 Ryan D. Jones
Ryan D. Jones Jie Liao
Jie Liao Guang-Yu Yang
Guang-Yu Yang- Department of Pathology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
Polyunsaturated fatty acids (PUFAs) including epoxide-modified ω-3 and ω-6 fatty acids are made via oxidation to create highly polarized carbon-oxygen bonds crucial to their function as signaling molecules. A critical PUFA, arachidonic acid (ARA), is metabolized to a diverse set of lipids signaling molecules through cyclooxygenase (COX), lipoxygenase (LOX), cytochrome P450 epoxygenase, or cytochrome P450 hydroxylase; however, the majority of ARA is metabolized into anti-inflammatory epoxides via cytochrome P450 enzymes. These short-lived epoxide lipids are rapidly metabolized or inactivated by the soluble epoxide hydrolase (sEH) into diol-containing products. sEH inhibition or knockout has been a practical approach to study the biology of the epoxide lipids, and has been shown to effectively treat inflammatory conditions in the preclinical models including gastrointestinal ulcers and colitis by shifting oxylipins to epoxide profiles, inhibiting inflammatory cell infiltration and activation, and enhancing epithelial cell defense via increased mucin production, thus providing further evidence for the role of sEH as a pro-inflammatory protein. Non-steroidal anti-inflammatory drugs (NSAIDs) with COX-inhibitor activity are among the most commonly used analgesics and have demonstrated applications in the management of cardiovascular disease and intriguingly cancer. Major side effects of NSAIDs however are gastrointestinal ulcers which frequently precludes their long-term application. In this review, we hope to bridge the gap between NSAID toxicity and sEH-mediated metabolic pathways to focus on the role of epoxy fatty acid metabolic pathway of PUFAs in NSAIDS-ulcer formation and healing as well as inflammation-related carcinogenesis. Specifically we address the potential application of sEH inhibition to enhance ulcer healing at the site of inflammation via their activity on altered lipid signaling, mitochondrial function, and diminished reactive oxygen species, and further discuss the significance of dual COX and sEH inhibitor in anti-inflammation and carcinogenesis.
Introduction
Inflammation is a diverse and complex series of pathogenetic processes, which act to protect host organisms against infectious pathogens and damaged cells or tissues. Inflammation is initiated via cellular and molecular signaling, and it is necessary for clearance of infections and tissue damage and for tissue repair. On the other hand, the inflammatory process itself can cause significant harm to the host (Jaeschke and Smith, 1997). One such family of molecular mediators or signaling of inflammation is arachidonic acid (ARA) and its metabolites.
ARA is a pivotal molecule in inflammation, which when released in response to tissue injury can be metabolized into three broad pathways governed by cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 enzymes (including epoxygenase and hydroxylase) (as outlined in Figure 1) (Morisseau and Hammock, 2012). Downstream active molecules from ARA metabolism include prostaglandins (PGs), leukotrienes, epoxyeicosanoids, and hydroxyeicosatetraenoic acid. In addition to ARA, other polyunsaturated fatty acids (PUFAs) including eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are also substrates for these same enzymes (Hiesinger et al., 2019). Of particular interest are the epoxyeicosatrienoic acids (EETs), the metabolites of cytochrome p450 epoxygenase. EETs play important roles in gastrointestinal (GI) epithelial integrity and wound healing, and are key negative regulators of inflammation (Zhang et al., 2012; Zhang et al., 2013b; Zhang et al., 2013c). The effects of EETs have been extensively reviewed by Spector and Kim (2015).
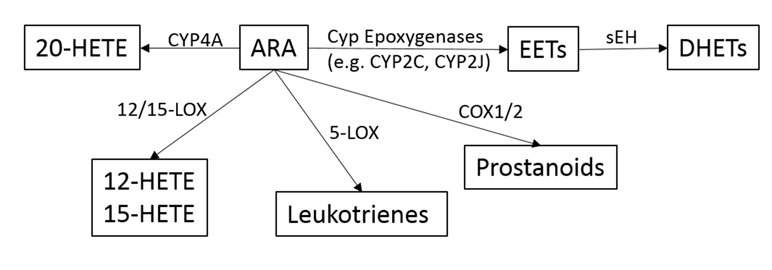
Figure 1 Overview of the metabolic pathways of the arachidonic acid (ARA) cascade.
EETs are substrates for the enzyme soluble epoxide hydrolase (sEH), which rapidly converts EETs to dihydroxyeicosatrienoic acids (DHETs) (Capdevila et al., 2000; Spector et al., 2004). The epoxide fatty acids generated from EPA and ARA are also physiologically active, and are substrates of sEH (Morisseau and Hammock, 2012). These diol-containing DHETs have drastically reduced biologic activity. Thus, inhibition of sEH has been extensively studied as a mechanism to increase the longevity of anti-inflammatory EETs via the discovery of sEH inhibitors, particularly in combination with other inhibitors as multi-target therapies, which were recently reviewed (Hiesinger et al., 2019). sEH inhibitors have been shown to robustly decrease sEH activity with little to no toxicity in animal models. Further, these compounds have proven effective to 1) decrease GI ulcers induced by non-steroidal anti-inflammatory drugs (NSAIDs) (Goswami et al., 2016; Goswami et al., 2017; Yang, 2018), 2) prevent carcinogenesis in murine models of colorectal and pancreatic tumors (Liao et al., 2016a; Liao et al., 2016b; Wang et al., 2018b; Yang, 2018), and 3) decrease chronic inflammation in mouse models for both colitis and pancreatitis (Zhang et al., 2012; Zhang et al., 2013b; Zhang et al., 2013c; Goswami et al., 2016; Liao et al., 2016a; Goswami et al., 2017; Wang et al., 2018b; Yang, 2018).
Prostaglandins (PGs), produced through oxygenation of ARA via COX enzymes, result in a diverse family of structures that modulate many functions including vascular tone, platelet aggregation, and inflammation (Ricciotti and FitzGerald, 2011). The NSAID family of drugs act through inhibition of COX-1 and/or COX-2. Specific inhibitors against COX enzymes have also been developed. In addition to NSAIDs anti-inflammatory properties, they have been shown to decrease the risk of several cancers including colorectal adenocarcinomas (Cao et al., 2016; Chan and Ladabaum, 2016; Tsoi et al., 2018). Long-term NSAID use, however, often leads to severe GI tract ulcers and potentially life-threatening bleeding precluding their widespread use in chemoprevention (Sostres et al., 2010).
This review aims to highlight a potential strategy combining sEH and COX inhibition for chemoprevention and inflammatory conditions while also mitigating the adverse side effects of single agent sEH inhibitors and NSAIDs.
Polyunsaturated Fatty Acid Metabolism and EETs
The 20-carbon ω-6 PUFA ARA is a lipid signaling molecule that resides among phospholipids and is a key substrate for a variety of downstream cell signaling mediators known as eicosanoids. ARA is broadly metabolized into one of three main pathways: 1) by COX into prostaglandins; 2) by 5-LOX into leukotrienes; and 3) by cytochrome P450 (CYP 450) epoxygenase and hydroxylase to form epoxyeicosanoids and hydroxyeicosatetraenoic acid. The EETs are signaling molecules formed within various types of cells by the metabolism of ARA (Capdevila et al., 2000; Capdevila and Falck, 2001). Several CYP 450 enzymes are involved in EET production, and include CYP2C and CYP2J, which epoxidize ARA at one of the four double bonds resulting in two different enantiomers for each EET regioisomer (Capdevila et al., 2000).
EETs can act as both autocrine and paracrine mediators of inflammation, angiogenesis, apoptosis, fibrinolysis, mitogenesis, vasodilation, and bronchodilation (Karara et al., 1990; Karara et al., 1991; Karara et al., 1992; Weintraub et al., 1999). The mechanism of their activity is not fully known, but likely involves two possible mechanisms. One postulated mechanism is that EETs function through membrane receptors such as G protein-coupled receptors or tyrosine kinase receptors to activate intracellular signaling cascades (Li and Campbell, 1997; Hoebel and Graier, 1998; Chen et al., 2000; Fukao et al., 2001; Node et al., 2001; Seubert et al., 2004; Wang et al., 2005). The other possible mechanism is that EETs directly interact with transcriptions factors, ion channels, or intermediary signal transduction proteins. But, the definitive EET-binding receptor/s or signaling proteins are not yet identified.
The catabolism of EETs can occur through several pathways including the main pathway of epoxide hydrolysis by sEH into DHETs, but can also occur through β-oxidation (Fang et al., 2000), fatty acid chain elongation (Fang et al., 1995; Fang et al., 1996; Fang et al., 2001), and rarely through CYP 450 ω-oxidases (Cowart et al., 2002), COX (Zhang et al., 1992; Carroll et al., 1993), LOX (Pace-Asciak et al., 1983), and glutathione-S-transferase (Spearman et al., 1985). Recently, COX-derived proangiogenic metabolites of EETs were identified (Rand et al., 2017). EETs have much higher anti-inflammatory and promoting tissue repair activities compared with DHETs produced by sEH, providing a rationale for sEH inhibition (Inceoglu et al., 2007).
Non-Steroidal Anti-Inflammatory Drugs, Side Effects, and Their Role in Chemoprevention
Acetylsalicylic acid (aspirin) was the first NSAID synthesized and developed for commercial use in 1897 by Felix Hoffman, and it became widely used for its analgesic and anti-inflammatory effects (Sostres et al., 2010). It took approximately 40 years before it was definitively linked to GI injury. It was not until the 1970s that it was discovered that aspirin acts to inhibit prostaglandin production. Today, over 20 NSAIDs are commercially available with varied toxicities and pharmacokinetics and include non-selective COX-1/COX-2 inhibitors (e.g., aspirin, ibuprofen, naproxen, diclofenac) and COX-2 selective inhibitors (coxibs, e.g., celecoxib).
NSAIDs are among the most commonly administered drugs worldwide, as they are available by prescription and over-the-counter. Excitingly, they are also among the most promising agents for prevention of cardiovascular diseases and cancer including colorectal cancer (CRC) (Fischer et al., 2011; Wongrakpanich et al., 2018). One of the major adverse effects of NSAIDs therapy is GI injury, including ulceration, bleeding, inflammation, and even perforation (Wolfe and Singh, 1999; Sudano et al., 2012; Tsoi, 2018). This is a serious clinical challenge causing a major burden on the health care system (specifically, in the US, >100,000 patients are hospitalized with serious NSAID-related GI complications annually, including 16,500 deaths) (Wolfe and Singh, 1999; Bjarnason et al., 2018). In addition to NSAIDs toxicity in the stomach, novel imaging techniques (e.g., capsule endoscopy) have identified the small intestine as a major target organ of toxicity, and lower GI bleeding events may even appear more frequently than upper GI events (Maiden, 2009; Sostres and Lanas, 2011). The local topical effect of NSAIDs is partially responsible for the GI-tract ulceration and bleeding, as co-prescription of aspirin and proton pump inhibitors (PPIs) or enteric-coating of NSAIDs both provide some protection against NSAID-induced gastritis and gastric ulcers; however, these treatment strategies actually increase the incidence of small-bowel ulcers. The non-topical systemic effect of NSAID use appears to be key, as enteric coating, parenteral, and rectal administration all continue to result in gastroenteropathies (Sostres et al., 2010). At least two-thirds of both long-term (>3 months) and short-term (>1 week) NSAID/coxibs users exhibit ulcers in the jejunum and ileum (Maiden, 2009). To date there is no therapy or preventive treatment for NSAIDs-induced GI ulcers available.
In contrast to the adverse ulcer-causing effects, several clinical trials and epidemiological and animal studies have clearly demonstrated that aspirin or NSAIDs actually decrease the risk and mortality of CRC and prevent colon adenoma formation (Dubois, 2009; Burn et al., 2011; Fischer et al., 2011; Cao et al., 2016). To acknowledge this supporting evidence, in 2016 the U.S. Preventive Services Task Force (USPSTF) recommended the use of aspirin to prevent CRC and cardiovascular disease for adults aged between 50 and 70 in the U.S. This recommendation distinguishes aspirin as the first pharmacologic agent to be endorsed by the USPSTF for chemoprevention of a cancer in a population not characterized as high risk (Cao et al., 2016; Chan and Ladabaum, 2016). In addition, notable chemopreventive clinical trials studied the use of aspirin for the prevention of adenomas and carcinomas in patients with Lynch syndrome (Burn et al., 2011) and the use of celecoxib for the reduction of sporadic colorectal adenoma (Dubois, 2009). To achieve significant chemoprevention while also reducing the adverse effects of GI ulcers, alternative approaches with either low doses of NSAIDs or in combination with other chemopreventive agents have been tried with little success (Fischer et al., 2011).
Soluble Epoxide Hydrolase
Epoxides are cyclic three atom rings which are highly reactive and are metabolized by epoxide hydrolases by adding water forming a diol group in a dihydroxylation reaction. The epoxide hydrolase family is comprised of numerous enzymes including: microsomal epoxide hydrolase (mEH), sEH, leukotriene A4 hydrolase, cholesterol 5,6-oxide hydrolase, and epoxide hydrolase 3 (Fretland and Omiecinski, 2000; Decker et al., 2012). The major epoxide hydrolase involved in EET metabolism is sEH, encoded by the EPHX-2 gene located on chromosome 8 (Larsson et al., 1995; Sandberg and Meijer, 1996). The 60-kDa sEH forms a homodimer with each monomer containing N-terminal domain lipid phosphatase activity with unknown physiologic significance, as well as a functionally independent C-terminal domain epoxide hydrolase activity (Argiriadi et al., 1999; Cronin et al., 2003; Newman et al., 2003).
In the early 1990s, concerns about the potential adverse effects of a chronic loss of sEH have centered around the clearance of xenobiotics, particularly the potential loss of function to clear epoxide-containing carcinogens. These concerns have been well studied and demonstrated that these xenobiotics are not the substrates of sEH (Seidegard and Ekstrom, 1997; Fretland and Omiecinski, 2000; Morisseau and Hammock, 2005; Shimada, 2006; Morisseau and Hammock, 2007; Decker et al., 2009). Studies of evolution of the microsomal EH (mEH) and sEH show that although they have common origins, they diverge at the level of prokaryotes and their substrate selectivity is vastly different (Fretland and Omiecinski, 2000). mEH rapidly degrades carcinogenic epoxides on cyclic systems (e.g., aflatoxin epoxide and benzo[a]pyrene epoxide) (Shimada, 2006). To date, sEH has not been shown to metabolize a single carcinogenic epoxide and endogenous eicosanoids are identified as its only substrates (Seidegard and Ekstrom, 1997; Decker et al., 2009). sEH inhibitors have been cross-tested on the mEH, which show a much greater selectivity for sEH (Morisseau and Hammock, 2005; Morisseau and Hammock, 2007). Most importantly, the fatty epoxides stabilized through sEH inhibition display a strong anti-inflammatory action (Norwood et al., 2010).
sEH shows high tissue expression in liver, intestine, kidneys, brain, and vasculature, with lower expression in spleen, lung, and testes (VanRollins et al., 1993; Fang et al., 1995; Johansson et al., 1995; Yu et al., 2000). The subcellular localization is variable; however, sEH is mostly localized to the cytosol with peroxisomal involvement in hepatocytes and renal proximal tubule epithelial cells (Hollinshead and Meijer, 1988; Eriksson et al., 1991; Mullen et al., 1999; Enayetallah et al., 2006). The biological activity and significance of different cell types and different subcellular localization of sEH have not been fully elucidated.
Development and Application of Selective sEH Inhibitors
Inhibitors have been developed for sEH, which have been instrumental in understanding the biology of EETs and sEH, as well as for testing their use in practical applications of disease. Importantly, through decades of studies, inhibitors have been selected for their stability, high selectivity against sEH, and high potency. The first iterations of sEH inhibitors were epoxide-containing trans-3-phenylglycidol and chalcone oxide molecules, and while these inhibitors are selective and potent, they are unstable with only temporary effects on sEH (Mullin and Hammock, 1982; Dietze et al., 1991; Dietze et al., 1993; Morisseau et al., 1998).
Newer classes of sEH inhibitors include urea- and carbamate-based molecules, which are more stable than previous inhibitors, and are also highly selective and potent (Morisseau et al., 1999). One inhibitor in this class is N,N’-dicyclohexylurea (DCU). DCU is the basis for several urea-based inhibitors with increased water solubility, and which bind to the active site of sEH via the urea subgroup with hydrogen bonds and salt bridges (Argiriadi et al., 2000; Morisseau et al., 2002; Kim et al., 2004).
The most recent and well-studied sEH inhibitors are 12-(3-adamantyl-ureido)-dodecanoic acid (AUDA) compounds. These AUDA compounds have been modified to bind the catalytic site of sEH via hydrogen bonds and have physical properties ideal for pharmacologic use including high water solubility, oral bioavailability, low melting point, increased stability, and high potency. Trans-4-[4-(3-adamantan-1-ylureido)cyclohexyl oxy]benzoic acid (t-AUCB) is another highly promising sEH inhibitor. Thus far, sEH inhibitors have been demonstrated to be safe, with very few side effects. The effective dosages show very little toxicity.
sEH Inhibition Increases EETs, Promotes Ulcer Healing, and Inhibits Pancreatitis, Colitis, and Inflammation-Associated Cancer in Mice
Chronic inflammation has been demonstrated to be among the most important factors leading to carcinogenesis. Obesity, inflammatory bowel disease (IBD), and chronic colitis and pancreatitis are some specific examples of inflammatory conditions that are known risk factors for cancer with shared mechanisms involving altered epoxide fatty acid metabolites and sEH (Zhang et al., 2013; Liao et al., 2016a; Wang et al., 2018b; Yang, 2018). As a result of chronic inflammation there is overproduction of reactive oxygen and nitrogen species (RONS), as well as dysregulation of ARA metabolites. Inflammatory cell infiltration driven by neutrophils, macrophages, and subsequently lymphocytes alters tissue microenvironments with an enhanced production of RONS (e.g., superoxide) and ARA metabolites (e.g., PGE2 and LTB4). Leukocytes also act to modify vasculature through effects on endothelial cells (Yang et al., 2009).
Mounting evidence links EETs and sEH inhibition to treatment for chronic inflammatory conditions and inflammation-associated cancer. First, sEH has been shown to be overexpressed in human colitis and CRC (Zhang et al., 2013b). Second, recent studies demonstrated that sEH bridges obesity to colonic inflammation and potentially to colorectal carcinogenesis (Wang et al., 2018b; Yang, 2018; Yang et al., 2018). And finally, sEH inhibition blocks colitis and its associated carcinogenesis (Zhang et al., 2012; Zhang et al., 2013b; Zhang et al., 2013c) and NSAID-induced GI ulcers (Goswami et al., 2016; Goswami et al., 2017). However, the relative contribution and mechanism of sEH in sporadic colon carcinogenesis is not fully known.
Initial studies regarding the anti-inflammatory effects of EETs showed that physiologic concentration of EETs or overexpression of CYP2J2 decreases cytokine-mediated endothelial cell adhesion molecule expression, thus preventing leukocyte recruitment via a mechanism involving NF-κB (Node et al., 1999). Inhibition of EET catabolism through sEH inhibitors results in a significant increase in their anti-inflammatory properties, and effectively treat mouse models of inflammatory diseases.
EETs cause an increased activity and expression of peroxisome proliferator-activated receptor-gamma (PPAR-γ), a low-affinity nuclear hormone receptor involved in fatty acid metabolism, and inhibition of the NF-κB pathway (Stienstra et al., 2007; Ward and Tan, 2007). It is likely that at least a portion of the anti-inflammatory effects of EETs act through PPAR-γ as cell culture models show that inhibition of this nuclear hormone receptor diminishes this effect. The role of PPAR-γ in inflammation is through inhibition of activity of pro-inflammatory transcription factors activator protein-1 (AP-1), signal transducers and activators of transcription (STAT), and nuclear factor κB (NF-κB). Thus, in addition to increasing EETs, ligand activation of PPAR-γ is another promising strategy for anti-inflammatory activity (Dubuquoy et al., 2006).
sEH Inhibition Can Block NSAID-Induced Gastrointestinal Ulcers
The underlying mechanisms of NSAID-induced GI ulcers are not fully understood; however, they likely involve endoplasmic reticulum (ER)/mitochondrial stress and mucinous epithelia-microvascular barrier breakdown (Hawkey, 1996; Wolfe and Singh, 1999; Cheng et al., 2015). The effects of NSAIDs/aspirin on enterocytes and on vascular endothelial cells in the lamina propria are considered toxic/pathogenic based to the findings of i) the relatively high concentrations (mM) of conjugated NSAIDs through the enterohepatic circulation (Seitz and Boelsterli, 1998; Treinen-Moslen and Kanz, 2006), ii) direct toxic effects on intestinal mucosal epithelial and endothelial cells (break down the mucosal-vascular barrier), iii) effects on mitochondria (Somasundaram et al., 1997) and ER stress (Tsutsumi et al., 2004), and iv) mucosal injury related to active inflammation.
NSAID-induced GI ulcers can be counter-balanced by epoxides through direct administration of EETs or indirectly by increasing EETs through sEH inhibition (Goswami et al., 2016; Goswami et al., 2017; Yang, 2018). sEH inhibition increases EET bioavailability and yields significant anti-inflammatory effects (Node et al., 1999; Schmelzer et al., 2005; Imig and Hammock, 2009; Norwood et al., 2010). In addition to increasing EET availability, a partial mechanism for the anti-inflammatory effect of sEH inhibition is through a decrease of its product DHET, which has been shown to be important for monocyte chemoattractant protein-1 (MCP-1)-mediated monocyte chemotaxis (Kundu et al., 2013). Thus, co-administration of NSAIDs and a sEH inhibitor has potential synergistic effects of modulating oxylipin profile toward the resolution of inflammation (Schmelzer et al., 2006; Zhang et al., 2014a; Zhang et al., 2014b). The potential mechanistic role of sEH inhibition against NSAID-induced GI ulcers is likely either through decreasing monocyte recruitment and inflammation (Rainsford, 1993; Kundu et al., 2013), blocking NSAID-induced ER/mitochondrial stress (Hawkey, 1996; Inceoglu et al., 2017) to reduce epithelial-vascular barrier injury (Rainsford, 1993), or through enhancing the tissue repair process and angiogenesis (Panigrahy et al., 2013) as outlined in Figure 2.
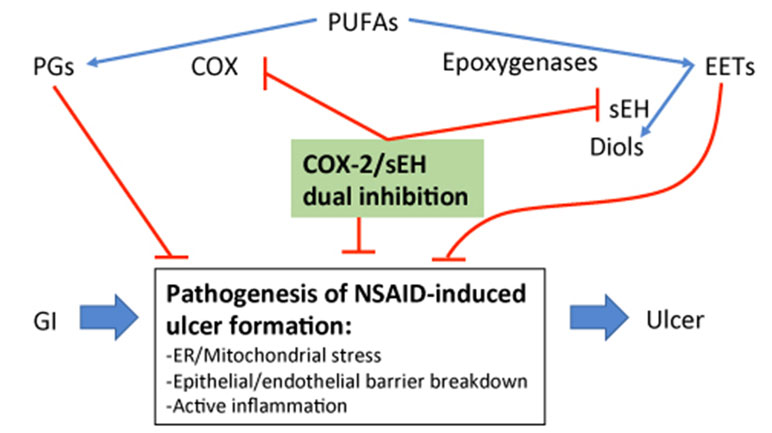
Figure 2 Outline of the potential mechanism of sEH inhibition-stabilized EETs in blocking NSAID-induced GI ulcers.
Chronic Pancreatitis and Carcinogenesis
One of the most common conditions leading to pancreatic tumorigenesis is the inflammatory condition chronic pancreatitis. In the development of chronic pancreatitis aberrant ARA metabolism partially mediated by sEH acts to promote inflammation with a decrease of the anti-inflammatory EETs. Inhibition of sEH has been demonstrated as a successful strategy in murine models to inhibit chronic pancreatitis and the development of pancreatic intraepithelial neoplasms (PanIN) using trans-4-{4-[3-(4-chloro-3-trifluoromethyl-phenyl)-ureido]-cyclohexyloxy}-pyridine-2-carboxylic acid methylamide (t-CUPM) in KRAS-mutant mice (Liao et al., 2016a; Liao et al., 2016b). t-CUPM is a novel molecule with dual inhibition of sEH and RAF1 proto-oncogene serine/threonine kinase (c-RAF). This effect was due to both the anti-inflammatory effects of sEH inhibition and the blockade of constitutive KRAS signaling through c-RAF inhibition.
Inflammatory Bowel Disease, Colorectal Carcinogenesis, and the Role of sEH and COX2
A well-recognized risk factor for sporadic colon cancer is chronic inflammation, in which aberrant PUFA metabolism, particularly ARA, is believed to be the key inflammatory mediator contributing to carcinogenesis (Ding et al., 2003; Hermanova et al., 2008). Using IBD as an example model, studies have shown that increased prostaglandins, prostaglandin synthase activity, and mucosal expression of COX-2 all correlate with the severity of IBD activity. NSAIDs are especially promising in chemoprevention for IBD, where there is a direct relationship between inflammation and carcinogenesis. Unfortunately, the clinical application of NSAIDs with COX inhibitory activity tends to exacerbate IBD. These findings are likely multifactorial related to both delayed ulcer healing, a known side effect of NSAIDs, as well as a shunt of ARA substrates to the LOX pathway leading to overproduction of the inflammatory 5-LOX-LTB4 pathway (Goswami et al., 2016).
Extensive epidemiological and experimental reports show COX-2 has a prominent role in carcinogenesis. Much of these observations come from the effects of aspirin and other NSAIDs resulting in the decreased rate of CRCs, including notable chemopreventive clinical trials testing the use of aspirin in the prevention of GI adenomas and carcinomas in patients with Lynch syndrome (Burn et al., 2011), and celecoxib for the reduction of colon adenomas (Dubois, 2009). COX-2 has been shown to be overexpressed by colon tumors, and COX-2 deficient mice show a decrease in APC-associated carcinogenesis. Further, under the inflammatory conditions of ulcerative colitis (UC), COX-2 is commonly overexpressed in the sequence of UC progression to carcinoma, as well as in non-dysplastic epithelium. The increased expression of COX-2 in UC has been shown to be associated with carcinogenesis rather than the inflammatory activity. PGE2 production secondary to COX-2 activity promotes epithelial proliferation and crypt stem cell survival. Because of this, COX-2 selective inhibitors were developed with the hopes of anti-cancer effects without the development of GI ulcers; however, these agents showed significant cardiovascular risks in clinical trials, and one trial showed a continued side effect of GI ulcers (Dubois, 2009).
Considering that sEH is overexpressed in human colitis and CRC (Zhang et al., 2013b), and that sEH inhibition blocks colonic inflammation and carcinogenesis (Wang et al., 2018b; Yang, 2018; Yang et al., 2018), sEH inhibition is a promising agent for the treatment of IBD. sEH inhibition markedly inhibits the progression of inflammation and dysplasia in mouse models of UC (Yang, 1996; Yang et al., 2008; Zhang, 2012). This is in part due to the regulation of cytokines and chemokines, but they also regulate other key enzymatic pathways in ARA metabolism. By increasing EETs, there is a reduction of COX-2 and 5-LOX and their downstream metabolites leading to a reduction of symptoms associated with severe inflammation (Schmelzer et al., 2005; Zhang, 2012). sEH inhibition can be simplistically viewed as shifting ARA metabolism to an EET-rich profile and restoring an anti-inflammatory state.
sEH inhibition—Potent Anti-Inflammatory Activity vs Potential Angiogenesis/Tumor Progression Action, Is It Double Edge Sword?
It has been well documented that sEH inhibition has high potential for anti-inflammatory, anti-hypertension, anti-diabetes, and anti-hyperalgesic activities. In addition to the anti-inflammatory activity, sEH inhibition is also associated with an indirect reduction of endotoxin-induced COX and LOX metabolite production in vivo, and EETs can transcriptionally inhibit COX-2 expression (Schmelzer et al., 2005; Wang et al., 2018a). However, one of the main concerns of sEH inhibition is related to the observations that EETs derived from the cytochrome p450 pathway actually promote angiogenesis and tumor progression (Elson and Yu, 1994; Panigrahy et al., 2012). This may be partly due to the specific fatty acids, which are epoxidized, as it appears that ω-3 epoxy fatty acid derivatives (e.g., epoxydocosapentaenoic acids) when stabilized by sEH inhibitors are anti-angiogenic and block tumor growth and metastasis, contrary to ω-6 ARA metabolites such as EETs (Zhang et al., 2013a). Furthermore, sEH inhibitors have been shown to promote tumor escape from dormancy and led to extensive metastases in multiple cancer models as a result of increased endothelium-derived EET concentration (Panigrahy et al., 2012). Another study aimed to reduce EETs through downregulation of the cytochrome p450 enzyme Cyp2c44 using PPARα agonists, and demonstrated a reduction in metastatic non-small cell lung cancer growth and angiogenesis (Skrypnyk et al., 2014). In spite of the association of EETs with angiogenesis and cancer progression, there is recent evidence that COX-derived products of EET metabolism may also contribute to angiogenesis, and the dual inhibition of sEH and COX results in a robust inhibition of tumor growth and a net negative effect on angiogenesis (Rand et al., 2017). This would further establish the COX-mediated EET metabolism as physiologically relevant and provides additional rationale for the sEH/COX dual inhibitor strategy. Two dual therapy cancer models of sEH inhibition combined with either COX-2 inhibition or c-RAF inhibition show robust tumor growth and metastasis inhibition, and in the case of the dual sEH/COX-2 inhibitor (discussed below), both bladder cancers and glioblastomas were found to be inhibited with significantly decreased angiogenesis (Liao et al., 2016b; Li et al., 2017; Wang et al., 2018a).
Dual COX2/sEH Inhibition
To minimize the angiogenic effect of sEH inhibitor, a novel COX-2/sEH dual inhibitor has been synthesized, 4-(5-phenyl-3-{3-[3-(4-trifluoromethyl-phenyl)-ureido]-propyl}-pyrazol-1-yl)-benzenesulfonamide (PTUPB), based on the structures of celecoxib and sEH inhibitor trans-4-[4-(3-adamantan-1-ylureido)cyclohexyloxy]benzoic acid (t-AUCB) (Hwang et al., 2011), to circumvent problems associated with formulation of two drugs for co-administration (Morphy and Rankovic, 2005). Studies related to the pharmacokinetics and safety profile have demonstrated favorable characteristics (Hwang et al., 2011; Hye Khan et al., 2016). PTUPB is potent in inhibiting sEH (IC50, 0.9nM) and COX2 (IC50, 1.26uM), but not COX-1 (IC50, > 100uM), and is effective at suppressing inflammation (Hwang et al., 2011; Zhang et al., 2014b).
PTUPB’s effects are more potent than celecoxib or t-AUCB alone and in combination (Hwang et al, 2011; Zhang et al., 2014b). PTUPB has been demonstrated to protect against metabolic abnormalities and retain kidney function in the Zucker diabetic fatty rat model (Hye Khan et al., 2016). In mouse models of cancer, PTUPB has been shown to be effective to inhibit tumor growth, metastasis, and angiogenesis, and to potentiate cisplatin-based therapies without increased toxicity (Zhang et al., 2014b; Li et al., 2017; Wang et al., 2018a). Thus, a novel strategy of co-targeting both COX2 and sEH via the dual inhibitor PTUPB is particularly intriguing for chemoprevention in the colorectum, especially in patients with IBD. This strategy would provide an environment enriched with EETs, with the chemopreventive benefits of COX-2 inhibition while minimizing side effects including GI tract ulceration, as outlined in Figure 3. Taken together, these preclinical studies highly support multi-target agents against sEH and COX2, and their potential for translating into clinical trial, and toward this direction, application for clinical trials is ongoing.
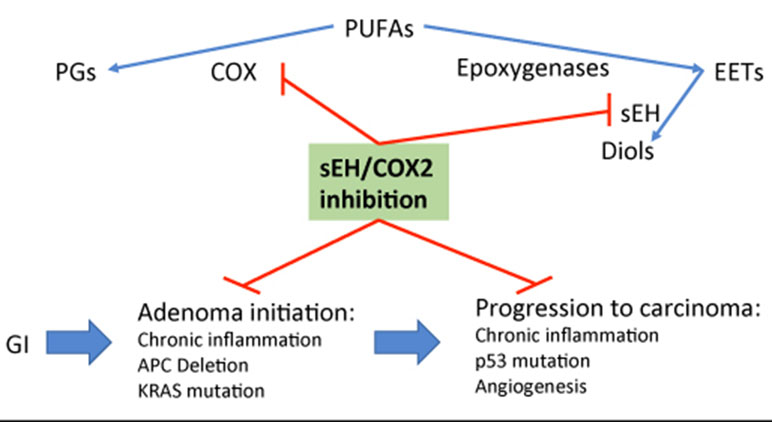
Figure 3 Outline of potential mechanism of sEH inhibition-stabilized EETs in inhibiting colonic adenoma—carcinoma sequence via targeting inflammation and its activated Wnt/mutant p53 signals.
Summary of sEH and COX2 Inhibition on Anti-Inflammatory and Carcinogenesis
The decades of studies of ARA metabolism and the sEH point to the potential of sEH inhibition and subsequent enrichment of EETs as an effective strategy for therapies in inflammatory conditions. This is particularly promising in colon and pancreas where chronic inflammatory conditions are well-documented risk factors for carcinogenesis, and therefore sEH inhibition may be ideal as a chemopreventive agent alone or in combination with other strategies. These effects have been robustly studied in mouse models, which show significant reductions in inflammation and cancer development with little to no side effects. Additionally, studies have shown that sEH inhibition can dramatically reduce the side effects of NSAIDs when given together. Thus, we believe that sEH should be further investigated in clinical trials for inflammatory conditions and inflammation-induced cancers.
One such strategy of sEH treatment involves their co-administration with NSAIDs to further modulate ARA metabolism. NSAIDs including aspirin and celecoxib are among the most widely administered medications worldwide. While they are promising for chemoprevention and in cardiovascular diseases, they also cause significant morbidity through their side effects of gastroenteropathies. These NSAID-related ulcers and bleeding are potentially life-threatening; thus, their use in chemoprevention is not practical. This emphasizes the necessity for a novel approach to harness the beneficial aspects of NSAIDs without the side effects, and one such approach may be through the dual inhibition of COX and sEH. PTUPB is one such agent, although single agent NSAIDs combined with sEH inhibitors may also be effective. This approach has the potential to revolutionize cancer treatment and prevention, and also has broad implications for the millions of people using NSAIDs chronically for pain and rheumatologic diseases.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was supported by NIH R01 DK10776, CA172431, and CA164041 to G-YY.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Argiriadi, M. A., Morisseau, C., Goodrow, M. H., Dowdy, D. L., Hammock, B. D., Christianson, D. W. (2000). Binding of alkylurea inhibitors to epoxide hydrolase implicates active site tyrosines in substrate activation. J. Biol. Chem. 275 (20), 15265–15270. doi: 10.1074/jbc.M000278200
Argiriadi, M. A., Morisseau, C., Hammock, B. D., Christianson, D. W. (1999). Detoxification of environmental mutagens and carcinogens: structure, mechanism, and evolution of liver epoxide hydrolase. Proc. Natl. Acad. Sci. U. S. A. 96 (19), 10637–10642. doi: 10.1073/pnas.96.19.10637
Bjarnason, I., Scarpignato, C., Holmgren, E., Olszewski, M., Rainsford, K. D., Lanas, A. (2018). Mechanisms of damage to the gastrointestinal tract from nonsteroidal anti-inflammatory drugs. Gastroenterology 154 (3), 500–514. doi: 10.1053/j.gastro.2017.10.049
Burn, J., Gerdes, A. M., Macrae, F., Mecklin, J. P., Moeslein, G., Olschwang, S., et al. (2011). Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 378 (9809), 2081–2087. doi: 10.1016/S0140-6736(11)61049-0
Cao, Y., Nishihara, R., Wu, K., Wang, M., Ogino, S., Willett, W. C., et al. (2016). Population-wide impact of long-term use of aspirin and the risk for cancer. JAMA Oncol. 2 (6), 762–769. doi: 10.1001/jamaoncol.2015.6396
Capdevila, J. H., Falck, J. R. (2001). The CYP P450 arachidonic acid monooxygenases: from cell signaling to blood pressure regulation. Biochem. Biophys. Res. Commun. 285 (3), 571–576. doi: 10.1006/bbrc.2001.5167
Capdevila, J. H., Falck, J. R., Harris, R. C. (2000). Cytochrome P450 and arachidonic acid bioactivation. J. Lipid. Res. 41 (2), 163–181.
Carroll, M. A., Balazy, M., Margiotta, P., Falck, J. R., McGiff, J. C. (1993). Renal vasodilator activity of 5,6-epoxyeicosatrienoic acid depends upon conversion by cyclooxygenase and release of prostaglandins. J. Biol. Chem. 268 (17), 12260–12266.
Chan, A. T., Ladabaum, U. (2016). Where do we stand with aspirin for the prevention of colorectal cancer? The USPSTF recommendations. Gastroenterology 150 (1), 14–18. doi: 10.1053/j.gastro.2015.11.018
Chen, J. K., Capdevila, J., Harris, R. C. (2000). Overexpression of C-terminal Src kinase blocks 14, 15-epoxyeicosatrienoic acid-induced tyrosine phosphorylation and mitogenesis. J. Biol. Chem. 275 (18), 13789–13792. doi: 10.1074/jbc.275.18.13789
Cheng, Y., Lin, J., Liu, J., Wang, Y., Yan, W., Zhang, M. (2015). Decreased vascular endothelial growth factor expression is associated with cell apoptosis in low-dose aspirin-induced gastric mucosal injury. Am. J. Med. Sci. 349 (2), 110–116. doi: 10.1097/MAJ.0000000000000409
Cowart, L. A., Wei, S., Hsu, M. H., Johnson, E. F., Krishna, M. U., Falck, J. R., et al. (2002). The CYP4A isoforms hydroxylate epoxyeicosatrienoic acids to form high affinity peroxisome proliferator-activated receptor ligands. J. Biol. Chem. 277 (38), 35105–35112. doi: 10.1074/jbc.M201575200
Cronin, A., Mowbray, S., Durk, H., Homburg, S., Fleming, I., Fisslthaler, B., et al. (2003). The N-terminal domain of mammalian soluble epoxide hydrolase is a phosphatase. Proc. Natl. Acad. Sci. U. S. A. 100 (4), 1552–1557. doi: 10.1073/pnas.0437829100
Decker, M., Arand, M., Cronin, A. (2009). Mammalian epoxide hydrolases in xenobiotic metabolism and signalling. Arch. Toxicol. 83 (4), 297–318. doi: 10.1007/s00204-009-0416-0
Decker, M., Adamska, M., Cronin, A., Di Giallonardo, F., Burgener, J., Marowsky, A., et al. (2012). EH3 (ABHD9): the first member of a new epoxide hydrolase family with high activity for fatty acid epoxides. J. Lipid. Res. 53 (10), 2038–2045. doi: 10.1194/jlr.M024448
Dietze, E. C., Casas, J., Kuwano, E., Hammock, B. D. (1993). Inhibition of epoxide hydrolase from human, monkey, bovine, rabbit and murine liver by trans-3-phenylglycidols. Comp. Biochem. Physiol. B. 104 (2), 309–314. doi: 10.1016/0305-0491(93)90373-D
Dietze, E. C., Kuwano, E., Casas, J., Hammock, B. D. (1991). Inhibition of cytosolic epoxide hydrolase by trans-3-phenylglycidols. Biochem. Pharmacol. 42 (6), 1163–1175. doi: 10.1016/0006-2952(91)90250-9
Ding, X. Z., Hennig, R., Adrian, T. E. (2003). Lipoxygenase and cyclooxygenase metabolism: new insights in treatment and chemoprevention of pancreatic cancer. Mol. Cancer 2, 10. doi: 10.1186/1476-4598-2-10
Dubois, R. N. (2009). New, long-term insights from the adenoma prevention with celecoxib trial on a promising but troubled class of drugs. Cancer Prev. Res. (Phila) 2 (4), 285–287. doi: 10.1158/1940-6207.CAPR-09-0038
Dubuquoy, L., Rousseaux, C., Thuru, X., Peyrin-Biroulet, L., Romano, O., Chavatte, P., et al. (2006). PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut 55 (9), 1341–1349. doi: 10.1136/gut.2006.093484
Elson, C. E., Yu, S. G. (1994). The chemoprevention of cancer by mevalonate-derived constituents of fruits and vegetables. J. Nutr. 124 (5), 607–614. doi: 10.1093/jn/124.5.607
Enayetallah, A. E., French, R. A., Barber, M., Grant, D. F. (2006). Cell-specific subcellular localization of soluble epoxide hydrolase in human tissues. J. Histochem. Cytochem. 54 (3), 329–335. doi: 10.1369/jhc.5A6808.2005
Eriksson, A. M., Zetterqvist, M. A., Lundgren, B., Andersson, K., Beije, B., DePierre, J. W. (1991). Studies on the intracellular distributions of soluble epoxide hydrolase and of catalase by digitonin-permeabilization of hepatocytes isolated from control and clofibrate-treated mice. Eur. J. Biochem. 198 (2), 471–476. doi: 10.1111/j.1432-1033.1991.tb16037.x
Fang, X., Kaduce, T. L., VanRollins, M., Weintraub, N. L., Spector, A. A. (2000). Conversion of epoxyeicosatrienoic acids (EETs) to chain-shortened epoxy fatty acids by human skin fibroblasts. J. Lipid. Res. 41 (1), 66–74. doi: 10.1074/jbc.M011761200
Fang, X., Kaduce, T. L., Weintraub, N. L., VanRollins, M., Spector, A. A. (1996). Functional implications of a newly characterized pathway of 11,12-epoxyeicosatrienoic acid metabolism in arterial smooth muscle. Circ. Res. 79 (4), 784–793. doi: 10.1161/01.RES.79.4.784
Fang, X., VanRollins, M., Kaduce, T. L., Spector, A. A. (1995). Epoxyeicosatrienoic acid metabolism in arterial smooth muscle cells. J. Lipid. Res. 36 (6), 1236–1246.
Fang, X., Kaduce, T. L., Weintraub, N. L., Harmon, S., Teesch, L. M., Morisseau, C., et al. (2001). Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. J. Biol. Chem. 276 (18), 14867–14874. doi: 10.1074/jbc.M011761200
Fischer, S. M., Hawk, E. T., Lubet, R. A. (2011). Coxibs and other nonsteroidal anti-inflammatory drugs in animal models of cancer chemoprevention. Cancer Prev. Res. (Phila) 4 (11), 1728–1735. doi: 10.1158/1940-6207.CAPR-11-0166
Fretland, A. J., Omiecinski, C. J. (2000). Epoxide hydrolases: biochemistry and molecular biology. Chem. Biol. Interact. 129 (1–2), 41–59. doi: 10.1016/S0009-2797(00)00197-6
Fukao, M., Mason, H. S., Kenyon, J. L., Horowitz, B., Keef, K. D. (2001). Regulation of BK(Ca) channels expressed in human embryonic kidney 293 cells by epoxyeicosatrienoic acid. Mol. Pharmacol. 59 (1), 16–23. doi: 10.1124/mol.59.1.16
Goswami, S. K., Rand, A. A., Wan, D., Yang, J., Inceoglu, B., Thomas, M., et al. (2017). Pharmacological inhibition of soluble epoxide hydrolase or genetic deletion reduces diclofenac-induced gastric ulcers. Life Sci. 180, 114–122. doi: 10.1016/j.lfs.2017.05.018
Goswami, S. K., Wan, D., Yang, J., Trindade da Silva, C. A., Morisseau, C., Kodani, S. D., et al. (2016). Anti-ulcer efficacy of soluble epoxide hydrolase inhibitor tppu on diclofenac-induced intestinal ulcers. J. Pharmacol. Exp. Ther. 357 (3), 529–536. doi: 10.1124/jpet.116.232108
Hawkey, C. J. (1996). Non-steroidal anti-inflammatory drug gastropathy: causes and treatment. Scand. J. Gastroenterol. Suppl. 220, 124–127. doi: 10.3109/00365529609094763
Hermanova, M., Trna, J., Nenutil, R., Dite, P., Kala, Z. (2008). Expression of COX-2 is associated with accumulation of p53 in pancreatic cancer: analysis of COX-2 and p53 expression in premalignant and malignant ductal pancreatic lesions. Eur. J. Gastroenterol. Hepatol. 20 (8), 732–739. doi: 10.1097/MEG.0b013e3282f945fb
Hiesinger, K., Wagner, K. M., Hammock, B. D., Proschak, E., Hwang, S. H. (2019). Development of multitarget agents possessing soluble epoxide hydrolase inhibitory activity. Prostaglandins. Other Lipid Mediat. 140, 31–39. doi: 10.1016/j.prostaglandins.2018.12.003
Hoebel, B. G., Graier, W. F. (1998). 11,12-Epoxyeicosatrienoic acid stimulates tyrosine kinase activity in porcine aortic endothelial cells. Eur. J. Pharmacol. 346 (1), 115–117. doi: 10.1016/S0014-2999(98)00118-6
Hollinshead, M., Meijer, J. (1988). Immunocytochemical analysis of soluble epoxide hydrolase and catalase in mouse and rat hepatocytes demonstrates a peroxisomal localization before and after clofibrate treatment. Eur. J. Cell Biol. 46 (3), 394–402.
Hwang, S. H., Wagner, K. M., Morisseau, C., Liu, J. Y., Dong, H., Wecksler, A. T., et al. (2011). Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J. Med. Chem. 54 (8), 3037–3050. doi: 10.1021/jm2001376
Hye Khan, M. A., Hwang, S. H., Sharma, A., Corbett, J. A., Hammock, B. D., Imig, J. D. (2016). A dual COX-2/sEH inhibitor improves the metabolic profile and reduces kidney injury in Zucker diabetic fatty rat. Prostaglandins Other Lipid Mediat. 125, 40–47. doi: 10.1016/j.prostaglandins.2016.07.003
Imig, J. D., Hammock, B. D. (2009). Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 8 (10), 794–805. doi: 10.1038/nrd2875
Inceoglu, B., Bettaieb, A., Haj, F. G., Gomes, A. V., Hammock, B. D. (2017). Modulation of mitochondrial dysfunction and endoplasmic reticulum stress are key mechanisms for the wide-ranging actions of epoxy fatty acids and soluble epoxide hydrolase inhibitors. Prostaglandins Other Lipid Mediat. 133, 68–78. doi: 10.1016/j.prostaglandins.2017.08.003
Inceoglu, B., Schmelzer, K. R., Morisseau, C., Jinks, S. L., Hammock, B. D. (2007). Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs). Prostaglandins. Other Lipid Mediat. 82 (17–4), 42–49. doi: 10.1016/j.prostaglandins.2006.05.004
Jaeschke, H., Smith, C. W. (1997). Mechanisms of neutrophil-induced parenchymal cell injury. J. Leukoc. Biol. 61 (6), 647–653. doi: 10.1002/jlb.61.6.647
Johansson, C., Stark, A., Sandberg, M., Ek, B., Rask, L., Meijer, J. (1995). Tissue specific basal expression of soluble murine epoxide hydrolase and effects of clofibrate on the mRNA levels in extrahepatic tissues and liver. Arch. Toxicol. 70 (1), 61–63. doi: 10.1007/s002040050250
Karara, A., Dishman, E., Falck, J. R., Capdevila, J. H. (1991). Endogenous epoxyeicosatrienoyl-phospholipids. J. Biol. Chem. 266 (12), 7561–7569.
Karara, A., Dishman, E., Jacobson, H., Falck, J. R., Capdevila, J. H. (1990). Arachidonic acid epoxygenase. FEBS Lett. 268 (1), 227–230. doi: 10.1016/0014-5793(90)81014-F
Karara, A., Wei, S., Spady, D., Swift, L., Capdevila, J. H., Falck, J. R. (1992). Arachidonic acid epoxygenase: structural characterization and quantification of epoxyeicosatrienoates in plasma. Biochem. Biophys. Res. Commun. 182 (3), 1320–1325. doi: 10.1016/0006-291X(92)91877-S
Kim, I. H., Morisseau, C., Watanabe, T., Hammock, B. D. (2004). Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J. Med. Chem. 47 (8), 2110–2122. doi: 10.1021/jm030514j
Kundu, S., Roome, T., Bhattacharjee, A., Carnevale, K. A., Yakubenko, V. P., Zhang, R., et al. (2013). Metabolic products of soluble epoxide hydrolase are essential for monocyte chemotaxis to MCP-1 in vitro and in vivo. J. Lipid. Res. 54 (2), 436–447. doi: 10.1194/jlr.M031914
Larsson, C., White, I., Johansson, C., Stark, A., Meijer, J. (1995). Localization of the human soluble epoxide hydrolase gene (EPHX2) to chromosomal region 8p21-p12. Hum. Genet. 95 (3), 356–358. doi: 10.1007/BF00225209
Li, P. L., Campbell, W. B. (1997). Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circ. Res. 80 (6), 877–884. doi: 10.1161/01.RES.80.6.877
Li, J., Zhou, Y., Wang, H., Gao, Y., Li, L., Hwang, S. H., et al. (2017). COX-2/sEH dual inhibitor PTUPB suppresses glioblastoma growth by targeting epidermal growth factor receptor and hyaluronan mediated motility receptor. Oncotarget 8 (50), 87353–87363. doi: 10.18632/oncotarget.20928
Liao, J., Hwang, S. H., Li, H., Liu, J. Y., Hammock, B. D., Yang, G. Y. (2016a). Inhibition of Chronic Pancreatitis and Murine Pancreatic Intraepithelial Neoplasia by a Dual Inhibitor of c-RAF and Soluble Epoxide Hydrolase in LSL-KrasG(1)(2)D/Pdx-1-Cre Mice. Anticancer Res. 36 (1), 27–37.
Liao, J., Hwang, S. H., Li, H., Yang, Y., Yang, J., Wecksler, A. T., et al. (2016b). Inhibition of mutant KrasG12D-initiated murine pancreatic carcinoma growth by a dual c-Raf and soluble epoxide hydrolase inhibitor t-CUPM. Cancer Lett. 371 (2), 187–193. doi: 10.1016/j.canlet.2015.11.042
Maiden, L. (2009). Capsule endoscopic diagnosis of nonsteroidal antiinflammatory drug-induced enteropathy. J. Gastroenterol. 44 Suppl 19, 64–71. doi: 10.1016/S0006-2952(02)00952-8
Morisseau, C., Hammock, B. D. (2005). Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu. Rev. Pharmacol. Toxicol. 45, 311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920
Morisseau, C., Hammock, B. D. (2007). Measurement of soluble epoxide hydrolase (sEH) activity. Curr. Protoc. Toxicol. Chapter 4, Unit 4, 23. doi: 10.1002/0471140856.tx0423s33
Morisseau, C., Hammock, B. D. (2012). Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 2013 (53), 37–58. doi: 10.1146/annurev-pharmtox-011112-140244
Morisseau, C., Du, G., Newman, J. W., Hammock, B. D. (1998). Mechanism of mammalian soluble epoxide hydrolase inhibition by chalcone oxide derivatives. Arch. Biochem. Biophys. 356 (2), 214–228. doi: 10.1006/abbi.1998.0756
Morisseau, C., Goodrow, M. H., Newman, J. W., Wheelock, C. E., Dowdy, D. L., Hammock, B. D. (2002). Structural refinement of inhibitors of urea-based soluble epoxide hydrolases. Biochem. Pharmacol. 63 (9), 1599–1608. doi: 10.1016/S0006-2952(02)00952-8
Morisseau, C., Goodrow, M. H., Dowdy, D., Zheng, J., Greene, J. F., Sanborn, J. R., et al. (1999). Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc. Natl. Acad. Sci. U. S. A. 96 (16), 8849–8854. doi: 10.1073/pnas.96.16.8849
Morphy, R., Rankovic, Z. (2005). Designed multiple ligands. J. Med. Chem. 48 (21), 6523–6543. doi: 10.1021/jm058225d
Mullen, R. T., Trelease, R. N., Duerk, H., Arand, M., Hammock, B. D., Oesch, F., et al. (1999). Differential subcellular localization of endogenous and transfected soluble epoxide hydrolase in mammalian cells: evidence for isozyme variants. FEBS Lett. 445 (2–3), 301–305. doi: 10.1016/S0014-5793(99)00142-8
Mullin, C. A., Hammock, B. D. (1982). Chalcone oxides–potent selective inhibitors of cytosolic epoxide hydrolase. Arch. Biochem. Biophys. 216 (2), 423–439. doi: 10.1016/0003-9861(82)90231-4
Newman, J. W., Morisseau, C., Harris, T. R., Hammock, B. D. (2003). The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc. Natl. Acad. Sci. U. S. A. 100 (4), 1558–1563. doi: 10.1073/pnas.0437724100
Node, K., Huo, Y., Ruan, X., Yang, B., Spiecker, M., Ley, K., et al. (1999). Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285 (5431), 1276–1279. doi: 10.1126/science.285.5431.1276
Node, K., Ruan, X. L., Dai, J., Yang, S. X., Graham, L., Zeldin, D. C., et al. (2001). Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J. Biol. Chem. 276 (19), 15983–15989. doi: 10.1074/jbc.M100439200
Norwood, S., Liao, J., Hammock, B. D., Yang, G. Y. (2010). Epoxyeicosatrienoic acids and soluble epoxide hydrolase: potential therapeutic targets for inflammation and its induced carcinogenesis. Am. J. Transl. Res. 2 (4), 447–457.
Pace-Asciak, C. R., Granstrom, E., Samuelsson, B. (1983). Arachidonic acid epoxides. J. Biol. Chem. 258 (11), 6835–6840.
Panigrahy, D., Edin, M. L., Lee, C. R., Huang, S., Bielenberg, D. R., Butterfield, C. E., et al. (2012). Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Invest. 122 (1), 178–191. doi: 10.1172/JCI58128
Panigrahy, D., Kalish, B. T., Huang, S., Bielenberg, D. R., Le, H. D., Yang, J., et al. (2013). Epoxyeicosanoids promote organ and tissue regeneration. Proc. Natl. Acad. Sci. U. S. A. 110 (33), 13528–13533. doi: 10.1073/pnas.1311565110
Rainsford, K. D. (1993). Mechanisms of gastrointestinal damage by NSAIDS. Agents Actions Suppl. 44, 59–64.
Rand, A. A., Barnych, B., Morisseau, C., Cajka, T., Lee, K. S. S., Panigrahy, D., et al. (2017). Cyclooxygenase-derived proangiogenic metabolites of epoxyeicosatrienoic acids. Proc. Natl. Acad. Sci. U. S. A. 114 (17), 4370–4375. doi: 10.1073/pnas.1616893114
Ricciotti, E., FitzGerald, G. A. (2011). Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31 (5), 986–1000. doi: 10.1161/ATVBAHA.110.207449
Sandberg, M., Meijer, J. (1996). Structural characterization of the human soluble epoxide hydrolase gene (EPHX2). Biochem. Biophys. Res. Commun. 221 (2), 333–339. doi: 10.1006/bbrc.1996.0596
Schmelzer, K. R., Kubala, L., Newman, J. W., Kim, I. H., Eiserich, J. P., Hammock, B. D. (2005). Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl. Acad. Sci. U. S. A. 102 (28), 9772–9777. doi: 10.1073/pnas.0503279102
Schmelzer, K. R., Inceoglu, B., Kubala, L., Kim, I. H., Jinks, S. L., Eiserich, J. P., et al. (2006). Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 103 (37), 13646–13651. doi: 10.1073/pnas.0605908103
Seidegard, J., Ekstrom, G. (1997). The role of human glutathione transferases and epoxide hydrolases in the metabolism of xenobiotics. Environ. Health Perspect. 105 Suppl 4, 791–799. doi: 10.1289/ehp.97105s4791
Seitz, S., Boelsterli, U. A. (1998). Diclofenac acyl glucuronide, a major biliary metabolite, is directly involved in small intestinal injury in rats. Gastroenterology 115 (6), 1476–1482. doi: 10.1016/S0016-5085(98)70026-5
Seubert, J., Yang, B., Bradbury, J. A., Graves, J., Degraff, L. M., Gabel, S., et al. (2004). Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ. Res. 95 (5), 506–514. doi: 10.1161/01.RES.0000139436.89654.c8
Shimada, T. (2006). Xenobiotic-metabolizing enzymes involved in activation and detoxification of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab. Pharmacokinet. 21 (4), 257–276. doi: 10.2133/dmpk.21.257
Skrypnyk, N., Chen, X., Hu, W., Su, Y., Mont, S., Yang, S., et al. (2014). PPARalpha activation can help prevent and treat non-small cell lung cancer. Cancer Res. 74 (2), 621–631. doi: 10.1158/0008-5472.CAN-13-1928
Somasundaram, S., Rafi, S., Hayllar, J., Sigthorsson, G., Jacob, M., Price, A. B., et al. (1997). Mitochondrial damage: a possible mechanism of the “topical”. Gut 41 (3), 344–353. doi: 10.1136/gut.41.3.344
Sostres, C., Lanas, A. (2011). Gastrointestinal effects of aspirin. Nat. Rev. Gastroenterol. Hepatol. 8 (7), 385–394. doi: 10.1038/nrgastro.2011.97
Sostres, C., Gargallo, C. J., Arroyo, M. T., Lanas, A. (2010). Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 24 (2), 121–132. doi: 10.1016/j.bpg.2009.11.005
Spearman, M. E., Prough, R. A., Estabrook, R. W., Falck, J. R., Manna, S., Leibman, K. C., et al. (1985). Novel glutathione conjugates formed from epoxyeicosatrienoic acids (EETs). Arch. Biochem. Biophys. 242 (1), 225–230. doi: 10.1016/0003-9861(85)90496-5
Spector, A. A., Kim, H. Y. (2015). Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim. Biophys. Acta 1851 (4), 356–365. doi: 10.1016/j.bbalip.2014.07.020
Spector, A. A., Fang, X., Snyder, G. D., Weintraub, N. L. (2004). Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog. Lipid Res. 43 (1), 55–90. doi: 10.1016/S0163-7827(03)00049-3
Stienstra, R., Duval, C., Muller, M., Kersten, S. (2007). PPARs, Obesity, and Inflammation. PPAR Res. 2007, 95974. doi: 10.1155/2007/95974
Sudano, I., Flammer, A. J., Roas, S., Enseleit, F., Noll, G., Ruschitzka, F. (2012). Nonsteroidal antiinflammatory drugs, acetaminophen, and hypertension. Curr. Hypertens. Rep. 14 (4), 304–309. doi: 10.1007/s11906-012-0274-7
Treinen-Moslen, M., Kanz, M. F. (2006). Intestinal tract injury by drugs: importance of metabolite delivery by yellow bile road. Pharmacol. Ther. 112 (3), 649–667. doi: 10.1016/j.pharmthera.2006.05.007
Tsoi, K. K., Chan, F. C., Hirai, H. W., Sung, J. J. (2018). Risk of gastrointestinal bleeding and benefit from colorectal cancer reduction from long-term use of low-dose aspirin: a retrospective study of 612 509 patients. J. Gastroenterol. Hepatol. 33 (10), 1728–1736. doi: 10.1111/jgh.14261
Tsutsumi, S., Gotoh, T., Tomisato, W., Mima, S., Hoshino, T., Hwang, H. J., et al. (2004). Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 11 (9), 1009–1016. doi: 10.1038/sj.cdd.4401436
VanRollins, M., Kaduce, T. L., Knapp, H. R., Spector, A. A. (1993). 14,15-Epoxyeicosatrienoic acid metabolism in endothelial cells. J. Lipid. Res. 34 (11), 1931–1942.
Wang, F., Zhang, H., Ma, A. H., Yu, W., Zimmermann, M., Yang, J., et al. (2018a). COX-2/sEH dual inhibitor PTUPB potentiates the antitumor efficacy of cisplatin. Mol. Cancer Ther. 17 (2), 474–483. doi: 10.1158/1535-7163.MCT-16-0818
Wang, W., Yang, J., Zhang, J., Wang, Y., Hwang, S. H., Qi, W., et al. (2018b). Lipidomic profiling reveals soluble epoxide hydrolase as a therapeutic target of obesity-induced colonic inflammation. Proc. Natl. Acad. Sci. U. S. A. 115 (20), 5283–5288. doi: 10.1073/pnas.1721711115
Wang, Y., Wei, X., Xiao, X., Hui, R., Card, J. W., Carey, M. A., et al. (2005). Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J. Pharmacol. Exp. Ther. 314 (2), 522–532. doi: 10.1124/jpet.105.083477
Ward, J. E., Tan, X. (2007). Peroxisome proliferator activated receptor ligands as regulators of airway inflammation and remodelling in chronic lung disease. PPAR Res. 2007, 14983. doi: 10.1155/2007/14983
Weintraub, N. L., Fang, X., Kaduce, T. L., VanRollins, M., Chatterjee, P., Spector, A. A. (1999). Epoxide hydrolases regulate epoxyeicosatrienoic acid incorporation into coronary endothelial phospholipids. Am. J. Physiol. 277 (5), H2098–2108. doi: 10.1152/ajpheart.1999.277.5.H2098
Wolfe, M. M. L., Singh, D. R. (1999). Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N. Engl. J. Med. 340 (24), 1888–1899. doi: 10.1056/NEJM199906173402407
Wongrakpanich, S. W., Melhado, A., Rangaswami, K. (2018). A comprehensive review of non-steroidal anti-inflammatory drug use in the elderly. Aging Dis. 9 (1), 143–150. doi: 10.14336/AD.2017.0306
Yang, V. W. (1996). Eicosanoids and inflammatory bowel disease. Gastroenterol. Clin. North. Am. 25 (2), 317–332. doi: 10.1016/S0889-8553(05)70249-1
Yang, G. Y. (2018). Proinflammatory enzyme soluble epoxide hydrolase bridges obesity to colonic inflammation and potential carcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 115 (23), 5827–5828. doi: 10.1073/pnas.1807520115
Yang, J., Schmelzer, K., Georgi, K., Hammock, B. D. (2009). Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Anal. Chem. 81 (19), 8085–8093. doi: 10.1021/ac901282n
Yang, G.-Y., Zhang, M., Chung, Y. T., Lou, C., Tsai, C., Hammock, B. D., Liao, J. (2008). Inhibition of spontaneous colitis development in interleukin-10 knockout mice by soluble epoxide hydrolase inhibitor. FASEB J. 22, 479.29-479.29. doi: 10.1096/fasebj.22.1_supplement.479.29
Yang, H., Wang, W., Romano, K. A., Gu, M., Sanidad, K. Z., Kim, D., et al. (2018). A common antimicrobial additive increases colonic inflammation and colitis-associated colon tumorigenesis in mice. Sci. Transl. Med. 10 (443), eaan4116. doi: 10.1126/scitranslmed.aan4116
Yu, Z., Huse, L. M., Adler, P., Graham, L., Ma, J., Zeldin, D. C., et al. (2000). Increased CYP2J expression and epoxyeicosatrienoic acid formation in spontaneously hypertensive rat kidney. Mol. Pharmacol. 57 (5), 1011–1020.
Zhang, G., Kodani, S., Hammock, B. D. (2014a). Stabilized epoxygenated fatty acids regulate inflammation, pain, angiogenesis and cancer. Prog. Lipid Res. 53, 108–123. doi: 10.1016/j.plipres.2013.11.003
Zhang, J. Y., Prakash, C., Yamashita, K., Blair, I. A. (1992). Regiospecific and enantioselective metabolism of 8,9-epoxyeicosatrienoic acid by cyclooxygenase. Biochem. Biophys. Res. Commun. 183 (1), 138–143. doi: 10.1016/0006-291X(92)91619-2
Zhang, W., Li, H., Dong, H., Liao, J., Hammock, B. D., Yang, G. Y. (2013b). Soluble epoxide hydrolase deficiency inhibits dextran sulfate sodium-induced colitis and carcinogenesis in mice. Anticancer Res. 33 (12), 5261–5271.
Zhang, W., Liao, J., Li, H., Dong, H., Bai, H., Yang, A., et al. (2013c). Reduction of inflammatory bowel disease-induced tumor development in IL-10 knockout mice with soluble epoxide hydrolase gene deficiency. Mol. Carcinog. 52 (9), 726–738. doi: 10.1002/mc.21918
Zhang, G., Panigrahy, D., Hwang, S. H., Yang, J., Mahakian, L. M., Wettersten, H. I., et al. (2014b). Dual inhibition of cyclooxygenase-2 and soluble epoxide hydrolase synergistically suppresses primary tumor growth and metastasis. Proc. Natl. Acad. Sci. U. S. A. 111 (30), 11127–11132. doi: 10.1073/pnas.1410432111
Zhang, G., Panigrahy, D., Mahakian, L. M., Yang, J., Liu, J. Y., Stephen Lee, K. S., et al. (2013a). Epoxy metabolites of docosahexaenoic acid (DHA) inhibit angiogenesis, tumor growth, and metastasis. Proc. Natl. Acad. Sci. U. S. A. 110 (16), 6530–6535. doi: 10.1073/pnas.1304321110
Keywords: oxylipin, soluble epoxide hydrolase, non-steroidal anti-inflammatory drug, inflammation, carcinogenesis
Citation: Jones RD, Liao J, Tong X, Xu D, Sun L, Li H and Yang G-Y (2019) Epoxy-Oxylipins and Soluble Epoxide Hydrolase Metabolic Pathway as Targets for NSAID-Induced Gastroenteropathy and Inflammation-Associated Carcinogenesis. Front. Pharmacol. 10:731. doi: 10.3389/fphar.2019.00731
Received: 18 December 2018; Accepted: 05 June 2019;
Published: 25 June 2019.
Edited by:
John D. Imig, Medical College of Wisconsin, United StatesReviewed by:
Jessica Roos, University Hospital Frankfurt, GermanyGuodong Zhang, University of Massachusetts Amherst, United States
Copyright © 2019 Jones, Liao, Tong, Xu, Sun, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang-Yu Yang, g-yang@northwestern.edu