 Tian-Tian Xu
Tian-Tian Xu Xiao-Wen Zeng
Xiao-Wen Zeng Xin-Hong Wang
Xin-Hong Wang Lu-Xi Yang
Lu-Xi Yang Gang Luo
Gang Luo Ting Yu
Ting Yu- 1Department of Periodontics, Affiliated Stomatology Hospital of Guangzhou Medical University, Guangzhou Key Laboratory of Basic and Applied Research of Oral Regenerative Medicine, Guangzhou, China
- 2Department of Oral Pathology and Medicine, Affiliated Stomatology Hospital of Guangzhou Medical University, Guangzhou Key Laboratory of Basic and Applied Research of Oral Regenerative Medicine, Guangzhou, China
Disturbance in the proteolytic process is one of the malignant signs of tumors. Proteolysis is highly orchestrated by cysteine cathepsin and its inhibitors. Cystatin-B (CSTB) is a general cysteine cathepsin inhibitor that prevents cysteine cathepsin from leaking from lysosomes and causing inappropriate proteolysis. Our study found that CSTB was downregulated in both oral squamous cell carcinoma (OSCC) tissues and cells compared with normal controls. Immunohistochemical analysis showed that CSTB was mainly distributed in the epithelial structure of OSCC tissues, and its expression intensity was related to the grade classification. A correlation analysis between CSTB and clinical prognosis was performed using gene expression data and clinical information acquired from The Cancer Genome Atlas (TCGA) database. Patients with lower expression levels of CSTB had shorter disease-free survival times and poorer clinicopathological features (e.g., lymph node metastases, perineural invasion, low degree of differentiation, and advanced tumor stage). OSCC cell models overexpressing CSTB were constructed to assess the effects of CSTB on malignant biological behaviors and upregulation of CSTB inhibited cell proliferation, migration, and invasion in vitro. Weighted gene correlation network analysis (WGCNA) and gene set enrichment analysis (GSEA) were performed based on the TCGA data to explore potential mechanisms, and CSTB appeared to correlate with squamous epithelial proliferation-differentiation processes, such as epidermal cell differentiation and keratinization. Moreover, in WGCNA, the gene module most associated with CSTB expression (i.e., the brown module) was also the one most associated with grade classification. Upregulation of CSTB promoted the expression levels of markers (LOR, IVL, KRT5/14, and KRT1/10), reflecting a tendency for differentiation and keratinization in vitro. Gene expression profile data of the overexpressed CSTB cell line were obtained by RNA sequencing (RNA-seq) technology. By comparing the GSEA enrichment results of RNA-seq data (from the OSCC models overexpressing CSTB) and existing public database data, three gene sets (i.e., apical junction, G2/M checkpoint, etc.) and six pathways (e.g., NOTCH signaling pathway, glycosaminoglycan degradation, mismatch repair, etc.) were enriched in the data from both sources. Overall, our study shows that CSTB is downregulated in OSCC and might regulate the malignant characteristics of OSCC via the epithelial proliferation/differentiation program.
Introduction
As the sixth most common cancer in the world, head and neck squamous cell carcinoma (HNSCC) continues to rise yearly, and oral squamous cell carcinoma (OSCC) accounts for 90% of HNSCC cases in the region (1–4). The survival of OSCC patients, especially those with distant metastases, remains low, at approximately 39% (1, 4, 5). Moreover, due to the special anatomical location of the oral cavity, OSCC patients often experience great psychological stress and compromised quality of life (6). To date, the potential pathogenesis, including the progression mechanism, of OSCC is not fully understood. It is generally accepted that the initiation and development of OSCC is a complex process requiring the accumulation of genomic alterations, which is modified by individual genetic predisposition and environmental carcinogenic risk factors (7–9). It is necessary and challenging for clinical oncology and precision medicine science to understand the carcinogenic and progression mechanisms of OSCC, to identify biomarkers for early screening and to establish an accurate prognostic evaluation system, all of which would contribute to the reduction in the incidence and improvement of both the survival rate and living quality of tumor-bearing patients (10–12).
Proteolysis has a vital role in the normal life activities of organisms. Disorders between the enzymatic reaction and the inhibitory reaction of the proteolytic cascade process are one of the malignant characteristics of tumors, and they also lead to tumor invasion and metastasis (13–16). Numerous evidences have indicated that cysteine cathepsins play a crucial role in these processes (17–20). Cysteine cathepsin can degrade the proteins of the extracellular matrix (ECM) and reshape the tumor microenvironment by regulating a variety of cytokines and thus participate in tumor growth, invasion, angiogenesis, and metastasis (21). Compared with the fully elucidated role of the cysteine protease family in tumors, research on its inhibitors in tumors is relatively limited. Most studies only focus on certain types of inhibitors and are not in-depth enough. As an inhibitor of cysteine cathepsin, cystatin-B (coded by Cstb gene) is considered a general cysteine cathepsin inhibitor in mammalian cells, preventing cysteine cathepsin from leaking from lysosomes and causing inappropriate proteolysis (18, 22–24). CSTB plays a bimodal role in cancer. Recent studies have associated CSTB with various cancers [e.g., hepatocellular carcinoma (HCC) (25–28), epithelial ovarian tumors (29, 30), breast cancer (31), laryngeal squamous cell carcinoma, and esophageal squamous cell carcinoma, and esophageal squamous cell carcinoma (ESCC) (32, 33)], in which the expression, in which the expression of CSTB is changed in different directions. For instance, CSTB was increased in HCC (25–28), epithelial ovarian tumors (29, 30), and breast cancer (31), while decreased in laryngeal squamous cell carcinoma and ESCC (32, 33). Some studies have indicated that increased CSTB is related to a poorer prognosis in bladder cancer (34) and a higher risk of tumor metastasis in HCC (27). However, most of the existing studies on the correlation between CSTB and tumors just provide observational evidence and whether CSTB has a causal role in cancers (including OSCC) is unclear. Only a few studies found that CSTB may participate in tumors by regulating cell apoptosis and oxidative stress (31). Only one noninterventional study based on human OSCC tissue specimens reported that low expression of CSTB in the invasive tumor front was correlated with local tumor recurrence. In addition, CSTB-specific peptides in saliva were associated with lymph node metastasis (35). However, the single study reached a conclusion based only on OSCC specimens but lacked a nontumor (normal control) group. Moreover, there is a lack of in-depth and comprehensive mechanistic studies on the role of CSTB in the progression of OSCC. Based on the above evidence, we hypothesized that CSTB may play a role in OSCC. Our research aimed to identify the role of CSTB in OSCC and to explore the relevant mechanisms.
Materials And Methods
Cell Line Acquisition and Culture
The human tongue squamous cell carcinoma cell lines SCC9 and SCC25 (ATCC, ATCC® CRL-1629™ and CRL-1628™, Manassas, VA, USA) and the normal oral epithelial keratinocyte line HOK (AULU, Guangdong, China) were purchased and cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Gibco, Waltham, MA, USA) and DMEM, respectively. The complete cell culture medium contained 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (Gibco), while the medium for carcinoma cells was supplemented with an additional 400-ng/ml hydrocortisone (MedChemExpress, Monmouth Junction, NJ, USA). The serum-free cell culture medium for carcinoma cells was prepared as DMEM/F12 containing 1% penicillin/streptomycin and 400 ng/ml hydrocortisone for a number of subsequent experiments (e.g., enzyme-linked immunosorbent assay, wound-healing assay, cell migration, and invasion assays). The cells were cultured in a 37°C, 5% CO2 incubator.
Clinical Patient Specimen Collection and Immunohistochemistry Staining for Cystatin-B
The studies involving human participants were approved by the Ethics Committee of the Stomatology Hospital of Guangzhou Medical University. Twenty-three primary OSCC specimens and fifteen normal oral tissues were obtained from the Department of Periodontics and Oral Mucosal Diseases, Stomatology Hospital of Guangzhou Medical University. All OSCC specimens were from samples pathologically diagnosed as OSCC. Normal specimens were excess tissues that needed to be removed due to tooth extractions or oral surgeries, which were also examined as normal oral mucosal epithelium by pathology. The pathological grading (36) of tissue sections was classified blindly by a single pathologist (Xin-Hong Wang). Immunohistochemistry (IHC) staining for cystatin-B was performed in the above tissues (Materials and Methods S1.1). After staining, five fields of view were randomly selected to take images under an upright optical microscope. The integrated optical density (IOD) value of each point on the image was collected by ImageJ and then the average density (%, IOD/target distribution area) was calculated, representing the expression level of CSTB in certain specimens. Immunostaining was analyzed by a researcher who was blinded to the pathological grade of the samples. Correlation analysis between CSTB expression and the degree of pathological differentiation was conducted.
Upregulation of CSTB in OSCC Cell Lines by Lentiviral Transfection
The commercial recombinant lentivirus (OBIO, Shanghai, China) named lenti-CSTB was utilized to overexpress CSTB, and an empty carrier lentivirus named lenti-NC was used as a negative control. SCC9 and SCC25 cells were infected with the above lentivirus (MOI = 30) and screened with puromycin (3 µg/ml) for 15 days. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and Western blotting were used to validate the overexpression of CSTB at the mRNA and protein levels, respectively. After successful transfection, the cell lines (i.e., SCC9/25-lenti-CSTB and SCC9/25-lenti-NC) were cultured with complete medium containing puromycin (1 µg/ml) for subsequent analysis.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was extracted from the cells using an RNA extraction kit (AG21017, Accurate Biology, Hunan, China). Extracted RNA was analyzed for quantity and quality by measuring A260/A280 with a spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific, Waltham, MA, USA). RNA integrity was confirmed by 1.5% agarose gel electrophoresis. For qRT-PCR, 1 μg of total RNA was used to synthesize cDNA (AG11706, Accurate Biology, China). qRT-PCR was performed using a SYBR Green qPCR kit (AG11718, Accurate Biology, China). Relative mRNA expression was normalized to that of the internal GAPDH control. The primer sequences used are listed in the Table S1. The relative expression of targeted genes was calculated using the 2−△△Ct method (37). Each test was repeated at least three times.
Western Blotting Analysis
Total protein was isolated from cell samples using precooled cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) with protease inhibitor (RayBiotech, Guangzhou, China) and quantified using a BCA protein assay kit (Beyotime, Shanghai, China). Equal protein extracts (20 µg) were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Burlington, MA, USA). After successive incubations with the primary antibody and the secondary antibody, the target protein was visualized by chemiluminescence using an ECL kit (Beyotime). The following antibodies were used in the Western blot assay: anti-loricrin polyclonal antibody (1:1,000, 55439-1-AF, Proteintech, Wuhan, China), anti-involucrin polyclonal antibody (1:1,000, 55328-1-AP, Proteintech), anti-cystatin-B monoclonal antibody (1:1,000, 66712-1-Ig, Proteintech), anti-beta-actin monoclonal antibody (1:10,000, 66009-1-Ig, Proteintech), anti-GAPDH antibody (1:10,000, EPR16891, Abcam, Cambridge, UK), goat anti-mouse IgG (H+L) antibody (HRP) (1:10,000, SA00001-1, Proteintech), and goat anti-rabbit IgG H&L (HRP) antibody (1:10,000, ab205718, Abcam). GAPDH or beta-actin served as the internal controls to calculate the relative expression of the targeted proteins.
Enzyme-Linked Immunosorbent Assay
Cells were inoculated at a density of 1 × 106 in complete medium, which was replaced by serum-free medium after 2 days of culture. After another 2 days, the supernatant was collected, centrifuged (4°C, 2,000 rpm, 10 min), and stored at −80°C. The protein levels of CSTB in cell supernatants were examined by an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, DCYB00) according to the manufacturer’s instructions. Each test was repeated at least three times.
Cell Proliferation Assay
Transfected SCC9 and SCC25 cells were inoculated with complete medium in 96-well plates at a density of 3,000 cells per well. Cell proliferation was detected using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) at 1, 2, 3, and 5 days according to the manufacturer’s instructions. Each test was repeated at least three times.
Colony Formation Assay
Three thousand transfected SCC9 cells and SCC25 cells were seeded respectively in six-well plates and cultured for 10 days in complete medium. Afterward, the colonies were fixed with 4% paraformaldehyde and stained with crystal violet. The colonies that contained more than 50 cells were counted. Each test was repeated at least three times.
Wound-Healing Assay
For the wound-healing assay, transfected SCC9 and SCC25 cells were first incubated and cultured with complete medium. A scratch was made with a sterile pipette tip after a confluent monolayer of cells was formed. Afterward, the cells were washed with phosphate-buffered saline (PBS) and cultured with serum-free medium. Images were taken at 0, 24, and 36 h postwounding. The wound-healing areas were assessed by ImageJ to calculate the wound-healing rate. Wound-healing rate% = [Areat0 − Areat1]/Areat0 × 100% (Areat0 is the area of the wound measured immediately after scratching, and Areat1 is the area of the wound measured t1 hours after scratching). In our research, the area of the wound was measured at 0, 24, and 36 h. Each test was repeated at least three times.
Cell Migration and Invasion Assays
Cells were seeded at a density of 1 × 105 (a density of 5 × 104 for transfected SCC9 cells for the migration assay) in serum-free DMEM/F12 medium in the upper wells of Transwell chambers (8 μm pore size, Corning, New York, NY, USA), while the lower wells were filled with complete medium containing 10% FBS. Chambers for the invasion assay were coated with 100 µl of Matrigel (200 μg/µl, Corning) and incubated for 2 h at 37°C. Cells in the upper layer were removed with a swab after 24 h of culture, and cells on the bottom membrane were fixed with 4% paraformaldehyde and stained with crystal violet. The results of each group were photographed at five randomized visual fields, and the experiments were repeated three times.
Bioinformatics Analysis for Identifying CSTB Expression
Five expression microarray series containing OSCC tumor and normal samples were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) (Table S2). One expression microarray series containing OSCC tumor and oral leucoplakia samples was also downloaded from GEO (Table S2). The Cancer Genome Atlas (TCGA) OSCC mRNA normalized count data and clinicopathological information of 329 OSCC tissues and 32 matched normal oral mucosal epithelial tissues were downloaded from the Genomic Data Commons Data Portal (https://cancergenome.nih.gov/). The Cstb expression levels of both the OSCC and control groups in all datasets were extracted and compared using R Studio software.
Survival Analysis and Clinicopathological Correlation Analysis
TCGA patients with clinicopathological information were used for clinicopathological correlation analysis. Among the patients, 291 OSCC patients with complete clinicopathological and survival data were selected for survival analysis. Next, these 291 patients were divided into high- and low-expression groups based on the median expression level of Cstb for weighted gene correlation network analysis (WGCNA) and gene set enrichment analysis (GSEA).
Weighted Gene Coexpression Network Analysis
The gene expression data and sample clinical information were downloaded from TCGA. First, variance analysis of 22,862 protein-coding genes (PCGs) was performed, the top 25% (5,716 genes) of which were selected for WGCNA. The WGCNA package in R Studio software was applied to construct a gene coexpression network. The soft threshold power (β = 6) was selected to ensure a scale-free network. The adjacency matrix was transformed into a topological overlap matrix (ToM), and the corresponding dissimilarity was calculated. The module eigengenes were calculated to identify modules that were significantly associated with the clinical feature information. In this process, the mRNA expression level of Cstb was also regarded as a feature and was inputted into the correlation analysis, aiming to identify the functional modules related to its expression. Modules with a high correlation coefficient were considered candidate modules related to clinical features and were selected for subsequent analysis. Finally, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed to reveal the functions of the genes in the target modules. p-Value ≤0.05 and q-value ≤0.05 were considered statistically significant. The detailed method for WGCNA is shown in the Materials and Methods S1.2.
mRNA Sequencing of the Transfected Cell Line
The total RNA of transfected SCC25 cells (lenti-CSTB/NC-SCC25) was extracted for further RNA-seq analysis (n = 3). High-throughput sequencing was conducted by SEQHEALTH (Wuhan, China). The raw reads of samples were obtained. The clean reads were mapped to the reference genome of Homo sapiens (Homo_sapiens, GRCh38) using STRA software (version 2.5.3a) with default parameters. The reads mapped to the exon regions of each gene were counted by FeatureCounts (Subread-1.5.1; Bioconductor), and then the reads per kilobase of exon model per million mapped reads (RPKM) were calculated. Genes differentially expressed between groups were identified using the edgeR package. A p-value cutoff of 0.05 and fold-change cutoff of 1.5 were used to judge the statistical significance of gene expression differences. Afterwards, the differentially expressed genes (DEGs) were compared with the mutational cancer driver gene set published in IntOGen (https://www.intogen.org/search#driver-genes:table) (38). Detailed methods for mRNA sequencing are shown in the Materials and Methods S1.3. Details of the mutational cancer driver genes are shown in the Supplementary Data Sheet 1.
Gene Set Enrichment Analysis
First, samples obtained from TCGA were divided into high- and low-expression groups based on the median expression level of Cstb. A similar analysis was also performed based on the RNA-seq data acquired from mRNA Sequencing of the Transfected Cell Line. KEGG and GO analyses were used to explore potential cancer-related biological pathways, while Hallmark GSEA was used to correlate the expression level of Cstb with the biological functions of oncogenes. p-Value ≤0.05 and FDR ≤25% were considered statistically significant.
Statistical Analysis
The data were analyzed using data statistics software (GraphPad Prism 7.0 and SPSS Statistics 16.0). The data are presented as the means and standard deviations. Student’s t-test was performed for two independent samples, while analysis of variance (ANOVA) was used for multiple independent samples, and post-hoc comparisons were made. The correlation between CSTB expression and the degree of pathological differentiation in IHC analysis was determined using Fisher’s exact test. Logistic regression analysis was performed to analyze the relationship between the expression of CSTB and the clinicopathological characteristics of OSCC patients. The correlation between genes in WGCNA was analyzed using Pearson’s correlation. p-Value <0.05 was considered statistically significant.
A snapshot of the entire experimental process is shown in Figure 1.
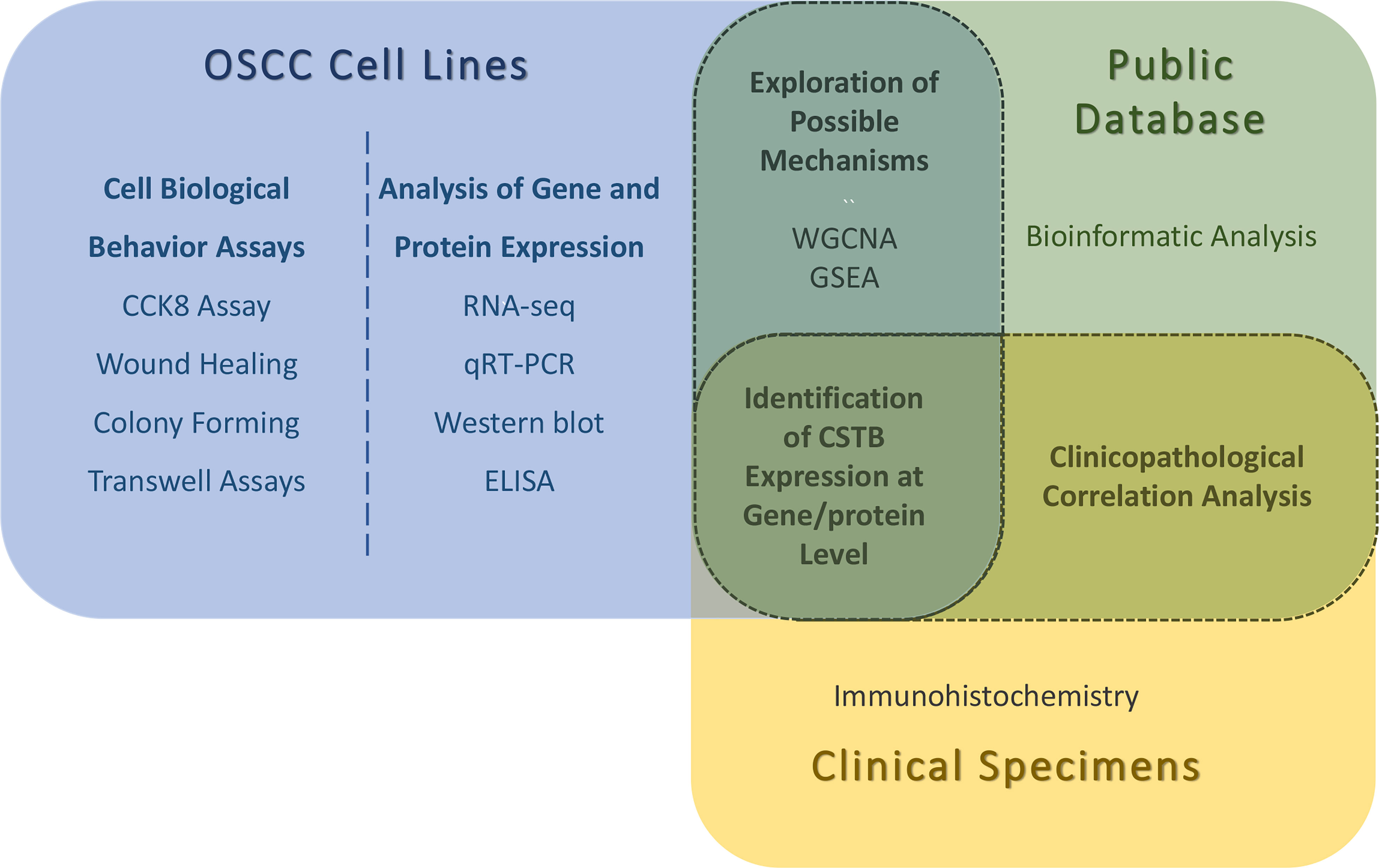
Figure 1 Snapshot of the entire experimental process. WGCNA, weighted gene correlation network analysis; GSEA, gene set enrichment analysis. The identifications of CSTB expression at gene/protein levels are obtained by in vitro (OSCC cell lines) and in vivo (public database/clinical specimens) experiments. Possible mechanisms for the involvement of CSTB in OSCC were explored using OSCC cell lines and public database data together. Using OSCC tissue samples (from public databases and independent clinical specimens) for clinicopathological correlation analysis.
Results
CSTB Is Downregulated in OSCC Both In Vivo and In Vitro
Based on the data from public databases, the expression of Cstb was lower in OSCC tissues than in normal oral mucosal epithelial tissues (Figures 2A–F) and potentially malignant disorder tissues (i.e., oral leucoplakia tissues, Figure 2G). IHC analysis of tissue samples (independently collected from OSCC patients by the coauthors) also showed a downregulation of protein CSTB expression in OSCC tissues (Figure 2H). CSTB was located in the epithelial structure of OSCC tissues, similar to the distribution of CSTB in the epithelial layer of normal oral mucosal epithelial tissues. The staining of CSTB was lighter in OSCC and showed preferential localizations in well-differentiated structures (i.e., cancer nests and keratinized pearls). There was little staining of CSTB in either the tumor stroma in OSCC or subepithelial tissues in control tissues (Figure 2I). Correlation analysis revealed that the levels of CSTB were positively correlated with the degree of tissue differentiation (p < 0.05, Table 1). For subcellular localization, positive CSTB staining was mainly distributed in the cytoplasm and occasionally distributed in the nucleus in both OSCC and control tissues (Figure 2I).
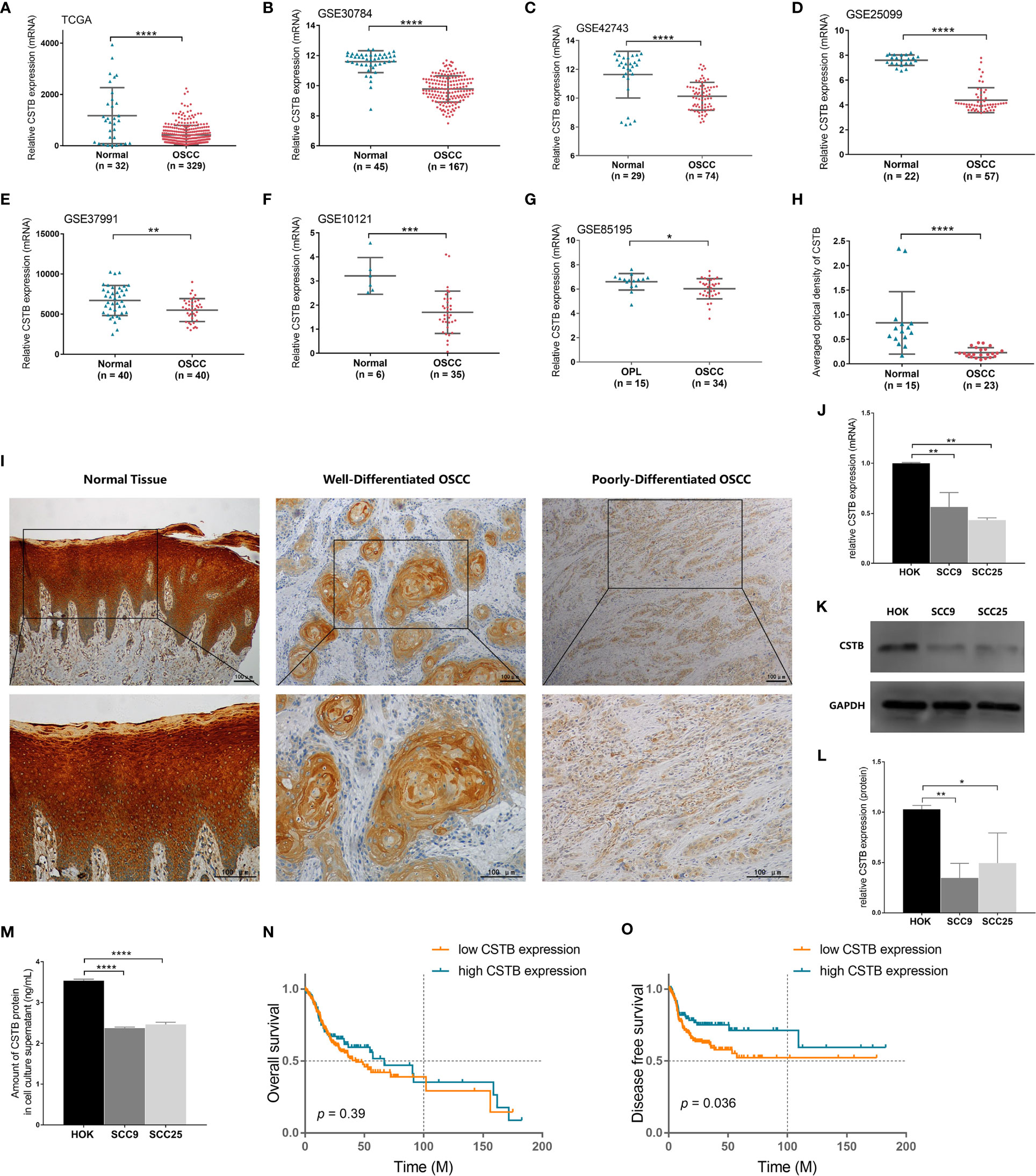
Figure 2 CSTB is downregulated in OSCC and is associated with poor prognosis. Relative expression of CSTB mRNA in OSCC tissues (n = 329) and matched normal tissues (n = 32) from the TCGA database. (B–F) Relative expression of CSTB mRNA in OSCC tissues and normal tissues from the GEO database. The sample size of each group is shown in the figures. (G) Relative expression of CSTB mRNA in OSCC tissues (n = 34) and oral leucoplakia (OPL) tissues (n = 15) from the GEO database. (H) Average optical density of CSTB in normal (n = 15) and OSCC (n = 23) tissues in IHC analysis. (I) IHC staining of CSTB in normal and OSCC tissues. Magnification at ×100 (upper panel) and ×200 (lower panel); scale bars, 200 μm. (J) The relative expression of CSTB mRNA in the oral epithelial keratinocyte line and OSCC cell lines was quantified by RT-qPCR. (K, L) CSTB protein expression in the oral epithelial keratinocyte line and OSCC cell lines was identified by Western blot analysis (K) and quantitatively analyzed (L). (M) The content of CSTB in the culture supernatant in the oral epithelial keratinocyte line and OSCC cell lines was quantified by ELISA. (N, O) Kaplan-Meier survival curves of overall survival and disease-free survival in the high and low CSTB expression groups. Error bars represent the standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
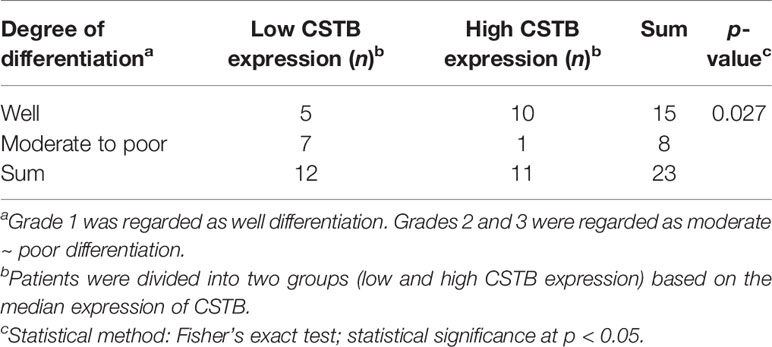
Table 1 Association between CSTB expression and tissue differentiation.
The results of the in vitro experiments were consistent with those of the in vivo experiments. CSTB was downregulated intracellularly in common OSCC cell lines (i.e., SCC9 and SCC25) compared with the normal cell line (i.e., HOK) (Figures 2J–L). Interestingly, extracellular CSTB (i.e., in the cell culture supernatant) was also downregulated in OSCC cell lines (Figure 2M).
Downregulation of CSTB in Tissues Is Associated With Poor Prognosis in OSCC Patients
To explore whether there was a link between CSTB expression and the clinical prognosis of OSCC patients, a correlation analysis between them was conducted based on the data extracted from the TCGA database. There was no significant difference in the overall survival rates between the two groups (Figure 2N). However, the disease-free survival (DFS) rate (an indirect indicator of cancer recurrence and metastasis) of the low Cstb expression group was lower than that of the high Cstb expression group (Figure 2O). Logistic regression analysis indicated that patients with lower expression levels of Cstb had poorer clinicopathological features (e.g., lymph node metastases, perineural invasion, low degree of differentiation, and advanced tumor stage). Patients with lymphovascular invasion tended to have lower Cstb expression (p = 0.06). No association was found between the abundance of Cstb and demographic characteristics, including age and sex (Table 2).
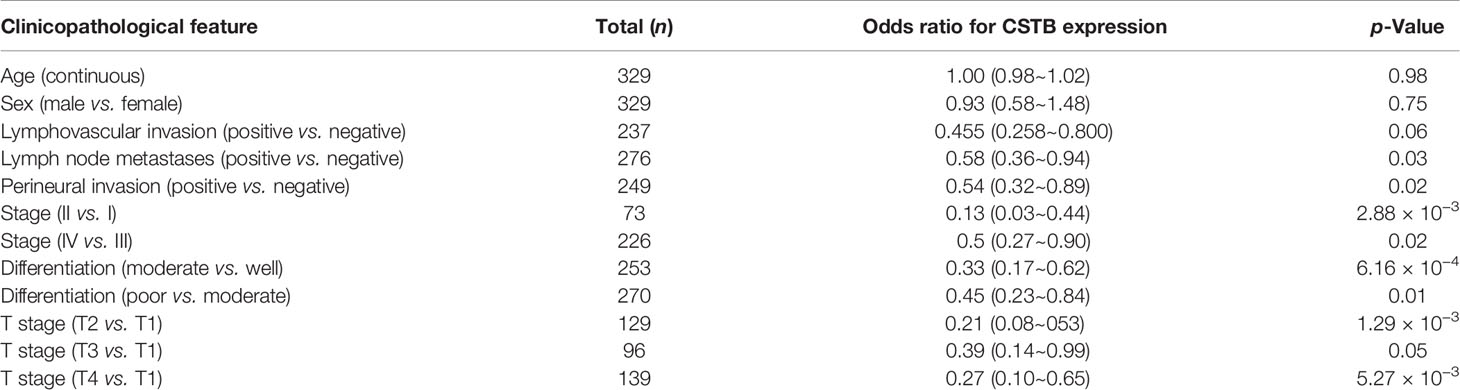
Table 2 Association between CSTB expression and clinicopathological features.
Upregulation of CSTB Inhibits Malignant Biological Behaviors of OSCC Cell Lines
Next, the regulatory role of CSTB in the malignant behaviors of OSCC was explored in vitro. CSTB was successfully transfected into OSCC cell lines (SCC9 and SCC25), and its content was upregulated both intracellularly and extracellularly (Figures 3A–D). Overexpression of CSTB suppressed the proliferation of SCC9 and SCC25 cells (Figures 3E, F) and caused a reduction in colony formation by 17% to 18% (Figures 3G, H). Compared with the NC groups, the wound-healing rate was decreased by 5% to 31% in SCC9/25 cells (Figures 3I–K). Regarding the invasive characteristics, overexpression of CSTB reduced the migration ability by ~37% and the invasion ability by 23%–38% in OSCC cell lines (Figures 3L–N).
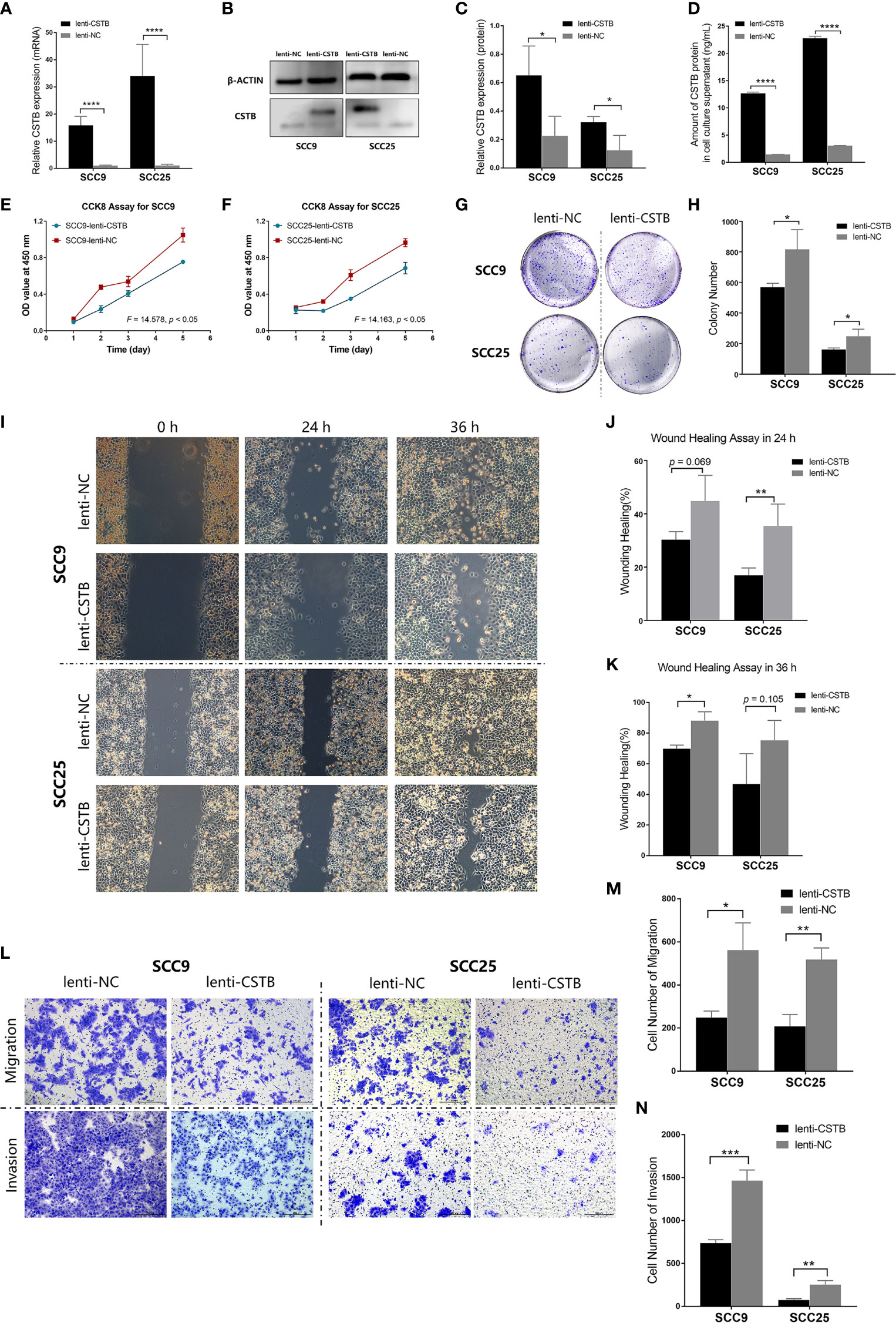
Figure 3 Upregulation of CSTB inhibits the proliferation, migration, invasion and adhesion of OSCC cell lines. (A) Relative expression of CSTB mRNA in OSCC cell lines after lentivirus transfection. (B, C) CSTB protein expression in OSCC cell lines after lentivirus transfection was identified by Western blot analysis (B) and quantitatively analyzed (C). (D) The content of CSTB in the culture supernatant of OSCC cell lines after lentivirus transfection was quantified by ELISA. (E, F) CCK8 assays for transfected OSCC cell lines. (G, H) Colony formation assays for transfected OSCC cell lines. (G) The representative staining images of each group; (H) the number of clones formed in each group. (I–K) Wound-healing assays for transfected OSCC cell lines. Representative images in (I) and statistical results in (J, K) at 0, 24, and 36 h in each group. (L–N) Transwell assays for transfected OSCC cell lines. Representative images for cell migration and invasion of each group are shown in (L). Statistical results for cell migration and invasion are shown in (M, N), respectively. Error bars represent the standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
WGCNA to Identify the Target Module
To explore the relevant mechanism of CSTB in OSCC, WGCNA was used as a nontargeted method to identify gene sets that were highly synergistic with CSTB. In WGCNA, the top 25% variant PCGs (a total of 5,716 genes) were selected as the input in the analysis. Thirteen outlier samples were removed, and the remaining 278 samples were clustered with clinical features and Cstb expression (Figure S1). A soft threshold power of β = 6 was selected to establish a ToM and further construct a scale-free network (Figure S2). The clustering tree was divided into 18 modules using dynamic shearing, and the modules were merged according to the coefficient of dissimilarity of <0.25 (Figure S3). The relationships between the input features and the module eigengenes are shown in Figure 4A. Interestingly, the module most associated with Cstb expression (i.e., the brown module, r = 0.83, p = 4 × 10−113, Figures 4A, B) was also the one most associated with grade classification (r = 0.73, p = 2.1 × 10−74, Figures 4A, C). This result suggests that the gene set highly coexpressed with Cstb may have a certain effect on the grade classification phenotype, which indicated that CSTB may play a regulatory role in the phenotype of grade classification.
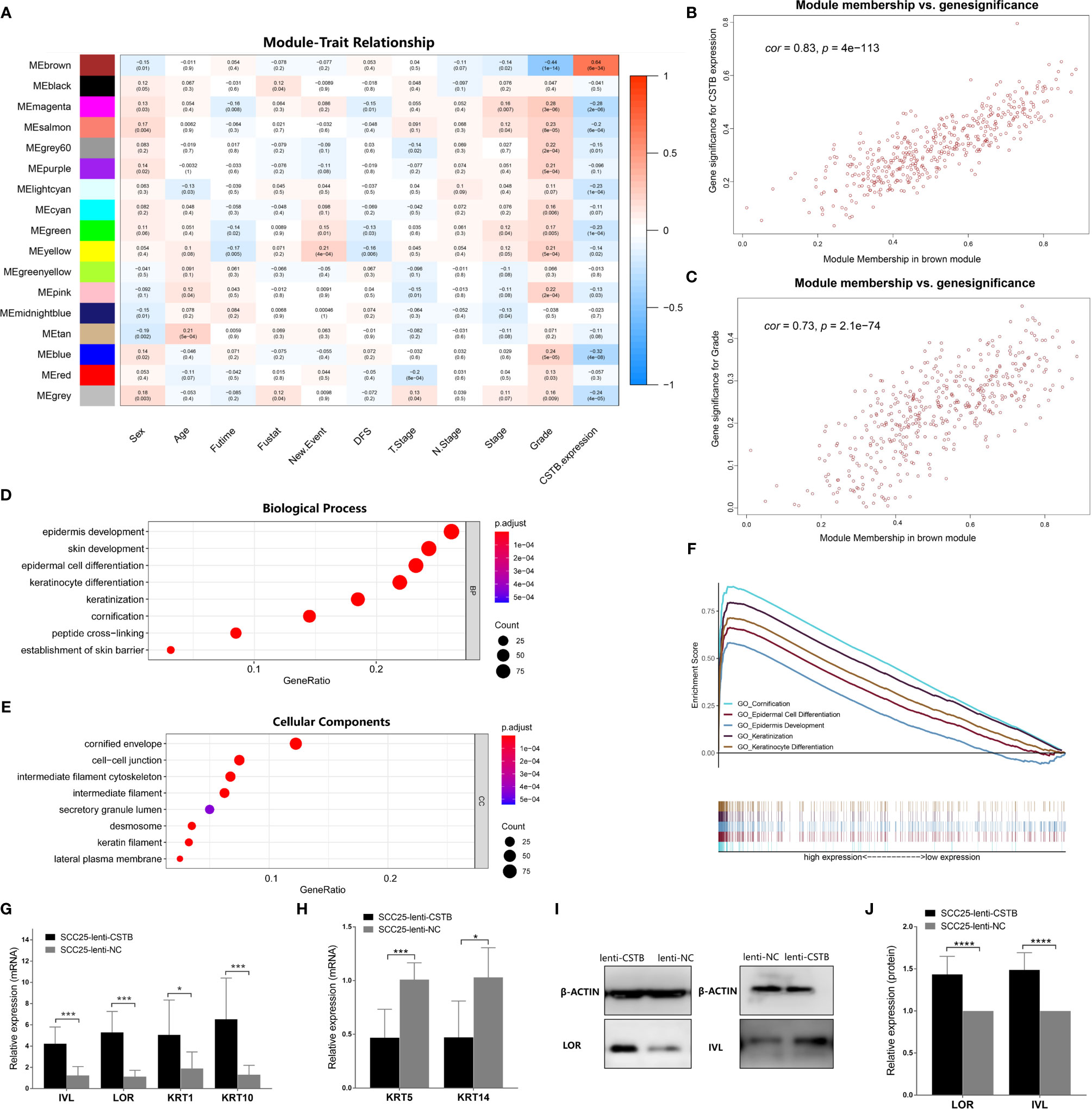
Figure 4 CSTB may regulate epithelial cell differentiation and keratinization in OSCC. (A) Heatmap of the correlation between module eigengenes (vertical axis) and clinical features (horizontal axis). The correlation coefficient (upper row) and p-value (lower row) are presented in each block. The color of each block represents the correlation coefficient according to the legend axis on the right. (B) Scatter diagram for the correlation between module membership and the gene significance of CSTB expression in the brown module. Cor, correlation coefficient; p, p-value. (C) Scatter diagram for the correlation between module membership and gene significance of grade classification in the brown module. Cor, correlation coefficient; p, p-value. (D, E) GO enrichment analysis of the brown module genes. (F) Squamous epithelial keratinization- and differentiation-related pathways with statistically significant differences in GSEA using KEGG enrichment analysis. (G, H) Relative mRNA expression of cell differentiation and keratinization-related markers in OSCC cell lines after lentivirus transfection. (I, J) Protein expression of cell differentiation and keratinization-related markers in OSCC cell lines after lentivirus transfection. Error bars represent the standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
CSTB Regulates Neoplastic Epithelial Differentiation and Keratinization in OSCC
Given the strong association of the brown module with the two traits (i.e., CSTB expression and grade classification), it was regarded as the target module for further GO enrichment analysis (Supplementary Data Sheet 2). The top 10 enriched biological processes (BPs) mainly included pathways associated with the epithelial proliferation/differentiation program, such as epidermal development, epidermal cell differentiation, and keratinocyte differentiation (Figure 4D). The top 10 enriched cellular components (CCs) consisted of cellular structures associated with epithelial proliferation/differentiation, including cornified envelopes, intermediate filament cytoskeleton, intermediate filament, etc. (Figure 4E). Moreover, GSEA was conducted as a targeted method to verify whether the pathways related to epithelial differentiation and keratinization showed differences between the high and low Cstb expression groups. The results showed that the high Cstb expression group activated more epithelial differentiation and keratinization processes (e.g., cornification, epidermal cell differentiation epidemics development, etc.) (Figure 4F).
Finally, cell differentiation/keratinization-related markers (i.e., IVL, LOR, KRT1, KRT5, KRT10, and KRT14) were validated at the protein and gene levels in OSCC models overexpressing CSTB (Figures 4G–J). It was found that upregulation of CSTB promoted the expression of these markers, reflecting a tendency for differentiation and keratinization. In summary, CSTB might regulate the epithelial proliferation/differentiation program in OSCC.
RNA Sequencing Derived Pathways/Genes of Interest Associated With CSTB in OSCC
In addition to keratinization- and differentiation-related pathways, other pathways of interest that CSTB may participate in were also identified. A comparison was made between the GSEA enrichment results of RNA-seq data (from the OSCC models overexpressing CSTB) and existing public database data (Supplementary Data Sheets 3–4). Three gene sets (i.e., apical junction, G2/M checkpoint and mitotic spindle) and six pathways (i.e., glycosaminoglycan degradation, homologous recombination, mismatch repair, NOTCH signaling pathway, nucleotide excision repair, and steroid biosynthesis) were enriched in the data from both sources (Figures 5A, C–F). These nine pathways/gene sets were named as shared pathways/gene sets. In the GSEA results for the cell line RNA-seq data, the core enrichment genes in the above shared pathways/gene sets were used to compare with 256 DEGs in order to obtain crossover genes (Table 3, No. 1–12), suggesting that overexpression of CSTB may be involved in the above shared pathways by regulating these 12 crossover genes.
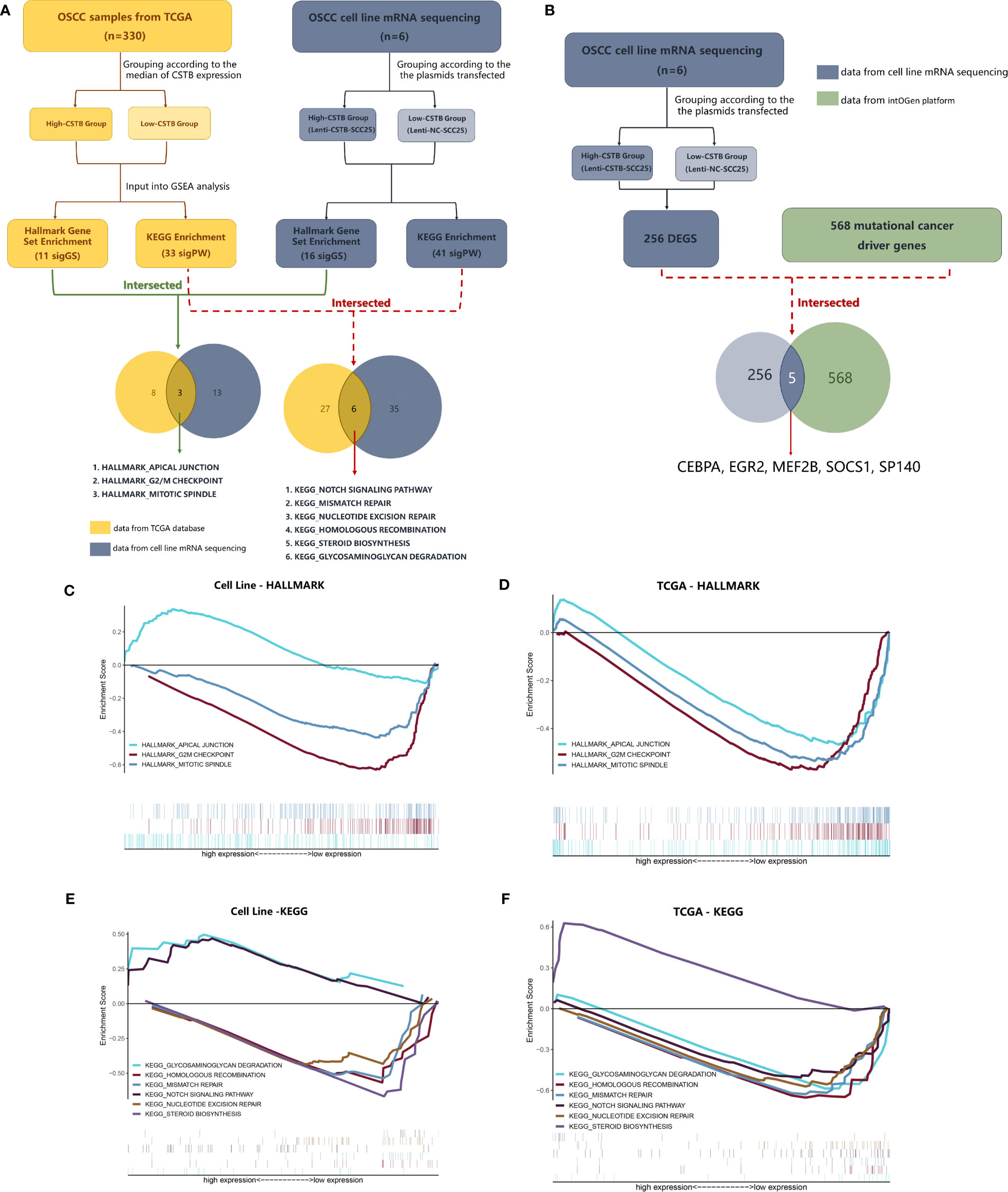
Figure 5 CSTB may participate in OSCC through other tumor-related pathways. (A) The process and results of GSEA based on data from the TCGA database/cell lines mRNA sequencing. (B) Comparison between mutational cancer driver genes and DEGs obtained from cell line mRNA sequencing. (C, D) GSEA using Hallmark gene sets. Gene sets with statistically significant differences based on data from the TCGA database/cell line mRNA sequencing. (E, F) GSEA using KEGG enrichment analysis. Pathways with both statistically significant differences based on data from the TCGA database/cell line mRNA sequencing.
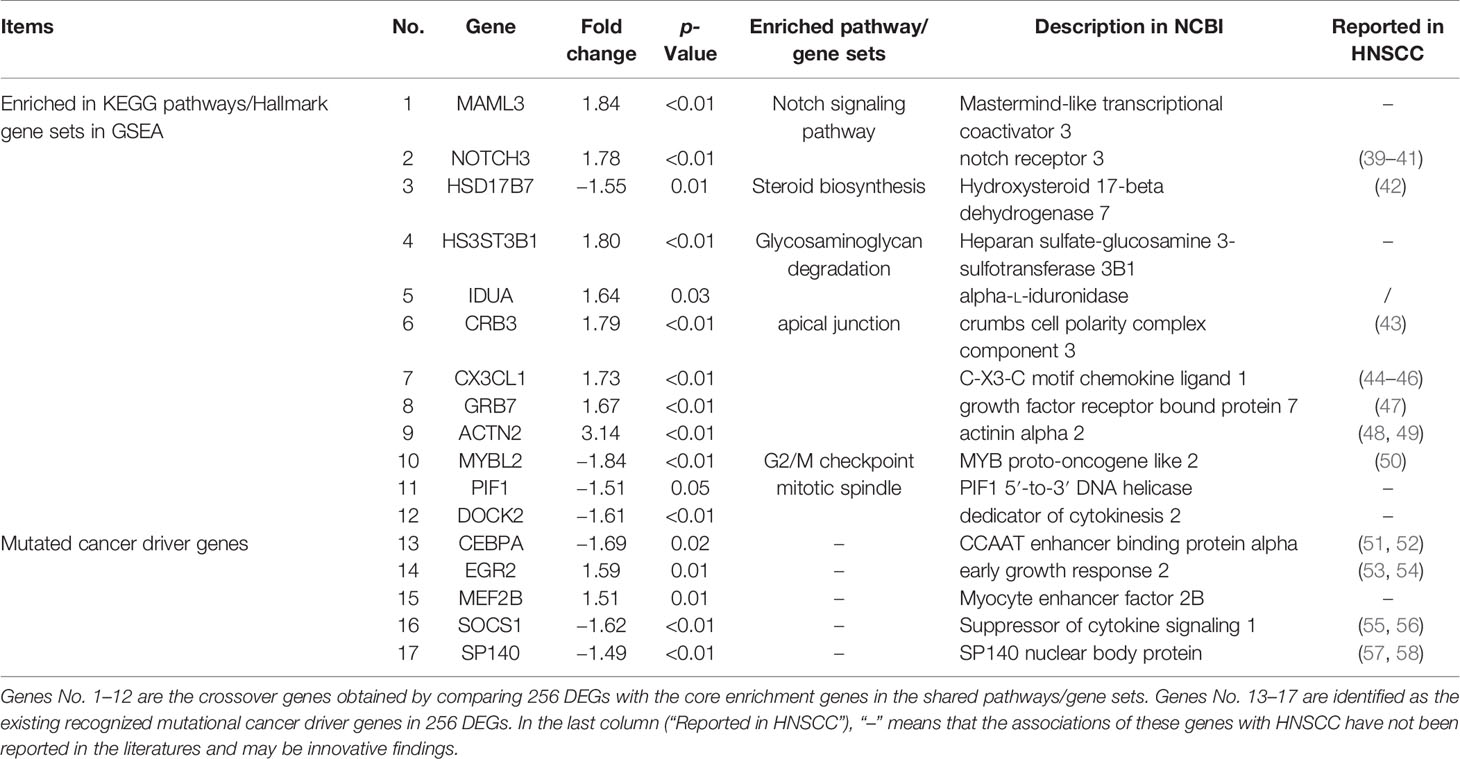
Table 3 Highlighted genes out of 256 differentially expressed genes.
In addition to exploration at the pathway level, RNA-seq data were used for comparisons with the existing recognized mutational cancer driver genes. Among the 256 DEGs, five genes (i.e., CEBPA, EGR2, MEF2B, SOCS1, and SP140) were recognized as mutated cancer driver genes (Figure 5B). Overexpression of CSTB can upregulate EGR2 and MEF2B while downregulating CEBPA, SOCS1, and SP140 in OSCC cell lines (Table 3, No. 13–17).
Discussion
The roles of CSTB, a cysteine cathepsin inhibitor associated with various cancers, have been poorly understood, and few studies have focused on its association with HNSCC. The present study focused on OSCC and explored the regulatory roles of CSTB in vivo and in vitro with both experimental and bioinformatic data. The findings supported our hypothesis and showed that CSTB was downregulated in OSCC compared with normal tissues, and this downregulation was related to worse clinical outcomes or signs of tumor malignancy. The promoting role of CSTB in the OSCC proliferation-differentiation program was unprecedentedly proposed. In addition, some relevant mechanisms by which CSTB might participate in OSCC were suggested at both the pathway and gene levels.
In OSCC, we found that CSTB was downregulated both in vivo and in vitro, both at the protein and gene levels. CSTB has been reported to have abnormal expression in various types of tumors, in which the variation trend of CSTB was inconsistent between different tumors. For instance, CSTB seems to be upregulated in HCC (25–28), epithelial ovarian tumors (29, 30), and breast cancer (31), but downregulated in ESCC (32, 59) and laryngeal squamous cell carcinoma (33) (another certain kind of HNSCC). To systematically compare the expression of CSTB in common tumor types, the GEPIA database (60) was used to show the difference in CSTB expression between the tumor and normal groups (Figure S4): (1). CSTB was downregulated in HNSCC, which was consistent with the findings of our study and a previous study on HNSCC (i.e., laryngeal squamous cell carcinoma) (33) (2). CSTB was upregulated in most carcinomas (i.e., malignant tumors of epithelial origin), except for HNSCC and ESCC. This radically different trend of expression may be partly explained by the different histological origins of these epithelial-derived tumors. Among these carcinomas, HNSCC and ESCC share a common histological origin (i.e., stratified squamous epithelium), while other carcinomas originate from simple columnar epithelium (e.g., cholangiocarcinoma, colon adenocarcinoma, cervical squamous cell carcinoma, and uterine corpus endometrial carcinoma) or glandular epithelium (e.g., pancreatic adenocarcinoma, rectum adenocarcinoma, and stomach adenocarcinoma). In addition, the pleiotropic roles of CSTB in tumors may also contribute to this inconsistent trend. For instance, knockdown of CSTB in an epithelial ovarian cancer cell line inhibited cell proliferation and promoted apoptosis (29), whereas the opposite result was found in a gastric cancer cell line (61) (3). The same expression trend of CSTB in HNSCC and ESCC could be attributed to the similarity of these two tumors. First, they are both located in the proximal digestive tract and originate from stratified squamous epithelium, sharing some similar histological characteristics under physiological and pathological (i.e., appearance of keratin pearls in tumor status) conditions (62). Second, the histological distributions of CSTB in HNSCC and ESCC are also consistent, that is, it is located in the epithelial structure of carcinoma tissues (59) (Figure 2I). Under physiological conditions, CSTB is located in the epithelial layer of both normal oral cavity tissues and esophageal tissues (Figures 2I and S5). Third, similarities in genomic characterization between ESCC and HNSCC may also lead to the same expression trend of CSTB (7, 63–67). There are some groups of gene sets that mutate in both ESCC and HNSCC, which indicates that common mechanisms can regulate the initiation of squamous cell carcinoma (SCC) across different tissues (7, 8, 62, 64, 68–72).
As the expression trend of CSTB differs from tumor to tumor, the relationship between CSTB and prognosis is also inconsistent in different tumor types. Elevated levels of CSTB in tumor samples were related to a higher risk of recurrence and advanced tumor stages in bladder cancer (34) and were linked to a higher risk of tumor metastasis in HCC (27). In our study on OSCC, CSTB expression was inversely related to the risk of aggressive tumor characteristics (i.e., lymph node metastasis, perineural infiltration, and lymphovascular invasion). These characteristics favor tumor recurrence and metastasis (73) and pose great challenges to the tumor-free goal of radical surgery (73–79). Enhanced cancer cell migration and the capacity of stromal infiltration are essential steps of OSCC invasion and metastasis, as is the proliferation of cancer cells in metastatic foci (80). These three abilities of OSCC cells were regulated by CSTB in vitro in the present experiments. Upregulation of CSTB inhibited the proliferation of OSCC cells, regardless of whether proliferation was initiated by a group of cells (i.e., CCK-8 assays) or by a single cell (i.e., colony formation assays). The inhibitory effect of CSTB on cell migration and invasion seems to be more obvious, whether it was simple cell migration movement or invasion requiring the degradation of ECM. These results might partly explain the lower DFS rate of OSCC patients in the low CSTB expression group, given that DFS is an indicator of tumor recurrence and metastasis. In addition, in the early stages of OSCC (i.e., lower clinical stage or pathological grade), the expression changes of CSTB seemed to be more obvious, and the correlation with the corresponding clinical features (stages I–II or grades 1–2) was higher. This result indicated that CSTB might have more dramatic changes in the early stage of disease and might be better at distinguishing and predicting early OSCC. CSTB had no obvious relationship with demographic characteristics, including age and sex, suggesting a relatively stable expression of CSTB in the physiological state. Instead, CSTB tends to be changed specifically under pathological conditions [e.g., cancer (21), viral infection (81–83), and neurodegenerative diseases (84, 85)], which suggests that it is a potential pathological marker. The relationship between CSTB and the prognosis of OSCC in our study was also consistent with previous research reporting that low expression of CSTB in neoplastic islands from the invasive tumor front (ITF) was related to local recurrence in OSCC (35). Other studies of HNSCC/ESCC also reported that a lower concentration of CSTB increased the risks of lymph node metastasis (35, 59) and local tumor recurrence (35) and may thus result in a shorter DFS (35, 86). The different expression patterns (i.e., different expression trends and inconsistent relationships with prognosis) of CSTB in tumors imply that CSTB could be a tumor-specific whistleblower. For instance, when tumoral diseases are speculated to have tissular or humoral changes in CSTB, the types or primary focus of tumors could be predicted according to the change modes of CSTB.
Another major finding was that CSTB was involved in the squamous epithelium proliferation-differentiation program. This finding was supported by several lines of evidence, as follows (1). In WGCNA, the gene modules most associated with CSTB were also closely related to epithelial development, differentiation and keratinization (2). In GSEA, epithelial differentiation and keratinization processes were more active in the high CSTB expression group (3). The expression of CSTB was positively related to the degree of tumor differentiation at the protein level (IHC). In WGCNA based on TCGA data, CSTB and tumor grade classification showed an indirect relationship at the gene level through the bridging role of the brown gene module; that is, the gene module most associated with CSTB expression was also the one most closely related to the phenotype of grade (4). Upregulation of CSTB increased epithelial differentiation/keratinization markers [i.e., IVL (87, 88), LOR (87, 89), and KRT1/10 (90, 91)] and decreased epithelial basal-like markers [i.e., KRT5/14 (90, 91)]. While our study provided a direct evidence on the association between CSTB and squamous epithelium differentiation, previous studies on other diseases have implied a possible link between CSTB and epithelial differentiation. In ESCC, CSTB always exists in the clearly differentiated cells rather than basal-like cells, and the expression of CSTB disappears after malignant transformation of keratinocytes (92). Moreover, in psoriasis, a condition that manifests as uncontrolled keratinocyte proliferation and epithelial hyperplasia (93, 94), the amount of CSTB is increased, which is usually not seen in healthy epidermis (92) (i.e., normal skin). The regulatory mechanisms of CSTB in squamous epithelial keratinization and differentiation programs are not yet clear. There might be at least two possible mechanisms, i.e., by affecting the formation of cornified cell envelopes (CE) or by regulating the NOTCH signaling pathway. First, the terminal differentiation of squamous epithelium is a process accompanied by the formation of CE (95), which is mediated and regulated by TGM1 [transglutaminase-1 (TGase-1)] (96, 97). In our study, the CE was enriched as a cell component in GO analysis in the brown module in WGCNA (Figure 4E) and TGM1 was a significant hub gene in the brown module (module membership = 0.85, module eigengene = 0.65). Given that the brown module was a gene module containing genes coexpressed with CSTB, these results suggested that CSTB might regulate the TGM1-mediated CE formation process and ultimately regulate the terminal differentiation of squamous epithelium. Second, NOTCH is mostly regarded as a tumor suppressor in HNSCC, and loss-of-function mutations in NOTCH1 are common events in HNSCC. The NOTCH signaling pathway plays an important role in the normal functioning of squamous epithelium development and differentiation (7, 62, 67, 98–100), acting as a promoter of keratinocyte differentiation (101, 102). Conditional deletion of NOTCH1 in the mouse epidermis can lead to basal hyperplasia and basal cell carcinoma (103). Our GSEA results based on cell line RNA-seq data were consistent with this downregulation trend of NOTCH signaling. The NOTCH signaling pathway became more active after CSTB overexpression, which was also in line with the differentiation-promoting effect of CSTB that we have observed. However, this alteration in the NOTCH signaling pathway showed an opposite trend (i.e., more active in the low CSTB expression group) based on patient RNA-seq data from TCGA. This could be explained by the contextual and bimodal role of NOTCH in cancers, including HNSCC (64, 67, 104–106). NOTCH can act as an oncogene and tumor suppressor gene in different cell populations within the same tumor (103), which was a possible reason for the difference in NOTCH signals in the data from two sources (TCGA data and cell line RNA seq data). The cell line RNA-seq data came from a single type of tumor cell (OSCC cell line), while the tissue sequencing data of TCGA were composed of a variety of cells including tumor cells, stromal cells, immune cells, etc. Another possible reason for the different NOTCH signals is that the involved NOTCH paralogues and ligands vary in different disease contexts, resulting in different biological outcomes (107). For instance, in GSEA, NOTCH1, and NOTCH3 were enriched in the high CSTB expression group based on cell line RNA-seq data, while NOTCH1 and NOTCH4 were enriched in the low CSTB expression group based on TCGA data (Supplementary Data Sheet 4). Studies have identified a bimodal role for NOTCH3 in HNSCC (39), while MAML3 is essential for the operation of NOTCH signaling (108). In the RNA-seq data, we found that overexpression of CSTB could upregulate the expression of NOTCH3 and MAML3 in vitro (Table 3), suggesting that CSTB may be involved in the NOTCH signaling pathway by regulating these two genes, but additional validation is required.
An interesting phenomenon was found in our research: the content of extracellular/intracellular CSTB showed a consistent change trend in the in vitro experiment (Figures 2M, D). CSTB is regarded as a protease inhibitor that is localized in the intracellular region (18, 23, 24). However, many studies have shown that CSTB and its changes can be detected in the body fluids of cancer patients [e.g., serum (28), ascites (109), and urine (34)] and are related to the clinical characteristics or prognosis. Given the consistency of the anatomical location, saliva as a body fluid may be more representative for reflecting the disease state of OSCC. A study of OSCC emphasized the potential of CSTB-specific peptides in saliva to reflect the status of lymph node metastasis in tumor-bearing patients (35). Although our study found differences in the content of CSTB in normal/OSCC cell culture supernatants, whether CSTB could be used to specifically identify tumor patients from the normal population is worthy of further research. Furthermore, a study showed the presence of CSTB in oral acquired enamel pellicles (110, 111). Future research may consider the changes in CSTB in plaque as a novel research direction in terms of the potential of CSTB as a biomarker, given that plaque could be regarded as a more stable form reflecting the biochemical composition of saliva. Other nontumor studies have identified certain extracellular effects of CSTB. CSTB in mouse synaptosomes can be secreted into cerebral spinal fluid in a depolarization-controlled manner and is involved in synaptic plasticity (84, 112). HIV-infected microglia secret more CSTB, participating in the neurotoxicity induced by cathepsin B (113). These studies suggest that pathological signals (tumor, virus, and abnormal nerve signals) may cause CSTB to translocate and participate in extracellular biological processes, while its specific extracellular role in tumors needs to be clarified by more tracer studies.
Other roles of CSTB in the context of OSCC were explored at both the pathway and gene levels in our study, which might provide a comprehensive perspective for further elucidating its mechanism. First, pathways related to the DNA damage repair mechanism (G2/M checkpoint, mitotic spindle, homologous recombination, mismatch repair, and nucleotide excision repair) were more active in the low CSTB expression group. This finding indicated that downregulation of CSTB might act as a predisposing factor of DNA damage in OSCC, thereby activating the pathways related to DNA damage repair pathways. Many studies have emphasized the protective effect of CSTB against oxidative stress (31, 114, 115). CSTB deficiency increases the sensitivity of cells to oxidative stress in cerebellar granule neurons (114) or breast cancer primary cells (31). Given that oxidative stress is a common factor that causes DNA damage (116, 117), we speculate that the effect of CSTB on the DNA damage repair mechanism in OSCC might be related to the loss of the protective role of CSTB in oxidative stress. Second, the identification of all the mutated genes capable of driving tumors is a landmark achievement towards tumor research (38). To explore the correlation between CSTB and the known cancer driver genes, we compared the DEGs obtained by cell line mRNA sequencing with the cancer driver gene set. As a result, five genes, CEBPA, EGR2, MEF2B, SOCS1, and SP140, were identified. CEBPA was reported to be mutated in HNSCC (51) and to play an important role in regulating the epidermal differentiation program (51, 52, 118–121). SP140 is an immune-related gene mutated in OSCC cell lines (57, 58, 122, 123). SOCS1, a negative regulator of cytokine signaling pathways (55, 124–126), was reported to regulate epigenetic modification in head and neck cancer (56, 125, 127). Cumulative studies have suggested the role of CSTB in immunomodulation, especially the relationship between CSTB and macrophages (128, 129). The lack of CSTB could transform macrophages to a proinflammatory phenotype by regulating related cytokines such as IL-10 (128). Meanwhile, some immune-related pathways (e.g., antigen processing and presentation and primary immunodeficiency) were enriched by GSEA based on cell RNA-seq data in our study (Supplementary Data Sheet 3). Both EGR2 and MEF2B are regulators of cell transcription (130, 131), and the former was reported to be involved in the epithelial-mesenchymal transition (EMT) pathway (131, 132), while the latter was differentially expressed between primary tumors and nodal metastasis tumor in HNSCC (53). Our experiments did not identify genes that are mutated due to changes in the expression level of CSTB, although changes in the expression level of the transcriptome are a common consequence of gene mutations. However, our aim was to interpret the possible role of CSTB in cancer at the genetic level, and the results still provide innovative insights while exploring the functions of CSTB in OSCC.
There were some limitations in our study. First, the correlations between Cstb and clinical characteristics found through data mining methods have not been confirmed by additional independent clinical data. The findings should be validated at the protein levels in tissues and body fluid (e.g., saliva and blood). Second, the regulatory role of CSTB in the epithelium differentiation phenotype was mainly recognized through bioinformatic analysis and in vitro experiments and needs in vivo confirmation in future studies. Third, the regulated pathways/genes identified by GSEA provided a relatively comprehensive perspective in clarifying the role of CSTB in OSCC. However, the relevant pathways/genes lacked targeted validation, so they should be interpreted with caution. Fourth, the sample size of OSCC tissue for IHC was relatively small, which was why we used Fisher’s exact test. In the subgroup analysis, the subgroup satisfying both high CSTB expression and moderate/poor differentiation included only one sample, which may be partly due to our relatively small overall sample size. Given the relationship of CSTB and differentiation in our study on OSCC, samples that meet both the phenotypes of high CSTB expression and low differentiation were relatively rare, which may be another reason for the small sample size of this subgroup. However, this state [i.e., high CSTB expression and poor tissue differentiation) is not uncommon in some other tumor types (e.g., HCC (27) and bladder cancer (34)], where the degree of tumor differentiation and CSTB expression show a negative correlation. This finding may further indicate that, compared with other tumor types, CSTB has a special additional role in the differentiation process of OSCC, but it is not the only factor determining the degree of tumor differentiation.
In conclusion, our study identified the modulatory role of CSTB in the malignant characteristics of OSCC for the first time and proposed some new relevant mechanisms, namely, CSTB may participate in OSCC by promoting the differentiation process of squamous epithelium. Our research provides a new perspective for interpreting the role of CSTB in tumors, especially in SCC. The specific role of CSTB in OSCC and its fine regulation of the squamous epithelial differentiation program still need to be explored in-depth by functional experiments and mechanistic research.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
Studies involving human participants were reviewed and approved by the Ethics Committee of the Stomatology Hospital of Guangzhou Medical University (LCYJ2021013). The patients/participants provided their written informed consent to participate in this study. No potentially identifiable human images or data are presented in this study.
Author Contributions
TX contributed to the conception and design of the study. TX and LY performed the experiments. TX, XZ, XW, and LY performed the statistical analysis. TX wrote the first draft of the manuscript. TX, TY, and GL critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Health Committee of Guangdong Province (Grant No. B2020209), Guangzhou Medical University (Grant No. C195015024), and the National Natural Science Foundation of China (Grant No. 81700985).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.707066/full#supplementary-material
References
1. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and Neck Squamous Cell Carcinoma. Nat Rev Dis Primers (2020) 6:92. doi: 10.1038/s41572-020-00224-3
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
3. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods. Int J Cancer (2019) 144:1941–53. doi: 10.1002/ijc.31937
4. Chi AC, Day TA, Neville BW. Oral Cavity and Oropharyngeal Squamous Cell Carcinoma–An Update. CA Cancer J Clin (2015) 65:401–21. doi: 10.3322/caac.21293
5. National Cancer Institute (NCI) Website. Available at: https://www.cancer.gov.
6. Osazuwa-Peters N, Simpson MC, Zhao L, Boakye EA, Olomukoro SI, Deshields T, et al. Suicide Risk Among Cancer Survivors: Head and Neck Versus Other Cancers. Cancer (2018) 124:4072–9. doi: 10.1002/cncr.31675
7. Dotto GP, Rustgi AK. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell (2016) 29:622–37. doi: 10.1016/j.ccell.2016.04.004
8. Leemans CR, Snijders PJF, Brakenhoff RH. The Molecular Landscape of Head and Neck Cancer. Nat Rev Cancer (2018) 18:269–82. doi: 10.1038/nrc.2018.11
9. Choi S, Myers JN. Molecular Pathogenesis of Oral Squamous Cell Carcinoma: Implications for Therapy. J Dent Res (2008) 87:14–32. doi: 10.1177/154405910808700104
10. Thomson PJ. Perspectives on Oral Squamous Cell Carcinoma Prevention—Proliferation, Position, Progression and Prediction. J Oral Pathol Med (2018) 47:803–7. doi: 10.1111/jop.12733
11. Pai SI, Westra WH. Molecular Pathology of Head and Neck Cancer: Implications for Diagnosis, Prognosis, and Treatment. Annu Rev Pathol (2009) 4:49–70. doi: 10.1146/annurev.pathol.4.110807.092158
12. Umelo IA, Costanza B, Castronovo V. Innovative Methods for Biomarker Discovery in the Evaluation and Development of Cancer Precision Therapies. Cancer Metastasis Rev (2018) 37:125–45. doi: 10.1007/s10555-017-9710-0
13. Ji K, Mayernik L, Moin K, Sloane BF. Acidosis and Proteolysis in the Tumor Microenvironment. Cancer Metastasis Rev (2019) 38:103–12. doi: 10.1007/s10555-019-09796-3
14. Osuala KO, Ji K, Mattingly RR, Sloane BF. Breast Cancer: Proteolysis and Migration. Adv Exp Med Biol (2019) 1152:401–11. doi: 10.1007/978-3-030-20301-6_21
15. Ni J, Zhang L. Cancer Cachexia: Definition, Staging, and Emerging Treatments. Cancer Manag Res (2020) 12:5597–605. doi: 10.2147/cmar.S261585
16. Li X, Song Y. Proteolysis-Targeting Chimera (PROTAC) for Targeted Protein Degradation and Cancer Therapy. J Hematol Oncol (2020) 13:50. doi: 10.1186/s13045-020-00885-3
17. Rudzińska M, Parodi A, Soond SM, Vinarov AZ, Korolev DO, Morozov AO, et al. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int J Mol Sci (2019) 20(14):3602. doi: 10.3390/ijms20143602
18. Pogorzelska A, Żołnowska B, Bartoszewski R. Cysteine Cathepsins as a Prospective Target for Anticancer Therapies-Current Progress and Prospects. Biochimie (2018) 151:85–106. doi: 10.1016/j.biochi.2018.05.023
19. Shamsi A, Bano B. Journey of Cystatins From Being Mere Thiol Protease Inhibitors to at Heart of Many Pathological Conditions. Int J Biol Macromol (2017) 102:674–93. doi: 10.1016/j.ijbiomac.2017.04.071
20. Olson OC, Joyce JA. Cysteine Cathepsin Proteases: Regulators of Cancer Progression and Therapeutic Response. Nat Rev Cancer (2015) 15:712–29. doi: 10.1038/nrc4027
21. Breznik B, Mitrović A, Lah TT, Kos J. Cystatins in Cancer Progression: More Than Just Cathepsin Inhibitors. Biochimie (2019) 166:233–50. doi: 10.1016/j.biochi.2019.05.002
22. Alakurtti K, Weber E, Rinne R, Theil G, de Haan GJ, Lindhout D, et al. Loss of Lysosomal Association of Cystatin B Proteins Representing Progressive Myoclonus Epilepsy, EPM1, Mutations. Europ J Hum Genet (2005) 13:208–15. doi: 10.1038/sj.ejhg.5201300
23. Turk V, Stoka V, Turk D. Cystatins: Biochemical and Structural Properties, and Medical Relevance. Front Biosci (2008) 13:5406–20. doi: 10.2741/3089
24. Kos J, Werle B, Lah T, Brunner N. Cysteine Proteinases and Their Inhibitors in Extracellular Fluids: Markers for Diagnosis and Prognosis in Cancer. Int J Biol Markers (2000) 15:84–9. doi: 10.1177/172460080001500116
25. Zhou X, Wang X, Huang K, Liao X, Yang C, Yu T, et al. Investigation of the Clinical Significance and Prospective Molecular Mechanisms of Cystatin Genes in Patients With Hepatitis B Virus−Related Hepatocellular Carcinoma. Oncol Rep (2019) 42:189–201. doi: 10.3892/or.2019.7154
26. Liu GM, Zeng HD, Zhang CY, Xu JW. Identification of a Six-Gene Signature Predicting Overall Survival for Hepatocellular Carcinoma. Cancer Cell Int (2019) 19:138. doi: 10.1186/s12935-019-0858-2
27. Lin YY, Chen ZW, Lin ZP, Lin LB, Yang XM, Xu LY, et al. Tissue Levels of Stefin A and Stefin B in Hepatocellular Carcinoma. Anat Rec (Hoboken) (2016) 299:428–38. doi: 10.1002/ar.23311
28. Lee MJ, Yu GR, Park SH, Cho BH, Ahn JS, Park HJ, et al. Identification of Cystatin B as a Potential Serum Marker in Hepatocellular Carcinoma. Clin Cancer Res (2008) 14:1080–9. doi: 10.1158/1078-0432.Ccr-07-1615
29. Guan W, Wang X, Lin Q, Zhang J, Ren W, Xu G. Transforming Growth Factor−β/Mir−143−3p/Cystatin B Axis Is a Therapeutic Target in Human Ovarian Cancer. Int J Oncol (2019) 55:267–76. doi: 10.3892/ijo.2019.4815
30. Wang X, Gui L, Zhang Y, Zhang J, Shi J, Xu G. Cystatin B Is a Progression Marker of Human Epithelial Ovarian Tumors Mediated by the TGF-β Signaling Pathway. Int J Oncol (2014) 44:1099–106. doi: 10.3892/ijo.2014.2261
31. Butinar M, Prebanda MT, Rajković J, Jerič B, Stoka V, Peters C, et al. Stefin B Deficiency Reduces Tumor Growth via Sensitization of Tumor Cells to Oxidative Stress in a Breast Cancer Model. Oncogene (2014) 33:3392–400. doi: 10.1038/onc.2013.314
32. Zhang J, Wang K, Zhang J, Liu SS, Dai L, Zhang JY. Using Proteomic Approach to Identify Tumor-Associated Proteins as Biomarkers in Human Esophageal Squamous Cell Carcinoma. J Proteome Res (2011) 10(6):2863–72. doi: 10.1021/pr200141c
33. Colombo J, Fachel AA, De Freitas Calmon M, Cury PM, Fukuyama EE, Tajara EH, et al. Gene Expression Profiling Reveals Molecular Marker Candidates of Laryngeal Squamous Cell Carcinoma. Oncol Rep (2009) 21:649–63.
34. Feldman AS, Banyard J, Wu CL, McDougal WS, Zetter BR. Cystatin B as a Tissue and Urinary Biomarker of Bladder Cancer Recurrence and Disease Progression. Clin Cancer Res (2009) 15:1024–31. doi: 10.1158/1078-0432.Ccr-08-1143
35. Carnielli CM, Macedo CCS, De Rossi T, Granato DC, Rivera C, Domingues RR, et al. Combining Discovery and Targeted Proteomics Reveals a Prognostic Signature in Oral Cancer. Nat Commun (2018) 9:3598. doi: 10.1038/s41467-018-05696-2
36. EI-Naggar AK, Chan JKC, G randis JR, Takata T, Slootweg PJ. WHO Classification of Head and Neck Tumours. 4th edition. IA RC: Lyon (2017).
37. Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (2001) 25:402–8. doi: 10.1006/meth.2001.1262
38. Martinez-Jimenez F, Muinos F, Sentis I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, et al. A Compendium of Mutational Cancer Driver Genes. Nat Rev Cancer (2020) 20:555–72. doi: 10.1038/s41568-020-0290-x
39. Fukusumi T, Califano JA. The NOTCH Pathway in Head and Neck Squamous Cell Carcinoma. J Dent Res (2018) 97(6):645–53. doi: 10.1177/0022034518760297
40. Shah PA, Huang C, Li Q, Kazi SA, Byers LA, Wang J, et al. NOTCH1 Signaling in Head and Neck Squamous Cell Carcinoma. Cells (2020) 9(12):2677. doi: 10.3390/cells9122677
41. Zhao YY, Yu GT, Xiao T, Hu J. The Notch Signaling Pathway in Head and Neck Squamous Cell Carcinoma: A Meta-Analysis. Adv Clin Exp Med (2017) 26(5):881–7. doi: 10.17219/acem/64000
42. Xu X, Tassone B, Ostano P, Katarkar A, Proust T, Joseph JM, et al. HSD17B7 Gene in Self-Renewal and Oncogenicity of Keratinocytes From Black Versus White Populations. EMBO Mol Med (2021) 13:e14133. doi: 10.15252/emmm.202114133
43. Tilston-Lünel AM, Haley KE, Schlecht NF, Wang Y, Chatterton ALD, Moleirinho S, et al. Crumbs 3b Promotes Tight Junctions in an Ezrin-Dependent Manner in Mammalian Cells. J Mol Cell Biol (2016) 8:439–55. doi: 10.1093/jmcb/mjw020
44. Son SH, Park J, Jung MJ, Lee SK, Kim H, Kim KR, et al. Transforming Growth Factor-β-Regulated Fractalkine as a Marker of Erosive Bone Invasion in Oral Squamous Cell Carcinoma. Eur J Oral Sci (2021) 129:e12750. doi: 10.1111/eos.12750
45. Merhi M, Raza A, Inchakalody VP, Nashwan AJJ, Allahverdi N, Krishnankutty R, et al. Squamous Cell Carcinomas of the Head and Neck Cancer Response to Programmed Cell Death Protein-1 Targeting and Differential Expression of Immunological Markers: A Case Report. Front Immunol (2018) 9:1769. doi: 10.3389/fimmu.2018.01769
46. Gyanchandani R, Ortega Alves MV, Myers JN, Kim S. A Proangiogenic Signature Is Revealed in FGF-Mediated Bevacizumab-Resistant Head and Neck Squamous Cell Carcinoma. Mol Cancer Res (2013) 11:1585–96. doi: 10.1158/1541-7786.Mcr-13-0358
47. Khanal S, Strickley JD, Ha T, Demehri S, Ghim SJ, Jenson AB, et al. Human Papillomavirus-Positivity Is Associated With EREG Down-Regulation and Promoter Hypermethylation in Head and Neck Squamous Cell Carcinoma. Exp Mol Pathol (2020) 117:104549. doi: 10.1016/j.yexmp.2020.104549
48. Xu Y, Xu F, Lv Y, Wang S, Li J, Zhou C, et al. A ceRNA-Associated Risk Model Predicts the Poor Prognosis for Head and Neck Squamous Cell Carcinoma Patients. Sci Rep (2021) 11:6374. doi: 10.1038/s41598-021-86048-x
49. Shaikh I, Ansari A, Ayachit G, Gandhi M, Sharma P, Bhairappanavar S, et al. Differential Gene Expression Analysis of HNSCC Tumors Deciphered Tobacco Dependent and Independent Molecular Signatures. Oncotarget (2019) 10:6168–83. doi: 10.18632/oncotarget.27249
50. Wang L, Jia Y, Jiang Z, Gao W, Wang B. FSCN1 Is Upregulated by SNAI2 and Promotes Epithelial to Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma. Cell Biol Int (2017) 41:833–41. doi: 10.1002/cbin.10786
51. Gaykalova DA, Mambo E, Choudhary A, Houghton J, Buddavarapu K, Sanford T, et al. Novel Insight Into Mutational Landscape of Head and Neck Squamous Cell Carcinoma. PloS One (2014) 9:e93102. doi: 10.1371/journal.pone.0093102
52. Bennett KL, Hackanson B, Smith LT, Morrison CD, Lang JC, Schuller DE, et al. Tumor Suppressor Activity of CCAAT/enhancer Binding Protein Alpha Is Epigenetically Down-Regulated in Head and Neck Squamous Cell Carcinoma. Cancer Res (2007) 67:4657–64. doi: 10.1158/0008-5472.Can-06-4793
53. Liu CJ, Liu TY, Kuo LT, Cheng HW, Chu TH, Chang KW, et al. Differential Gene Expression Signature Between Primary and Metastatic Head and Neck Squamous Cell Carcinoma. J Pathol (2008) 214:489–97. doi: 10.1002/path.2306
54. Liu CJ, Lin SC, Chen YJ, Chang KM, Chang KW. Array-Comparative Genomic Hybridization to Detect Genomewide Changes in Microdissected Primary and Metastatic Oral Squamous Cell Carcinomas. Mol Carcinog (2006) 45:721–31. doi: 10.1002/mc.20213
55. Yao Y, Yan Z, Lian S, Wei L, Zhou C, Feng D, et al. Prognostic Value of Novel Immune-Related Genomic Biomarkers Identified in Head and Neck Squamous Cell Carcinoma. J Immunother Cancer (2020) 8(2):e000444. doi: 10.1136/jitc-2019-000444
56. Lee TL, Yeh J, Van Waes C, Chen Z. Epigenetic Modification of SOCS-1 Differentially Regulates STAT3 Activation in Response to Interleukin-6 Receptor and Epidermal Growth Factor Receptor Signaling Through JAK and/or MEK in Head and Neck Squamous Cell Carcinomas. Mol Cancer Ther (2006) 5:8–19. doi: 10.1158/1535-7163.Mct-05-0069
57. Mo BY, Li GS, Huang SN, Wei ZX, Su YS, Dai WB, et al. Laryngeal Squamous Cell Carcinoma: Potential Molecular Mechanism and Prognostic Signature Based on Immune-Related Genes. Med Sci Monit (2020) 26:e928185. doi: 10.12659/MSM.928185
58. Song Y, Pan Y, Liu J. The Relevance Between the Immune Response-Related Gene Module and Clinical Traits in Head and Neck Squamous Cell Carcinoma. Cancer Manag Res (2019) 11:7455–72. doi: 10.2147/cmar.S201177
59. Shiraishi T, Mori M, Tanaka S, Sugimachi K, Akiyoshi T. Identification of Cystatin B in Human Esophageal Carcinoma, Using Differential Displays in Which the Gene Expression Is Related to Lymph-Node Metastasis. Int J Cancer (1998) 79:175–8. doi: 10.1002/(sici)1097-0215(19980417)79:2<175::aid-ijc13>3.0.co;2-9
60. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res (2017) 45:W98–W102. doi: 10.1093/nar/gkx247
61. Zhang J, Shi Z, Huang J, Zou X. CSTB Downregulation Promotes Cell Proliferation and Migration and Suppresses Apoptosis in Gastric Cancer SGC-7901 Cell Line. Oncol Res (2016) 24:487–94. doi: 10.3727/096504016x14685034103752
62. Sanchez-Danes A, Blanpain C. Deciphering the Cells of Origin of Squamous Cell Carcinomas. Nat Rev Cancer (2018) 18:549–61. doi: 10.1038/s41568-018-0024-5
63. Grandis JR. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Clin Cancer Res (2017) 23. doi: 10.1158/1557-3265.AACRAHNS17-IA09
64. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome Sequencing of Head and Neck Squamous Cell Carcinoma Reveals Inactivating Mutations in NOTCH1. Science (2011) 333:1154–7. doi: 10.1126/science.1206923
65. Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, et al. Genetic Landscape of Esophageal Squamous Cell Carcinoma. Nat Genet (2014) 46:1097–102. doi: 10.1038/ng.3076
66. Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, et al. Genomic and Molecular Characterization of Esophageal Squamous Cell Carcinoma. Nat Genet (2014) 46:467–73. doi: 10.1038/ng.2935
67. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science (2011) 333(6046):1157–60. doi: 10.1126/science.1208130
68. Cancer Genome Atlas Research Network, Analysis Working Group: Asan University, BC Cancer Agency, Brigham and Women’s Hospital, Broad Institute, Brown University. Integrated Genomic Characterization of Oesophageal Carcinoma. Nature (2017) 541(7636):169–75. doi: 10.1038/nature20805
69. Cancer Genome Atlas Network, Lawrence MS, Sougnez C, Lichtenstein L, Cibulskis K, Lander E, et al. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature (2015) 517:576–82. doi: 10.1038/nature14129
70. Chen YP, Wang YQ, Lv JW, Li YQ, Chua MLK, Le QT, et al. Identification and Validation of Novel Microenvironment-Based Immune Molecular Subgroups of Head and Neck Squamous Cell Carcinoma: Implications for Immunotherapy. Ann Oncol (2019) 30:68–75. doi: 10.1093/annonc/mdy470
71. Song Y, Li L, Ou Y, Gao Z, Li E, Li X, et al. Identification of Genomic Alterations in Oesophageal Squamous Cell Cancer. Nature (2014) 509:91–5. doi: 10.1038/nature13176
72. Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, et al. Integrative Genomic Characterization of Oral Squamous Cell Carcinoma Identifies Frequent Somatic Drivers. Cancer Discovery (2013) 3:770–81. doi: 10.1158/2159-8290.CD-12-0537
73. Bakst RL, Glastonbury CM, Parvathaneni U, Katabi N, Hu KS, Yom SS. Perineural Invasion and Perineural Tumor Spread in Head and Neck Cancer. Perineural Invasion Perineural Tumor Spread Head (2019) 103:1109–24. doi: 10.1016/j.ijrobp.2018.12.009
74. Mupparapu M, Shanti RM. Evaluation and Staging of Oral Cancer. Dent Clin North Am (2018) 62:47–58. doi: 10.1016/j.cden.2017.08.003
75. Obinu A, Gavini E, Rassu G, Maestri M, Bonferoni MC, Giunchedi P. Lymph Node Metastases: Importance of Detection and Treatment Strategies. Expert Opin Drug Deliv (2018) 15:459–67. doi: 10.1080/17425247.2018.1446937
76. Schmitd LB, Scanlon CS, D'Silva NJ. Perineural Invasion in Head and Neck Cancer. J Dent Res (2018) 97:742–50. doi: 10.1177/0022034518756297
77. Binmadi NO, Basile JR. Perineural Invasion in Oral Squamous Cell Carcinoma: A Discussion of Significance and Review of the Literature. Oral Oncol (2011) 47:1005–10. doi: 10.1016/j.oraloncology.2011.08.002
78. Chinn SB, Spector ME, Bellile EL, McHugh JB, Gernon TJ, Bradford CR, et al. Impact of Perineural Invasion in the Pathologically N0 Neck in Oral Cavity Squamous Cell Carcinoma. Otolaryngol Head Neck Surg (2013) 149:893–9. doi: 10.1177/0194599813506867
79. Zanoni DK, Montero PH, Migliacci JC, Shah JP, Wong RJ, Ganly I, et al. Survival Outcomes After Treatment of Cancer of the Oral Cavity (1985-2015). Oral Oncol (2019) 90:115–21. doi: 10.1016/j.oraloncology.2019.02.001
80. Tian C, Öhlund D, Rickelt S, Lidström T, Huang Y, Hao L, et al. Cancer Cell-Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Res (2020) 80:1461–74. doi: 10.1158/0008-5472.Can-19-2578
81. Luciano-Montalvo C, Meléndez LM. Cystatin B Associates With Signal Transducer and Activator of Transcription 1 in Monocyte-Derived and Placental Macrophages. Placenta (2009) 30:464–7. doi: 10.1016/j.placenta.2009.03.003
82. Rivera-Rivera L, Perez-Laspiur J, Colón K, Meléndez LM. Inhibition of Interferon Response by Cystatin B: Implication in HIV Replication of Macrophage Reservoirs. J Neurovirol (2012) 18:20–9. doi: 10.1007/s13365-011-0061-2
83. Luciano-Montalvo C, Ciborowski P, Duan F, Gendelman HE, Meléndez LM. Proteomic Analyses Associate Cystatin B With Restricted HIV-1 Replication in Placental Macrophages. Placenta (2008) 29:1016–23. doi: 10.1016/j.placenta.2008.09.005
84. Di Matteo F, Pipicelli F, Kyrousi C, Tovecci I, Penna E, Crispino M, et al. Cystatin B Is Essential for Proliferation and Interneuron Migration in Individuals With EPM1 Epilepsy. EMBO Mol Med (2020) 12:e11419. doi: 10.15252/emmm.201911419
85. Genton P. Unverricht-Lundborg Disease (EPM1). Epilepsia (2010) 511(Suppl):37–9. doi: 10.1111/j.1528-1167.2009.02441.x
86. Strojan P, Anicin A, Svetic B, Pohar M, Smid L, Kos J. Stefin a and Stefin B: Markers for Prognosis in Operable Squamous Cell Carcinoma of the Head and Neck. Int J Radiat Oncol Biol Phys (2007) 68:1335–41. doi: 10.1016/j.ijrobp.2007.02.004
87. Rao RS, Patil S, Ganavi BS. Oral Cytokeratins in Health and Disease. J Contemp Dent Pract (2014) 15:127–36. doi: 10.5005/jp-journals-10024-1502
88. Crish JF, Bone F, Banks EB, Eckert RL. The Human Involucrin Gene Contains Spatially Distinct Regulatory Elements That Regulate Expression During Early Versus Late Epidermal Differentiation. Oncogene (2002) 21:738–47. doi: 10.1038/sj.onc.1205038
89. Ishitsuka Y, Roop DR. Loricrin: Past, Present, and Future. Int J Mol Sci (2020) 21(7):2271. doi: 10.3390/ijms21072271
90. Dmello C, Srivastava SS, Tiwari R, Chaudhari PR, Sawant S, Vaidya MM. Multifaceted Role of Keratins in Epithelial Cell Differentiation and Transformation. J Biosci (Bangalore) (2019) 44(2):33. doi: 10.1007/s12038-019-9864-8
91. Karantza V. Keratins in Health and Cancer: More Than Mere Epithelial Cell Markers. Oncogene (2011) 30:127–38. doi: 10.1038/onc.2010.456
92. Järvinen M, Rinne A, Hopsu-Havu VK. Human Cystatins in Normal and Diseased Tissues–a Review. Acta Histochem (1987) 82:5–18. doi: 10.1016/s0065-1281(87)80043-0
94. Rendon A, Schäkel K. Psoriasis Pathogenesis and Treatment. Int J Mol Sci (2019) 20(6):1475. doi: 10.3390/ijms20061475
95. Candi E, Schmidt R, Melino G. The Cornified Envelope: A Model of Cell Death in the Skin. Nat Rev Mol Cell Biol (2005) 6:328–40. doi: 10.1038/nrm1619
96. Zeeuwen PL. Epidermal Differentiation: The Role of Proteases and Their Inhibitors. Eur J Cell Biol (2004) 83:761–73. doi: 10.1078/0171-9335-00388
97. Brocklehurst K, Philpott MP. Cysteine Proteases: Mode of Action and Role in Epidermal Differentiation. Cell Tissue Res (2013) 351:237–44. doi: 10.1007/s00441-013-1557-2
98. Brooks YS, Ostano P, Jo SH, Dai J, Getsios S, Dziunycz P, et al. Multifactorial ERbeta and NOTCH1 Control of Squamous Differentiation and Cancer. J Clin Invest (2014) 124:2260–76. doi: 10.1172/JCI72718
99. Nowell CS, Radtke F. Notch as a Tumour Suppressor. Nat Rev Cancer (2017) 17:145–59. doi: 10.1038/nrc.2016.145
100. Koch U, Lehal R, Radtke F. Stem Cells Living With a Notch. Development (2013) 140:689–704. doi: 10.1242/dev.080614
101. Lefort K, Dotto GP. Notch Signaling in the Integrated Control of Keratinocyte Growth/Differentiation and Tumor Suppression. Semin Cancer Biol (2004) 14:374–86. doi: 10.1016/j.semcancer.2004.04.017
102. Dotto GP. Notch Tumor Suppressor Function. Oncogene (2008) 27:5115–23. doi: 10.1038/onc.2008.225
103. Majumder S, Crabtree JS, Golde TE, Minter LM, Osborne BA, Miele L. Targeting Notch in Oncology: The Path Forward. Nat Rev Drug Discovery (2021) 20:125–44. doi: 10.1038/s41573-020-00091-3
104. Rettig EM, Chung CH, Bishop JA, Howard JD, Sharma R, Li RJ, et al. Cleaved NOTCH1 Expression Pattern in Head and Neck Squamous Cell Carcinoma Is Associated With NOTCH1 Mutation, HPV Status, and High-Risk Features. Cancer Prev Res (Phila) (2015) 8:287–95. doi: 10.1158/1940-6207.CAPR-14-0366
105. Song XM, Xia RH, Li J, Long ZF, Ren HN, Chen WT, et al. Common and Complex Notch1 Mutations in Chinese Oral Squamous Cell Carcinoma. Clin Cancer Res (2014) 20:701–10. doi: 10.1158/1078-0432.CCR-13-1050
106. Joo YH, Jung CK, Kim MS, Sun DI. Relationship Between Vascular Endothelial Growth Factor and Notch1 Expression and Lymphatic Metastasis in Tongue Cancer. Otolaryngol Head Neck Surg (2009) 140:512–8. doi: 10.1016/j.otohns.2008.12.057
107. Misiorek JO, Przybyszewska-Podstawka A, Kalafut J, Paziewska B, Rolle K, Rivero-Muller A, et al. Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis. Cells (2021) 10(1):94. doi: 10.3390/cells10010094
108. Oyama T, Harigaya K, Sasaki N, Okamura Y, Kokubo H, Saga Y, et al. Mastermind-Like 1 (MamL1) and Mastermind-Like 3 (MamL3) are Essential for Notch Signaling In Vivo. Development (2011) 138:5235–46. doi: 10.1242/dev.062802
109. Lah TT, Kokalj-Kunovar M, Kastelic L, Babnik J, Stolfa A, Rainer S, et al. Cystatins and Stefins in Ascites Fluid From Ovarian Carcinoma. Cancer Lett (1992) 61:243–53. doi: 10.1016/0304-3835(92)90295-7
110. Delecrode TR, Siqueira WL, Zaidan FC, Bellini MR, Moffa EB, Mussi MC, et al. Identification of Acid-Resistant Proteins in Acquired Enamel Pellicle. J Dent (2015) 43:1470–5. doi: 10.1016/j.jdent.2015.10.009
111. Santiago AC, Khan ZN, Miguel MC, Gironda CC, Soares-Costa A, Pelá VT, et al. A New Sugarcane Cystatin Strongly Binds to Dental Enamel and Reduces Erosion. J Dent Res (2017) 96:1051–7. doi: 10.1177/0022034517712981
112. Penna E, Cerciello A, Chambery A, Russo R, Cernilogar FM, Pedone EM, et al. Cystatin B Involvement in Synapse Physiology of Rodent Brains and Human Cerebral Organoids. Front Mol Neurosci (2019) 12:195. doi: 10.3389/fnmol.2019.00195
113. Zenón F, Cantres-Rosario Y, Adiga R, Gonzalez M, Rodriguez-Franco E, Langford D, et al. HIV-Infected Microglia Mediate Cathepsin B-Induced Neurotoxicity. J Neurovirol (2015) 21:544–58. doi: 10.1007/s13365-015-0358-7
114. Lehtinen MK, Tegelberg S, Schipper H, Su H, Zukor H, Manninen O, et al. Cystatin B Deficiency Sensitizes Neurons to Oxidative Stress in Progressive Myoclonus Epilepsy, EPM1. J Neurosci (2009) 29:5910–5. doi: 10.1523/jneurosci.0682-09.2009
115. Žerovnik E. Possible Mechanisms by Which Stefin B Could Regulate Proteostasis and Oxidative Stress. Cells (2019) 8(1):70. doi: 10.3390/cells8010070
116. Barnes RP, Fouquerel E, Opresko PL. The Impact of Oxidative DNA Damage and Stress on Telomere Homeostasis. Mech Ageing Dev (2019) 177:37–45. doi: 10.1016/j.mad.2018.03.013
117. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA Damage: Mechanisms, Mutation, and Disease. FASEB J (2003) 17:1195–214. doi: 10.1096/fj.02-0752rev
118. Maytin EV, Habener JF. Transcription Factors C/EBP Alpha, C/EBP Beta, and CHOP (Gadd153) Expressed During the Differentiation Program of Keratinocytes In Vitro and In Vivo. J Invest Dermatol (1998) 110:238–46. doi: 10.1046/j.1523-1747.1998.00123.x
119. Lopez RG, Garcia-Silva S, Moore SJ, Bereshchenko O, Martinez-Cruz AB, Ermakova O, et al. C/EBPalpha and Beta Couple Interfollicular Keratinocyte Proliferation Arrest to Commitment and Terminal Differentiation. Nat Cell Biol (2009) 11:1181–90. doi: 10.1038/ncb1960
120. Schrem H, Klempnauer J, Borlak J. Liver-Enriched Transcription Factors in Liver Function and Development. Part II: The C/EBPs and D Site-Binding Protein in Cell Cycle Control, Carcinogenesis, Circadian Gene Regulation, Liver Regeneration, Apoptosis, and Liver-Specific Gene Regulation. Pharmacol Rev (2004) 56:291–330. doi: 10.1124/pr.56.2.5
121. Balasubramanian S, Agarwal C, Efimova T, Dubyak GR, Banks E, Welter J, et al. Thapsigargin Suppresses Phorbol Ester-Dependent Human Involucrin Promoter Activity by Suppressing CCAAT-Enhancer-Binding Protein Alpha (C/EBPalpha) DNA Binding. Biochem J (2000) 350 Pt 3:791–6. doi: 10.1042/bj3500791
122. Chang KW, Lin CE, Tu HF, Chung HY, Chen YF, Lin SC. Establishment of a P53 Null Murine Oral Carcinoma Cell Line and the Identification of Genetic Alterations Associated With This Carcinoma. Int J Mol Sci (2020) 21(24):9354. doi: 10.3390/ijms21249354
123. Mehta S, Cronkite DA, Basavappa M, Saunders TL, Adiliaghdam F, Amatullah H, et al. Maintenance of Macrophage Transcriptional Programs and Intestinal Homeostasis by Epigenetic Reader SP140. Sci Immunol (2017) 2(9):eaag3160. doi: 10.1126/sciimmunol.aag3160
124. Bhatt S, Matthews J, Parvin S, Sarosiek KA, Zhao D, Jiang X, et al. Direct and Immune-Mediated Cytotoxicity of Interleukin-21 Contributes to Antitumor Effects in Mantle Cell Lymphoma. Blood (2015) 126:1555–64. doi: 10.1182/blood-2015-01-624585
125. Yasmin R, Siraj S, Hassan A, Khan AR, Abbasi R, Ahmad N. Epigenetic Regulation of Inflammatory Cytokines and Associated Genes in Human Malignancies. Mediators Inflamm (2015) 2015:201703. doi: 10.1155/2015/201703
126. Tiacci E, Ladewig E, Schiavoni G, Penson A, Fortini E, Pettirossi V, et al. Pervasive Mutations of JAK-STAT Pathway Genes in Classical Hodgkin Lymphoma. Blood (2018) 131:2454–65. doi: 10.1182/blood-2017-11-814913
127. Weber A, Hengge UR, Bardenheuer W, Tischoff I, Sommerer F, Markwarth A, et al. SOCS-3 Is Frequently Methylated in Head and Neck Squamous Cell Carcinoma and Its Precursor Lesions and Causes Growth Inhibition. Oncogene (2005) 24:6699–708. doi: 10.1038/sj.onc.1208818
128. Maher K, Završnik J, Jerič-Kokelj B, Vasiljeva O, Turk B, Kopitar-Jerala N. Decreased IL-10 Expression in Stefin B-Deficient Macrophages Is Regulated by the MAP Kinase and STAT-3 Signaling Pathways. FEBS Lett (2014) 588:720–6. doi: 10.1016/j.febslet.2014.01.015
129. Trstenjak Prebanda M, Završnik J, Turk B, Kopitar Jerala N. Upregulation of Mitochondrial Redox Sensitive Proteins in LPS-Treated Stefin B-Deficient Macrophages. Cells (2019) 8(12):1476. doi: 10.3390/cells8121476
130. Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr Protoc Bioinf (2016) 54:1.30.1–1.3. doi: 10.1002/cpbi.5
131. Pon JR, Marra MA. MEF2 Transcription Factors: Developmental Regulators and Emerging Cancer Genes. Oncotarget (2016) 7:2297–312. doi: 10.18632/oncotarget.6223
Keywords: oral squamous cell carcinoma, head and neck squamous cell carcinoma, cysteine cathepsin, cystatin-B, epithelial proliferation/differentiation, WGCNA, GSEA
Citation: Xu T-T, Zeng X-W, Wang X-H, Yang L-X, Luo G and Yu T (2021) Cystatin-B Negatively Regulates the Malignant Characteristics of Oral Squamous Cell Carcinoma Possibly Via the Epithelium Proliferation/Differentiation Program. Front. Oncol. 11:707066. doi: 10.3389/fonc.2021.707066
Received: 08 May 2021; Accepted: 30 July 2021;
Published: 24 August 2021.
Edited by:
Eva Csosz, University of Debrecen, HungaryReviewed by:
Csongor Kiss, University of Debrecen, HungaryGergely Nagy, University of Debrecen, Hungary
Copyright © 2021 Xu, Zeng, Wang, Yang, Luo and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Luo, gangluo@139.com; Ting Yu, dent_yu@163.com