 Valentina Masola1*
Valentina Masola1* Marco Franchi2
Marco Franchi2 Gianluigi Zaza3
Gianluigi Zaza3 Francesca Mansa Atsina4
Francesca Mansa Atsina4 Giovanni Gambaro5
Giovanni Gambaro5 Maurizio Onisto1*
Maurizio Onisto1*- 1Department of Biomedical Sciences, University of Padova, Padova, Italy
- 2Department of Life Quality Sciences, University of Bologna, Rimini, Italy
- 3Department of Medical and Surgical Sciences, University of Foggia, Foggia, Italy
- 4University Hospital of Verona, Verona, Italy
- 5Renal Unit, Department of Medicine, University Hospital of Verona, Verona, Italy
Prostate cancer displays a certain phenotypic plasticity that allows for the transition of cells from the epithelial to the mesenchymal state. This process, known as epithelial–mesenchymal transition (EMT), is one of the factors that give the tumor cells greater invasive and migratory capacity with subsequent formation of metastases. In addition, many cancers, including prostate cancer, are derived from a cell population that shows the properties of stem cells. These cells, called cancer stem cells (CSCs) or tumor-initiating cells, not only initiate the tumor process and growth but are also able to mediate metastasis and drug resistance. However, the impact of EMT and CSCs in prostate cancer progression and patient survival is still far from fully understood. Heparanase (HPSE), the sole mammalian endoglycosidase capable of degrading heparan sulfate (HS), is also involved in prostate cancer progression. We had previously proved that HPSE regulates EMT in non-cancerous pathologies. Two prostate cancer cell lines (DU145 and PC3) were silenced and overexpressed for HPSE. Expression of EMT and stemness markers was evaluated. Results showed that the expression of several EMT markers are modified by HPSE expression in both the prostate cancer cell lines analyzed. In the same way, the stemness markers and features are also modulated by HPSE expression. Taken together, the present findings seem to prove a new mechanism of action of HPSE in sustaining prostate cancer growth and diffusion. As for other tumors, these results highlight the importance of HPSE as a potential pharmacological target in prostate cancer treatment.
Introduction
Of all the existing cancers, prostate cancer is the one that has globally recorded the greatest growth in cases over the last 30 years in both developing and developed countries. Being a male cancer that occurs mainly in old age, the aging of the population is one of the factors that have contributed most to the increase in cases (1). It is estimated that prostate cancer will increase to nearly 2.3 million new cases and 740,000 deaths per year by 2040 simply as a result of population growth and aging (2, 3). The severity of the disease is conferred by the ability of cancer cells to disseminate and metastasize, affecting various organs and tissues—specifically, bone tissue in 84% of cases, distant lymph nodes in 10.6%, the liver in 10.2%, and the thorax in 9.1% (4).
Two phenomena that contribute to the progression and metastasis of this neoplasm are the epithelium–mesenchymal transition process (EMT) and the presence of tumor stem cells. The EMT is a reversible cell-differentiation process during which the morphological and phenotypic conversion of polarized epithelial cells into mesenchymal cells occurs. These cells have a greater migratory capacity, greater resistance to apoptosis, increased expression of mesenchymal markers, and resistance to senescence. This change involves the loss of the junction systems that hold epithelial cells together and the loss of baso-apical polarity and various rearrangements of the cytoskeletal apparatus (5, 6).
The EMT process has also been shown to be present in prostate cancer, and among the various factors that seem to be involved in promoting this change, androgens and estrogens with their related signaling, hypoxia, transforming growth factor beta (TGF-β), and epidermal growth factor (EGF) must be mentioned (7).
These factors promote the activation of the PI3K/AKT and MAPK signaling pathway—thus activating the downstream effectors such as GSK3β and NF-kB, which increase the activity of SNAI-1 and Twist and consequently induce the expression of mesenchymal proteins (8).
Cancer stem cells, according to the American Association for Cancer Research, are described as “a cell within a tumor that possesses the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumor” (9).
Consequently, in line with this definition, CSCs possess both the ability to expand the population of cancer stem cells and, after differentiation, to give rise to other types of neoplastic cells that will make up a large part of the tumor mass. Since non-stem cells in cancer have a limited capacity for proliferation, the only cells with unlimited potential are cancer stem cells, which are thus able to guide the growth and metastatic process (10). For this reason, CSCs that have very long division times, and which are relatively insensitive to drug therapies aimed at targeting rapidly proliferating cells, may be one of the sources of cancer recurrence (11). Over the last few years, experimental evidence has accumulated in favor of the existence of CSC in prostate cancer and its role in tumor and metastatic progression (12). CSCs of the prostate can originate from basal or luminal-type progenitor/stem cells that will develop into tumors with markedly different biological and clinical characteristics in terms of aggressiveness and response to chemotherapy treatments and androgen-deprivation therapy (ADT) (13, 14). Since prostatic CSCs play a fundamental role in ADT resistance, it is currently considered essential to develop new anti-CSC strategies to compensate for treatment failure and disease recurrence (15).
More recently, it has been suggested that the expression of HPSE, the only enzyme capable of degrading the heparan sulfate (HS) chains of HS-proteoglycans (HSPGs), is associated with the characteristics of CSCs in Hodgkin’s lymphoma cells (16) and in myeloma (17).
HS is a highly sulfated linear polysaccharide, attached to the core protein of heparan sulfate proteoglycans (18, 19). HS proteoglycans are ubiquitously found both at the cell surface (i.e., syndecans and glypicans) and in the extracellular matrix (ECM) (18) where they regulate ECM structure and cell–ECM interaction (20, 21). In addition, HS regulates the activity of several molecules (cytokines, growth factors, etc.) (22–25). Currently, heparanase (HPSE) is defined as a multitasking protein capable of performing enzymatic-degradative activity towards HS chains, but which, at the same time, also manifests non-enzymatic activities (26). Through its cutting activity of the side chains of heparane sulfates (HS), it contributes both to the remodeling of the extracellular matrix and to the release and diffusion of various bioactive molecules linked to HS such as growth factors, cytokines, and enzymes. Considering that heparanase is not only produced and secreted by cancer cells but also by endothelial cells and activated immune cells and platelets, it is not surprising that its activity has a strong impact on the tumor microenvironment, thanks to those factors linked to HS, which, once released, promote tumor growth, neo-angiogenesis, and the formation of a metastatic niche (27). As a proof of concept, it has been shown that heparanase overexpression in transgenic mice (Hpa-Tg) makes the tumor microenvironment more conducive to neoplastic development in various experimental models of in vivo tumorigenesis (28, 29).
Since prostate cancer also has an increased expression of HPSE (30), we decided to verify whether this increase could be able to regulate EMT and cancer stem cells properties of prostate cancer.
Methods
Cell lines
DU145 (ATCC® HTB-81™) and PC3 (ATCC® CRL-1435™) prostate cancer cell lines were cultured in Roswell Park Memorial Institute (RPMI) containing 10% fetal bovine serum and supplemented with 1% penicillin/streptomycin. Cells were maintained in a humidified environment containing 5% CO2 at 37°C, and the culture medium was replaced every 2 days.
Transfection of HPSE overexpressing and shRNA plasmid
In order to stably obtain HPSE-overexpressing DU145 cells, we used a plasmid-coding HPSE ORF purchased from OriGene, and as a negative control, we used the corresponding empty vector.
To stably obtain HPSE-silenced PC3 cells, we used four different shRNAs targeting human heparanase (NM_006665) purchased from OriGene as described earlier (31, 32). As a negative control, we used an shRNA pRS non-effective GFP plasmid (TR30003).
DU145 and PC3 cells were seeded in six-well plates and when they reached 70%–80% of confluence, they were transfected with Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. Forty-eight hours after transfection, DU145 cells overexpressing HPSE were selected with 500 μg/ml G418 (Sigma), and PC3 cells silenced for HPSE were selected with 0.75 μg/ml of puromycin (Sigma). Single clones were isolated and analyzed for HPSE expression. The ones with the highest overexpression/silencing rate were used in the subsequent experiments.
RNA isolation and real-time qPCR analysis
Total RNA was extracted from cells by Trizol reagent (Invitrogen) according to the manufacturer’s instructions (33). RNA yield and purity were checked using a Nanodrop spectrophotometer (EuroClone), and total RNA from each sample was reverse transcribed into cDNA using Moloney Murine Leukemia Virus Reverse Transcriptase (Sigma-Aldrich). Real-time PCR was performed on a StepOne™ Real-Time PCR System (Thermo Fisher) using SensiFAST SYBR Hi-Rox (Bioline). The comparative Ct method (ΔΔCt) was used to quantify gene expression, and the relative quantification was calculated as 2−ΔΔCt. The presence of non-specific amplification products was excluded by melting curve analysis. Statistical analyses on real-time PCR data were performed using the Relative Expression Software Tool (REST) (34). The forward and reverse primer sequences were reported in Table 1.
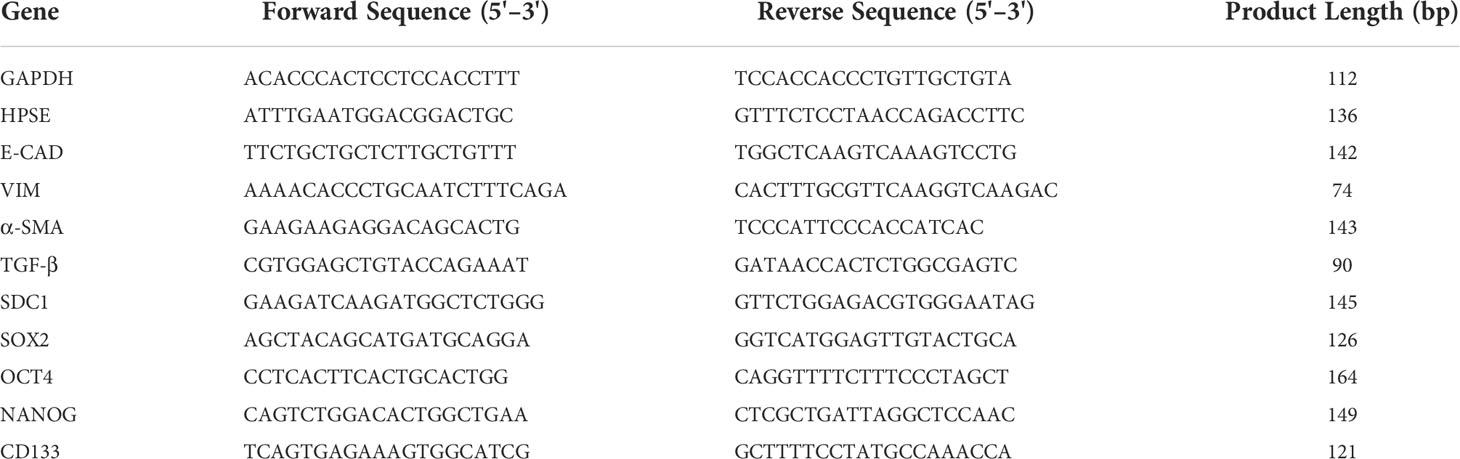
Table 1 Primer sequences used for real-time PCR .
Western blotting and immunofluorescence
Cells were lysed in radioimmunoprecipitation assay buffer (RIPA) buffer composed of 150 mM NaCl, 50 mM TRIS_HCl (pH 8), 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 1% Triton-X with Complete Protease Inhibitor Mixture (Roche Applied Science, Penzberg, Germany). In brief, equal amounts of proteins were treated in reducing sample buffer and denatured for 10 min at 100°C. Protein samples were then resolved in 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and electrotransferred to nitrocellulose membranes. Non-specific binding was blocked for 1 h at room temperature with non-fat milk (5%) in TBST buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl and 0.1% Tween 20). Membranes were exposed to primary antibodies GAPDH (sc-47778 Santa Cruz), HPSE (MA1-83806 HP3/17, Thermo Fisher), E-cadherin (E-CAD) (GTX10443 GeneTex), vimentin (VIM) (sc-7557 Santa Cruz), α-SMA (A5228 Sigma), SOX2 (GTX101507 GeneTex), OCT4 (GTX627419 GeneTex), and NANOG (GTX100863 GeneTex), overnight at 4°C and incubated with a secondary peroxidase-conjugated antibody for 1 h at room temperature. The signal was detected with Luminata™ Forte Western HRP Substrate (Millipore) according to the manufacturer’s instructions, and the signal was acquired with Mini HD9 (UVItec, Cambridge, UK). Immunofluorescence cells were fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS) for 5 min, and blocked with 3% bovine serum albumin (BSA) in PBS at room temperature for 30 min. Cells were then incubated overnight at 4°C with the primary antibodies in PBS with 1% BSA and then washed three times for 5 min with PBS before incubation for 1 h at 37°C with the secondary antibody, again in PBS with 1% BSA. Cell nuclei were visualized with a Hoechst 33258. Images were obtained with a confocal LeicaSP5 microscope.
Colony formation assay
WT and HPSE-silenced/overexpressing prostate cancer cells were seeded in 35-mm culture dishes (1,000 cells per well) and incubated with RPMI supplemented with 10% FBS for 7–10 days (35, 36). The media was renewed every 2 days. The colonies were fixed using paraformaldehyde and stained with 0.1% crystal violet. Cell colony-forming ability was assessed by counting the number of colonies. A colony was defined when the number of cells was more than 50.
Hanging drop assay
To assess the spheroid-formation capacity and compare the spheroid size of the cells, 500 cells in DMEM-F12 complete medium were placed as drops (20 μl each) into the lid of a Petri dish. The lid was then rapidly re-inverted onto the culture dish that was filled with 10 ml of sterile PBS to prevent evaporation of the drops. The hanging drop cultures were incubated at 37°C in a humidified atmosphere with 5% CO2 for 1 week. Pictures of the spheroids inside the drop were taken using a Leica MZ16F stereomicroscope, and their comparative size was obtained measuring the area occupied by the spheres using the software NIH ImageJ.
Results
Establishment of HPSE overexpressing/silenced prostate cancer cell lines
In order to investigate the function of HPSE in prostate cancer, we choose to use two prostate cancer cell lines because they display very different morphological aspects related to EMT. DU145 has a more epithelial phenotype, whereas PC3 cells are elongated and spindle-shaped like mesenchymal cells. Starting from this evidence, we decided to overexpress HPSE in DU145 and to silence it in PC3 (Supplementary Figure S1). HPSE was overexpressed in DU145 cells and silenced in PC3 cells. Results confirmed a significant HPSE upregulation in DU145 cells both at gene and protein level confirmed by WB and immunofluorescence (Figure 1) with respect to DU145 control cells transfected with the empty vector (CTR). CTR cells displayed HPSE expression levels comparable to wt DU145 cells (data not shown). Furthermore, HPSE was silenced by shRNA in PC3 cells by more than 50% both at gene and protein levels (Figure 1).
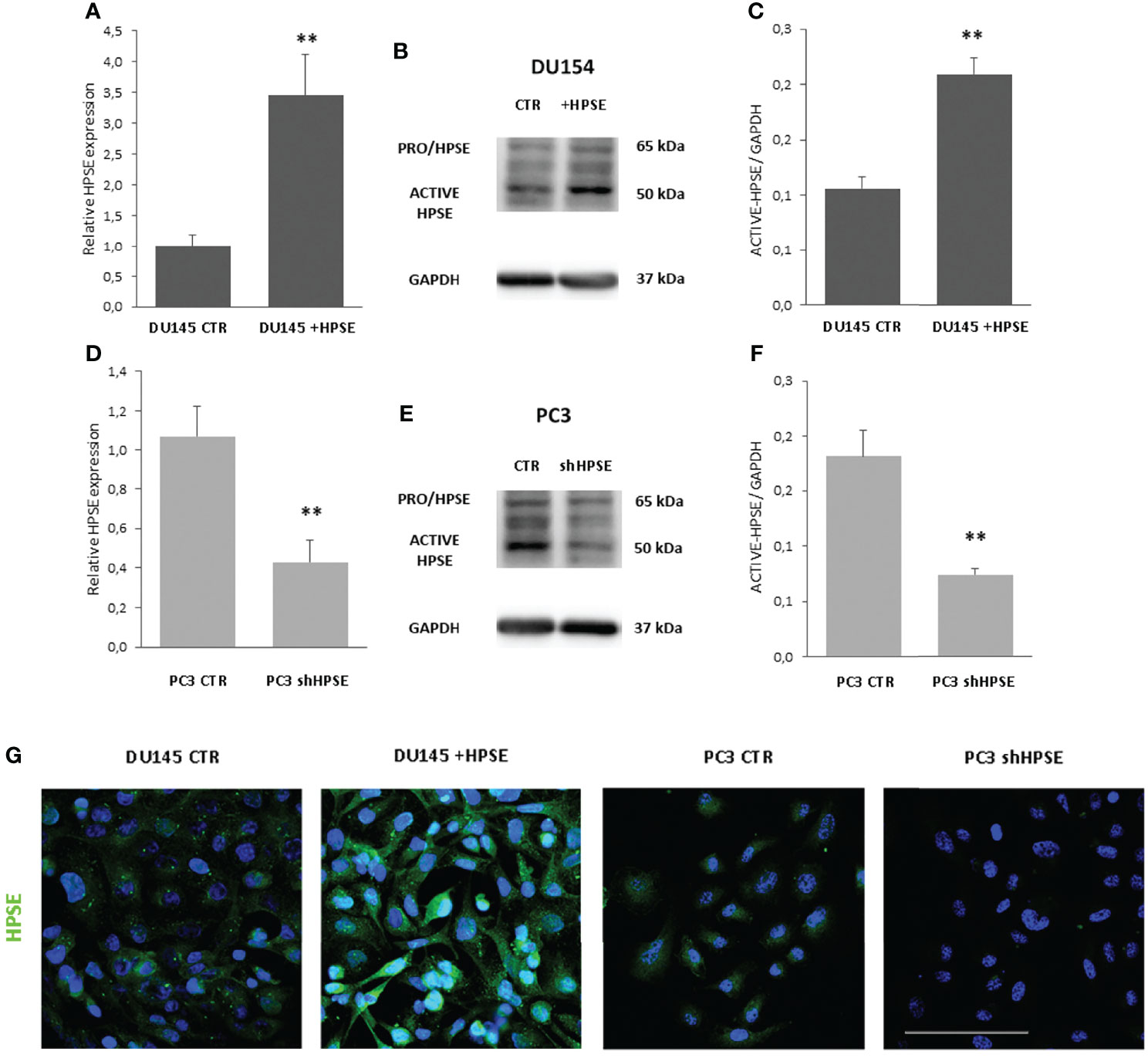
Figure 1 HPSE overexpression and silencing in prostate cancer cells. HPSE gene expression was evaluated by real-time PCR on DU145 (A) and PC3 (D) cells. Data were normalized to GAPDH expression. Means ± SD (error bars), n=6. **p < 0.001 vs. CTR. WB analysis of HPSE in DU145 (B) and PC3 (E) and relative quantification (C, F). (G) HPSE immunofluorescence in DU145 and PC3 cells (green). Nuclei were counterstained with Hoechst 33342. Scale bar = 100 μm.
HPSE regulates EMT in prostate cancer cells
In order to investigate whether HPSE is able to modulate EMT, the expression levels of epithelial and mesenchymal markers were measured in DU145 and PC3 cells. Gene expression analyses indicated a decrease in epithelial marker E-cadherin in HPSE overexpressing DU145 cells compared with the control. Also observed was a marked increase in the expression levels of vimentin, α-SMA, SNAI1, and TGF-β compared with the control (Figure 2A). By contrast, in HPSE-silenced PC3 cells, E-cadherin gene expression was increased, and the expression of mesenchymal markers was reduced (Figure 2C). The reduction in E-cadherin and the increase in vimentin and a-SMA in HPSE-overexpressing DU145 cells were also confirmed at protein level by WB and by immunofluorescence (Figures 2B, E). On the other hand, the reduction in vimentin and a-SMA in HPSE-silenced PC3 cells was also confirmed at protein level (Figures 2D, E). These results indicate that HPSE likely promotes EMT in prostate cancer cells.
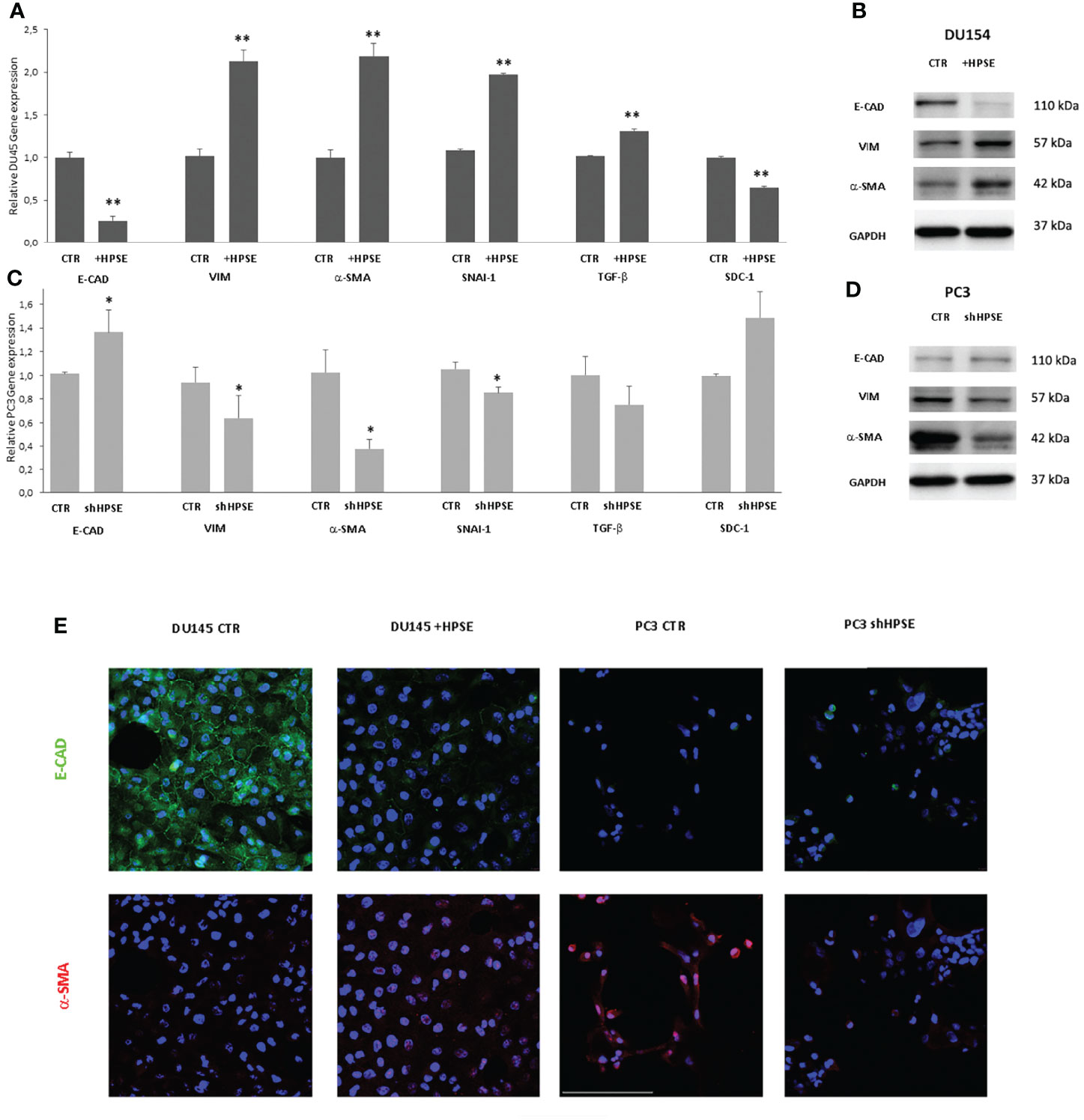
Figure 2 EMT markers expression in prostate cancer cells. E-CAD, VIM, α-SMA, TGF-β, and SDC-1 gene expression was evaluated by real-time PCR in DU145 (A) and PC3 (C) cells. Data were normalized to GAPDH expression. Means ± SD (error bars), n=6. **p < 0.001, *p<0.05 vs. CTR. E-CAD, VIM, a-SMA, and protein level were quantified by WB in DU145 (B) and PC3 (D) cells. GAPDH was included as loading control. (E) E-CAD (green) and α-SMA (red) immunofluorescence in DU145 and PC3 cells. Nuclei were counterstained with Hoechst 33342. Scale bar = 100 μm.
HS proteoglycans has an important role on prostate epithelium–stroma architecture (34). In particular, syndecans, a family of heparan sulfate proteoglycans that are present on the cell surface, are involved in the control of cell proliferation, apoptosis, and transformation. In prostate cancer, the expression of syndecan-1 in epithelial cells decreases when cells are transformed and acquire invasive properties. This decreased expression is associated with a bad prognosis (37–39).
In addition, HPSE cleaves HS chains on syndecan-1, and a tight relationship between HPSE and syndecan-1 has been documented in tumor and non-tumor models (19, 32, 40, 41).
Results showed that syndecan-1 gene expression was reduced in HPSE-overexpressing DU145 cells, and, in contrast, syndecan-1 expression was increased in HPSE-silenced PC3 cells.
HPSE regulates stemness features in prostate cancer cells
Numerous studies have shown that the key regulators in maintaining the stemness of embryonic stem cells, including Oct4, Sox2, and Nanog, along with their activation targets, are commonly overexpressed in cancer stem cells in several malignancies (42–44). In addition, a series of molecules, including CD44, CD133, integrin α2β1, ALDH1A1, and Bmi1, involved in the regulation of cancer stem cell self-renewal, metastasis, and drug resistance, have also been confirmed in prostate cancer (45, 46). Results showed that the stemness markers SOX2, OCT4, NANOG, and CD133 were upregulated both at gene and protein level in HPSE-overexpressing DU145 cells compared with the control. By contrast, the same markers were significantly reduced in HPSE-silenced PC3 cells compared with the control (Figure 3).
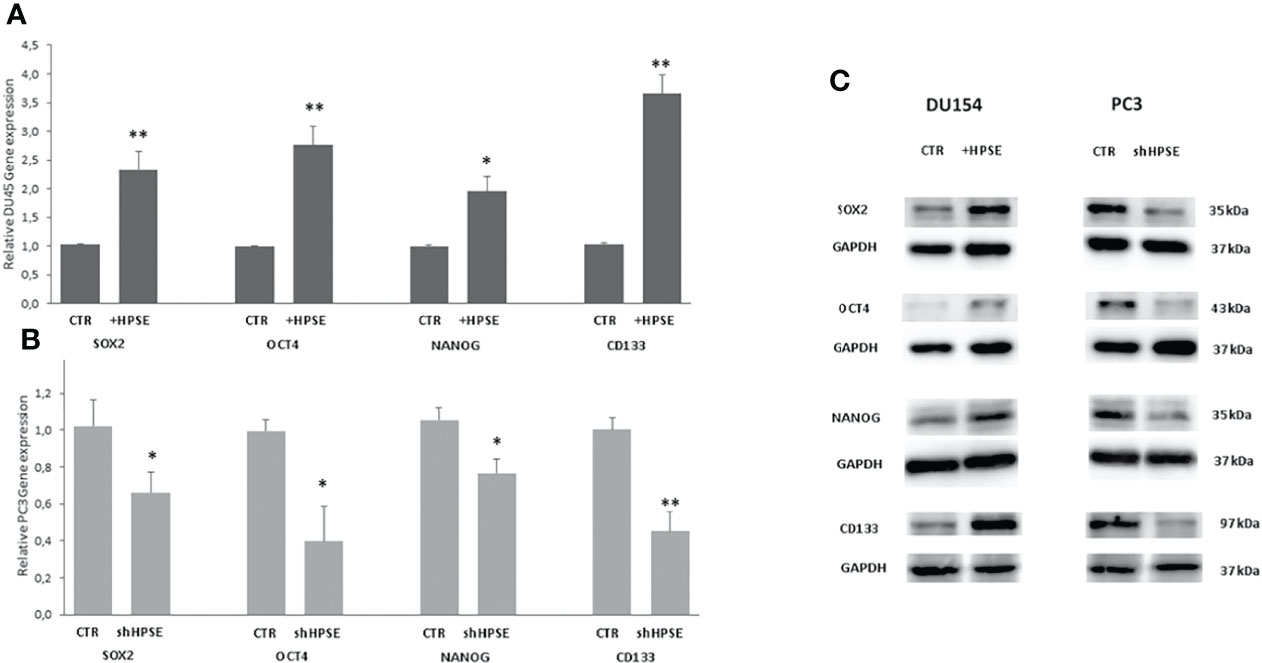
Figure 3 Stemness markers expression in prostate cancer cells. SOX2, OCT4, NANOG, and CD133 gene expression were evaluated by real-time PCR in DU145 (A) and PC3 (B) cells. Data were normalized to GAPDH expression. Means ± SD (error bars), n=6. **p < 0.001, *p<0.05 vs. (C) SOX2, OCT4, and NANOG protein level were quantified by WB in DU145 and PC3 cells. GAPDH was included as loading control.
Two important characteristics of cancer stem cells are the capacity to grow starting from a single cell and to form a sphere in an independent anchoring system.
Colony formation is the ability of cancer stem cells to form colonies when seeded on cell culture dishes at very low concentrations after limiting dilutions (47). Results showed that HPSE overexpression in DU145 cells increases the number of colonies compared to control cells and that HPSE silencing in PC3 significantly limited the ability to form colonies (Figures 4A, B).
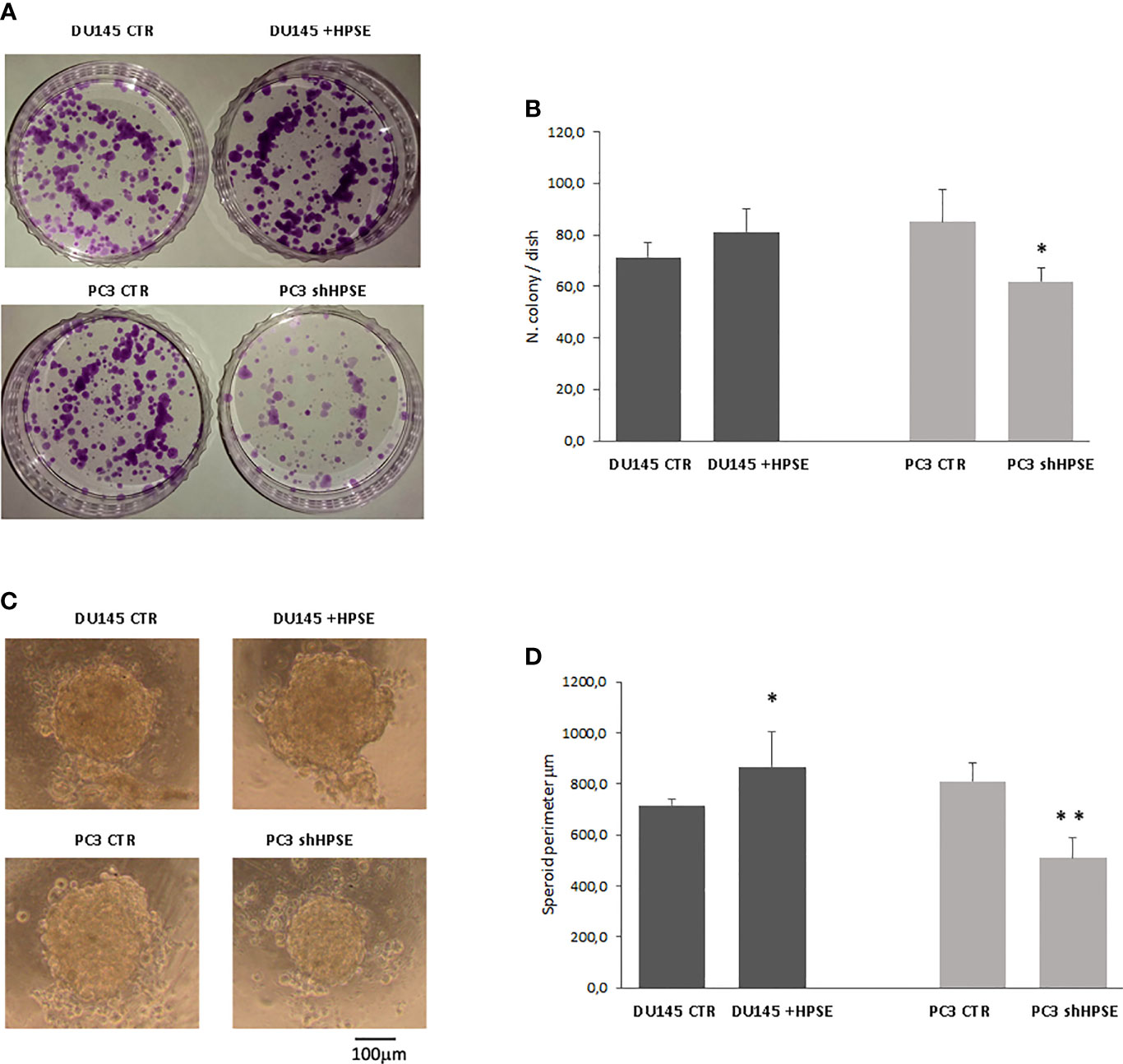
Figure 4 Stemness properties in prostate cancer cells. Representative images of colony (A) and sphere (C) assays evaluated in DU145 and PC3 cells. (B, D) Bars represent the quantification of colony and sphere assay respectively and are expressed as mean ± SD values; n=8. **p < 0.001, *p<0.05 vs. CTR.
To assess the spontaneous sphere formation efficiency of cancer stem cells, we used the hanging drop method (48, 49). Both cell lines were able to form similar circular spheres. HPSE overexpression in DU145 cells increased the sphere perimeter, whereas HPSE silencing in PC3 cells exerted the opposite effects (Figures 4C, D).
Discussion
The malignant growth and progression of tumor disease are supported by key features of cancer cells that have collectively been referred to as “hallmarks of cancer.” To the initial six hallmarks (supporting proliferative signaling, evading growth suppressors, replicative immortality, resistance to cell death, inducing angiogenesis, and activating invasion and metastasis), four other “enabling features” have more recently been added, such as instability and mutation of the genome, tumor-promoting inflammation, deregulated energy metabolism, and escape from immune destruction (50). Since it is now consolidated evidence that all tumors examined to date overexpress HPSE, some authors have proposed that with its multiple roles within the tumor microenvironment, heparanase can regulate each of these distinctive characteristics of cancer and, in turn, highlight the need for therapies aimed at its inhibition (51).
Starting from this evidence, our goal was to evaluate how the overexpression and silencing of heparanase in prostate cancer cells affect stemness characteristics and epithelium–mesenchymal transition (EMT)—two of the classic tumor “hallmarks.”
EMT is thought to be activated in cancer cells, linked to their dissociation from the primary tumor and their intravasation into blood vessels (52). However, the impact of EMT in cancer progression and patient survival is still far from fully understood.
EMT was originally described during morphogenesis and later was observed in several pathological events, including fibrosis and cancer metastasis (53). During EMT, epithelial cells lose adherence junctions and (54) apical–basal polarity and acquire a mesenchymal phenotype with an enhanced motility. In response to various signals (55–58), epithelial cells upregulate a group of transcription factors to orchestrate EMT, and the main ones are SNAI-1 and TWIST. EMT can be considered to be a continuum process (59), and cells with this hybrid phenotype have been referred to as “metastable” (60), reflecting the flexibility of these cells in inducing or reversing the EMT process (61).
Heparanase is the sole mammalian endoglycosidase capable of degrading glycosaminoglycan HS. The enzymatic cleavage of HS by HPSE results in ECM remodeling and releasing these bioactive mediators, producing a rapid tissue response to local or systemic stimuli. These effects profoundly affect multiple pathophysiological processes such as tumor progression, inflammation, and fibrosis (62–65). Since uncontrolled HS degradation results in significant tissue damage, HPSE expression is tightly regulated (19, 66, 67), whereas it is overexpressed in malignant tumors (66, 68).
Prostate cancer is responsible for more gender-specific cancer-related deaths in men than any other cancer (1). Zhou et al. (69) have shown that HPSE overexpression can facilitate tumor invasion and accelerate bone destruction caused by prostate cancer metastasis. Expression of HPSE has also been evaluated in prostate neoplasia; its malignant transformation has been shown to be associated with heparanase-1 increased expression at both mRNA and protein levels (70). These authors have also correlated HPSE expression with the degree of metastasizing tumors and suggested its use as a potential marker for diagnosis of the prostate metastatic process (70).
We and other previous literature support the contention that heparanase is needed for pathological organ fibrosis, and this action is exerted thorough the regulation of EMT. In vitro and in vivo studies have shown that HPSE regulates renal EMT induced by FGF-2, TGF-beta, and hypoxia (71–74). We have also proved that HPSE regulates high glucose-induced EMT of mesothelial cells (75). In addition, it has been proven that HPSE participates in lung fibrosis by regulating EMT (76), and it probably also has a role in the liver (33).
We induced HPSE overexpression in DU145 cell line and its silencing, by shRNA, in PC3 cell line. The silencing/overexpression rate was confirmed both at gene and protein levels. Results showed that HPSE is an EMT inducer in prostate cancer. Indeed, the epithelial marker E-cadherin was reduced in HPSE-overexpressing DU145 cells and upregulated in HPSE-silenced PC3 cells with respect to control. In contrast, the expression of mesenchymal markers a-SMA and vimentin was increased in HPSE-overexpressing DU45 cells and reduced in HPSE-silenced PC3 cells with respect to control.
Moreover, we have proved that HPSE regulates the expression of TGF-β (one of the principal EMT activators) and the levels of its associated transcription factor SNAI-1. Specifically, TGF-b and SNAI-1 were increased in HPSE-overexpressing DU145 cells and reduced in HPSE-silenced PC3 cells with respect to control.
During malignant transformation depletion of epithelial cell surface, syndecan-1 profoundly alters cell morphology and anchorage-dependent growth: thus, syndecan-1 is necessary in maintaining the epithelial phenotype. TGF-β can induce EMT by activating SNAI-1, which in turn, represses the expression of syndecan-1. A coordinated loss of syndecan-1 and E-cadherin has been documented in many epithelial malignancies compared to their benign counterparts. In the prostate, changes in syndecan-1 expression are linked to EMT (77). It has been described that syndecan-1 expression is lower in PC3 and DU145 prostate cancer cell lines than in normal prostate epithelial cells (78).
Here, we have shown that HPSE expression modulates syndecan-1: HPSE overexpression reduced syndecan-1 in DU145 cells, and HPSE silencing increased syndecan-1 expression in PC3 cell line.
Recently, EMT has been linked to stem cell phenotype (79), since cancer cells with EMT characteristics acquires stem-cell-like features, such as self-renewal and slow proliferation (80, 81). Cancer stem cells acquire more complete EMT molecular characteristics and exhibit more aggressive abilities. Specifically, prostate cancer stem cells display increased EMT markers and increased tumorigenesis, migration, and invasion ability (82). Cancer stem cells have a specific gene signature, and the principal markers are SOX2, OCT4, and NANOG. Additionally, other genes involved in maintaining self-renewal capacity in prostate cancer include CD-133 and CD-44 (83, 84).
Here, we have proved that HPSE expression modulates prostate CSCs. Specifically, the stemness markers SOX2, OCT4, NANOG, and CD133 were upregulated both at gene and protein levels in HPSE-overexpressing DU145 cells and reduced in HPSE-silenced PC3 cells compared with the control. In addition, functional assays confirmed a role of HPSE in prostate cancer stemness: HPSE increases the capacity to grow starting from a single cell and to form a sphere in an independent anchoring system. Future studies could also characterize the potential role of HPSE in self-renewal capacity of prostate CSCs.
Here, we report that the expression of several EMT markers is controlled by HPSE expression in prostate cancer. Moreover, stemness markers and features of CSCs are also modulated by HPSE. Collectively, these results proved an additional mechanism by means of which HPSE can contribute to prostate cancer progression and metastasis, and further studies will be necessary to clarify its potential as a pharmacological goal.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
VM, MO, and GG conceptualized this study. GZ and MF contributed to the design and implementation of the research. VM and FA carried out the experiment. VM and MO wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant provided by the University of Padova (BIRD192859/19).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor AP declared a past co-authorship with the authors MF, MO, VM.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.918419/full#supplementary-material
References
1. Dy GW, Gore JL, Forouzanfar MH, Naghavi M, Fitzmaurice C. Global burden of urologic cancers, 1990-2013. Eur Urol (2017) 71(3):437–46. doi: 10.1016/j.eururo.2016.10.008
2. Zhou CK, Check DP, Lortet-Tieulent J, Laversanne M, Jemal A, Ferlay J, et al. Prostate cancer incidence in 43 populations worldwide: An analysis of time trends overall and by age group. Int J Cancer (2016) 138(6):1388–400. doi: 10.1002/ijc.29894
3. Culp MB, Soerjomataram I, Efstathiou JA, Bray F, Jemal A. Recent global patterns in prostate cancer incidence and mortality rates. Eur Urol (2020) 77(1):38–52. doi: 10.1016/j.eururo.2019.08.005
4. Gandaglia G, Abdollah F, Schiffmann J, Trudeau V, Shariat SF, Kim SP, et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate (2014) 74(2):210–6. doi: 10.1002/pros.22742
5. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi: 10.1038/nrm3758
6. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119(6):1420–8. doi: 10.1172/JCI39104
7. Montanari M, Rossetti S, Cavaliere C, D’Aniello C, Malzone MG, Vanacore D, et al. Epithelial-mesenchymal transition in prostate cancer: an overview. Oncotarget (2017) 8(21):35376–89. doi: 10.18632/oncotarget.15686
8. Di Zazzo E, Galasso G, Giovannelli P, Di Donato M, Bilancio A, Perillo B, et al. Estrogen receptors in epithelial-mesenchymal transition of prostate cancer. Cancers (Basel) (2019) 11(10):1418. doi: 10.3390/cancers11101418
9. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CHM, Jones DL, et al. Cancer stem cells–perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res (2006) 66(19):9339–44. doi: 10.1158/0008-5472.CAN-06-3126
10. Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell (2006) 124(6):1111–5. doi: 10.1016/j.cell.2006.03.011
11. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer (2005) 5(4):275–84. doi: 10.1038/nrc1590
12. Li JJ, Shen MM. Prostate stem cells and cancer stem cells. Cold Spring Harb Perspect Med (2019) 9(6):a030395. doi: 10.1101/cshperspect.a030395
13. Wang ZA, Shen MM. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene (2011) 30(11):1261–71. doi: 10.1038/onc.2010.530
14. Shen MM, Wang X, Economides KD, Walker D, Abate-Shen C. Progenitor cells for the prostate epithelium: roles in development, regeneration, and cancer. Cold Spring Harb Symp Quant Biol (2008) 73:529–38. doi: 10.1101/sqb.2008.73.050
15. Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol (2011) 8(1):12–23. doi: 10.1038/nrclinonc.2010.136
16. Troschel FM, Linsenmaier M, Borrmann K, Eich HT, Götte M, Greve B. Heparanase expression is associated with cancer stem cell features and radioresistance in hodgkin’s lymphoma cells. Anticancer Res (2021) 41(7):3299–308. doi: 10.21873/anticanres.15117
17. Tripathi K, Ramani VC, Bandari SK, Amin R, Brown EE, Ritchie JP, et al. Heparanase promotes myeloma stemness and in vivo tumorigenesis. Matrix Biol (2020) 88:53–68. doi: 10.1016/j.matbio.2019.11.004
18. Parish CR, Freeman C, Hulett MD. Heparanase: a key enzyme involved in cell invasion. Biochim Biophys Acta (2001) 1471(3):M99–108. doi: 10.1016/S0304-419X(01)00017-8
19. Ramani VC, Purushothaman A, Stewart MD, Thompson CA, Vlodavsky I, Au JL-S, et al. The heparanase/syndecan-1 axis in cancer: mechanisms and therapies. FEBS J (2013) 280(10):2294–306. doi: 10.1111/febs.12168
20. Theocharis AD, Skandalis SS, Tzanakakis GN, Karamanos NK. Proteoglycans in health and disease: novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J (2010) 277(19):3904–23. doi: 10.1111/j.1742-4658.2010.07800.x
21. Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature (2007) 446(7139):1030–7. doi: 10.1038/nature05817
22. Kato M, Wang H, Kainulainen V, Fitzgerald ML, Ledbetter S, Ornitz DM, et al. Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nat Med (1998) 4(6):691–7. doi: 10.1038/nm0698-691
23. Rider CC, Mulloy B. Heparin, heparan sulphate and the TGF-β cytokine superfamily. Molecules (2017) 22(5):458–60. doi: 10.3390/molecules22050713
24. Meneghetti MCZ, Hughes AJ, Rudd TR, Nader HB, Powell AK, Yates EA, et al. Heparan sulfate and heparin interactions with proteins. J R Soc Interface (2015) 12(110):0589. doi: 10.1098/rsif.2015.0589
25. Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol (2006) 6(9):633–43. doi: 10.1038/nri1918
26. Masola V, Bellin G, Gambaro G, Onisto M. Heparanase: A multitasking protein involved in extracellular matrix (ECM) remodeling and intracellular events. Cells (2018) 7(12):236. doi: 10.3390/cells7120236
27. Masola V, Zaza G, Gambaro G, Franchi M, Onisto M. Role of heparanase in tumor progression: Molecular aspects and therapeutic options. Semin Cancer Biol (2020) 62:86–98. doi: 10.1016/j.semcancer.2019.07.014
28. Boyango I, Barash U, Naroditsky I, Li J-P, Hammond E, Ilan N, et al. Heparanase cooperates with ras to drive breast and skin tumorigenesis. Cancer Res (2014) 74(16):4504–14. doi: 10.1158/0008-5472.CAN-13-2962
29. Weissmann M, Arvatz G, Horowitz N, Feld S, Naroditsky I, Zhang Y, et al. Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proc Natl Acad Sci U S A (2016) 113(3):704–9. doi: 10.1073/pnas.1519453113
30. Barbosa GO, Cervigne NK, Carvalho HF, Augusto TM. Heparanase 1 involvement in prostate physiopathology. Cell Biol Int (2017) 41(11):1194–202. doi: 10.1002/cbin.10748
31. Masola V, Gambaro G, Tibaldi E, Onisto M, Abaterusso C, Lupo A. Regulation of heparanase by albumin and advanced glycation end products in proximal tubular cells. Biochim Biophys Acta (2011) 1813(8):1475–82. doi: 10.1016/j.bbamcr.2011.05.004
32. Masola V, Gambaro G, Tibaldi E, Brunati AM, Gastaldello A, D’Angelo A, et al. Heparanase and syndecan-1 interplay orchestrates fibroblast growth factor-2-induced epithelial-mesenchymal transition in renal tubular cells. J Biol Chem (2012) 287(2):1478–88. doi: 10.1074/jbc.M111.279836
33. Secchi MF, Crescenzi M, Masola V, Russo FP, Floreani A, Onisto M. Heparanase and macrophage interplay in the onset of liver fibrosis. Sci Rep (2017) 7(1):14956. doi: 10.1038/s41598-017-14946-0
34. Masola V, Bonomini M, Onisto M, Ferraro PM, Arduini A, Gambaro G. Biological effects of XyloCore, a glucose sparing PD solution, on mesothelial cells: Focus on mesothelial-mesenchymal transition, inflammation and angiogenesis. Nutrients (2021) 13(7):2282. doi: 10.3390/nu13072282
35. Zhang Y, Jin Z, Zhou H, Ou X, Xu Y, Li H, et al. Suppression of prostate cancer progression by cancer cell stemness inhibitor napabucasin. Cancer Med (2016) 5(6):1251–8. doi: 10.1002/cam4.675
36. Wang M, Ren D, Guo W, Huang S, Wang Z, Li Q, et al. N-cadherin promotes epithelial-mesenchymal transition and cancer stem cell-like traits via ErbB signaling in prostate cancer cells. Int J Oncol (2016) 48(2):595–606. doi: 10.3892/ijo.2015.3270
37. Contreras HR, Ledezma RA, Vergara J, Cifuentes F, Barra C, Cabello P, et al. The expression of syndecan-1 and -2 is associated with Gleason score and epithelial-mesenchymal transition markers, e-cadherin and beta-catenin, in prostate cancer. Urol Oncol ottobre (2010) 28(5):534–40. doi: 10.1016/j.urolonc.2009.03.018
38. Barbosa GO, Biancardi MF, Carvalho HF. Heparan sulfate fine-tunes stromal-epithelial communication in the prostate gland. Dev Dyn (2021) 250(5):618–28. doi: 10.1002/dvdy.281
39. Contreras HR. [Syndecans in the diagnosis and prognosis of prostate cancer]. Rev Med Chil Jan (2010) 138(1):95–101. doi: 10.4067/S0034-98872010000100014
40. Rangarajan S, Richter JR, Richter RP, Bandari SK, Tripathi K, Vlodavsky I, et al. Heparanase-enhanced shedding of syndecan-1 and its role in driving disease pathogenesis and progression. J Histochem Cytochem (2020) 68(12):823–40. doi: 10.1369/0022155420937087
41. Teixeira FCOB, Götte M. Involvement of syndecan-1 and heparanase in cancer and inflammation. Adv Exp Med Biol (2020) 1221:97–135. doi: 10.1007/978-3-030-34521-1_4
42. Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, et al. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene (2011) 30(36):3833–45. doi: 10.1038/onc.2011.114
43. Chiou S-H, Wang M-L, Chou Y-T, Chen C-J, Hong C-F, Hsieh W-J, et al. Coexpression of Oct4 and nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res (2010) 70(24):10433–44. doi: 10.1158/0008-5472.CAN-10-2638
44. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature (2014) 511(7508):246–50. doi: 10.1038/nature13305
45. Lin X, Farooqi AA, Qureshi MZ, Romero MA, Tabassum S, Ismail M. Prostate cancer stem cells: Viewing signaling cascades at a finer resolution. Arch Immunol Ther Exp (Warsz) (2016) 64(3):217–23. doi: 10.1007/s00005-016-0383-0
46. Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, et al. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab Invest (2010) 90(2):234–44. doi: 10.1038/labinvest.2009.127
47. Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc (2006) 1(5):2315–9. doi: 10.1038/nprot.2006.339
48. Chen K, Li X, Li N, Dong H, Zhang Y, Yoshizawa M, et al. Spontaneously formed spheroids from mouse compact bone-derived cells retain highly potent stem cells with enhanced differentiation capability. Stem Cells Int (2019) 2019:8469012. doi: 10.1155/2019/8469012
49. Foty R. A simple hanging drop cell culture protocol for generation of 3D spheroids. J Vis Exp (2011) 51):2720. doi: 10.3791/2720
50. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
51. Jayatilleke KM, Hulett MD. Heparanase and the hallmarks of cancer. J Transl Med (2020) 18(1):453. doi: 10.1186/s12967-020-02624-1
52. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139(5):871–90. doi: 10.1016/j.cell.2009.11.007
53. Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol (2017) 11(1):28–39. doi: 10.1002/1878-0261.12017
54. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science (2013) 342(6159):1234850. doi: 10.1126/science.1234850
55. Strutz F, Zeisberg M, Ziyadeh FN, Yang C-Q, Kalluri R, Müller GA, et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int (2002) 61(5):1714–28. doi: 10.1046/j.1523-1755.2002.00333.x
56. Moustakas A, Heldin C-H. Mechanisms of TGFβ-induced epithelial-mesenchymal transition. J Clin Med (2016) 5(7):63. doi: 10.3390/jcm5070063
57. Lo H-W, Hsu S-C, Xia W, Cao X, Shih J-Y, Wei Y, et al. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res (2007) 67(19):9066–76. doi: 10.1158/0008-5472.CAN-07-0575
58. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal (2014) 7(344):re8. doi: 10.1126/scisignal.2005189
59. Nieto MA, Huang RY-J, Jackson RA, Thiery JP. EMT: 2016. Cell (2016) 166(1):21–45. doi: 10.1016/j.cell.2016.06.028
60. Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol (2006) 172(7):973–81. doi: 10.1083/jcb.200601018
61. Rosanò L, Spinella F, Di Castro V, Decandia S, Nicotra MR, Natali PG, et al. Endothelin-1 is required during epithelial to mesenchymal transition in ovarian cancer progression. Exp Biol Med (Maywood) (2006) 231(6):1128–31. doi: 10.3181/00379727-232-2311128
62. Secchi MF, Masola V, Zaza G, Lupo A, Gambaro G, Onisto M. Recent data concerning heparanase: focus on fibrosis, inflammation and cancer. Biomol Concepts (2015) 6(5–6):415–21. doi: 10.1515/bmc-2015-0021
63. Lv Q, Zeng J, He L. The advancements of heparanase in fibrosis. Int J Mol Epidemiol Genet (2016) 7(4):137–40.
64. Sanderson RD, Elkin M, Rapraeger AC, Ilan N, Vlodavsky I. Heparanase regulation of cancer, autophagy and inflammation: new mechanisms and targets for therapy. FEBS J (2017) 284(1):42–55. doi: 10.1111/febs.13932
65. Masola V, Zaza G, Onisto M, Lupo A, Gambaro G. Impact of heparanase on renal fibrosis. J Transl Med (2015) 13:181. doi: 10.1186/s12967-015-0538-5
66. Meirovitz A, Goldberg R, Binder A, Rubinstein AM, Hermano E, Elkin M. Heparanase in inflammation and inflammation-associated cancer. FEBS J (2013) 280(10):2307–19. doi: 10.1111/febs.12184
67. Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol (2006) 38(12):2018–39. doi: 10.1016/j.biocel.2006.06.004
68. Nadir Y, Brenner B. Heparanase multiple effects in cancer. Thromb Res (2014) 133 Suppl 2:S90–94. doi: 10.1016/S0049-3848(14)50015-1
69. Zhou Y, Song B, Qin W-J, Zhang G, Zhang R, Luan Q, et al. Heparanase promotes bone destruction and invasiveness in prostate cancer. Cancer Lett (2008) 268(2):252–9. doi: 10.1016/j.canlet.2008.04.008
70. Kutsenko OS, Kovner AV, Mostovich LA, Kunin IS, Nepomnyashchikh RD, Prudnikova TY, et al. Expression of heparanase-1 in prostate gland tumors. Bull Exp Biol Med (2012) 152(3):344–7. doi: 10.1007/s10517-012-1524-z
71. Masola V, Onisto M, Zaza G, Lupo A, Gambaro G. A new mechanism of action of sulodexide in diabetic nephropathy: inhibits heparanase-1 and prevents FGF-2-induced renal epithelial-mesenchymal transition. J Transl Med (2012) 10:213. doi: 10.1186/1479-5876-10-213
72. Masola V, Zaza G, Secchi MF, Gambaro G, Lupo A, Onisto M. Heparanase is a key player in renal fibrosis by regulating TGF-β expression and activity. Biochim Biophys Acta (2014) 1843(9):2122–8. doi: 10.1016/j.bbamcr.2014.06.005
73. Masola V, Zaza G, Gambaro G, Onisto M, Bellin G, Vischini G, et al. Heparanase: A potential new factor involved in the renal epithelial mesenchymal transition (EMT) induced by Ischemia/Reperfusion (I/R) injury. PLoS One (2016) 11(7):e0160074. doi: 10.1371/journal.pone.0160074
74. Abassi Z, Hamoud S, Hassan A, Khamaysi I, Nativ O, Heyman SN, et al. Involvement of heparanase in the pathogenesis of acute kidney injury: nephroprotective effect of PG545. Oncotarget (2017) 8(21):34191–204. doi: 10.18632/oncotarget.16573
75. Masola V, Granata S, Bellin G, Gambaro G, Onisto M, Rugiu C, et al. Specific heparanase inhibition reverses glucose-induced mesothelial-to-mesenchymal transition. Nephrol Dial Transplant (2017) 32(7):1145–54. doi: 10.1093/ndt/gfw403
76. He L, Sun F, Wang Y, Zhu J, Fang J, Zhang S, et al. HMGB1 exacerbates bronchiolitis obliterans syndrome via RAGE/NF-κB/HPSE signaling to enhance latent TGF-β release from ECM. Am J Transl Res (2016) 8(5):1971–84.
77. Szatmári T, Ötvös R, Hjerpe A, Dobra K. Syndecan-1 in cancer: Implications for cell signaling, differentiation, and prognostication. Dis Markers (2015) 2015:796052. doi: 10.1155/2015/796052
78. Hu Y, Sun H, Owens RT, Gu Z, Wu J, Chen YQ, et al. Syndecan-1-dependent suppression of PDK1/Akt/bad signaling by docosahexaenoic acid induces apoptosis in prostate cancer. Neoplasia (2010) 12(10):826–36. doi: 10.1593/neo.10586
79. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133(4):704–15. doi: 10.1016/j.cell.2008.03.027
80. Pirozzi G, Tirino V, Camerlingo R, Franco R, La Rocca A, Liguori E, et al. Epithelial to mesenchymal transition by TGFβ-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS One (2011) 6(6):e21548. doi: 10.1371/journal.pone.0021548
81. Bisson I, Prowse DM. WNT signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res (2009) 19(6):683–97. doi: 10.1038/cr.2009.43
82. Luo Y, Cui X, Zhao J, Han Y, Li M, Lin Y, et al. Cells susceptible to epithelial-mesenchymal transition are enriched in stem-like side population cells from prostate cancer. Oncol Rep (2014) 31(2):874–84. doi: 10.3892/or.2013.2905
83. Desai B, Ma T, Zhu J, Chellaiah MA. Characterization of the expression of variant and standard CD44 in prostate cancer cells: identification of the possible molecular mechanism of CD44/MMP9 complex formation on the cell surface. J Cell Biochem (2009) 108(1):272–84. doi: 10.1002/jcb.22248
Keywords: prostate cancer, heparanase, epithelial to mesenchymal transition, cancer stem cells, in vitro
Citation: Masola V, Franchi M, Zaza G, Atsina FM, Gambaro G and Onisto M (2022) Heparanase regulates EMT and cancer stem cell properties in prostate tumors. Front. Oncol. 12:918419. doi: 10.3389/fonc.2022.918419
Received: 12 April 2022; Accepted: 27 June 2022;
Published: 27 July 2022.
Edited by:
Alberto Passi, University of Insubria, ItalyReviewed by:
Taize M. Augusto, Faculty of Medicine of Jundiai, BrazilPaul J. Higgins, Albany Medical College, United States
Neta Ilan, Technion Israel Institute of Technology, Israel
Copyright © 2022 Masola, Franchi, Zaza, Atsina, Gambaro and Onisto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurizio Onisto, maurizio.onisto@unipd.it; Valentina Masola, valentina.masola@unipd.it