 Chih-Chung Lin1†
Chih-Chung Lin1† Wei-Ning Lin
Wei-Ning Lin Rou-Ling Cho
Rou-Ling Cho Chen-yu Wang
Chen-yu Wang Li-Der Hsiao
Li-Der Hsiao Chuen-Mao Yang
Chuen-Mao Yang- 1Department of Anesthetics, Chang Gung Memorial Hospital at Linkou and College of Medicine, Chang Gung University, Tao-Yuan, Taiwan
- 2Graduate Institute of Basic Medicine, Fu Jen Catholic University, New Taipei City, Taiwan
- 3Department of Physiology and Pharmacology and Health Aging Research Center, College of Medicine, Chang Gung University, Tao-Yuan, Taiwan
- 4Research Center for Industry of Human Ecology, Research Center for Chinese Herbal Medicine, and Graduate Institute of Health Industry Technology, Chang Gung University of Science and Technology, Tao-Yuan, Taiwan
Tumor necrosis factor-α (TNF-α) triggers activation of cytosolic phospholipase A2 (cPLA2) and then enhancing the synthesis of prostaglandin (PG) in inflammatory diseases. However, the detailed mechanisms of TNF-α induced cPLA2 expression were not fully defined in human pulmonary alveolar epithelial cells (HPAEpiCs). We found that TNF-α-stimulated increases in cPLA2 mRNA (5.2 folds) and protein (3.9 folds) expression, promoter activity (4.3 folds), and PGE2 secretion (4.7 folds) in HPAEpiCs, determined by Western blot, real-time PCR, promoter activity assay and PGE2 ELISA kit. These TNF-α-mediated responses were abrogated by the inhibitors of NADPH oxidase [apocynin (APO) and diphenyleneiodonium chloride (DPI)], ROS [N-acetyl cysteine, (NAC)], NF-κB (Bay11-7082) and transfection with siRNA of ASK1, p47phox, TRAF2, NIK, IKKα, IKKβ, or p65. TNF-α markedly stimulated NADPH oxidase activation and ROS including superoxide and hydrogen peroxide production which were inhibited by pretreatment with a TNFR1 neutralizing antibody, APO, DPI or transfection with siRNA of TRAF2, ASK1, or p47phox. In addition, TNF-α also stimulated p47phox phosphorylation and translocation in a time-dependent manner. On the other hand, TNF-α induced TNFR1, TRAF2, ASK1, and p47phox complex formation in HPAEpiCs, which were attenuated by a TNF-α neutralizing antibody. We found that pretreatment with NAC, DPI, or APO also attenuated the TNF-α-stimulated IKKα/β and NF-κB p65 phosphorylation, NF-κB (p65) translocation, and NF-κB promoter activity in HPAEpiCs. Finally, we observed that TNF-α-stimulated NADPH oxidase activation and ROS generation activates NF-κB through the NIK/IKKα/β pathway. Taken together, our results demonstrated that in HPAEpiCs, up-regulation of cPLA2 by TNF-α is, at least in part, mediated through the cooperation of TNFR1, TRAF2, ASK1, and NADPH oxidase leading to ROS generation and ultimately activates NF-κB pathway.
Introduction
The occurrence and exacerbation of lung diseases, including chronic obstructive pulmonary disease (COPD) and asthma, is dependent on the severity of lung inflammation (Lee and Yang, 2012). Eicosanoids, one of lipid mediators generating from conversion of arachidonic acid (AA), have been found in situ in airway secretion of asthmatics (Barnes, 1989; Henderson et al., 2002). Phospholipase A2 (PLA2) enzymes catalyze the hydrolysis of membrane phospholipids resulting in the release of AA (Borsch-Haubold et al., 1998). The constitutive enzyme cyclooxygenase (COX)-1 or the inducible COX-2 then converts AA to prostaglandins (PGs), such as PGE2 (Yang et al., 2002; Hsieh et al., 2006). Three PLA2 have been identified including secretory PLA2, the 85 kDa cytosolic group IV PLA2 (cPLA2), and a calcium-independent group VI PLA2 in mammalian cells (Six and Dennis, 2000). cPLA2 plays a major role in agonist-induced AA release and eicosanoid production (Leslie, 1997). Involvement of cPLA2 in sepsis-related acute lung injury (Nagase et al., 2000) and anaphylaxis-associated bronchial reactivity has been proved (Uozumi et al., 1997). Furthermore, PGE2 synthesis increases are dependent on upregulation of cPLA2 activity in various cell types (Dieter et al., 2002; Gilroy et al., 2004). Elevated levels of TNF-α have been detected in the bronchoalveolar lavage fluid of asthmatic patients. TNF-α could exaggerate inflammatory responses through up-regulation of inflammatory genes, such as cPLA2 (Hulkower et al., 1994; Van Putten et al., 2001). Up-regulation of cPLA2 further catalyzes the hydrolysis of membrane phospholipids and releases AA served as a substrate for PGs synthesis (i.e., PGE2) that augments lung inflammation. Moreover, our previous findings also provided insights into the correlation between COX-2 and cPLA2 expression in ATPγS-stimulated vascular smooth muscle cells (VSMCs) with similar molecular mechanisms and functional coupling to amplify the occurrence of vascular inflammation (Lin et al., 2009). Therefore, the synthesis of PGE2 could be a good index of AA release that is more sensitive than [3H]AA mobilization (Berenbaum et al., 2003). In this study, although the effect of TNF-α on COX-2 expression was not investigated, we tested the effect of TNF-α on PGE2 synthesis as a parameter of cPLA2 activity in human pulmonary alveolar epithelial cells (HPAEpiCs). Therefore, up-regulation of cPLA2 may play a key role in local and systemic inflammation in airway diseases. However, the molecular mechanisms by which TNF-α induces cPLA2 expression and PGE2 synthesis in HPAEpiCs are not completely understood.
Previous report indicates that TNF-α binds to distinct receptors, TNFR1 and TNFR2, and triggers various inflammatory responses (Lee et al., 2009). The association of TNF-α and TNFR1 modulates the severity of tissue injury via activation of proinflammatory or programmed cell death pathway (van Vliet et al., 2005; Lee et al., 2009). TNF receptor associated factor 2 (TRAF2) plays an important role in innate immune and inflammatory responses. However, the interaction among TNF-α, TNFR1, TRAF2 and downstream components leading to cPLA2 expression is still unknown in HPAEpiCs.
Reactive oxygen species (ROS) are products of normal cellular metabolism acting as second messengers (Lee and Yang, 2012). However, either reduced nicotinamide adenine dinucleotide phosphate (NADPH) by pro-inflammatory cytokines such as TNF-α or the mitochondrial electron transport chain and xanthine oxidase leads to increased production of ROS and unbalance of cellular oxidative stress, which are causes of airway/lung damages and subsequently respiratory inflammatory diseases/injuries (Lee and Yang, 2012). Apoptosis signal-regulating kinase 1 (ASK1), a mitogen-activated protein kinase kinase kinase, participates in regulating stress and immune responses. ASK1 is activated by cytokines and various environmental and cellular stresses. Hsu et al. indicated that peptidoglycan (PGN) induced COX-2 expression via an ASK1 signaling in A549 cells (Hsu et al., 2010). Therefore, we explored whether TNFR1, TRAF2, ASK1, and NADPH oxidase/ROS are involved in TNF-α-induced cPLA2 expression and PGE2 release.
NF-κB plays major roles not only in the evolution but also in the resolution of inflammatory responses. A wide spectrum of biological effects including immune and stress-induced responses, proliferation, differentiation, tumorigenesis, apoptosis, and tissue remodeling are all controlled by activated NF-κB (Lee and Yang, 2012). The activation of NF-κB can be regulated by various extracellular stimuli, including cytokines and oxidative stress (Lee and Yang, 2012). We noticed that ROS generation can impact NF-κB signaling pathways (Morgan and Liu, 2011). In addition, NF-κB modulates cPLA2 gene activity in various cell types (Luo et al., 2006; Huwiler et al., 2012; Chi et al., 2014). Therefore, we examined whether TNF-α regulates cPLA2 expression via ROS-dependent NF-κB activation in HPAEpiCs.
In addressing these questions, the experiments were performed to investigate the mechanisms underlying TNF-α-induced cPLA2 expression and PGE2 synthesis in HPAEpiCs. These findings suggested that TNF-α-induced cPLA2-expression associated PGE2 release is, at least in part, mediated through a TNFR1/TRAF2/ASK1/p47phox/NADPH oxidase/ROS/NIK/IKKα/β/NF-κB pathway in these cells.
Materials and Methods
Materials
Recombinant human TNF-α was from R&D System (Minneapolis, MN, USA). Anti-cPLA2 (sc-454), anti-p47phox (sc-14015), anti-Gαs (sc-823), anti-TRAF2 (sc-7346), anti-ASK1 (sc-5294), anti-TNFR1 (sc-52739), anti-NIK (sc7211), anti-IKKα (sc7218), anti-IKKβ (sc8014), anti-p65 (sc-7151) and anti-phospho-serine (sc-81515) antibodies were from Santa Cruz (Santa Cruz, CA). An anti-GAPDH antibody (#MCA-1D4) was from Encor (Gainesville, FL, USA). Human TNF-α neutralizing antibody (#7321), anti-phospho-tyrosine (#9411), anti-phospho-ASK1 (#3765), anti-phospho-p65 (#3031), and anti-phospho-IKKα/β (#2697) antibodies were from Cell Signaling (Danvers, MA, USA). N-Acetyl cysteine (NAC), diphenyleneiodonium chloride (DPI), apocynin (APO), and Bay11-7082 were from Biomol (Plymouth Meeting, PA, USA). Dihydroethidium (DHE) and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) were from Molecular Probes (Eugene, OR, USA). SDS-PAGE supplies were from MDBio Inc (Taipei, Taiwan). All other reagents were from Sigma (St. Louis, MO, USA).
Cell Culture and Treatment
Human pulmonary alveolar epithelial cells (HPAEpiCs) were ordered from ScienCell Research Lab (San Diego, CA, USA). The passages 4–7 were used throughout this study. HPAEpiCs were cultured in DMEM/F12 medium containing 10% FBS, as previously described Lee et al. (2008). The growth medium was changed after 48 h and then every 3 days. The viability of HPAEpiCs after treatment with DMSO or the pharmacological inhibitors alone was determined by an XTT [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide] assay, which showed no significant differences (data not shown).
Western Blot Analysis
Serum-starved HPAEpiCs were incubated with TNF-α at 37°C for the various time points. At the end of treatment, the cells were harvested and centrifuged at 45000 × g at 4°C for 1 h to prepare the whole cell lysate, as previously described (Chi et al., 2012). The denatured samples were analyzed by 10% SDS-PAGE gels and transferred to nitrocellulose membrane. The Western blot was performed by incubation membrane with an anti-cPLA2, anti-p47phox, anti-Gαs, anti-TRAF2, anti-ASK1, anti-TNFR1, anti-NIK, anti-IKKα, anti-IKKβ, anti-p65, anti-phospho-serine or anti-GAPDH (1:1000) antibody for 24 h, and then incubated with an anti-mouse horseradish peroxidase antibody (1:2000) for 1 h. ECL reagents and UVP BioSpectrum 500 Imaging System (Upland, CA, USA) were used to detect and capture the immunoreactive bands. The image densitometry of each immunoreactive band was analyzed and quantified by the UN-SCAN-IT gel software (Orem, UT, USA).
Real-Time PCR Analysis
Total RNA of HPAEpiCs was extracted using TRIzol reagent and reverse-transcribed into cDNA. Real-time PCR using SYBR Green PCR reagents (Applied Biosystems, Branchburg, NJ, USA) and primers specific for cPLA2 and GAPDH genes were used, as previously described (Chi et al., 2012). The real-time primers were as follows: cPLA2α, forward primer: 5′-ATGATAGCTCGGACAGTGATGATGA-3′; reverse primer: 5′-CATACGATGAATCCAACTTGCTTGA-3′ and GAPDH, forward primer: 5′-CTCTGCTCCTCCTGTTCGAC-3′; reverse primer: 5′-TTAAAAGCAGCCCTGGTGAC-3′. The expression of cPLA2 was quantified by normalization to the GAPDH expression.
Measurement of Intracellular ROS Accumulation
The intracellular H2O2 levels were determined by measuring fluorescence of 2′,7′-dichloro fluorescein diacetate (DCF-DA) and the levels were determined by measuring the level of dihydroethidium (DHE), as previously described Hsu et al. (2014). The fluorescence for DCF and DHE staining was detected at 495/529 and 518/605 nm, respectively, using a fluorescence microscope (Zeiss, Axiovert 200M). For the purpose of these experiments, HPAEpiCs were washed with warm HBSS and incubated in HBSS or cell medium containing 10 μM DCFH-DA or DHE at 37°C for 45 min. Subsequently, HBSS or medium containing DCFH-DA or DHE was removed and replaced with fresh medium. HPAEpiCs were then incubated with TNF-α. Cells were washed twice with PBS and detached with trypsin/EDTA, and the fluorescence intensity of the cells was analyzed using a multi-technology reader (Thermo, Appliskan) at Ex/Em: 485/530 nm.
Determination of NADPH Oxidase Activity by Chemiluminescence Assay
HPAEpiCs (2 × 106 cells/ml) were cultured in 6-well plates and then incubated with TNF-α after growth to confluence and serum-starved. At the end of incubation, cells were harvested and centrifuged at 400 × g for 10 min at 4°C. The cell pellet was kept on ice after re-suspended by 35 μl/per well of ice-cold RPMI-1640 medium. The complex including NADPH (1 μM) or lucigenin (20 μM), 5 μl of cell suspension (0.2 × 105 cells) and a final 200 μl volume of pre-warmed (37°C) RPMI-1640 medium were prepared and the chemiluminescence was immediate recorded with an Appliskan luminometer (Thermo®) in out-of-coincidence mode. Appropriate blanks and controls were used. There was no background chemiluminescence of lucigenin in NADPH or NADH alone group (30–40 counts per min), as previously described Hsu et al. (2014). The signals of chemiluminescence were continuously measured for 12 min, and the activity of NADPH oxidase was expressed as counts per million cells.
Measurement of cPLA2 Luciferase Activity
Human cPLA2 promoter spanning - 2375 to +75 bp as cloned into pGL3-basic vector (Promega, Madison, WI, USA) as cPLA2-luc plasmid. The activity of cPLA2-luc was detected using a luciferase assay system (Promega, Madison, WI, USA), as previously described Chi et al. (2012). The detected luciferase activities were standardized with the activity of β-gal.
Measurement of PGE2 Generation
The serum-starved cells were treated with TNF-α for the different time points. At the end of treatment, culture media were collected and stored at -80°C. The concentrations of PGE2 were detected by a PGE2 enzyme immunoassay kit (Cayman) according to the manufacturer’s instructions, as previously described Lee et al. (2008).
Transient Transfection with siRNAs
All human ASK1, p47phox, TRAF2, NIK, IKKα, IKKβ, and p65 siRNA together with scramble siRNA were purchased from Sigma (St. Louis, MO, USA). LipofectamineTM RNAiMAX reagents were used to prepare siRNA liposome complexes (100 nM of siRNAs) according to the manufacturer’s instructions, as previously described Lee et al. (2008).
Co-immunoprecipitation Assay
Cell lysates containing 1 mg of protein were incubated with 2 μg of an anti-p47phox, anti-TNFR1, anti-TRAF2, or anti-ASK1 antibody at 4°C for 24 h, and then 10 μl of 50% protein A-agarose beads was added and mixed at 4°C for 24 h. The immunoprecipitates were collected and washed thrice with a lysis buffer without Triton X-100. 5x Laemmli buffer was added and subjected to electrophoresis on SDS-PAGE, and then blotted using an anti-phospho-tyrosine, anti-phospho-serine, anti-p47phox, anti-TRAF2, anti-TNFR1, or anti-ASK1 antibody, as previously described (Yang et al., 2014).
Immunofluorescence Staining
Growth-arrested HPAEpiCs were incubated with TNF-α for the indicated time intervals. After washing twice with ice-cold PBS, cells were fixed, permeabilized and stained using an anti-p65 antibody, as previously described (Chi et al., 2011). A fluorescence microscope (Zeiss, Axiovert 200M) were used to observe images.
Cell Fractions Isolation
The cell lysates were sonicated for 5 s at output 1.5 using a sonicator (Misonix, Farmingdale, NY, USA) and then centrifuged at 6800 × g for 15 min at 4°C, as previously described Lee et al. (2008). The pellet was collected as the nuclear fraction. The supernatant was further centrifuged at 20,000 × g at 4°C for 60 min to yield the pellet (membrane fraction) and the supernatant (cytosolic fraction).
Statistical Analysis of Data
Data were showed as the mean or mean ± SEM of five individual experiments and estimated using GraphPad Prism Program (GraphPad, San Diego, CA, USA). All the data were analyzed by paired two-tailed Student’s t-test or by one-way analysis of variance (ANOVA) followed with Tukey’s post hoc test at p < 0.05 level of significance.
Results
TNF-α Induces cPLA2 Expression via NADPH Oxidase and ROS
Reactive oxygen species play both deleterious and beneficial roles. Accumulation of ROS and cellular oxidative stress trigger expression of inflammatory genes and result in tissue damages and various diseases (Lee and Yang, 2012). Thus, we attempted to investigate the roles of NADPH Oxidase and ROS in cPLA2 expression. Here we reported that TNF-α-induced cPLA2 protein levels were significantly reduced by pretreatment with a ROS scavenger (NAC) or the inhibitors of NADPH oxidase (APO and DPI) (Figure 1A). In addition, pretreatment with NAC, DPI, or APO also attenuated the TNF-α-stimulated cPLA2 mRNA expression and promoter activity (Figure 1B). The p47phox protein, one of cytosolic subunits of NADPH oxidase, contributed to acute NADPH oxidase activation via being phosphorylated and binding to p22phox (Lee et al., 2009). Thus, we confirmed the role of p47phox in cPLA2 expression by transfection with p47phox siRNA which knocked down protein expression of p47phox and then markedly inhibited TNF-α-induced cPLA2 protein expression in these cells (Figure 1C). To confirm this cPLA2 expression is mediated through TNF-α-dependent induction, HPAEpiCs were pretreated with a human TNF-α neutralizing antibody followed by TNF-α treatment. We found that TNF-α neutralizing antibody significantly blocked the cPLA2 induction in a dose-dependent manner (Figure 1D). These results indicated that TNF-α induces cPLA2 expression via NADPH oxidase and ROS in HPAEpiCs.
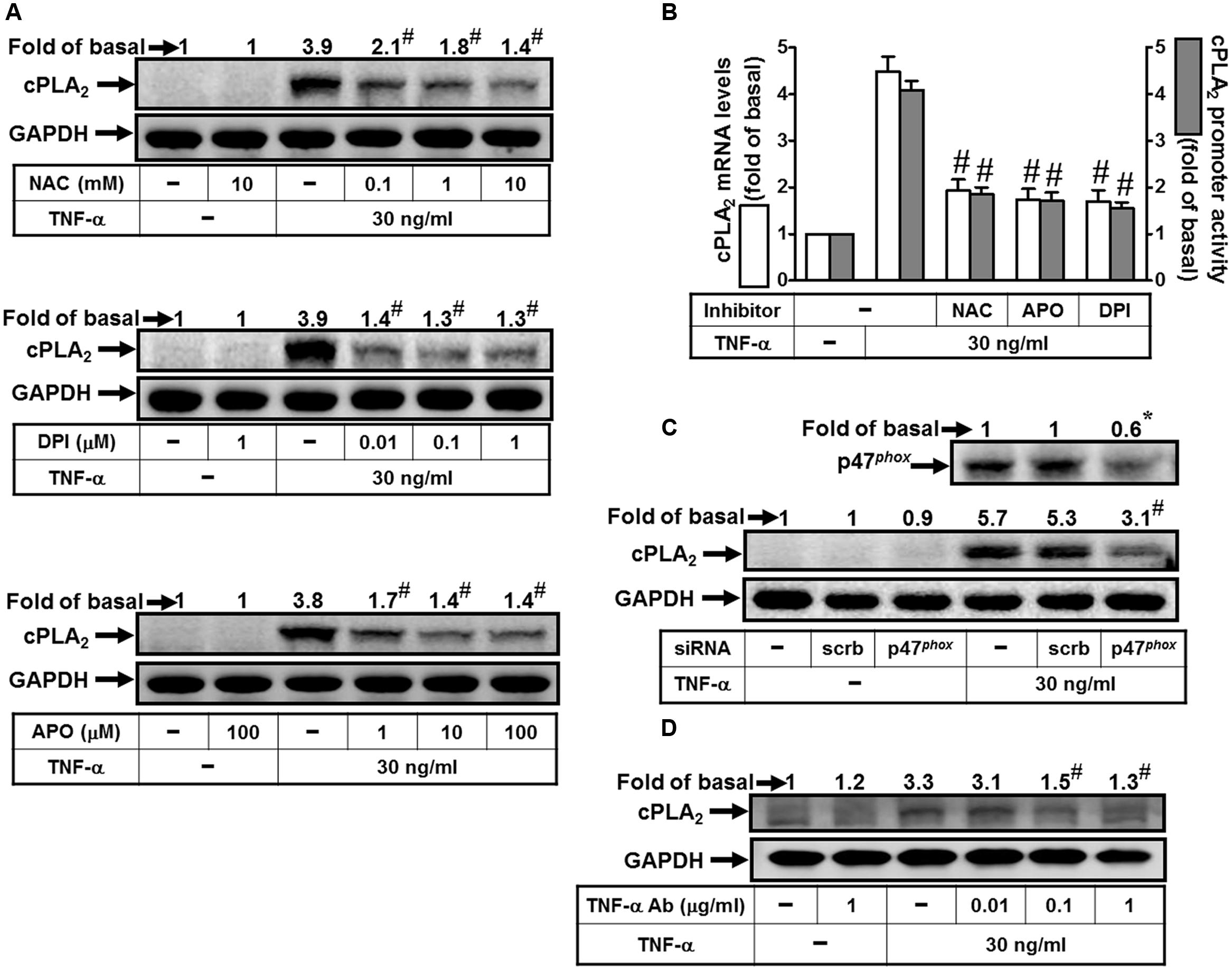
FIGURE 1. TNF-α induces NADPH oxidase- and ROS-dependent cPLA2 expression. (A) HPAEpiCs were pretreated with NAC, DPI, or APO for 1 h, and then incubated with TNF-α for 24 h. (B) Cells were pretreated with NAC (10 mM), DPI (1 μM), or APO (100 μM) for 1 h, and then incubated with TNF-α for 6 h. cPLA2 mRNA levels and promoter activity were determined. (C) Cells were transfected with scrambled or p47phox siRNA, and then incubated with TNF-α for 24 h. (D) HPAEpiCs were pretreated with human TNF-α neutralizing antibody (TNF-α nAb: 0.01, 0.1 and 1 μg/ml) for 1 h, and then incubated with TNF-α for 24 h. (A,C,D) The protein levels of cPLA2 and p47phox were determined by Western blot. Data are expressed as mean ± SEM of three independent experiments. ∗P < 0.05, #P < 0.01 as compared with the cells exposed to TNF-α alone.
TNF-α Induces NADPH Oxidase-Dependent Superoxide and Hydrogen Peroxide Production
TNF-α may stimulate ROS production by several sources, such as mitochondria, but recent studies have strongly suggested that a major source of ROS is a phagocyte-type NADPH oxidase. Several reports also demonstrate that TNF-α triggers several signal transduction pathways to activate the NOX activity and enhances intracellular ROS generation leading to expression of inflammatory genes (Rahman et al., 1998; Hashimoto et al., 2001; Li et al., 2005; Lee et al., 2013; Yang et al., 2014). Therefore, we investigated whether TNF-α-induced cPLA2 expression is due to activation of NADPH oxidase and ROS generation. Here, we found that TNF-α markedly induced superoxide and hydrogen peroxide production, determined by using DHE or DCF under a fluorescence microscope (Figure 2A). On the other hand, we also observed that TNF-α time-dependently induces NADPH oxidase activation (Figure 2B), which was reduced by TNFR1 neutralizing antibody, APO, or DPI (Figure 2C). These results suggested that TNF-α induced ROS generation via NADPH oxidase activation. The p47phox is phosphorylated on several serine residues within the polybasic region of the protein, and these multiple phosphorylation events induce conformational changes that permit p47phox to interact with the cytoplasmic tail of p22phox and to initiate the formation of the active NADPH oxidase complex (Dang et al., 2006). Previous report also indicates that Src modulates tyrosine phosphorylation of p47phox in hyperoxia-induced activation of NADPH oxidase and generation of ROS in lung endothelial cells (Chowdhury et al., 2005). Thus, we investigated whether TNF-α stimulates the phosphorylation of tyrosine or serine on p47phox. As shown in Figure 2D, TNF-α increased tyrosine and serine phosphorylation of p47phox in a time-dependent manner. Moreover, p47phox was translocated from the cytosol to the membrane fractions in TNF-α-stimulated cells (Figure 2E). Finally, we investigated whether TNF-α-induced superoxide and hydrogen peroxide production are mediated through NADPH oxidase activation. Here, we observed that TNF-α-induced superoxide and hydrogen peroxide production were inhibited by TNFR1 neutralizing antibody, APO, DPI and NAC (Figure 2F). These results suggested that TNF-α induces ROS generation via p47phox translocation and NADPH oxidase activation in HPAEpiCs.
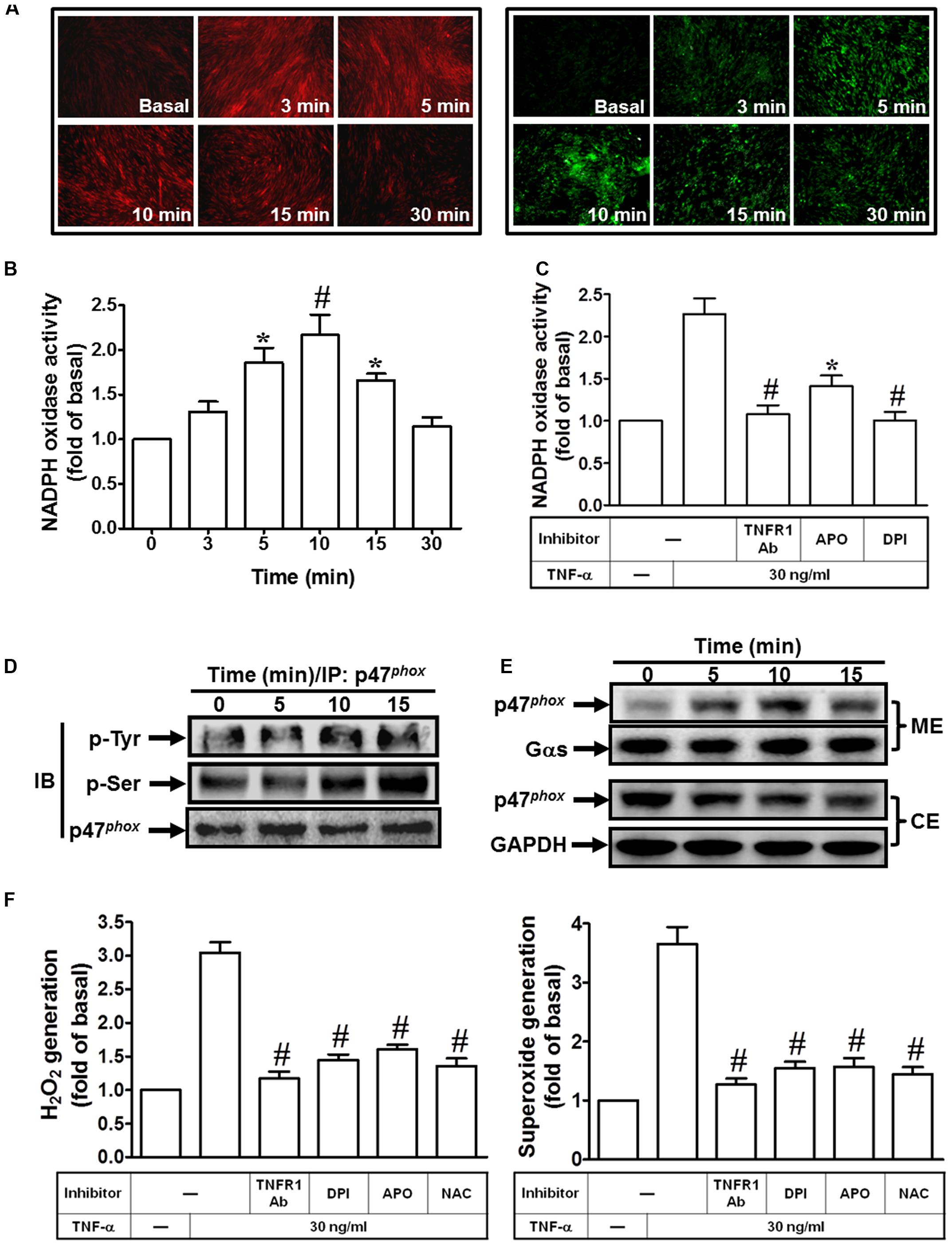
FIGURE 2. TNF-α induces NADPH oxidase-dependent ROS generation. (A) HPAEpiCs were treated with TNF-α for the indicated time intervals. DHE (red) or DCF (green) fluorescence image was observed. Images shown are representative of five independent experiments with similar results. Cells were (B) treated with TNF-α for the indicated time intervals or (C) pretreated with TNFR1 neutralizing antibody (10 μg/ml), APO (100 μM), or DPI (1 μM) for 1 h, and then treated with TNF-α for 10 min. NADPH oxidase activity was measured. (D) Cells were treated with TNF-α for the indicated time intervals. The cell lysates were subjected to immunoprecipitation using an anti-p47phox antibody, and then the immunoprecipitates were analyzed by Western blot using an anti-phospho-tyrosine, anti-phospho-serine, or anti-p47phox antibody. (E) Cells were treated with TNF-α for the indicated time intervals. The cytosolic and membrane fractions were prepared and analyzed by Western blot using an anti-p47phox, anti-Gαs, or anti-GAPDH antibody. GAPDH and Gαs were used as a marker protein for cytosolic and membrane fractions, respectively. (F) Cells were pretreated with TNFR1 neutralizing antibody, NAC, APO, or DPI for 1 h, and then incubated with TNF-α for 10 min. H2O2 and superoxide generation were measured. Data are expressed as mean ± SEM of three independent experiments. ∗P < 0.05, #P < 0.01 as compared with the cells exposed to vesicle alone (B) or TNF-α alone (C,F).
TNF-α Induces TNFR1, TRAF2, ASK1, and p47phox Complex Formation
Both tissue injury-related proinflammatory and programmed cell death pathways are activated by TNF-α via binding to TNFR1 (van Vliet et al., 2005; Lee et al., 2009). TRAF2 plays an important role in innate immune and inflammatory responses. Thus, we determine whether TRAF2 is a downstream component of TNF-α/TNFR1 complex in cPLA2 expression. Here, we demonstrated that TNF-α induced cPLA2 expression via TRAF2 by transfection with TRAF2 siRNA (Figure 3A). We found that transfection with TRAF2 siRNA reduced TRAF2 protein expression by about 50% and significantly attenuated the TNF-α-induced cPLA2 expression from 5.4-fold to 2.2-fold. In addition, we also showed that TNF-α stimulated TRAF2 and TNFR1 complex formation and its downstream components (Figure 3B). Therefore, we further studied whether TNF-α promoted the association of TNFR1, TRAF2, ASK1, and p47phox in HPAEpiCs. Data in Figure 3B showed that TNF-α time-dependently induced TNFR1, TRAF2, ASK1, and p47phox complex formation. Importantly, this interaction was blocked by pretreatment with TNF-α neutralizing antibody.
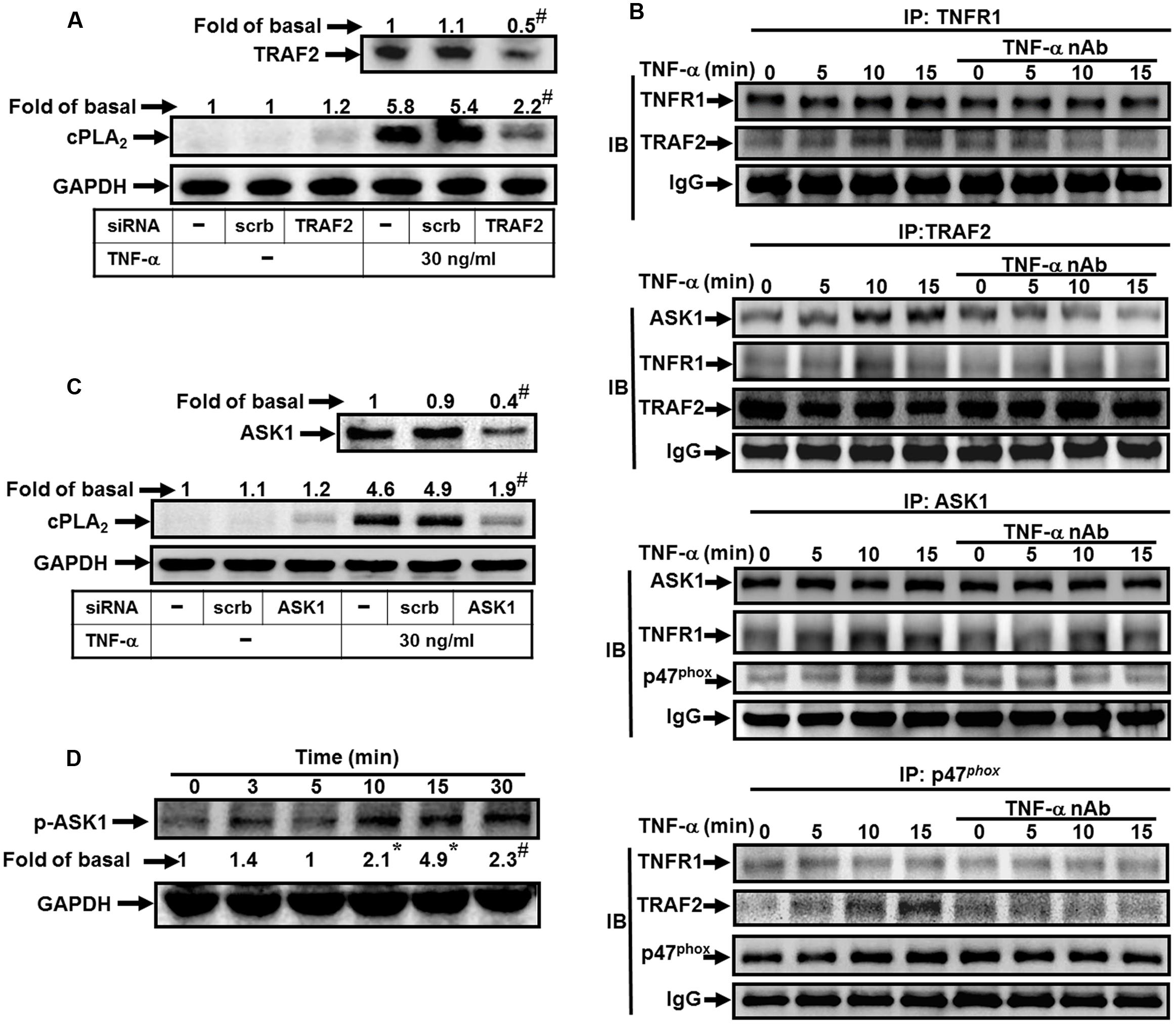
FIGURE 3. TNF-α induces TNFR1/TRAF2/ASK1/p47phox complex formation. (A) HPAEpiCs were transfected with scrambled or TRAF2 siRNA, and then incubated with TNF-α for 24 h. The protein levels of TRAF2 and cPLA2 were determined. (B) Cells were pretreated without or with human TNF-α neutralizing antibody (TNF-α nAb:1 μg/ml) for 1 h and then incubated with TNF-α for the indicated time intervals. The cell lysates were subjected to immunoprecipitation using an anti-TNFR1, anti-TRAF2, anti-ASK1, or anti-p47phox antibody, and then the immunoprecipitates were analyzed by Western blot by using an anti-TNFR1, anti-TRAF2, anti-ASK1, anti-p47phox or anti-IgG antibody. The results of IgG were used to be the loading control. (C) Cells were transfected with scrambled or ASK1 siRNA, and then incubated with TNF-α for 24 h. The protein levels of ASK1 and cPLA2 were determined. (D) Cells were treated with TNF-α for the indicated time intervals. The levels of phospho-ASK1 were determined by Western blot. Data are representative of three independent experiments with similar results. ∗P < 0.05, #P < 0.01 as compared with the basal group.
Various cell stresses, including ROS, TNF-α, lipopolysaccharide (LPS), and ER stress, activate ASK1 and resulting in apoptosis, differentiation and inflammation (Soga et al., 2012). However, whether ASK1 involved in TNF-α-mediated responses was still unknown in HPAEpiCs. Here, we found that transfection with ASK1 siRNA knocked down ASK1 protein level by about 50% and then significantly attenuated the TNF-α-induced cPLA2 expression from 4.9-fold to 1.9-fold (Figure 3C). ASK1 plays a pivotal role in ROS generation. In resting cells, endogenous ASK1 constitutively associated with thioredoxin (Trx) forming an inactive high-molecular-mass complex, known as ASK1 signalosome. Once cellular ROS increased, Trx dissociated from ASK1 signalosome and resulted in full activation of higher-molecular-mass complex by the recruitment of TRAF2 and TRAF6 (Fujino et al., 2007). Thus, we further investigated whether p47phox was recruited to the complex of TNF-α/TNFR1/TRAF2/ASK1. We observed that TNF-α time-dependently induced TNFR1, TRAF2, ASK1, and p47phox complex formation determined by immunoprecipitation (Figure 3B). Consistently, this interaction was also blocked by pretreatment with a TNF-α neutralizing antibody. To confirm whether ASK1 phosphorylation is necessary for TNF-α-induced cPLA2 expression, activation of the ASK1 was assayed by Western blot using an antibody specific for the phosphorylated form of ASK1. We found that TNF-α significantly stimulated ASK1 phosphorylation in a time-dependent manner (Figure 3D). These results suggested that TNF-α stimulates TNFR1, TRAF2, ASK1, and p47phox complex formation leading to NADPH oxidase activation and ROS generation in HPAEpiCs.
TNF-α Induces NADPH Oxidase/ROS Generation and PGE2 Release via TRAF2, ASK1, and p47phox
Next, we investigated whether TRAF2, ASK1 and p47phox are involved in TNF-α-induced ROS generation. As shown in Figure 4A, transfection with TRAF2, ASK1, or p47phox siRNA markedly inhibited TNF-α-induced superoxide and hydrogen peroxide production. In addition, these siRNAs also inhibited the TNF-α-induced NADPH oxidase activation (Figure 4B). Moreover, our previous findings also provided the correlation between COX-2 and cPLA2 expression in ATPγS-stimulated VSMCs with similar molecular mechanisms and functional coupling to amplify the occurrence of vascular inflammation (Lin et al., 2009). Therefore, the synthesis of PGE2 could be a good index of AA release that is more sensitive than [3H]AA mobilization (Berenbaum et al., 2003). In this study, we tested the effect of TNF-α on PGE2 synthesis as a parameter of cPLA2 activity in HPAEpiCs. Here, we observed that TNF-α-induced PGE2 release was reduced by transfection with TRAF2, ASK1, or p47phox siRNA (Figure 4C). These results suggested that TNF-α induces ROS generation and PGE2 release via TRAF2, ASK1, and p47phox in HPAEpiCs.
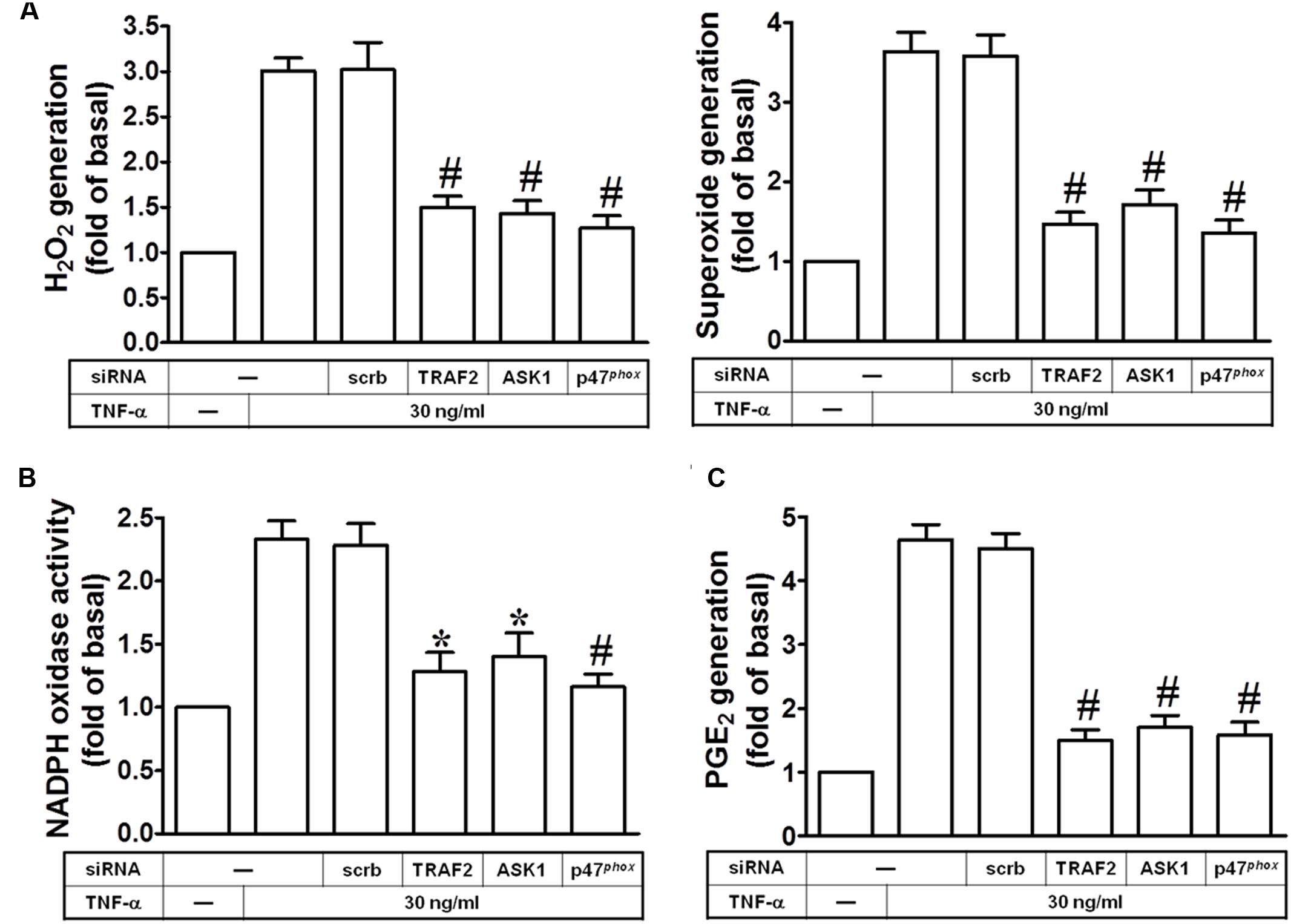
FIGURE 4. TNF-α induces NADPH oxidase/ROS generation and PGE2 release via TRAF2, ASK1, and p47phox. HPAEpiCs were transfected with scrambled, ASK1, p47phox, or TRAF2 siRNA, and then incubated with TNF-α for (A,B) 10 min or (C) 24 h. (A) H2O2 and superoxide generation were measured. (B) NADPH oxidase activity was measured. (C) PGE2 generation was measured. Data are expressed as mean ± SEM of three independent experiments. ∗P < 0.05, #P < 0.01 as compared with the cells exposed to TNF-α + scrambled siRNA.
TNF-α Induces cPLA2 Expression via NIK, IKKα/β, and NF-κB
NF-κB is mainly involved in stress-induced immune and inflammatory responses. Activities of NF-κB are regulated by various inflammatory cytokines such as TNF-α (Lee et al., 2007). The signaling mechanisms mediated activation of NF-κB includes canonical and non-canonical pathways (Tak and Firestein, 2001). The canonical pathway has been well documented to regulate the pathophysiological functions; however, the non-canonical NF-κB-inducing kinase (NIK) pathway is not well understood in the expression of inflammatory genes (Uno et al., 2014). NIK plays central roles in the activation of non-canonical NF-κB pathway (Tak and Firestein, 2001; Uno et al., 2014). NIK was first identified as a TRAF2 interacting protein. In addition, IκBs associate with NF-κB being inactive forms. IκBs are phosphorylated by IKKα and IKKβ then degradation via ubiquitination to release NF-κB and nuclear translocation (Sun, 2011). Therefore, we investigated whether TNF-α induced cPLA2 expression via NIK, IKKα, and IKKβ by transfection with respective siRNAs. Transfection with NIK, IKKα, or IKKβ siRNAs knocked down their own protein levels and subsequently attenuated the TNF-α-induced cPLA2 expression (Figure 5A). To ascertain whether NF-κB participated in TNF-α-induced cPLA2 expression, a selective pharmacological inhibitor of NF-κB, Bay11-7082, was used for this purpose. Pretreatment with Bay11-7082 significantly reduced the TNF-α-induced cPLA2 protein, mRNA, and promoter activity (Figures 5B,C). Moreover, p65 siRNA was used to confirm the role of NF-κB in TNF-α-mediated effects. In addition, transfection with p65 siRNA also reduced TNF-α-induced cPLA2 expression (Figure 5D) and PGE2 secretion (Figure 5E) in these cells. The non-canonical NF-κB pathway is activated through particular TNF-α receptors that bind to the TRAF2 which leads to translocation of NF-κB into nucleus and induction of target gene expression (Tak and Firestein, 2001). Therefore, the translocation of NF-κB p65 was observed by an immunofluorescence microscope. Our findings showed that TNF-α time-dependently stimulated NF-κB p65 translocation from the cytosol into the nucleus fractions (Figure 5F) together with enhancing NF-κB promoter activity (Figure 5G). These results suggested that TNF-α-induced cPLA2 expression is mediated through NIK, IKKα/β, and NF-κB in these cells.
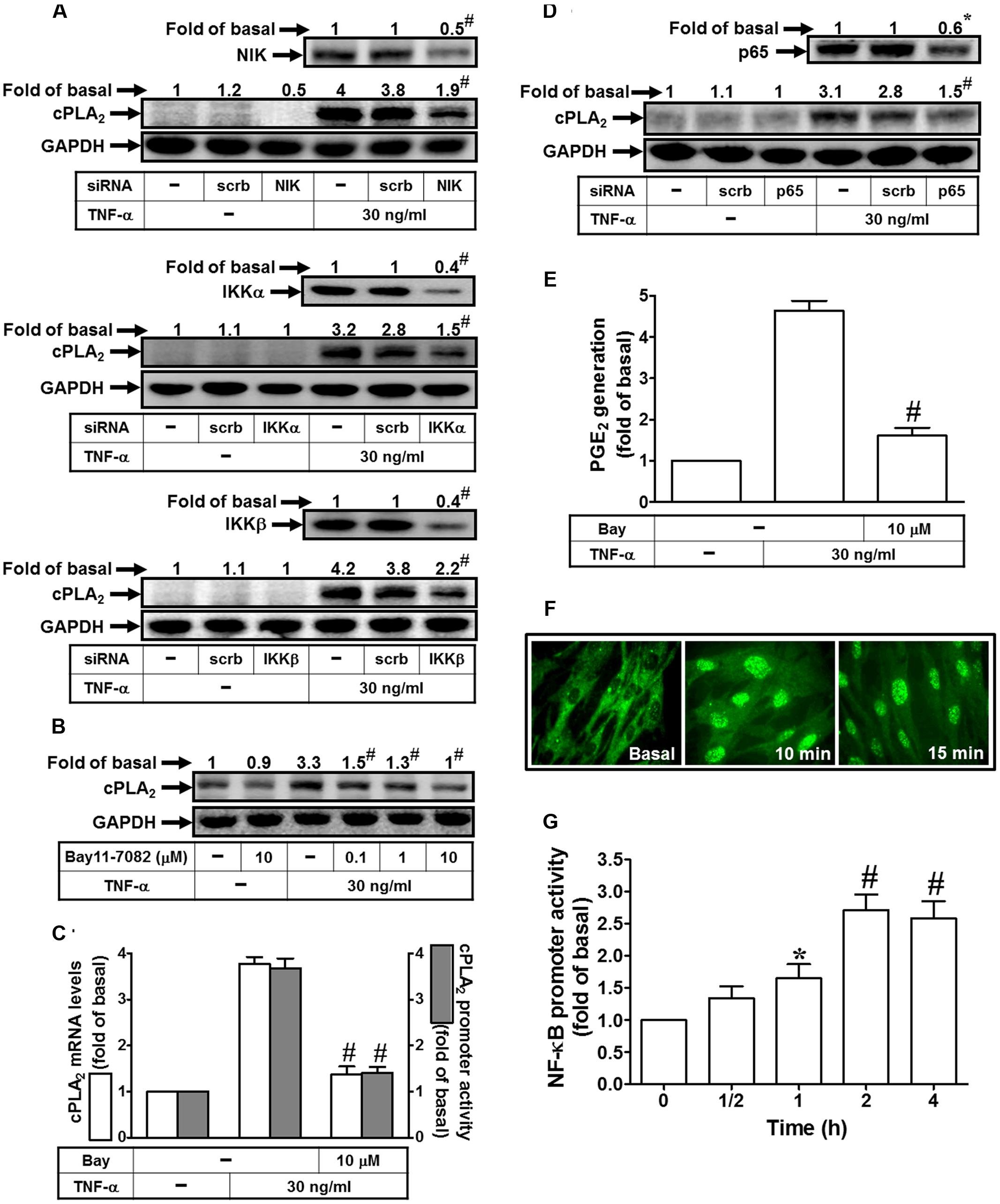
FIGURE 5. TNF-α induces cPLA2 expression via NIK, IKKα/β, and NF-κB. (A) HPAEpiCs were transfected with scrambled, NIK, IKKα, or IKKβ siRNA, and then incubated with TNF-α for 24 h. The protein levels of NIK, IKKα, IKKβ, and cPLA2 were determined by Western blot. (B) Cells were pretreated with Bay11-7082 for 1 h, and then incubated with TNF-α for 24 h. The protein levels of cPLA2 were determined by Western blot. (C) Cells were pretreated with Bay11-7082 for 1 h, and then incubated with TNF-α for 6 h. cPLA2 mRNA levels and promoter activity were determined. (D) Cells were transfected with scrambled or p65 siRNA, and then incubated with TNF-α for 24 h. The protein levels of p65 and cPLA2 were determined. (E) Cells were pretreated with Bay11-7082 for 1 h, and then incubated with TNF-α for 24 h. PGE2 generation was measured. (F) Cells were incubated with TNF-α for the indicated time intervals. Cells were fixed, labeled with an anti-p65 antibody, and then FITC-conjugated secondary antibody. Individual cells were imaged. (G) Cells were incubated with TNF-α for the indicated time intervals. NF-κB promoter activity was determined. Data are expressed as mean ± SEM of three independent experiments. ∗P < 0.05, #P < 0.01 as compared with the cells exposed to TNF-α alone (C,E) or vesicle alone (G).
TNF-α Induces NIK/IKKα/β-Dependent NF-κB Activation
In this study, we investigated whether TNF-α stimulates NF-κB p65 activation via an NADPH oxidase/ROS-dependent pathway. As shown in Figure 6A, TNF-α markedly stimulated NF-κB p65 phosphorylation which was inhibited by NAC, DPI, APO or Bay11-7082. In addition, pretreatment with NAC, DPI, or APO also inhibited TNF-α-induced NF-κB promoter activity and NF-κB p65 translocation in HPAEpiCs (Figures 6B,C). On the other hand, we also observed that TNF-α significantly stimulated IKKα/β phosphorylation in a time-dependent manner, which was reduced by NAC, DPI or APO (Figure 6D). Finally, we investigated whether TNF-α stimulates NF-κB p65 phosphorylation via an NIK/IKKα/β pathway. As shown in Figure 6E, transfection with NIK siRNA attenuated the TNF-α-induced IKKα/β and NF-κB p65 activation. In addition, transfection with siRNA of IKKα or IKKβ markedly inhibited TNF-α-induced NF-κB p65 phosphorylation (Figure 6E). These results suggested that TNF-α stimulates NF-κB p65 activation through an NIK/IKKα/β-dependent cascade in HPAEpiCs.
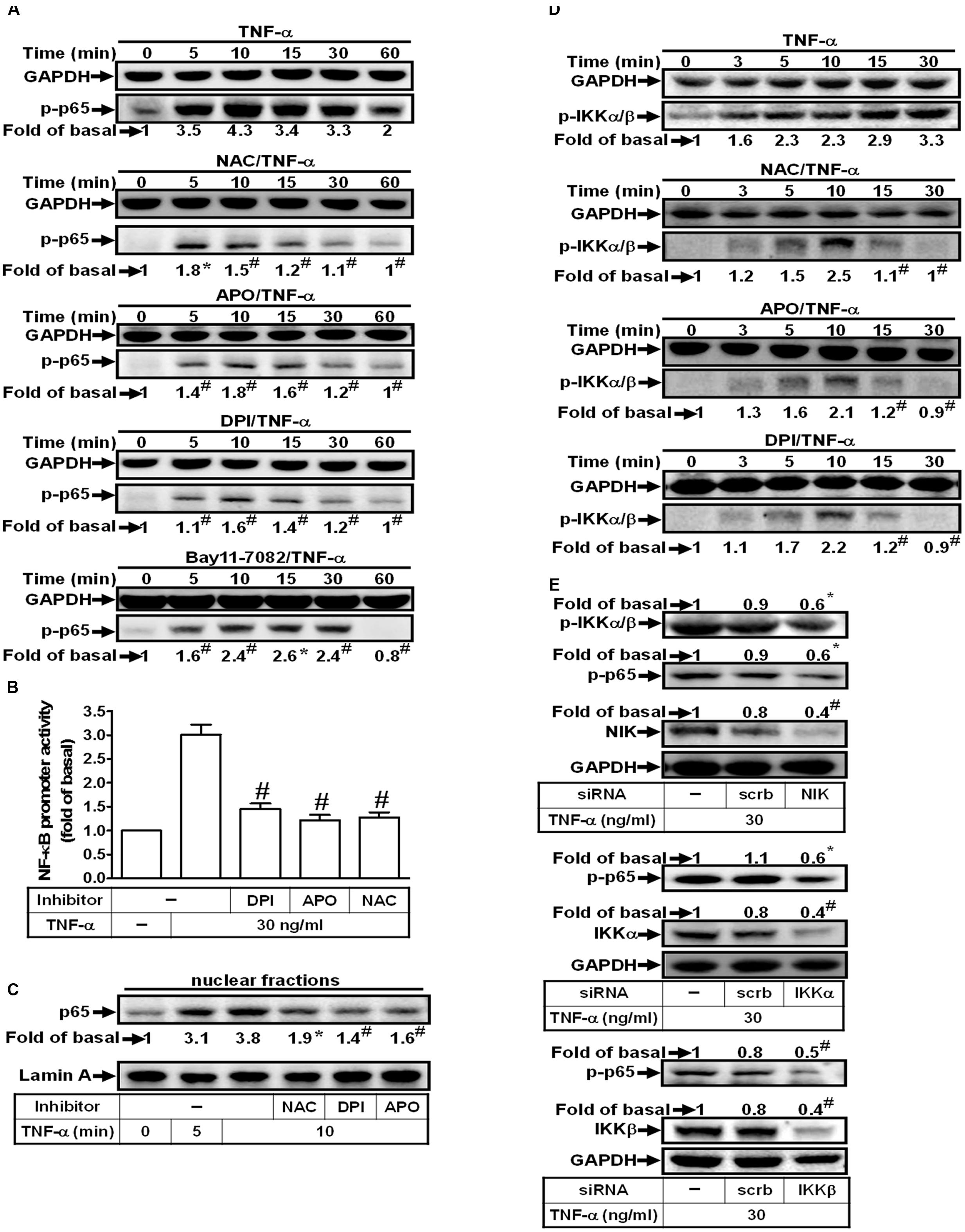
FIGURE 6. TNF-α induces NIK/IKKα/β-dependent NF-κB activation. (A) HPAEpiCs were pretreated with NAC (10 mM), DPI (1 μM), APO (100 μM) or Bay11-7082 (10 μM) for 1 h, and then treated with TNF-α for the indicated time intervals. The levels of p65 phosphorylation were determined by Western blot. The raw data of the GAPDH of (A) were provided with a supplementary data (Supplementary Figure S2). (B) Cells were pretreated with NAC, DPI, or APO for 1 h, and then treated with TNF-α for 2 h. NF-κB promoter activity was determined. (C) Cells were pretreated with NAC, DPI, or APO for 1 h, and then treated with TNF-α for the indicated time intervals. The nuclear fractions were prepared and analyzed by Western blot using an anti-p65 antibody. Lamin A was used as a marker protein for nuclear fractions. (D) Cells were pretreated with NAC, APO, or DPI for 1 h, and then treated with TNF-α for the indicated time intervals. The levels of IKKα/β phosphorylation were determined by Western blot. (E) Cells were transfected with scrambled, NIK, IKKα, or IKKβ siRNA, and then incubated with TNF-α for 10 min. The protein levels of phospho-IKKα/β, phospho-p65, NIK, IKKα, and IKKβ were determined. Data are expressed as mean ± SEM of three independent experiments. ∗P < 0.05, #P < 0.01 as compared with the cells exposed to TNF-α alone.
Discussion
Various degrees of inflammation and tissue remodeling are characteristics of different pulmonary disorders including asthma and COPD. Expression of cPLA2 by mesenchymal cells in several extra-pulmonary sites contributes to the production of PGE2 which functions as biologically active lipid mediators in inflammatory responses (Khanapure et al., 2007). TNF-α has been shown to activate cPLA2 gene and involves in the late-phase airway hyperresponsiveness and inflammation (Choi et al., 2005), but few studies address the intracellular signaling pathways leading to its expression. It is showed that TNF-α activates ASK1, NADPH oxidase/ROS, IKKα/β, and NF-κB pathways in several cell types (Lee et al., 2009, 2011; Byeon et al., 2012). However, whether these signaling molecules participated in cPLA2 expression in TNF-α-treated HPAEpiCs was not completely defined. In this study, Figure 7 addressed that pretreatment with the inhibitors of NADPH oxidase (APO and DPI), ROS (NAC), or NF-κB (Bay11-7082) or transfection with siRNA of ASK1, TRAF2, p47phox, NIK, IKKα, IKKβ, or p65 attenuated TNF-α-induced cPLA2 expression and PGE2 production in HPAEpiCs. Our studies confirmed that activation of NADPH oxidase, ROS, NIK, IKKα/β, and NF-κB via association of TNFR1, TRAF2, ASK1, and p47phox may be essential for cPLA2 expression in TNF-α-stimulated HPAEpiCs. cPLA2 induction and TNF-α-induced complex formation of TNFR1, TRAF2, ASK1, and p47phox were blocked by pretreatment with TNF-α neutralizing antibody. These results suggested that up-regulation of cPLA2 by TNF-α is, at least in part, mediated through the cooperation of TNFR1, TRAF2, ASK1, and NADPH oxidase leading to ROS generation and ultimately activates NF-κB pathway.
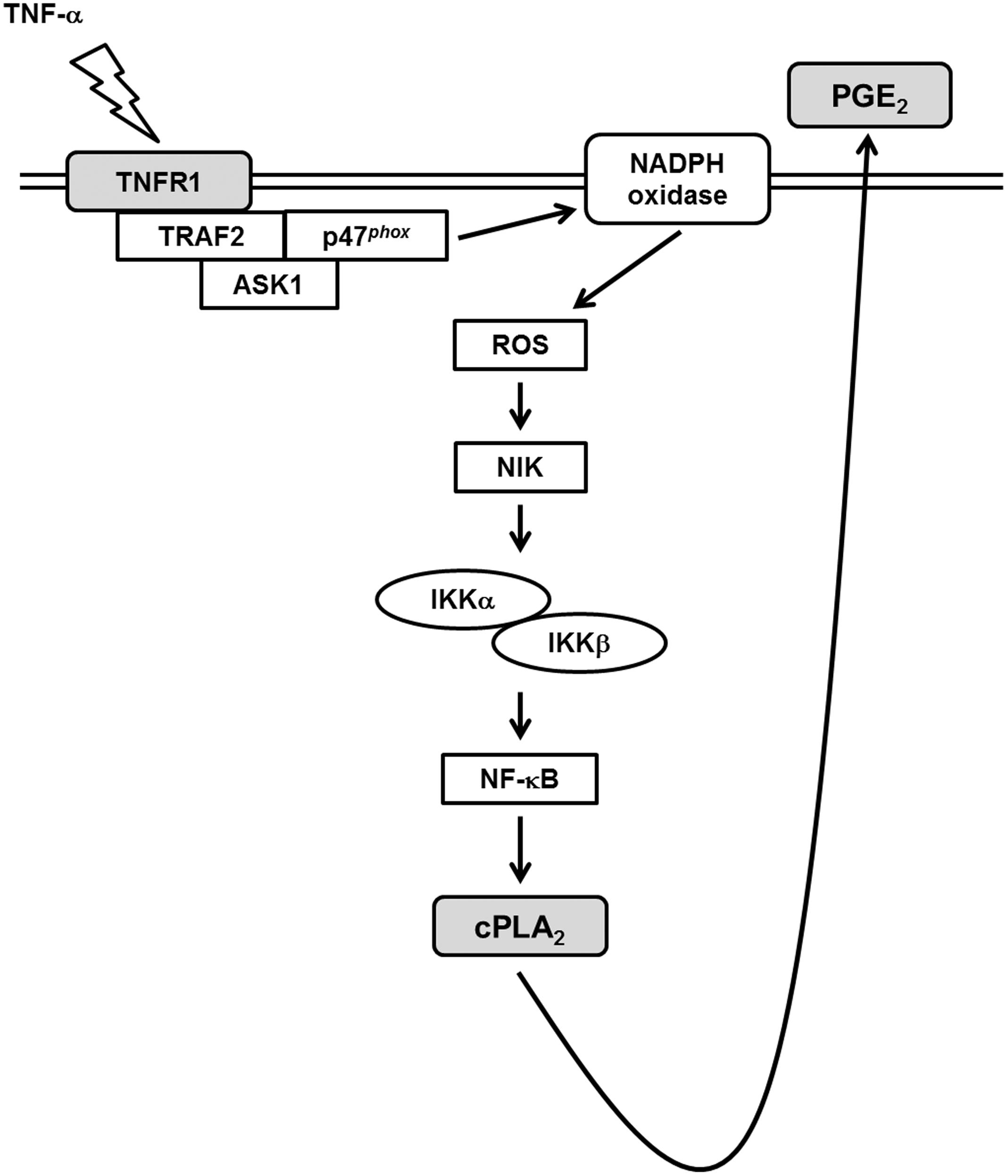
FIGURE 7. Schematic representation of the signaling pathways involved in the TNF-α-induced cPLA2 expression in HPAEpiCs. The mechanisms underlying TNF-α-mediated activation of TNFR1/TRAF2/ASK1/p47phox-dependent NADPH oxidase is required for the expression of cPLA2 in HPAEpiCs. Finally, the activation of ROS/NIK/IKKα/β/NF-κB pathway led to cPLA2 gene transcription and PGE2 release.
Several reports indicate that TNF-α may regulate inflammatory protein expression via activating various downstream protein kinases (Lin et al., 2004; Lee et al., 2009). Effects of TNF-α are achieved by binding to one of two distinct receptors, known as TNFR1 and TNFR2 (Lee et al., 2009). However, it is reported that TNF-α activates the proinflammatory or the programmed cell death pathways leading to tissue injury via binding to TNFR1 (van Vliet et al., 2005; Lee et al., 2009). In contrast, TNFR2 is involved in promoting tissue repair and angiogenesis (Bradley, 2008). Indeed, transfection with TNFR1 siRNA attenuated NADPH oxidase and ROS generation, revealing that TNFR1 plays a key role in modulating inflammatory responses in TNF-α-stimulated HPAEpiCs.
Reactive oxygen species are generated by various enzymatic reactions and chemical processes or can directly be inhaled from environment (Lee and Yang, 2012). Formation of ROS takes place constantly in every cell during normal metabolic processes. Activated phagocytic cells could produce large amounts of ROS induced by inhaled particles, microorganisms, or other mediators leading to the activation of the membrane-bound NADPH oxidase complex and the generation of superoxide anion (Lee and Yang, 2012). NADPH oxidase is recognized to be a key player in the generation of ROS when the cells or tissues exposed to various insults. Upon activation of NADPH oxidase by various stimuli, the complex of catalytic subunit (gp91phox) and p22phox is anchored in plasma membrane which assembles with the regulatory subunits (p47phox, p40phox, p67phox, and small GTPase Rac) distributed in cytoplasm and leading to ROS generation (El-Benna et al., 2008). The initiation of the assembly NADPH oxidase is first dependent on the phosphorylation of p47phox triggered by various activated protein kinases. Thus, p47phox plays a role in the membrane translocation and ROS generation. Indeed, our results confirmed that blockage ROS accumulation either by a ROS scavenger (NAC), the inhibitors of NADPH oxidase (DPI and APO) or transfection with p47phox siRNA significantly attenuated TNF-α-induced cPLA2 expression and PGE2 synthesis. DPI or APO pretreatment reduced TNF-α-stimulated ROS generation. These results revealed that TNF-α increased cPLA2/PGE2 expression via NADPH oxidase-dependent ROS generation. p47phox is phosphorylated on several serine residues within the polybasic region of the protein, and these multiple phosphorylation events induce conformational changes that permit p47phox to interact with the cytoplasmic tail of p22phox and to initiate the formation of the active oxidase (Dang et al., 2006). In lung endothelial cells, Src regulates tyrosine phosphorylation of p47phox in hyperoxia-induced activation of NADPH oxidase and generation of ROS (Chowdhury et al., 2005). Moreover, in HPAEpiCs, we found that TNF-α could stimulate both serine and tyrosine phosphorylation of p47phox, and then promotes p47phox translocation from the cytosol to the membrane.
TRAF2 plays a central role in the cellular responses to stress and cytokines via their regulation of stress kinases, resulting in the activation of key transcription factors, including NF-κB, c-Jun, and ATF2. Upon exposure to TNF-α, TRAF2 is recruited directly to TNFR2 or via TRADD to TNFR1, which results in the activation of JNK1/2, p38 MAPK and NF-κB (Cabal-Hierro and Lazo, 2012). However, our previous report indicated that Jak2 also mediates the TNF-α-induced cPLA2 expression (Yang et al., 2014). To evaluate the relationship between Jak2 and MAPKs pathways, we have performed some more experiments to determine whether there is any connection between MAPKs and Jak2 using pharmacological inhibitors (AG490, SB202190, SP600125 and U0126). We found that pretreatment with AG490 had no effect on TNF-α-stimulated MAPKs phosphorylation, or with MAPK inhibitors (SB202190, SP600125 and U0126) failed to inhibit Jak2 phosphorylation (Supplementary Figure S1). These results suggested that Jak2 and MAPKs are independent pathways to mediate the TNF-α-induced cPLA2 expression. Here, we also found that TNF-α-induced cPLA2 expression and PGE2 release were markedly inhibited by blockage TRAF2. In addition, we also observed that TNF-α induced TNFR1 and TRAF2 complex formation. Previous study indicated that TNF-α stimulated the formation of a TNFR1/c-Src/p47phox complex in human airway smooth muscle cells (Lee et al., 2009). Here, we demonstrated that TNF-α induced TNFR1, TRAF2, and p47phox complex formation which was blocked by pretreatment with TNF-α neutralizing antibody, and leading to cPLA2 expression in HPAEpiCs.
ASK1, a member of the MAP3K family, regulates the activation of MAPK kinase 4 (MKK4)/MKK7-JNK and MKK3/6-p38 pathways. ASK1 can be activated by various types of stresses, such as ROS, TNF-α, and ER stress, and exerts pivotal roles in regulating cell apoptosis, differentiation, and inflammation (Fujino et al., 2007). Therefore, unregulated ASK1 activation is tightly related to various diseases. Moreover, we found that inhibition of ASK1 markedly reduced TNF-α-induced cPLA2 expression and PGE2 release. TNF-α also stimulated ASK1 phosphorylation in these cells. Upon exposure to ROS, Trx dissociated from the N-terminal of ASK1, which then became fully activated by recruitment of TRAF2 and TRAF6 (Fujino et al., 2007). Indeed, we found consistent results that TNF-α induces TRAF2 and ASK1 complex formation which is also blocked by pretreatment with TNF-α neutralizing antibody. In this study, we are the first to show a novel role of TNFR1/TRAF2/ASK1/p47phox complex formation in TNF-α-induced NADPH oxidase activation and ROS production in HPAEpiCs. In the future, we will further determine which domains of TNFR1, TRAF2, ASK1, and p47phox are involved in protein-protein interactions caused by TNF-α. Although ROS have been shown to regulate ASK1 activation (Fujino et al., 2007), in this study, we emphasized the critical role of ASK1 in TNF-α-induced ROS generation. Thus, in addition to the role of ROS in ASK1 activation, our results supported that the opposite hierarchical relationship exists between ROS and ASK1. Therefore, NADPH oxidase/ROS may act both as upstream regulators and downstream effectors of ASK1 in various cell types.
The NF-κB/Rel family complex, a redox-sensitive transcription factor, participates in controlling expression of many inflammatory genes (Sun, 2011). NF-κB usually forms herterodimer by p50 and p65/RelA subunits. In resting cells, nuclear translocation signal of NF-κB is masked by binding with an inhibitor protein called inhibitory κB (IκB) as an inactive non-DNA-binding form. Upon the stimulation by various NF-κB inducers, two serine residues of IκBα is phosphorylated, which then being ubiquinated by E3 ubiquitin-ligases (E3RSIκB) and subsequently degraded by the 26S proteasome (Sun, 2011). The released NF-κB dimers can then translocate into the nucleus and bind to the κB elements on the promoter of target genes. NIK was first identified as a TRAF2 interacting protein (Tak and Firestein, 2001). In inactive form, NF-κB transcription factors binds with the inhibitory proteins IκBs. IKKα and IKKβ regulates the phosphorylation of IκB proteins, which then being ubiquitination and degradation leading to nuclear localization of NF-κB transcription factors (Sun, 2011). In this study, inhibition of NIK, IKKα, IKKβ, and NF-κB markedly inhibited TNF-α-induced cPLA2 expression. Various extracellular stimuli such as TNF-α and IL-1β, viruses and environmental particulates (PM10s), and oxidative stress regulate the activation of NF-κB (Sun, 2011). Here, we reported that TNF-α time-dependently induced phosphorylation and translocation of NF-κB p65 and NF-κB promoter activity via an NADPH oxidase/ROS pathway. Otherwise, we also proved that TNF-α significantly induced IKKα/β phosphorylation via an NIK-dependent signaling, and then promoted NF-κB activation. Thus, we recognized that TNF-α-induced ROS generation may promote activation of the NIK/IKKα/β/NF-κB pathway in HPAEpiCs.
COX-2 and cPLA2 are tightly regulated by various mediators in several species (Beasley, 1999; Ali et al., 2008; Pavicevic et al., 2008). cPLA2 hydrolyzes the membrane phospholipids, resulting in the release of AA, which is further converted by the constitutive enzyme COX-1 or by the inducible COX-2, and PG synthases to biologically active PGs (DeWitt, 1999). On the other hand, several reports indicated that the levels of PGE2 are also degraded by an important enzyme, 15-hydroxyprostaglandin dehydrogenase (15-PGDH) to regulate the levels of PGEs (Pomini et al., 1999; Otani et al., 2006; Thiel et al., 2009). In our previous study, up-regulation of COX-2 in TSMCs has been shown to enhance PGE2 synthesis induced by LPS (Luo et al., 2003). Moreover, we also provided insights into the correlation between COX-2 and cPLA2 expression in ATPγS-stimulated VSMCs with similar molecular mechanisms and functional coupling to amplify the occurrence of vascular inflammation (Lin et al., 2009). PGE2 treatment also induced cPLA2 expression in VSMCs, which exerted as a positive feedback regulator in this response (Lin et al., 2009). Therefore, the synthesis of PGE2 could be a good index of AA release that is more sensitive than [3H]AA mobilization (Berenbaum et al., 2003) and PGE2 may further induce cPLA2 expression to amplify the occurrence of pulmonary inflammation. In this study, although the effect of PGE2 on COX-2 expression was not investigated, we tested the effect of TNF-α on PGE2 synthesis as a parameter of cPLA2 activity which may be an important issue for further study in HPAEpiCs.
Conclusion
According to the literature reports and our results, Figure 7 addresses a model for the molecular mechanisms of TNF-α-stimulated cPLA2 expression and PGE2 release in HPAEpiCs. To our knowledge, this is the first study to indicate that in HPAEpiCs, TNF-α-regulated activation of TNFR1/TRAF2/ASK1/p47phox-dependent NADPH oxidase is required for the expression of cPLA2. Finally, activation of the ROS/NIK/IKKα/β/NF-κB pathway leads to cPLA2 gene activation and expression. It is an important link for TNF-α-regulated cPLA2 expression in the pathogenesis of lung inflammatory diseases. Therefore, uncovering the signaling components in TNF-α-mediated cPLA2 expression in HPAEpiCs is important to develop new therapeutic strategies in pulmonary diseases.
Author Contributions
C-CL, W-NL, R-LC, C-yW, L-DH, and C-MY substantially contributed to the conception or design of the work, the acquisition, analysis, and interpretation of data for the work. C-CL, W-NL, R-LC, C-yW, L-DH, and C-MY drafted the work and revised it critically for important intellectual content. C-CL, W-NL, R-LC, CyW, L-DH, and C-MY finally approved the version to be published. C-CL, W-NL, R-LC, C-yW, L-DH, and C-MY agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was supported by the Ministry of Education, Taiwan, Grant number: EMRPD1E1641; the Ministry of Science and Technology, Taiwan, Grant number: MOST104-2320-B-182A-003-MY3, MOST104-2320-B-182-010 and MOST105-2320-B-182-005-MY3; Chang Gung Medical Research Foundation, Grant number: CMRPD1C0103, CMRPD1C0563, CMRPD1B0332, CMRPD1F0021, CMRPG3B1093, CMRPG3C1303, CMRPG3A0051, and CMRPG3E2231; and Fu Jen Catholic University Research Foundation, Grant number: 9991A01.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer HH and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We thank Ms. Yu-Wen Chen for her technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphar.2016.00447/full#supplementary-material
References
Ali, K., Lund-Katz, S., Lawson, J., Phillips, M. C., and Rader, D. J. (2008). Structure function properties of the apoE-dependent COX-2 pathway in vascular smooth muscle cells. Atherosclerosis 196, 201–209. doi: 10.1016/j.atherosclerosis.2007.03.038
Barnes, P. J. (1989). New concepts in the pathogenesis of bronchial hyperresponsiveness and asthma. J. Allergy Clin. Immunol. 83, 1013–1026. doi: 10.1016/0091-6749(89)90441-7
Beasley, D. (1999). COX-2 and cytosolic PLA2 mediate IL-1β-induced cAMP production in human vascular smooth muscle cells. Am. J. Physiol. 276, H1369–H1378.
Berenbaum, F., Humber, L., Bereziat, G., and Thirion, S. (2003). Concomitant recruitment of ERK1/2 and p38 MAPK signalling pathway is required for activation of cytoplasmic phospholipase A2 via ATP in articular chondrocytes. J. Biol. Chem. 278, 13680–13687. doi: 10.1074/jbc.M211570200
Borsch-Haubold, A. G., Bartoli, F., Asselin, J., Dudler, T., Kramer, R. M., Apitz-Castro, R., et al. (1998). Identification of the phosphorylation sites of cytosolic phospholipase A2 in agonist-stimulated human platelets and HeLa cells. J. Biol. Chem. 273, 4449–4458. doi: 10.1074/jbc.273.8.4449
Bradley, J. R. (2008). TNF-mediated inflammatory disease. J. Pathol. 214, 149–160. doi: 10.1002/path.2287
Byeon, H. E., Um, S. H., Yim, J. H., Lee, H. K., and Pyo, S. (2012). Ohioensin F suppresses TNF-α-induced adhesion molecule expression by inactivation of the MAPK, Akt and NF-κB pathways in vascular smooth muscle cells. Life Sci. 90, 396–406. doi: 10.1016/j.lfs.2011.12.017
Cabal-Hierro, L., and Lazo, P. S. (2012). Signal transduction by tumor necrosis factor receptors. Cell. Signal. 24, 1297–1305. doi: 10.1016/j.cellsig.2012.02.006
Chi, P. L., Chen, Y. W., Hsiao, L. D., Chen, Y. L., and Yang, C. M. (2012). Heme oxygenase-1 attenuates IL-1β-induced cPLA2 expression via a decrease in NADPH oxidase/ROS/AP-1 activation in human rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 64, 2114–2125. doi: 10.1002/art.34371
Chi, P. L., Liu, C. J., Lee, I. T., Chen, Y. W., Hsiao, L. D., and Yang, C. M. (2014). HO-1 induction by CO-RM2 attenuates TNF-α-induced cytosolic phospholipase A2 expression via inhibition of PKCα-dependent NADPH oxidase/ROS and NF-κB. Mediators Inflamm. 2014:279171. doi: 10.1155/2014/279171
Chi, P. L., Luo, S. F., Hsieh, H. L., Lee, I. T., Hsiao, L. D., Chen, Y. L., et al. (2011). cPLA2 induction and PGE2 release by IL-1β via the MyD88-dependent and cooperation of p300, Akt, and NF-κB pathway in human rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 63, 2905–2917. doi: 10.1002/art.30504
Choi, I. W., Sun, K., Kim, Y. S., Ko, H. M., Im, S. Y., Kim, J. H., et al. (2005). TNF-α induces the late-phase airway hyperresponsiveness and airway inflammation through cytosolic phospholipase A2 activation. J. Allergy Clin. Immunol. 116, 537–543. doi: 10.1016/j.jaci.2005.05.034
Chowdhury, A. K., Watkins, T., Parinandi, N. L., Saatian, B., Kleinberg, M. E., Usatyuk, V., et al. (2005). Src-mediated tyrosine phosphorylation of p47phox in hyperoxia-induced activation of NADPH oxidase and generation of reactive oxygen species in lung endothelial cells. J. Biol. Chem. 280, 20700–20711. doi: 10.1074/jbc.M411722200
Dang, P. M., Elbim, C., Marie, J. C., Chiandotto, M., Gougerot-Pocidalo, M. A., and El-Benna, J. (2006). Anti-inflammatory effect of interleukin-10 on human neutrophil respiratory burst involves inhibition of GM-CSF-induced p47phox phosphorylation through a decrease in ERK1/2 activity. FASEB J. 20, 1504–1506. doi: 10.1096/fj.05-5395fje
DeWitt, D. L. (1999). Cox-2-selective inhibitors: the new super aspirins. Mol. Pharmacol. 55, 625–631.
Dieter, P., Kolada, A., Kamionka, S., Schadow, A., and Kaszkin, M. (2002). Lipopolysaccharide-induced release of arachidonic acid and prostaglandins in liver macrophages: regulation by Group IV cytosolic phospholipase A2, but not by Group V and Group IIA secretory phospholipase A2. Cell. Signal. 14, 199–204. doi: 10.1016/S0898-6568(01)00243-1
El-Benna, J., Dang, P. M. C., and Gougerot-Pocidalo, M. A. (2008). Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin. Immunopathol. 30, 279–289. doi: 10.1007/s00281-008-0118-3
Fujino, G., Noguchi, T., Matsuzawa, A., Yamauchi, S., Saitoh, M., Takeda, K., et al. (2007). Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol. Cell. Biol. 27, 8152–8163. doi: 10.1128/MCB.00227-07
Gilroy, D. W., Newson, J., Sawmynaden, P., Willoughby, D. A., and Croxtall, J. D. (2004). A novel role for phospholipase A2 isoforms in the checkpoint control of acute inflammation. FASEB J. 18, 489–498. doi: 10.1096/fj.03-0837com
Hashimoto, S., Gon, Y., Matsumoto, K., Takeshita, I., and Horie, T. (2001). N-acetylcysteine attenuates TNF-α-induced p38 MAP kinase activation and p38 MAP kinase-mediated IL-8 production by human pulmonary vascular endothelial cells. Br. J. Pharmacol. 132, 270–276. doi: 10.1038/sj.bjp.0703787
Henderson, W. R. Jr., Tang, L. O., Chu, S. J., Tsao, S. M., Chiang, G. K., Jones, F., et al. (2002). A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am. J. Respir. Crit. Care Med. 165, 108–116. doi: 10.1164/ajrccm.165.1.2105051
Hsieh, H. L., Wu, C. Y., Hwang, T. L., Yen, M. H., Parker, P., and Yang, C. M. (2006). BK-induced cytosolic phospholipase A2 expression via sequential PKC-α, p42/p44 MAPK, and NF-κB activation in rat brain astrocytes. J. Cell. Physiol. 206, 246–254. doi: 10.1002/jcp.20457
Hsu, C. K., Lee, I. T., Lin, C. C., Hsiao, L. D., and Yang, C. M. (2014). Nox2/ROS-dependent human antigen R translocation contributes to TNF-α-induced SOCS-3 expression in human tracheal smooth muscle cells. Am. J. Physiol. Lung Mol. Cell. Biol. 306, L521–L533. doi: 10.1152/ajplung.00274.2013
Hsu, M. J., Chang, C. K., Chen, M. C., Chen, B. C., Ma, H. P., Hong, C. Y., et al. (2010). Apoptosis signal-regulating kinase 1 in peptidoglycan-induced COX-2 expression in macrophages. J. Leukoc. Biol. 87, 1069–1082. doi: 10.1189/jlb.1009668
Hulkower, K. I., Wertheimer, S. J., Levin, W., Coffey, J. W., Anderson, C. M., Chen, T., et al. (1994). Interleukin-1β induces cytosolic phospholipase A2 and prostaglandin H synthase in rheumatoid synovial fibroblasts. Evidence for their roles in the production of prostaglandin E2. Arthritis Rheum. 37, 653–661. doi: 10.1002/art.1780370508
Huwiler, A., Feuerherm, A. J., Sakem, B., Pastukhov, O., Filipenko, I., Nguyen, T., et al. (2012). The omega3-polyunsaturated fatty acid derivatives AVX001 and AVX002 directly inhibit cytosolic phospholipase A2 and suppress PGE2 formation in mesangial cells. Br. J. Pharmacol. 167, 1691–1701. doi: 10.1111/j.1476-5381.2012.02114.x
Khanapure, S. P., Garvey, D. S., Janero, D. R., and Letts, L. G. (2007). Eicosanoids in inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr. Top. Med. Chem. 7, 311–340. doi: 10.2174/156802607779941314
Lee, C. W., Lin, C. C., Lee, I. T., Lee, H. C., and Yang, C. M. (2011). Activation and induction of cytosolic phospholipase A2 by TNF-α mediated through Nox2, MAPKs, NF-κB, and p300 in human tracheal smooth muscle cells. J. Cell. Physiol. 226, 2103–2114. doi: 10.1002/jcp.22537
Lee, C. W., Lin, C. C., Lin, W. N., Liang, K. C., Luo, S. F., Wu, C. B., et al. (2007). TNF-α induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/ Akt cascade and promotion of NF-κB/p300 binding in human tracheal smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L799–L812. doi: 10.1152/ajplung.00311.2006
Lee, I. T., Lin, C. C., Lee, C. Y., Hsieh, P. W., and Yang, C. M. (2013). Protective effects of (-)-epigallocatechin-3-gallate against TNF-α-induced lung inflammation via ROS-dependent ICAM-1 inhibition. J. Nutr. Biochem. 24, 124–136. doi: 10.1016/j.jnutbio.2012.03.009
Lee, I. T., Luo, S. F., Lee, C. W., Wang, S. W., Lin, C. C., Chang, C. C., et al. (2009). Overexpression of HO-1 protects against TNF-α-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am. J. Pathol. 175, 519–532. doi: 10.2353/ajpath.2009.090016
Lee, I. T., Wang, S. W., Lee, C. W., Chang, C. C., Lin, C. C., Luo, S. F., et al. (2008). Lipoteichoic acid induces HO-1 expression via the TLR2/MyD88/c-Src/NADPH oxidase pathway and Nrf2 in human tracheal smooth muscle cells. J. Immunol. 181, 5098–5110. doi: 10.4049/jimmunol.181.7.5098
Lee, I. T., and Yang, C. M. (2012). Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem. Pharmacol. 84, 581–590. doi: 10.1016/j.bcp.2012.05.005
Leslie, C. C. (1997). Properties and regulation of cytosolic phospholipase A2. J. Biol. Chem. 272, 16709–16712. doi: 10.1074/jbc.272.27.16709
Li, J. M., Fan, L. M., Christie, M. R., and Shah, A. M. (2005). Acute tumor necrosis factor α a signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol. Cell. Biol. 25, 2320–2330. doi: 10.1128/MCB.25.6.2320-2330.2005
Lin, C. C., Hsiao, L. D., Chien, C. S., Lee, C. W., Hsieh, J. T., and Yang, C. M. (2004). Tumor necrosis factor-α-induced cyclooxygenase-2 expression in human tracheal smooth muscle cells: involvement of p42/p44 and p38 mitogen-activated protein kinases and nuclear factor-κB. Cell. Signal. 16, 597–607. doi: 10.1016/j.cellsig.2003.10.002
Lin, C. C., Lin, W. N., Wang, W. J., Sun, C. C., Tung, W. H., Wang, H. H., et al. (2009). Functional coupling expression of COX-2 and cPLA2 induced by ATP in rat vascular smooth muscle cells: role of ERK1/2, p38 MAPK, and NF-κB. Cardiovasc. Res. 82, 522–531. doi: 10.1093/cvr/cvp069
Luo, S. F., Lin, W. N., Yang, C. M., Lee, C. W., Liao, C. H., Leu, Y. L., et al. (2006). Induction of cytosolic phospholipase A2 by lipopolysaccharide in canine tracheal smooth muscle cells: involvement of MAPKs and NF-κB pathways. Cell. Signal. 18, 1201–1211. doi: 10.1016/j.cellsig.2005.09.011
Luo, S. F., Wang, C. C., Chien, C. S., Hsiao, L. D., and Yang, C. M. (2003). Induction of cyclooxygenase-2 by lipopolysaccharide in canine tracheal smooth muscle cells: involvement of p42/p44 and p38 mitogen-activated protein kinases and nuclear factor-κB pathways. Cell. Signal. 15, 497–509. doi: 10.1016/S0898-6568(02)00135-3
Morgan, M. J., and Liu, Z. G. (2011). Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 21, 103–115. doi: 10.1038/cr.2010.178
Nagase, T., Uozumi, N., Ishii, S., Kume, K., Izumi, T., Ouchi, Y., et al. (2000). Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat. Immunol. 1, 42–46. doi: 10.1038/76897
Otani, T., Yamaguchi, K., Scherl, E., Du, B., Tai, H. H., Greifer, M., et al. (2006). Levels of NAD+-dependent 15-hydroxyprostaglandin dehydrogenase are reduced in inflammatory bowel disease: evidence for involvement of TNF-α. Am. J. Physiol Gastrointest. Liver Physiol. 290, G361–G368. doi: 10.1152/ajpgi.00348.2005
Pavicevic, Z., Leslie, C. C., and Malik, K. U. (2008). cPLA2 phosphorylation at S515 and S505 is required for arachidonic acid release in vascular smooth muscle cell. J. Lipid Res. 49, 724–737. doi: 10.1194/jlr.M700419-JLR200
Pomini, F., Caruso, A., and Challis, J. R. G. (1999). Interleukin-10 modifies the effects of interleukin-1β and tumor necrosis factor-α on the activity and expression of prostaglandin H synthase-2 and the NAD+-dependent 15-hydroxyprostaglandin dehydrogenase in cultured term human villous trophoblast and chorion trophoblast cells. J. Clin. Endocrinol. Metab. 84, 4645–4651.
Rahman, A., Kefer, J., Bando, M., Niles, W. D., and Malik, A. B. (1998). E-selectin expression in human endothelial cells by TNF-α-induced oxidant generation and NF-κB activation. Am. J. Physiol. 275, L533–L544.
Six, D. A., and Dennis, E. A. (2000). The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim. Biophys. Acta 1488, 1–19. doi: 10.1016/S1388-1981(00)00105-0
Soga, M., Matsuzawa, A., and Ichijo, H. (2012). Oxidative stress-induced diseases via the ASK1 signaling pathway. Int. J. Cell Biol. 2012:439587. doi: 10.1155/2012/439587
Sun, S. C. (2011). Non-canonical NF-kappaB signaling pathway. Cell Res. 21, 71–85. doi: 10.1038/cr.2010.177
Tak, P. P., and Firestein, G. S. (2001). NF-κB: a key role in inflammatory diseases. J. Clin. Invest. 107, 7–11. doi: 10.1172/JCI11830
Thiel, A., Ganesan, A., Mrena, J., Junnila, S., Nykänen, A., Hemmes, A., et al. (2009). 15-hydroxyprostaglandin dehydrogenase is down-regulated in gastric cancer. Clin. Cancer Res. 15, 4572–4580. doi: 10.1158/1078-0432.CCR-08-2518
Uno, M., Saitoh, Y., Mochida, K., Tsuruyama, E., Kiyono, T., Imoto, I., et al. (2014). NF-κB inducing kinase, a central signaling component of the non-canonical pathway of NF-κB, contributes to ovarian cancer progression. PLoS ONE 9:e88347. doi: 10.1371/journal.pone.0088347
Uozumi, N., Kume, K., Nagase, T., Nakatani, N., Ishii, S., Tashiro, F., et al. (1997). Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 390, 618–622. doi: 10.1038/37622
Van Putten, V., Refaat, Z., Dessev, C., Blaine, S., Wick, M., Butterfield, L., et al. (2001). Induction of cytosolic phospholipase A2 by oncogenic Ras is mediated through the JNK and ERK pathways in rat epithelial cells. J. Biol. Chem. 276, 1226–1232. doi: 10.1074/jbc.M003581200
van Vliet, C., Bukczynska, P. E., Puryer, M. A., Sadek, C. M., Shields, B. J., Tremblay, M. L., et al. (2005). Selective regulation of tumor necrosis factor-induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat. Immunol. 6, 253–260. doi: 10.1038/ni1169
Yang, C. M., Chien, C. S., Hsiao, L. D., Luo, S. F., and Wang, C. C. (2002). Interleukin-1β-induced cyclooxygenase-2 expression is mediated through activation of p42/44 and p38 MAPKS, and NF-κB pathways in canine tracheal smooth muscle cells. Cell. Signal. 14, 899–911. doi: 10.1016/S0898-6568(02)00037-2
Yang, C. M., Lee, I. T., Chi, P. L., Cheng, S. E., Hsiao, L. D., and Hsu, C. K. (2014). TNF-α induces cytosolic phospholipase A2 expression via Jak2/PDGFR-dependent Elk-1/p300 activation in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Biol. 306, L543–L551. doi: 10.1152/ajplung.00320.2013
Keywords: ASK1, cytokines, cytosolic phospholipase A2, lung inflammation, signaling transduction
Citation: Lin C-C, Lin W-N, Cho R-L, Wang C-y, Hsiao L-D and Yang C-M (2016) TNF-α-Induced cPLA2 Expression via NADPH Oxidase/Reactive Oxygen Species-Dependent NF-κB Cascade on Human Pulmonary Alveolar Epithelial Cells. Front. Pharmacol. 7:447. doi: 10.3389/fphar.2016.00447
Received: 28 July 2016; Accepted: 08 November 2016;
Published: 25 November 2016.
Edited by:
Wenliang Song, Vanderbilt University Medical Center, USAReviewed by:
Aida Habib, American University of Beirut, LebanonHui Huang, Vanderbilt University, USA
Copyright © 2016 Lin, Lin, Cho, Wang, Hsiao and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuen-Mao Yang, Y2h1ZW5tYW9AbWFpbC5jZ3UuZWR1LnR3
†These authors have contributed equally to this work.