 Erika S. Dahl
Erika S. Dahl Katherine M. Aird
Katherine M. Aird- Department of Cellular and Molecular Physiology, Penn State College of Medicine, Hershey, PA, United States
The ataxia-telangiectasia mutated (ATM) protein kinase has been extensively studied for its role in the DNA damage response and its association with the disease ataxia telangiectasia. There is increasing evidence that ATM also plays an important role in other cellular processes, including carbon metabolism. Carbon metabolism is highly dysregulated in cancer due to the increased need for cellular biomass. A number of recent studies report a non-canonical role for ATM in the regulation of carbon metabolism. This review highlights what is currently known about ATM’s regulation of carbon metabolism, the implication of these pathways in cancer, and the development of ATM inhibitors as therapeutic strategies for cancer.
Introduction
Ataxia-Telangiectasia Mutated (ATM)
Ataxia-telangiectasia mutated is a serine/threonine kinase that is recruited to sites of DNA double-strand breaks and signals to various downstream targets to initiate cell cycle arrest and DNA repair (1). Although mainly nuclear, ATM is also found in the cytoplasm and mitochondria (2, 3). In the phosphatidylinositol kinase-related family, ATM consists of many conserved domains and is a tumor suppressor (4). Its kinase domain is flanked by a FAT (FRAT, ATM, and TRRAP) and FATC (C-terminus) domain (5, 6). The function of the FAT domain has yet to be elucidated; however, the FATC domain is essential for kinase activity (7, 8). In addition, ATM has a leucine zipper domain, which is important for its kinase function but not required for dimerization (9). Finally, the N-terminus of ATM encompasses HEAT (huntingtin, elongation factor 3, A subunit of protein phosphatase 2A, and TOR1) repeats, which form helices that interact with various macromolecules and play a role in ATM’s kinase function (10, 11).
The activity of ATM in response to DNA damage has been extensively studied as ATM is known as the central regulator of the DNA damage response (DDR). During induction of DNA double-strand breaks, the MRN complex, containing Mre11, Rad50, and Nbs1, binds to the damage site (1). ATM is then activated and autophosphorylates its inactive dimer at serine 1981 (12). Monomeric, active ATM is then recruited to the damage site, where it phosphorylates downstream targets including SMC1, Nbs1, Chk2, BRCA1, and histone H2AX (13, 14). In addition, ATM phosphorylates p53 at serine 15 (15, 16). Activation and repression of ATM’s downstream targets ultimately leads to senescence, genome repair, or apoptosis (17).
ATM is the primary gene mutation in ataxia telangiectasia (A-T) (18, 19). A-T is primarily documented as an immunodeficiency and neuronal degeneration disorder affecting 1:40,000–1:100,000 people worldwide (18, 20). Inherited in an autosomal recessive manner, patients typically produce symptoms of delayed development due to neurodegeneration, deficient immune response, and predisposition to cancer. Approximately 10–15% of ATM null A-T patients develop childhood leukemia and lymphoma, specifically T-cell prolymphocytic leukemia (21, 22). In addition, patients are predisposed to breast cancer, pancreatic cancer, and melanoma (23). Renwick et al. conducted an unbiased screen in familial breast cancer patients and identified a number of premature truncations and missense variants in ATM that predispose patients to cancer (24). Furthermore, immunohistochemical staining of ATM and p53 in pancreatic tumor samples reveal that tumoral loss of ATM with wild-type p53 correlates with a decrease in patient survival, especially in families with a history of pancreatic cancer (25). Finally, somatic ATM mutations are implicated in increased melanoma risk (26). Moreover, ATM repairs mitochondrial genome defects, and loss of ATM leads to mitochondrial dysregulation (27). A-T patients have alterations in metabolism, including fluctuations in glucose metabolism (28). In addition, low NAD+ and SIRT1 levels are observed in rat models of A-T (29). These observations lead to the investigation of the role of ATM in metabolism.
Carbon Metabolism in Cancer
Carbon metabolism is defined as the breakdown of carbon sources, such as glucose and amino acids, to be utilized for cellular energy. Alteration in carbon metabolism is a hallmark of cancer (30). Highly proliferative cancer cells predominantly proceed through aerobic glycolysis rather than the TCA cycle, termed the Warburg effect, requiring high intake of glucose and glutamine (31). This allows cancer cells to compete in a nutrient depleted environment to reduce reactive oxygen species (ROS), generate ATP, and produce dNTPs for proliferation (32, 33). This emphasizes the importance in studying carbon metabolism in cancer and using this knowledge to discover novel, metabolic-based therapeutics.
Metabolic Roles of ATM
ATM and ROS
Apart from its role in the DDR, ATM has more recently been implicated in sensing ROS. The role of ATM in ROS sensing has been extensively reviewed (34, 35). Here, we will focus on the coupling of ATM-mediated ROS sensing in cellular metabolism.
In 2011, Cosentino et al. published a pivotal paper linking ROS and the pentose phosphate pathway (PPP) (36). The PPP acts as the de novo pathway for deoxyribonucleotide (dNTP) synthesis, important for proliferation and DDR of cancer cells. ATM activates glucose-6-phosphate dehydrogenase (G6PD) through phosphorylation of heat shock protein 27 (Hsp27), which promotes shunting of glycolytic intermediates into the PPP to increase nucleotide synthesis. Furthermore, stimulation of the PPP increases NADPH production, which acts as a cofactor for antioxidants. Together, these data suggest the important role of ATM in the production of dNTPs and NADPH in the proliferation of cancer cells and protection against ROS.
Loss of ATM increases mitochondrial dysregulation, mitochondrial number, and ROS (3). A fraction of ATM localizes to the mitochondria, suggesting that A-T should be further classified as a mitochondrial disorder. Interestingly, this study suggested that the tumor predisposition of A-T patients may be in part due to the mitochondrial dysfunction observed.
Overall, ATM plays a key role in ROS prevention and sensing. The ability of cancer cells to sense ROS through ATM and reprogram metabolism by increasing PPP activity allows for cancer cell survival and resistance to therapy. Cells lacking wild-type ATM are prone to ROS accumulation and oxidative stress. However, the full mechanistic pathway for ATM activation after ROS accumulation is currently unclear.
ATM and Insulin Signaling
Although beyond the scope of this review, it is important to recognize the evident role of ATM in insulin signaling. The purpose of insulin is to reduce the amount of glucose circulating in the blood and promote cellular uptake of glucose (37). Insulin binds to its respective receptor and recruits GLUT4, a central regulator in glucose homeostasis, to the membrane. GLUT4 transports glucose into the cell where it is used for various processes including glycolysis. A-T patients have an increased risk of developing insulin resistance and type 2 diabetes. Early studies found that A-T patient monocytes have a decreased binding affinity for insulin when compared to unaffected controls (38). Furthermore, ATM signaling through p53 is vital to glucose homeostasis and insulin resistance. Together, these data suggest that ATM regulates glucose homeostasis in part through insulin signaling. Additional information on ATM and insulin signaling can be obtained in several excellent reviews (39–42).
ATM and Glycolysis
Glycolysis is the main carbon metabolism pathway occurring in the cytosol in which glucose is catabolized into pyruvate through a series of biochemical reactions. Importantly, glycolysis does not require oxygen to proceed and produces a net gain of two ATP molecules and two NADH molecules. Subsequently, in the presence of oxygen, pyruvate enters the mitochondria in the form of acetyl CoA and proceeds through the TCA cycle and oxidative phosphorylation. Conversely, pyruvate is converted to lactic acid in the absence of oxygen or in highly proliferative cancer cells as described above as the Warburg effect (31). ATM phosphorylates and activates the tumor suppressor p53 to regulate cell cycle arrest, apoptosis, senescence, and metabolism (43). p53 suppresses glycolysis through a number of pathways. Interestingly, p53 transcriptionally regulates metabolic genes, including glucose transporters SLC2A and SLC2A4 (encoding for GLUT1 and GLUT4, respectively) (44). p53 also inhibits kinase IKK and targets NFκB, effectively suppressing glycolysis (45). In addition, p53 targets TIGAR, which reduces glycolysis by acting as a fructose-2,6-bisphosphotase (46). It is tempting to speculate that ATM activates p53 to modulate glycolysis through these pathways. Indeed, various DDR proteins are connected to mitochondrial signaling, as discussed in a recent excellent review (47).
ATM and the PPP
Metabolism is altered in cancer mainly due to the need for nutrients and essential macromolecules in a competing and proliferative environment (32). The PPP is a key pathway in the breakdown of glucose and diverges from glycolysis at glucose-6-phosphate (G6P) (48). Indeed, the increase in proliferation of cancer cells requires the biosynthesis of dNTPs in order to faithfully replicate the genome and repair DNA damage (49, 50). The PPP is essential for de novo dNTP synthesis. The PPP produces ribose-5-phosphate, the sugar backbone precursor for purine and pyrimidine synthesis (51). The PPP is divided into the oxidative and non-oxidative pathways. The first irreversible step of the PPP converts NAD+ to NADPH during the conversion of G6P to 6-phosphate-gluconolactone (6PG). The production of NADPH acts as an antioxidant cofactor, protecting the cell from ROS and oxidative stress (52). Together these data suggest an important role of the PPP in the proliferation and reduction of ROS for cancer cell survival.
In response to DNA double-strand breaks, ATM activates Hsp27 and G6PD (36). This interaction increases the flux of G6P to enter the PPP, which increases dNTPs and NADPH to aid DNA repair and reduce ROS, respectively. Conversely, other groups found that ATM negatively regulates the PPP through p53 (52, 53). It is interesting to speculate that there is a balance between positive and negative regulation of the PPP downstream of ATM. It is possible that the amount of DNA damage differentially modulates PPP activity. Under low amounts of DNA damage, Hsp27 is activated to increase dNTP synthesis for DNA repair; however, significant DNA damage accumulation may hyperactivate p53 to inhibit the PPP to fully shut down biosynthetic pathways. Nevertheless, these data support the notion that ATM regulates the PPP to affect dNTP synthesis and NADPH production in cancer cells.
ATM and Cancer
Tumor Suppressive Role of ATM in Senescence
Cellular senescence is defined as a stable cell cycle arrest (54) and is, therefore, a potent inhibitor of transformation (55). Senescence also plays a role in aging and is increased in age-related pathologies (56, 57). Senescence occurs due to multiple cellular insults, including telomere shortening, oncogene activation, termed oncogene-induced senescence (OIS), oxidative stress, and DNA damage (54). Senescence is characterized in part by alterations in metabolism (58). Senescence is now considered a reversible process (49, 53, 59–62). Therefore, dissecting how cells escape senescence is critical for understanding the earliest events in tumorigenesis.
One of the underlying mechanisms of OIS is increased replication stress, leading to DNA damage accumulation and cell cycle arrest (63, 64). Replication stress is due to a decrease in dNTP production via suppression of ribonucleotide reductase subunit 2 (RRM2), the rate-limiting enzyme in de novo dNTP synthesis (49). Replication stress due to decreased dNTPs activates ATM, correlating with senescence induction (53). Loss of ATM rescues senescence through restoration of dNTP levels. This is mediated by a p53-dependent modulation of PPP activity and increased c-myc stability to increase glucose and glutamine consumption. Consistently, a recent study found that pharmacological inhibition of ATM suppresses senescence (65). In this study, pharmacological ATM inhibition also modulated glucose consumption. Together, these data suggest that ATM functions in metabolic regulation and reprogramming in senescent cells.
Oxidative stress induced by ROS can also cause premature senescence in part through DNA damage accumulation. As discussed above, ATM senses and is activated by DNA damage (66). ATM signals through the AKT/p53/p21 pathway to induce senescence in human umbilical vein endothelial cells after oxidative stress (67). In addition, ATM activation is necessary for senescence due to nitric oxide (68). Finally, recent evidence suggests that loss of ATM in A-T mice increases NADPH oxidase 4 (NOX4) expression, leading to increased ROS and senescence (69). Together, these data demonstrate the importance of ATM signaling to induce senescence and suggest that ATM’s role in modulating senescence status offers the possibility of a future therapeutic target in the fields of both aging and cancer.
ATM Suppresses c-myc
Many cancers upregulate oncogenes that modulate metabolism, including the well-known transcription factor c-myc (70, 71). Specifically, c-myc transcriptionally regulates various enzymes related to metabolic pathways (70, 71). In relation to cancer, c-myc increases the Warburg Effect through upregulation of lactate dehydrogenase, glucose transporters, and pyruvate dehydrogenase kinase. The regulation of c-myc by ATM has just begun to be elucidated. Loss of ATM increases c-myc protein stability, which in turn increases glucose and glutamine consumption (53). Consistently, ATM partially suppresses c-myc-induced lymphomagenesis in mouse models (72, 73). It is interesting to speculate whether this is due to suppression of pro-tumorigenic metabolism. Loss of ATM and c-myc amplification/overexpression are often mutually exclusive in multiple cancer types, suggesting a redundancy in the pathway. Altogether, this suggests an interplay between ATM and c-myc in cancer metabolism.
ATM Activates AKT
AKT is a well-known serine/threonine kinase that is activated by phosphatidylinositol-3-kinase (PI3K) and regulates many cellular processes related to cancer, including survival, cellular metabolism, and DNA repair (74, 75). ATM activates AKT in response to DNA damage (76–78). Activated AKT then promotes DNA repair (79) and inhibition of AKT decreases DNA repair (80, 81). Consistently, pharmacological inhibition of ATM inhibits AKT phosphorylation and survival in multiple cancer types (82–84). These findings suggest a vital role for AKT in the maintenance of genome integrity, and inhibition of this DNA repair function may result in accumulation of DNA damage and cell death.
AKT also modulates cancer metabolism (85–89). Active AKT increases glucose uptake by recruiting GLUT4 to the plasma membrane (90). In addition, pharmacological inhibition of AKT in primary effusion lymphoma decreases the rate of aerobic glycolysis (91). This suggests that ATM-mediated regulation of AKT activity in cancer reprograms metabolism by increasing glucose uptake and potentially shifting metabolism from aerobic glycolysis to oxidative phosphorylation. It is particularly interesting that ATM-mediated AKT activation may be a double-edged sword, both increasing DNA repair to promote genomic integrity while at the same time increasing pro-tumorigenic metabolism. These data suggest that ATM inhibitors may both alleviate the metabolic changes induced by activated AKT and lead to DNA damage-induced death of cancer cells.
ATM Regulates p53
p53 is defined as the “guardian of the genome” as it serves to regulate genome stability as a tumor suppressor (92). TP53 is one of the most mutated genes among all cancers. p53 is a transcription factor that can be activated by ATM (10). Activation of p53 by ATM was originally shown to be important for the regulation of genes essential in apoptosis and DNA repair (93). Further investigation into the interplay between ATM and p53 has revealed its importance in cancer metabolism. p53 regulates many pathways in cellular metabolism, including GLUT recruitment, glycolysis, and oxidative phosphorylation (94). Mutations in p53 lead to metabolic reprogramming in a cancer cells, allowing increased glucose intake through GLUT recruitment to the cell membrane, increased aerobic glycolysis, and decreased oxidative phosphorylation (94, 95). In addition, ATM directly impacts p53-mediated PPP metabolism as discussed above (53). Moreover, ATM loss and p53 mutation are often mutually exclusive in cancer, suggesting that these proteins act in the same pathway to promote cancer cell survival.
ATM Inhibitors for Cancer Therapy
A variety of ATM inhibitors are currently in pre-clinical and clinical trials for multiple cancer types. ATM inhibitors sensitize various cancer cell lines and tumors in vitro and in vivo to radiation treatment (83, 96–98). In addition, a phase I clinical trial is currently ongoing with an ATM inhibitor in combination with a PARP inhibitor in advanced cancer patients who are resistant to the standard-of-care (99). Together, these studies have found that cancer cells may be sensitized to DNA damage through inhibition of ATM.
As discussed throughout this review, ATM modulates metabolism through various pathways, proteins, and enzymes (Figure 1). Thus, ATM inhibitors may offer a promising way to reprogram the metabolism of cancer cells to make them more vulnerable to anti-metabolic strategies. It will be important to dissect the role of metabolism in pre-clinical and clinical trials using ATM inhibitors.
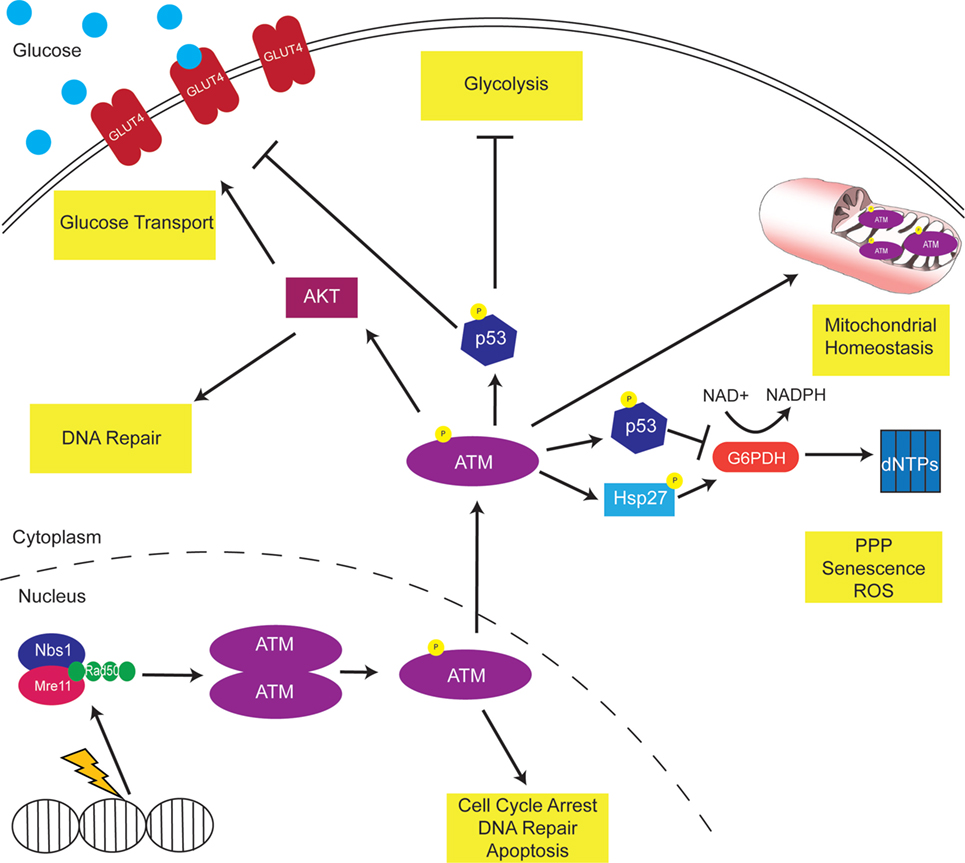
Figure 1. Ataxia-telangiectasia mutated (ATM) modulates cellular metabolism. DNA damage activates ATM to phosphorylate multiple downstream proteins regulate cell cycle arrest, DNA repair, and apoptosis pathways. A non-canonical function of ATM is the regulation of cellular metabolism. Mitochondrial ATM acts to regulate mitochondrial homeostasis by repairing mitochondrial genome defects. ATM activates the tumor suppressor p53, which inhibits GLUT recruitment, glycolysis, and dNTP production. Consistently, p53 targets the oncogene c-myc, inhibiting the TCA cycle and increasing the Warburg effect. In addition, ATM activates AKT to increase GLUT recruitment to the membrane.
Conclusion
Proliferation of cancer cells requires a metabolic shift allowing for an increase in cellular biomass in a highly competitive and nutrient-deprived environment. Although extensively studied for its role in the DDR, non-canonical roles of ATM in metabolic reprogramming have recently been elucidated. ATM modulates carbon metabolism through many pathways that are essential for cancer development, survival, and therapeutic response. Due to their radio- and chemo-sensitizing effects, ATM inhibitors are in pre-clinical and clinical trials as anti-cancer therapeutics. We suggest that ATM inhibitors may also be used to identify metabolic vulnerabilities that could be therapeutically exploited.
Author Contributions
ED and KA jointly came up with the topic for this mini-review. Both ED and KA wrote and edited the text.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AT and handling editor declared their shared affiliation.
Acknowledgments
We will like to thank the members of the Aird lab for their thoughtful comments. In addition, we would like to thank Benjamin G. Bitler for generously providing his mitochondria graphic. ED is supported by the Biomedical Sciences Graduate Program at Pennsylvania State College of Medicine. KA is supported by an NIH/NCI grant (R00CA194309).
References
1. Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J (2003) 22(20):5612–21. doi:10.1093/emboj/cdg541
2. Lim D-S, Kirsch DG, Canman CE, Ahn J-H, Ziv Y, Newman LS, et al. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc Natl Acad Sci U S A (1998) 95:10146–51. doi:10.1073/pnas.95.17.10146
3. Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, et al. Mitochondrial dysfunction in ataxia-telangiectasia. Blood (2012) 119(6):1490–501. doi:10.1182/blood-2011-08-373639.The
4. Overstreet JM, Samarakoon R, Cardona-Grau D, Goldschmeding R, Higgins PJ. Tumor suppressor ataxia telangiectasia mutated functions downstream of TGF-β in orchestrating profibrotic responses. FASEB J (2017) 29(4):1258–68. doi:10.1096/fj.14-262527
5. Khalil HS, Tummala H, Zhelev N. ATM in focus: a damage sensor and cancer target. Biodiscovery (2012) 5:1–60. doi:10.7750/BioDiscovery.2012.5.1
6. Bosotti R, Isacchi A. FAT: a novel domain in PIK-related kinases. Trends Biochem Sci (2000) 3:225–7. doi:10.1016/S0968-0004(00)01563-2
7. Jiang X, Sun Y, Chen S, Roy K, Price BD. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J Biol Chem (2006) 281(23):15741–6. doi:10.1074/jbc.M513172200
8. Vermezovic J, Adamowicz M, Santarpia L, Rustighi A, Forcato M, Lucano C, et al. Notch is a direct negative regulator of the DNA-damage response. Nature (2015) 22(5):417–24. doi:10.1038/nsmb.3013
9. Chen S, Paul P, Price BD. ATM’s leucine-rich domain and adjacent sequences are essential for ATM to regulate the DNA damage response. Oncogene (2003) 22:6332–9. doi:10.1038/sj.onc.1206760
10. Turenne GA, Paul P, La L, Price BD. Activation of p53 transcriptional activity requires ATM’S kinase domain and multiple N-terminal serine residues of p53. Oncogene (2001) 2:5100–10. doi:10.1038/sj.onc.1204665
11. Perry J, Kleckner N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell (2003) 112:151–5. doi:10.1016/S0092-8674(03)00033-3
12. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature (2003) 421:499–506. doi:10.1038/nature01368
13. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature (2005) 434:605–11. doi:10.1038/nature03442
14. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev (2000) 14:927–39. doi:10.1101/gad.14.8.927
15. Banin S, Moyal L, Shieh S-Y, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science (1998) 281:1674–8. doi:10.1126/science.281.5383.1674
16. Canman CE, Lim D-S, Cimprich KA, Taya Y, Tamai K, Sakaguchi K. Activation of the ATM Kinase by ionizing radiation and phosphorylation of p53. Science (1998) 281:1996–9. doi:10.1126/science.281.5383.1677
17. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer (2003) 3:155–68. doi:10.1038/nrc1011
18. Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Tagle DA, et al. A Single Ataxia Telangiectasia Gene with a Product Similar to PI-3 Kinase. American Association for the Advancement of Science (2017). Available from: http://www.jstor.org/stable/2887809
19. Humar B, Müller H, Scott RJ. Short reports cell cycle dependent dna break increase in ataxia telangiectasia lymphoblasts after radiation exposure. J Clin Pathol (2001) 54:347–50. doi:10.1136/mp.54.5.347
20. National Cancer Institute. Ataxia Telangiectasia. (2017). Available from: https://www.cancer.gov/about-cancer/causes-prevention/genetics/ataxia-fact-sheet
21. Boultwood J. Ataxia telangiectasia gene mutations in leukaemia and lymphoma. J Clin Pathol (2001) 54:512–6. doi:10.1136/jcp.54.7.512
22. Gumy-Pause F, Wacker P, Sappino A-P. ATM gene and lymphoid malignancies. Nature (2004) 18:238–42. doi:10.1038/sj.leu.2403221
23. Swift M, Reitnauer PJ, Morrell D, Chase CL. Breast and other cancer in families with ataxia-telangiectasia. N Engl J Med (1987) 316(21):1289–94. doi:10.1056/NEJM198705213162101
24. Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet (2006) 38(8):873–6. doi:10.1038/ng1837
25. Kim H, Saka B, Knight S, Borges M, Childs E, Wolfgang C, et al. Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin Cancer Res (2014) 20(7):1865–72. doi:10.1158/1078-0432.CCR-13-1239.Having
26. Bhandaru M, Martinka M, Mcelwee KJ, Rotte A. Prognostic significance of nuclear phospho-ATM expression in melanoma. PLoS One (2015) 10(8):1–15. doi:10.1371/journal.pone.0134678
27. Sharma NK, Lebedeva M, Thomas T, Kovalenko OA, Stumpf JD, Shadel S, et al. Intrinsic mitochondiral DNA repair defects in ataxia telangiectasia. DNA Repair (2014) 13:22–31. doi:10.1016/j.dnarep.2013.11.002
28. Volkow ND, Tomasi D, Wang GJ, Studentsova Y, Margus B, Crawford TO. Brain glucose metabolism in adults with ataxia-telangiectasia and their asymptomatic relatives. Brain (2014) 137:1753–61. doi:10.1093/brain/awu092
29. Fang EF, Kassahun H, Croteau DL, Mattson MP, Nilsen H, Fang EF, et al. NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab (2016) 24(4):566–81. doi:10.1016/j.cmet.2016.09.004
30. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
31. Warburg O, Posener K, Negelein E. Uber Den Stoffwechsel Der Carcinomzelle. Z Biochem (1924) 152:319.
32. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell (2017) 168(4):657–69. doi:10.1016/j.cell.2016.12.039
33. Pavlova NN, Thompson CB. Perspective the emerging hallmarks of cancer metabolism. Cell Metab (2016) 23(1):27–47. doi:10.1016/j.cmet.2015.12.006
34. Ditch S, Paull TT. The ATM protein kinase and cellular redox signalling: beyond the DNA damage response. Trends Biochem Sci (2013) 37(1):15–22. doi:10.1021/nl061786n.Core-Shell
35. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol (2013) 14:197–210. doi:10.1038/nrm3546
36. Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J (2011) 30(3):546–55. doi:10.1038/emboj.2010.330
37. Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab (2007) 5:237–52. doi:10.1016/j.cmet.2007.03.006
38. Bar RS, Levis WR, Rechler MM, Harrison LC, Siebert C, Podskalny J, et al. Extreme insulin resistance in ataxia telangiectasia. N Engl J Med (1978) 298(21):1164–71. doi:10.1056/NEJM197805252982103
39. Ambrose M, Gatti RA. Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood (2013) 121(20):4036–46. doi:10.1182/blood-2012-09-456897.ATX/SMG
40. Espach Y, Lochner A, Strijdom H, Huisamen B. ATM protein kinase signaling, type 2 diabetes and cardiovascular disease. Cardiovasc Drugs Ther (2015) 1:51–8. doi:10.1007/s10557-015-6571-z
41. Choy KR, Watters DJ. Neurodegeneration in ataxia-telangiectasia: Multiple roles of ATM kinase in cellular homeostasis. Dev Dyn (2017). doi:10.1002/DVDY.24522
42. Yang DQ, Halaby MJ, Li Y, Hibma JC, Burn P. Cytoplasmic ATM protein kinase: an emerging therapeutic target for diabetes, cancer and neuronal degeneration. Drug Discov Today (2011) 16(7–8):332–8. doi:10.1016/j.drudis.2011.02.001
43. Rozan LM, El-Deiry WS. p53 downstream target genes and tumor suppression: a classical view in evolution. Cell Death Differ (2007) 14:3–9. doi:10.1038/sj.cdd.4402058
44. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res (2004) 64:2627–33. doi:10.1158/0008-5472.CAN-03-0846
45. Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK–NF-κB pathway and inhibits cell transformation. Nat Cell Biol (2008) 10(5):611–8. doi:10.1038/ncb1724
46. Bensaad K, Tsuruta A, Selak MA, Nieves Calvo Vidal M, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell (2006) 126:107–20. doi:10.1016/j.cell.2006.05.036
47. Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol (2016) 17(5):308–21. doi:10.1038/nrm.2016.14.Nuclear
48. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell (2014) 5(8):592–602. doi:10.1007/s13238-014-0082-8
49. Aird KM, Zhang G, Li H, Tu Z, Bitler B, Garipov A, et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep (2013) 3(4):1252–65. doi:10.1016/j.celrep.2013.03.004.Suppression
50. Aye Y, Li M, Long MJC, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene (2015) 34:2011–21. doi:10.1038/onc.2014.155
51. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci (2014) 39(8):347–54. doi:10.1016/j.tibs.2014.06.005.The
52. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol (2011) 13(3):310–6. doi:10.1038/ncb2172.p53
53. Aird KM, Worth AJ, Snyder NW, Lee JV, Sivanand S, Liu Q, et al. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep (2015) 11(6):893–901. doi:10.1016/j.celrep.2015.04.014
54. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell (2005) 120:513–22. doi:10.1016/j.cell.2005.02.003
55. Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, et al. PRAK is essential for Ras-induced senescence and tumor suppression. Cell (2007) 1:295–308. doi:10.1016/j.cell.2006.11.050
56. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med (2015) 21(12):1424–35. doi:10.1038/nm.4000.Cellular
57. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer (2015) 15:397–408. doi:10.1038/nrc3960
58. Aird KM, Zhang R. Metabolic alterations accompanying oncogene-induced senescence. Mol Cell Oncol (2014) 1(3):1–8. doi:10.4161/23723548.2014.963481
59. Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J (2003) 22(16):4212–22. doi:10.1093/emboj/cdg417
60. Dirac AMG, Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral. J Biol Chem (2003) 278(14):11731–5. doi:10.1074/jbc.C300023200
61. Aird KM, Zhang R. ATM in senescence. Oncotarget (2015) 6(17):14729–30. doi:10.18632/oncotarget.4411
62. Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol (2016) 215(3):325–34. doi:10.1083/jcb.201608026
63. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature (2006) 444:638–42. doi:10.1038/nature05327
64. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature (2006) 444:633–7. doi:10.1038/nature05268
65. Kang HT, Park JT, Choi K, Kim Y, Choi HJC, Jung CW, et al. Chemical screening identifies ATM as a target for alleviating senescence. Nat Chem Biol (2017) 13(6):616–23. doi:10.1038/nchembio.2342
66. Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science (2010) 330:517–21. doi:10.1126/science.1192912
67. Zhan H, Suzuki T, Aizawa K, Miyagawa K, Nagai R. Ataxia telangiectasia mutated (ATM)-mediated DNA damage response in oxidative stress-induced vascular endothelial cell senescence. J Biol Chem (2010) 285(38):29662–70. doi:10.1074/jbc.M110.125138
68. Bagheri M, Nair RR, Kumar K, Kumar D. ATM-ROS-iNOS axis regulates nitric oxide mediated cellular senescence. In: Chishti A, van der Klei I, Schmitz L editors. BBA – Molecular Cell Research 1864. Cambridge, MA: Elsevier B.V. (2016). p. 177–90.
69. Weyemi U, Redon CE, Aziz T, Choudhuri R, Maeda D, Parekh PR, et al. NADPH oxidase 4 is a critical mediator in ataxia telangiectasia disease. Proc Natl Acad Sci U S A (2015) 112(7):2121–6. doi:10.1073/pnas.1418139112
70. Jones NP, Schulze A. Targeting cancer metabolism – aiming at a tumour’s sweet-spot. Drug Discov Today (2012) 17(5–6):232–41. doi:10.1016/j.drudis.2011.12.017
72. Maclean KH, Kastan MB, Cleveland JL. Atm deficiency affects both apoptosis and proliferation to augment Myc-induced lymphomagenesis. Mol Cancer Res (2007) 5:705–12. doi:10.1158/1541-7786.MCR-07-0058
73. Pusapati RV, Rounbehler RJ, Hong S, Powers JT, Yan M, Kiguchi K, et al. ATM promotes apoptosis and suppresses tumorigenesis in response to myc. Proc Natl Acad Sci U S A (2006) 103(5):1451–66. doi:10.1073/pnas.0507367103
74. Vivanco I, Sawyers CL. The phophatidylinositol-3-kinase-AKT pathways in human cancer. Nat Rev Cancer (2002) 2:489–501. doi:10.1038/nrc839
75. Xu N, Lao Y, Zhang Y, Gillespie DA. AKT: a double-edged sword in cell proliferation and genome stability. J Oncol (2012) 2012:1–15 doi:10.1155/2012/951724
76. Yang D-Q, Kastan MB. Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nat Cell Biol (2000) 2:893–8. doi:10.1038/35046542
77. Viniegra JG, Martinez N, Modirassari P, Losa JH, Cobo CP, Sa J, et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem (2005) 280(6):4029–36. doi:10.1074/jbc.M410344200
78. Fraser M, Harding SM, Zhao H, Coackley C, Durocher D, Bristow RG. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle (2011) 10(13):2218–32. doi:10.4161/cc.10.13.16305
79. Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBα/AKT1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell (2008) 30(2):203–13. doi:10.1016/j.molcel.2008.02.024
80. Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A. Inhibition of phosphatidylinositol-3-OH kinase/ASK signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem (2007) 282(29):21206–12. doi:10.1074/jbc.M703042200
81. Holler M, Grottke A, Mueck K, Manes J, Jücker M, Rodemann HP, et al. Dual targeting of Akt and mTORC1 impairs repair of DNA double-strand breaks and increases radiation sensitivity of human tumor cells. PLoS One (2016) 11(5):e0154745. doi:10.1371/journal.pone.0154745
82. Li Y, Yang D-Q. The ATM inhibitor KU-55933 suppresses cell proliferation and induces apoptosis by blocking Akt in cancer cells with overactivated Akt. Mol Cancer Ther (2010) 7:113–26. doi:10.1158/1535-7163.MCT-08-1189
83. Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Clin Cancer Res (2009) 8(10):2894–903. doi:10.1158/1535-7163.MCT-09-0519
84. Fujimaki S, Matsuda Y, Wakai T, Sanpei A, Kubota M, Takamura M, et al. Blockade of ataxia telangiectasia mutated sensitizes hepatoma cell lines to sorafenib by interfering with Akt signaling. Cancer Lett (2012) 319:98–108. doi:10.1016/j.canlet.2011.12.043
85. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res (2004) 473:3892–9. doi:10.1158/0008-5472.CAN-03-2904
86. Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem (1996) 271(49):31372–8. doi:10.1074/jbc.271.49.31372
87. Robey RB, Hay N. Is Akt the ‘Warburg kinase’?—Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol (2010) 19(1):1–15. doi:10.1016/j.semcancer.2008.11.010.Is
88. Gonzalez E, Mcgraw TE. The Akt kinases: isoform specificity in metabolism and cancer the Akt kinases isoform specificity in metabolism and cancer. Cell Cycle (2017) 8(16):2502–8. doi:10.4161/cc.8.16.9335
89. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet (2006) 7:606–19. doi:10.1038/nrg1879
90. Thong, Farah SL, Chandrasagar B, Dugani, Amira Klip. Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. Physiology (2005) 20:271–84. doi:10.1152/physiol.00017.2005
91. Mediani L, Gibellini F, Bertacchini J, Frasson C, Bosco R, Accordi B, et al. Reversal of the glycolytic phenotype of primary effusion lymphoma cells by combined targeting of cellular metabolism and PI3K/Akt/mTOR signaling. Oncotarget (2015) 7(5):5521–37. doi:10.18632/oncotarget.6315
93. Saito S, Goodarzi AA, Higashimotot Y, Noda Y, Lees-miller SP, Appella E, et al. ATM mediates phosphorylation at multiple p53 sites, including ser46, in response to ionizing radiation. J Biol Chem (2002) 277(15):12491–5. doi:10.1074/jbc.C200093200
94. Puzio-Kuter AM. The role of p53 in metabolic regulation. Genes Cancer (2011) 2(4):385–91. doi:10.1177/1947601911409738
95. Shen L, Sun X, Fu Z, Yang G, Li J, Yao L. The fundamental role of the p53 pathway in tumor metabolism and its implication in tumor therapy. Clin Cancer Res (2012) 18(6):1561–8. doi:10.1158/1078-0432.CCR-11-3040
96. Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res (2008) 68(18):7466–74. doi:10.1158/0008-5472.CAN-08-0763.Transient
97. Batey MA, Zhao Y, Kyle S, Richardson C, Slade A, Martin NMB, et al. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol Cancer Ther (2013) 12(6):959–68. doi:10.1158/1535-7163.MCT-12-0707
98. Biddlestone-Thorpe L, Sajjad M, Rosenberg E, Jason M, Valerie NCK, Tokarz M, et al. ATM kinase inhibition preferentially sensitizes p53 mutant glioma to ionizing radiation. Clin Cancer Res (2013) 19(12):3189–200. doi:10.1158/1078-0432.CCR-12-3408.ATM
99. ClinicalTrials.Gov. Study to Assess the Safety and Preliminary Efficacy of AZD0156 at Increasing Doses Alone or in Combination with Other Anti-Cancer Treatment in Patients with Advanced Cancer (AToM). (2017). Available from: https://clinicaltrials.gov/ct2/show/NCT02588105?term=azd0156&rank=1
Keywords: ataxia-telangiectasia mutated, cellular metabolism, cancer, reactive oxygen species, senescence, p53, AKT, c-myc
Citation: Dahl ES and Aird KM (2017) Ataxia-Telangiectasia Mutated Modulation of Carbon Metabolism in Cancer. Front. Oncol. 7:291. doi: 10.3389/fonc.2017.00291
Received: 11 October 2017; Accepted: 14 November 2017;
Published: 29 November 2017
Edited by:
Sergio Giannattasio, Istituto di Biomembrane, Bioenergetica e Biotecnologie Molecolari (IBIOM), ItalyReviewed by:
Ferdinando Chiaradonna, Università degli studi di Milano Bicocca, ItalyApollonia Tullo, Istituto di Biomembrane, Bioenergetica e Biotecnologie Molecolari (IBIOM), Italy
Copyright: © 2017 Dahl and Aird. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine M. Aird, kaird@psu.edu