 Christopher Chin
Christopher Chin Emma S. Lunking
Emma S. Lunking Macarena de la Fuente2,3
Macarena de la Fuente2,3 Nagi G. Ayad
Nagi G. Ayad- 1Department of Psychiatry and Behavioral Sciences, Center for Therapeutic Innovation, Sylvester Comprehensive Cancer Center, Miami Project to Cure Paralysis, University of Miami Miller School of Medicine, Miami, FL, United States
- 2Department of Neurology, University of Miami, Miami, FL, United States
- 3Sylvester Comprehensive Cancer Center, University of Miami, Miami, FL, United States
Glioblastoma Multiforme (GBM) is the most common malignant primary brain tumor. Despite aggressive multimodality treatment it remains one of the most challenging and intractable cancers (1). While current standard of care treatment for GBM is maximal safe surgical resection, systemic chemotherapy with Temozolimide (TMZ), and radiation therapy, the current prognosis of GBM patients remains poor, with a median overall survival of 12–15 months (2, 3). Therefore, other treatments are needed to provide better outcomes for GBM patients. Immunotherapy is one of the most promising new cancer treatment approaches. Immunotherapy drugs have obtained regulatory approval in a variety of cancers including melanoma (4), Hodgkin lymphoma (5), and non-small cell lung cancer (6). The basis of immunotherapy in cancer treatment is linked to stimulating the immune system to recognize cancer cells as foreign, thereby leading to the eventual elimination of the tumor. One form of immunotherapy utilizes vaccines that target tumor antigens (7), while other approaches utilize T-cells in patients to stimulate them to attack tumor cells (8). Despite intensive efforts all approaches have not been overtly successful (9), suggesting that we need to better understand the underlying biology of tumor cells and their environment as they respond to immunotherapy. Recent studies have elucidated epigenetic pathway regulation of GBM tumor expansion (10), suggesting that combined epigenetic pathway inhibition with immunotherapy may be feasible. In this review, we discuss current GBM clinical trials and how immune system interactions with epigenetic pathways and signaling nodes can be delineated to uncover potential combination therapies for this incurable disease.
Review of Clinical Trials Using Combination Immunotherapy
Multiple immunotherapy clinical trials in GBM have been initiated although few have reached completion. And of those few, the results observed were limited. For example, an upfront (newly diagnosed GBM) phase 2 trial tested the efficacy of a patient specific dendritic cell vaccine termed ICT-107, by observing its ability to significantly change the median survival rate of newly diagnosed GBM patients. In the ICT-107 study, patients in the experimental arm as well as control arm underwent surgical resection followed by radiation and TMZ treatment for six weeks. Subsequently, white blood cells (WBCs) from GBM patients were extracted from both experimental and control groups and cultured with antigens found in the GBM experimental group and not in the control group. Over the period of several months, the WBCs pulsed with GBM antigen for the experimental arm and WBCs not pulsed with GBM antigen for the control arm were reintroduced as vaccines to patients in the respective groups. The results showed that when compared to patients in the control group, whose extracted WBC were not cultured with GBM antigens, the GBM cultured WBCs increased survival in ICT-107 treated patients by only less than 2 months [Table 1, (23)].
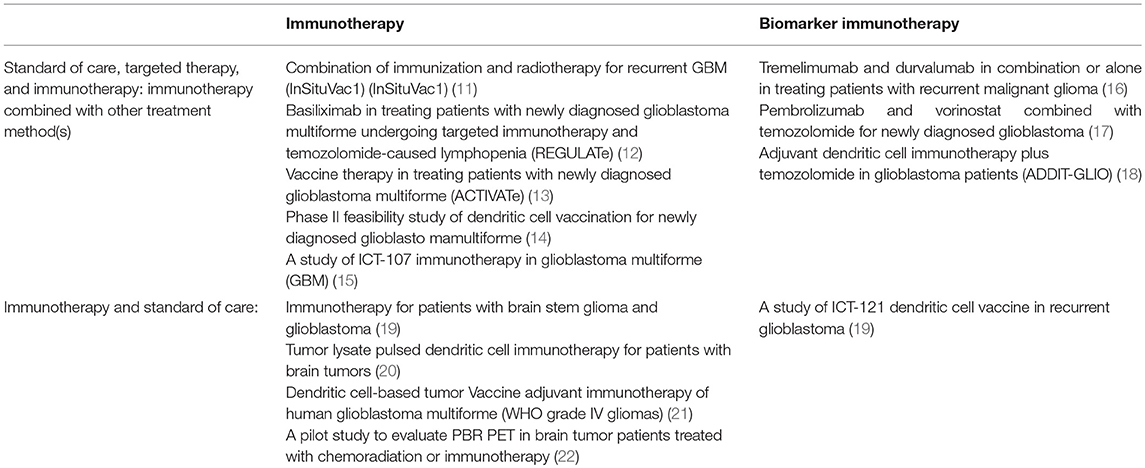
Table 1. A summary of clinical trials utilizing standard of care for GBM in addition to immunotherapy and biomarker immunotherapy in combination with and without targeted therapy.
Other combination trials using immunotherapy with chemotherapy and/or radiotherapy have been performed in the hopes of attaining statistically significant results, but most have fallen short. Another upfront phase 2 immunotherapy combination trial investigated the effect of immunotherapy with radiation and chemotherapy to determine the efficacy of combination therapy as treatment for GBM with the PEP-3-KLH vaccine for newly diagnosed EGFRvIII-expressing GBM patients. The PEP-3-KLH vaccine is a synthetic peptide derived from a mutated segment of the epidermal growth factor type vIII (EGFRvIII), which is overexpressed in some patients with malignant glioma (24). This mutated segment is then conjugated onto the adjuvant keyhole limpet hemocyanin (KLH), a respiratory protein that is similar to some GBM antigens. The researchers in this study used this vaccine to assess its synergistic ability to elicit an immune response in conjunction with radio- and chemo-therapies when compared to patients who only received radio- and chemotherapies alone. In the first of three arms of this study, the PEP-3-KLH vaccine was administered to the GBM patients after they had completed radiation treatment. In the second and third arms of this study, the PEP-3 vaccine was used after radiation in combination with the oral chemotherapy drug, TMZ (25). The patients in this study who received TMZ, radiotherapy, and vaccine were compared to a matched cohort who were only treated with radiation therapy and TMZ. When compared to the median overall survival (OS) of the matched cohort, which was 15 months (95% CI 11.4 to 19.7 months), the vaccinated patients had an increased median OS of 26 months (95% CI, 21.0 to 47.7 months) (26). While these results are promising, it should be noted that only a subset of GBM patients have the EGFRvIII antigen on their GBM tumor cells, which was present in all participants studied (26). In fact, a study discussing the prognostic significance of EGFRvIII antigen found that only 14 of 73 (19.2%) patients evaluated with primary GBM expressed the EGFRvIII antigen (27). Collectively, these findings suggest that only a select few GBM patients have the potential to benefit from this treatment.
A second upfront combination trial analyzed the efficacy of immunotherapy in conjunction with radiotherapy and chemotherapy. In this trial, patients who had recently undergone tumor resection for their newly diagnosed GBM were first treated with radiotherapy and concurrent chemotherapy, TMZ. After completion of both radiotherapy and chemotherapy, WBCs were collected from these patients and cultured along with dendritic cells with each individual patient's GBM tumor antigens (Figure 1). This autologous dendritic cell vaccine was reintroduced to each patient whose GBM antigens were used to culture the vaccine (28). This study showed that only 50% of this carefully selected patient population mounted an immune response resulting in improved survival. In addition, the study noted that there was vast heterogeneity in the immune response (29), meaning that the immune reaction to treatment is different in each individual.
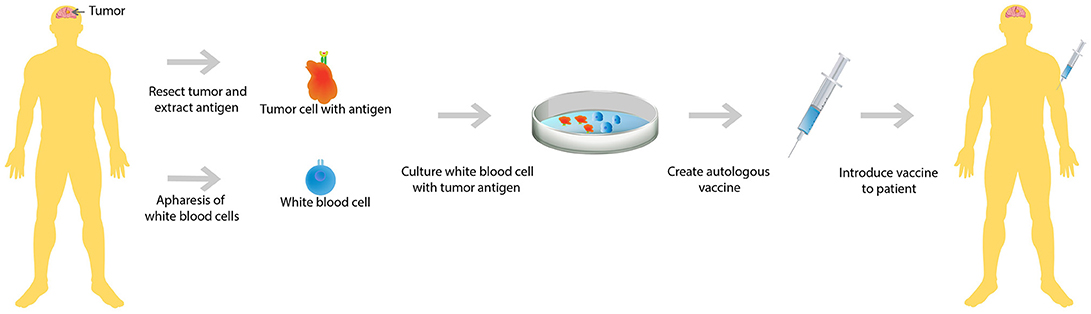
Figure 1. Autologous vaccine therapy. Both GBM tumor antigen and white blood cells are extracted from the patient. Subsequently, the extracted white blood cells are cultured with the GBM tumor antigens. A vaccine is created that is specific to each individual GBM patient's tumor antigen and then the cultured white blood cells are reintroduced to the patient.
The latter two upfront GBM trials mentioned above suggest a common theme in the ability to properly treat GBM. Patient outcome is highly variable, which could possibly be due to the heterogeneity of GBM tumors. In 2010, research utilizing data from The Cancer Genome Atlas developed a catalog of genomic abnormalities correlated with GBM tumors and helped to categorize four different GBM tumor subtypes: Classical, Proneural, Mesenchymal, and Neural (30), which has been more recently reclassified into Classical, Proneural, and Mesenchymal as the 3 main the subtypes of GBM (31). The significance of GBM subtype lies within the discovery that each subtype holds distinct genomic abnormalities, tumor microenvironments, and most importantly in terms of patient outcome, treatment response. Furthermore, GBM subtype switching has been observed upon disease recurrence, such as a Proneural to Mesenchymal transition, which been implicated in treatment resistance GBM (32). Thus, GBM subclasses have the potential to influence patient treatment, though the clinical relevance of this proposed classification remains to be determined. This is especially true given recent single cell sequencing of GBM tumors demonstrating that the even within classes of tumors vast heterogeneity exists (33–35). Future single cell sequencing studies will delineate the cell populations that remain after immunotherapy in order to better design combination therapies.
In comparison to upfront GBM trials for combination immunotherapy, recurrent GBM trials have even fewer results. These trials include single therapy dendritic cell vaccines in which patient WBCs are cultured and reintroduced (similar to the ICT-107 trial mentioned above) (19), treatment with immune adjuvants with radiation (11), and combination treatments utilizing checkpoint inhibitors (discussed below) (16). Overall, while there are some other recurrent GBM clinical trials, few have reported results.
PD-L1 Immune System Blockade and Checkpoint Inhibition
The programmed death pathway's role in GBM tissue immunity has been examined and preclinical studies suggest that inhibition of this pathway, termed “checkpoint inhibition,” has high therapeutic potential. This pathway involves down regulation of the immune response after it has run its course (36). Specifically, in a healthy individual, it stimulates the inhibition of function in T-cells, and thus prevents T-cells from being overactive in an individual whose immune response needs to be stopped after an infection (37). In the case of tumors, activation of this pathway prevents T-cells, which express the PD-1 receptor, from accessing and acting on tumors. In mice transplanted with human GBM tumors, anti-PD-1 therapy completely eradicated GBM in 44.4% of mice and combination anti-PD-1 therapy with the chemotherapy drug TMZ completely eradicated GBM tissue in all mice. Combined therapy also reduces the frequency of exhausted tumor infiltrating lymphocytes (TIL), or T-cells that have lost their functional ability to develop immune responses (38), without affecting the non-exhausted TIL load that penetrate the BBB and enter GBM tissue. In other words, after combined therapy TIL (CD8+ T-cells) do not highly express the PD-1 receptor and therefore, cannot bind the PD-L1 (ligand) on GBM tissue resulting in the high success rate of this combined therapy (39). However, while these results boast an extremely potent immunotherapy regimen in mice, they have not translated well into efficacy in GBM clinical trials.
One issue that needs to be addressed in immunotherapy trials is the immunosuppressive microenvironment of GBM (40), which can induce angiogenesis leading to tumor growth (41). The PD-L1 ligand can induce and upregulate T-reg cells (42), which are immunosuppressive cells that protect the GBM tumor from the body's immune system (43). T-reg cells are involved in the inhibition of T-cells that recognize and attack self-antigen that are present on both normal tissue and tumor tissue (44). In a normal setting, this prevents a host's immune system from developing autoimmunity, or the condition in which the immune system attacks its own cells. In the case of GBM, induction of T-reg cells prevents the destruction of the tumor tissue due to presence of self-antigen on tumor cells.
The first large scale anti-PD-L1 therapy clinical trial in GBM was conducted using a drug called Nivolumab. Unfortunately, unlike the mouse studies mentioned above, the results were not promising. Specifically, nivolumab was determined to be no more effective at increasing overall survival than the anti-VEGF cancer drug, bevacizumab (37). But this lack of success was not a failure because it highlighted a few key points about the immunosuppression of GBM. It appears anti-PD-L1 therapy may not be sufficient to alleviate immunosuppression of GBM patients. In other words, other immunosuppressive factors within the microenvironment of GBM may render T-cells anergic, or unable to produce a functional response, which would result in a failed immune response despite PD-1 pathway inhibition. In addition, this highlights the blood brain barrier (BBB) as a significant limiting factor in GBM treatment. Because nivolumab is too large to cross the BBB, this study supports the assumption that anti-PD-L1 antibody therapy exerts its effect outside of the BBB. The therapeutic effect of anti-PD-1 treatment on T-cells occurs before they cross the BBB and enter the tumor microenvironment. However, if there is an inadequate population of T-cells in the periphery and/or the T-cells have already crossed the BBB and have been rendered anergic by the microenvironment of the GBM tumor, then PD-L1 therapy may not be effective, as was exemplified in this trial (37).
Epigenetics
As discussed above, immunotherapy and combined immunotherapy are intensively studied in the treatment of GBM, although they do not seem to be wholly effective in treating GBM nor do they encompass the entire picture of GBM treatment. Recent discoveries have identified epigenetic pathway specific GBM biomarkers, which have enormous potential in terms of treatment because they can be used as targets for therapy. This epigenetic focus encompasses genes and gene regulators that are not dependent on the DNA sequence yet can be inherited and modified by endogenous enzymes. The influence of these epigenetic enzymes facilitates DNA modifications like methylation, acetylation, phosphorylation, and ubiquitination, all of which alter gene expression and change the state of cells within the body. Ultimately, dysfunction in these enzymes can lead to modifications within the cell that can lead to the development of cancer (45).
However, there is hope regarding epigenetic modifications that result in cancer. The good news is that modification of the DNA sequence by epigenetic enzymes is reversible. In fact, it appears as though a key factor in epigenetic regulation are long noncoding RNA transcripts (lncRNAs) that facilitate the molecular processes of epigenetic regulation (46). lncRNAs have been proposed to control activation and modulation of epigenetic enzymes (46, 47), and lncRNAs have been shown to be involved in cancer resistance to immune reaction through antigen release, antigen presentation, immune activation, and immune cell migration and infiltration (45, 48). Additionally, nine lncRNAs as prognostic markers for GBM patient outcome (49). Therefore, inhibition of epigenetic changes by lncRNAs has immense potential as a GBM therapy (50).
Several lncRNAs are differentially expressed in GBM relative to normal brain tissue (51). For instance, the HOX Transcript Antisense Intergenic RNA (HOTAIR) is completely undetectable in normal brain but is overexpressed in GBM tumors. HOTAIR is a lncRNA from the homeobox super family on the HOXC locus on chromosome 12q13.13 (52). It was the first transregulation lncRNA to be found and has been linked to osteoarthritis and cardiovascular disease in addition to multiple cancers (53). HOTAIR interacts with chromatin modeling complexes, which consequently leads to gene regulation and the promotion of tumor cell invasion, metastasis, and maintenance of stemness in cancer cells (54). However, in specific cancers like colorectal cancer (CRC) its exact method of action is unclear. What is known is that knockdown of HOTAIR in CRC cell lines drastically reduces CRC cell proliferation, which has also been observed in mouse GBM models as well (55, 56). Additionally, HOTAIR also plays a role in drug sensitivity in CRC cells. Knockdown of HOTAIR demonstrates that CRC cells display increased sensitivity to Cisplatin, a chemotherapy agent, and HOTAIR was observed to be upregulated in drug resistant CRC cell lines (57).
We and others determined whether HOTAIR levels were significantly correlated with GBM tumors and GBM serum (52, 54, 58). Quantitative real-time-PCR (qRT-PCR) was conducted to detect HOTAIR levels in 15 pairs of GBM tissue and GBM serum samples. The result of the study demonstrated that not only is HOTAIR dysregulated and that the dysregulated lncRNA facilitates GBM proliferation, but that there are also higher levels of HOTAIR in serum exosomes of GBM patients (52). This association between HOTAIR and GBM proliferation and expansion has been demonstrated by others as well in GBM patients in vivo (58). Additionally, it was discovered that HOTAIR mediates the ability of GBM cells to migrate and invade through membranes in vitro (56). Therefore, there is considerable evidence that not only is HOTAIR related to cancer proliferation, but that it is also an independent negative prognostic marker in GBM (54, 58).
We demonstrated that HOTAIR is part of a proliferative pathway controlled by the bromodomain and extra-terminal domain (BET) epigenetic reader proteins (Figure 2). One such protein, Bromodomain Containing 4 (BRD4), was shown to bind to the HOTAIR promoter and in doing so, controlled HOTAIR levels. Specifically, BRD4 binding and activation led to increased levels in HOTAIR (52). As expected then, the use of a Bromodomain and Extraterminal (BET) inhibitor, I-BET151, that reduces BRD4 binding at the HOTAIR promoter was observed to cause a consequential decrease in the expression of HOTAIR in GBM cells (52, 59). Another BET inhibitor, JQ1, was observed to induce G1 cell cycle arrest and apoptosis, which led to reduction of significant GBM genes (c-MYC, hTERT, Bcl-2, Bcl-xL, and P21CiP1/WAF1) (60). JQ1 reduces tumor growth via reduction of PD-L1 expression on tumor cells leading to less T-cell death induced by the PD-L1 pathway (36). This idea of BET inhibitors promoting T-cell immune reactions against cancer cells has been further supported by research that has shown that BET inhibitors promote T-cell infiltration in mouse models. Moreover, it was discovered that epigenetic inhibitors can even rejuvenate the ability of exhausted T-cells to infiltrate tumor cells once again (61). This further exemplifies the ability of epigenetic pathways to control neoplastic activity and highlights the therapeutic potential of epigenetic pathway modulation in GBM.
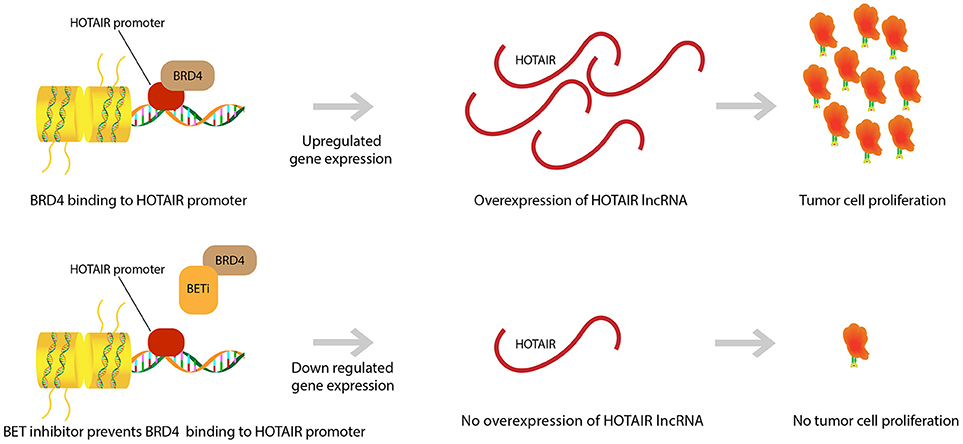
Figure 2. BET inhibitor drugs and HOTAIR level regulation. BRD4 binding to HOTAIR promoter region causes an upregulation in gene expression for the lncRNA HOTAIR. This overexpression in HOTAIR is associated with proliferation and expansion of GBM tumor cells. BET inhibitors prevent binding of BRD4 to the HOTAIR promoter and yield a down regulation in HOTAIR expression. Subsequently, there is prevention of tumor cell proliferation.
Another study involving lncRNAs looked at lncRNA LINC00470, which like HOTAIR, is overexpressed in GBM when compared to normal brain tissue. Moreover, it was reported that patients with higher levels of LINC00470 had poorer prognoses in terms of survival time when compared with patients with lower levels of LINC00470 (62). Researchers in this study investigated LINC00470 and its interaction with AKT, a serine/threonine kinase related to cell proliferation, autophagy, and survival. What was discovered was that interaction between LINC00470 and AKT caused an upregulation of AKT activation thus leading to cell proliferation and GBM tumorigenesis (62). Thus, there is strong evidence for the role of lncRNA and cancer proliferation through epigenetic interactions.
Research looking into epigenetic management of cancer has shown that epigenetic inhibitors are safe and may be effective in treating certain neoplasms. For example, Vorinostat, a histone deacetylase (HDAC) inhibitor that works by preventing the action of histone deacetylases (HDACs), has been approved for treatment of Cutaneous T-cell Lymphoma and may benefit patients suffering from prostate cancer, breast cancer, and lung cancer (63). 5-Azacytidine (5-AZA), another epigenetic inhibitor that works through prevention of the enzyme DNA methyltransferase, has been shown to be effective in treating patients with myelodysplastic syndromes (64). Additionally when used in combination with chemotherapy agents, epigenetic inhibitors, such as the DNA methyltransferase inhibitor Decitabine, and chemotherapy drugs, such as DNA synthesis inhibitors Mitoxantrone Hydrochloride, Cytarabine and Etoposide, were effective in increasing overall survival in patients suffering from relapsed or refractory acute myeloid leukemia or high-risk myelodysplastic syndromes (65). Regarding epigenetic and immunotherapy combination trials, researchers are currently assessing the viability and safety of epigenetic inhibitors, like 5-AZA, with the checkpoint inhibitor, like pembrolizumab, in combinations such as 5-AZA with pembrolizumab, entinostat (HDAC inhibitor) with pembrolizumab, and vorinostat with pembrolizumab (66).
Conclusions and Future Perspectives
GBM remains an incurable disease despite increased understanding of the genetic and epigenetic pathways dysregulated in these tumors. While immunotherapy trials have shown minor improvements in overall survival, the actual increase in time for the patient is still only months and treatment is highly limited to certain subtypes of GBM. Importantly, lncRNAs can be detected in patient serum and can be used as biomarkers for efficacy of drugs in patients. Thus, it seems that the pursuit of other treatment options such as combining epigenetic pathway inhibitors along with immunotherapy treatment could bring medicine closer to treating GBM. Though it may be difficult to create a blanket treatment for all types of GBM, it is worth dedicating resources into future research of individualized patient treatment.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge funding from the NIH via grants NS067289, R56102590 to NA. We also acknowledge support from the Epigenetics Program of the Sylvester Comprehensive Cancer Center at the University of Miami. Additionally, we acknowledge a grant from the Sylvester Comprehensive Cancer Center (M.I.D.) Body, brain and petri dish images Created by Brgfx—Freepik.com. Syringe image Created by Ikaika—Freepik.com. String image created by Layerace—Freepik.com. Cylinder image Created by Alvaro_cabrera—Freepik.com. DNA images Created by Macrovector—Freepik.com.
References
1. Thomas AA, Brennan CW, DeAngelis LM, Omuro AM. Emerging therapies for glioblastoma. JAMA Neurol. (2014) 71:1437–44. doi: 10.1001/jamaneurol.2014.1701
2. Davis ME. Glioblastoma: overview of disease and treatment. Clin J Oncol Nurs. (2016) 20:S2–8. doi: 10.1188/16.CJON.S1.2-8
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
4. Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ, Dudley ME, Liu F, et al. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood (2012) 119:5688–96. doi: 10.1182/blood-2011-10-386482
5. Castello, Grizzi F, Qehajaj D, Rahal D, Lutman F, Lopci E. (18)F-FDG PET/CT for response assessment in Hodgkin lymphoma undergoing immunotherapy with checkpoint inhibitors. Leuk Lymphoma (2018) 1–9. doi: 10.1080/10428194.2018.1488254
6. Butts, Maksymiuk A, Goss G, Soulieres D, Marshall E, Cormier Y, et al. Updated survival analysis in patients with stage IIIB or IV non-small-cell lung cancer receiving BLP25 liposome vaccine (L-BLP25): phase IIB randomized, multicenter, open-label trial. J Cancer Res Clin Oncol. (2011) 137:1337–42. doi: 10.1007/s00432-011-1003-3
7. Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science (2018) 359:1355–60. doi: 10.1126/science.aar7112
8. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev. (2014) 14:135–46. doi: 10.1038/nrc3670
9. Reardon DA, Wucherpfennig K, Chiocca EA. Immunotherapy for glioblastoma:on the sidelines or in the game? Discov Med. (2017) 24:201–8.
10. Lee DH, Ryu HW, Won HR, Kwon SH. Advances in epigenetic glioblastoma therapy. Oncotarget (2017) 8:18577–89. doi: 10.18632/oncotarget.14612
11. ClinicalTrials.gov. Combination of Immunization and Radiotherapy for Recurrent GBM (InSituVac1). ClinicalTrials.gov Identifier: NCT03392545.
12. ClinicalTrials.gov. Basiliximab in Treating Patients with Newly Diagnosed Glioblastoma Multiforme Undergoing Targeted Immunotherapy and Temozolomide-Caused Lymphopenia. ClinicalTrials.gov Identifier: NCT00626483.
13. ClinicalTrials.gov. Vaccine Therapy in Treating Patients with Newly Diagnosed Glioblastoma Multiforme. ClinicalTrials.gov Identifier: NCT00643097.
14. ClinicalTrials.gov. Phase II Feasibility Study of Dendritic Cell Vaccination for Newly Diagnosed Glioblastoma Multiforme. ClinicalTrials.gov Identifier: NCT00323115.
15. ClinicalTrials.gov. A Study of ICT-107 Immunotherapy in Glioblastoma Multiforme (GBM). ClinicalTrials.gov Identifier: NCT01280552.
16. ClinicalTrials.gov. Tremelimumab and Durvalumabin Combination or Alonein Treating Patients with Recurrent Malignant Glioma. ClinicalTrials.gov Identifier: NCT02794883.
17. ClinicalTrials.gov. Pembrolizumab and Vorinostat Combined with Temozolomide for Newly Diagnosed Glioblastoma. ClinicalTrials.gov Identifier: NCT03426891.
18. ClinicalTrials.gov. Adjuvant Dendritic Cell-immunotherapy Plus Temozolomide in Glioblastoma Patients. ClinicalTrials.gov Identifier: NCT02649582.
19. ClinicalTrials.gov. A Study of ICT-121 Dendritic Cell Vaccine in Recurrent Glioblastoma. ClinicalTrials.gov Identifier: NCT02049489.
20. ClinicalTrials.gov. Tumor Lysate Pulsed Dendritic Cell Immunotherapy for Patients with Brain Tumors. ClinicalTrials.gov Identifier: NCT00576537.
21. ClinicalTrials.gov. Dendritic Cell-Based Tumor Vaccine Adjuvant Immunotherapy of Human Glioblastoma Multiforme (WHO Grade IV Gliomas). ClinicalTrials.gov Identifier: NCT02772094.
22. ClinicalTrials.gov. A Pilot Study to Evaluate PBR PET in Brain Tumor Patients Treated with Chemoradiation or Immunotherapy. ClinicalTrials.gov Identifier: NCT02431572.
24. Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res. (2003) 9:4247–54.
26. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. (2010) 28:4722–9. doi: 10.1200/JCO.2010.28.6963
27. Montano N, Cenci T, Martini M, D'Alessandris QG, Pelacchi F, Ricci-Vitiani L, et al. Expression of EGFRvIII in glioblastoma: prognostic significance revisited. Neoplasia (2011) 13:1113–21. doi: 10.1593/neo.111338
28. Phase II Feasibility Study of Dendritic Cell Vaccination for Newly Diagnosed Glioblastoma Multiforme.
29. Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. (2011) 34:382–9. doi: 10.1097/CJI.0b013e318215e300
30. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. N. Cancer genome atlas research, integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
31. Sidaway P. Glioblastoma subtypes revisited. Nat Rev Clin Oncol. (2017) 14:587. doi: 10.1038/nrclinonc.2017.122
32. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell (2017) 32:42–56.e6. doi: 10.1016/j.ccell.2017.06.003
33. Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science (2017) 355:eaai8478. doi: 10.1126/science.aai8478
34. Lee E, Yong RL, Paddison P, Zhu J. Comparison of glioblastoma (GBM) molecular classification methods. Semin Cancer Biol. (2018). doi: 10.1016/j.semcancer.2018.07.006. [Epub ahead of print].
35. Burbulis IE, Wierman MB, Wolpert M, Haakenson M, Lopes MB, Schiff D, et al. Improved molecular karyotyping in glioblastoma. Mutat Res. (2018) 811:16–26. doi: 10.1016/j.mrfmmm.2018.06.002
36. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. (2016) 16:2829–37. doi: 10.1016/j.celrep.2016.08.032
37. Filley AC, Henriquez M, Dey M. Recurrent glioma clinical trial, CHECKMATE-143: the game is not over yet. Oncotarget (2017) 8:91779–94. doi: 10.18632/oncotarget.21586
38. Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology (2010) 129:474–81. doi: 10.1111/j.1365-2567.2010.03255.x
39. Park J, Kim CG, Shim J-K, Lee JE, Chang JH, Shin E-C, et al. IMMU-16. Effect of Anti-PD-1 therapy combination on Temozolomide in mouse glioblastom. Neuro Oncol. (2017) 19:vi116. doi: 10.1093/neuonc/nox168.475
40. Luoto S, Hermelo I, Vuorinen EM, Hannus P, Kesseli J, Nykter M, et al. Computational characterization of suppressive immune microenvironments in glioblastoma. Cancer Res. (2018) 78:5574–85. doi: 10.1158/0008-5472.CAN-17-3714
41. Adriana O, Aldape KD. Using the molecular classification of glioblastoma to inform personalized treatment. J Pathol. (2014) 232:165–77. doi: 10.1002/path.4282
42. DiDomenico J, Lamano JB, Oyon D, Li Y, Veliceasa D, Kaur G, et al. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology (2018) 7:e1448329. doi: 10.1080/2162402X.2018.1448329
43. Humphries W, Wei J, Sampson JH, Heimberger AB. The role of tregs in glioma-mediated immunosuppression: potential target for intervention. Neurosurg Clin N Am. (2010) 21:125–37. doi: 10.1016/j.nec.2009.08.012
44. Houghton AN, Guevara-Pati JA. Immune recognition of self in immunity against cancer. J Clin Invest. (2004) 114:468–71. doi: 10.1172/JCI22685
45. Heerboth S, Lapinska K, Snyder N, Leary M, Rollinson S, Sarkar S. Use of epigenetic drugs in disease: an overview. Genet Epigenet. (2014) 6:9–19. doi: 10.4137/GEG.S12270
46. Wang C, Wang L, Ding Y, Lu X, Zhang G, Yang J, et al. lncrna structural characteristics in epigenetic regulation. Int J Mol Sci. (2017) 18:E2659. doi: 10.3390/ijms18122659
47. Saxena, Carninci P. Long non-coding RNA modifies chromatin. BioEssays (2011) 33:830–9. doi: 10.1002/bies.201100084
48. Yu W-D, Wang H, He Q-F, Xu Y, Wang X-C. Long noncoding RNAs in cancer-immunity cycle. J Cell Physiol. (2018) 233:6518–23. doi: 10.1002/jcp.26568
49. Lei B, Yu L, Jung TA, Deng Y, Xiang W, Liu Y, et al. Prospective series of nine long noncoding rnas associated with survival of patients with glioblastoma. J Neurol Surg A Cent Eur Neurosurg. (2018) 79:471–8. doi: 10.1055/s-0038-1655549
50. Balci T, Yilmaz Susluer S, Kayabasi C, Ozmen Yelken B, Biray Avci C, Gunduz C. Analysis of dysregulated long non-coding RNA expressions in glioblastoma cells. Gene (2016) 590:120–2. doi: 10.1016/j.gene.2016.06.024
51. Kiang KM, Zhang XQ, Leung GK. Long non-coding RNAs: the key players in glioma pathogenesis. Cancers (2015) 7:1406–24. doi: 10.3390/cancers7030843
52. Tan SK, Pastori C, Penas C, Komotar RJ, Ivan ME, Wahlestedt C, et al. Serum long noncoding RNA HOTAIR as a novel diagnostic and prognostic biomarker in glioblastoma multiforme. Mol Cancer (2018) 17:74. doi: 10.1186/s12943-018-0822-0
53. Jiang Y, Mo H, Luo J, Zhao S, Liang S, Zhang M, et al. HOTAIR Is a potential novel biomarker in patients with congenital heart diseases. Biomed Res Int. (2018) 2018:2850657. doi: 10.1155/2018/2850657
54. Shen J, Hodges TR, Song R, Gong Y, Calin GA, Heimberger AB, et al. Serum HOTAIR and GAS5 levels as predictors of survival in patients with glioblastoma. Mol Carcinog. (2018) 57:137–41. doi: 10.1002/mc.22739
55. Huang K, Sun J, Yang C, Wang Y, Zhou B, Kang C, et al. HOTAIR upregulates an 18-gene cell cycle-related mRNA network in glioma. Int J Oncol. (2017). doi: 10.3892/ijo.2017.3901. [Epub ahead of print].
56. Zhou X, Ren Y, Zhang J, Zhang C, Zhang K, Han L, et al. HOTAIR is a therapeutic target in glioblastoma. Oncotarget (2015) 6:8353–65. doi: 10.18632/oncotarget.3229
57. Xiao Z, Qu Z, Chen Z, Fang Z, Zhou K, Huang Z, et al. LncRNA HOTAIR is a prognostic biomarker for the proliferation and chemoresistance of colorectal cancer via MiR-203a-3p-mediated Wnt/Ss-catenin signaling pathway. Cell Physiol Biochem. (2018) 46:1275–85. doi: 10.1159/000489110
58. Xavier-Magalhaes, Goncalves CS, Fogli A, Lourenco T, Pojo M, Pereira B, et al. The long non-coding RNA HOTAIR is transcriptionally activated by HOXA9 and is an independent prognostic marker in patients with malignant glioma. Oncotarget (2018) 9:15740–56. doi: 10.18632/oncotarget.24597
59. Pastori, Kapranov P, Penas C, Peschansky V, Volmar C-H, Sarkaria JN, et al. The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc Natl Acad Sci USA. (2015) 112:8326–31. doi: 10.1073/pnas.1424220112
60. Wadhwa E, Nicolaides T. bromodomain inhibitor review: bromodomain and extra-terminal family protein inhibitors as a potential new therapy in central nervous system tumors. Cureus (2016) 8:e620. doi: 10.7759/cureus.620
61. Aspeslagh S, Morel D, Soria JC, Postel-Vinay S. Epigenetic modifiers as new immunomodulatory therapies in solid tumours. Ann Oncol. (2018) 29:812–24. doi: 10.1093/annonc/mdy050
62. Liu C, Zhang Y, She X, Fan L, Li P, Feng J, et al. A cytoplasmic long noncoding RNA LINC00470 as a new AKT activator to mediate glioblastoma cell autophagy. J Hematol Oncol. (2018) 11:77. doi: 10.1186/s13045-018-0619-z
63. Bubna AK. Vorinostat-an overview. Indian J Dermatol. (2015) 60:419. doi: 10.4103/0019-5154.160511
64. Raj K, John A, Ho A, Chronis C, Khan S, Samuel J, et al. CDKN2B methylation status and isolated chromosome 7 abnormalities predict responses to treatment with 5-azacytidine. Leukemia (2007) 21:1937–44. doi: 10.1038/sj.leu.2404796
65. ClinicalTrials.gov. Decitabine Followed by Mitoxantrone Hydrochloride, Etoposide, and Cytarabine in Treating Patients with Relapsed or Refractory Acute Myeloid Leukemia or High-Risk Myelodysplastic Syndromes. ClinicalTrials.gov Identifier: NCT01729845.
Keywords: glioblastoma, immunotherapeutic, clinical trial, epigenetic, long non coding RNA
Citation: Chin C, Lunking ES, de la Fuente M and Ayad NG (2018) Immunotherapy and Epigenetic Pathway Modulation in Glioblastoma Multiforme. Front. Oncol. 8:521. doi: 10.3389/fonc.2018.00521
Received: 03 September 2018; Accepted: 24 October 2018;
Published: 13 November 2018.
Edited by:
Shiv K. Gupta, Mayo Clinic, United StatesReviewed by:
Karishma Rajani, Mayo Clinic, United StatesAntonio Rozzi, INI, Istituto Neurotraumatologico Italiano, Italy
Copyright © 2018 Chin, Lunking, de la Fuente and Ayad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nagi G. Ayad, nayad@miami.edu