 Takashi Toya
Takashi Toya Hironori Harada
Hironori Harada Yuka Harada3
Yuka Harada3- 1Hematology Division, Tokyo Metropolitan Cancer and Infectious Diseases Center, Komagome Hospital, Tokyo, Japan
- 2Laboratory of Oncology, School of Life Sciences, Tokyo University of Pharmacy & Life Sciences, Tokyo, Japan
- 3Clinical Research Support Center, Tokyo Metropolitan Cancer and Infectious Diseases Center, Komagome Hospital, Tokyo, Japan
Hereditary myeloid malignancies, especially in adults or elderly persons, had been considered quite rare before the next-generation sequencing era; however, increased usage of clinical sequencing has revealed much higher prevalence of inherited myeloid malignancies. DDX41 and various pathogenic germline mutations have newly been recognized as the cause of adult-onset familial leukemia and myeloid malignancies. Although germline predisposition to myeloid neoplasms had been categorized as a provisional entity in the World Health Organization classification of hematopoietic neoplasms in 2016, methodology for the identification of hereditary myeloid malignancies has not been fully established yet. In addition, many unresolved problems, such as epidemiology, the exact pathogenic mechanisms, and ideal treatment strategy, including indications of allogeneic hematopoietic stem cell transplantation, still remain. Related donor selection for stem cell transplant is a particularly sensitive issue due to the possibility of germline mutation of the candidate relatives and the risk of donor cell leukemia after transplantation. Here, we reviewed the current evidence regarding epidemiology, diagnosis, mechanisms of progression, and transplantation strategy for hereditary myeloid malignancies.
Introduction
Hereditary myeloid malignancies (HMMs) are myeloid neoplasms that arise in individuals with germline mutations associated with increased risk of myeloid malignancies. The first report of HMMs were published in 1861, although the causality remained unclear (1); HMMs had been considered an extremely rare disease for a long time. In 1999, germline RUNX1 mutation was identified, for the first time, as the genetic background of familial platelet disorder with predisposition to myeloid malignancy (FPD-MM). Subsequent advent of next-generation sequencing (NGS) era unraveled HMMs as much more manifold and common diseases than considered earlier (2–6). In 2016, HMMs were defined as “myeloid neoplasms with germline predisposition” in the revised fourth edition of the World Health Organization (WHO) classification of myeloid neoplasms (7), and were renamed as “myeloid neoplasms associated with germline predisposition” in the fifth edition of WHO classification (8). Candidate genes associated with HMMs, such as GATA2, CEBPA, ETV6, ANKRD26, SAMD9, and SAMD9L, have been discovered subsequently (9). Recent reports have shown that approximately 5–10% of patients with hematological malignancy carry a germline variant (10). However, the penetrance is not 100% in HMMs, since not all patients with a germline variant develop hematological malignancy. Moreover, de novo germline variant may occur where an obvious family history could be lacking. Some cases with HMMs are often elderly-onset, especially in individuals with germline DDX41 mutations (11). Therefore, the accurate and prompt diagnosis of HMMs is often challenging.
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative intervention for HMMs. However, the optimal strategy for HSCT, including indication, timing, donor selection, conditioning regimen, and toxicity management has not been fully elucidated yet. Although many guidelines recommend HSCT for HMMs, data regarding transplant outcome are highly limited (12). In this manuscript, we reviewed the pathophysiology, clinical characteristics, and recent evidence regarding HSCT for HMMs. The review focused on adult-onset HMMs, which often get involved with solitary adult myeloid malignancies in clinical practice. Some child-onset hereditary disorders are beyond the scope of the current review, and the relevant guidelines should be perused in that regard.
Clinical features corresponding to each mutation
In general, HMMs are classified into three groups (Table 1), namely (1) myeloid neoplasms with germline predisposition and pre-existing platelet disorder (RUNX1, ANKRD26, and ETV6), (2) myeloid neoplasms with germline predisposition and potential organ dysfunction (GATA2, SAMD9, SAMD9L, etc.), and (3) myeloid neoplasms with germline predisposition without a preexisting platelet disorder or organ dysfunction (CEBPA and DDX41). Four representative genes responsible for adult-onset HMMs, namely RUNX1, GATA2, CEBPA, and DDX41, are described in detail below.

Table 1 Major subtypes of myeloid neoplasms associated with germline predisposition8.
RUNX1
RUNX1 is a critical transcription factor for hematopoiesis; RUNX1 mutation is recurrently detected and is a poor prognostic marker in myeloid malignancies (24). FPD-MM is the first HMMs whose genetic causality was recognized in 1999 (20). A germline RUNX1 mutation is associated with predisposition to mainly acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) while some cases with various lymphoid malignancies have also been reported (25–28). FPD-MM accounts for 8–30% of AML cases with RUNX1 mutations (29–33).
Chronic thrombocytopenia and/or bleeding tendency is a characteristic of FPD-MM pedigree. Some pedigrees may be considered as immune thrombocytopenic purpura. Approximately 40% of the family members with blood relationship develop myeloid malignancy during their lifetime (34), although significant heterogeneity is evident across families (35). Churpek et al. had reported that cumulative risk of developing clonal hematopoiesis by 50 years of age is > 80% (36). Heterozygous RUNX1 germline mutation alone is not sufficient for leukemogenesis in this context, and additionally acquired mutations, such as gene mutations in ASXL1, FLT3, WT1 and CDC25C, may also be important for malignant transformation (37–39).
GATA2
MonoMAC syndrome is characterized by monocytopenia, B lymphocytopenia, pulmonary alveolar proteinosis (PAP), and frequent M. avium complex infection. Patients with Emberger syndrome exhibit primary lymphoedema, cutaneous warts, and sensorineural deafness. Dendritic cell, monocyte, and B- and NK-lymphoid deficiencies occur with DCML deficiency. The patients frequently develop familial MDS/AML, although the pathogenesis remains to be clarified. In 2011, Hahn et al. revealed that loss-of-function germline GATA-binding protein 2 (GATA2) mutations are associated with all the above-mentioned syndromes (22). Currently, the syndromes are collectively called GATA2 deficiency. Opportunistic viral and mycobacterial infections are frequently observed in patients with GATA2 deficiency. Interestingly, interindividual variation of clinical phenotypes is significant even among patients with the same germline mutation (21).
GATA2 deficiency is the most common cause of childhood MDS, and monosomy 7 and ASXL1 mutation are often accompanied by the development of MDS and AML in cases with germline GATA2 mutation (40). Donadieu et al. analyzed 79 patients with GATA2 deficiency and reported 92% of them to develop some symptom at 40 years of age (41).
CEBPA
Germline mutations in CCAAT enhancer binding protein alpha (CEBPA) that predispose an individual to AML were first reported in 2004 (42). Among the patients with AML having CEBPA mutations, approximately 7–11% had those of germline origin (43, 44). Individuals with a germline CEBPA mutation do not have specific phenotype before leukemia onset. In general, the onset age of leukemic progression in patients with germline CEBPA mutations is lower than that in patients with germline RUNX1 and GATA2 mutations (13). Importantly, CEBPA germline mutations are autosomal dominant inheritance and the penetrance of AML is reported to be nearly 100% (14).
Most patients with familial AML having germline CEBPA mutations have been reported to possess both the N-terminal germline mutation and the C-terminal acquired mutation (14, 45). N-terminal mutations are known to generate p30 isoforms, which have dominant negative effect (46). Most sporadic AML cases with CEBPA mutations were reported to possess C-terminal mutations or both (14), suggesting the importance of secondary C-terminal somatic mutations.
DDX41
Germline mutations in DEAD/H-box helicase gene (DDX41) were reported for the first time in 2015 (15). Despite being a recent discovery, DDX41 mutation is the most common genetic predisposition to MDS/AML, representing 1–5% of myeloid malignancies (11, 16, 17). Many HMMs cases with germline DDX41 mutations were accompanied by somatic DDX41 mutations (16, 47). In addition, remarkable ethnic deviation was found in the mutation sites in DDX41; however, second hit was predominantly R525H, irrespective of ethnicity (47–49). TP53 and ASXL1 somatic mutations are recurrently detected (48), although the prognostic impact of concomitant mutations is limited (50).
AML with germline DDX41 mutations has unique clinical characteristics, such as approximately 3:1 male predominance, frequent absence of family history, and indolent clinical course (18, 50, 51). Makishima et al. reported that the age of progression in individuals with germline DDX41 mutation was solely greater than 40 years, and penetrance of pathogenic DDX41 germline variants was estimated to be 38.5% at the age of 85. In addition, they also suggested outstanding efficacy of hypomethylating agents in patients with HMMs having DDX41 mutations (52).
Diagnosis
Considering the low prevalence of hematological neoplasms, family history of hematological disorder is a key circumstantial evidence of HMMs, and mutation analysis should be done; however, it should be noted that lack of family history cannot deny the possibility of HMMs completely (53). Patients with signs/symptoms indicative of HMMs and those younger than 50 years should undergo genetic testing (12). However, considering the frequent absence of family history and the elderly onset in patients with germline DDX41 mutations (51), genetic tests can be also applied for patients older than 50 years old.
Genetic testing of non-hematological tissue is the standard option to confirm the diagnosis of HMMs, and the most authorized method is a culture of fibroblasts obtained from a skin biopsy (54, 55). Although the technique can avoid contamination of blood cells, it is complicated, and buccal swab, nails, or hair roots are often used as alternatives (21).
In addition, if NGS panel in clinical sequencing detects a suspected mutation with variant allele frequency close to 50 or 100%, further testing should be done. Mutations remaining in hematologic complete remission (CR) also suggested the possibility of germline origin (37), although clonal hematopoiesis (CH) can as well persist (56, 57). The NGS panel approaches are able to efficiently identify single nucleotide variants and small insertion/deletion in target region, but can overlook copy number variants or loss of heterozygosity (58–60). Especially when the patient could be a candidate for HSCT, early diagnosis of HMMs is important, since the diagnosis can affect donor selection, as described below.
First-line treatment for patients with myeloid malignancies
There is mostly no evidence about first-line therapy about HMMs and the optimal treatment strategies for HMMs have not been sufficiently established. However, in general, patients with HMMs, who developed myeloid malignancies, are treated as the patients with sporadic myeloid malignancies, i.e., intensive chemotherapy for fit patients with AML and using demethylating agents for patients with MDS (21). Targeted therapy can also be administered when adequate molecular target is available, although specific data for HMMs are not yet available. Whether chemosensitivity in patients with HMMs is different from that in sporadic cases is unclear, except that CR rate of patients with AML having germline DDX41 mutations after induction chemotherapy was higher than that of patients with wild-type DDX41 (94% vs. 69%) (16). More vigorous studies to explore the suitable initial treatment strategies are warranted.
Indication and timing of transplantation
The suitable indication and timing of HSCT is still unclear. However, in general, long-term remission can only be obtained by allogeneic HSCT, and HSCT should be considered soon after diagnosis of myeloid malignancy, except for cases with germline CEBPA mutations (61).
For patients with AML having a germline CEBPA mutation, HSCT in CR1 is not routinely recommended because of modest survival outcome after chemotherapy and the risk of morbidity and mortality after HSCT (12, 21, 45). However, the appropriateness of refraining from HSCT in CR1 has not been sufficiently validated yet, because prospective and/or controlled studies are still lacking due to the rarity of the disease. Considering the high relapse rate and/or secondary leukemia development after chemotherapy (14), availability of a suitable donor, low dose intensity/density of chemotherapy due to side effects, and development of safer HSCT strategy may rationalize HSCT in CR1. In addition, prognostic impact of the mutation site has been reported in sporadic AML with CEBPA mutation (62, 63). Clinical consequences of CEBPA mutation site in AML with germline mutation should be clarified, although it is difficult due to the remarkable rarity of C-terminal CEBPA germline mutation.
Prophylactic HSCT before transformation is another strategy, but it is not commonly performed due to the risk of morbidity and mortality. However, different from other HMMs, GATA2 deficiency can be treated with HSCT before transformation. Although optimum timing and indication of HSCT in GATA2 deficiency is still unclear, frequent/severe infection and serious organ damage, such as pulmonary function, can be a trigger for launching HSCT (64). Norwegian nationwide survey revealed that approximately 80% of patients with symptomatic GATA2 deficiency need HSCT (65).
In cases with a germline variant but without cytopenia and/or dysplasia, bone marrow should be assessed at the time of diagnosis, and be followed every six months or every year (53).
Donor selection and risk of donor cell leukemia
HSCT from a donor with predisposition to myeloid malignancy should be withheld due to the risk of donor cell leukemia (DCL) (66–68). DCL is a rare post-HSCT complication and cumulative incidence has been estimated to be approximately 0.16-0.70% 15 years after HSCT (69, 70). The exact incidence of DCL in HMMs setting is unknown, but Williams et al. reviewed 19 DCL cases with genetic sequencing results and at least three of them had germline predisposing mutations (71); it suggested significant proportion of DCLs to have been derived from donors with germline mutation.
Related donor candidates of patients with HMMs should undergo genetic testing as well as HLA typing; importantly, genetic counseling before testing is also essential (12). When only a matched related donor with predisposing germline mutation is available and the relapse risk without HSCT is very high, there is no consensus about necessary and sufficient condition of the donor because the quantitative risk of DCL has not been clarified.
Genetic testing of a donor might add fuel to the problems. Gibson et al. had shown donor CH to be associated with improved recipient survival due to reduced relapse risk (70). They also showed donor DNMT3A-CH to be associated with lower relapse risk and superior survival only when post-transplant cyclophosphamide (PT-CY) GVHD prophylaxis was not used. The situation is more complicated, since Crysandt et al. suggested germline predisposition to be extended to polymorphisms (21). Inamoto et al. had reported the association between donor/recipient polymorphisms and the risk of sclerotic GVHD after HSCT (72). Should a donor candidate test HLA, germline predisposition, CH, and polymorphisms? The obligation of the bona fide donor is large, and exploration of the “perfect” donor can reduce donor availability. Suitable biomarkers for quantitative evaluation of the relevant risks are highly required.
Among alternative donors, comparison between umbilical cord blood (UCB) and haploidentical donor remains a matter of debate (73). In most patients with HMMs, little evidence exists about alternative donor selection. However, in patients with GATA2 deficiency, Grossman et al. reported poor outcome of UCB transplantation (73) and Nichols-Vinueza et al. reported excellent outcome of haploidentical transplantation with PT-CY GVHD prophylaxis (74). At least in HSCT for GATA2 deficiency, haploidentical transplantation with PT-CY may be more suitable compared with UCB transplant.
Conditioning regimen
There is no recommended conditioning regimen yet specific for patients with HMMs. In general, adult patients with HMMs are younger than patients with sporadic myeloid malignancies, and myeloablative conditioning can be applied in more cases than in sporadic cases. However, reduced intensity conditioning is preferred in patients with GATA2 deficiency due to comorbidities such as concomitant infections and PAP (73).
High-dose total body irradiation (TBI)-regimens are avoided in cases with germline DNA damage response gene mutations due to the risk of second cancer after HSCT. For example, in patients with Fanconi anemia, non-TBI- or low dose-TBI (4 Gy)-containing regimens are usually employed (75). Although some categories of HMMs, such as Fanconi anemia and Li-Fraumeni syndrome, indicate high-risk feature (19, 76), the risk of second cancer after HSCT in most patients with HMMs is unclear, and further accumulation of cases would be necessary to answer these questions.
Complications after transplantation
In patients with myeloid neoplasms and germline predisposition without potential organ dysfunction (i.e., patients with germline mutation in RUNX1, DDX41, CEBPA and so on), no specific complication after HSCT has been reported till date. However, there is no prospective as well as large-scale retrospective study which focus on transplant complications in patients with HMMs; therefore, we have limited data about post-HSCT complications in HMMs and future studies in these settings are necessary.
Adult patients with germline predisposition and potential organ dysfunction practically refer to patients with GATA2 deficiency, since nearly all patients with SAMD9/SAMD9L germline mutations, i.e. MIRAGE syndrome and Ataxia pancytopenia syndrome, are diagnosed and treated in their childhood (23). For HSCT in patients with GATA2 deficiency, variable comorbidities could complicate the transplantation procedures. For example, many patients with GATA2 deficiency suffer from nontuberculous mycobacteria (NTM) and human papillomavirus (HPV) infections in their clinical courses. However, previous reports had suggested that reactivation of NTM during HSCT could be quite rare under appropriate antimycobacterial therapy up to at least three months after transplantation (73). Drug interaction between calcineurin inhibitors (CIs) and anti-NTM drugs might affect the concentration of CIs (77), and hepatic toxicity due to anti-NTM drugs should also be noted. Reactivation of HPV after HSCT seems to be also rare (73). Patients with GATA2 deficiency frequently develop PAP, which can cause severe respiratory failure. Early HSCT may be better for lower post-HSCT morbidity and mortality in patients with PAP, although pulmonary dysfunction can also be corrected to some extent after HSCT (73, 78).
Nichols-Vinueza et al. reported the outcome of 59 HSCT recipients with GATA2 deficiency and compared transplant outcome based on GVHD prophylaxis (PT-CY vs. tacrolimus and methotrexate-based prophylaxis). PT-CY resulted in a significantly reduced incidence of grade III–IV acute GVHD (0 vs. 32%) and moderate-to-severe chronic GVHD (9 vs. 42%) without increase of relapse (0 vs. 5.2%) compared to that in patients who were administered tacrolimus and methotrexate-based prophylaxis (74). PT-CY may be a promising HSCT strategy in patients with GATA2 deficiency, although the exact mechanism of deteriorating GVHD is still unclear. Hofmann et al. comparatively analyzed the transplant outcomes of pediatric patients with GATA2 deficiency with those of patients without, and revealed that neurologic toxicities (six of 15 cases) and thrombotic events (eight out of 15 cases) were more common in patients with GATA2 deficiency, although the underlying mechanism is uncertain (79). The risk of acute and chronic GVHD was not different based on the existence of germline GATA2 mutation, and second malignancy was rare with a median follow up of nearly six years after HSCT.
Transplantation outcome
Most reports about HMMs are case reports and case series, and large-scale prospective study of HSCT outcome for HMMs is lacking. Even retrospective analysis is highly limited due to its rarity (Table 2).
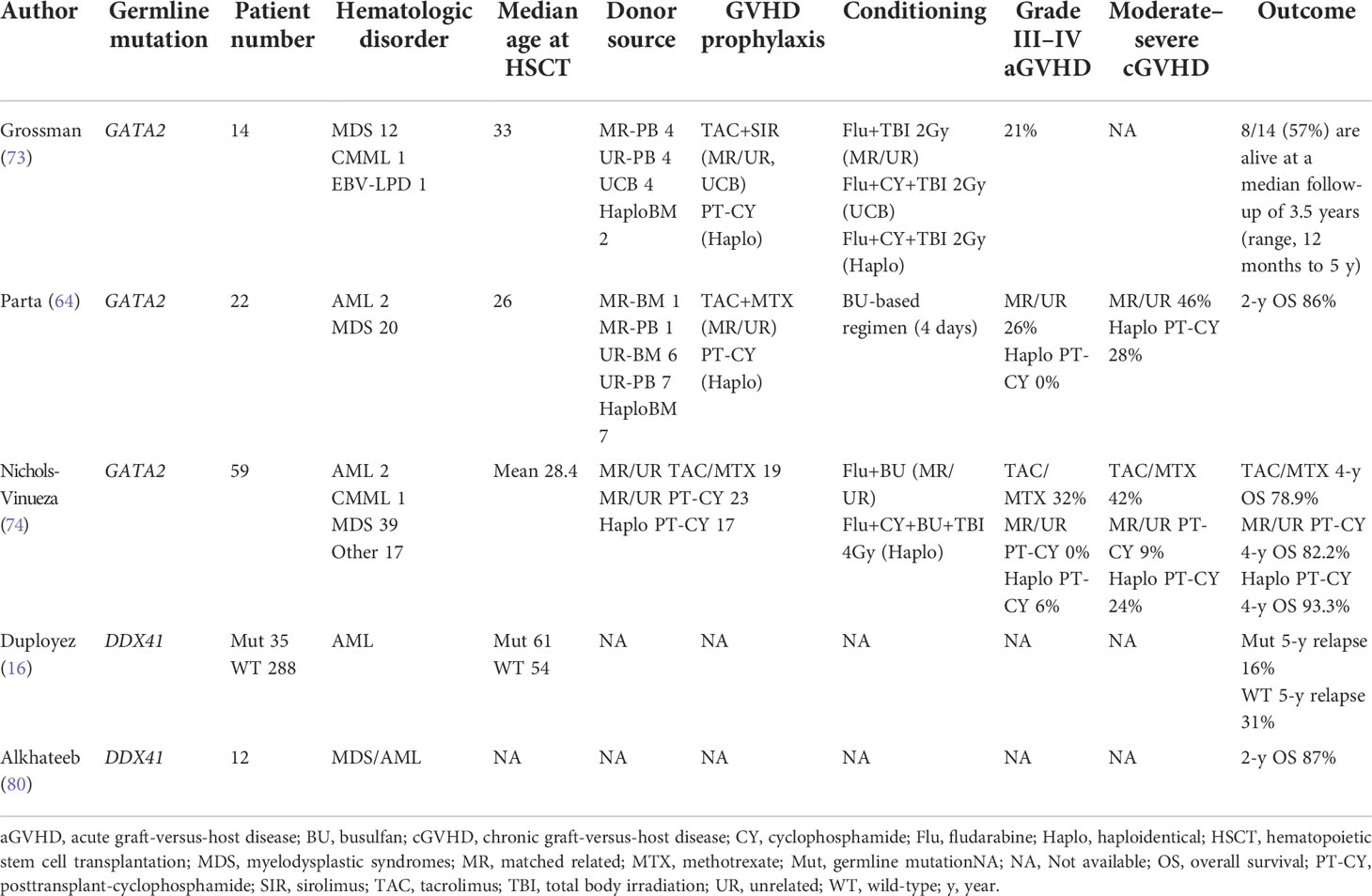
Table 2 Previous reports about HSCT for HMMs.
Since the prevalence of patients with germline DDX41 mutations is relatively high among patients with HMMs, retrospective comparison of patients with and without germline DDX41 mutations using large-scale data of some clinical trials was recently reported by Duployez et al.; 191 patients with AML having DDX41 germline mutations and 1,604 DDX41 wild-type patients with AML were compared (16). Interestingly, AML with germline DDX41 mutation displayed a specific relapse kinetics, with a lower short-term relapse rate and higher late relapse risk. Some previous reports had suggested apparently favorable outcomes in AML with germline DDX41 mutations (11, 51, 80); however, long-term follow up may be necessary to evaluate the genuine prognosis of AML with germline DDX41 mutations. Regarding the role of HSCT, Duployez et al. showed that HSCT in first CR for AML with germline DDX41 mutation may contribute to the suppression of late relapse, although OS was not significantly different (16).
Transplantation outcomes in GATA2 deficiency have been prospectively analyzed in some clinical trials (73, 74, 78), perhaps because the unique clinical features promoted participation in clinical trials. Many of the studies on GATA2 deficiency included tens of patients, mostly children, adolescents, or young adults. In children and adolescents, OS and disease-free survival (DFS) after HSCT in bone marrow failure, MDS, or AML in patients with GATA2 deficiency were 65% and 51%, and those were comparable to the values in patients without GATA2 deficiency (81). Other studies also reported good survival rate after HSCT; four- or five-year OS was 78–93% (64, 74). However, reported HSCT outcomes for GATA2 deficiency should be interpreted with caution, since the proportion of MDS with high-risk features, such as monosomy 7, and severity of concomitant organ dysfunction can affect the outcomes.
Transplant outcomes for FPD-MM, AML with germline CEBPA mutations, and other HMMs have not been reported yet, and HSCT outcomes of patients with sporadic relevant mutations still need to be extrapolated. Waidhauser et al. analyzed 674 patients with AML, who underwent HSCT in first CR (RUNX1 mutation; positive 183, negative 491). Two-year overall survival (OS) was not significantly different between RUNX1 mutation-positive and -negative (67.7 vs 66.1%, p = 0.7). Considering the poor prognosis of AML with RUNX1 mutation (33), HSCT in first CR was considered to overcome the unfavorability. In patients with AML having somatic biallelic CEBPA mutations and having underwent HSCT, five-year OS was 71.8%, which was not significantly different from that in patients with AML having somatic biallelic CEBPA mutations and receiving consolidation chemotherapy alone (82).
Concluding remarks
We summarized the concurrent available reports about HSCT in patients with HMMs. Although HMMs have recently been noticed more widely, there is too little evidence about recipients as well as donors. Increasing awareness of HMMs would ensure better management by physicians, which could surely improve patient outcomes.
Author contributions
TT wrote the manuscript. HH and YH critically revised the paper and approved the final version. ND supervised the study. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Takaoka K, Koya J, Yoshimi A, Toya T, Kobayashi T, Nannya Y, et al. Nationwide epidemiological survey of familial myelodysplastic Syndromes/Acute myeloid leukemia in Japan: A multicenter retrospective study. Leuk Lymphoma (2020) 61(7):1688–94. doi: 10.1080/10428194.2020.1734595
3. Yoshimi A, Toya T, Nannya Y, Takaoka K, Kirito K, Ito E, et al. Spectrum of clinical and genetic features of patients with inherited platelet disorder with suspected predisposition to hematological malignancies: A nationwide survey in Japan. Ann Oncol (2016) 27(5):887–95. doi: 10.1093/annonc/mdw066
4. Holme H, Hossain U, Kirwan M, Walne A, Vulliamy T, Dokal I. Marked genetic heterogeneity in familial Myelodysplasia/Acute myeloid leukaemia. Br J Haematol (2012) 158(2):242–8. doi: 10.1111/j.1365-2141.2012.09136.x
5. Yasuda T, Sanada M, Nishijima D, Kanamori T, Iijima Y, Hattori H, et al. Clinical utility of target capture-based panel sequencing in hematological malignancies: A multicenter feasibility study. Cancer Sci (2020) 111(9):3367–78. doi: 10.1111/cas.14552
6. Martin ES, Ferrer A, Mangaonkar AA, Khan SP, Kohorst MA, Joshi AY, et al. Spectrum of hematological malignancies, clonal evolution and outcomes in 144 Mayo clinic patients with germline predisposition syndromes. Am J Hematol (2021) 96(11):1450–60. doi: 10.1002/ajh.26321
7. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
8. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: Myeloid and Histiocytic/Dendritic neoplasms. Leukemia (2022) 36(7):1703–19. doi: 10.1038/s41375-022-01613-1
9. Roloff GW, Drazer MW, Godley LA. Inherited susceptibility to hematopoietic malignancies in the era of precision oncology. JCO Precis Oncol (2021) 5:107–22. doi: 10.1200/PO.20.00387
10. Kim B, Yun W, Lee ST, Choi JR, Yoo KH, Koo HH, et al. Prevalence and clinical implications of germline predisposition gene mutations in patients with acute myeloid leukemia. Sci Rep (2020) 10(1):14297. doi: 10.1038/s41598-020-71386-z
11. Sebert M, Passet M, Raimbault A, Rahme R, Raffoux E, Sicre de Fontbrune F, et al. Germline DDX41 mutations define a significant entity within adult Mds/Aml patients. Blood (2019) 134(17):1441–4. doi: 10.1182/blood.2019000909
12. Baliakas P, Tesi B, Wartiovaara-Kautto U, Stray-Pedersen A, Friis LS, Dybedal I, et al. Nordic Guidelines for germline predisposition to myeloid neoplasms in adults: Recommendations for genetic diagnosis, clinical management and follow-up. Hemasphere (2019) 3(6):e321. doi: 10.1097/HS9.0000000000000321
13. Brown AL, Hahn CN, Scott HS. Secondary leukemia in patients with germline transcription factor mutations (RUNX1, GATA2, CEBPA). Blood (2020) 136(1):24–35. doi: 10.1182/blood.2019000937
14. Tawana K, Wang J, Renneville A, Bodor C, Hills R, Loveday C, et al. Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood (2015) 126(10):1214–23. doi: 10.1182/blood-2015-05-647172
15. Polprasert C, Schulze I, Sekeres MA, Makishima H, Przychodzen B, Hosono N, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell (2015) 27(5):658–70. doi: 10.1016/j.ccell.2015.03.017
16. Duployez N, Largeaud L, Duchmann M, Kim R, Rieunier J, Lambert J, et al. Prognostic impact of DDX41 germline mutations in intensively treated acute myeloid leukemia patients: An ALFA-FILO study. Blood (2022) 140(7):756–68. doi: 10.1182/blood.2021015328
17. Bannon SA, Routbort MJ, Montalban-Bravo G, Mehta RS, Jelloul FZ, Takahashi K, et al. Next-generation sequencing of DDX41 in myeloid neoplasms leads to increased detection of germline alterations. Front Oncol (2020) 10:582213. doi: 10.3389/fonc.2020.582213
18. Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer (2021) 21(2):122–37. doi: 10.1038/s41568-020-00315-z
19. Hendrickson PG, Luo Y, Kohlmann W, Schiffman J, Maese L, Bishop AJ, et al. Radiation therapy and secondary malignancy in Li-fraumeni syndrome: A hereditary cancer registry study. Cancer Med (2020) 9(21):7954–63. doi: 10.1002/cam4.3427
20. Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet (1999) 23(2):166–75. doi: 10.1038/13793
21. Crysandt M, Brings K, Beier F, Thiede C, Brummendorf TH, Jost E. Germ line predisposition to myeloid malignancies appearing in adulthood. Expert Rev Hematol (2018) 11(8):625–36. doi: 10.1080/17474086.2018.1494566
22. Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet (2011) 43(10):1012–7. doi: 10.1038/ng.913
23. Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet (2016) 48(7):792–7. doi: 10.1038/ng.3569
24. Hayashi Y, Harada Y, Harada H. Myeloid neoplasms and clonal hematopoiesis from the Runx1 perspective. Leukemia (2022) 36(5):1203–14. doi: 10.1038/s41375-022-01548-7
25. Nishimoto N, Imai Y, Ueda K, Nakagawa M, Shinohara A, Ichikawa M, et al. T Cell acute lymphoblastic leukemia arising from familial platelet disorder. Int J Hematol (2010) 92(1):194–7. doi: 10.1007/s12185-010-0612-y
26. Li Y, Yang W, Devidas M, Winter SS, Kesserwan C, Yang W, et al. Germline RUNX1 variation and predisposition to childhood acute lymphoblastic leukemia. J Clin Invest (2021) 131(17):e147898. doi: 10.1172/JCI147898
27. Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood (2017) 129(15):2070–82. doi: 10.1182/blood-2016-10-687830
28. Toya T, Yoshimi A, Morioka T, Arai S, Ichikawa M, Usuki K, et al. Development of hairy cell leukemia in familial platelet disorder with predisposition to acute myeloid leukemia. Platelets (2014) 25(4):300–2. doi: 10.3109/09537104.2013.818636
29. Ernst MPT, Kavelaars FG, Lowenberg B, Valk PJM, Raaijmakers M. RUNX1 germline variants in RUNX1-mutant AML: How frequent? Blood (2021) 137(10):1428–31. doi: 10.1182/blood.2020008478
30. Simon L, Spinella JF, Yao CY, Lavallee VP, Boivin I, Boucher G, et al. High frequency of germline RUNX1 mutations in patients with RUNX1-mutated AML. Blood (2020) 135(21):1882–6. doi: 10.1182/blood.2019003357
31. Drazer MW, Kadri S, Sukhanova M, Patil SA, West AH, Feurstein S, et al. Prognostic tumor sequencing panels frequently identify germ line variants associated with hereditary hematopoietic malignancies. Blood Adv (2018) 2(2):146–50. doi: 10.1182/bloodadvances.2017013037
32. Gaidzik VI, Teleanu V, Papaemmanuil E, Weber D, Paschka P, Hahn J, et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia (2016) 30(11):2282. doi: 10.1038/leu.2016.207
33. Mendler JH, Maharry K, Radmacher MD, Mrozek K, Becker H, Metzeler KH, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and microrna expression signatures. J Clin Oncol (2012) 30(25):3109–18. doi: 10.1200/JCO.2011.40.6652
34. Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia–a review. Br J Haematol (2008) 140(2):123–32. doi: 10.1111/j.1365-2141.2007.06909.x
35. Brown AL, Arts P, Carmichael CL, Babic M, Dobbins J, Chong CE, et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv (2020) 4(6):1131–44. doi: 10.1182/bloodadvances.2019000901
36. Churpek JE, Pyrtel K, Kanchi KL, Shao J, Koboldt D, Miller CA, et al. Genomic analysis of germ line and somatic variants in familial Myelodysplasia/Acute myeloid leukemia. Blood (2015) 126(22):2484–90. doi: 10.1182/blood-2015-04-641100
37. Yoshimi A, Toya T, Kawazu M, Ueno T, Tsukamoto A, Iizuka H, et al. Recurrent CDC25C mutations drive malignant transformation in Fpd/Aml. Nat Commun (2014) 5:4770. doi: 10.1038/ncomms5770
38. Forster A, Decker M, Schlegelberger B, Ripperger T. Beyond pathogenic RUNX1 germline variants: The spectrum of somatic alterations in RUNX1-familial platelet disorder with predisposition to hematologic malignancies. Cancers (Basel) (2022) 14(14):3431. doi: 10.3390/cancers14143431
39. Ripperger T, Steinemann D, Gohring G, Finke J, Niemeyer CM, Strahm B, et al. A novel pedigree with heterozygous germline RUNX1 mutation causing familial MDS-related AML: Can these families serve as a multistep model for leukemic transformation? Leukemia (2009) 23(7):1364–6. doi: 10.1038/leu.2009.87
40. Wlodarski MW, Hirabayashi S, Pastor V, Stary J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood (2016) 127(11):1387–97. doi: 10.1182/blood-2015-09-669937
41. Donadieu J, Lamant M, Fieschi C, de Fontbrune FS, Caye A, Ouachee M, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica (2018) 103(8):1278–87. doi: 10.3324/haematol.2017.181909
42. Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med (2004) 351(23):2403–7. doi: 10.1056/NEJMoa041331
43. Pabst T, Eyholzer M, Haefliger S, Schardt J, Mueller BU. Somatic CEBPA mutations are a frequent second event in families with germline CEBPA mutations and familial acute myeloid leukemia. J Clin Oncol (2008) 26(31):5088–93. doi: 10.1200/JCO.2008.16.5563
44. Taskesen E, Bullinger L, Corbacioglu A, Sanders MA, Erpelinck CA, Wouters BJ, et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: Further evidence for CEBPA double mutant aml as a distinctive disease entity. Blood (2011) 117(8):2469–75. doi: 10.1182/blood-2010-09-307280
45. Su L, Shi YY, Liu ZY, Gao SJ. Acute myeloid leukemia with CEBPA mutations: Current progress and future directions. Front Oncol (2022) 12:806137. doi: 10.3389/fonc.2022.806137
46. Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/Enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet (2001) 27(3):263–70. doi: 10.1038/85820
47. Choi EJ, Cho YU, Hur EH, Jang S, Kim N, Park HS, et al. Unique ethnic features of DDX41 mutations in patients with idiopathic cytopenia of undetermined significance, myelodysplastic syndrome, or acute myeloid leukemia. Haematologica (2022) 107(2):510–8. doi: 10.3324/haematol.2020.270553
48. Quesada AE, Routbort MJ, DiNardo CD, Bueso-Ramos CE, Kanagal-Shamanna R, Khoury JD, et al. DDX41 mutations in myeloid neoplasms are associated with Male gender, TP53 mutations and high-risk disease. Am J Hematol (2019) 94(7):757–66. doi: 10.1002/ajh.25486
49. Makishima H. [Genomic aberrations in myelodysplastic syndromes and related disorders]. Rinsho Ketsueki (2019) 60(6):600–9. doi: 10.11406/rinketsu.60.600
50. Li P, Brown S, Williams M, White T, Xie W, Cui W, et al. The genetic landscape of germline DDX41 variants predisposing to myeloid neoplasms. Blood (2022) 140(7):716–55. doi: 10.1182/blood.2021015135
51. Li P, White T, Xie W, Cui W, Peker D, Zeng G, et al. Aml with germline DDX41 variants is a clinicopathologically distinct entity with an indolent clinical course and favorable outcome. Leukemia (2022) 36(3):664–74. doi: 10.1038/s41375-021-01404-0
52. Makishima H, Saiki R, Nannya Y, Korotev S, Gurnari C, Takeda J, et al. P741: DDX41 mutations define a unique subtype of myeloid neoplasms. HemaSphere (2022) 6:636–7. doi: 10.1097/01.HS9.0000845848.97346.3e
53. Schlegelberger B, Mecucci C, Wlodarski M. Review of guidelines for the identification and clinical care of patients with genetic predisposition for hematological malignancies. Fam Cancer (2021) 20(4):295–303. doi: 10.1007/s10689-021-00263-z
54. Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood (2017) 129(4):424–47. doi: 10.1182/blood-2016-08-733196
55. Churpek JE, Lorenz R, Nedumgottil S, Onel K, Olopade OI, Sorrell A, et al. Proposal for the clinical detection and management of patients and their family members with familial myelodysplastic Syndrome/Acute leukemia predisposition syndromes. Leuk Lymphoma (2013) 54(1):28–35. doi: 10.3109/10428194.2012.701738
56. Morita K, Kantarjian HM, Wang F, Yan Y, Bueso-Ramos C, Sasaki K, et al. Clearance of somatic mutations at remission and the risk of relapse in acute myeloid leukemia. J Clin Oncol (2018) 36(18):1788–97. doi: 10.1200/JCO.2017.77.6757
57. Tanaka T, Morita K, Loghavi S, Wang F, Furudate K, Sasaki Y, et al. Clonal dynamics and clinical implications of postremission clonal hematopoiesis in acute myeloid leukemia. Blood (2021) 138(18):1733–9. doi: 10.1182/blood.2020010483
58. Tawana K, Brown AL, Churpek JE. Integrating germline variant assessment into routine clinical practice for myelodysplastic syndrome and acute myeloid leukaemia: Current strategies and challenges. Br J Haematol (2022) 196(6):1293–310. doi: 10.1111/bjh.17855
59. Engvall M, Karlsson Y, Kuchinskaya E, Jornegren A, Mathot L, Pandzic T, et al. Familial platelet disorder due to germline exonic deletions in RUNX1: A diagnostic challenge with distinct alterations of the transcript isoform equilibrium. Leuk Lymphoma (2022) 1-10. doi: 10.1080/10428194.2022.2067997
60. Feurstein S, Drazer M, Godley LA. Germline predisposition to hematopoietic malignancies. Hum Mol Genet (2021) 30(20):R225–R35. doi: 10.1093/hmg/ddab141
61. Kohlmann W, Schiffman JD. Discussing and managing hematologic germ line variants. Blood (2016) 128(21):2497–503. doi: 10.1182/blood-2016-06-716704
62. Wakita S, Sakaguchi M, Oh I, Kako S, Toya T, Najima Y, et al. Prognostic impact of CEBPA bZIP domain mutation in acute myeloid leukemia. Blood Adv (2022) 6(1):238–47. doi: 10.1182/bloodadvances.2021004292
63. Tarlock K, Lamble AJ, Wang YC, Gerbing RB, Ries RE, Loken MR, et al. CEBPA-bZIP mutations are associated with favorable prognosis in de novo AML: A report from the children’s oncology group. Blood (2021) 138(13):1137–47. doi: 10.1182/blood.2020009652
64. Parta M, Shah NN, Baird K, Rafei H, Calvo KR, Hughes T, et al. Allogeneic hematopoietic stem cell transplantation for GATA2 deficiency using a busulfan-based regimen. Biol Blood Marrow Transplant (2018) 24(6):1250–9. doi: 10.1016/j.bbmt.2018.01.030
65. Jorgensen SF, Buechner J, Myhre AE, Galteland E, Spetalen S, Kulseth MA, et al. A nationwide study of GATA2 deficiency in Norway-the majority of patients have undergone allo-HSCT. J Clin Immunol (2022) 42(2):404–20. doi: 10.1007/s10875-021-01189-y
66. Galera P, Hsu AP, Wang W, Droll S, Chen R, Schwartz JR, et al. Donor-derived MDS/AML in families with germline GATA2 mutation. Blood (2018) 132(18):1994–8. doi: 10.1182/blood-2018-07-861070
67. Berger G, van den Berg E, Sikkema-Raddatz B, Abbott KM, Sinke RJ, Bungener LB, et al. Re-emergence of acute myeloid leukemia in donor cells following allogeneic transplantation in a family with a germline DDX41 mutation. Leukemia (2017) 31(2):520–2. doi: 10.1038/leu.2016.310
68. Kobayashi S, Kobayashi A, Osawa Y, Nagao S, Takano K, Okada Y, et al. Donor cell leukemia arising from preleukemic clones with a novel germline DDX41 mutation after allogenic hematopoietic stem cell transplantation. Leukemia (2017) 31(4):1020–2. doi: 10.1038/leu.2017.44
69. Kato M, Yamashita T, Suzuki R, Matsumoto K, Nishimori H, Takahashi S, et al. Donor cell-derived hematological malignancy: A survey by the Japan society for hematopoietic cell transplantation. Leukemia (2016) 30(8):1742–5. doi: 10.1038/leu.2016.23
70. Gibson CJ, Kim HT, Zhao L, Murdock HM, Hambley B, Ogata A, et al. Donor clonal hematopoiesis and recipient outcomes after transplantation. J Clin Oncol (2022) 40(2):189–201. doi: 10.1200/JCO.21.02286
71. Williams L, Doucette K, Karp JE, Lai C. Genetics of donor cell leukemia in acute myelogenous leukemia and myelodysplastic syndrome. Bone Marrow Transplant (2021) 56(7):1535–49. doi: 10.1038/s41409-021-01214-z
72. Inamoto Y, Martin PJ, Flowers ME, Lee SJ, Carpenter PA, Warren EH, et al. Genetic risk factors for sclerotic graft-Versus-Host disease. Blood (2016) 128(11):1516–24. doi: 10.1182/blood-2016-05-715342
73. Grossman J, Cuellar-Rodriguez J, Gea-Banacloche J, Zerbe C, Calvo K, Hughes T, et al. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biol Blood Marrow Transplant (2014) 20(12):1940–8. doi: 10.1016/j.bbmt.2014.08.004
74. Nichols-Vinueza DX, Parta M, Shah NN, Cuellar-Rodriguez JM, Bauer TR Jr., West RR, et al. Donor source and post-transplantation cyclophosphamide influence outcome in allogeneic stem cell transplantation for GATA2 deficiency. Br J Haematol (2022) 196(1):169–78. doi: 10.1111/bjh.17840
75. Sakaguchi H, Yoshida N. Recent advances in hematopoietic cell transplantation for inherited bone marrow failure syndromes. Int J Hematol (2022) 116(1):16–27. doi: 10.1007/s12185-022-03362-4
76. Savage SA, Walsh MF. Myelodysplastic syndrome, acute myeloid leukemia, and cancer surveillance in fanconi anemia. Hematol Oncol Clin North Am (2018) 32(4):657–68. doi: 10.1016/j.hoc.2018.04.002
77. Ibrahim RB, Abella EM, Chandrasekar PH. Tacrolimus-clarithromycin interaction in a patient receiving bone marrow transplantation. Ann Pharmacother (2002) 36(12):1971–2. doi: 10.1345/aph.1C117
78. van Lier YF, de Bree GJ, Jonkers RE, Roelofs J, Ten Berge IJM, Rutten CE, et al. Allogeneic hematopoietic cell transplantation in the management of GATA2 deficiency and pulmonary alveolar proteinosis. Clin Immunol (2020) 218:108522. doi: 10.1016/j.clim.2020.108522
79. Hofmann I, Avagyan S, Stetson A, Guo D, Al-Sayegh H, London WB, et al. Comparison of outcomes of myeloablative allogeneic stem cell transplantation for pediatric patients with bone marrow failure, myelodysplastic syndrome and acute myeloid leukemia with and without germline GATA2 mutations. Biol Blood Marrow Transplant (2020) 26(6):1124–30. doi: 10.1016/j.bbmt.2020.02.015
80. Alkhateeb HB, Nanaa A, Viswanatha D, Foran JM, Badar T, Sproat L, et al. Genetic features and clinical outcomes of patients with isolated and comutated DDX41-mutated myeloid neoplasms. Blood Adv (2022) 6(2):528–32. doi: 10.1182/bloodadvances.2021005738
81. Bortnick R, Wlodarski M, de Haas V, De Moerloose B, Dworzak M, Hasle H, et al. Hematopoietic stem cell transplantation in children and adolescents with GATA2-related myelodysplastic syndrome. Bone Marrow Transplant (2021) 56(11):2732–41. doi: 10.1038/s41409-021-01374-y
Keywords: hereditary myeloid malignancy, germline mutation, genetic testing, allogeneic stem cell transplantation, donor cell leukemia
Citation: Toya T, Harada H, Harada Y and Doki N (2022) Adult-onset hereditary myeloid malignancy and allogeneic stem cell transplantation. Front. Oncol. 12:997530. doi: 10.3389/fonc.2022.997530
Received: 19 July 2022; Accepted: 31 August 2022;
Published: 16 September 2022.
Edited by:
Makiko Mochizuki-Kashio, Tokyo Women’s Medical University, JapanReviewed by:
Amina Metidji, St. Jude Children’s Research Hospital, United StatesShunsuke Kimura, St. Jude Children’s Research Hospital, United States
Copyright © 2022 Toya, Harada, Harada and Doki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hironori Harada, hharada@toyaku.ac.jp