 Rahul Mittal
Rahul Mittal Danay Saavedra
Danay Saavedra Mannat Mittal1
Mannat Mittal1 Khemraj Hirani
Khemraj Hirani- 1Diabetes Research Institute, University of Miami Miller School of Medicine, Miami, Florida, FL, United States
- 2Division of Endocrinology, Diabetes, and Metabolism, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida, FL, United States
Autoimmune diseases, particularly those with early onset such as systemic lupus erythematosus, juvenile idiopathic arthritis, and type 1 diabetes, are paradoxically characterized by molecular and cellular features typically associated with aging. These include telomere shortening, mitochondrial dysfunction, epigenetic alterations, and skewed immune cell phenotypes, which are considered hallmarks of immunosenescence. This perspective explores the hypothesis that aberrant inflammasome activation, particularly of the NLRP3 complex, serves as a key upstream driver of premature immune aging in autoimmunity. We examine how chronic inflammasome signaling induces senescence through pro-inflammatory cytokine production and oxidative stress, reinforces the senescence-associated secretory phenotype (SASP), and perpetuates immune dysregulation. By reframing autoimmunity as a disorder of accelerated immune aging, we highlight emerging opportunities for therapeutic intervention using senolytics, inflammasome inhibitors, and lifestyle modifications. In addition, incorporating biomarkers of immune aging into clinical assessment may enable precision immunogerontology, particularly in pediatric populations where biological and chronological age may be dissociated. Elucidating the relationship between inflammasome signaling and immune senescence provides a critical framework for understanding autoimmune pathogenesis and for developing interventions that modify disease course by targeting age-associated mechanisms.
1 Introduction
Autoimmune diseases are commonly characterized by a breakdown in immune tolerance and persistent activation of inflammatory pathways (Hoi et al., 2024; DiMeglio et al., 2018; Siegel and Sammaritano, 2024; Quattrin et al., 2023; Mittal et al., 2024; Mittal et al., 2025). Although these conditions can affect individuals across the lifespan, a significant proportion of cases, including systemic lupus erythematosus (SLE), juvenile idiopathic arthritis (JIA), and type 1 diabetes (T1D), begin early in life (Pan et al., 2023; Boteanu et al., 2025). This early onset is paradoxical, as affected individuals often exhibit molecular and cellular features typically associated with an aged immune system (van den Hoogen et al., 2015; Tsai et al., 2019; Handono et al., 2021; Mayerl and Prelog, 2012; Shapiro et al., 2023).
In physiological aging, the immune system undergoes a process known as immunosenescence, which encompasses several functional and structural alterations. These include reduced thymic output of naïve T cells, accumulation of terminally differentiated and exhausted lymphocytes, impaired antigen responsiveness, and a chronic low-grade inflammatory state referred to as “inflammaging” (Figure 1) (Teissier et al., 2022; Liao et al., 2022; Li X. et al., 2023). Surprisingly, many of these hallmarks are also observed in young individuals with autoimmune disease. Pediatric and adolescent patients with SLE have been shown to exhibit shortened telomeres, mitochondrial dysfunction, epigenetic alterations, and elevated expression of cell cycle inhibitors such as p16INK4a and p21CIP1 (Tsai et al., 2019; Melk et al., 2005; Yang et al., 2018; Gao et al., 2019; Kurosaka et al., 2006; Haque et al., 2013). Additionally, immune cell compartments in these individuals often display skewing toward memory and senescent phenotypes, resembling the profile seen in elderly populations (Li et al., 2022; Tan et al., 2024; Rother and van der Vlag, 2015; Kalim et al., 2021). Functionally, immune senescence results in impaired immune surveillance, reduced vaccine efficacy, and heightened susceptibility to infections (Allen et al., 2020). Importantly, senescent cells, despite being metabolically active, are often dysfunctional, contributing to tissue damage and perpetuating systemic inflammation (Tripathi et al., 2021; Palmer et al., 2022).
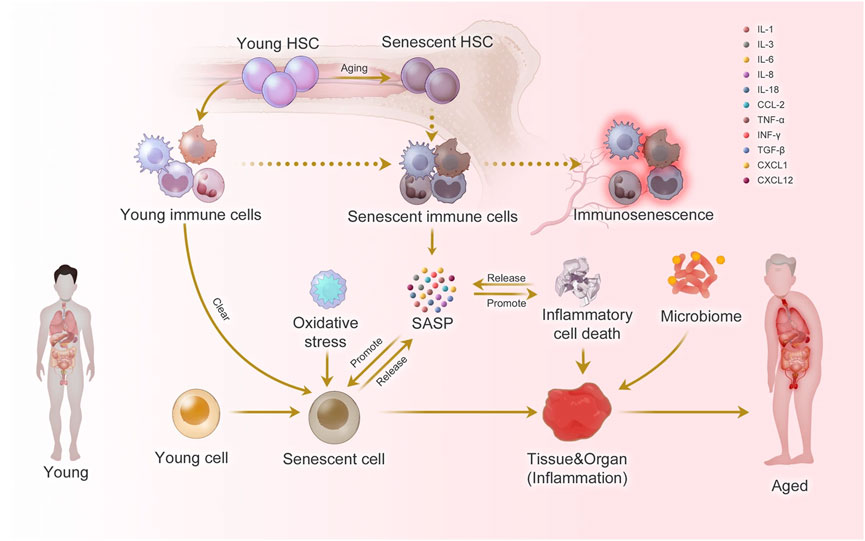
Figure 1. Inflammaging at the molecular, cellular, and organ levels. During the aging process, almost all cells in the body undergo senescence, a state characterized by a dysfunctional state and senescence-associated secretory phenotype (SASP). While immune cells play a crucial role in recognizing and eliminating these senescent cells, they are also affected by SASP, leading to a phenomenon called immunosenescence. Immunosenescence can impair the immunity to respond to infections and diseases, making the organism more vulnerable to illnesses. Moreover, the accumulation of senescent cells can trigger inflammation in organs, leading to organ damage and an increased risk of age-related diseases. This process is exacerbated by positive feedback loops that drive the accumulation of inflammation and organ damage, leading to further inflammation and an even higher risk of aging-related diseases. Taken from (Li X. et al., 2023) under a Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
These observations suggest that premature immune aging may be a fundamental feature of autoimmunity, rather than a secondary consequence of chronic inflammation or immunosuppressive therapy. However, the mechanistic basis for this accelerated senescence remains incompletely understood. Among the potential upstream drivers is aberrant activation of the inflammasome complex, particularly the NLRP3 inflammasome (Figure 2) (Kelley et al., 2019; Paik et al., 2021; Seoane et al., 2020). Inflammasome activation results in the maturation and secretion of interleukin-1β (IL-1β) and interleukin-18 (IL-18), cytokines known to induce oxidative stress, DNA damage, and pro-senescent signaling pathways in both immune and non-immune cells (Yao et al., 2024; Li H. et al., 2023).
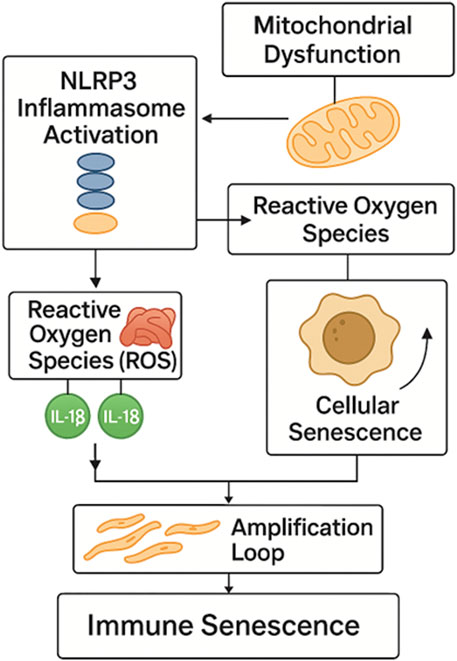
Figure 2. NLRP3 inflammasome-driven pathway to immune senescence. Activation of the NLRP3 inflammasome induces mitochondrial dysfunction and ROS generation, leading to IL-1β and IL-18 release. These cytokines promote cellular senescence and SASP amplification, establishing a feed-forward loop that drives premature immune aging in autoimmunity. NLRP3 - NOD-like receptor family pyrin domain containing 3; ROS - Reactive Oxygen Species; IL-1β - Interleukin-1 beta; IL-18 - Interleukin-18; SASP - Senescence-Associated Secretory P.henotype.
Despite advances in immunosenescence research, direct, within-patient evidence linking NLRP3 inflammasome activation with cellular senescence in autoimmune diseases remains limited. Most available data do not derive from paired analyses that simultaneously assess inflammasome activation and senescence markers within the same cellular populations or across the same individuals. Instead, the proposed link between NLRP3 activity and senescence in human autoimmunity is largely inferred. Current understanding is primarily based on converging but separate lines of evidence, such as animal models, mechanistic cell-based studies, and independent clinical observations of senescence-associated signatures and inflammasome activity. As a result, the hypothesized NLRP3–senescence axis remains a compelling but as yet unproven framework in the context of robust human clinical cohorts, highlighting the need for further longitudinal and mechanistically integrated studies to establish causality.
This perspective article aims to explore the hypothesis that inflammasome activation may act as a central mechanism linking autoimmunity to accelerated immune aging. We synthetize current evidence linking chronic inflammasome signaling to the emergence of senescence-associated phenotypes in young patients, emphasizing mechanistic pathways and feedback loops that sustain immune dysfunction. Furthermore, we discuss the implications of this relationship for understanding disease pathogenesis and for developing therapeutic strategies that target the intersection of inflammation and immune aging, with particular attention to early-onset autoimmunity.
2 Inflammasomes as accelerators of immune senescence
2.1 Mechanistic link
Inflammasomes are cytosolic multiprotein complexes that serve as innate immune sensors, detecting pathogen-associated molecular patterns and damage-associated molecular patterns. Among them, the NLRP3 inflammasome is the most extensively studied and is activated by a wide range of stimuli, including mitochondrial dysfunction, extracellular ATP, crystalline substances, and various environmental stressors (Kelley et al., 2019; Paik et al., 2021; Seoane et al., 2020).
Upon activation, the NLRP3 inflammasome facilitates the cleavage of pro–caspase-1 into its active form, caspase-1 (Kelley et al., 2019; Paik et al., 2021; Seoane et al., 2020). Active caspase-1 subsequently processes pro–interleukin-1β (IL-1β) and pro–interleukin-18 (IL-18) into their mature, secreted forms (Kelley et al., 2019; Paik et al., 2021; Seoane et al., 2020). Sustained production of IL-1β and IL-18 results in chronic inflammatory signaling, which promotes the generation of reactive oxygen species (ROS), loss of mitochondrial membrane potential, and accumulation of nuclear DNA damage (Harijith et al., 2014; Patergnani et al., 2021; Hong et al., 2024; Checa and Aran, 2020). These intracellular stressors contribute directly to the induction of cellular senescence pathways, including the activation of p53/p21 and p16/Rb signaling cascades, which enforce irreversible cell cycle arrest.
Mitochondrial dysfunction, a central feature of senescent cells, also serves to propagate inflammasome activity (Wei et al., 2025; Vasileiou et al., 2019; Picca et al., 2017). Oxidized mitochondrial DNA released into the cytosol can further stimulate NLRP3, thereby creating a feed-forward loop that perpetuates inflammasome activation and inflammatory cytokine production (Zhang X. et al., 2025). This bidirectional relationship establishes a cellular environment highly conducive to senescence induction, particularly within immune and barrier tissue compartments.
Pharmacological or genetic inhibition of NLRP3 or downstream cytokine pathways has been shown to attenuate these senescence-associated features in multiple experimental models (Marín-Aguilar et al., 2020; Cañadas-Lozano et al., 2020; Navarro-Pando et al., 2021; Quijano et al., 2022). These findings suggest a causal role for inflammasome signaling in promoting and maintaining the senescent phenotype.
2.2 SASP and feedback loops
Senescent cells develop a complex pro-inflammatory secretome known as the senescence-associated secretory phenotype, or SASP (Wang B. et al., 2024; Kuma et al., 2021; Roger et al., 2021). This secretory profile includes a broad spectrum of cytokines, chemokines, matrix metalloproteinases, and growth factors, many of which are directly or indirectly regulated by IL-1β signaling (Lopes-Paciencia et al., 2019). Inflammasome activation amplifies the SASP through autocrine and paracrine mechanisms (Yue et al., 2022). IL-1β produced via inflammasome signaling activates NF-κB and C/EBPβ transcriptional programs, which in turn promote the expression of additional SASP components such as IL-6 and IL-8 (Kelley et al., 2019; Paik et al., 2021; Seoane et al., 2020). This creates a self-reinforcing loop in which inflammasome-derived cytokines potentiate further SASP production and sustain the senescent state (Fu et al., 2025). SASP factors exert significant bystander effects, as they can induce DNA damage, oxidative stress, and inflammatory signaling in neighboring cells, thereby spreading senescence beyond the initially affected population (Han et al., 2024; Ohtani, 2022). This propagation of the senescent phenotype contributes to tissue dysfunction and systemic inflammation, hallmarks of immune aging.
In immune cells, the persistent activation of inflammasomes and sustained exposure to SASP components alter cell fate and function (Kuma et al., 2021; Sebastian-Valverde and Pasinetti, 2020; Latz and Duewell, 2018; Meyers and Zhu, 2020; Muela-Zarzuela et al., 2024). Naïve and memory T-cell pools become skewed toward terminal differentiation and exhaustion, B cells exhibit reduced antigen specificity and increased autoreactivity, and myeloid cells adopt a pro-inflammatory, senescent-like phenotype (Gritsenko et al., 2020; Liu et al., 2023; Zheng et al., 2023; Zhao et al., 2022; Camell et al., 2019; Ma et al., 2019; Wang L. et al., 2024; Zhu et al., 2023; Behmoaras and Gil, 2021; Hu et al., 2019). Within the stromal and epithelial compartments, SASP and inflammasome activity disrupt tissue architecture and impair regenerative capacity (Wan et al., 2021; Paez-Ribes et al., 2019).
Together, these mechanisms illustrate how inflammasome activation not only induces cellular senescence but also stabilizes and expands it through SASP amplification and inflammatory feedback. The integration of these pathways is a central feature of accelerated immune aging in the context of chronic inflammation and autoimmunity.
3 Early-onset inflammaging in autoimmunity
The term “inflammaging” refers to a chronic, low-grade, sterile inflammatory state that gradually develops with advancing age and is considered a hallmark of immunosenescence (Franceschi et al., 2018; Fulop et al., 2023; Franceschi and Campisi, 2014). It is characterized by persistent elevation of pro-inflammatory mediators such as interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and C-reactive protein (CRP), occurring in the absence of overt infection (Fülöp et al., 2019). This inflammatory environment is sustained by lifelong exposure to antigenic stimuli, cumulative tissue damage, impaired autophagy, mitochondrial dysfunction, and the accumulation of senescent cells that secrete pro-inflammatory factors (Ajoolabady et al., 2024).
Although inflammaging was originally conceptualized in the context of geriatric biology, emerging evidence indicates that many of its immunologic, molecular, and metabolic features are present in young individuals with autoimmune diseases (Franceschi et al., 2025). Early-onset autoimmune conditions such as SLE, JIA, and T1D display an inflammatory milieu that phenocopies the immune landscape of aged individuals (Frazzei et al., 2022; Goronzy and Weyand, 2012). In these patients, inflammatory biomarkers remain elevated even during periods of clinical remission, suggesting a constitutive and potentially irreversible rewiring of immune regulation toward a pro-inflammatory state. This early-onset inflammaging is not merely a reflection of disease activity but may instead constitute a foundational process driving disease pathogenesis and progression.
3.1 Immune cell rewiring
One of the central consequences of early inflammaging in autoimmunity is the reprogramming of immune cell lineages toward pro-inflammatory, senescent-like phenotypes (Alexander et al., 2024). Chronic activation of inflammasome complexes, particularly NLRP3, promotes sustained production of IL-1β and IL-18, which act on both adaptive and innate immune compartments (Kelley et al., 2019). In T cells, prolonged exposure to inflammatory cytokines alters differentiation trajectories, favoring the expansion of effector memory (Tem) and terminally differentiated effector memory RA (TEMRA) populations at the expense of the naïve T-cell pool (Kim and Harty, 2014; Raeber et al., 2018; Fritsch et al., 2006; Skapenko et al., 1999). These terminal subsets exhibit reduced proliferative potential, shortened telomeres, impaired synapse formation, and a biased cytokine profile skewed toward interferon-gamma and cytotoxic granule production (Ohmes et al., 2022).
CD8+ T cells lacking the co-stimulatory molecule CD28, a well-established marker of replicative senescence, are frequently expanded in young autoimmune patients and contribute to tissue damage through cytotoxic and inflammatory mechanisms (Żabińska et al., 2016; Minning et al., 2019). Similarly, CD4+ T cells demonstrate increased expression of PD-1 and other exhaustion markers, consistent with chronic antigen stimulation and senescence-associated dysfunction (Fischer et al., 2022; Saggau et al., 2024; Koutsonikoli et al., 2024).
B cells are also affected by this rewiring. Chronic inflammatory signaling, particularly via IL-1β and IL-6, disrupts central and peripheral tolerance checkpoints, promoting the survival of autoreactive clones (Maeda et al., 2010; Nicholas et al., 2025; Canny and Jackson, 2021; Arkatkar et al., 2017; Nagafuchi et al., 1993). These B cells often acquire a hyperactive phenotype, with increased spontaneous antibody production, elevated class switching, and loss of regulatory B-cell subsets (Iwata et al., 2023). In parallel, age-associated B cells (ABCs), a population initially characterized in aged mice and humans, are found at higher frequencies in young individuals with autoimmune disease (Mouat et al., 2022; Li Z. Y. et al., 2023; Wehr et al., 2004; Cancro, 2020; Xie et al., 2025; Nickerson et al., 2023; Su et al., 2025; Miranda-Prieto et al., 2025; Sachinidis et al., 2025). These ABCs exhibit pro-inflammatory characteristics and are potent antigen-presenting cells, further perpetuating autoreactivity (Ratliff et al., 2013; Riley et al., 2017; Qin et al., 2022).
3.2 Epigenetic and metabolic signatures
In addition to phenotypic and functional changes, early-onset inflammaging in autoimmunity is accompanied by alterations in the epigenetic and metabolic landscapes of immune cells (Nardini et al., 2018). Epigenetic aging, assessed through DNA methylation clocks, has been shown to be accelerated in patients with autoimmune diseases (Somers et al., 2024; Pérez et al., 2020). These findings suggest that chronic immune activation may impose an age-related epigenetic program on hematopoietic and immune progenitors. Methylation changes are particularly enriched in promoters of genes involved in cytokine signaling, antigen presentation, and lymphocyte differentiation (Morales-Nebreda et al., 2019; Calle-Fabregat et al., 2020). These modifications may serve as both biomarkers and mediators of inflammaging, reinforcing transcriptional programs that sustain inflammatory signaling and senescence.
Metabolic reprogramming is another key feature of early inflammaging. Immune cells from autoimmune patients often shift from oxidative phosphorylation to lactate-producing glycolysis, even in the presence of oxygen, a phenomenon known as the Warburg effect (Granados et al., 2017; Mocholi et al., 2025; Takeshima et al., 2019). This shift is typically associated with activation and effector differentiation, but when sustained, it contributes to mitochondrial dysfunction, excessive ROS production, and metabolic stress (Palmer et al., 2015; Yennemadi et al., 2023; Gergely et al., 2002). Furthermore, impaired mitophagy and accumulation of dysfunctional mitochondria serve as endogenous activators of inflammasomes, creating a positive feedback loop between metabolic stress and inflammatory signaling (Yuk et al., 2020; Mishra et al., 2021; Kim et al., 2016; Liu et al., 2018).
Together, these epigenetic and metabolic signatures reflect a biologically aged immune phenotype in individuals with early-onset autoimmunity. Importantly, they support the hypothesis that immune aging processes, once thought to be restricted to the elderly, are operational much earlier in life in the context of chronic auto-inflammatory disease.
4 Clinical and translational implications
Conventional clinical paradigms often use chronological age as a primary determinant of immune status, disease prognosis, and therapeutic strategy. However, in the context of autoimmune diseases that manifest during childhood or early adulthood, this approach may be inadequate. Evidence of premature immune senescence in young patients suggests that biological age, rather than chronological age, may be a more accurate indicator of disease severity, immunologic competence, and long-term outcomes. Recognition of this discrepancy has important implications for personalized medicine, as it calls for an age-stratified model based not solely on calendar age but also on molecular and cellular indicators of immune aging. Integrating biomarkers of senescence and inflammaging into clinical practice could enable earlier identification of high-risk patients and guide the selection of targeted interventions aimed at modulating the underlying age-related mechanisms contributing to autoimmunity.
4.1 Therapeutic targeting opportunities
The identification of senescence and inflammasome activation as key drivers of early-onset autoimmunity opens new avenues for therapeutic intervention (Supplementary Table 1). One promising approach involves the use of senolytic agents, which selectively eliminate senescent cells, and SASP modulators, which suppress the deleterious secretory phenotype associated with these cells (Shao et al., 2021; Chen et al., 2025; Atlante et al., 2025; Zhang M. et al., 2025; Kim et al., 2025). Although originally developed for geriatric applications, these compounds may have utility in younger patients with autoimmune disease who exhibit premature immune aging.
Another class of therapeutic agents with potential relevance is inflammasome inhibitors. Pharmacological blockade of the NLRP3 inflammasome or its downstream effectors, such as interleukin-1β and interleukin-18, has demonstrated efficacy in preclinical models of autoimmune and inflammatory disease. These agents may act not only to suppress acute inflammation but also to decelerate immune aging processes by disrupting the feed-forward loops that maintain chronic inflammasome activation and cellular senescence.
In addition to pharmacologic strategies, lifestyle-based interventions may offer adjunctive benefit by modulating immune aging. Regular physical activity, dietary interventions, and caloric restriction mimetics have been shown to attenuate systemic inflammation and support mitochondrial function (Abad-Jiménez et al., 2024; Vitetta and Anton, 2007; Gabandé-Rodríguez et al., 2019; Kyriazis et al., 2022). When combined with molecularly targeted therapies, these interventions may produce synergistic effects, improving immune resilience and altering disease trajectory in individuals with early signs of immune senescence.
While the targeting of senescent cells holds promise for mitigating age-related dysfunction and chronic inflammation, it is important to recognize potential risks. Senescent cells may also play context dependent roles in tissue repair, remodeling, and the maintenance of homeostasis (Faust et al., 2020; Gadecka et al., 2025; Demaria et al., 2014). Broad elimination or functional modulation of these cells could therefore disrupt beneficial processes, impair regenerative capacity, or compromise immune surveillance. These considerations highlight the need for approaches that achieve sufficient selectivity, whether by context, timing, or cell type, to minimize unintended consequences while preserving physiological functions.
4.2 Biomarker development for risk stratification and monitoring
The integration of biomarkers that reflect immune aging into clinical assessment could transform the management of autoimmune disease (Supplementary Table 2). Telomere length, expression of senescence-associated genes such as p16INK4a and p21CIP1, and levels of SASP components including IL-6, IL-1β, and matrix metalloproteinases offer potential as diagnostic and prognostic tools. Additionally, quantification of inflammasome components, including NLRP3 expression and caspase-1 activity, may provide insight into the inflammatory status of immune cells and their propensity to enter senescence.
These biomarkers could be employed to stratify patients based on biological age and inflammaging burden, enabling more precise prediction of disease course and responsiveness to immune-modifying therapies. Longitudinal monitoring of these parameters could also help assess therapeutic efficacy, particularly in interventions aimed at mitigating immune senescence or targeting the inflammasome-senescence axis.
4.3 Age-specific considerations
Age-specific considerations are critical when evaluating inflammasome-targeted therapies, as efficacy and safety profiles established in adults cannot be assumed to directly translate to pediatric populations. Importantly, therapeutic targeting of the inflammasome pathway is already feasible in pediatric autoimmunity. IL-1 blockers, such as anakinra, have demonstrated efficacy and safety in children with systemic juvenile idiopathic arthritis, including robust long-term data (Quartier et al., 2011; Giancane et al., 2022) Canakinumab has induced sustained remissions in pediatric CAPS with favorable multi-year safety outcomes (Kuemmerle-Deschner et al., 2011; Brogan et al., 2019). By contrast, direct oral NLRP3 inhibitors (dapansutrile/OLT1177) remain limited to adult clinical data in conditions such as gout and heart failure (Klughammer et al., 2023; Terkeltaub et al., 2019; Wohlford et al., 2020). Thus, pediatric translation of such agents will require dedicated studies addressing dosing, growth, and infection risk.
To date, no clinical trials have evaluated senolytic therapies such as dasatinib plus quercetin or fisetin in pediatric populations for the management of immune aging or autoimmunity. Cellular senescence contributes to developmental processes, tissue repair, and neurodevelopment, while preservation of vaccine responsiveness and normal growth remains critical in children. These considerations argue against indiscriminate senescent cell clearance in this age group. Accordingly, senescence-targeted interventions should be restricted to rigorously controlled research settings. Within such contexts, current best practice favors reversible modulation of SASP rather than broad elimination of senescent cells.
5 Future directions
5.1 Integrated profiling of immune senescence and inflammasome activity
A major priority for advancing the understanding of early-onset autoimmunity involves the implementation of longitudinal studies that systematically characterize immune aging across disease progression. These studies should incorporate multi-parametric immune phenotyping, quantitative assessment of senescence-associated biomarkers, and detailed profiling of inflammasome activity. Pediatric and adolescent populations represent a critical cohort for such investigations, as early disease stages may reveal mechanistic insights into the initial triggers of immune senescence and chronic inflammation. The integration of single-cell transcriptomics, epigenetic landscape mapping, proteomics, and metabolomics will provide a comprehensive framework to define immune trajectories and identify inflection points predictive of disease acceleration or remission.
By correlating molecular signatures with clinical phenotypes, it may become possible to identify distinct biological aging patterns that underlie disease heterogeneity, inform therapeutic response, and facilitate the development of predictive tools for early intervention.
5.2 Toward precision immunogerontology
The concept of precision immunogerontology emphasizes the need to tailor interventions based on biological immune age rather than relying solely on chronological metrics. In the context of autoimmune disease, this approach requires stratification of patients according to validated biomarkers of immune senescence, inflammaging intensity, and functional immune reserve. Personalized immunogerontologic strategies could involve dynamic modulation of senescence-associated signaling pathways, selective depletion of senescent cells, or restoration of mitochondrial and epigenetic integrity in immune progenitors.
Advancing this framework will require the development of clinical algorithms that integrate molecular diagnostics with real-time immune monitoring. These tools will support individualized treatment decisions and enable risk-adjusted strategies to prevent disease progression or recurrence. Ultimately, precision immunogerontology may redefine therapeutic windows and optimize long-term outcomes for patients with early-onset autoimmune disorders.
5.3 Bridging the fields of immunology and aging research
The intersection between immunology and aging biology remains underexplored, particularly in the setting of autoimmunity. Promoting interdisciplinary collaboration between researchers in cellular senescence, mitochondrial biology, epigenetics, and immune regulation is essential to uncover convergent mechanisms that drive immune dysfunction across the lifespan. Shared platforms for data integration and cross-disciplinary communication will accelerate the translation of basic science discoveries into therapeutic applications.
Additionally, fostering collaborative consortia that include pediatric rheumatologists, geroscientists, computational biologists, and clinical trialists will enhance the design of studies that address the complex interplay between immune development, senescence, and inflammation. Such efforts will be crucial to identify early biomarkers of immune aging, establish causal relationships between inflammasome activity and senescence, and validate age-modifying interventions for autoimmune disease.
6 Discussion
The convergence of autoimmunity and premature immune aging represents a paradigm shift in our understanding of disease initiation and progression in young patients. While traditionally framed as pathologies of immune hyperactivity, autoimmune diseases are increasingly recognized to embody features of immune senescence typically reserved for advanced age. The co-occurrence of telomere attrition, diminished thymic output, mitochondrial dysfunction, and a proinflammatory cytokine milieu in young autoimmune cohorts highlight a biologically aged immune landscape occurring decades earlier than anticipated. At the center of this shift lies inflammasome biology. The NLRP3 inflammasome, in particular, emerges not merely as a mediator of acute inflammation but as a pivotal orchestrator of immune aging. Its activation instigates a cascade of intracellular events including mitochondrial impairment, reactive oxygen species accumulation, and DNA instability that converge on classical senescence pathways. The downstream elaboration of interleukin 1 beta and interleukin 18 consolidates this trajectory by reinforcing the senescence associated secretory phenotype, which extends proaging signals across tissue compartments through paracrine amplification. Thus, inflammasome signaling constitutes both a precipitant and perpetuator of immune dysfunction.
Bidirectional interactions between mitochondrial dysfunction and NLRP3 inflammasome activation are now well-established (Qiu et al., 2022; Pan et al., 2018; Billingham et al., 2022; Bronner et al., 2015; He et al., 2025; Ning et al., 2025; Wu et al., 2025; Huang et al., 2025). Damaged or oxidized mitochondrial DNA (ox-mtDNA) released into the cytosol activates NLRP3, while inhibition of mitophagy or augmented mitochondrial ROS amplifies inflammasome assembly and IL-1β secretion (Xian et al., 2025; Shimada et al., 2012; Zhong et al., 2018; Nakahira et al., 2011; Zhou et al., 2011; Iyer et al., 2013; Heid et al., 2013). Studies have shown that ox-mtDNA can exit via mitochondrial permeability transition pore (mPTP) or VDAC channels, activating both NLRP3 and interferon responses (Xian et al., 2022; Kim et al., 2019; Baik et al., 2023). Conversely, inflammasome activation can exacerbate mitochondrial injury (Yu et al., 2014). Gasdermin-D, downstream of inflammasome activation, forms pores that compromise mitochondrial integrity, contributing to mitochondrial dysfunction during pyroptosis (Miao et al., 2023; Tang et al., 2024; de Torre-Minguela et al., 2021). Thus, a feed-forward loop exists in which mitochondrial stress primes NLRP3 activation, and inflammasome-mediated pyroptotic processes in turn worsen mitochondrial health, consistent with mechanisms underlying “inflammaging” in autoimmunity.
Crucially, the immunosenescent profile observed in youth is not merely epiphenomenal. Mounting data implicates these cellular alterations in the etiology of autoimmune pathogenesis itself. The persistence of senescent T and B cell subsets, impaired tolerance checkpoints, and the emergence of proinflammatory myeloid phenotypes reflect a systemic remodeling of immunity that precedes and sustains autoimmunity. In this light, biological aging of the immune system is not an outcome but a driver of disease. This framework mandates a reassessment of how age is conceptualized in autoimmune disease. Chronological age is no longer a sufficient proxy for immune competence or resilience. Instead, biologically grounded metrics including markers of cellular senescence, inflammasome activity, and epigenetic age offer a more precise approach through which to interpret disease severity, progression, and therapeutic responsiveness.
Therapeutic implications are equally profound. Targeting the inflammasome senescence axis may yield interventions that not only suppress inflammation but recalibrate immune aging itself. Agents that abrogate inflammasome activation, modulate senescence associated secretory activity, or selectively eliminate senescent immune populations represent rational strategies for disrupting the self-amplifying cycle of immune decay. When complemented by metabolic and lifestyle interventions aimed at restoring mitochondrial function and reducing oxidative load, these strategies offer a multifaceted approach to restoring immune homeostasis. Future studies should focus on delineating the temporal sequence linking inflammasome activity with senescence onset in autoimmunity. Disentangling cause from consequence will require longitudinal designs and integrated omics profiling, particularly in early disease stages. Collaborative efforts spanning immunology, geroscience, and systems biology will be essential to operationalize biological age as a clinically actionable variable.
Reframing autoimmunity as a disorder of accelerated immune aging opens a new frontier in which pediatric and adolescent patients are not exceptions to the aging paradigm but key to understanding its pathological extremes. This reconceptualization holds promise for transforming how autoimmune diseases are detected, stratified, and treated across the lifespan.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
RM: Formal Analysis, Visualization, Data curation, Project administration, Validation, Methodology, Writing – review and editing, Supervision, Investigation, Writing – original draft, Conceptualization. DS: Investigation, Writing – original draft, Formal Analysis, Validation, Methodology, Conceptualization, Writing – review and editing. MM: Conceptualization, Investigation, Data curation, Validation, Writing – review and editing, Writing – original draft. JL: Conceptualization, Data curation, Formal Analysis, Validation, Methodology, Writing – review and editing, Investigation, Writing – original draft. KH: Methodology, Writing – review and editing, Investigation, Writing – original draft, Supervision, Conceptualization, Validation, Resources, Formal Analysis, Project administration, Data curation.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We are grateful to Valerie Gramling for critical reading of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fragi.2025.1688060/full#supplementary-material
References
Abad-Jiménez, Z., López-Domènech, S., Pelechá, M., Perea-Galera, L., Rovira-Llopis, S., Bañuls, C., et al. (2024). Calorie restriction modulates mitochondrial dynamics and autophagy in leukocytes of patients with obesity. Free Radic. Biol. Med. 225, 677–686. doi:10.1016/j.freeradbiomed.2024.10.295
Ajoolabady, A., Pratico, D., Tang, D., Zhou, S., Franceschi, C., and Ren, J. (2024). Immunosenescence and inflammaging: mechanisms and role in diseases. Ageing Res. Rev. 101, 102540. doi:10.1016/j.arr.2024.102540
Alexander, M., Cho, E., Gliozheni, E., Salem, Y., Cheung, J., and Ichii, H. (2024). Pathology of diabetes-induced immune dysfunction. Int. J. Mol. Sci. 25 (13), 7105. doi:10.3390/ijms25137105
Allen, J. C., Toapanta, F. R., Chen, W., and Tennant, S. M. (2020). Understanding immunosenescence and its impact on vaccination of older adults. Vaccine 38 (52), 8264–8272. doi:10.1016/j.vaccine.2020.11.002
Arkatkar, T., Du, S. W., Jacobs, H. M., Dam, E. M., Hou, B., Buckner, J. H., et al. (2017). B cell-derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J. Exp. Med. 214 (11), 3207–3217. doi:10.1084/jem.20170580
Atlante, S., Gottardi Zamperla, M., Cis, L., Farsetti, A., and Gaetano, C. (2025). Senolytic therapies for cardiovascular aging: tackling fibrosis and metabolic dysfunction. Eur. J. Intern Med., 106413. doi:10.1016/j.ejim.2025.07.009
Baik, S. H., Ramanujan, V. K., Becker, C., Fett, S., Underhill, D. M., and Wolf, A. J. (2023). Hexokinase dissociation from mitochondria promotes oligomerization of VDAC that facilitates NLRP3 inflammasome assembly and activation. Sci. Immunol. 8 (84), eade7652. doi:10.1126/sciimmunol.ade7652
Behmoaras, J., and Gil, J. (2021). Similarities and interplay between senescent cells and macrophages. J. Cell. Biol. 220 (2), e202010162. doi:10.1083/jcb.202010162
Billingham, L. K., Stoolman, J. S., Vasan, K., Rodriguez, A. E., Poor, T. A., Szibor, M., et al. (2022). Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat. Immunol. 23 (5), 692–704. doi:10.1038/s41590-022-01185-3
Boteanu, A., Bethencourt, J. J., Calzada-Hernández, J., Clemente, D., Nieto-González, J. C., López, C., et al. (2025). Management of childhood-onset systemic lupus erythematosus (cSLE) over the last two decades in Spain. Pediatr. Rheumatol. Online J. 23 (1), 59. doi:10.1186/s12969-025-01113-3
Brogan, P. A., Hofer, M., Kuemmerle-Deschner, J. B., Koné-Paut, I., Roesler, J., Kallinich, T., et al. (2019). Rapid and sustained long-term efficacy and safety of canakinumab in patients with cryopyrin-associated periodic syndrome ages five years and younger. Arthritis Rheumatol. 71 (11), 1955–1963. doi:10.1002/art.41004
Bronner, D. N., Abuaita, B. H., Chen, X., Fitzgerald, K. A., Nuñez, G., He, Y., et al. (2015). Endoplasmic reticulum stress activates the inflammasome via NLRP3- and Caspase-2-Driven mitochondrial damage. Immunity 43 (3), 451–462. doi:10.1016/j.immuni.2015.08.008
Calle-Fabregat, C., Morante-Palacios, O., and Ballestar, E. (2020). Understanding the relevance of DNA methylation changes in immune differentiation and disease. Genes. (Basel) 11 (1), 110. doi:10.3390/genes11010110
Camell, C. D., Günther, P., Lee, A., Goldberg, E. L., Spadaro, O., Youm, Y. H., et al. (2019). Aging induces an Nlrp3 inflammasome-dependent expansion of adipose B cells that Impairs Metabolic Homeostasis. Cell. Metab. 30 (6), 1024–1039.e6. doi:10.1016/j.cmet.2019.10.006
Cañadas-Lozano, D., Marín-Aguilar, F., Castejón-Vega, B., Ryffel, B., Navarro-Pando, J. M., Ruiz-Cabello, J., et al. (2020). Blockade of the NLRP3 inflammasome improves metabolic health and lifespan in obese mice. Geroscience 42 (2), 715–725. doi:10.1007/s11357-019-00151-6
Cancro, M. P. (2020). Age-Associated B cells. Annu. Rev. Immunol. 38, 315–340. doi:10.1146/annurev-immunol-092419-031130
Canny, S. P., and Jackson, S. W. (2021). B cells in systemic lupus erythematosus: from disease mechanisms to targeted therapies. Rheum. Dis. Clin. North Am. 47 (3), 395–413. doi:10.1016/j.rdc.2021.04.006
Checa, J., and Aran, J. M. (2020). Reactive oxygen species: drivers of physiological and pathological processes. J. Inflamm. Res. 13, 1057–1073. doi:10.2147/JIR.S275595
Chen, Y., Huang, H., Luo, Y., Wu, H., Deng, W., Min, X., et al. (2025). Senolytic treatment alleviates cochlear senescence and delays age-related hearing loss in C57BL/6J mice. Phytomedicine 142, 156772. doi:10.1016/j.phymed.2025.156772
de Torre-Minguela, C., Gómez, A. I., Couillin, I., and Pelegrín, P. (2021). Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. Faseb J. 35 (8), e21757. doi:10.1096/fj.202100085R
Demaria, M., Ohtani, N., Youssef, S. A., Rodier, F., Toussaint, W., Mitchell, J. R., et al. (2014). An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell. 31 (6), 722–733. doi:10.1016/j.devcel.2014.11.012
DiMeglio, L. A., Evans-Molina, C., and Oram, R. A. (2018). Type 1 diabetes. Lancet 391 (10138), 2449–2462. doi:10.1016/S0140-6736(18)31320-5
Faust, H. J., Zhang, H., Han, J., Wolf, M. T., Jeon, O. H., Sadtler, K., et al. (2020). IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J. Clin. Investig. 130 (10), 5493–5507. doi:10.1172/JCI134091
Fischer, J., Dirks, J., Klaussner, J., Haase, G., Holl-Wieden, A., Hofmann, C., et al. (2022). Effect of clonally expanded PD-1(high) CXCR5-CD4+ peripheral T helper cells on B cell differentiation in the joints of patients with antinuclear antibody-positive juvenile idiopathic arthritis. Arthritis Rheumatol. 74 (1), 150–162. doi:10.1002/art.41913
Franceschi, C., and Campisi, J. (2014). Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 69 (Suppl. 1), S4–S9. doi:10.1093/gerona/glu057
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C., and Santoro, A. (2018). Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14 (10), 576–590. doi:10.1038/s41574-018-0059-4
Franceschi, C., Olivieri, F., Moskalev, A., Ivanchenko, M., and Santoro, A. (2025). Toward precision interventions and metrics of inflammaging. Nat. Aging 5 (8), 1441–1454. doi:10.1038/s43587-025-00938-7
Frazzei, G., van Vollenhoven, R. F., de Jong, B. A., Siegelaar, S. E., and van Schaardenburg, D. (2022). Preclinical autoimmune disease: a comparison of Rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis and type 1 diabetes. Front. Immunol. 13, 899372. doi:10.3389/fimmu.2022.899372
Fritsch, R. D., Shen, X., Illei, G. G., Yarboro, C. H., Prussin, C., Hathcock, K. S., et al. (2006). Abnormal differentiation of memory T cells in systemic lupus erythematosus. Arthritis Rheum. 54 (7), 2184–2197. doi:10.1002/art.21943
Fu, Y., Wang, B., Alu, A., Hong, W., Lei, H., He, X., et al. (2025). Immunosenescence: signaling pathways, diseases and therapeutic targets. Signal Transduct. Target Ther. 10 (1), 250. doi:10.1038/s41392-025-02371-z
Fülöp, T., Larbi, A., and Witkowski, J. M. (2019). Human inflammaging. Gerontology 65 (5), 495–504. doi:10.1159/000497375
Fulop, T., Larbi, A., Pawelec, G., Khalil, A., Cohen, A. A., Hirokawa, K., et al. (2023). Immunology of aging: the birth of inflammaging. Clin. Rev. Allergy Immunol. 64 (2), 109–122. doi:10.1007/s12016-021-08899-6
Gabandé-Rodríguez, E., Gómez de Las Heras, M. M., and Mittelbrunn, M. (2019). Control of inflammation by calorie restriction mimetics: on the crossroad of autophagy and Mitochondria. Cells 9 (1), 82. doi:10.3390/cells9010082
Gadecka, A., Nowak, N., Bulanda, E., Janiszewska, D., Dudkowska, M., Sikora, E., et al. (2025). The senolytic cocktail, dasatinib and quercetin, impacts the chromatin structure of both young and senescent vascular smooth muscle cells. Geroscience 47 (3), 3907–3925. doi:10.1007/s11357-024-01504-6
Gao, L., Slack, M., McDavid, A., Anolik, J., and Looney, R. J. (2019). Cell senescence in lupus. Curr. Rheumatol. Rep. 21 (2), 1. doi:10.1007/s11926-019-0800-6
Gergely, P., Grossman, C., Niland, B., Puskas, F., Neupane, H., Allam, F., et al. (2002). Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 46 (1), 175–190. doi:10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
Giancane, G., Papa, R., Vastert, S., Bagnasco, F., Swart, J. F., Quartier, P., et al. (2022). Anakinra in patients with systemic juvenile idiopathic arthritis: Long-term safety from the pharmachild registry. J. Rheumatol. 49 (4), 398–407. doi:10.3899/jrheum.210563
Goronzy, J. J., and Weyand, C. M. (2012). Immune aging and autoimmunity. Cell. Mol. Life Sci. 69 (10), 1615–1623. doi:10.1007/s00018-012-0970-0
Granados, H. M., Draghi, A., Tsurutani, N., Wright, K., Fernandez, M. L., Sylvester, F. A., et al. (2017). Programmed cell death-1, PD-1, is dysregulated in T cells from children with new onset type 1 diabetes. PLoS One 12 (9), e0183887. doi:10.1371/journal.pone.0183887
Gritsenko, A., Green, J. P., Brough, D., and Lopez-Castejon, G. (2020). Mechanisms of NLRP3 priming in inflammaging and age related diseases. Cytokine Growth Factor Rev. 55, 15–25. doi:10.1016/j.cytogfr.2020.08.003
Han, Z., Wang, K., Ding, S., and Zhang, M. (2024). Cross-talk of inflammation and cellular senescence: a new insight into the occurrence and progression of osteoarthritis. Bone Res. 12 (1), 69. doi:10.1038/s41413-024-00375-z
Handono, K., Wahono, C. S., Pratama, M. Z., and Kalim, H. (2021). Association of the premature immunosenescence with the presence and severity of anemia among patients with systemic lupus erythematosus. Lupus 30 (12), 1906–1914. doi:10.1177/09612033211038057
Haque, S., Rakieh, C., Marriage, F., Ho, P., Gorodkin, R., Teh, L. S., et al. (2013). Shortened telomere length in patients with systemic lupus erythematosus. Arthritis Rheum. 65 (5), 1319–1323. doi:10.1002/art.37895
Harijith, A., Ebenezer, D. L., and Natarajan, V. (2014). Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 5, 352. doi:10.3389/fphys.2014.00352
He, S. Y., Bu, Y. R., Xu, J., Wang, Y. M., Feng, T. X., Li, P. J., et al. (2025). Acute high-altitude hypoxia induced NLRP3 inflammasome activation in pulmonary artery smooth muscle cells by BMAL1 targeting mitochondrial VDAC1-mediated MtDNA leakage. Apoptosis. doi:10.1007/s10495-025-02138-5
Heid, M. E., Keyel, P. A., Kamga, C., Shiva, S., Watkins, S. C., and Salter, R. D. (2013). Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 191 (10), 5230–5238. doi:10.4049/jimmunol.1301490
Hoi, A., Igel, T., Mok, C. C., and Arnaud, L. (2024). Systemic lupus erythematosus. Lancet 403 (10441), 2326–2338. doi:10.1016/S0140-6736(24)00398-2
Hong, Y., Boiti, A., Vallone, D., and Foulkes, N. S. (2024). Reactive oxygen species signaling and oxidative stress: transcriptional regulation and evolution. Antioxidants (Basel) 13 (3), 312. doi:10.3390/antiox13030312
Hu, M. Y., Lin, Y. Y., Zhang, B. J., Lu, D. L., Lu, Z. Q., and Cai, W. (2019). Update of inflammasome activation in microglia/macrophage in aging and aging-related disease. CNS Neurosci. Ther. 25 (12), 1299–1307. doi:10.1111/cns.13262
Huang, J., Dai, M., Huang, X., Qi, W., Jing, J., He, J., et al. (2025). Shikonin inhibits NLRP3 inflammasome activation and controls inflammatory disease. Sci. Rep. 15 (1), 19037. doi:10.1038/s41598-025-03512-8
Iwata, S., Hajime Sumikawa, M., and Tanaka, Y. (2023). B cell activation via immunometabolism in systemic lupus erythematosus. Front. Immunol. 14, 1155421. doi:10.3389/fimmu.2023.1155421
Iyer, S. S., He, Q., Janczy, J. R., Elliott, E. I., Zhong, Z., Olivier, A. K., et al. (2013). Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39 (2), 311–323. doi:10.1016/j.immuni.2013.08.001
Kalim, H., Wahono, C. S., Permana, B. P. O., Pratama, M. Z., and Handono, K. (2021). Association between senescence of T cells and disease activity in patients with systemic lupus erythematosus. Reumatologia 59 (5), 292–301. doi:10.5114/reum.2021.110318
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20 (13), 3328. doi:10.3390/ijms20133328
Kim, M. T., and Harty, J. T. (2014). Impact of inflammatory cytokines on effector and memory CD8+ T cells. Front. Immunol. 5, 295. doi:10.3389/fimmu.2014.00295
Kim, M. J., Yoon, J. H., and Ryu, J. H. (2016). Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep. 49 (10), 529–535. doi:10.5483/bmbrep.2016.49.10.115
Kim, J., Gupta, R., Blanco, L. P., Yang, S., Shteinfer-Kuzmine, A., Wang, K., et al. (2019). VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366 (6472), 1531–1536. doi:10.1126/science.aav4011
Kim, J. C., Kim, N. Y., Kim, Y., Baek, D. J., Park, T. J., and Kang, H. Y. (2025). Senolytic targeting of anti-apoptotic bcl family increases cell death in UV-Irradiated senescent melanocytes: search for senolytics. Exp. Dermatol 34 (1), e70037. doi:10.1111/exd.70037
Klughammer, B., Piali, L., Nica, A., Nagel, S., Bailey, L., Jochum, C., et al. (2023). A randomized, double-blind phase 1b study evaluating the safety, tolerability, pharmacokinetics and pharmacodynamics of the NLRP3 inhibitor selnoflast in patients with moderate to severe active ulcerative colitis. Clin. Transl. Med. 13 (11), e1471. doi:10.1002/ctm2.1471
Koutsonikoli, A., Taparkou, A., Pratsidou-Gertsi, P., Sgouropoulou, V., and Trachana, M. (2024). A Study on the immunoregulatory role of the PD1 pathway in juvenile idiopathic arthritis. Mediterr. J. Rheumatol. 35 (1), 134–142. doi:10.31138/mjr.140523.aso
Kuemmerle-Deschner, J. B., Ramos, E., Blank, N., Roesler, J., Felix, S. D., Jung, T., et al. (2011). Canakinumab (ACZ885, a fully human IgG1 anti-IL-1β mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS). Arthritis Res. Ther. 13 (1), R34. doi:10.1186/ar3266
Kumari, R., and Jat, P. (2021). Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front. Cell. Dev. Biol. 9, 645593. doi:10.3389/fcell.2021.645593
Kurosaka, D., Yasuda, J., Yoshida, K., Yoneda, A., Yasuda, C., Kingetsu, I., et al. (2006). Abnormal telomerase activity and telomere length in T and B cells from patients with systemic lupus erythematosus. J. Rheumatol. 33 (6), 1102–1107.
Kyriazis, I. D., Vassi, E., Alvanou, M., Angelakis, C., Skaperda, Z., Tekos, F., et al. (2022). The impact of diet upon mitochondrial physiology (Review). Int. J. Mol. Med. 50 (5), 135. doi:10.3892/ijmm.2022.5191
Latz, E., and Duewell, P. (2018). NLRP3 inflammasome activation in inflammaging. Semin. Immunol. 40, 61–73. doi:10.1016/j.smim.2018.09.001
Li, H., Boulougoura, A., Endo, Y., and Tsokos, G. C. (2022). Abnormalities of T cells in systemic lupus erythematosus: new insights in pathogenesis and therapeutic strategies. J. Autoimmun. 132, 102870. doi:10.1016/j.jaut.2022.102870
Li, X., Li, C., Zhang, W., Wang, Y., Qian, P., and Huang, H. (2023a). Inflammation and aging: signaling pathways and intervention therapies. Signal Transduct. Target Ther. 8 (1), 239. doi:10.1038/s41392-023-01502-8
Li, H., Wang, X., Pan, H., Xiao, C., Wang, C., Guo, S., et al. (2023b). The mechanisms and functions of IL-1β in intervertebral disc degeneration. Exp. Gerontol. 177, 112181. doi:10.1016/j.exger.2023.112181
Li, Z. Y., Cai, M. L., Qin, Y., and Chen, Z. (2023c). Age/autoimmunity-associated B cells in inflammatory arthritis: an emerging therapeutic target. Front. Immunol. 14, 1103307. doi:10.3389/fimmu.2023.1103307
Liao, S., Ning, Q., Chen, Y., Zhao, X., and Tang, S. (2022). Interaction of aging and Immunosenescence: new therapeutic targets of aging. Int. Immunopharmacol. 113 (Pt A), 109397. doi:10.1016/j.intimp.2022.109397
Liu, Q., Zhang, D., Hu, D., Zhou, X., and Zhou, Y. (2018). The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 103, 115–124. doi:10.1016/j.molimm.2018.09.010
Liu, Q., Zheng, Y., Goronzy, J. J., and Weyand, C. M. (2023). T cell aging as a risk factor for autoimmunity. J. Autoimmun. 137, 102947. doi:10.1016/j.jaut.2022.102947
Lopes-Paciencia, S., Saint-Germain, E., Rowell, M. C., Ruiz, A. F., Kalegari, P., and Ferbeyre, G. (2019). The senescence-associated secretory phenotype and its regulation. Cytokine 117, 15–22. doi:10.1016/j.cyto.2019.01.013
Ma, S., Wang, C., Mao, X., and Hao, Y. (2019). B cell dysfunction associated with aging and autoimmune diseases. Front. Immunol. 10, 318. doi:10.3389/fimmu.2019.00318
Maeda, K., Mehta, H., Drevets, D. A., and Coggeshall, K. M. (2010). IL-6 increases B-cell IgG production in a feed-forward proinflammatory mechanism to skew hematopoiesis and elevate myeloid production. Blood 115 (23), 4699–4706. doi:10.1182/blood-2009-07-230631
Marín-Aguilar, F., Lechuga-Vieco, A. V., Alcocer-Gómez, E., Castejón-Vega, B., Lucas, J., Garrido, C., et al. (2020). NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell. 19 (1), e13050. doi:10.1111/acel.13050
Mayerl, C., and Prelog, M. (2012). Immunosenescence and juvenile idiopathic arthritis. Autoimmun. Rev. 11 (5), 297–300. doi:10.1016/j.autrev.2010.02.015
Melk, A., Schmidt, B. M. W., Vongwiwatana, A., Rayner, D. C., and Halloran, P. F. (2005). Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am. J. Transpl. 5 (6), 1375–1382. doi:10.1111/j.1600-6143.2005.00846.x
Meyers, A. K., and Zhu, X. (2020). The NLRP3 inflammasome: metabolic regulation and contribution to inflammaging. Cells 9 (8), 1808. doi:10.3390/cells9081808
Miao, R., Jiang, C., Chang, W. Y., Zhang, H., An, J., Ho, F., et al. (2023). Gasdermin D permeabilization of mitochondrial inner and outer membranes accelerates and enhances pyroptosis. Immunity 56 (11), 2523–2541.e8. doi:10.1016/j.immuni.2023.10.004
Minning, S., Xiaofan, Y., Anqi, X., Bingjie, G., Dinglei, S., Mingshun, Z., et al. (2019). Imbalance between CD8(+)CD28(+) and CD8(+)CD28(-) T-cell subsets and its clinical significance in patients with systemic lupus erythematosus. Lupus 28 (10), 1214–1223. doi:10.1177/0961203319867130
Miranda-Prieto, D., Alperi-López, M., Pérez-Álvarez, Á. I., Alonso-Castro, S., Suárez, A., and Rodríguez-Carrio, J. (2025). Higher frequencies of age-associated B-cells in early arthritis are linked to atherosclerosis and immune circuits-a potential role as a biomarker for risk stratification. Rheumatol. Oxf., keaf318. doi:10.1093/rheumatology/keaf318
Mishra, S. R., Mahapatra, K. K., Behera, B. P., Patra, S., Bhol, C. S., Panigrahi, D. P., et al. (2021). Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell. Biol. 136, 106013. doi:10.1016/j.biocel.2021.106013
Mittal, R., Camick, N., Lemos, J. R. N., and Hirani, K. (2024). Gene-environment interaction in the pathophysiology of type 1 diabetes. Front. Endocrinol. (Lausanne) 15, 1335435. doi:10.3389/fendo.2024.1335435
Mittal, R., Lemos, J. R. N., Chapagain, P., and Hirani, K. (2025). Interplay of hypoxia, immune dysregulation, and metabolic stress in pathophysiology of type 1 diabetes. Front. Immunol. 16, 1599321. doi:10.3389/fimmu.2025.1599321
Mocholi, E., Corrigan, E., Chalkiadakis, T., Gulersonmez, C., Stigter, E., Vastert, B., et al. (2025). Glycolytic reprogramming shapes the histone acetylation profile of activated CD4(+) T cells in juvenile idiopathic arthritis. Cell. Rep. 44 (2), 115287. doi:10.1016/j.celrep.2025.115287
Morales-Nebreda, L., McLafferty, F. S., and Singer, B. D. (2019). DNA methylation as a transcriptional regulator of the immune system. Transl. Res. 204, 1–18. doi:10.1016/j.trsl.2018.08.001
Mouat, I. C., Goldberg, E., and Horwitz, M. S. (2022). Age-associated B cells in autoimmune diseases. Cell. Mol. Life Sci. 79 (8), 402. doi:10.1007/s00018-022-04433-9
Muela-Zarzuela, I., Suarez-Rivero, J. M., Gallardo-Orihuela, A., Wang, C., Izawa, K., de Gregorio-Procopio, M., et al. (2024). NLRP1 inflammasome promotes senescence and senescence-associated secretory phenotype. Inflamm. Res. 73 (8), 1253–1266. doi:10.1007/s00011-024-01892-7
Nagafuchi, H., Suzuki, N., Mizushima, Y., and Sakane, T. (1993). Constitutive expression of IL-6 receptors and their role in the excessive B cell function in patients with systemic lupus erythematosus. J. Immunol. 151 (11), 6525–6534.
Nakahira, K., Haspel, J. A., Rathinam, V. A. K., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12 (3), 222–230. doi:10.1038/ni.1980
Nardini, C., Moreau, J. F., Gensous, N., Ravaioli, F., Garagnani, P., and Bacalini, M. G. (2018). The epigenetics of inflammaging: the contribution of age-related heterochromatin loss and locus-specific remodelling and the modulation by environmental stimuli. Semin. Immunol. 40, 49–60. doi:10.1016/j.smim.2018.10.009
Navarro-Pando, J. M., Alcocer-Gómez, E., Castejón-Vega, B., Navarro-Villarán, E., Condés-Hervás, M., Mundi-Roldan, M., et al. (2021). Inhibition of the NLRP3 inflammasome prevents ovarian aging. Sci. Adv. 7 (1), eabc7409. doi:10.1126/sciadv.abc7409
Nicholas, C. A., Tensun, F. A., Evans, S. A., Toole, K. P., Prendergast, J. E., Broncucia, H., et al. (2025). Activated polyreactive B cells are clonally expanded in autoantibody positive and patients with recent-onset type 1 diabetes. Cell. Rep. 44 (4), 115425. doi:10.1016/j.celrep.2025.115425
Nickerson, K. M., Smita, S., Hoehn, K. B., Marinov, A. D., Thomas, K. B., Kos, J. T., et al. (2023). Age-associated B cells are heterogeneous and dynamic drivers of autoimmunity in mice. J. Exp. Med. 220 (5), e20221346. doi:10.1084/jem.20221346
Ning, C., Gao, F., Wang, Z., An, H., Liu, P., Sun, Y., et al. (2025). LBH589 reduces oxidized mitochondrial DNA and suppresses NLRP3 inflammasome activation to relieve pulmonary inflammation. PLoS One 20 (8), e0328522. doi:10.1371/journal.pone.0328522
Ohmes, J., Comdühr, S., Akbarzadeh, R., Riemekasten, G., and Humrich, J. Y. (2022). Dysregulation and chronicity of pathogenic T cell responses in the pre-diseased stage of lupus. Front. Immunol. 13, 1007078. doi:10.3389/fimmu.2022.1007078
Ohtani, N. (2022). The roles and mechanisms of senescence-associated secretory phenotype (SASP): can it be controlled by senolysis? Inflamm. Regen. 42 (1), 11. doi:10.1186/s41232-022-00197-8
Paez-Ribes, M., González-Gualda, E., Doherty, G. J., and Muñoz-Espín, D. (2019). Targeting senescent cells in translational medicine. EMBO Mol. Med. 11 (12), e10234. doi:10.15252/emmm.201810234
Paik, S., Kim, J. K., Silwal, P., Sasakawa, C., and Jo, E. K. (2021). An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 18 (5), 1141–1160. doi:10.1038/s41423-021-00670-3
Palmer, C. S., Ostrowski, M., Balderson, B., Christian, N., and Crowe, S. M. (2015). Glucose metabolism regulates T cell activation, differentiation, and functions. Front. Immunol. 6, 1. doi:10.3389/fimmu.2015.00001
Palmer, A. K., Tchkonia, T., and Kirkland, J. L. (2022). Targeting cellular senescence in metabolic disease. Mol. Metab. 66, 101601. doi:10.1016/j.molmet.2022.101601
Pan, J., Ou, Z., Cai, C., Li, P., Gong, J., Ruan, X. Z., et al. (2018). Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell. Immunol. 332, 111–120. doi:10.1016/j.cellimm.2018.08.006
Pan, L., Liu, J., Liu, C., Guo, L., Punaro, M., and Yang, S. (2023). Childhood-onset systemic lupus erythematosus: characteristics and the prospect of glucocorticoid pulse therapy. Front. Immunol. 14, 1128754. doi:10.3389/fimmu.2023.1128754
Patergnani, S., Bouhamida, E., Leo, S., Pinton, P., and Rimessi, A. (2021). Mitochondrial oxidative stress and “Mito-Inflammation”: actors in the diseases. Biomedicines 9 (2), 216. doi:10.3390/biomedicines9020216
Pérez, R. F., Fernandez-Morera, J. L., Romano-Garcia, J., Menendez-Torre, E., Delgado-Alvarez, E., Fraga, M. F., et al. (2020). DNA methylomes and epigenetic Age acceleration associations with poor metabolic control in T1D. Biomedicines 9 (1), 13. doi:10.3390/biomedicines9010013
Picca, A., Lezza, A. M. S., Leeuwenburgh, C., Pesce, V., Calvani, R., Landi, F., et al. (2017). Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int. J. Mol. Sci. 18 (5), 933. doi:10.3390/ijms18050933
Qin, Y., Cai, M. L., Jin, H. Z., Huang, W., Zhu, C., Bozec, A., et al. (2022). Age-associated B cells contribute to the pathogenesis of rheumatoid arthritis by inducing activation of fibroblast-like synoviocytes via TNF-α-mediated ERK1/2 and JAK-STAT1 pathways. Ann. Rheum. Dis. 81 (11), 1504–1514. doi:10.1136/ard-2022-222605
Qiu, Y., Huang, Y., Chen, M., Yang, Y., Li, X., and Zhang, W. (2022). Mitochondrial DNA in NLRP3 inflammasome activation. Int. Immunopharmacol. 108, 108719. doi:10.1016/j.intimp.2022.108719
Quartier, P., Allantaz, F., Cimaz, R., Pillet, P., Messiaen, C., Bardin, C., et al. (2011). A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann. Rheum. Dis. 70 (5), 747–754. doi:10.1136/ard.2010.134254
Quattrin, T., Mastrandrea, L. D., and Walker, L. S. K. (2023). Type 1 diabetes. Lancet 401 (10394), 2149–2162. doi:10.1016/S0140-6736(23)00223-4
Quijano, A., Diaz-Ruiz, C., Lopez-Lopez, A., Villar-Cheda, B., Muñoz, A., Rodriguez-Perez, A. I., et al. (2022). Angiotensin Type-1 receptor inhibition reduces NLRP3 inflammasome upregulation induced by aging and neurodegeneration in the Substantia Nigra of Male rodents and primary mesencephalic cultures. Antioxidants (Basel) 11 (2), 329. doi:10.3390/antiox11020329
Raeber, M. E., Zurbuchen, Y., Impellizzieri, D., and Boyman, O. (2018). The role of cytokines in T-cell memory in health and disease. Immunol. Rev. 283 (1), 176–193. doi:10.1111/imr.12644
Ratliff, M., Alter, S., Frasca, D., Blomberg, B. B., and Riley, R. L. (2013). In senescence, age-associated B cells secrete TNFα and inhibit survival of B-cell precursors. Aging Cell. 12 (2), 303–311. doi:10.1111/acel.12055
Riley, R. L., Khomtchouk, K., and Blomberg, B. B. (2017). Age-associated B cells (ABC) inhibit B lymphopoiesis and alter antibody repertoires in old age. Cell. Immunol. 321, 61–67. doi:10.1016/j.cellimm.2017.04.008
Roger, L., Tomas, F., and Gire, V. (2021). Mechanisms and regulation of cellular senescence. Int. J. Mol. Sci. 22 (23), 13173. doi:10.3390/ijms222313173
Rother, N., and van der Vlag, J. (2015). Disturbed T cell signaling and altered Th17 and regulatory T cell subsets in the pathogenesis of systemic lupus erythematosus. Front. Immunol. 6, 610. doi:10.3389/fimmu.2015.00610
Sachinidis, A., Trachana, M., Taparkou, A., Gavriilidis, G., Vasileiou, V., Keisaris, S., et al. (2025). Characterization of T-bet expressing B cells in lupus patients indicates a putative prognostic and therapeutic value of these cells for the disease. Clin. Exp. Immunol. 219 (1), uxaf008. doi:10.1093/cei/uxaf008
Saggau, C., Bacher, P., Esser, D., Rasa, M., Meise, S., Mohr, N., et al. (2024). Autoantigen-specific CD4(+) T cells acquire an exhausted phenotype and persist in human antigen-specific autoimmune diseases. Immunity 57 (10), 2416–2432.e8. doi:10.1016/j.immuni.2024.08.005
Sebastian-Valverde, M., and Pasinetti, G. M. (2020). The NLRP3 inflammasome as a critical actor in the inflammaging process. Cells 9 (6), 1552. doi:10.3390/cells9061552
Seoane, P. I., Lee, B., Hoyle, C., Yu, S., Lopez-Castejon, G., Lowe, M., et al. (2020). The NLRP3-inflammasome as a sensor of organelle dysfunction. J. Cell. Biol. 219 (12), e202006194. doi:10.1083/jcb.202006194
Shao, Z., Wang, B., Shi, Y., Xie, C., Huang, C., Chen, B., et al. (2021). Senolytic agent Quercetin ameliorates intervertebral disc degeneration via the Nrf2/NF-κB axis. Osteoarthr. Cartil. 29 (3), 413–422. doi:10.1016/j.joca.2020.11.006
Shapiro, M. R., Dong, X., Perry, D. J., McNichols, J. M., Thirawatananond, P., Posgai, A. L., et al. (2023). Human immune phenotyping reveals accelerated aging in type 1 diabetes. JCI Insight 8 (17), e170767. doi:10.1172/jci.insight.170767
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36 (3), 401–414. doi:10.1016/j.immuni.2012.01.009
Siegel, C. H., and Sammaritano, L. R. (2024). Systemic Lupus erythematosus: a review. Jama 331 (17), 1480–1491. doi:10.1001/jama.2024.2315
Skapenko, A., Wendler, J., Lipsky, P. E., Kalden, J. R., and Schulze-Koops, H. (1999). Altered memory T cell differentiation in patients with early rheumatoid arthritis. J. Immunol. 163 (1), 491–499.
Somers, E. C., Goodrich, J. M., Wang, L., Harlow, S. D., Marder, W., Hassett, A. L., et al. (2024). Associations between CD70 methylation of T cell DNA and age in adults with systemic lupus erythematosus and population controls: the Michigan Lupus Epidemiology & Surveillance (MILES) Program. J. Autoimmun. 142, 103137. doi:10.1016/j.jaut.2023.103137
Su, Q. Y., Zheng, X. X., Han, X. T., Li, Q., Gao, Y. R., Zhang, S. X., et al. (2025). The role of age-associated B cells in systemic lupus erythematosus. J. Autoimmun. 154, 103433. doi:10.1016/j.jaut.2025.103433
Takeshima, Y., Iwasaki, Y., Fujio, K., and Yamamoto, K. (2019). Metabolism as a key regulator in the pathogenesis of systemic lupus erythematosus. Semin. Arthritis Rheum. 48 (6), 1142–1145. doi:10.1016/j.semarthrit.2019.04.006
Tan, Y. N., Jiang, G. G., Meng, X. W., Lu, Z. Y., Yan-Ma, , Li, J., et al. (2024). CMPK2 promotes CD4(+) T cell activation and apoptosis through modulation of mitochondrial dysfunction in systemic lupus erythematosus. Cell. Biochem. Biophys. 82 (4), 3547–3557. doi:10.1007/s12013-024-01443-1
Tang, Y., Wu, J., Sun, X., Tan, S., Li, W., Yin, S., et al. (2024). Cardiolipin oxidized by ROS from complex II acts as a target of gasdermin D to drive mitochondrial pore and heart dysfunction in endotoxemia. Cell. Rep. 43 (5), 114237. doi:10.1016/j.celrep.2024.114237
Teissier, T., Boulanger, E., and Cox, L. S. (2022). Interconnections between inflammageing and immunosenescence during ageing. Cells 11 (3), 359. doi:10.3390/cells11030359
Terkeltaub, R., Sundy, J. S., Schumacher, H. R., Murphy, F., Bookbinder, S., Biedermann, S., et al. (2009). The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann. Rheum. Dis. 68 (10), 1613–1617. doi:10.1136/ard.2009.108936
Tripathi, U., Misra, A., Tchkonia, T., and Kirkland, J. L. (2021). Impact of senescent cell subtypes on tissue dysfunction and repair: importance and research questions. Mech. Ageing Dev. 198, 111548. doi:10.1016/j.mad.2021.111548
Tsai, C. Y., Shen, C. Y., Liao, H. T., Li, K. J., Lee, H. T., Lu, C. S., et al. (2019). Molecular and cellular bases of immunosenescence, inflammation, and cardiovascular complications mimicking “Inflammaging” in patients with systemic lupus erythematosus. Int. J. Mol. Sci. 20 (16), 3878. doi:10.3390/ijms20163878
van den Hoogen, L. L., Sims, G. P., van Roon, J. A. G., and Fritsch-Stork, R. D. E. (2015). Aging and systemic lupus Erythematosus - immunosenescence and beyond. Curr. Aging Sci. 8 (2), 158–177. doi:10.2174/1874609808666150727111904
Vasileiou, P. V. S., Evangelou, K., Vlasis, K., Fildisis, G., Panayiotidis, M. I., Chronopoulos, E., et al. (2019). Mitochondrial homeostasis and cellular senescence. Cells 8 (7), 686. doi:10.3390/cells8070686
Vitetta, L., and Anton, B. (2007). Lifestyle and nutrition, caloric restriction, mitochondrial health and hormones: scientific interventions for anti-aging. Clin. Interv. Aging 2 (4), 537–543. doi:10.2147/cia.s866
Wan, M., Gray-Gaillard, E. F., and Elisseeff, J. H. (2021). Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 9 (1), 41. doi:10.1038/s41413-021-00164-y
Wang, B., Han, J., Elisseeff, J. H., and Demaria, M. (2024a). The senescence-associated secretory phenotype and its physiological and pathological implications. Nat. Rev. Mol. Cell. Biol. 25 (12), 958–978. doi:10.1038/s41580-024-00727-x
Wang, L., Hong, W., Zhu, H., He, Q., Yang, B., Wang, J., et al. (2024b). Macrophage senescence in health and diseases. Acta Pharm. Sin. B 14 (4), 1508–1524. doi:10.1016/j.apsb.2024.01.008
Wehr, C., Eibel, H., Masilamani, M., Illges, H., Schlesier, M., Peter, H. H., et al. (2004). A new CD21low B cell population in the peripheral blood of patients with SLE. Clin. Immunol. 113 (2), 161–171. doi:10.1016/j.clim.2004.05.010
Wei, P., Zhang, X., Yan, C., Sun, S., Chen, Z., and Lin, F. (2025). Mitochondrial dysfunction and aging: multidimensional mechanisms and therapeutic strategies. Biogerontology 26 (4), 142. doi:10.1007/s10522-025-10273-4
Wohlford, G. F., Van Tassell, B. W., Billingsley, H. E., Kadariya, D., Canada, J. M., Carbone, S., et al. (2020). Phase 1B, randomized, Double-Blinded, dose escalation, Single-Center, repeat dose safety and pharmacodynamics Study of the oral NLRP3 inhibitor dapansutrile in subjects with NYHA II-III systolic heart failure. J. Cardiovasc Pharmacol. 77 (1), 49–60. doi:10.1097/FJC.0000000000000931
Wu, M., Yu, C., Wen, F., Li, Y., Zhang, X., Wang, Y., et al. (2025). NLRP3 inflammasome inhibits mitophagy during the progression of temporal lobe epilepsy. Sci. Rep. 15 (1), 16341. doi:10.1038/s41598-025-01087-y
Xian, H., Watari, K., Sanchez-Lopez, E., Offenberger, J., Onyuru, J., Sampath, H., et al. (2022). Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 55 (8), 1370–1385.e8. doi:10.1016/j.immuni.2022.06.007
Xian, H., Watari, K., Ohira, M., Brito, J. S., He, P., Onyuru, J., et al. (2025). Mitochondrial DNA oxidation propagates autoimmunity by enabling plasmacytoid dendritic cells to induce T(FH) differentiation. Nat. Immunol. 26 (7), 1168–1181. doi:10.1038/s41590-025-02179-7
Xie, G., Chen, X., Gao, Y., Yang, M., Zhou, S., Lu, L., et al. (2025). Age-Associated B cells in autoimmune diseases: pathogenesis and clinical implications. Clin. Rev. Allergy Immunol. 68 (1), 18. doi:10.1007/s12016-025-09021-w
Yang, C., Xue, J., An, N., Huang, X. J., Wu, Z. H., Ye, L., et al. (2018). Accelerated glomerular cell senescence in experimental lupus nephritis. Med. Sci. Monit. 24, 6882–6891. doi:10.12659/MSM.909353
Yao, J., Sterling, K., Wang, Z., Zhang, Y., and Song, W. (2024). The role of inflammasomes in human diseases and their potential as therapeutic targets. Signal Transduct. Target Ther. 9 (1), 10. doi:10.1038/s41392-023-01687-y
Yennemadi, A. S., Keane, J., and Leisching, G. (2023). Mitochondrial bioenergetic changes in systemic lupus erythematosus immune cell subsets: contributions to pathogenesis and clinical applications. Lupus 32 (5), 603–611. doi:10.1177/09612033231164635
Yu, J., Nagasu, H., Murakami, T., Hoang, H., Broderick, L., Hoffman, H. M., et al. (2014). Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. U. S. A. 111 (43), 15514–15519. doi:10.1073/pnas.1414859111
Yue, Z., Nie, L., Zhao, P., Ji, N., Liao, G., and Wang, Q. (2022). Senescence-associated secretory phenotype and its impact on oral immune homeostasis. Front. Immunol. 13, 1019313. doi:10.3389/fimmu.2022.1019313
Yuk, J. M., Silwal, P., and Jo, E. K. (2020). Inflammasome and mitophagy connection in health and disease. Int. J. Mol. Sci. 21 (13), 4714. doi:10.3390/ijms21134714
Żabińska, M., Krajewska, M., Kościelska-Kasprzak, K., and Klinger, M. (2016). CD3(+)CD8(+)CD28(-) T lymphocytes in patients with lupus nephritis. J. Immunol. Res. 2016, 1058165. doi:10.1155/2016/1058165
Zhang, X., Gao, Y., zhang, S., Wang, Y., Pei, X., Chen, Y., et al. (2025a). Mitochondrial dysfunction in the regulation of aging and aging-related diseases. Cell. Commun. Signal 23 (1), 290. doi:10.1186/s12964-025-02308-7
Zhang, M., Lou, P., Shou, D., Tong, P., and Zhang, Y. (2025b). Research progress of senolytic drugs in the treatment of orthopedic diseases. Gerontology 71 (3), 221–238. doi:10.1159/000543386
Zhao, T. V., Sato, Y., Goronzy, J. J., and Weyand, C. M. (2022). T-Cell aging-associated phenotypes in autoimmune disease. Front. Aging 3, 867950. doi:10.3389/fragi.2022.867950
Zheng, Y., Liu, Q., Goronzy, J. J., and Weyand, C. M. (2023). Immune aging - a mechanism in autoimmune disease. Semin. Immunol. 69, 101814. doi:10.1016/j.smim.2023.101814
Zhong, Z., Liang, S., Sanchez-Lopez, E., He, F., Shalapour, S., Lin, X. J., et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560 (7717), 198–203. doi:10.1038/s41586-018-0372-z
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469 (7329), 221–225. doi:10.1038/nature09663
Keywords: cellular senescence, senescence-associated secretory phenotype (SASP), inflammaging, immune senescence, autoimmunity
Citation: Mittal R, Saavedra D, Mittal M, Lemos JRN and Hirani K (2025) Inflammasome activation and accelerated immune aging in autoimmune disorders. Front. Aging 6:1688060. doi: 10.3389/fragi.2025.1688060
Received: 18 August 2025; Accepted: 17 September 2025;
Published: 30 September 2025.
Edited by:
Matthew Yousefzadeh, Columbia University, United StatesReviewed by:
Rituparna Ghosh, Columbia University, United StatesCopyright © 2025 Mittal, Saavedra, Mittal, Lemos and Hirani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rahul Mittal, ci5taXR0YWwxMUBtZWQubWlhbWkuZWR1; Khemraj Hirani, a2hpcmFuaUBtZWQubWlhbWkuZWR1