 The 3xTg-AD model drives EndoMT, cell death, and fibrosis in brain and brain cells through the upregulation of catabolic pathways
Single-cell RNA seq datasets from ischemic stroke, hemorrhagic stroke, and AD models have been integrated to construct metabolic flux profiles at the single-cell level. These three disorders induce shared metabolic shifts in ECs, with altered metabolic modules primarily enriched in transporter-related pathways. These shifts are predicted to potentially reduce metabolites such as pyruvate and fumarate. Additionally, Lef1 (lymphoid enhancer binding factor 1, Elk3 (ETS transcription factor), and Fosl1 (FOS like 1, AP-1 transcription factor subunit) may function as upstream transcriptional regulators driving these metabolic changes (Guo et al., 2023). To explore the potential catabolic reprogramming mechanisms underlying EndoMT, cell death, and fibrosis in the 3xTg-AD brain and brain cells, we examined transcriptomic reprogramming of catabolic process induced by the 3xTg-AD model. As shown in Table 4, we identified the upregulation of 63 catabolic genes and the downregulation of 27 catabolic genes. Metascape pathway analysis of the upregulated catabolic genes revealed the key enriched pathways, including lipid catabolic process, regulation of small molecule metabolic process, glycerolipid catabolic process, PPAR signaling pathway, regulation of lipid localization, positive regulation of cold-induced thermogenesis, mitochondrial FAO of unsaturated fatty acids (FAs), FAO using acyl-CoA dehydrogenase, regulation of sequestering of triglyceride, and regulation of ATP metabolic process. Metascape pathway analysis of the downregulated catabolic genes identified the top five pathways, including carbohydrate derivative catabolic process, small molecule catabolic process, positive regulation of catabolic process, regulation of type II interferon production, and carboxylic acid transport. Notably, six AD-upregulated catabolic genes, including DBH (dopamine beta-hydroxylase), ADA (adenosine deaminase), FHIT (fragile histidine triad diadenosine triphosphatase), HSPA1B (heat shock protein family A (Hsp70) member 1B), and VIP (vasoactive intestinal peptide) were upregulated across multiple cell types in AD brain (Supplementary Figure 6).10 In conclusion, our findings demonstrate that the 3xTg-AD brain upregulates EndoMT, cell death, and fibrosis pathways in brain and brain cells by enhancing the expressions of catabolic genes to a greater extent than downregulating them.
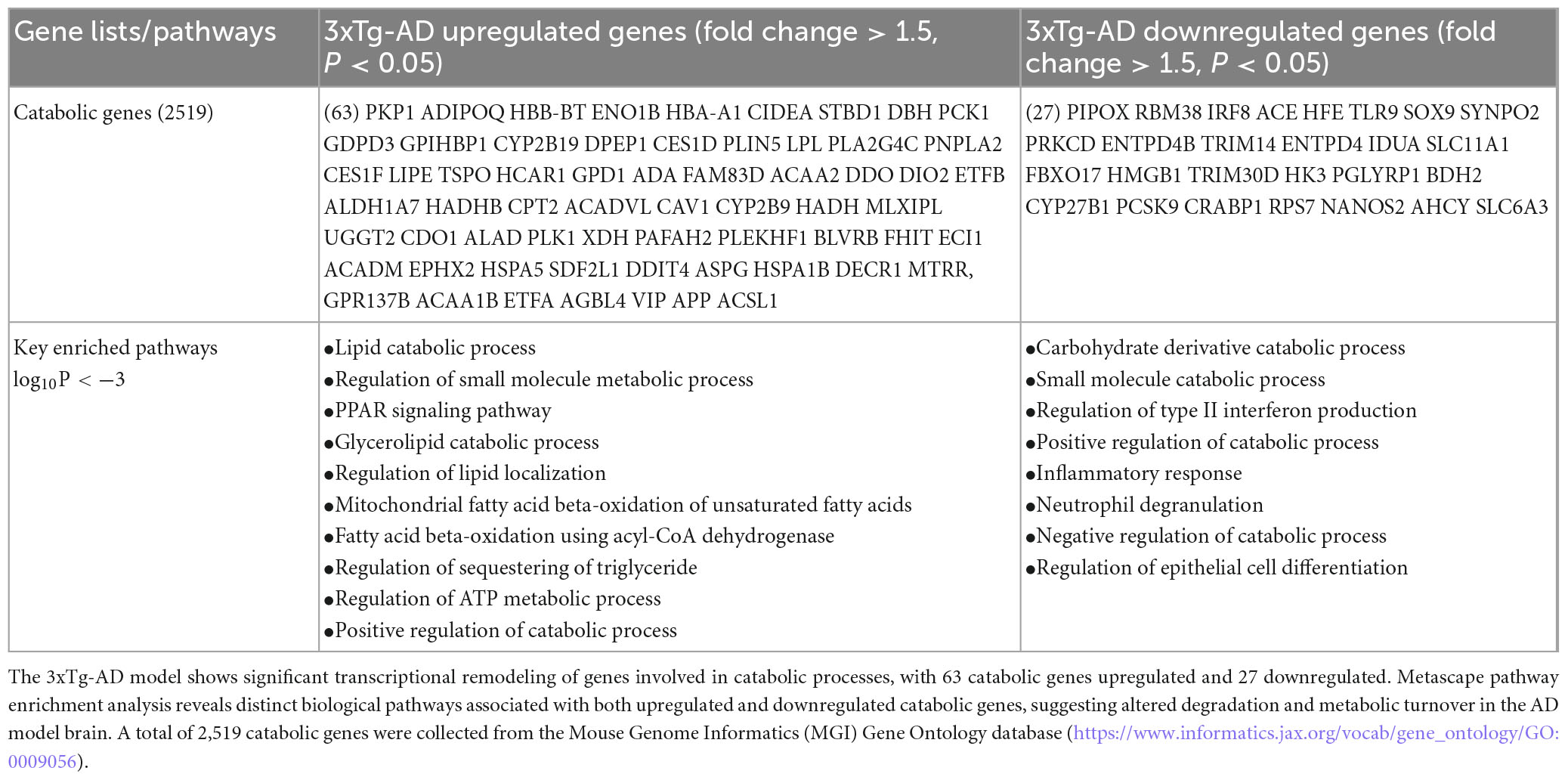
Table 4. Differential regulation of catabolic pathways in the 3xTg-AD mouse brain.
4 Discussion
Despite significant advancement in AD research, current therapies offer no cure and provide only limited palliative benefits against disease progression. AD is characterized by two hallmark pathologies: extracellular plaques composed primarily of Aβ and intraneuronal neurofibrillary tangles (NFTs) primarily composed of hyperphosphorylated Tau protein (DeTure and Dickson, 2019; Trejo-Lopez et al., 2022). However, several pathogenic processes—such as EC activation for inflammatory cell recruitment and EndoMT, cell death, and fibrosis in AD and brain ECs—remain poorly understood.
Using our novel knowledge-based transcriptomic analysis approach (Xu et al., 2021), single-cell RNA-seq data from the Single Cell Portal database at the MIT-Broad Institute, along with our unpublished data, we found compelling evidences that the 3xTg-AD brain induces EndoMT, activates 14 types of cell death, and promotes fibrosis and fibroblast generation in the brain and brain ECs. In addition, we identified five potential mechanisms underlying EndoMT, cell death, and fibrosis, including mitochondrial stress, ER stress, nuclear stress, metabolic reprogramming, and catabolic stress.
In AD, elevated levels of Aβ and p-tau can induce ROS production, leading to excessive mitochondrial fragmentation, fission (Zhu et al., 2013), and defective mitophagy. Five types of mitophagy—primarily focused on neurons—have been reported: (1) Aβ and p-tau-induced mitophagy, (2) stress-induced mitophagy, (3) receptor-mediated mitophagy, (4) ubiquitin-mediated mitophagy, and (5) basal mitophagy (Pradeepkiran and Reddy, 2020). Additionally, sporadic AD exhibits slower mitochondrial dynamics and accumulation of aged mitochondria (Martín-Maestro et al., 2017). Reducing oxidative stress, restoring mitochondrial energetics, and lifestyle interventions (such as diet and exercise) have been shown to decrease Aβ and ameliorate learning and memory impairments in AD (Johri, 2021). Various mitochondria-targeted antioxidants are currently under development for AD (Johri, 2021).
Endoplasmic reticulum stress also plays a distinct role in AD pathogenesis (Ajoolabady et al., 2022), and misfolded proteins and cellular stressors can cause significant structural and molecular alterations in the nucleus (Iatrou et al., 2021). Metabolic dysregulations in AD (Polis and Samson, 2020), including dysfunctional glucose metabolism (Butterfield and Halliwell, 2019), insulin resistance (Pansuria et al., 2012; Sedzikowska and Szablewski, 2021), lipid metabolism abnormalities (Yin, 2023), energy metabolism issues (Yin et al., 2016), gut microbiota dysbiosis (Liu et al., 2020), and hypometabolism (Correas et al., 2024), further contribute to disease progression (Yan et al., 2020). Amino acid oxidation can temporarily compensate for decreased glucose metabolism via glutaminolysis-fed tricarboxylic acid (TCA) cycle(Xu et al., 2023; Xu et al., 2024), but altered levels of amino acids and their catabolites(Sun et al., 2018) may eventually lead to toxicities that exacerbate AD progression (Griffin and Bradshaw, 2017). Collectively, these findings strongly support our identification of five mechanisms underlying EndoMT, cell death, and fibrosis in the AD brain and brain ECs.
Based on our results, we propose a new working model (Figure 4). First, the 3xTg-AD brain induces expression of genes associated with several types of cell death, exceeding those previously reported (Goel et al., 2022). This observation aligns with findings that neurons in AD brains undergo a programmed form of cell death known as necroptosis in response to amyloid plaques and tau tangles, which are misfolded proteins (Goel et al., 2022). Second, the 3xTg-AD brain promotes EndoMT in brain ECs and fibroblasts, as well as fibrosis in the AD brain. In line with this, our previous report identified AD as an auto-innate immune/auto-inflammatory and autoimmune disease (Saaoud et al., 2025). Our findings on increased EndoMT in AD brain ECs support current reports suggesting that neuro-inflammatory diseases disrupt the blood-brain/CNS barrier (BBB) through crosstalk between pro-inflammatory and EndoMT signaling pathways (Sun et al., 2022), and that microvascular contributions to AD pathogenesis and endotheliopathy are significant features of the disease (Tarawneh, 2023). Third, leveraging our knowledge-based transcriptomic analysis approach (Xu et al., 2021), single-cell RNA-seq data, and our unpublished data, we found robust evidence of five mechanisms underlying EndoMT, cell death, and fibrosis in AD brain and brain ECs. These mechanisms include mitochondrial stress, ER stress, nuclear stress, metabolic reprogramming, and catabolic stress. Fourth, mechanistic analysis revealed that genes upregulated by 3xTg-AD were also upregulated by FOSB overexpression while downregulated by deficiencies in MEOX1 and ER stress kinase EIF2AK3 (PERK) knockdown, suggesting that these master genes may drive AD pathology and associated mechanisms. Overall, our panoramic and mechanistic findings provide novel insights into the roles of 3xTg AD-induced endotheliopathy, EndoMT, cell death, and fibrosis in promoting AD cerebrovascular damage and disease progression via organelle stresses and bioenergetic mechanisms.
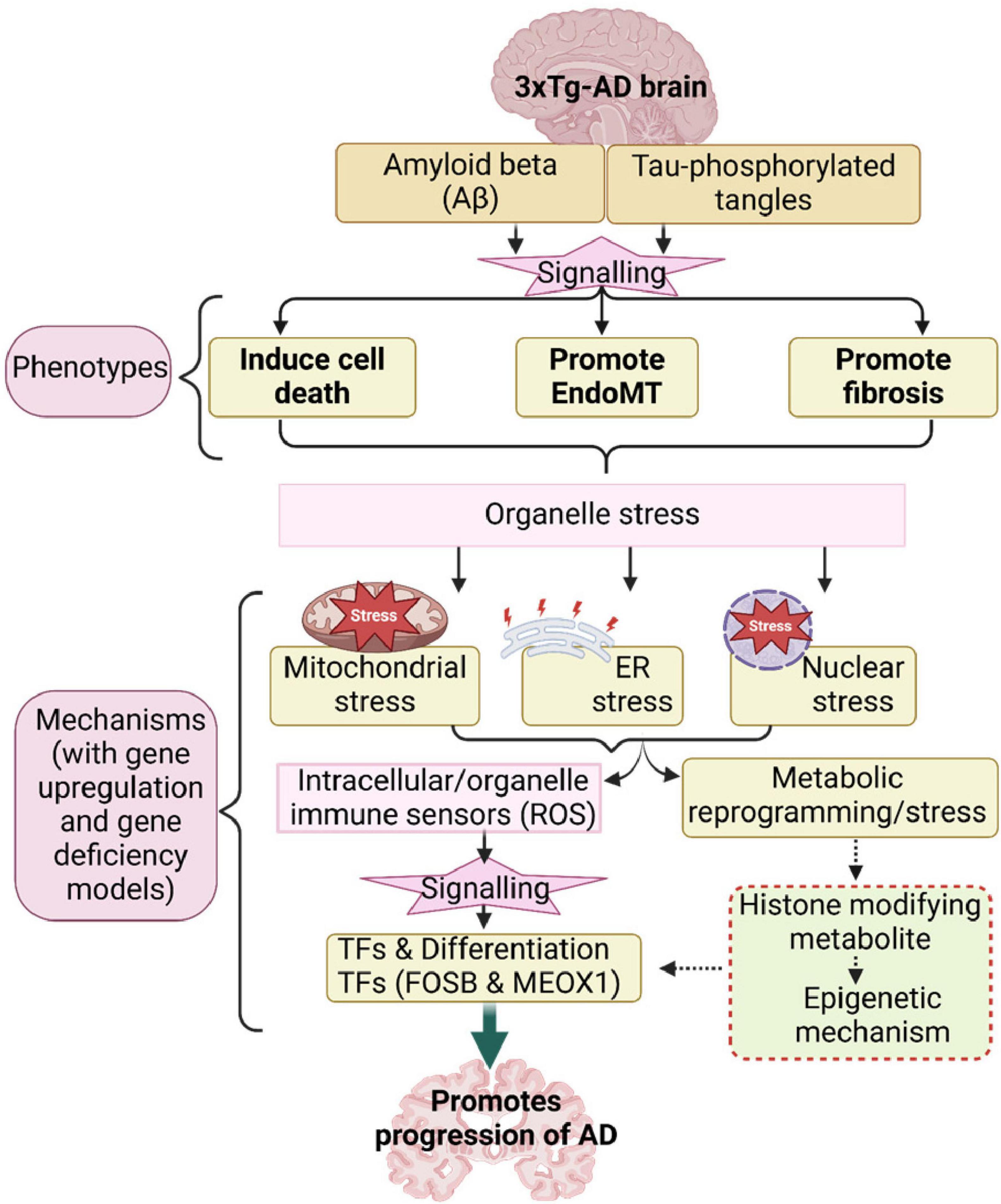
Figure 4. Our working model.
Data availability statement
Original datasets are available in a publicly accessible repository: GSE305112. The original contributions presented in the study are publicly available. This data can be found here: [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE305112].
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee (IACUC) of Temple University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
FS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. MB: Conceptualization, Formal analysis, Investigation, Methodology, Writing – review & editing. LL: Conceptualization, Formal analysis, Writing – review & editing. KX: Conceptualization, Formal analysis, Writing – review & editing. YL: Conceptualization, Formal analysis, Writing – review & editing. YS: Conceptualization, Formal analysis, Writing – review & editing. BH: Conceptualization, Formal analysis, Writing – review & editing. XJ: Conceptualization, Formal analysis, Writing – review & editing. XL: Conceptualization, Formal analysis, Writing – review & editing. AG: Conceptualization, Formal analysis, Writing – review & editing. JL: Conceptualization, Formal analysis, Writing – review & editing. LM: Conceptualization, Formal analysis, Writing – review & editing. RV-P: Conceptualization, Formal analysis, Writing – review & editing. SM: Conceptualization, Formal analysis, Writing – review & editing. BK: Conceptualization, Formal analysis, Writing – review & editing. HW: Conceptualization, Formal analysis, Supervision, Writing – review & editing. SF: Conceptualization, Formal analysis, Supervision, Writing – review & editing. XY: Conceptualization, Data curation, Formal analysis, Funding acquisition, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Institute of Health (R01 HL163570-01A1 and 1R01HL147565-01).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2025.1605012/full#supplementary-material
Footnotes
1. ^https://www.alz.org/alzheimer_s_dementia
2. ^https://www.ncbi.nlm.nih.gov/gds
4. ^https://www.proteinatlas.org/
5. ^https://singlecell.broadinstitute.org/single_cell/study/SCP1375/integrative-in~situ-mapping-of-single-cell-transcriptional-states-and-tissue-histopathology-in-an-alzheimer-disease-model
6. ^https://www.ncbi.nlm.nih.gov/gene/28984
7. ^https://singlecell.broadinstitute.org/single_cell
8. ^https://informatics.jax.org/
9. ^http://www.zhounan.org/ferrdb/current/
10. ^https://singlecell.broadinstitute.org/single_cell/study/SCP1375/integrative-in-situ-mapping-of-single-cell-transcriptional-states-and-tissue-histopathology-in-an-alzheimer-disease-model#study-visualize
11. ^https://maayanlab.cloud/Harmonizome/gene_set/AP-1/MotifMap+Predicted+Transcription+Factor+Targets
References
Ajoolabady, A., Lindholm, D., Ren, J., and Pratico, D. (2022). ER stress and UPR in Alzheimer’s disease: Mechanisms, pathogenesis, treatments. Cell. Death Dis. 13:706. doi: 10.1038/s41419-022-05153-5
An, X., Jin, Y., Guo, H., Foo, S., Cully, B., Wu, J., et al. (2009). Response gene to complement 32, a novel hypoxia-regulated angiogenic inhibitor. Circulation 120, 617–627. doi: 10.1161/CIRCULATIONAHA.108.841502
Baik, S., Kang, S., Lee, W., Choi, H., Chung, S., Kim, J., et al. (2019). A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell. Metab. 30, 493–507.e6. doi: 10.1016/j.cmet.2019.06.005
Beatty, A., Singh, T., Tyurina, Y., Tyurin, V., Samovich, S., Nicolas, E., et al. (2021). Ferroptotic cell death triggered by conjugated linolenic acids is mediated by ACSL1. Nat. Commun. 12:2244. doi: 10.1038/s41467-021-22471-y
Bell, M., Meier, S., Ingram, A., and Abisambra, J. F. (2016). PERK-opathies: An Endoplasmic Reticulum Stress Mechanism Underlying Neurodegeneration. Curr. Alzheimer Res. 13, 150–163. doi: 10.2174/1567205013666151218145431
Brokaw, D., Piras, I., Mastroeni, D., Weisenberger, D., Nolz, J., Delvaux, E., et al. (2020). Cell death and survival pathways in Alzheimer’s disease: An integrative hypothesis testing approach utilizing -omic data sets. Neurobiol. Aging 95, 15–25. doi: 10.1016/j.neurobiolaging.2020.06.022
Butterfield, D., and Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. doi: 10.1038/s41583-019-0132-6
Chan, M., Yang, X., Wang, H., Saaoud, F., Sun, Y., and Fong, D. (2019). The Microbial metabolite trimethylamine N-oxide links vascular dysfunctions and the autoimmune disease rheumatoid arthritis. Nutrients 11:1821. doi: 10.3390/nu11081821
Chen, S., Henderson, A., Petriello, M., Romano, K., Gearing, M., Miao, J., et al. (2019). Trimethylamine N-oxide binds and activates PERK to promote metabolic dysfunction. Cell. Metab. 30, 1141–1151.e5. doi: 10.1016/j.cmet.2019.08.021
Chen, S., Tian, D., Petersen, L., Cao, S., Quinn, Z., Kan, J., et al. (2022). Prognostic value of GIMAP4 and its role in promoting immune cell infiltration into tumor microenvironment of lung adenocarcinoma. Biomed. Res. Int. 2022:7440189. doi: 10.1155/2022/7440189
Chen, X., and Holtzman, D. (2022). Emerging roles of innate and adaptive immunity in Alzheimer’s disease. Immunity 55, 2236–2254. doi: 10.1016/j.immuni.2022.10.016
Chu, B., Kon, N., Chen, D., Li, T., Liu, T., Jiang, L., et al. (2019). ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell. Biol. 21, 579–591. doi: 10.1038/s41556-019-0305-6
Correas, A., Olaso-Gonzalez, G., Roca, M., Blanco-Gandía, M., Nascimento, C., Lahoz, A., et al. (2024). Glucose 6 phosphate dehydrogenase overexpression rescues the loss of cognition in the double transgenic APP/PS1 mouse model of Alzheimer’s disease. Redox Biol. 75:103242. doi: 10.1016/j.redox.2024.103242
D’Ambrosi, N., and Apolloni, S. (2020). Fibrotic scar in neurodegenerative diseases. Front. Immunol. 11:1394. doi: 10.3389/fimmu.2020.01394
Derada Troletti, C., de Goede, P., Kamermans, A., and de Vries, H. (2016). Molecular alterations of the blood-brain barrier under inflammatory conditions: The role of endothelial to mesenchymal transition. Biochim. Biophys. Acta 1862, 452–460. doi: 10.1016/j.bbadis.2015.10.010
DeTure, M., and Dickson, D. (2019). The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14:32. doi: 10.1186/s13024-019-0333-5
Drummer, C., Saaoud, F., Jhala, N., Cueto, R., Sun, Y., Xu, K., et al. (2023). Caspase-11 promotes high-fat diet-induced NAFLD by increasing glycolysis, OXPHOS, and pyroptosis in macrophages. Front. Immunol. 14:1113883. doi: 10.3389/fimmu.2023.1113883
Drummer, C., Saaoud, F., Shao (邵颖), Y., Sun (孙宇), Y., Xu (徐克曼), K., Lu (路一凡), Y., et al. (2021a). Trained immunity and reactivity of macrophages and endothelial cells. Arterioscler. Thromb. Vasc. Biol. 41, 1032–1046. doi: 10.1161/ATVBAHA.120.315452
Drummer, C., Saaoud, F., Sun, Y., Atar, D., Xu, K., Lu, Y., et al. (2021b). Hyperlipidemia may synergize with hypomethylation in establishing trained immunity and promoting inflammation in NASH and NAFLD. J. Immunol. Res. 2021:3928323. doi: 10.1155/2021/3928323
Duran-Aniotz, C., Cornejo, V., Espinoza, S., Ardiles, ÁO., Medinas, D. B., Salazar, C., et al. (2017). IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 134, 489–506. doi: 10.1007/s00401-017-1694-x
Emmerzaal, T., Rodenburg, R., Tanila, H., Verweij, V., Kiliaan, A., and Kozicz, T. (2018). Age-dependent decrease of mitochondrial complex II Activity in a familial mouse model for Alzheimer’s disease. J. Alzheimers Dis. 66, 75–82. doi: 10.3233/JAD-180337
Engel, D., Beckers, L., Wijnands, E., Seijkens, T., Lievens, D., Drechsler, M., et al. (2011). Caveolin-1 deficiency decreases atherosclerosis by hampering leukocyte influx into the arterial wall and generating a regulatory T-cell response. FASEB J. 25, 3838–3848. doi: 10.1096/fj.11-183350
Fang, Y., Hsieh, Y., Hu, C., and Tu, Y. (2023). Endothelial dysfunction in neurodegenerative diseases. Int. J. Mol. Sci. 24:2909. doi: 10.3390/ijms24032909
Feng, W., Chen, L., Nguyen, P., Wu, S., and Li, G. (2019). Single cell analysis of endothelial cells identified organ-specific molecular signatures and heart-specific cell populations and molecular features. Front. Cardiovasc. Med. 6:165. doi: 10.3389/fcvm.2019.00165
Feng, Z., Meng, F., Huo, F., Zhu, Y., Qin, Y., Gui, Y., et al. (2024). Inhibition of ferroptosis rescues M2 macrophages and alleviates arthritis by suppressing the HMGB1/TLR4/STAT3 axis in M1 macrophages. Redox Biol. 75:103255. doi: 10.1016/j.redox.2024.103255
Frazier, A., Thorburn, D., and Compton, A. (2019). Mitochondrial energy generation disorders: Genes, mechanisms, and clues to pathology. J. Biol. Chem. 294, 5386–5395. doi: 10.1074/jbc.R117.809194
Galluzzi, L., Vitale, I., Aaronson, S., Abrams, J., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell. Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Goel, P., Chakrabarti, S., Goel, K., Bhutani, K., Chopra, T., and Bali, S. (2022). Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Front. Mol. Neurosci. 15:937133. doi: 10.3389/fnmol.2022.937133
Griffin, J., and Bradshaw, P. (2017). Amino acid catabolism in Alzheimer’s disease brain: Friend or Foe? Oxid. Med. Cell. Longev. 2017:5472792. doi: 10.1155/2017/5472792
Gu, C., Shi, X., Dang, X., Chen, J., Chen, C., Chen, Y., et al. (2020). Identification of common genes and pathways in eight fibrosis diseases. Front. Genet. 11:627396. doi: 10.3389/fgene.2020.627396
Guo, G., Fan, L., Yan, Y., Xu, Y., Deng, Z., Tian, M., et al. (2023). Shared metabolic shifts in endothelial cells in stroke and Alzheimer’s disease revealed by integrated analysis. Sci. Data 10:66. doi: 10.1038/s41597-023-02512-5
Hagar, J., and Miao, E. (2014). Detection of cytosolic bacteria by inflammatory caspases. Curr. Opin. Microbiol. 17, 61–66. doi: 10.1016/j.mib.2013.11.008
Iatrou, A., Clark, E., and Wang, Y. (2021). Nuclear dynamics and stress responses in Alzheimer’s disease. Mol. Neurodegener. 16:65. doi: 10.1186/s13024-021-00489-6
Jiang, X., Stockwell, B., and Conrad, M. (2021). Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell. Biol. 22, 266–282. doi: 10.1038/s41580-020-00324-8
Johri, A. (2021). Disentangling mitochondria in Alzheimer’s disease. Int. J. Mol. Sci. 22:11520. doi: 10.3390/ijms222111520
Knopman, D., Amieva, H., Petersen, R., Chételat, G., Holtzman, D., Hyman, B., et al. (2021). Alzheimer disease. Nat. Rev. Dis. Primers 7:33. doi: 10.1038/s41572-021-00269-y
Kovacic, J., Dimmeler, S., Harvey, R., Finkel, T., Aikawa, E., Krenning, G., et al. (2019). Endothelial to mesenchymal transition in cardiovascular disease: Jacc state-of-the-art review. J. Am. Coll. Cardiol. 73, 190–209. doi: 10.1016/j.jacc.2018.09.089
Krishnan, S., Manoharan, J., Wang, H., Gupta, D., Fatima, S., Yu, Y., et al. (2023). CD248 induces a maladaptive unfolded protein response in diabetic kidney disease. Kidney Int. 103, 304–319. doi: 10.1016/j.kint.2022.09.024
Krshnan, L., van de Weijer, M., and Carvalho, P. (2022). Endoplasmic reticulum-associated protein degradation. Cold Spring Harb. Perspect. Biol. 14:a041247. doi: 10.1101/cshperspect.a041247
Lagrange, J., Lecompte, T., Knopp, T., Lacolley, P., and Regnault, V. (2022). Alpha-2-macroglobulin in hemostasis and thrombosis: An underestimated old double-edged sword. J. Thromb. Haemost. 20, 806–815. doi: 10.1111/jth.15647
Li, J., Cao, F., Yin, H., Huang, Z., Lin, Z., Mao, N., et al. (2020). Ferroptosis: Past, present and future. Cell. Death Dis. 11:88. doi: 10.1038/s41419-020-2298-2
Liu, M., Wu, N., Xu, K., Saaoud, F., Vasilopoulos, E., Shao, Y., et al. (2021). Organelle crosstalk regulators are regulated in diseases, tumors, and regulatory T cells: Novel classification of organelle crosstalk regulators. Front. Cardiovasc. Med. 8:713170. doi: 10.3389/fcvm.2021.713170
Liu, S., Gao, J., Zhu, M., Liu, K., and Zhang, H. (2020). Gut microbiota and dysbiosis in Alzheimer’s disease: Implications for pathogenesis and treatment. Mol. Neurobiol. 57, 5026–5043. doi: 10.1007/s12035-020-02073-3
Lopez Sanchez, M., van Wijngaarden, P., and Trounce, I. (2019). Amyloid precursor protein-mediated mitochondrial regulation and Alzheimer’s disease. Br. J. Pharmacol. 176, 3464–3474. doi: 10.1111/bph.14554
Lu, Y., Nanayakkara, G., Sun, Y., Liu, L., Xu, K., Drummer, C., et al. (2021). Procaspase-1 patrolled to the nucleus of proatherogenic lipid LPC-activated human aortic endothelial cells induces ROS promoter CYP1B1 and strong inflammation. Redox Biol. 47:102142. doi: 10.1016/j.redox.2021.102142
Lu, Y., Sun, Y., Drummer, C., Nanayakkara, G., Shao, Y., Saaoud, F., et al. (2019). Increased acetylation of H3K14 in the genomic regions that encode trained immunity enzymes in lysophosphatidylcholine-activated human aortic endothelial cells - Novel qualification markers for chronic disease risk factors and conditional DAMPs. Redox Biol. 24:101221. doi: 10.1016/j.redox.2019.101221
Lu, Y., Sun, Y., Saaoud, F., Shao, Y., Xu, K., Jiang, X., et al. (2023). ER stress mediates Angiotensin II-augmented innate immunity memory and facilitates distinct susceptibilities of thoracic from abdominal aorta to aneurysm development. Front. Immunol. 14:1268916. doi: 10.3389/fimmu.2023.1268916
Maezawa, I., Jin, L., Woltjer, R., Maeda, N., Martin, G., Montine, T., et al. (2004). Apolipoprotein E isoforms and apolipoprotein AI protect from amyloid precursor protein carboxy terminal fragment-associated cytotoxicity. J. Neurochem. 91, 1312–1321. doi: 10.1111/j.1471-4159.2004.02818.x
Martín-Maestro, P., Gargini, R., García, E., Perry, G., Avila, J., and García-Escudero, V. (2017). slower dynamics and aged mitochondria in sporadic Alzheimer’s disease. Oxid. Med. Cell. Longev. 2017:9302761. doi: 10.1155/2017/9302761
Morais, F., Ribeiro, A., Moreira, F., and Silva, P. (2021). Systematic review and meta-analysis on the role of mitochondrial cytochrome c oxidase in Alzheimer’s disease. Acta Neuropsychiatr. 33, 55–64. doi: 10.1017/neu.2020.43
Ng, A., Khoshakhlagh, P., Rojo Arias, J., Pasquini, G., Wang, K., Swiersy, A., et al. (2021). A comprehensive library of human transcription factors for cell fate engineering. Nat. Biotechnol. 39, 510–519. doi: 10.1038/s41587-020-0742-6
Ogawa, H., Oohashi, T., Sata, M., Bekku, Y., Hirohata, S., Nakamura, K., et al. (2004). Lp3/Hapln3, a novel link protein that co-localizes with versican and is coordinately up-regulated by platelet-derived growth factor in arterial smooth muscle cells. Matrix Biol. 23, 287–298. doi: 10.1016/j.matbio.2004.07.001
Pansuria, M., Xi, H., Li, L., Yang, X., and Wang, H. (2012). Insulin resistance, metabolic stress, and atherosclerosis. Front. Biosci. 4:916–931. doi: 10.2741/s308
Pardali, E., Sanchez-Duffhues, G., Gomez-Puerto, M., and Ten Dijke, P. (2017). TGF-β-Induced endothelial-mesenchymal transition in fibrotic diseases. Int. J. Mol. Sci. 18:2157. doi: 10.3390/ijms18102157
Piera-Velazquez, S., and Jimenez, S. (2019). Endothelial to mesenchymal transition: Role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 99, 1281–1324. doi: 10.1152/physrev.00021.2018
Polis, B., and Samson, A. (2020). Role of the metabolism of branched-chain amino acids in the development of Alzheimer’s disease and other metabolic disorders. Neural Regen. Res. 15, 1460–1470. doi: 10.4103/1673-5374.274328
Pradeepkiran, J., and Reddy, P. (2020). Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 64:101191. doi: 10.1016/j.arr.2020.101191
Ricciarelli, R., and Fedele, E. (2017). The amyloid cascade hypothesis in Alzheimer’s Disease: It’s time to change our mind. Curr. Neuropharmacol. 15, 926–935. doi: 10.2174/1570159X15666170116143743
Richard, R., and Mousa, S. (2022). Necroptosis in Alzheimer’s disease: Potential therapeutic target. Biomed. Pharmacother. 152:113203. doi: 10.1016/j.biopha.2022.113203
Saaoud, F., Liu, L., Xu, K., Cueto, R., Shao, Y., Lu, Y., et al. (2023). Aorta- and liver-generated TMAO enhances trained immunity for increased inflammation via ER stress/mitochondrial ROS/glycolysis pathways. JCI Insight 8:e158183. doi: 10.1172/jci.insight.158183
Saaoud, F., Liu, L., Xu, K., Lu, Y., Shao, Y., Ben Issa, M., et al. (2025). Alzheimer’s disease as an auto-innate immune pathology with potential cell trans-differentiation and enhanced trained immunity in 3xTg-AD mouse model. J. Alzheimers Dis. 105, 550–572. doi: 10.1177/13872877251329583
Saaoud, F., Lu, Y., Xu, K., Shao, Y., Praticò, D., Vazquez-Padron, R., et al. (2024). Protein-rich foods, sea foods, and gut microbiota amplify immune responses in chronic diseases and cancers - Targeting PERK as a novel therapeutic strategy for chronic inflammatory diseases, neurodegenerative disorders, and cancer. Pharmacol. Ther. 255:108604. doi: 10.1016/j.pharmthera.2024.108604
Sato, Y., Kawashima, K., Fukui, E., Matsumoto, H., Yoshizawa, F., and Sato, Y. (2022). Functional analysis reveals that Tinagl1 is required for normal muscle development in mice through the activation of ERK signaling. Biochim. Biophys. Acta Mol. Cell. Res. 1869:119294. doi: 10.1016/j.bbamcr.2022.119294
Sedzikowska, A., and Szablewski, L. (2021). Insulin and insulin resistance in Alzheimer’s disease. Int. J. Mol. Sci. 22:9987. doi: 10.3390/ijms22189987
Selkoe, D. (2001). Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 81, 741–766. doi: 10.1152/physrev.2001.81.2.741
Song, N., Scholtemeijer, M., and Shah, K. (2020). Mesenchymal stem cell immunomodulation: Mechanisms and therapeutic potential. Trends Pharmacol. Sci. 41, 653–664. doi: 10.1016/j.tips.2020.06.009
Su, H., Liu, Y., and Huang, J. (2023). Ferroptosis-related gene SLC1A5 is a novel prognostic biomarker and correlates with immune microenvironment in HBV-related HCC. J. Clin. Med. 12:1715. doi: 10.3390/jcm12051715
Sun, Y., Johnson, C., Zhou, J., Wang, L., Li, Y., Lu, Y., et al. (2018). Uremic toxins are conditional danger- or homeostasis-associated molecular patterns. Front. Biosci. 23:348–387. doi: 10.2741/4595
Sun, Y., Lu, Y., Liu, L., Saaoud, F., Shao, Y., Xu, K., et al. (2024). Caspase-4/11 promotes hyperlipidemia and chronic kidney disease-accelerated vascular inflammation by enhancing trained immunity. JCI Insight 9:e177229. doi: 10.1172/jci.insight.177229
Sun, Z., Zhao, H., Fang, D., Davis, C., Shi, D., Lei, K., et al. (2022). Neuroinflammatory disease disrupts the blood-CNS barrier via crosstalk between proinflammatory and endothelial-to-mesenchymal-transition signaling. Neuron 110, 3106–3120.e7. doi: 10.1016/j.neuron.2022.07.015
Tai, Y., Ou, C., Chiang, Y., Chang, C., Chen, C., and Cheng, W. (2022). Overexpression of transmembrane protein 102 implicates poor prognosis and chemoresistance in epithelial ovarian carcinoma patients. Am. J. Cancer Res. 12, 4211–4226.
Tarawneh, R. (2023). Microvascular contributions to alzheimer disease pathogenesis: Is Alzheimer disease primarily an endotheliopathy? Biomolecules 13:830. doi: 10.3390/biom13050830
Tatomir, A., Cuevas, J., Badea, T., Muresanu, D., Rus, V., and Rus, H. (2022). Role of RGC-32 in multiple sclerosis and neuroinflammation - few answers and many questions. Front. Immunol. 13:979414. doi: 10.3389/fimmu.2022.979414
Thal, D., Gawor, K., and Moonen, S. (2024). Regulated cell death and its role in Alzheimer’s disease and amyotrophic lateral sclerosis. Acta Neuropathol. 147:69. doi: 10.1007/s00401-024-02722-0
Trejo-Lopez, J., Yachnis, A., and Prokop, S. (2022). Neuropathology of Alzheimer’s disease. Neurotherapeutics 19, 173–185. doi: 10.1007/s13311-021-01146-y
Tretina, K., Park, E., Maminska, A., and MacMicking, J. (2019). Interferon-induced guanylate-binding proteins: Guardians of host defense in health and disease. J. Exp. Med. 216, 482–500. doi: 10.1084/jem.20182031
Vestweber, D. (2021). Vascular endothelial protein tyrosine phosphatase regulates endothelial function. Physiology 36, 84–93. doi: 10.1152/physiol.00026.2020
von Mässenhausen, A., Zamora Gonzalez, N., Maremonti, F., Belavgeni, A., Tonnus, W., Meyer, C., et al. (2022). Dexamethasone sensitizes to ferroptosis by glucocorticoid receptor-induced dipeptidase-1 expression and glutathione depletion. Sci. Adv. 8:eabl8920. doi: 10.1126/sciadv.abl8920
Wandel, M., Kim, B., Park, E., Boyle, K., Nayak, K., Lagrange, B., et al. (2020). Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat. Immunol. 21, 880–891. doi: 10.1038/s41590-020-0697-2
Wang, J., Lai, B., Nanayakkara, G., Yang, Q., Sun, Y., Lu, Y., et al. (2019). Experimental data-mining analyses reveal new roles of low-intensity ultrasound in differentiating cell death regulatome in cancer and non-cancer cells via potential modulation of chromatin long-range interactions. Front. Oncol. 9:600. doi: 10.3389/fonc.2019.00600
Wang, Z., Yemanyi, F., Blomfield, A., Bora, K., Huang, S., Liu, C., et al. (2022). Amino acid transporter SLC38A5 regulates developmental and pathological retinal angiogenesis. Elife 11:e73105. doi: 10.7554/eLife.73105
Wight, T., Kang, I., Evanko, S., Harten, I., Chang, M., Pearce, O., et al. (2020). Versican-A critical extracellular matrix regulator of immunity and inflammation. Front. Immunol. 11:512. doi: 10.3389/fimmu.2020.00512
Wojahn, I., Lüdtke, T., Christoffels, V., Trowe, M., and Kispert, A. (2019). TBX2-positive cells represent a multi-potent mesenchymal progenitor pool in the developing lung. Respir. Res. 20:292. doi: 10.1186/s12931-019-1264-y
Wójcik, P., Jastrzêbski, M., Ziêba, A., Matosiuk, D., and Kaczor, A. (2024). Caspases in Alzheimer’s disease: Mechanism of activation, role, and potential treatment. Mol. Neurobiol. 61, 4834–4853. doi: 10.1007/s12035-023-03847-1
Xiong, J., Kawagishi, H., Yan, Y., Liu, J., Wells, Q., Edmunds, L., et al. (2018). A metabolic basis for endothelial-to-mesenchymal transition. Mol. Cell. 69, 689–698.e7. doi: 10.1016/j.molcel.2018.01.010
Xu, K., Saaoud, F., Shao, Y., Lu, Y., Wu, S., Zhao, H., et al. (2023). Early hyperlipidemia triggers metabolomic reprogramming with increased SAH, increased acetyl-CoA-cholesterol synthesis, and decreased glycolysis. Redox Biol. 64:102771. doi: 10.1016/j.redox.2023.102771
Xu, K., Saaoud, F., Shao, Y., Lu, Y., Yang, Q., Jiang, X., et al. (2024). A new paradigm in intracellular immunology: Mitochondria emerging as leading immune organelles. Redox Biol. 76:103331. doi: 10.1016/j.redox.2024.103331
Xu, K., Shao, Y., Saaoud, F., Gillespie, A., Drummer, C., Liu, L., et al. (2021). Novel knowledge-based transcriptomic profiling of lipid lysophosphatidylinositol-induced endothelial cell activation. Front. Cardiovasc. Med. 8:773473. doi: 10.3389/fcvm.2021.773473
Yan, X., Hu, Y., Wang, B., Wang, S., and Zhang, X. (2020). Metabolic dysregulation contributes to the progression of Alzheimer’s disease. Front. Neurosci. 14:530219. doi: 10.3389/fnins.2020.530219
Yang, A., Vest, R., Kern, F., Lee, D., Agam, M., Maat, C., et al. (2022). A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892. doi: 10.1038/s41586-021-04369-3
Yang, Q., Saaoud, F., Lu, Y., Pu, Y., Xu, K., Shao, Y., et al. (2023). Innate immunity of vascular smooth muscle cells contributes to two-wave inflammation in atherosclerosis, twin-peak inflammation in aortic aneurysms and trans-differentiation potential into 25 cell types. Front. Immunol. 14:1348238. doi: 10.3389/fimmu.2023.1348238
Yang, W., Ben Issa, M., Saaoud, F., Xu, K., Shao, Y., Lu, Y., et al. (2024). Perspective: Pathological transdifferentiation-a novel therapeutic target for cardiovascular diseases and chronic inflammation. Front. Cardiovasc. Med. 11:1500775. doi: 10.3389/fcvm.2024.1500775
Yasuma, T., and Gabazza, E. (2024). Cell death in acute organ injury and fibrosis. Int. J. Mol. Sci. 25:3930. doi: 10.3390/ijms25073930
Yin, F. (2023). Lipid metabolism and Alzheimer’s disease: Clinical evidence, mechanistic link and therapeutic promise. FEBS J. 290, 1420–1453. doi: 10.1111/febs.16344
Yin, F., Sancheti, H., Patil, I., and Cadenas, E. (2016). Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 100, 108–122. doi: 10.1016/j.freeradbiomed.2016.04.200
Yoshimatsu, Y., and Watabe, T. (2022). Emerging roles of inflammation-mediated endothelial-mesenchymal transition in health and disease. Inflamm. Regen. 42:9. doi: 10.1186/s41232-021-00186-3
Zeng, H., Nanayakkara, G., Shao, Y., Fu, H., Sun, Y., Cueto, R., et al. (2018). DNA checkpoint and repair factors are nuclear sensors for intracellular organelle stresses-inflammations and cancers can have high genomic risks. Front. Physiol. 9:516. doi: 10.3389/fphys.2018.00516
Zhang, J., Ma, S., Liu, X., Du, Y., Zhu, X., Liu, Y., et al. (2022). Activating transcription factor 6 regulates cystathionine to increase autophagy and restore memory in Alzheimer’ s disease model mice. Biochem. Biophys. Res. Commun. 615, 109–115. doi: 10.1016/j.bbrc.2022.05.053
Zhao, D., Yang, K., Guo, H., Zeng, J., Wang, S., Xu, H., et al. (2023). Mechanisms of ferroptosis in Alzheimer’s disease and therapeutic effects of natural plant products: A review. Biomed. Pharmacother. 164:114312. doi: 10.1016/j.biopha.2023.114312
Zhao, J., Liu, X., Xia, W., Zhang, Y., and Wang, C. (2020). Targeting amyloidogenic processing of APP in Alzheimer’s disease. Front. Mol. Neurosci. 13:137. doi: 10.3389/fnmol.2020.00137
Zhong, C., Yang, X., Feng, Y., and Yu, J. (2020). Trained immunity: An underlying driver of inflammatory atherosclerosis. Front. Immunol. 11:284. doi: 10.3389/fimmu.2020.00284
Keywords: Alzheimer’s disease, endothelial-to-mesenchymal transition (EndoMT), fibrosis, endoplasmic reticulum (ER) stress, cellular stress, metabolic reprogramming
Citation: Saaoud F, Ben Issa M, Liu L, Xu K, Lu Y, Shao Y, Han B, Jiang X, Liu X, Gillespie A, Luo JJ, Martinez L, Vazquez-Padron R, Mohsin S, Kosmider B, Wang H, Fossati S and Yang X (2025) Organelle stresses and energetic metabolisms promote endothelial–to–mesenchymal transition and fibrosis via upregulating FOSB and MEOX1 in Alzheimer’s disease. Front. Mol. Neurosci. 18:1605012. doi: 10.3389/fnmol.2025.1605012
Received: 03 April 2025; Accepted: 31 July 2025;
Published: 22 August 2025.
Edited by:
Sudhanshu P. Raikwar, Barrow Neurological Institute (BNI), United StatesReviewed by:
Erik J. Behringer, Loma Linda University, United StatesLe Wang, University of California, San Diego, United States
Amanda Alves Marcelino Da Silva, Universidade de Pernambuco, Brazil
Copyright © 2025 Saaoud, Ben Issa, Liu, Xu, Lu, Shao, Han, Jiang, Liu, Gillespie, Luo, Martinez, Vazquez-Padron, Mohsin, Kosmider, Wang, Fossati and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Yang, eGZ5YW5nQHRlbXBsZS5lZHU=
†These authors share first authorship