 Yiran Wang
Yiran Wang Changxiong Wang2†
Changxiong Wang2† Shuanghu Wang
Shuanghu Wang Quan Zhou
Quan Zhou- 1Department of Gastroenterology, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China
- 2Department of Gastroenterology, The Sixth Affiliated Hospital of Wenzhou Medical University, The People's Hospital of Lishui, Lishui, China
- 3The Laboratory of Clinical Pharmacy, The Sixth Affiliated Hospital of Wenzhou Medical University, The People's Hospital of Lishui, Lishui, China
- 4The Key Laboratory of Geriatrics, Beijing Institute of Geriatrics, Beijing Hospital, National Center of Gerontology, National Health Commission, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China
Background: Vonoprazan fumarate is a potassium-competitive acid blocker that was developed as a novel acid-suppressing drug for multiple indications. As a potential alternative to proton-pump inhibitors, the determination of the drug-drug interactions is vital for further applications. Probe drug cocktails are a type of rapid, economical, and efficient approach for evaluating cytochrome P450 enzyme activities. Since vonoprazan is metabolized partly by cytochrome P450, cocktails were used to study CYP-based drug-drug interactions.
Methods: This study was conducted both in vitro and in vivo. In the in vitro study of rat liver microsomes, ultra-performance liquid chromatography coupled to tandem mass spectrometry was utilized to assess the reversible inhibition of cytochrome P450 by vonoprazan by determining the concentration of probe drugs (phenacetin, bupropion, tolbutamide, dextromethorphan, midazolam, chlorzoxazone). The differences in the levels of probe drugs between the rat groups with or without vonoprazan administration were also tested in the rats.
Results: In vitro analysis revealed that the IC50 values of midazolam, tolbutamide, dextromethorphan, and bupropion in rat microsomes were 22.48, 18.34, 3.62, and 3.68 μM, respectively, while chlorzoxazone and phenacetin displayed no inhibition. In vivo analysis revealed that midazolam, bupropion, dextromethorphan, and tolbutamide showed significant (P < 0.05) differences in distinct pharmacokinetic parameters after vonoprazan administration, while those of chlorzoxazone and phenacetin were not significantly different.
Conclusion: The in vitro and in vivo results indicated that vonoprazan can inhibit CYP3A4, CYP2C9, CYP2D6, and CYP2B6, suggesting that the coadministration of vonoprazan with cytochrome P450 substrates should be performed cautiously in clinical settings.
Introduction
Vonoprazan fumarate (TAK-438) is a potassium-competitive acid blocker (P-CAB) that was developed as a novel acid-suppressing drug and launched in 2015 (Garnock-Jones, 2015). As a potential alternative to proton-pump inhibitors (PPIs), vonoprazan has been verified to be superior to conventional PPIs or even more effective in distinct gastric acid-related clinical indications. A phase III multicenter study demonstrated that there was no significant difference between vonoprazan and lansoprazole in treating erosive esophagitis (Xiao et al., 2020). In terms of preventing the recurrence of low-dose or long-term aspirin-associated ulcers, vonoprazan has proven to be as effective as lansoprazole (Kawai et al., 2018). The superior effect to rabeprazole was observed in healing artificial ulcer after endoscopic submucosal dissection (Yamasaki et al., 2018). Since PPIs play a vital role in helping to eradicate Helicobacter pylori infection, vonoprazan has been applied in similar trails and is considered to be a substitute for PPIs in resistant groups (O'Connor et al., 2019).
Additionally, regarding the pharmacokinetic profile, vonoprazan exhibits some advantages over PPIs e.g., it takes effect more quickly, suppresses acid secretory more potently, and exhibits better tolerability (Jenkins et al., 2015). Specifically, 20 mg of vonoprazan once daily equals 60 mg of omeprazole b.i.d., which is also equivalent to esomeprazole 40 mg b.i.d. (Graham and Tansel, 2018). It has been reported that vonoprazan is metabolized in two ways: the oxidative part by cytochrome P450 (CYP) enzyme isoforms and the nonoxidative part by sulfotransferase (SULT2A1) (Yamasaki et al., 2017).
Cytochrome P450 (CYP), which represents a diverse group of enzymes found in liver microsomes, is significantly indispensable in biological metabolism (Wilkinson, 2005) by metabolizing a large group of clinically used drugs (van Dyk et al., 2018). Because these enzymes can facilitate the elimination of various drugs, or modify their pharmacologic activities, the inhibition of these enzymes caused by drug coadministration or drug abuse can account for the increasing risk of adverse reactions (Vazquez, 2018). Human CYP includes 18 families and 44 subfamily members, which are categorized by amino acid similarities. Although the functions of genes in human cytochrome clusters contained differ from those of mice (Barzi et al., 2017), human CYP have their functional counterparts in mice, providing us with efficient approaches for further drug-drug interaction (DDI) studies.
Thus, probe drug cocktails were developed to evaluate CYP activities and the potential of DDIs (Frye et al., 1997). After years of modification, the use of these cocktails is now a rapid, economical, and efficient approach for evaluating different CYP enzymes simultaneously but independently (Rowland et al., 2016). To our best of our knowledge, no comprehensive DDI study of vonoprazan both in vitro and in vivo using the cocktails approach. Additionally, the existing results of vonoprazan DDIs were constrained to limited types of CYPs or were contradictory (Kagami and Furuta, 2018). Vonoprazan has not yet been launched in many countries, however, considering its effectiveness and safety, it has substantial potential to be widely utilized. Therefore, determining its DDIs will be beneficial for future clinical applications. Hence, in the present study, we explored the latent drug-drug interactions of vonoprazan. We chose phenacetin (CYP1A2), bupropion (CYP2B6), tolbutamide (CYP2C9), dextromethorphan (CYP2D6), midazolam (CYP3A), and chlorzoxazone (CYP2E1) as the core cocktail probe drugs, and ultrahigh-performance liquid chromatography coupled with triple quadrupole electrospray tandem mass spectrometry (UPLC-MS/MS) was performed to determine the results sensitively and reliably.
Materials and Methods
Chemicals and Reagents
Phenacetin, bupropion, tolbutamide, dextromethorphan, midazolam, chlorzoxazone, and the diazepam (all purity> 98%) that used as internal standards (ISs) were purchased from J&K Scientific Ltd. (Beijing, China). Vonoprazan was purchased from Beijing Sunflower Scientific Ltd. (Beijing, China). Hydroxybupropion and hydroxymidazolam were purchased from Sigma-Aldrich (St Louis, USA). Dextrorphan, hydroxytolbutamide, hydroxychlorzoxazone, and 4-acetamidophenol were purchased from Toronto Research Chemicals (Toronto, Canada). Reduced nicotinamide adenine dinucleotide phosphate (NADPH) was acquired from Roche Pharmaceuticals Ltd. (Basel, Switzerland). Ultra-pure water was produced by Milli-Q, a reagent-standard water purification system (Millipore, Bedford, USA). Acetonitrile and methanol that of high performance liquid chromatography (HPLC) grade were obtained from Merck Company (Darmstadt, Germany).
Instrumentation and Analytical Conditions
The UPLC-MS/MS conditions were established as described previously (Ma et al., 2015) and were modified by adding chlorzoxazone and replacing metoprolol with dextromethorphan in the probe drug system. Chromatographic separation was performed using the Acquity UPLC system (Waters Corp., Milford, MA, USA) and Acquity BEH C18 column (2.1 mm × 50 mm, 1.7 μM) at 40°C. The mobile phase comprising acetonitrile (A) and 0.1% formic acid water (B) was set with a 0.40-ml/min flow rate. The gradient program was applied as follows: 0–0.6 min, 10–50% A; 0.6–1 min, 50–80%; 1–2 min, 80–95% A; 2–2.5 min, 95% A; 2.5–2.6 min, 95–10% A; 2.6–3 min, 10% A. XEVO TQD triple quadrupole mass spectrometer was equipped with electrospray ionization (ESI), and multiple-reaction monitoring (MRM) mode was selected for quantitation. The transitions are shown in Table 1. MassLynx 4.1 software (Waters Corp., Milford, MA, USA) was used for data acquisition, and the UPLC-MS/MS chromatogram of blank plasma that was spiked with probe drugs and metabolites is shown below (Figure 1).
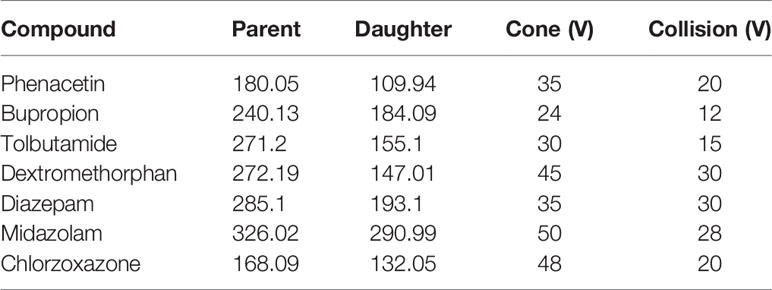
Table 1 The transitions of probe drugs.

Figure 1 UPLC-MS/MS chromatograms, blank plasma spiked with chlorzoxazone, midazolam, diazepam (IS), dextromethorphan, tolbutamide, bupropion, and phenacetin.
Preparation of Rat Liver Microsomes
The pooled rat liver microsomes (RLMs) obtained from eight rats, which were weighed and homogenized with cold 0.01 mM phosphate-buffered saline (PBS), containing 0.25 mM sucrose. After centrifugation for 15 min at 11,000 rpm, the supernatants were separated and transferred into new tubes for another 15-min centrifugation at 11,000 rpm. Next, ultracentrifugation was performed at 10,000 rpm at 4°C for 1 h., and the microsomal pellets were resuspended with cold 0.01 mM PBS and stored at −80°C (Wang et al., 2015). The protein concentration of RLMs was determined using the Bradford Protein Assay Kit (Thermo Scientific, Waltham, MA, USA).
In Vitro Interaction Studies in Rat Liver Microsomes
Next, vonoprazan was applied as the inhibitor to determine the half maximal inhibitory concentration (IC50). The incubation mixture in a total volume of 200 μl contained 0.5 mg/ml of RLMs, 1 mM potassium phosphate buffer (pH 7.4), the probe drug mixture that dissolved by dimethyl sulfoxide (40, 20, 10, 100, 20, and 5 μM for phenacetin, bupropion, tolbutamide, dextromethorphan, chlorzoxazone, and midazolam, respectively), vonoprazan (1, 2.5, 5, 10, 25, 50, and 100 μM) and 1 mM NADPH (Stoetzer et al., 2015). After preincubation in a shaking water bath at 37°C for 5 min, an NADPH-regenerating system was added to initiate the reaction in a final volume of 200 μl. The reaction was performed for 30 min, and was stopped by cooling to −80°C immediately. Next, 20 μl of internal standard (IS) working solution and 200 μl of acetonitrile were added. Centrifugation was performed at 13,000 rpm for 5 min after vortexing for 1 min. The supernatant mixture (2 μl) was injected into the UPLC–MS/MS system for analysis.
Pharmacokinetic Study of Vonoprazan
Ten specific pathogen-free (SPF) grade Sprague–Dawley rats (male, 220 ± 20 g) were provided by Wenzhou Medical University Laboratory Animal Research Center. This study was carried out in accordance with the principles of the Basel Declaration and recommendations of Wenzhou Medical University Administration Committee of Experimental Animals. The protocol was approved by the Wenzhou Medical University Administration Committee of Experimental Animals (ID Number: wydw2019-650). Ten rats were randomly divided into two groups: the vonoprazan group (n = 5) and control group (n = 5). Both groups were fasted overnight before experiments but were allowed water all the time. The six probe drug mixtures and vonoprazan were dissolved in 0.5% carboxy methyl cellulose sodium (CMC-Na), respectively. The vonoprazan group was administered 5 mg/kg of vonoprazan for 14 days. The control group was administered 0.5% CMC-Na. After 30 min of vonoprazan or 0.5% CMC-Na, on the 14th day, the two groups were treated by gavage at a single dosage of 10 mg/kg for bupropion, dextromethorphan, phenacetin, midazolam, and chlorzoxazone, and of 1 mg/kg for tolbutamide.
Sample Collection and Preparation
Blood samples (300 µl) were collected via the tail vein at the time points of 0.083, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h. after gavage. Next, the samples were collected into 1.5-ml centrifuge tubes and were immediately centrifuged for 10 min at 4,000 rpm speed. For every 100 µl of rat plasma was added with 200 µl of acetonitrile in 0.5 μg/ml of IS was added, followed by vortex mixing for one minute. After centrifugation at 13,000 rpm for 15 min, 5 µl of supernatant was prepared for UPLC-MS/MS system to analysis. In vitro DDI detection system, the pH was 7.4 and the component of buffer was consisted of 100 mM potassium phosphate.
Statistical Analysis
The GraphPad (version 7.0; GraphPad Software Inc., San Diego, CA, USA) was applied to calculate IC50 values and plot plasma concentration-time curves. The pharmacokinetic parameters using noncompartmental analysis was calculated by DAS (version 3.2.8; Wenzhou Medical University, China). Statistical comparisons within groups were conducted by SPSS (version 25.0; SPSS Inc., Chicago, IL, USA), using Student's t-test. A P-value <0.05 was considered statistically significant.
Results
Effects of Vonoprazan on the Metabolism of Probe Drugs In Vitro
Following vonoprazan addition, the metabolism of the four probe drugs midazolam, tolbutamide, dextromethorphan, and bupropion were inhibited to various degrees (Figure 2). Their IC50 values in rat microsomes were 22.48 µM (Figure 2A), 18.34 µM (Figure 2B), 3.62 Mm (Figure 2C), and 3.68 µM (Figure 2D), respectively. By contrast, chlorzoxazone (Figure 2E) and phenacetin (Figure 2F) displayed no inhibition. The results illustrated that vonoprazan can inhibit the metabolism of midazolam, tolbutamide, dextromethorphan, and bupropion in rat microsomes.
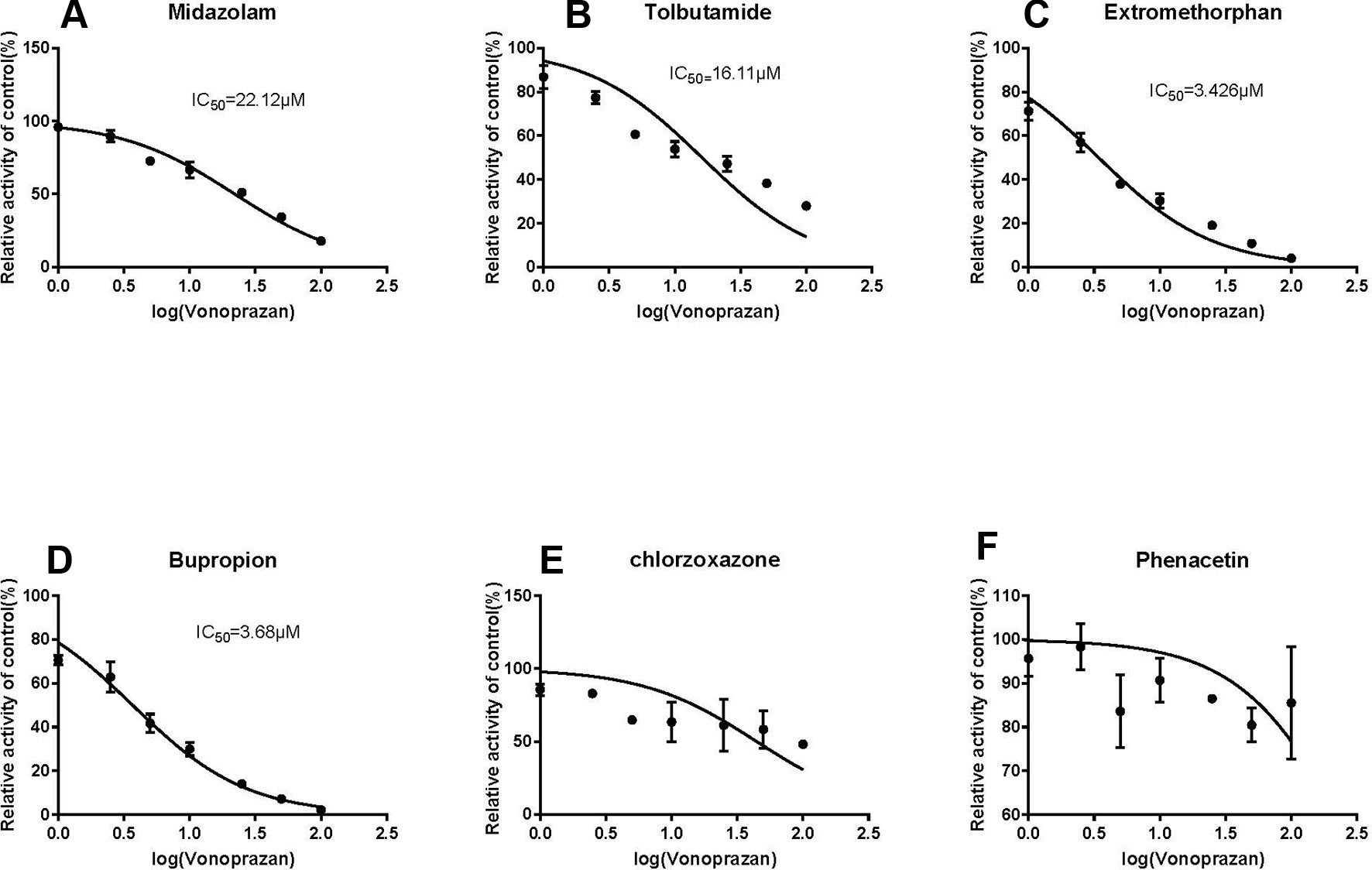
Figure 2 The inhibitory effect of vonoprazan on midazolam (A), tolbutamide (B), dextromethorphan (C), bupropion (D), chloraoxazone(E), and phenacetin (F) for IC50 values in RLMs (values are Mean ± SD, n = 3).
Effects of Vonoprazan on the Metabolism of Probe Drugs In Vivo
The statistical analysis results of the mean pharmacokinetic parameters are demonstrated in Tables 2–4, which were analyzed by DSA 3.2.8. The mean plasma concentration versus time curves of each probe drug in the vonoprazan and control groups are presented in Figure 3.
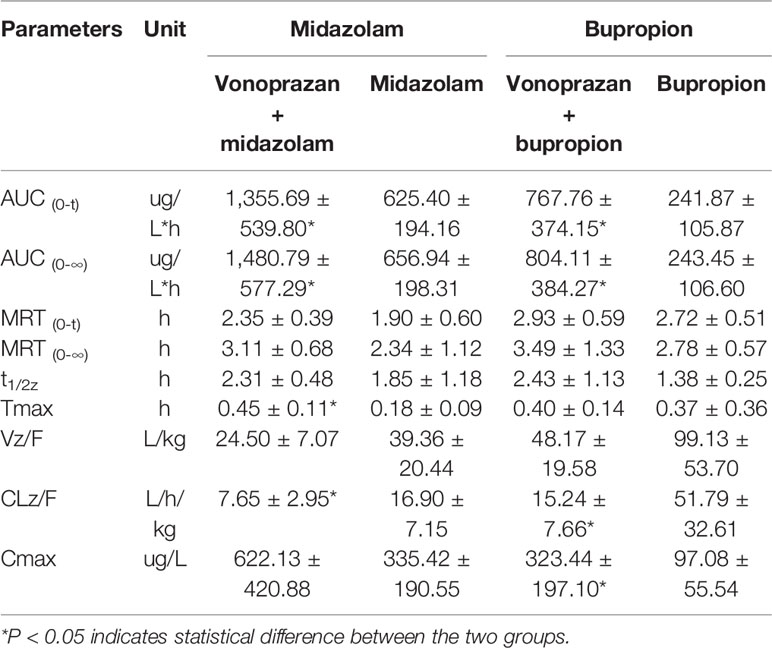
Table 2 Pharmacokinetic parameters of probe drugs (midazolam and bupropion) from control group and vonoprazan group rats (mean ± SD, n = 5).
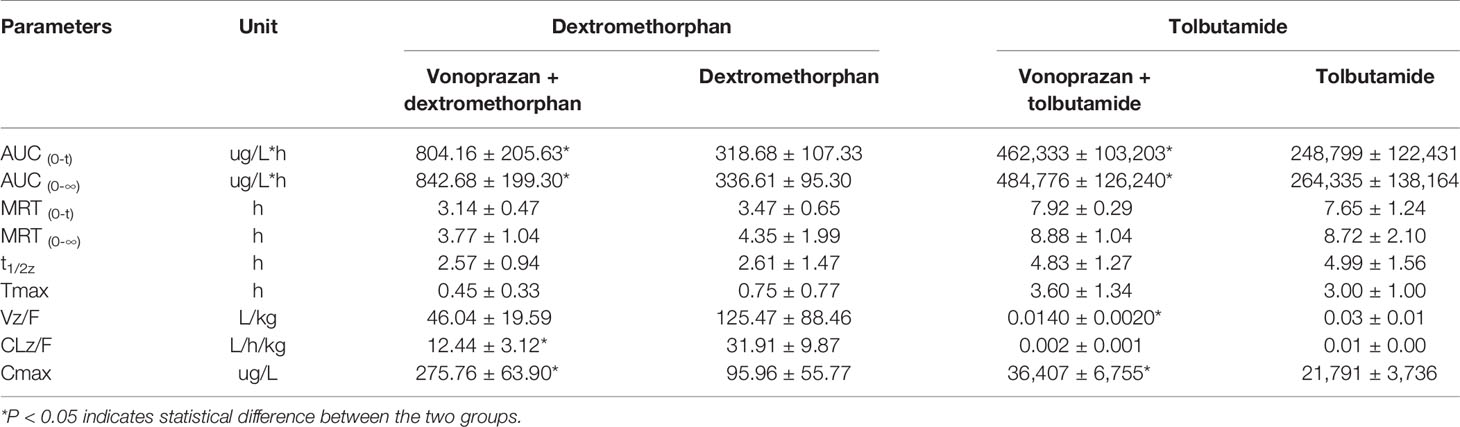
Table 3 Pharmacokinetic parameters of probe drugs (dextromethorphan and tolbutamide) from control group and vonoprazan group rats (mean ± SD, n = 5).
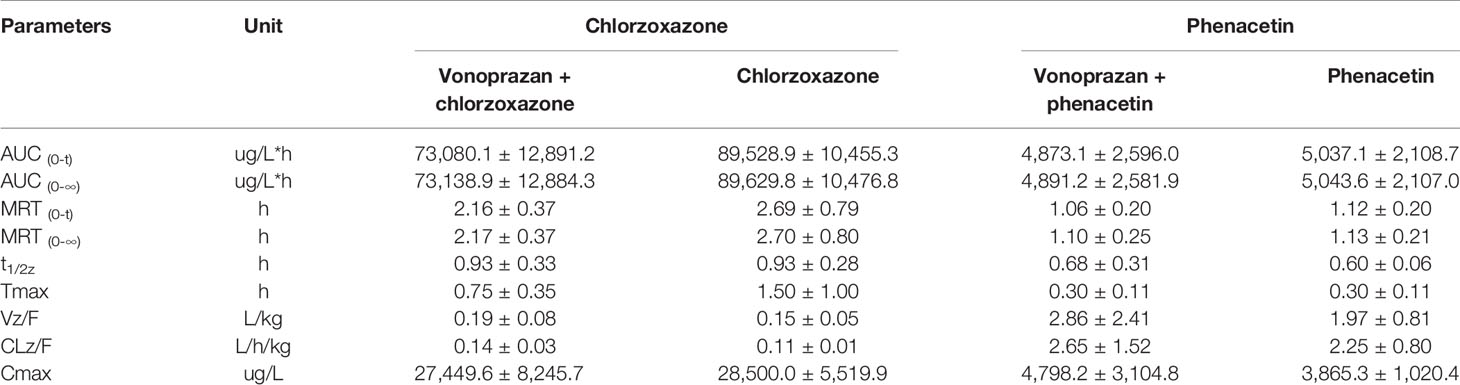
Table 4 Pharmacokinetic parameters of probe drugs (chlorzoxazone and phenacetin) from control group and vonoprazan group rats (mean ± SD, n = 5).
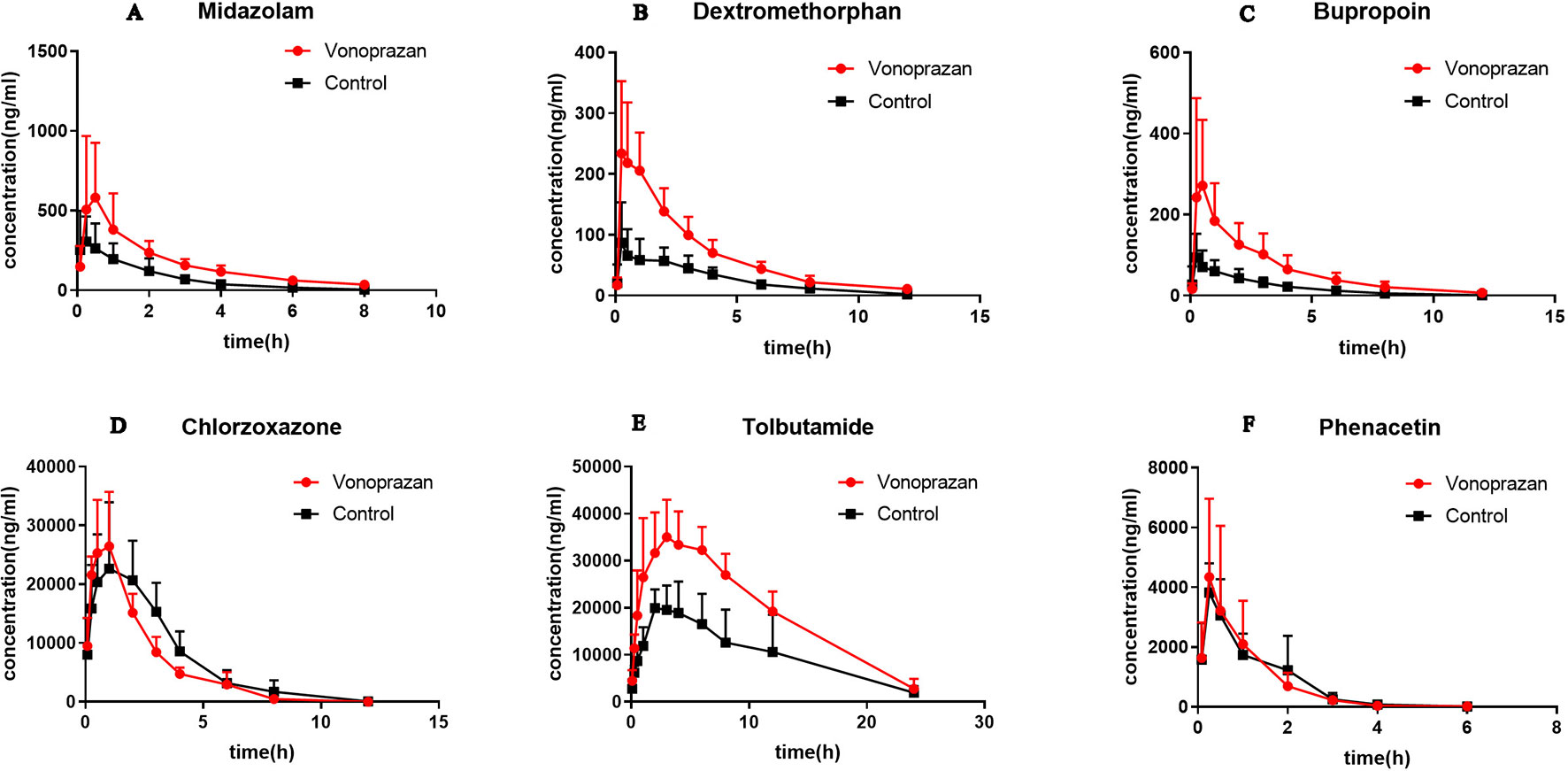
Figure 3 The pharmacokinetic profiles of midazolam (A), dextromethorphan (B), bupropion (C), chlorzoxazone (D), tolbutamide (E), and phenacetin (F) in control group and vonoprazan group rats (n = 5).
Midazolam, bupropion, dextromethorphan, and tolbutamide showed significant differences (P < 0.05) in distinct pharmacokinetic parameters compared with the control group, while the parameters of chlorzoxazone and phenacetin were not significantly different. The AUC0→t values of midazolam, bupropion, dextromethorphan, and tolbutamide were all increased greatly, by 1.17-fold, 2.17-fold, 1.52-fold, and 0.86-fold, respectively, compared with that of the control group. Additionally, the AUC0→∞ values were elevated by 1.25-fold, 2.30-fold, 1.50-fold, and 0.83-fold, respectively. Specifically, the addition of vonoprazan significantly increased the Tmax of midazolam and the Cmax values of bupropion, dextromethorphan, and tolbutamide by 1.50-fold, 2.33-fold, 1.87-fold, and 0.67-fold, respectively. Additionally, vonoprazan significantly decreased the CLz/F of midazolam, bupropion, dextromethorphan, and the Vz/F of bupropion by 54.7, 70.6, 61.0, and 53.3%, respectively.
These results indicate that vonoprazan has an inhibitory effect on the metabolism of midazolam, bupropion, dextromethorphan, and tolbutamide in rats, agreeing with the results obtained in vitro.
Discussion
Vonoprazan, a P-CAB drug, was approved for application in the treatment of acid-related diseases in Japan on December 26, 2014 (Takeda, 2014). Previously, proton pump inhibitors (PPIs) were considered the first-in-class available drug to treat acid-related diseases, such as gastroesophageal reflux disease and gastroduodenal ulcers (Spechler et al., 2019), and has been widely utilized in the eradication therapy of H. pylori (Crowe, 2019). The unmet clinical needs of PPIs promote new drug development. Because the high acidic environment in the stomach is established by the gastric H+ and K+-ATPase(Yamamoto et al., 2019), classical PPIs have been produced previously. P-CABs can both block the H+ and K+-ATPase enzyme and reversibly bind K+ ion (Abe et al., 2018), enabling vonoprazan to show long duration and good bioavailability that can be less affected by the pH in the stomach (Hori et al., 2011). The better are the physicochemical characteristics, the more frequent are well-designed controlled clinical trials conducted. Thus, vonoprazan has been gradually applied in triple or quadruple regimens as a substitute for PPIs to enhance the antimicrobial effect against H. pylori because drug resistance to classical strategies has increased annually (Fallone et al., 2019). Vonoprazan was confirmed to be superior in the secondary prevention of nonsteroidal anti-inflammatory drug-induced peptic ulcers (Mizokami et al., 2018). Vonoprazan is also thought to potentially show superiority over PPIs in patients who are difficult to treat, given that it can reliably achieve a therapeutically required intragastric pH (Tansel and Graham, 2017).
Concerning the pharmacodynamics of vonoprazan, before it entered the market, in vitro studies demonstrated that it is mainly metabolized by CYP3A4 and partially by CYP2C19, CYP2B6, CYP2D6, and SULT2A1 (Echizen, 2016). At that time, its inhibitory effect on any CYP isozymes were considered would not to emerge in clinical settings. However, though the distinct CYP2C19 genotypes showed no appreciable correlation with its metabolism in initial study (Kagami et al., 2016), subsequent trials exhibited dissimilar results (Funakoshi et al., 2019), indicating there may be other possible DDIs. Thus, intending to explore as much DDIs as possible, we chose those six probe drugs for wider coverage: phenacetin (CYP1A2), bupropion (CYP2B6), tolbutamide (CYP2C9), dextromethorphan (CYP2D6), midazolam (CYP3A), and chlorzoxazone (CYP2E1). Those similar enzymes also have been detected in other clinical trials (Gravel et al., 2019). We utilized the system we established before, which is more convincing and reliable. But as rats do not have CYP2C19, the results are limited and it should be paid attention to this, if the trials on human were to be conducted.
This study investigated the potential DDIs in vitro by using rat liver microsomes and in vivo using performing ultra-performance liquid chromatography-tandem mass spectrometry to detect the metabolites of probe drugs in rat plasma. The in vitro results indicated that vonoprazan has direct inhibitory effects on CYP3A4 (IC50 = 22.48 µM), CYP2C9 (IC50 = 18.34 µM), CYP2D6 (IC50 = 3.62 µM), and CYP2B6 (IC50 = 3.68 µM), but no effect on CYP1A2 and CYP2E1. These findings partially confirm the results produced using making use of human liver microsomes (HLMs) (Nishihara et al., 2019). Nevertheless, the IC50 values in previous studies via HLMs were much higher than the maximum plasma concentrations of vonoprazan after therapeutic oral doses; therefore, theoretically, few clinical restraint effects should be considered. However, actual situations are more complex than expected. A previous study reported a reference maximum plasma concentration level in rats of 17 ng/ml (Kogame et al., 2017), indicating that the threshold value of vonoprazan could be reached and that the metabolism of these four probe drugs could be extended in rats in vivo.
Therefore, to further consolidate our results, pharmacokinetic interaction studies in rats in vivo were implemented. The main pharmacokinetic parameters (AUC0→t and AUC0→∞) of midazolam, bupropion, dextromethorphan, and tolbutamide were elevated significantly following vonoprazan, indicating vonoprazan indeed constrained their metabolism. Specifically, the oral clearance (CLz/F) of midazolam was decreased while the Tmax was increased, indicating vonoprazan affected midazolam by postponing the exposure and reducing the clearance. Regarding bupropion and dextromethorphan, vonoprazan may lessen the clearance and resulted in a rise in the maximum plasma concentration (Cmax). Regarding tolbutamide, the apparent volume of distribution (Vz/F) declined, implying that the vonoprazan approximately can enhance its binding ability, thus increasing Cmax. Similar to the in vitro findings, chlorzoxazone and phenacetin was not affected by vonoprazan. Because the diverse probe drugs represent corresponding CYP enzymes, considering all of the data, the results in vitro and in vivo were coherent: the vonoprazan can inhibit CYP3A4, CYP2C9, CYP2D6, and CYP2B6 to a distinct extent.
Drug interactions of PPI have been studied a lot and results are quite specific (Xie et al., 2019). As a feasible substitute in future, the vonoprazan's DDIs deserve explorations. Thus far, the DDI investigation of vonoprazan mainly concerned CYP3A4 and CYP2C19, with unconvincing consequences. In the triple regimens to eradicate H. pylori infection, whether the coadministration of vonoprazan and clarithromycin (CYP3A4 inhibitor) would have a mutual effect remains debatable (Jenkins et al., 2017; Sugimoto and Yamaoka, 2018). Additionally, Kagami et al. (2018) observed that vonoprazan could attenuate the function of clopidogrel, not depending on the CYP2C19 or CYP3A4 genotypes. Other CYP types were also confirmed to not be associated with reducing the antiplatelet effectiveness of both clopidogrel (Nishihara and Czerniak, 2018) and prasugrel (Nishihara, 2019). It seems the relationship between anti-acid and antiplatelet agents is more intricate. The controversial findings could be due to the heterogeneity of different studies or influenced by other type enzymes noted in this study. The potential inhibitory effects of vonoprazan on CYP2C9, CYP2D6, and CYP2B6 have been revealed in our results but didn't be paid much attention in other studies. Considering various common drugs are metabolized by these pathways: warfarin by 3A4 and 2C9 (Vazquez, 2018), tricyclic antidepressants by 2D6 (Gaedigk et al., 2017), nicotine by 2B6 (Tomaz et al., 2019), it has significant value to verify the authentic DDIs in clinical settings.
Owing to limited condition, we have not applied it to human beings yet. But it is necessary for further study and we will consider to perform it in future, basing on the present results that we get from rats. Furthermore, other inhibitory mechanisms can be explored (Yasumuro et al., 2018). Thus, stricter clinical trials will be needed to further confirm these findings.
In summary, our study investigated CYP-based drug-drug interactions both in vitro and in vivo. The results demonstrated that the vonoprazan could inhibit CYP3A4, CYP2C9, CYP2D6, and CYP2B6, suggesting that the coadministration of vonoprazan and CYP substrates should be performed cautiously in clinical settings.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
The animal study was reviewed and approved by the Wenzhou Medical University Administration Committee of Experimental Animals.
Author Contributions
YW, CW, SW, and QZ contributed conception and design of the study. DD performed the statistical analysis. YW and CW wrote the first draft of the manuscript. SW, QZ, DD, JS, and XX wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version. QL are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This research was funded by 135 Major National Science and Technology Projects (No. 2017ZX09304026).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to express our thanks for the financial support of 135 Major National Science and Technology Projects and for the encouragement of the Beijing Hospital faculty.
References
Abe, K., Irie, K., Nakanishi, H., Suzuki, H., Fujiyoshi, Y. (2018). Crystal structures of the gastric proton pump. Nature 556 (7700), 214–218. doi: 10.1038/s41586-018-0003-8
Barzi, M., Pankowicz, F. P., Zorman, B., Liu, X., Legras, X., Yang, D., et al. (2017). A novel humanized mouse lacking murine P450 oxidoreductase for studying human drug metabolism. Nat. Commun. 8 (1), 39. doi: 10.1038/s41467-017-00049-x
Crowe, S. E. (2019). Helicobacter pylori infection. N. Engl. J. Med. 380 (12), 1158–1165. doi: 10.1056/NEJMcp1710945
Echizen, H. (2016). The first-in-class potassium-competitive acid blocker, vonoprazan fumarate: pharmacokinetic and pharmacodynamic considerations. Clin. Pharmacokinet 55 (4), 409–418. doi: 10.1007/s40262-015-0326-7
Fallone, C. A., Moss, S. F., Malfertheiner, P. (2019). Reconciliation of recent helicobacter pylori treatment guidelines in a time of increasing resistance to antibiotics. Gastroenterology 157 (1), 44–53. doi: 10.1053/j.gastro.2019.04.011
Frye, R. F., Matzke, G. R., Adedoyin, A., Porter, J. A., R.A., B. (1997). Validation of the five-drug “Pittsburgh cocktail” approach for assessment of selective regulation of drug-metabolizing enzymes. Clin. Pharmacol. Ther. 62 (4), 365–376. doi: 10.1016/S0009-9236(97)90114-4
Funakoshi, R., Tomoda, Y., Kudo, T., Furihata, K., Kusuhara, H., Ito, K. (2019). Effects of proton pump inhibitors, esomeprazole and vonoprazan, on the disposition of proguanil, a CYP2C19 substrate, in healthy volunteers. Br. J. Clin. Pharmacol. 85 (7), 1454–1463. doi: 10.1111/bcp.13914
Gaedigk, A., Sangkuhl, K., Whirl-Carrillo, M., Klein, T., Leeder, J. S. (2017). Prediction of CYP2D6 phenotype from genotype across world populations. Genet. Med. 19 (1), 69–76. doi: 10.1038/gim.2016.80
Garnock-Jones, K. P. (2015). Vonoprazan: first global approval. Drugs 75 (4), 439–443. doi: 10.1007/s40265-015-0368-z
Graham, D. Y., Tansel, A. (2018). Interchangeable use of proton pump inhibitors based on relative potency. Clin. Gastroenterol. Hepatol. 16 (6), 800–808.e807. doi: 10.1016/j.cgh.2017.09.033
Gravel, S., Chiasson, J. L., Turgeon, J., Grangeon, A., Michaud, V. (2019). Modulation of CYP450 activities in patients with type 2 diabetes. Clin. Pharmacol. Ther. 106 (6), 1280–1289. doi: 10.1002/cpt.1496
Hori, Y., Matsukawa, J., Takeuchi, T., Nishida, H., Kajino, M., Inatomi, N. (2011). A study comparing the antisecretory effect of TAK-438, a novel potassium-competitive acid blocker, with lansoprazole in animals. J. Pharmacol. Exp. Ther. 337 (3), 797–804. doi: 10.1124/jpet.111.179556
Jenkins, H., Sakurai, Y., Nishimura, A., Okamoto, H., Hibberd, M., Jenkins, R., et al. (2015). Randomised clinical trial: safety, tolerability, pharmacokinetics and pharmacodynamics of repeated doses of TAK-438 (vonoprazan), a novel potassium-competitive acid blocker, in healthy male subjects. Aliment Pharmacol. Ther. 41 (7), 636–648. doi: 10.1111/apt.13121
Jenkins, H., Jenkins, R., Patat, A. (2017). Effect of multiple oral doses of the potent CYP3A4 inhibitor clarithromycin on the pharmacokinetics of a single oral dose of vonoprazan: a phase I, open-label, sequential design study. Clin. Drug Invest. 37 (3), 311–316. doi: 10.1007/s40261-016-0488-6
Kagami, T., Furuta, T. (2018). Response to “CYP-mediated drug-drug interaction is not a major determinant of attenuation of antiplatelet function of clopidogrel by vonoprazan”. Clin. Pharmacol. Ther. 104 (1), 33–34. doi: 10.1002/cpt.1059
Kagami, T., Sahara, S., Ichikawa, H., Uotani, T., Yamade, M., Sugimoto, M., et al. (2016). Potent acid inhibition by vonoprazan in comparison with esomeprazole, with reference to CYP2C19 genotype. Aliment Pharmacol. Ther. 43 (10), 1048–1059. doi: 10.1111/apt.13588
Kagami, T., Yamade, M., Suzuki, T., Uotani, T., Hamaya, Y., Iwaizumi, M., et al. (2018). Comparative study of effects of vonoprazan and esomeprazole on antiplatelet function of clopidogrel or prasugrel in relation to CYP2C19 genotype. Clin. Pharmacol. Ther. 103 (5), 906–913. doi: 10.1002/cpt.863
Kawai, T., Oda, K., Funao, N., Nishimura, A., Matsumoto, Y., Mizokami, Y., et al. (2018). Vonoprazan prevents low-dose aspirin-associated ulcer recurrence: randomised phase 3 study. Gut. 67 (6), 1033–1041. doi: 10.1136/gutjnl-2017-314852
Kogame, A., Takeuchi, T., Nonaka, M., Yamasaki, H., Kawaguchi, N., Bernards, A., et al. (2017). Disposition and metabolism of TAK-438 (vonoprazan fumarate), a novel potassium-competitive acid blocker, in rats and dogs. Xenobiotica 47 (3), 255–266. doi: 10.1080/00498254.2016.1182667
Ma, J., Wang, S., Zhang, M., Zhang, Q., Zhou, Y., Lin, C., et al. (2015). Simultaneous determination of bupropion, metroprolol, midazolam, phenacetin, omeprazole and tolbutamide in rat plasma by UPLC-MS/MS and its application to cytochrome P450 activity study in rats. BioMed. Chromatogr. 29 (8), 1203–1212. doi: 10.1002/bmc.3409
Mizokami, Y., Oda, K., Funao, N., Nishimura, A., Soen, S., Kawai, T., et al. (2018). Vonoprazan prevents ulcer recurrence during long-term NSAID therapy: randomised, lansoprazole-controlled non-inferiority and single-blind extension study. Gut 67 (6), 1042–1051. doi: 10.1136/gutjnl-2017-314010
Nishihara, M., Czerniak, R. (2018). CYP-mediated drug-drug interaction is not a major determinant of attenuation of antiplatelet function of clopidogrel by vonoprazan. Clin. Pharmacol. Ther. 104 (1), 31–32. doi: 10.1002/cpt.1060
Nishihara, M., Yamasaki, H., Czerniak, R., Jenkins, H. (2019). In vitro assessment of potential for CYP-inhibition-based drug-drug interaction between vonoprazan and clopidogrel. Eur. J. Drug Metab. Pharmacokinet 44 (2), 217–227. doi: 10.1007/s13318-018-0521-7
Nishihara, M. (2019). Inhibitory effect of vonoprazan on the metabolism of [14C]Prasugrel in human liver microsomes. Eur. J. Drug Metab. Pharmacokinet. 44 (5), 713–717. doi: 10.1007/s13318-019-00554-y
O'Connor, A., Liou, J. M., Gisbert, J. P., O'Morain, C. (2019). Review: treatment of helicobacter pylori infection 2019. Helicobacter 24 Suppl 1, e12640. doi: 10.1111/hel.12640
Rowland, A., Mangoni, A. A., Hopkins, A., Sorich, M. J., Rowland, A. (2016). Optimized cocktail phenotyping study protocol using physiological based pharmacokinetic modeling and assessment of metabolic drug-drug interactions involving modafinil. Front. Pharmacol. 7 (undefined), 517. doi: 10.3389/fphar.2016.00517
Spechler, S. J., Hunter, J. G., Jones, K. M., Lee, R., Smith, B. R., Mashimo, H., et al. (2019). Randomized trial of medical versus surgical treatment for refractory heartburn. N. Engl. J. Med. 381 (16), 1513–1523. doi: 10.1056/NEJMoa1811424
Stoetzer, C., Kistner, K., Stüber, T., Wirths, M., Schulze, V., Doll, T., et al. (2015). Methadone is a local anaesthetic-like inhibitor of neuronal Na+ channels and blocks excitability of mouse peripheral nerves. Br. J. Anaesth 114 (1), 110–120. doi: 10.1093/bja/aeu206
Sugimoto, M., Yamaoka, Y. (2018). Role of vonoprazan in helicobacter pylori eradication therapy in Japan. Front. Pharmacol. 9, 1560. doi: 10.3389/fphar.2018.01560
Takeda (2014). New drug application approval of TAKECAB?for the treatment of acid-related diseases in Japan (media release) [Online]. http://www.takeda.com. [Accessed 26 Dec 2014].
Tansel, A., Graham, D. Y. (2017). New insight into an effective treatment of marginal ulceration after Roux-en-Y gastric bypass. Clin. Gastroenterol. Hepatol. 15 (4), 501–503. doi: 10.1016/j.cgh.2016.12.025
Tomaz, P. R. X., Kajita, M. S., Santos, J. R., Scholz, J., Abe, T. O., Gaya, P. V., et al. (2019). Cytochrome P450 2A6 and 2B6 polymorphisms and smoking cessation success in patients treated with varenicline. Eur. J. Clin. Pharmacol. 75 (11), 1541–1545. doi: 10.1007/s00228-019-02731-z
van Dyk, M., Marshall, J. C., Sorich, M. J., Wood, L. S., Rowland, A. (2018). Assessment of inter-racial variability in CYP3A4 activity and inducibility among healthy adult males of Caucasian and South Asian ancestries. Eur. J. Clin. Pharmacol. 74 (7), 913–920. doi: 10.1007/s00228-018-2450-4
Vazquez, S. R. (2018). Drug-drug interactions in an era of multiple anticoagulants: a focus on clinically relevant drug interactions. Blood 132 (21), 2230–2239. doi: 10.1182/blood-2018-06-848747
Wang, Z., Sun, W., Huang, C. K., Wang, L., Xia, M. M., Cui, X., et al. (2015). Inhibitory effects of curcumin on activity of cytochrome P450 2C9 enzyme in human and 2C11 in rat liver microsomes. Drug Dev. Ind. Pharm. 41 (4), 613–616. doi: 10.3109/03639045.2014.886697
Wilkinson, G. R. (2005). Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 352 (21), 2211–2221. doi: 10.1056/NEJMra032424
Xiao, Y., Zhang, S., Dai, N., Fei, G., Goh, K. L., Chun, H. J., et al. (2020). Phase III, randomised, double-blind, multicentre study to evaluate the efficacy and safety of vonoprazan compared with lansoprazole in Asian patients with erosive oesophagitis. Gut. 69 (2), 224–230. doi: 10.1136/gutjnl-2019-318365
Xie, Y., Bowe, B., Yan, Y., Xian, H., Li, T., Al-Aly, Z. (2019). Estimates of all cause mortality and cause specific mortality associated with proton pump inhibitors among US veterans: cohort study. BMJ Open Gastroenterol. 365, l1580. doi: 10.1136/bmj.l1580
Yamamoto, K., Dubey, V., Irie, K., Nakanishi, H., Khandelia, H., Fujiyoshi, Y., et al. (2019). A single K-binding site in the crystal structure of the gastric proton pump. Elife 8, undefined. doi: 10.7554/eLife.47701
Yamasaki, H., Kawaguchi, N., Nonaka, M., Takahashi, J., Morohashi, A., Hirabayashi, H., et al. (2017). In vitro metabolism of TAK-438, vonoprazan fumarate, a novel potassium-competitive acid blocker. Xenobiotica 47 (12), 1027–1034. doi: 10.1080/00498254.2016.1203505
Yamasaki, A., Yoshio, T., Muramatsu, Y., Horiuchi, Y., Ishiyama, A., Hirasawa, T., et al. (2018). Vonoprazan is superior to rabeprazole for healing endoscopic submucosal dissection: induced ulcers. Digestion 97 (2), 170–176. doi: 10.1159/000485028
Yasumuro, O., Uchida, S., Kashiwagura, Y., Suzuki, A., Tanaka, S., Inui, N., et al. (2018). Changes in gefitinib, erlotinib and osimertinib pharmacokinetics under various gastric pH levels following oral administration of omeprazole and vonoprazan in rats. Xenobiotica 48 (11), 1106–1112. doi: 10.1080/00498254.2017.1396379
Keywords: cytochrome P450, drug-drug interactions, cocktails, vonoprazan, drug metabolism
Citation: Wang Y, Wang C, Wang S, Zhou Q, Dai D, Shi J, Xu X and Luo Q (2020) Cytochrome P450-Based Drug-Drug Interactions of Vonoprazan In Vitro and In Vivo. Front. Pharmacol. 11:53. doi: 10.3389/fphar.2020.00053
Received: 06 November 2019; Accepted: 16 January 2020;
Published: 14 February 2020.
Edited by:
Jun Lu, Auckland University of Technology, New ZealandReviewed by:
Reza Nemati, Canterbury Health Laboratories, New ZealandZhang Baohong, Shanghai Jiao Tong University, China
Copyright © 2020 Wang, Wang, Wang, Zhou, Dai, Shi, Xu and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingfeng Luo, bHVvcWYyMDAwQDEyNi5jb20=
†These authors have contributed equally to this work