 Fang Dai1†Jizheng Guo2† Yang Wang2Tian Jiang1
Fang Dai1†Jizheng Guo2† Yang Wang2Tian Jiang1 Hongbo Chen3Ying Hu2
Hongbo Chen3Ying Hu2 Juan Du2Xianming Xia4Qiu Zhang1*
Juan Du2Xianming Xia4Qiu Zhang1* Bing Shen2*
Bing Shen2*- 1Department of Endocrinology, The First Affiliated Hospital of Anhui Medical University, Hefei, China
- 2School of Basic Medicine, Anhui Medical University, Hefei, China
- 3Department of Obstetrics and Gynecology, Maternal and Child Health Hospital Affiliated to Anhui Medical University, Hefei, China
- 4Digestive Medicine Center, Department of General Practice, The Seventh Affiliated Hospital, Sun Yat-sen University, Shenzhen, China
Aims: The underlying mechanism of diabetic enteropathy, a common complication of type 1 diabetes, remains unclear. Store-operated Ca2+ entry (SOCE) is a ubiquitous type of Ca2+ influx involved in various cellular functions. Here, we show that SOCE-related stromal interaction molecule 1 (STIM1) and Orai1 participate in inappropriate cellular Ca2+ homeostasis, augmenting agonist-induced small intestinal smooth muscle contraction and small bowel transit speed in a mouse model of type 1 diabetes.
Methods and Results: We used small interfering (si)RNA to suppress STIM1 and Orai1 proteins, and employed intracellular Ca2+, small intestinal contraction and intestinal transit speed measurement to investigate the functional change. We found that SOCE activity and Orai1 and STIM1 expression levels of small intestinal smooth muscle were significantly increased in cells cultured in high glucose medium or in diabetic mice. Gastrointestinal transit speed and SOCE-mediated contractions were markedly increased in diabetic mice; Knocking down Orai1 or STIM1 with siRNA rescued both alterations in diabetic mice. However, the Orai1-large conductance Ca2+-activated K+ (BKCa) channel interaction was decreased in diabetic mice, and suppressing Orai1 expression or inhibiting the BKCa channel increased agonist-induced small intestinal contractions in normal mice.
Conclusion: We concluded that the increased SOCE caused by excessive STIM1 and Orai1 expression and decreased Orai1-BKCa interaction augmented small intestinal smooth muscle contraction and accelerated small bowel transit speed in diabetic mice. This finding demonstrates a pathological role for SOCE in diabetic enteropathy and provides a potential therapeutic target for diabetic enteropathy.
Introduction
Type 1 diabetes is an immune-mediated metabolic disease characterized by insulin deficiency and glucose fluctuation accompanied by diverse complications (Cheng and Yilmaz, 2015). Diabetic gastrointestinal disorders are common complications, including diabetic gastroparesis, and intestinal enteropathy (Yarandi and Srinivasan, 2014). Many studies have reported that diabetic gastroparesis is caused by abnormal electrophysiological activity, impaired gastric enteric neurons, and incongruous muscle activity (Stevens et al., 2013; Koch and Calles-Escandon, 2015). However, studies examining diabetic enteropathy are relatively rare. Currently, diabetic enteric complications in type 1 diabetes, mainly constipation and diarrhea, are commonly considered a manifestation of symptomatic diabetic autonomic neuropathy (Yarandi and Srinivasan, 2014). Accumulated evidence shows that neurodegeneration of the myenteric plexus (Anitha et al., 2006) and loss of mucosal nerve fibers (Selim et al., 2010) are basic factors. Electric stimulation caused by luminal contents relay neuronal activity to generate muscle responses; however, nervous dysfunction results in a mild increase of cholinergic contractions, decrease of noradrenaline, and attenuated inhibitory neurotransmission in the ileum of mice with type 1 diabetes (Domenech et al., 2011; Uranga-Ocio et al., 2015). In addition, degeneration of the interstitial cells of Cajal and reduced association with enteric neurons lead to decreased electrical activity (Iwasaki et al., 2006). Although Ca2+ is an essential participant in smooth muscle contraction, the changes in cellular Ca2+ homeostasis of the small intestinal smooth muscle (SISM) in type 1 diabetes is still not entirely clear.
Ca2+ plays a critical role in numerous physiological functions. Store-operated Ca2+ entry (SOCE) is a ubiquitous mechanism of Ca2+ influx in many cell types (Putney, 2011). With the development of RNA interfering techniques, two major SOCE-related components were discovered: stromal interaction molecule 1 (STIM1) and Orai1 (Feske et al., 2006; Soboloff et al., 2006). The N-terminus of STIM1 acts as a Ca2+ sensor located in the lumen of the endoplasmic reticulum (ER)/sarcoplasmic reticulum (SR). Upon Ca2+ depletion of the ER/SR, STIM1 aggregates and translocates to ER-plasma membrane junction regions and then activates Orai1, which is an ion channel formed by four transmembrane spanning protein subunits (Liou et al., 2005; Prakriya et al., 2006; Soboloff et al., 2006). When agonists act on G protein-coupled receptors (GPCR), GPCR signaling through the Gaq activates phospholipase C (PLC) to drive both inositol triphosphate (IP3) and diacylglycerol (DAG) production, IP3 acts on IP3 receptors, causing Ca2+ release from the ER/SR and depletion of internal Ca2+ stores. Growing evidence indicates an association of altered SOCE with various diabetic complications. Enhanced SOCE in glomerular mesangial cells (Chaudhari et al., 2014; Chaudhari and Ma, 2016), vascular endothelium (Tamareille et al., 2006; Daskoulidou et al., 2015), and platelets (Chaudhari and Ma, 2016) contributes to diabetic nephropathy and vasculopathy. Attenuated SOCE, found in retinal microvascular smooth muscle (Curtis et al., 2003) of diabetic rats, reduces vessel contractile responses or may be a compensatory response to avoid over-reactive contraction in diabetes. However, the pathological role of SOCE in SISM in type 1 diabetes remains unknown.
Therefore, in the present study, we used a mouse SISM cell line (MUS-M1) to investigate changes of SOCE in a high glucose environment in vitro. We also examined the pathological role of the SOCE-mediated Ca2+ signal in small intestine transit speed in vivo in a mouse model of type 1 diabetes.
Materials and Methods
Materials
Thapsigargin (TG), carbachol (CCh), and adenosine triphosphate (ATP) were obtained from Calbiochem. Iberiotoxin (IbTX) and streptozotocin (STZ) were purchased from Sigma-Aldrich. Anti-STIM1 (sc-68897) and anti-Orai1 (sc-68895) primary antibodies were purchased from Santa Cruz Biotechnology. The anti-BKCa channel (APC-107) primary antibody was purchased from Alomone Lab. Specific small interfering (si)RNA for mouse STIM1 (5′-UACAGUGGCUCAUUACGUA-3′) and Orai1 (5′- GCCAUAAGACGGACCGGCA-3′) (Dietrich et al., 2007; Potier et al., 2009) and scrambled siRNA (5′-ACGCGUAACGCGGGAA UUU-3′) were designed and synthesized by Biomics Company.
Cell Culture and siRNA Transfection
MUS-M1, a mouse SISM cell line, was purchased from the Kunming Cell Bank of the Chinese Academy of Sciences and cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum. Only sub-passage 3–10 MUS-M1 cells were used in the present study. For the group with normal glucose (NG) levels, the culture medium contained 5.6 mM glucose and 20 mM α-mannitol as an osmotic control. In the group exposed to high glucose (HG), the medium contained 25 mM glucose (Chaudhari et al., 2014). The medium was replaced every other day. Specific siRNAs against mouse STIM1 and Orai1 or scrambled control siRNA were transiently transfected into MUS-M1 cells using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s protocol. Cells were harvested for the following experiments 36 h after siRNA transfection.
Intracellular Ca2+ Measurement
The cytosolic Ca2+ concentration ([Ca2+]i) was measured as described previously (Chen et al., 2016). Briefly, MUS-M1 cells were plated at 1 × 105 per well on glass coverslips in 12-well culture plates as described above. Cells were loaded with a fluorescent Ca2+ ion indicator Fluo-8/AM (6 μmol/L) and 0.02% pluronic F-127 at 37°C for 30 min. The Ca2+ stores in MUS-M1 cells were depleted using 2 μmol/L TG or 100 μmol/L CCh for 10 min in Ca2+-free saline solution, which contained (in mmol/L) 140 NaCl, 5 KCl, 1 MgCl2, 10 glucose, 0.2 EGTA, and 5 HEPES at pH 7.4. The Ca2+ influx was initiated by applying 2 mmol/L extracellular Ca2+. The fluorescence signal was recorded using a Nikon fluorescence microscope with 488-nm excitation and 515-nm long pass emission wavelengths. Changes in the peak value of cytosolic [Ca2+]i were displayed as the ratio of fluorescence intensities relative to the baseline intensity before the application of extracellular Ca2+ (F1/F0). Each data point is an average of 20–30 cells.
Western Blotting and Co-immunoprecipitation
Western blotting and co-immunoprecipitation assays were performed as described in our previous study (Chen et al., 2016). Proteins were extracted from the lysates of MUS-M1 cells and the SISM tissue from diabetic and normal mice with detergent extraction buffer, which contained 1% Nonidet P-40, 150 mmol/L NaCl, and 20 mmol/L Tris–HCl at pH 8.0, plus protease inhibitor cocktail tablets. For SISM tissue, the peripheral adipose tissue and intestinal villi was removed. The extracts were fractionated by 12% SDS-PAGE and then transferred to polyvinylidene difluoride membranes. The membrane was blocked with 5% non-fat milk diluted by PBST for 1 h at room temperature and incubated at 4°C overnight with anti-Orai1 and anti-STIM1 primary antibodies. The next day, the membrane was washed three times and then incubated with a horseradish peroxidase-conjugated secondary antibody. The signal for each protein was detected using an ECL system. The optical densities of the protein bands were normalized to that of α-actin analyzed within the same lane and presented as the percentage of the optical density.
Induction of Type 1 Diabetes
All animal experiments were performed according to the guidelines presented in NIH publication no. 8523 and approved by the Experimentation Ethics Committee of Anhui Medical University. Five-week-old Kunming mice (male) were provided by the Experimental Animal Center of Anhui Medical University and randomly divided into two groups: STZ and age-matched controls. Both groups were fed regular chow and given free access to tap water. STZ (6 mg/mL) was dissolved in 50 mM sodium citrate buffer (pH 4.4) before the injection. As previously described, mice in the STZ group were injected with STZ (40 mg/kg) intraperitoneally for five consecutive days to induce type 1 diabetes (Furman, 2015). Mice in the control group received injections of acetate buffer. Plasma glucose levels were determined with a One Touch Blood Glucose Monitoring System (LifeScan, Inc., United States). Mice with fasting plasma glucose levels higher than 11.1 mmol/L were selected for use.
In vivo siRNA Injections
Diabetic mice were randomly separated into three groups. Each mouse received two injections every 3 days of Orai1 siRNA, STIM1 siRNA, or scrambled control siRNA. Based on previous studies (Filleur et al., 2003; Sang et al., 2018), all siRNA (in 50 μL of saline in each case) injections were administered intraperitoneally (125 μg/kg/day). The mice were used 3 days after the second siRNA treatment. In our intestinal smooth muscle cell culture studies using MUS-M1 cells, siRNA-mediated suppression of either Orai1 or STIM1 by 36% was sufficient to inhibit SOCE significantly. Based on these findings, we set a threshold of a minimum of 36% reduction in Orai1 or STIM1 protein within the intestinal wall as an inclusion criterion when assessing mice treated with siRNA in vivo via i.p. injections. With our approach, 80% of the animals injected with siRNA met this criterion and were analyzed as described.
Intestinal Transit Speed
The small intestinal transit speed experiment was performed as described elsewhere (Yuece et al., 2007). Briefly, after an overnight fast (water ad libitum), a charcoal meal marker (10% charcoal suspension in 5% gum arabic, 0.1 mL per 10 g body weight) was freshly prepared by dispersion. The mixture was administered orally to assess upper gastrointestinal transit speed. After 20 min, the mice were euthanized using an overdose of CO2 gas, and the intestines were immediately isolated. The distance traveled by the marker was measured (in centimeters). The results of the intestinal transit speed experiment are shown as a percentage of the total length of the mouse small intestine, from the pylorus to the terminal ileum.
Preparation of Small Intestine Segment and Tension Measurement
Small intestine segment preparation and tension measurements were performed as described elsewhere (Montgomery et al., 2016). Briefly, the mice were killed by an overdose of CO2 gas. The small intestine was quickly dissected and placed in Krebs-Henseleit solution containing (in mmol/L) 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25.2 NaHCO3, and 11.1 glucose, pH 7.4 at room temperature. The peripheral adipose tissue was removed. The small intestine was cut into segments 2 cm in length. One end of the tissue was fastened to a hook on the bottom of a glass organ bath. The other end was connected to an isometric force transducer, which was connected to an amplifier. The bubbling O2 should mix the solution. The tension signal was recorded and analyzed by a data acquisition and analysis system (BL-420E+, Chengdu Technology & Market Corp.). The segments were placed in 5 mL organ baths containing Krebs solution at 37°C and continuously bubbled with a gas mixture of 95% O2 and 5% CO2 to maintain pH at 7.4. After equilibrating for 60 min with 1 g passive tension, the small intestine segments were activated by two applications of high-K+ solution, containing 60 mmol/L K+, prepared by replacing NaCl with an equimolar amount of KCl. Next, the contractile response to CCh (100 μmol/L) or Ca2+ (2 mmol/L) was determined. Detailed group treatments are described in the section “Results.” For the IbTX group, IbTX (50 nmol/L) was added to Krebs-Henseleit solution for 20 min pretreatment.
Statistical Analysis
Data are presented as means ± SEM. Student’s unpaired t-test was used to analyze the difference between two groups with SigmaPlot 12.5 software. A value of P < 0.05 was considered statistically significant.
Results
Effect of a High Glucose Environment on Store-Operated Ca2+ Entry in MUS-M1 Cells
Store-operated Ca2+ entry is a crucial Ca2+ signal that initiates many cellular biological processes (Putney, 2011; Chaudhari and Ma, 2016). Several cell types cultured in HG medium mimicking diabetic hyperglycemia in vitro show enhanced SOCE (Tamareille et al., 2006; Chaudhari et al., 2014; Daskoulidou et al., 2015). To determine the effect of an HG environment on SOCE in intestinal smooth muscle cells, we used Ca2+ imaging and measured the change in [Ca2+]i evoked by SOCE in NG- and HG-cultured MUS-M1 cells. The Ca2+ store in MUS-M1 cells was depleted by 2 μM TG (a Ca2+ pump inhibitor in the ER) and 100 μM CCh (a muscarinic receptor activator). SOCE was initiated by the re-addition of 2 mM Ca2+ using a classic “Ca2+ added back” protocol. MUS-M1 cells were cultured in NG and HG media for 1, 3, and 7 days. The SOCE evoked by TG and CCh was significantly increased in HG-cultured cells at 3 and 7 days, but not at 1 day, compared with that in NG-cultured cells at 1, 3, and 7 days (Figure 1). By contrast, TG- and CCh-induced Ca2+ transients in Ca2+-free solution were not significantly changed between NG- and HG-cultured cells (Supplementary Figure 1). These results indicate that a HG environment enhances SOCE in MUS-M1 cells.
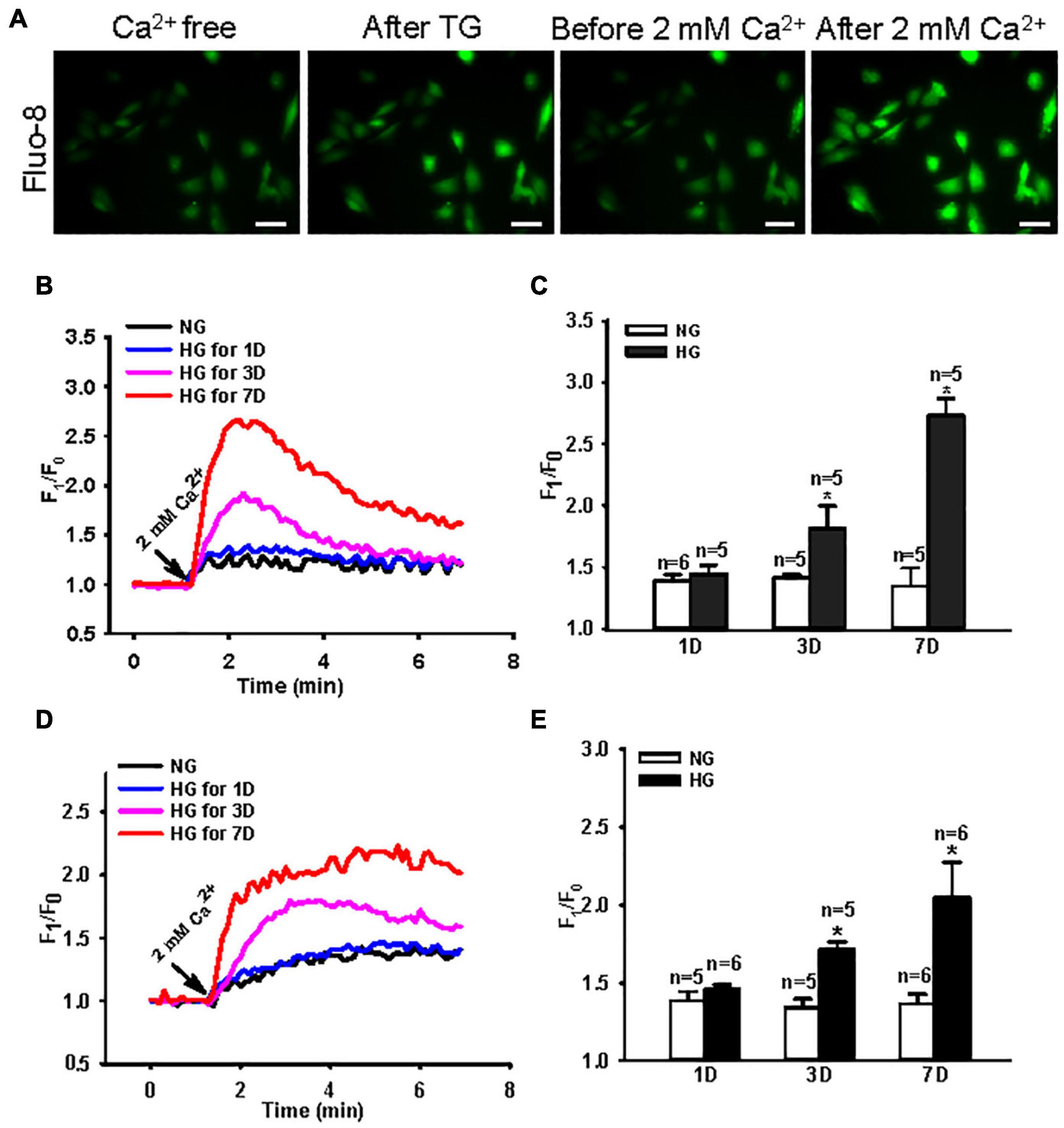
Figure 1. Effect of high glucose (HG) culture medium on store-operated Ca2+ entry (SOCE) in MUS-M1 cells. (A) SOCE was evoked by a classic “Ca2+ added back” protocol. Images showing fluorescence intensity indicating the change of transient intracellular Ca2+ concentration ([Ca2+]i). Scale bars, 10 μm. Representative traces (B,D) and summarized data (C,E) showing the change in [Ca2+]i (SOCE) evoked by extracellular application of 2 mM Ca2+ after the treatment of 2 μM thapsigargin (TG) (B,C) or 100 μM carbachol (D,E) for 20 min in Ca2+-free solution to deplete internal Ca2+ stores in MUS-M1 cells cultured in normal glucose (NG, 5.6 mM D-glucose + 20 mM α-mannitol) or HG (25 mM D-glucose) medium for 1, 3, and 7 days. Values are shown as the mean ± SEM (n = 5–6); *P < 0.05, NG vs. HG on the same day.
Role of Orai1 and Stromal Interaction Molecule 1 in Enhanced Store-Operated Ca2+ Entry in High Glucose-Cultured MUS-M1 Cells
Orai1 and STIM1 are two essential components in SOCE. Therefore, changes in Orai1 and STIM1 protein expression may affect the intensity of SOCE. Thus, we examined the expression profiles of Orai1 and STIM1 proteins in NG- and HG-cultured MUS-M1 cells. We found that Orai1 and STIM1 expression levels were significantly enhanced in HG-cultured cells on 3 and 7 days, but not 1 day, compared with those in NG-cultured cells on 1, 3, and 7 days (Figure 2).
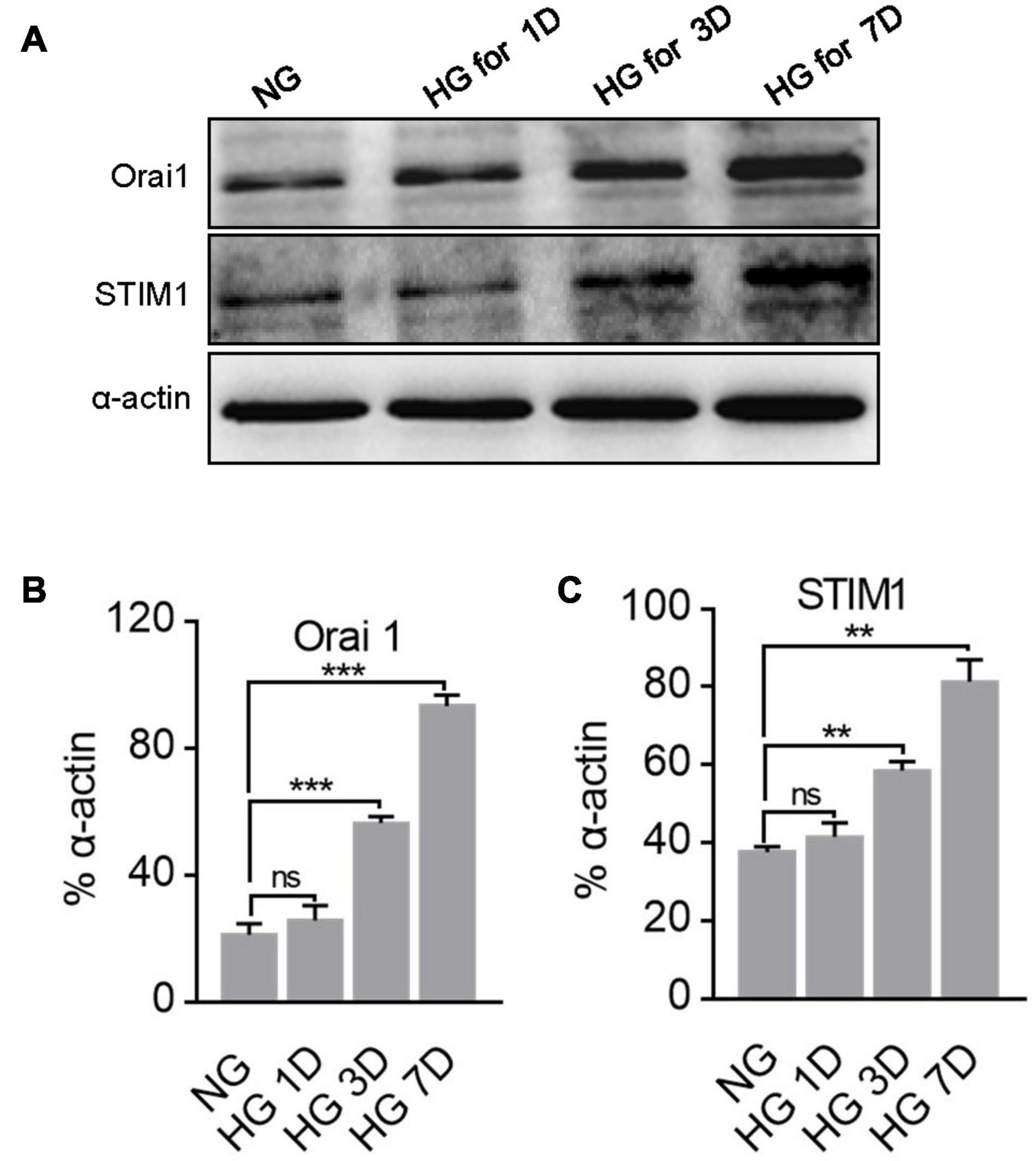
Figure 2. Effect of high glucose (HG) culture medium on Orai1 and STIM1 protein expression in MUS-M1 cells. Representative immunoblotting images (A) and summarized data (B,C) showing the expression levels of Orai1 (B) and STIM1 (C) proteins in MUS-M1 cells cultured in normal glucose (NG, 5.6 mM D-glucose + 20 mM α-mannitol) or HG (25 mM D-glucose) medium for 1, 3, and 7 days. Alpha-actin was used as a loading control. Values are shown as the mean ± SEM (n = 3); **, and *** denote P < 0.01, and P < 0.001, respectively. NS means non-significant, NG vs. HG on the same day.
To further identify the role of Orai1 and STIM1 in the SOCE of MUS-M1 cells, we used an RNA interfering technique to suppress Orai1 and STIM1 protein expression. We found that compared with the scrambled siRNA control, Orai1- and STIM1-specific siRNAs knocked down Orai1 and STIM1 expression, respectively, in MUS-M1 cells (Figures 3A,B). In addition, our [Ca2+]i measurement data indicated that compared with the scrambled siRNA control, Orai1- and STIM1-specific siRNAs significantly reduced CCh-evoked SOCE in NG- or HG-cultured (for 7 days) MUS-M1 cells (Figures 3C–F). However, CCh-induced Ca2+ transients in Ca2+-free solution were not significantly changed among scrambled, STIM1, and Orai1 siRNA-transfected cells (Supplementary Figures 2A,B). Pre-incubation with BTP-2 (20 μM) for 20 min significantly inhibited the SOCE of MUS-M1 cells on the 7 days in NG and HG-cultured (Supplementary Figures 2C,D). This result is consistent with the result using specific siRNA. Taken together, these data indicate that Orai1 and STIM1 are two important proteins participating in SOCE, and the increased Orai1 and STIM1 expression may amplify the SOCE intensity in HG-cultured MUS-M1 cells.
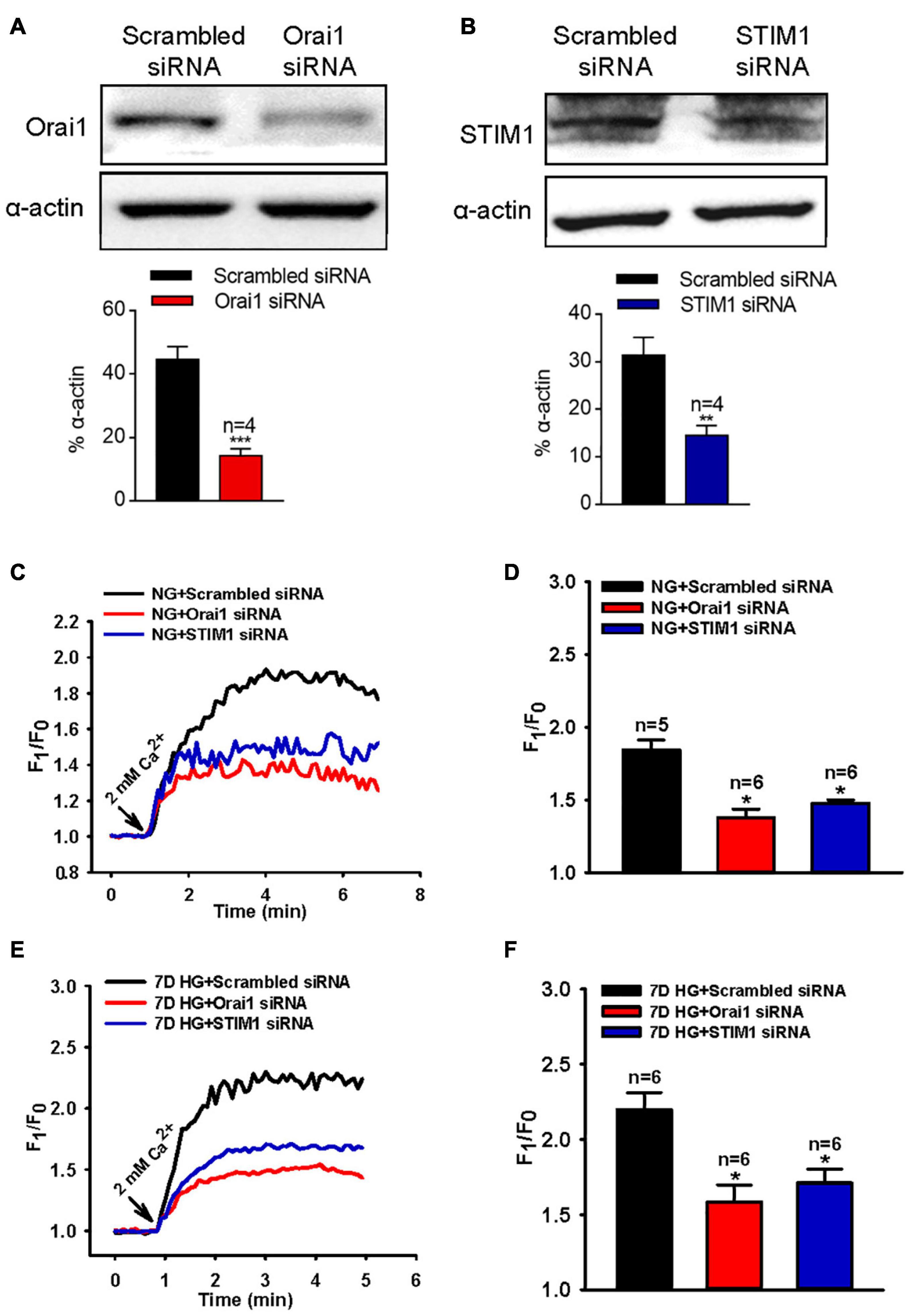
Figure 3. Effect of Orai1 and STIM1 siRNAs on store-operated Ca2+ entry (SOCE) in MUS-M1 cells. (A,B) Representative immunoblotting images showing Orai1 and STIM1 expression levels in MUS-M1 cells transfected with Orai1 siRNA (A), STIM1 siRNA (B), or scrambled control siRNA. **, and *** denote P < 0.01, and P < 0.001, respectively. Representative traces (C,E) and summarized data (D,F) showing the alterations in [Ca2+]i (SOCE) evoked by extracellular application of 2 mM Ca2+ after the treatment of 100 μM carbachol for 10 min in Ca2+-free solution to deplete internal Ca2+ stores in MUS-M1 cells cultured in normal glucose (NG, C,D, 5.6 mM D-glucose + 20 mM α-mannitol) or high glucose (HG, E,F, 25 mM D-glucose) medium for 7 days. Alpha-actin was used as a loading control. Values are shown as the mean ± SEM (n = 5–6); *P < 0.05, scrambled siRNA vs. Orai1 or STIM1 siRNA transfections.
Role of Orai1 and Stromal Interaction Molecule 1 in Small Intestine Contraction and Gastrointestinal Transit Speed in Diabetic Mice
Type 1 diabetes commonly induces gastrointestinal dysfunction, including constipation and diarrhea. The results above indicated that culturing cells in an HG environment increased Orai1 and STIM1 expression levels. Therefore, we generated type 1 diabetic mice by injecting them with STZ intraperitoneally, and investigated Orai1 and STIM1 protein expression in SISM. We found that Orai1 and STIM1 expression levels in the SISM of diabetic mice were significantly increased compared with those in age-matched controls (Figures 4A–C). Ca2+ is crucial for inducing smooth muscle contractions, and Orai1 and STIM1 mediate Ca2+ influx. Thus, altered Orai1 and STIM1 expression may affect the SOCE-mediated contraction of the small intestine. We next used 100 μM CCh to evoke SOCE in fresh-isolated small intestine segments bathed in Ca2+-free Krebs solution, in which 0.2 mM EGTA was used to chelate residual Ca2+ ions in the bath, and 1 μM verapamil to inhibit voltage-dependent Ca2+ channels (VDCCs). Re-adding 2 mM Ca2+ induced longitudinal contractions of the small intestine segment. Because VDCCs were inhibited by verapamil, any contraction induced by re-adding Ca2+ would be due mainly to the SOCE-mediated Ca2+ influx. These results indicated that SOCE-mediated small intestine segment contractions were significantly increased in diabetic mice compared with those in control mice (Figures 4D,E). However, 60 mM (high) K+-induced and CCh-induced contractions were not altered in diabetic mice (Supplementary Figures 3A,C). Additionally, hematoxylin and eosin (HE) staining showed no obvious difference in the thickness of the smooth muscle layer of the small intestine between the mouse groups (Supplementary Figure 4).
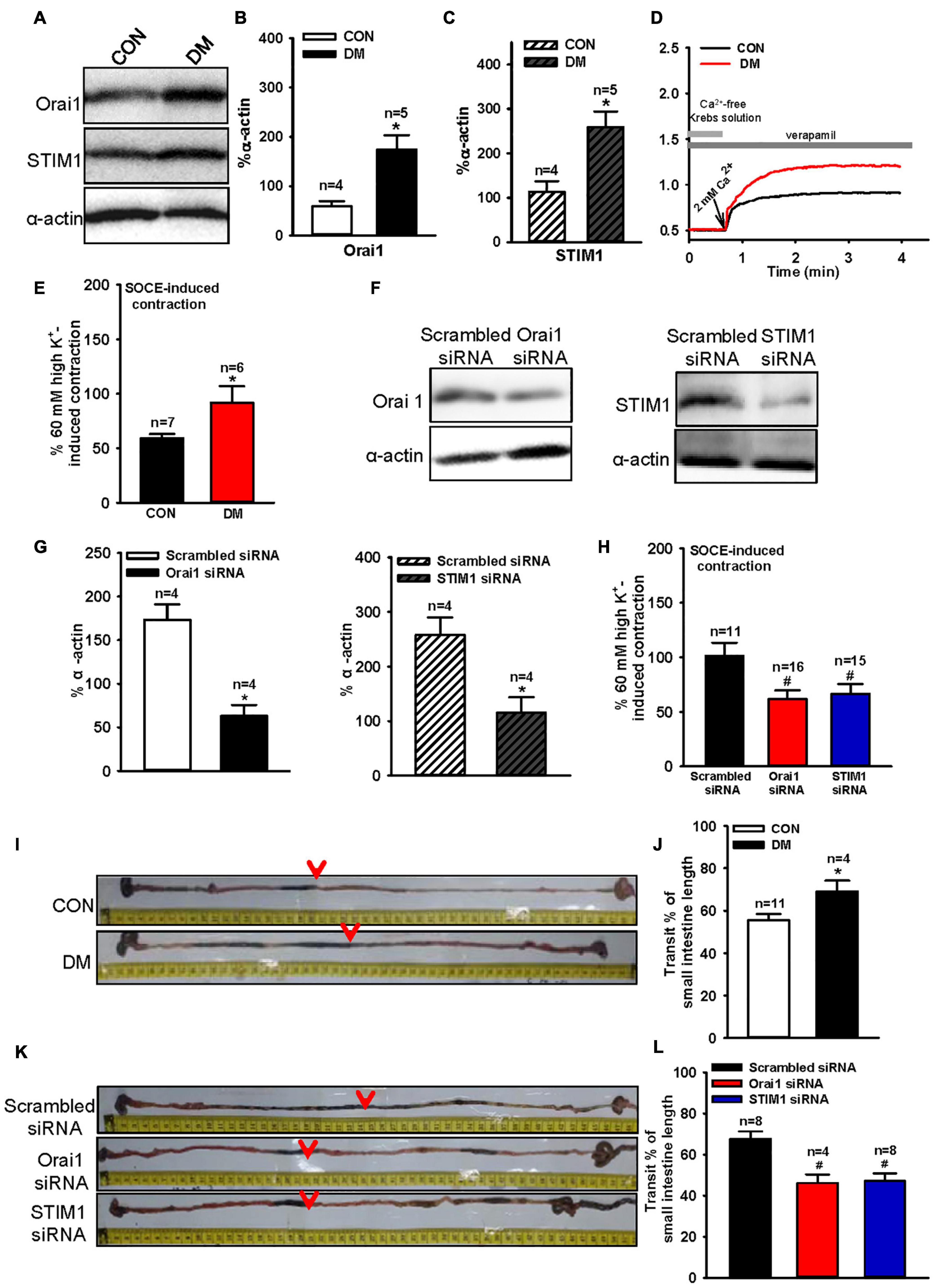
Figure 4. Orai1 and STIM1 protein expression levels, store-operated Ca2+ entry (SOCE)-mediated mouse small intestinal longitudinal contraction and intestinal transit time alterations in diabetic mice and the effect of Orai1 and STIM1 siRNA transfections. Representative immunoblotting images (A) and summarized data (B,C) showing the expression levels of Orai1 (A,B) and STIM1 (A,C) proteins in small intestinal smooth muscle cells of age-matched control (CON) and diabetic (DM) mice. Alpha-actin was used as a loading control. Values are shown as the mean ± SEM (n = 4–5); *P < 0.05, age-matched control vs. diabetic mice. Mouse small intestine segments were precontracted by the application of 100 μM carbachol in Ca2+-free Krebs solution to evoke SOCE. Then, re-addition of 2 mM Ca2+ induced longitudinal contractions of small intestine segments. Representative traces (D) and summarized data (E,F) showing SOCE-mediated small intestinal longitudinal contraction in age-matched control (CON) and diabetic (DM) mice (D,E). Representative immunoblotting images and summarized data showing Orai1 and STIM1 expression levels in diabetic mice small intestinal smooth muscle cells transfected with Orai1 siRNA or STIM1 siRNA or scrambled control siRNA. Alpha-actin was used as a loading control (F,G). Values are shown as the mean ± SEM (n = 4); *P < 0.05, scrambled siRNA vs. Orai1 or STIM1 siRNA transfection. SOCE-mediated small intestinal longitudinal contraction in diabetic mice transfected with scrambled, Orai1 or STIM1 siRNA (H). Values are shown as the mean ± SEM (n = 6–16); *P < 0.05, age-matched control vs. diabetic mice; #P < 0.05, scrambled siRNA vs. Orai1 or STIM1 siRNA. Representative images (I,K) and summarized data (J,L) showing intestinal transit distance in age-matched control (CON) and diabetic (DM) mice (I,K), or diabetic mice transfected with scrambled, Orai1, or STIM1 siRNA (J,L). The dark charcoal marker appeared as indicated (red arrows). Values are shown as the mean ± SEM (n = 4–11); *P < 0.05, age-matched control vs. diabetic mice; #P < 0.05, scrambled siRNA vs. Orai1 or STIM1 siRNA.
To verify the role of Orai1 and STIM1 in the enhanced small intestine contractions mediated by SOCE, we intraperitoneally injected Orai1- or STIM1-specific siRNA into diabetic mice to suppress Orai1 or STIM1 expression, respectively, in SISM cells. After siRNA treatment for 6 days (two injections every 3 days), western blotting analysis showed that Orai1 and STIM1 expression levels were significantly reduced in the SISM of diabetic mice (Figures 4F,G). Our contraction experiments indicated that compared with scrambled siRNA transfection, Orai1- or STIM1-specific siRNA transfection largely suppressed the SOCE-mediated small intestine contraction in diabetic mice (Figure 4H) but did not affect 60 mM (high) K+-induced contraction caused by voltage-dependent Ca2+ channel-mediated Ca2+ influx in Krebs solution or CCh-induced contraction in Ca2+-free solution, in which CCh-induced contraction is induced by Ca2+ release from Ca2+ store (Supplementary Figures 3B,D).
Small intestinal longitudinal contractions are correlated with gastrointestinal transit speed (Grider, 2003). Therefore, we investigated gastrointestinal transit speed in another group of diabetic mice. Gastrointestinal transit speed was significantly increased in diabetic mice compared with that in age-matched controls (Figures 4I,J). Additionally, transfections with Orai1- and STIM1-specific siRNAs significantly decreased the gastrointestinal transit speed of diabetic mice (Figures 4K,L). These results suggest that increased Orai1 and STIM1 expression may contribute to the enhancement of agonist-induced small intestine contraction and gastrointestinal transit speed.
Role of the Orai1-Large Conductance Ca2+-Activated K+ Channel Interaction in Agonist-Induced Small Intestine Contraction
Our recent studies have demonstrated that Orai1 associates with Ca2+-activated K+ channels (for example, the small conductance Ca2+-activated K+ channel 3 and BKCa channel) to form signal complexes regulating smooth muscle contraction (Song et al., 2015; Chen et al., 2016). Here, we also investigated the interaction of Orai1 and the BKCa channel in SISM. We first used co-immunoprecipitation assays followed by immunoblotting and found that Orai1 pulled down the BKCa channel from mouse SISM (Figure 5A). We then used tension measurements to examine the function of the Orai1-BKCa complex in SISM. In normal mice, our data showed that IbTX (a specific inhibitor of BKCa channels, 50 nM) and Orai1 siRNA transfection (Supplementary Figure 5) significantly increased 100 μM CCh-induced small intestinal longitudinal contractions (Figures 5D–G), but did not affect 60 mM (high) K+-induced contractions in Krebs solution without verapamil (Supplementary Figure 6), indicating that the Orai1-BKCa complex can negatively regulate agonist-induced small intestine contractions.
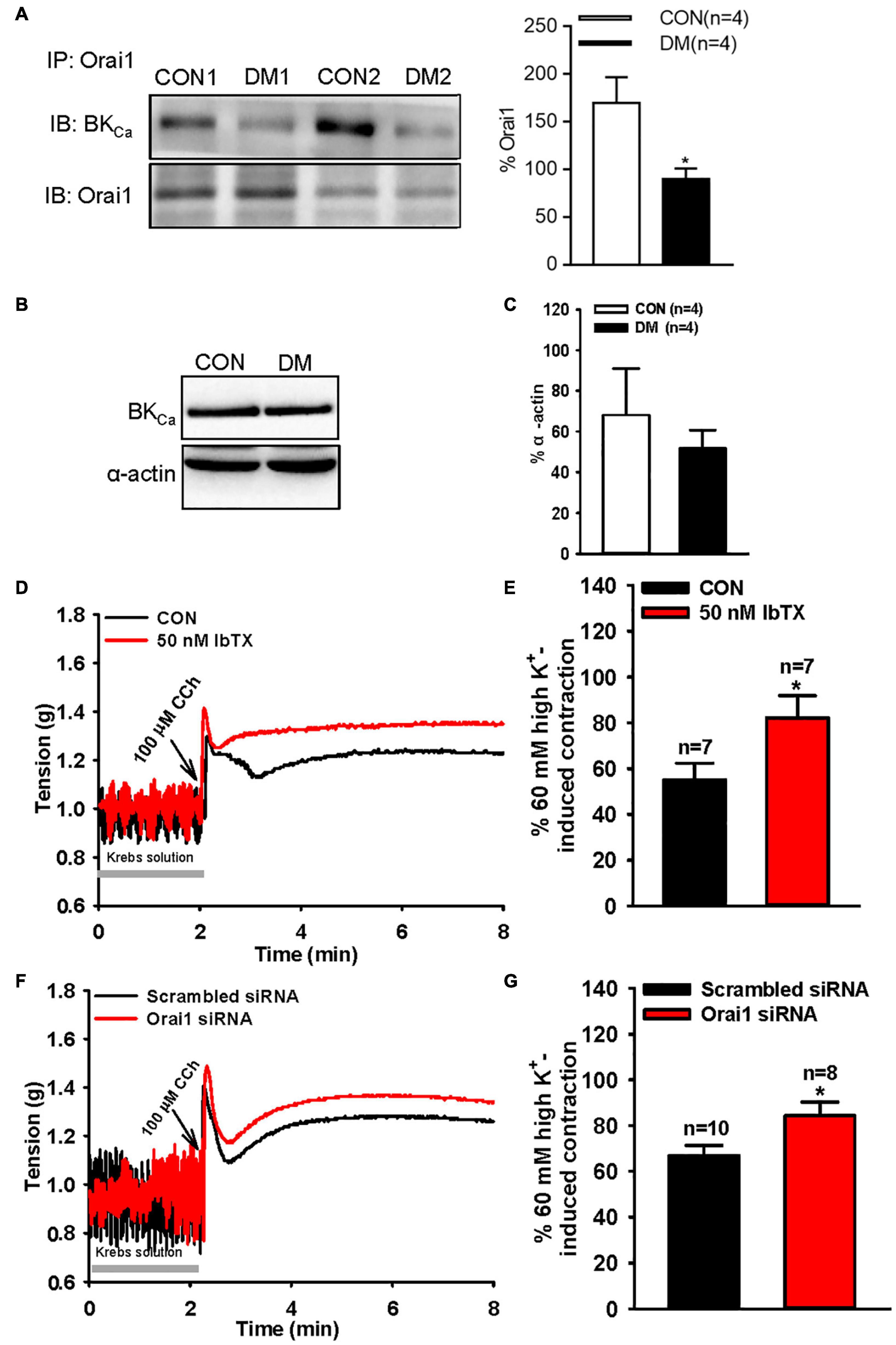
Figure 5. Orai1-BKCa interaction and BKCa channel protein expression level alterations in diabetic mouse small intestinal smooth muscle cells and role of store-operated Ca2+ entry (SOCE) and the BKCa channel in carbachol (CCh)-induced mouse small intestinal longitudinal contraction. (A) Representative paired images and summarized data showing co-immunoprecipitation followed by immunoblotting (IB) with anti-BKCa channel or anti-Orai1. Proteins from small intestinal smooth muscle of age-matched control (CON) or diabetic (DM) mice were immunoprecipitated with anti-Orai1 antibody; n = 4 experiments. Representative images (B) and summarized data (C) showing the expression level of the BKCa channel protein. Alpha-actin was used as a loading control. Values are shown as the mean ± SEM (n = 4); P > 0.05, age-matched control vs. diabetic mice. Representative traces (D,E) and summarized data (E,G) showing 100 μM CCh-induced mouse small intestine contractions. The segments of the small intestine were pretreated with 50 nM IbTX for 20 min (D,E) or transfected with scrambled or Orai1 siRNA (F,G) by intraperitoneal injections. Values are shown as the mean ± SEM (n = 7–10); *P < 0.05, control (CON) vs. IbTX treatment, or scrambled siRNA vs. Orai1 siRNA.
Orai1-BKCa Interaction Alteration in Small Intestinal Smooth Muscle of Diabetic Mice
To further investigate the pathological relevance of the Orai1-BKCa interaction, we used co-immunoprecipitation assays to examine changes in the association between Orai1 and the BKCa channel. Our data indicated that compared with that in age-matched controls, the amount of BKCa protein pulled down by Orai1 was markedly decreased in the SISM of diabetic mice (Figure 5A). Western blotting analysis showed that the BKCa protein expression has not changed (Figures 5B,C). These results suggest that compared with that in control mice, the Orai1 and BKCa channel interaction in the SISM of diabetic mice is weaker.
Discussion
Here, we examined SOCE in SISM cells cultured in HG medium or in a mouse model of type 1 diabetes. Our major findings are as follows: (1) SOCE activity and Orai1 and STIM1 expression levels in SISM cells were significantly increased in HG culture conditions or in diabetic mice compared with those in NG culture conditions or age-matched control mice, respectively; (2) gastrointestinal transit speed and SOCE-mediated contractions were markedly increased in diabetic mice; (3) siRNA knockdown of Orai1 or STIM1 rescued the augmented gastrointestinal transit speed and SOCE-mediated contractions in diabetic mice; (4) Orai1 pulled down the BKCa channel in SISM cells, and the interaction between Orai1 and the BKCa channel was decreased in diabetic mice; (5) suppressing Orai1 expression or inhibiting BKCa channel activity increased agonist-induced small intestine contractions. Thus, we demonstrated that SOCE activity and SOCE-mediated contraction were significantly increased in SISM cells cultured in HG medium or in mice with type 1 diabetes and that altered SOCE activity and Orai1-BKCa channel interactions might contribute to accelerated gastrointestinal transit speed.
Although diabetic enteropathy affects the life quality of patients with type 1 diabetes (Cheng and Yilmaz, 2015), compared with other more serious complications, patients often feel that the gastrointestinal symptoms are less likely to cause critical disabilities or death. Thus, owing to limited attention and research, the effect of type 1 diabetes on SISM is poorly understood. An imbalance in the Ca2+ homeostasis of muscle cells disrupts contractile function and gastrointestinal motility, and SOCE disorders may lead to many dysfunctions, ranging from immunodeficiency to myopathy and vascular diseases (Feske, 2007; Duke et al., 2010; Voelkers et al., 2010). To our knowledge, this study is the first to report the physiological mechanisms of SOCE in the SISM associated with type 1 diabetes. We found that the SOCE of MUS-M1 cells cultured in HG was increased without significantly altering Ca2+ store release. As expected, our western blot data showed that Orai1 and STIM1 protein expression was also significantly increased. In diabetic mice, Orai1 and STIM1 protein expression and SOCE-mediated small intestine contraction were also markedly increased, whereas high-K+- and CCh-induced contractions were not significantly altered. Moreover, HE staining showed that the thickness of the smooth muscle layer in the small intestine did not differ between age-matched control and diabetic mice. Suppressing Orai1 or STIM1 expression using specific siRNA did not significantly affect Ca2+ store release but significantly inhibited SOCE in MUS-M1 cells and SOCE-mediated small intestine contractions in diabetic and control mice without affecting high-K+- and CCh-induced contractions. Together these results suggest that a HG environment and diabetes enhance SOCE in SISM cells, leading to increased Ca2+ mobilization and small intestine contraction.
Recently, we and another group reported that Orai1 associates with the KCa channel to participate in smooth muscle contraction as well as cancer cell migration and bone metastasis (Chantome et al., 2013; Song et al., 2015; Chen et al., 2016). In the present study, we found that Orai1 also associated with the BKCa channel to regulate mouse small intestine contraction. In the flow chart of our study hypothesis, Orai1-mediated Ca2+ influx activates the BKCa channel, leading to membrane hyperpolarization (Supplementary Figure 7). The membrane hyperpolarization inhibits VDCCs to prevent agonist-induced small intestine contraction. Additionally, our results showed that Orai1 siRNA transfection to suppress Orai1 protein expression or IbTX administration to inhibit the BKCa channel significantly increased CCh-induced mouse small intestine contractions, but did not affect high-K+-induced contractions. Moreover, Orai1 protein expression and SOCE activity were increased in the SISM cells of diabetic mice. According to the working model of the Orai1-BKCa complex shown in Supplementary Figure 7, increasing Orai1 activity and SOCE should strongly activate BKCa channels, which in turn would inhibit small intestine contraction and motility largely by inhibiting VDCC opening. However, in contrast to this hypothetical model, our data indicated that small intestine transit speed was markedly increased in diabetic mice. To determine the reason for this acceleration, we investigated alterations in the Orai1-BKCa channel interaction in the SISM cells of diabetic mice. Our co-immunoprecipitation data showed that the Orai1-BKCa complex interaction was markedly weakened in the SISM cells of diabetic mice. Based on this finding, we speculate that although Orai1 protein expression and SOCE activity were increased, Orai1-mediated Ca2+ influx did not effectively activate BKCa channels but more likely induced muscle contraction and intestine motility. Therefore, increased Orai1 protein expression but decreased Orai1-BKCa interaction may reconcile this conflicting result. How best to alter this weakened Orai1-BKCa channel interaction in diabetic animals to achieve a therapeutic response will be examined in future studies.
Numerous previous studies have shown that the alteration of gastrointestinal tract motility in type 1 diabetes is controversial. Several studies have reported that gastrointestinal transit speed is faster in patients with type 1 diabetes than in controls (Rosa-e-Silva et al., 1996; Perano et al., 2015), but other reports indicate delayed gastrointestinal activity in these patients (Zhao et al., 2006; Faria et al., 2013). Conflicting results are also found in rodent models of type 1 diabetes (Martin et al., 2004; Izbeki et al., 2008; Umathe et al., 2009; Durmus-Altun et al., 2011; Hu and Feng, 2012). Our data indicated that gastrointestinal transit speed was markedly increased in a mouse model of type 1 diabetes. Suppressing Orai1 and STIM1 with siRNA-specific transfections reduced the gastrointestinal transit speed in diabetic mice to that of control mice. Zheng et al. (2018) reported that SOCE-mediated Ca2+ influx is necessary to maintain interstitial cells of Cajal (ICC) pacemaker activity. Besides, colonic pacemaker ICC-SM exhibit complex Ca2+-firing patterns and drive smooth muscle activity and overall colonic contractions (Baker et al., 2021). Maintenance and refilling of cellular Ca2+ stores by SOCE may also be important for mediation and shaping of Ca2+ signals in ICC-SM. Therefore, increased SOCE activity and Ca2+ mobilization in SISM, ICC cells and/or even nervous system may all contribute to the accelerated gastrointestinal transit speed and diabetes-associated diarrhea because in body Orai1 and STIM1 siRNAs may affect many cell types involving gastrointestinal transit speed. However, which cell is more important and why the gastrointestinal transit speed was shown to be faster in some studies but delayed in others in animal or patients with type 1 diabetes is unknown. Perhaps factors such as the stage of type 1 diabetes, meals, degree of blood glucose, etc., may differentially affect gastrointestinal transit speed. This issue should be addressed in future studies.
Store-operated Ca2+ entry regulates various cellular functions, including cell proliferation, contraction, and secretion (Parekh and Putney, 2005). In type 1 diabetes, morphological changes and biomechanical remodeling occur in the small intestine, manifesting as proliferation in all layers of gastrointestinal wall, increasing cross-section thickness and length and altering opening angles and residual strain (Domenech et al., 2011; Liu et al., 2012). In addition to muscle contraction, SOCE is also involved in muscle proliferation, including that of cardiomyocytes, aortic smooth muscle cells, and bronchial smooth muscle cells (Sweeney et al., 2002; Berra-Romani et al., 2008; Luo et al., 2012). To exclude this effect on the small intestine transit speed, we compared high-K+-induced small intestine contractions between diabetic and age-matched control mice and found no significant difference between them. Our HE staining results also showed that the SISM layer thickness was not altered in diabetic mice. Therefore, our results exclude the effect of proliferation or hypertrophy of SISM cells on enhanced small intestine transit speed.
In live cells, Ca2+ homeostasis regulation and Ca2+-mediated signal transduction are complicated. Although we demonstrated that the SOCE-mediated Ca2+ signal was increased in the SISM cells of mice with type 1 diabetes, another group found that Ca2+-activated Cl– channels also participate in glucose-induced [Ca2+]i increase in small intestine cells (Yin et al., 2014). In diabetes, the body is under a high oxidative state. Excessive reactive oxygen species can induce numerous cellular disorders, destroying Ca2+ homeostasis. Therefore, our finding may be only one of the pathways involved in type 1 diabetes pathology.
Conclusion
We demonstrated that SOCE activity was significantly increased in SISM cells cultured in HG medium or in mice with type 1 diabetes. Upregulated SOCE promoted small intestinal muscle contractions and gastrointestinal transit speed, which may be among the factors inducing diabetic diarrhea. Therefore, the SOCE pathway likely represents a potential therapeutic target for diabetic diarrhea.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by the Animal Ethics Committee of Anhui Medical University.
Author Contributions
FD and JG performed the experiments, analyzed the data, and co-wrote the manuscript. YW, TJ, HC, and YH conducted the experiments and analyzed the data. XX and JD designed a part of the study and helped to interpret the part of the data. BS and QZ conceived the idea, designed and supervised the study, analyzed the data, and revised the manuscript. All authors reviewed the report and approved the final version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 81570403, U1732157, 81970703, 81700283, and 82002598) and Anhui Provincial Natural Science Foundation (Grant No. 140808 5MH158).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are thankful to Zhang G. L. for constructive comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.691867/full#supplementary-material
References
Anitha, M., Gondha, C., Sutliff, R., Parsadanian, A., Mwangi, S., Sitaraman, S. V., et al. (2006). GDNF rescues hyperglycemia-induced diabetic enteric neuropathy through activation of the PI3K/Akt pathway. J. Clin. Invest. 116, 344–356. doi: 10.1172/JCI26295
Baker, S. A., Leigh, W. A., Del Valle, G., De Yturriaga, I. F., Ward, S. M., Cobine, C. A., et al. (2021). Ca(2+) signaling driving pacemaker activity in submucosal interstitial cells of Cajal in the murine colon. Elife 10:64099. doi: 10.7554/eLife.64099
Berra-Romani, R., Mazzocco-Spezzia, A., Pulina, M. V., and Golovina, V. A. (2008). Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am. J. Physiol. Cell Physiol. 295, C779–C790. doi: 10.1152/ajpcell.00173.2008
Chantome, A., Potier-Cartereau, M., Clarysse, L., Fromont, G., Marionneau-Lambot, S., Gueguinou, M., et al. (2013). Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 73, 4852–4861. doi: 10.1158/0008-5472.CAN-12-4572
Chaudhari, S., and Ma, R. (2016). Store-operated calcium entry and diabetic complications. Exp. Biol. Med. 241, 343–352. doi: 10.1177/1535370215609693
Chaudhari, S., Wu, P., Wang, Y., Ding, Y., Yuan, J., Begg, M., et al. (2014). High glucose and diabetes enhanced store-operated Ca(2+) entry and increased expression of its signaling proteins in mesangial cells. Am. J. Physiol. Renal Physiol. 306, F1069–F1080. doi: 10.1152/ajprenal.00463.2013
Chen, M., Li, J., Jiang, F., Fu, J., Xia, X., Du, J., et al. (2016). Orai1 forms a signal complex with BKCa channel in mesenteric artery smooth muscle cells. Physiol. Rep. 4:12682. doi: 10.14814/phy2.12682
Cheng, C. W., and Yilmaz, O. H. (2015). IGFBP3 and T1D: Systemic Factors in Colonic Stem Cell Function and Diabetic Enteropathy. Cell Stem Cell 17, 379–380. doi: 10.1016/j.stem.2015.09.008
Curtis, T. M., Major, E. H., Trimble, E. R., and Scholfield, C. N. (2003). Diabetes-induced activation of protein kinase C inhibits store-operated Ca2+ uptake in rat retinal microvascular smooth muscle. Diabetologia 46, 1252–1259. doi: 10.1007/s00125-003-1178-5
Daskoulidou, N., Zeng, B., Berglund, L. M., Jiang, H., Chen, G. L., Kotova, O., et al. (2015). High glucose enhances store-operated calcium entry by upregulating ORAI/STIM via calcineurin-NFAT signalling. J. Mol. Med. 93, 511–521. doi: 10.1007/s00109-014-1234-2
Dietrich, A., Kalwa, H., Storch, U., Mederos y Schnitzler, M., Salanova, B., Pinkenburg, O., et al. (2007). Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 455, 465–477. doi: 10.1007/s00424-007-0314-3
Domenech, A., Pasquinelli, G., De Giorgio, R., Gori, A., Bosch, F., Pumarola, M., et al. (2011). Morphofunctional changes underlying intestinal dysmotility in diabetic RIP-I/hIFNbeta transgenic mice. Int. J. Exp. Pathol. 92, 400–412. doi: 10.1111/j.1365-2613.2011.00789.x
Duke, A. M., Hopkins, P. M., Calaghan, S. C., Halsall, J. P., and Steele, D. S. (2010). Store-operated Ca2+ entry in malignant hyperthermia-susceptible human skeletal muscle. J. Biol. Chem. 285, 25645–25653. doi: 10.1074/jbc.M110.104976
Durmus-Altun, G., Vatansever, U., Arzu Vardar, S., Altaner, S., and Dirlik, B. (2011). Scintigraphic evaluation of small intestinal transit in the streptozotocin induced diabetic rats. Hippokratia 15, 262–264.
Faria, M., Pavin, E. J., Parisi, M. C., Lorena, S. L., Brunetto, S. Q., Ramos, C. D., et al. (2013). Delayed small intestinal transit in patients with long-standing type 1 diabetes mellitus: investigation of the relationships with clinical features, gastric emptying, psychological distress, and nutritional parameters. Diabetes Technol. Ther. 15, 32–38. doi: 10.1089/dia.2012.0158
Feske, S. (2007). Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 7, 690–702. doi: 10.1038/nri2152
Feske, S., Gwack, Y., Prakriya, M., Srikanth, S., Puppel, S. H., Tanasa, B., et al. (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. doi: 10.1038/nature04702
Filleur, S., Courtin, A., Ait-Si-Ali, S., Guglielmi, J., Merle, C., Harel-Bellan, A., et al. (2003). SiRNA-mediated inhibition of vascular endothelial growth factor severely limits tumor resistance to antiangiogenic thrombospondin-1 and slows tumor vascularization and growth. Cancer Res. 63, 3919–3922.
Furman, B. L. (2015). Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. Pharmacol. 70, 41–20. doi: 10.1002/0471141755.ph0547s70
Grider, J. R. (2003). Reciprocal activity of longitudinal and circular muscle during intestinal peristaltic reflex. Am. J. Physiol. Gastrointest. Liver Physiol. 284, G768–G775. doi: 10.1152/ajpgi.00384.1998
Hu, W., and Feng, P. (2012). Myosin light chain kinase is involved in the mechanism of gastrointestinal dysfunction in diabetic rats. Dig. Dis. Sci. 57, 1197–1202. doi: 10.1007/s10620-012-2041-7
Iwasaki, H., Kajimura, M., Osawa, S., Kanaoka, S., Furuta, T., Ikuma, M., et al. (2006). A deficiency of gastric interstitial cells of Cajal accompanied by decreased expression of neuronal nitric oxide synthase and substance P in patients with type 2 diabetes mellitus. J. Gastroenterol. 41, 1076–1087. doi: 10.1007/s00535-006-1909-8
Izbeki, F., Wittman, T., Rosztoczy, A., Linke, N., Bodi, N., Fekete, E., et al. (2008). Immediate insulin treatment prevents gut motility alterations and loss of nitrergic neurons in the ileum and colon of rats with streptozotocin-induced diabetes. Diabetes Res. Clin. Pract. 80, 192–198. doi: 10.1016/j.diabres.2007.12.013
Koch, K. L., and Calles-Escandon, J. (2015). Diabetic gastroparesis. Gastroenterol. Clin. North Am. 44, 39–57. doi: 10.1016/j.gtc.2014.11.005
Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E. Jr., et al. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241. doi: 10.1016/j.cub.2005.05.055
Liu, G. F., Zhao, J. B., Zhen, Z., Sha, H., Chen, P. M., Li, M., et al. (2012). Effect of tangweian jianji on upper gastrointestinal remodeling in streptozotocin-induced diabetic rats. World J. Gastroenterol. 18, 4875–4884. doi: 10.3748/wjg.v18.i35.4875
Luo, X., Hojayev, B., Jiang, N., Wang, Z. V., Tandan, S., Rakalin, A., et al. (2012). STIM1-dependent store-operated Ca(2)(+) entry is required for pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 52, 136–147. doi: 10.1016/j.yjmcc.2011.11.003
Martin, M. T., Azpiroz, F., and Malagelada, J. R. (2004). Ileal brake failure in streptozotocin-induced diabetic rat. Scand. J. Gastroenterol. 39, 423–427.
Montgomery, L. E., Tansey, E. A., Johnson, C. D., Roe, S. M., and Quinn, J. G. (2016). Autonomic modification of intestinal smooth muscle contractility. Adv. Physiol. Educ. 40, 104–109. doi: 10.1152/advan.00038.2015
Parekh, A. B., and Putney, J. W. Jr. (2005). Store-operated calcium channels. Physiol. Rev. 85, 757–810. doi: 10.1152/physrev.00057.2003
Perano, S. J., Rayner, C. K., Kritas, S., Horowitz, M., Donaghue, K., Mpundu-Kaambwa, C., et al. (2015). Gastric Emptying Is More Rapid in Adolescents With Type 1 Diabetes and Impacts on Postprandial Glycemia. J. Clin. Endocrinol. Metab. 100, 2248–2253. doi: 10.1210/jc.2015-1055
Potier, M., Gonzalez, J. C., Motiani, R. K., Abdullaev, I. F., Bisaillon, J. M., Singer, H. A., et al. (2009). Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J. 23, 2425–2437. doi: 10.1096/fj.09-131128
Prakriya, M., Feske, S., Gwack, Y., Srikanth, S., Rao, A., and Hogan, P. G. (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. doi: 10.1038/nature05122
Putney, J. W. (2011). Origins of the concept of store-operated calcium entry. Front. Biosci. 3, 980–984. doi: 10.2741/202
Rosa-e-Silva, L., Troncon, L. E., Oliveira, R. B., Foss, M. C., Braga, F. J., and Gallo Junior, L. (1996). Rapid distal small bowel transit associated with sympathetic denervation in type I diabetes mellitus. Gut 39, 748–756.
Sang, L. J., Ju, H. Q., Liu, G. P., Tian, T., Ma, G. L., Lu, Y. X., et al. (2018). LncRNA CamK-A Regulates Ca(2+)-Signaling-Mediated Tumor Microenvironment Remodeling. Mol. Cell 72, 71–83e77. doi: 10.1016/j.molcel.2018.08.014
Selim, M. M., Wendelschafer-Crabb, G., Redmon, J. B., Khoruts, A., Hodges, J. S., Koch, K., et al. (2010). Gastric mucosal nerve density: a biomarker for diabetic autonomic neuropathy? Neurology 75, 973–981. doi: 10.1212/WNL.0b013e3181f25f19
Soboloff, J., Spassova, M. A., Tang, X. D., Hewavitharana, T., Xu, W., and Gill, D. L. (2006). Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 281, 20661–20665. doi: 10.1074/jbc.C600126200
Song, K., Zhong, X. G., Xia, X. M., Huang, J. H., Fan, Y. F., Yuan, R. X., et al. (2015). Orai1 forms a signal complex with SK3 channel in gallbladder smooth muscle. Biochem. Biophys. Res. Commun. 466, 456–462. doi: 10.1016/j.bbrc.2015.09.049
Stevens, J. E., Jones, K. L., Rayner, C. K., and Horowitz, M. (2013). Pathophysiology and pharmacotherapy of gastroparesis: current and future perspectives. Expert Opin. Pharmacother. 14, 1171–1186. doi: 10.1517/14656566.2013.795948
Sweeney, M., McDaniel, S. S., Platoshyn, O., Zhang, S., Yu, Y., Lapp, B. R., et al. (2002). Role of capacitative Ca2+ entry in bronchial contraction and remodeling. J. Appl. Physiol. 92, 1594–1602. doi: 10.1152/japplphysiol.00722.2001
Tamareille, S., Mignen, O., Capiod, T., Rucker-Martin, C., and Feuvray, D. (2006). High glucose-induced apoptosis through store-operated calcium entry and calcineurin in human umbilical vein endothelial cells. Cell Calcium 39, 47–55. doi: 10.1016/j.ceca.2005.09.008
Umathe, S. N., Kochar, N. I., Jain, N. S., and Dixit, P. V. (2009). Gastrointestinal dysfunction in diabetic rats relates with a decline in tissue L-arginine content and consequent low levels of nitric oxide. Nitric Oxide 20, 129–133. doi: 10.1016/j.niox.2008.10.007
Uranga-Ocio, J. A., Bastus-Diez, S., Delkader-Palacios, D., Garcia-Cristobal, N., Leal-Garcia, M. A., and Abalo-Delgado, R. (2015). Enteric neuropathy associated to diabetes mellitus. Rev. Esp. Enferm Dig. 107, 366–373.
Voelkers, M., Salz, M., Herzog, N., Frank, D., Dolatabadi, N., Frey, N., et al. (2010). Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J. Mol. Cell Cardiol. 48, 1329–1334. doi: 10.1016/j.yjmcc.2010.01.020
Yarandi, S. S., and Srinivasan, S. (2014). Diabetic gastrointestinal motility disorders and the role of enteric nervous system: current status and future directions. Neurogastroenterol. Motil. 26, 611–624. doi: 10.1111/nmo.12330
Yin, L., Vijaygopal, P., MacGregor, G. G., Menon, R., Ranganathan, P., Prabhakaran, S., et al. (2014). Glucose stimulates calcium-activated chloride secretion in small intestinal cells. Am. J. Physiol. Cell Physiol. 306, C687–C696. doi: 10.1152/ajpcell.00174.2013
Yuece, B., Sibaev, A., Broedl, U. C., Marsicano, G., Goke, B., Lutz, B., et al. (2007). Cannabinoid type 1 receptor modulates intestinal propulsion by an attenuation of intestinal motor responses within the myenteric part of the peristaltic reflex. Neurogastroenterol. Motil. 19, 744–753. doi: 10.1111/j.1365-2982.2007.00975.x
Zhao, J., Frokjaer, J. B., Drewes, A. M., and Ejskjaer, N. (2006). Upper gastrointestinal sensory-motor dysfunction in diabetes mellitus. World J. Gastroenterol. 12, 2846–2857.
Keywords: type 1 diabetes, small bowel transit, store-operated Ca2+ entry, Orai1, Ca2+-activated K+ channel, small intestinal smooth muscle
Citation: Dai F, Guo J, Wang Y, Jiang T, Chen H, Hu Y, Du J, Xia X, Zhang Q and Shen B (2021) Enhanced Store-Operated Ca2+ Signal of Small Intestinal Smooth Muscle Cells Accelerates Small Bowel Transit Speed in Type 1 Diabetic Mouse. Front. Physiol. 12:691867. doi: 10.3389/fphys.2021.691867
Received: 07 April 2021; Accepted: 27 September 2021;
Published: 20 October 2021.
Edited by:
Sal A. Baker, University of Nevada, Reno, United StatesReviewed by:
Zui Pan, University of Texas at Arlington, United StatesScott D. Zawieja, University of Missouri, United States
Copyright © 2021 Dai, Guo, Wang, Jiang, Chen, Hu, Du, Xia, Zhang and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bing Shen, c2hlbmJpbmdAYWhtdS5lZHUuY24=; Qiu Zhang, YXluZm1rQDE2My5jb20=
†These authors have contributed equally to this work