 Alejandro Ramirez Garcia1,2†
Alejandro Ramirez Garcia1,2† Anna Greppi1†
Anna Greppi1† Florentin Constancias1
Florentin Constancias1 Hans-Joachim Ruscheweyh3
Hans-Joachim Ruscheweyh3 Julie Gasser1Katherine Hurley2Shana J. Sturla2
Julie Gasser1Katherine Hurley2Shana J. Sturla2 Clarissa Schwab1,4
Clarissa Schwab1,4 Christophe Lacroix1*
Christophe Lacroix1*- 1Laboratory of Food Biotechnology, Department of Health Sciences and Technology, ETH Zürich, Zürich, Switzerland
- 2Laboratory of Toxicology, Department of Health Sciences and Technology, ETH Zürich, Zürich, Switzerland
- 3Institute of Microbiology, Department of Biology, and Swiss Institute of Bioinformatics, ETH Zürich, Zürich, Switzerland
- 4Department of Biological and Chemical Engineering, Aarhus University, Aarhus, Denmark
Introduction: Anaerobutyricum hallii is a human gut commensal that transforms the heterocyclic amine 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine (PhIP), a carcinogen from cooked meat. The transformation mechanism involves the microbial production of acrolein from glycerol, and its conjugation with PhIP, thus blocking its mutagenic potential. A potential cancer prevention strategy could therefore involve supplementing complex human microbial communities with metabolically competent bacteria such as A. hallii that can deplete PhIP. However, it has not been established how the proportion of A. hallii in diverse healthy human gut microbial communities relates to functional capacity for PhIP transformation and, moreover, how supplementing microbiomes with A. hallii affects this function.
Methods: In this study, shotgun metagenomics was used to study taxonomic profiling, the abundance of glycerol/diol dehydratase (gdh)-harboring taxa, the proportion of resident A. hallii, and the reconstruction of A. hallii population genomes in the fecal samples of 20 healthy young adult donors. Furthermore, the influence of supplementing 106 cells/mL of A. hallii DSM 3353 with diluted fecal microbiota was characterized.
Results and discussion: Six microbiota were assigned to Bacteroides, nine to Prevotella, and five to Ruminococcus by enterotype-associated clustering. The total number of gdh copies in the 20 fecal microbiota expressed per 1010 bacterial cells ranged between 1.32 × 108 and 1.15 × 109. Eighteen out of the 20 donors were dominated by A. hallii, representing between 33% and 94% of the total gdh relative abundance of the samples. The microbiota with low A. hallii abundance (i.e., with a relative abundance < 1%) transformed less PhIP than the microbiota with high A. hallii abundance (i.e., with a relative abundance > 1%). Furthermore, supplementing the low-A. hallii-abundant microbiota with glycerol significantly increased the PhIP transformation capacity after 6 h while reducing total short-chain fatty acid (SCFA) levels, which is most likely due to acrolein production. Although acetate decreased in all microbiota with glycerol and with the combination of glycerol and A. hallii, for most of the microbiomes, butyrate production increased over time. Thus, for a significant number of diverse healthy human fecal microbiomes, and especially when they have little of the taxa to start with, supplementing A. hallii increases PhIP transformation. These findings suggest the need to test in vivo whether supplementing microbiomes with A. hallii reduces PhIP exposure.
1 Introduction
The human gut microbiota comprise a vast number of microorganisms that metabolize dietary substrates and produce numerous compounds, thus influencing host health (Ursell et al., 2012). Gut bacteria harboring cobalamin-dependent glycerol/diol dehydratases (GDHs) (EC: 4.2.1.28/30) metabolize dietary or endogenous glycerol to 3‐hydroxypropanal (3‐HPA), which can be transformed into 1,3-propanediol (1,3-PD) or can spontaneously degrade to acrolein in physiological conditions (Engels et al., 2016b). Enzymes responsible for glycerol transformation via GDH are encoded by the pdu-cob-cbi-hem operon, which contains the genes required for cobalamin biosynthesis (cob-cbi) and for GDH (pduCDE) (Morita et al., 2008; Santos et al., 2008; Sriramulu et al., 2008). Acrolein is a highly reactive unsaturated aldehyde that exhibits cytotoxic and broad-spectrum antimicrobial activities (Stevens and Maier, 2008; Engels et al., 2016b; Zhang and Schwab, 2022). In addition, acrolein has recently been reported to be involved in the detoxification of xenobiotic heterocyclic amines (HCAs) (Zhang et al., 2019a; Zhang et al., 2019b), which can be produced during the cooking of meat due to the high-temperature pyrolysis of amino acids and are suggested to be involved in the initiation of colorectal carcinogenesis (CRC) (Bouvard et al., 2015; Dolan et al., 2021; Kang et al., 2022). The transformation of HCA yields acrolein conjugates (Beer et al., 2017) with reduced cytotoxicity and mutagenicity, and may be beneficial for the host by decreasing HCA exposure and thus reducing CRC risk (Zhang et al., 2019b).
Anaerobutyricum is a commonly occurring genus within human gut microbiota (Almeida et al., 2021) and yet consists of only two species: Anaerobutyricum hallii (formerly Eubacterium hallii) and Anaerobutyricum soehngenii (Shetty et al., 2018). Both strains (i.e., DSM 33523 and L2-7, also known as DSM 17630) were isolated from human feces and showed GDH activity (Engels et al., 2016b). A. hallii is considered a candidate biotherapeutic due to its ability to produce the short-chain fatty acids (SCFAs) butyrate and propionate, which contribute to intestinal homeostasis (Duncan et al., 2004; Muñoz-Tamayo et al., 2011; Scott et al., 2014; Engels et al., 2016b). Moreover, among gut bacteria, A. hallii is reported to be one of the most efficient taxa in the transformation of the HCA 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine (PhIP) (Fekry et al., 2016). PhIP is produced by Maillard reactions between glucose, phenylalanine, and creatinine, and is the most abundant HCA produced during the cooking of meat and fish (Felton et al., 2000). On the basis of sufficient evidence in experimental animals, PhIP has been classified since 1993 as a possible human carcinogen (IARC Working Group et al., 1993). Since then, there have been numerous mechanistic data supporting its genotoxicity (Masumura et al., 1999; Tanaka et al., 2005; Bellamri et al., 2021). PhIP can be transformed by A. hallii into its acrolein conjugate 7-hydroxy-5-methyl-3-phenyl-6,7,8,9-tetrahydropyrido[3′,2′:4,5]imidazo[1,2-a]pyrimidin-5-ium chloride (PhIP-M1) (Fekry et al., 2016; Zhang et al., 2019b). On the basis of metagenomic predictions, we previously suggested A. hallii to be a key microbe in GDH-mediated glycerol metabolism in the gut, followed by Clostridiales spp. and Blautia spp., and that GDH activity could be enhanced in vitro by supplementing three adult fecal microbiota with A. hallii in batch fermentation (Ramirez Garcia et al., 2021).
In this study, we tested the hypothesis that PhIP transformation by human fecal microbiota correlates with the A. hallii proportion and supplementing various complex microbiomes with A. hallii can increase their capacity to transform PhIP. Thus, we used shotgun metagenomics to quantify resident A. hallii abundance and the diversity of A. hallii populations, and analyzed the gdh community in a broader panel composed of 20 diverse fecal microbiota from healthy adults. The fecal microbiota composition of 20 adult donors was characterized, and the microbiota were divided into two groups, high and low, based on the proportion of resident A. hallii. Next, we conducted batch fermentations of all 20 fecal microbiota in the presence of PhIP and glycerol and with or without A. hallii supplementation. The PhIP transformation capacity, glycerol metabolites, and SCFAs produced during batch fermentations were evaluated based on these two groups and related to the A. hallii resident population or supplementation.
2 Materials and methods
2.1 Fecal sample collection
The fecal samples were collected from 20 healthy adult individuals (median age 30 years) who had not received antibiotics in the previous 3 months. Informed written consent was obtained from each donor, and a donor number was randomly assigned for blinding. Fecal samples were collected by donors immediately after defecation using a sterile container. An atmosphere generator (AnaeroGen™; Thermo Fisher Scientific, Pratteln, Switzerland) was added into the container to produce anaerobic conditions during transportation. The samples were transferred within 2 hours into an anaerobic chamber (10% CO2, 5% H2, and 85% N2) (Coy Laboratories. Grass Lake, MI, USA), aliquots were stored at −80°C for DNA isolation, and the samples were used to immediately inoculate batch fermentations, as outlined below. The Ethics Committee of ETH Zürich exempted this study from review because the sample collection procedure was not performed under conditions of intervention and was done anonymously.
2.2 Fecal batch fermentation
A 10% w/v (weight/volume) fecal suspension was prepared inside an anaerobic chamber by resuspending 1 g of fresh feces in 10 mL of sterile peptone water in a 50 mL sterile Falcon™ tube with four sterile glass beads (5 mm). The tube was vortexed for 5 min and the mixture was left to stand for 5 min to allow the heavy particles to settle. The resulting supernatant was used to inoculate (0.1% w/v) serum flasks containing 20 mL of Macfarlane medium prepared according to Bircher et al. (2018b) and supplemented with 200 nM of PhIP (Chemie Brunschwig AG, Basel, Switzerland) and 100 mM of glycerol (Sigma-Aldrich, Buchs, Switzerland). This concentration of glycerol was chosen based on previous reports (from De Weirdt et al., 2010; Fekry et al., 2016; and Engels et al., 2016a), and was the minimum dose at which PhIP transformation was detected. The fermentations in the Macfarlane medium without added glycerol served as a control. The additional fermentations were supplemented with 1% of a 16-h overnight culture of A. hallii DSM 3353 [final concentration in the medium measured by quantitative PCR (qPCR) as 106 cells/mL] grown in a yeast extract—casein hydrolysate—fatty acid (YCFA) medium prepared as previously described (Bircher et al., 2018a). Triplicate fermentations were incubated at 37°C with magnetic stirring (120 rpm) and sampled and analyzed as previously described (Ramirez Garcia et al., 2021). Briefly, 1-mL samples were taken after 0 h, 6 h, and 24 h of fermentation using sterile syringes flushed with CO2 and centrifuged in sterile Eppendorf™ Tubes™ (14,000 × g for 10 min at 4°C).
2.3 DNA isolation, quantitative PCR, and shotgun metagenome sequencing
DNA was isolated from the pure culture of A. hallii inoculum, from the fecal samples, and from the fermentation samples using the FastDNA™ SPIN Kit for Soil (MP Biomedicals, Zurich, Switzerland), following the manufacturer’s protocol. The concentration of the isolated fecal DNA was assessed with a NanoDrop® 1000 instrument and a Qubit™ 4 fluorometer (Thermo Fisher Scientific), whereas the DNA integrity was verified via gel electrophoresis. A previously established qPCR assay was used to quantify the abundance of A. hallii in the overnight culture used for A. hallii supplementation, using specific primers for gdh (forward primer 5′-CGTTATGCTCCATTTAATGCT-3′ and reverse primer 5′-CCAAGGAGTATCATCACCATC-3′) (Ramirez Garcia et al., 2021). The reactions were performed using a Roche LightCycler® II device (Roche Diagnostics, Rotkreuz, Switzerland). For shotgun metagenome sequencing, all fecal samples were diluted to 25 ng/µL−1 and sent for sequencing (Novogene Co. Ltd., Cambridge, UK), with the number of reads/samples ranging from 405,268,068 to 620,549,190. The sequencing was performed using the Illumina NovaSeq 6000 platform in 150-bp paired-end mode. The number of generated raw read pairs ranged between 405 million and 620 million, with an average of 466 million.
2.4 Gene catalog annotation and quantification
The quality control of raw reads was performed with BBMap (v.38.71) by removing adapters, reads mapping to the human genome, and low-quality reads (Bushnell et al., 2017). The filtered reads were merged with a minimum overlap of 16 bases and were assembled using the SPAdes assembler (v3.14) in metagenomic mode (Bankevich et al., 2012). Gene calling on the assembled scaffolds was performed using Prodigal (Hyatt et al., 2010), and the genes were clustered using CD-HIT-EST (Li and Godzik, 2006) with a cutoff value of 95% identity and 90% overlap of the shorter gene. The reads from all samples were mapped against all representative genes. To account for the differences in gene size, the number of inserts mapped were normalized to represent 1,000-bp genes. The abundance of each gene in each sample was normalized using the median abundance of the 10 single-copy marker genes described by Milanese et al. (2019) to estimate gene copies per bacterial cells, which was expressed per 1010 cells in the manuscript.
2.5 Metagenome-based taxonomic profiling and identification of gdh-harboring taxa
The taxonomic profiles for each sample were generated by profiling quality-controlled metagenomic reads using the metagenomic-based operational taxonomic units (mOTUs) profiler version 3.0.1 using default parameters (Milanese et al., 2019).
The contigs harboring genes belonging to the pduC/gdh-annotated clusters from the gene catalog were identified. The collections of metagenome-assembled genomes (MAGs) that were specific for each donor (i.e., list of contigs belonging to a particular MAG) were then used to link pduC/gdh genes to bacterial populations and their Genome Taxonomy Database (GDTB) assignments. When the identified contigs were not assigned to any MAG, the taxonomy of the gene belonging to pduC-annotated clusters was determined by aligning the corresponding contig sequence against the National Center for Biotechnology Information (NCBI) non-redundant nucleotide database (nt).
2.6 Microbiota community analysis
The merged mOTU profiles were loaded into R (The R Foundation for Statistical Computing, Vienna, Austria) as phyloseq objects (McMurdie and Holmes, 2013), and data analysis was conducted using DivComAnalyses R functions (Constancias and Sneha-Sundar, 2022). The taxonomic structures were clustered to determine the enterotype, as previously defined (Arumugam et al., 2011). The principal coordinate analysis was used to depict similarity between donors’ microbiota. The correlation between family-level proportion and ordination pattern was evaluated, and the top 10 significantly correlated families were displayed (p < 0.05).
The association between microbiota and metadata (i.e., PhIP transformation, age, diet) was investigated at the alpha/beta and mOTU abundance levels. A permutational multivariate analysis was used to identify marginal associations between community structure and covariates. In a second step, distance-based partial redundancy analysis was used to evaluate the association between microbiota structure and PhIP transformation while controlling for differences in the microbiota associated with enterotypes.
2.7 Metagenome-assembled genome binning, quality filtering, and annotation
The assembled scaffolds were length filtered (≥ 1,000 bp), and quality-controlled metagenomic reads from all samples were individually mapped against the scaffolds of each sample using the Burrows–Wheeler aligner (v0.7.17-r1188) (Li and Durbin, 2009), allowing for secondary alignments. The alignments were filtered to > 45 bases, with an identity of ≥ 97% and covering ≥ 80% of the read sequence. To provide within- and between-sample coverages for each scaffold, the resulting binary alignment map (BAM) files from the previous step were processed using MetaBAT2 (v2.12.1) (Kang et al., 2019). The scaffolds on all samples were finally binned with MetaBAT, and the quality of the resulting bins was assessed using CheckM (v1.0.13) (Parks et al., 2015). Only the bins with a completeness level of ≥ 50% and a contamination level of < 10%, and which belonged to the domains of Bacteria or Archaea as reported by Anvi’o (v5.5.0) (Eren et al., 2015) were used as MAGs for further analysis. The taxonomic annotation of MAGs was performed using GTDB-Tk (v1.5.0, database R06-RS202) (Chaumeil et al., 2019). The MAGs assigned to the Anaerobutyricum genus were subjected to the Anvi’o pangenomic workflow, which identified two fragmentated MAGs in donors 2 and 15. The FASTA files corresponding to these MAGs were concatenated and subjected to CheckM and GToTree together with the other MAGs for final completeness and contamination estimation.
To identify the presence of the pdu-cob-cbi-hem operon in the MAGs assigned to the genus Anaerobutyricum, the MAGs were annotated using Prokka (Seemann, 2014) and the resulting annotation was manually screened for the pdu-cob-cbi-hem operon genes. Publicly available genomes of the strains A. hallii DSM 3353 and A. soehngenii L2-7 (DSM 17630) were annotated and used as reference strains.
2.8 Determination of glycerol consumption and metabolite production
The consumption of glycerol and the production of SCFA and 1,3-PD was monitored using high-pressure liquid chromatography—refractive index (HPLC-RI), as described before (Zhang et al., 2019b). In brief, culture supernatant was filtered with a 0.45-µm nylon filter (Phenomenex, Basel, Switzerland), diluted 1: 1 in H2O, and transferred into an HPLC vial with a 250-µL conical glass insert (BGB Analytik, Boeckten, Switzerland). The samples were eluted isocratically with 10 mM H2SO4 at 40°C and at a flow rate of 0.4 mL/min−1 using an Aminex HPX-87H column (300 mm × 7.8 mm, with a 9-µm particle size; Bio-Rad Laboratories, Reinach, Switzerland). Glycerol, 1,3-PD, and SCFA were quantified by independent calibration with external standards (all obtained from Sigma-Aldrich). The detection limits were 0.7 mM for acetate, propionate, and butyrate, 0.9 mM for glycerol, and 0.2 mM for 1,3-PD.
2.9 Quantification of PhIP transformation by nanospray liquid chromatography—tandem mass spectrometry
The transformation of PhIP into PhIP-M1 was determined via nanospray liquid chromatography coupled to tandem mass spectrometry (nanoLC-MS)/MS using a nanoACQUITY Ultra Performance LC™ (UPLC®) System (Waters Corporation, Milford, MA, USA) attached to a TSQ Vantage™ triple-stage quadrupole mass spectrometer (Thermo Fisher Scientific), as described previously (Ramirez Garcia et al., 2021).
The calibration curves were generated using the ratio between the peak area of 200 nM of the internal standard 5-amino-6-methylbenzamidazolone (AMBI) and of 5–250 nM PhIP and PhIP-M1. The transformation of PhIP was reported as a percentage of the initial PhIP transformed to PhIP-M1.
2.10 Statistical analyses
R (version 4.2.2) was used for the statistical analyses. Based on the Shapiro–Wilk test, the data displayed non-normal distribution. Therefore, the differences between the groups for PhIP transformation, glycerol consumption, SCFAs, 1,3-PD, and A. hallii abundance (low vs. high) were assessed using a two-sided non-parametric Wilcoxon test. Adjusted p-values of ≤ 0.05 were considered to be significant. Spearman-ranked correlations were used to investigate associations between (i) observed species (i.e., number of mOTUs) and Shannon and Simpson diversity indices (i.e., diversity of mOTUs in a community), or (ii) mOTU proportions of the top 200 mOTUs and PhIP transformation for which only correlations with adjusted p-values of ≤ 0.05 and |Spearman’s rho| ≥ 0.6 were considered. Computed p-values were false discovery rate (FDR)-adjusted for multiple comparisons.
2.11 Sequence deposits
Raw sequence data were deposited with NCBI (in the Sequence Read Archive) and are available under BioProject PRJNA720271.
3 Results
3.1 Microbial community structure of fecal microbiota
To understand the diversity of the microbiomes used for this study, the microbial composition of fecal samples collected from 20 young healthy adults was assessed by shotgun metagenomics (Figure 1). Collectively, we identified 1,839 individual mOTUs within all 20 fecal samples. In the samples, taxa belonging to the Firmicutes phylum were the most abundant of all microbiota, with relative abundances of between 48.4% and 90.5%. This was followed by taxa belonging to the Bacteroidetes phylum, which ranged between 1.9% and 38.4% (Figure 1A). With regard to community diversity values, these ranged from 2.9 to 4.9 by the Shannon index, and 0.91 to 0.98 by the Simpson index, and individual fecal microbiotas harbored from 162 (D.13) to 701 (D.08) mOTUs (Figure 1B). To identify enterotype-associated clusters of the microbiota, a beta-diversity analysis (i.e., comparison of samples based on overall microbiota similarity) based on the Jensen–Shannon divergence was computed at the genus level. The analysis assigned six microbiota to the Bacteroides enterotype, nine to Prevotella, and five to Ruminococcus (Figure 1C).

Figure 1 Microbial community structure of 20 human fecal donors (D) analyzed by shotgun metagenomics. Relative abundance at the phylum level (A). Shannon index, Simpson index, and the number of observed species (B). Principal coordinate analysis displaying enterotype clustering based on Jensen–Shannon divergence computed at the genus level, and the top seven significantly correlated families (adjusted p-value ≤ 0.05) (C).
3.2 Proportion and distribution of gdh in fecal microbial communities
As a next step, the proportion of taxa contributing to the gdh prevalence in each fecal microbiota was evaluated by creating a gene catalog from the shotgun metagenomes to quantify the proportion and distribution of gdh-harboring taxa in the fecal microbiota. In the 20 fecal microbiota, the total number of gdh copies expressed per 1010 bacterial cells ranged from 1.32 × 108 to 1.15 × 109 (Figure 2A). When looking into specific gdh-harboring taxa, a total of 17 taxa harboring gdh [i.e., the propanediol dehydratase large subunit (pduC)—functional annotation from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database K01699] were identified (Figure 2B). Specifically, the dominant gdh-harboring taxa in the microbiota of 18 donors was A. hallii, representing between 33% (D.20) and 94% (D.11) of the total gdh relative abundance of the samples. When the most abundant gdh-harboring taxon was not A. hallii, it was Faecalicatena gnavus (78%) for D.14 or a taxon that could not be assigned in D.18, accounting for 56% of the total gdh community (Figure 2B). To further explore such unassigned gdh-harboring taxa in D.18, it was BLASTed and could be identified with Alkalimarinus sp. SCSIO (84% identity and 13% electable coverage). Besides A. hallii and F. gnavus, other taxa that contributed significantly to the gdh communities in the fecal microbiomes were Blautia obeum (present in the feces of 18 donors, representing between 3% and 40% of the total gdh), an unidentified species of the Blautia genus (present in the feces of nine donors at between 0.6% and 9% of the total gdh), and Flavonifractor plautii (present in the feces of eight donors at between 1% and 11% of the total gdh) (Figure 2B).
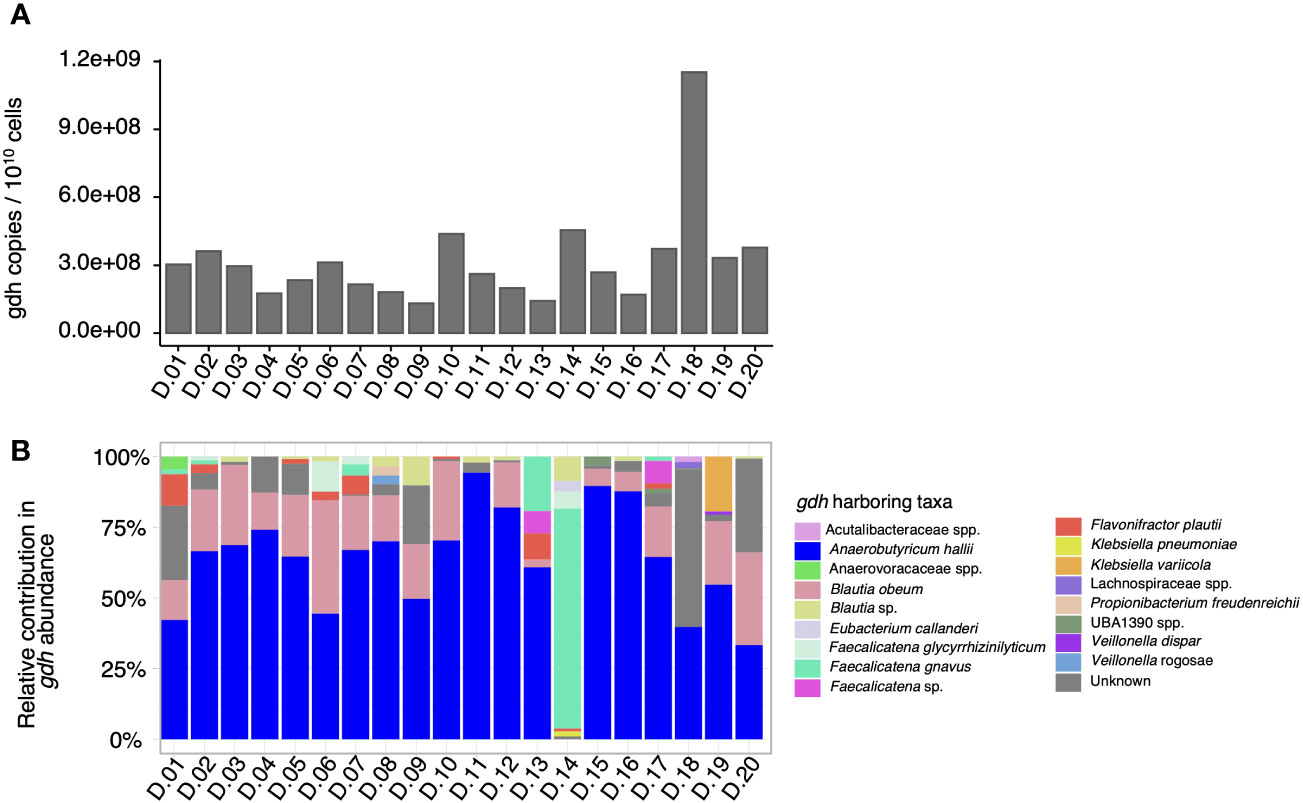
Figure 2 Total gdh abundance and gdh-contributing taxa quantified by shotgun metagenome sequencing in 20 adult human fecal microbiota donors (D). The total number of gdh copies per 1010 bacterial cells in the microbiota of each of the 20 donors, which was quantified using the gene catalog approach (A). The relative contribution of gdh-harboring taxa based on the MAG catalog and contig-level taxonomy (B).
3.3 Proportion of A. hallii population in fecal metagenomes
Given that A. hallii was the most abundant gdh-harboring taxa in the fecal microbiome, we further estimated its proportion on the basis of both a taxonomic marker (i.e., 16S rRNA) and the proportion of a functional gene of A. hallii (i.e., gdh) in the fecal samples. At first, we evaluated the coherence of these two approaches and found a significant correlation (p = 0.00043), thus validating the results previously obtained for A. hallii and gdh for all the fecal samples (Figure 3A). Based on the A. hallii proportion, 1% was arbitrarily chosen to stratify the fecal microbiota in two groups defined as “low” (A. hallii < 1%) and “high” (A. hallii > 1%), and 14 microbiota were assigned to the “low” group and six microbiota to the “high” group (Figure 3B). Importantly, A. hallii identified by (Eubacterium hallii) mOTU_v3_03632 was the only taxa significantly associated with PhIP transformation in the 20 fecal metagenomes (Supplementary Table 1).
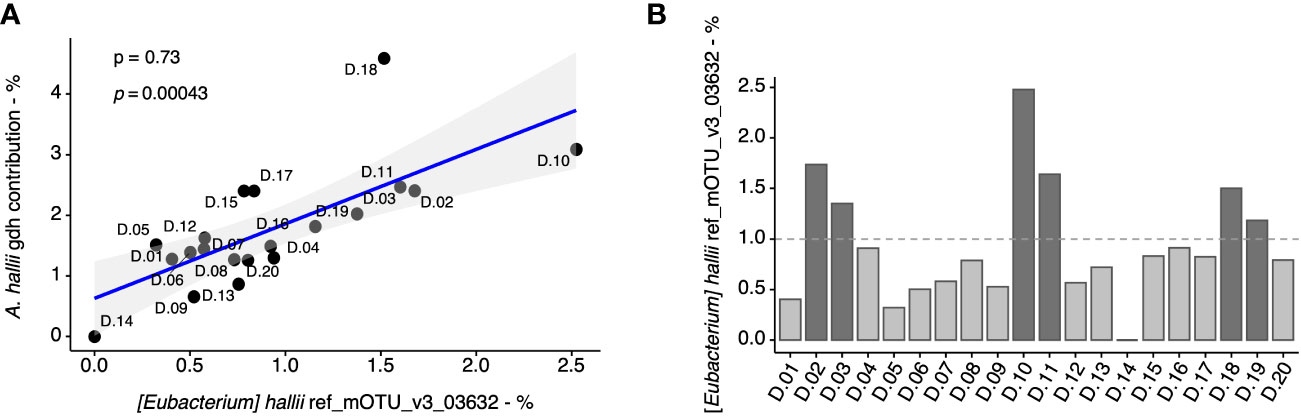
Figure 3 Quantification of A. hallii in fecal metagenomes of 20 adult donors. Correlation (Spearman) between the percentage of gdh assigned to A. hallii by metagenomics based on the gene catalog data, and the percentage of A. hallii mOTUs detected by metagenomics using mOTU-based taxonomic profiling in each donors’ sample (A). Spearman’s rho (ρ) and associated p-values are displayed. The relative abundance of A. hallii [(Eubacterium) hallii ref_mOTU_v3_03632] in the fecal metagenomes (B).
3.4 Presence of A. hallii populations and the pdu-cob-cbi-hem operon in fecal metagenomes
To further investigate A. hallii populations, we used genome-resolved MAGs to compare the composition of the cbi-cob-hem-pdu operon in each fecal sample (Figure S1). Overall, binning of the obtained assembled contigs from all samples resulted in 2,891 MAGs. These MAGs were clustered at 95% similarity for species identification, resulting in a collection of 850 species-level MAGs. In all microbiomes, we recovered one MAG belonging to the A. hallii species-level MAG cluster. The taxonomy of those MAGs matched with A. hallii, with the only exceptions being D.14 and D.03, in which one MAG assigned to an unidentified species of the genus Anaerobutyricum was recovered (Figure S1). Despite the methodological limitations of MAG reconstruction related to fragmented MAGs, we identified donor-specific A. hallii populations that differed in the content of the genes required for acrolein production. Specifically, nine A. hallii MAGs harbored a pdu-cob-cbi-hem operon with highly similar gene content and organization to A. hallii DSM 3553 and A. soehngenii L2-7. The cbi and hem genes were not recovered in MAGs from D.17, D.20, D.10, D.06, or D.19, whereas the same MAGs harbored only two cob genes (D.19 only one). (Figure S1).
3.4 PhIP transformation by human microbiota with glycerol and A. hallii
Once we identified A. hallii as being the main contributor of gdh activity for 18 fecal microbiota and confirmed that the 20 diverse fecal microbiota of healthy adults could be stratified by proportion of A. hallii mOTUs, we hypothesized that the groups would differ in their actual capacity to transform PhIP to PhIP-M1. Thus, fecal batch fermentations were conducted anaerobically in the presence of PhIP (200 mM) and glycerol (100 mM), with or without supplementing additional A. hallii, and samples were analyzed after 24 h and 48 h for PhIP transformation (Figure 4A). In the presence of glycerol, the microbiota with high A. hallii transformed more of the PhIP than the microbiota with low A. hallii did [after 6 h, median of 2% versus 0% (p = 0.17), and after 24 h, 9% versus 4% (p = 0.0508), respectively]. In the same low A. hallii group, when fecal microbiota were supplemented with A. hallii at a final concentration of 106 cells per mL a significant increase in PhIP transformation was observed after 6 h compared with glycerol alone [median of 4% compared with 0% transformation in the presence of glycerol only (p = 0.007)] (Figure 4A). Finally, when fecal microbiota were incubated for 24 h in controlled batch fermentations without glycerol, no PhIP transformation was detected (data not shown). In line with the batch fermentation results, PhIP was better depleted with increasing proportions of resident A. hallii in all 20 fecal microbiota, at both 6 h and 24 h (Figure S2A). This association was not observed for any other taxa. On the other hand, PhIP transformation could not be associated with either alpha- or beta-diversity metrics (Figure S3 and Table S1). Furthermore, no significant correlation between PhIP transformation and community structure was detected when we controlled for variations associated with enterotype (p = 0.45; Table S2).
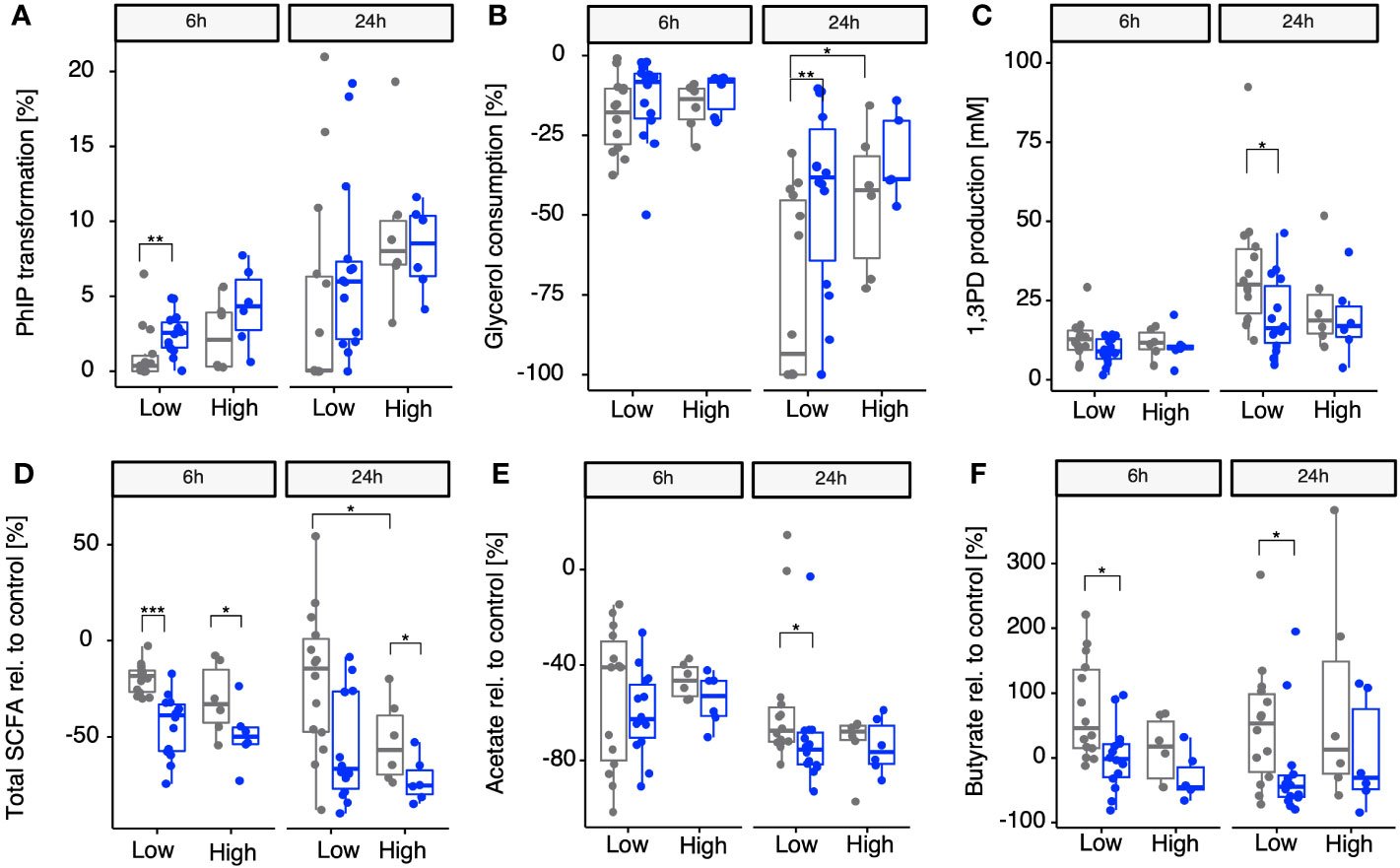
Figure 4 Impact of supplementing microbiomes with A. hallii on PhIP transformation, glycerol metabolism, and SCFAs in batch-fermented fecal microbiota. Gray points, no A. hallii supplement added. Blue points, A. hallii supplement added. Transformation of PhIP (%) by the low (< 1% abundance) and high (> 1% abundance) A. hallii abundance group (A), consumption of glycerol (B), and the production of 1,3-PD (C) and total SCFAs relative to the control condition, which was the condition in which no glycerol was added to the fermentation (D), acetate relative to the control (E), and butyrate relative to the control (F). Each box represents the median of all donors present in the low or high group. Non-parametric Wilcoxon tests were used to compare each group between A. hallii-supplemented and non-supplemented microbiota, as well as between non-supplemented microbiota in each A. hallii abundance group. Only significant comparisons are represented with adjusted p-values ≤ 0.05 (*), ≤ 0.01 (**), and ≤ 0.001 (***).
3.5 Impact of A. hallii on microbial glycerol metabolism
For PhIP transformation to occur, acrolein has to be produced from glycerol via the production of the intermediate 3-HPA, leading to the generation of 1,3-PD as the final metabolic product (Vollenweider and Lacroix, 2004). Therefore, besides PhIP transformation, the impact of A. hallii on the consumption of glycerol and the production of 1,3-PD was also assessed in the same batch fermentations after 6 h and 24 h. 1,3-PD production was used as a validation for the production of HPA in equilibrium with acrolein, both not detectable in complex cultures (Ramirez Garcia et al., 2021). When stratifying the microbiota in low and high resident A. hallii, consumption of added glycerol after 6 h was comparable for both groups. However, aligning with the previous observations, after 24 h, an increase in glycerol consumption was observed in both groups when the microbiota were supplemented with glycerol alone (median of 18% and 14% (p = 0.0495) in the low and high groups, respectively) (Figure 4B). On supplementing microbiomes with A. hallii, a significantly reduced glycerol consumption was observed in only the low A. hallii group compared with glycerol alone after 24 h (median of 8% consumed versus 18% consumed in the presence of glycerol alone, Figure 4B). In line with this, glycerol consumption significantly positively correlated with resident A. hallii proportion after 24 h (p = 0.59 and p = 0.006; Figure S2B). Concurrently, a decrease in 1,3PD production was observed (Figure 4C), which negatively correlated with the abundance of resident A. hallii in the microbiota (Figure S2C).
3.6 Impact of glycerol and A. hallii on SCFA production
To assess whether the abundance of resident A. hallii and supplementing with A. hallii played roles in the fermentation activity in the presence of glycerol, total SCFAs (as sums of acetate, propionate, and butyrate) and individual metabolites produced during the fecal fermentations were compared with control fermentations without glycerol. After 6 h, a significant decrease in total SCFAs relative to the control group was observed in both the low (median of −18% versus −39% without and with A. hallii, respectively) and high (median of −33% and −50% without and with A. hallii, respectively) groups when A. hallii was supplemented, as compared with glycerol alone (Figure 4D). The same trend was observed after 24 h, when a significant decrease of total SCFA production relative to the control was observed in the presence of glycerol and A. hallii in the high group (median of −75%), as compared with glycerol alone (median of −57%). Specifically, acetate production decreased in glycerol-supplemented microbiota of both low- and high-resident A. hallii groups after 6 h (median −40% and −46%, respectively) and 24 h (median of −67% and −65%, respectively), as compared with the control. However, when A. hallii was further supplemented, a significantly higher decrease in acetate was observed in the low group after 24 h (median of −75% versus 65%) as compared with glycerol alone (Figure 4E). Butyrate production increased in both the low and the high groups when glycerol was supplemented after both 6 h (+45% and +16% in the low and high groups, respectively) and 24 h (+53% and +12%, respectively), as compared with the control. However, on supplementing microbiomes with A. hallii, butyrate production relative to the control significantly decreased, as compared with glycerol alone, in the low group after 6 h (–2%) and 24 h (–45%) (Figure 4F). In alignment with previous observations, a negative correlation was observed between the proportion of resident A. hallii and total SCFA production relative to the control after 6 h and 24 h when glycerol was supplemented in the 20 fecal microbiota and the trend was confirmed when A. hallii was additionally supplemented after 24 h (Figure S2D).
4 Discussion
Intestinal microbiota have the potential to alter the harmful effects of food xenobiotics, such as HCA (e.g., PhIP). In this study, we characterized the influence of A. hallii on PhIP transformation efficiency and we demonstrated the key role of this taxon in the transformation capacity of 20 diverse human fecal microbiomes. By using shotgun metagenome sequencing, we identified a total of 1,839 species in 20 fecal microbial communities, 17 of which harbored gdh. Nevertheless, all tested microbiota produced 1,3PD in the presence of glycerol, the end metabolite of GDH glycerol metabolism. This confirmed previous findings (Zhang et al., 2019b; Ramirez Garcia et al., 2021) indicating that, even though a small number of taxa harbor gdh, GDH activity is still a common trait in microbiomes.
Among the gdh-harboring taxa identified in the fecal samples, and despite the differences in the fecal microbial composition observed, A. hallii was the most abundant in 95% of the samples. The MAG reconstructions further confirmed the persistence of A. hallii in all fecal microbiota, with differences in the content and organization of the pdu-cob-cbi-hem operon, possibly impacting microbial acrolein production. However, it is worth mentioning that the absence of a gene in the reconstructed MAGs does not necessarily mean that the gene is lacking in the genome of the populations because MAGs are assembled from short-read metagenomic reads, and technical aspects impact MAG binning (Chen et al., 2020).
We observed that PhIP transformation by fecal microbiota, in the presence of glycerol, was significantly correlated only with the proportion of resident A. hallii mOTUs and with none of the other 1,838 mOTUs identified. This association may be explained by the high capacity of A. hallii to produce acrolein in pure culture (Zhang et al., 2019b). Acrolein is a wide-spectrum antimicrobial, which is active against intestinal bacteria (Cleusix et al., 2007). It may provide a competitive advantage for the growth of A. hallii compared with other commensals in the gut when glycerol is present, including those that can metabolize glycerol via glycerol kinase and glycerol dehydrogenase.
Besides transforming PhIP, acrolein accumulation reduced the fermentation activity of all fecal microbiota with a stronger effect and higher acrolein accumulation being observed when the culture was supplemented with A. hallii. Likewise, for most microbiota, acrolein accumulation, detected indirectly by PhIP transformation, derived from glycerol metabolism resulted in a significant decrease of acetate and an increase in butyrate. This observation is consistent with the previous observations that supplementing glycerol to microbial communities leads to an increase in butyrate in batch fermentations (De Weirdt et al., 2010; Huber et al., 2020; Ramirez Garcia et al., 2021).
Previous in silico data showed decreased A. hallii abundance in fecal metagenomes from CRC patients, compared with healthy individuals (Zhang et al., 2019b; Ramirez Garcia et al., 2021). Therefore, A. hallii abundance might be a useful marker to estimate intestinal PhIP exposure and risk of CRC initiation. Supplementing fecal samples with A. hallii, together with glycerol, led to an overall increase in PhIP transformation, especially in the microbiota harboring an original resident A. hallii abundance of lower than 1%. This confirmed the possibility of increasing the GDH activity of complex microbiota by supplementing microbiomes with A. hallii, especially in microbiota with low resident A. hallii, as we previously suggested, with a limited number of three human fecal microbiota in vitro (Engels et al., 2016a; Ramirez Garcia et al., 2021). Overall, 60% of healthy fecal microbiota were capable of transforming PhIP by between 3% and 21% after 24 h in fecal batch fermentations, and 95% transformed PhIP when supplemented with A. hallii, indicating that acrolein was produced. However, the measured PhIP transformation and glycerol consumption by the microbiota especially after 24 h, suggested a remarkable donor-dependent response. Taken together, these observations suggest that A. hallii persistence in intestinal microbiota is linked to acrolein production and thus on PhIP to PhIP-M1 transformation by the microbiota. However, due to the very high reactivity of acrolein in the tested conditions, we could not quantify the actual production but only estimate it based on the production of 1,3-PD as a final metabolic product and indirectly prove PhIP transformation, as previously demonstrated (Engels et al., 2016b; Ramirez Garcia et al., 2021).
Besides transforming PhIP, acrolein accumulation detected indirectly by PhIP transformation reduced the fermentation activity of all fecal microbiota, with a stronger effect observed when A. hallii was further supplemented, increasing acrolein accumulation. Likewise, for most microbiota, acrolein accumulation derived from glycerol metabolism resulted in a significant decrease of acetate and to an increase in butyrate. This observation is consistent with the previous observations that supplementing glycerol to microbial communities leads to an increase in butyrate in batch fermentations (De Weirdt et al., 2010; Huber et al., 2020; Ramirez Garcia et al., 2021).
Despite the potentially beneficial effects of microbial metabolism that promotes PhIP consumption, generation of acrolein should be considered, as it can give rise to cytotoxicity and mutagenicity due to its high electrophilicity (Zhang et al., 2018; Zhang and Schwab, 2022). Its accumulation may damage the intestinal cells by degrading functional proteins (Stevens and Maier, 2008) or increasing intestinal barrier permeability (Chen et al., 2017). In the presence of high glycerol concentrations (up to 600 mM) and of specific single-producer strains, acrolein–DNA adducts can also be formed (Ramirez Garcia et al., 2022). However, such glycerol concentrations, as well as the 100 mM glycerol tested here, are unlikely to be physiologically available to gut bacteria. Furthermore, the added concentration of A. hallii (at a final concentration of 106 cells/mL) was high and in the same range as the inoculated fecal microbiota (10−4 dilution). It is still unclear what impact the concentration of acrolein, produced in the gut, has on the gut microbiota while keeping the HCA effects intact.
In conclusion, by assessing the transformation of PhIP, a carcinogen in cooked meat, of human fecal microbiota from 20 healthy individuals, we found that the proportion of A. hallii in the complex microbiota positively correlates with PhIP transformation. Furthermore, supplementing fecal microbiota with A. hallii increased PhIP transformation, indirectly indicating an accumulation of acrolein. Such accumulation resulted in altered microbial metabolic activity. Overall, the results provide a potential target for microbial manipulation by supplementing with A. hallii, which might decrease the host exposure to PhIP, although the observed effect was relatively small.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
ARG, SS, AG, CL, and CS conceived the research. ARG and JG performed the experiments. ARG, FC, KH, H-JR, and CS analyzed the data. ARG, SS, AG, CS, FC, and CL evaluated and interpreted the data. All authors contributed to manuscript writing and reviewing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the ETH Zürich (Grant ETH-41 16–1). The funding body was not involved in the study design, data collection, analysis, and interpretation, decision to publish, and preparation of the manuscript. Open access funding by ETH Zurich.
Acknowledgments
The authors would like to thank Alfonso Die, Nathalie Ziegler, Yu Wang, and Christine Dalla Pola for their technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors AG, FC, CS, CL declared that they were editorial board members of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frmbi.2023.1194516/full#supplementary-material
References
Almeida A., Nayfach S., Boland M., Strozzi F., Beracochea M., Shi Z. J., et al. (2021). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114. doi: 10.1038/s41587-020-0603-3
Arumugam M., Raes J., Pelletier E., Le Paslier D., Yamada T., Mende D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Beer F., Urbat F., Steck J., Huch M., Bunzel D., Bunzel M., et al. (2017). Metabolism of foodborne heterocyclic aromatic amines by Lactobacillus reuteri DSM 20016. J. Agric. Food Chem. 65, 6797–6811. doi: 10.1021/acs.jafc.7b01663
Bellamri M., Walmsley S. J., Turesky R. J. (2021). Metabolism and biomarkers of heterocyclic aromatic amines in humans. Genes Environ. 43, 29. doi: 10.1186/s41021-021-00200-7
Bircher L., Geirnaert A., Hammes F., Lacroix C., Schwab C. (2018a). Effect of cryopreservation and lyophilization on viability and growth of strict anaerobic human gut microbes. Microb. Biotechnol. 11, 721–733. doi: 10.1111/1751-7915.13265
Bircher L., Schwab C., Geirnaert A., Lacroix C. (2018b). Cryopreservation of artificial gut microbiota produced with in vitro fermentation technology. Microb. Biotechnol. 11, 163–175. doi: 10.1111/1751-7915.12844
Bouvard V., Loomis D., Guyton K. Z., Grosse Y., El Ghissassi F., Benbrahim-Tallaa L., et al. (2015). Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 16, 1599–1600. doi: 10.1016/S1470-2045(15)00444-1
Bushnell B., Rood J., Singer E. (2017). BBMerge – Accurate paired shotgun read merging via overlap. PloS One 12, e0185056. doi: 10.1371/journal.pone.0185056
Chaumeil P.-A., Mussig A. J., Hugenholtz P., Parks D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Chen L.-X., Anantharaman K., Shaiber A., Eren A. M., Banfield J. F. (2020). Accurate and complete genomes from metagenomes. Genome Res. 30, 315–333. doi: 10.1101/gr.258640.119
Chen W.-Y., Wang M., Zhang J., Barve S. S., McClain C. J., Joshi-Barve S. (2017). Acrolein disrupts tight junction proteins and causes endoplasmic reticulum stress-mediated epithelial cell death leading to intestinal barrier dysfunction and permeability. Am. J. Pathol. 187, 2686–2697. doi: 10.1016/j.ajpath.2017.08.015
Cleusix V., Lacroix C., Vollenweider S., Duboux M., Le Blay G. (2007). Inhibitory activity spectrum of reuterin produced by Lactobacillus reuteri against intestinal bacteria. BMC Microbiol. 7, 101. doi: 10.1186/1471-2180-7-101
De Weirdt R., Possemiers S., Vermeulen G., Moerdijk-Poortvliet T. C. W., Boschker H. T. S., Verstraete W., et al. (2010). Human faecal microbiota display variable patterns of glycerol metabolism. FEMS Microbiol. Ecol. 74, 601–611. doi: 10.1111/j.1574-6941.2010.00974.x
Dolan L., Smith K. S., Marlin M. B., Bell L. N., Blythe E., Greene M. W., et al. (2021). Food security, obesity, and meat-derived carcinogen exposure in US adults. Food Chem. Toxicol. 155, 112412. doi: 10.1016/J.FCT.2021.112412
Duncan S. H., Louis P., Flint H. J. (2004). Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl. Environ. Microbiol. 70, 5810–5817. doi: 10.1128/AEM.70.10.5810-5817.2004
Engels C., Ruscheweyh H.-J., Beerenwinkel N., Lacroix C., Schwab C. (2016a). The Common Gut Microbe Eubacterium hallii also Contributes to Intestinal Propionate Formation. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00713
Engels C., Schwab C., Zhang J., Stevens M. J. A., Bieri C., Ebert M.-O., et al. (2016b). Acrolein contributes strongly to antimicrobial and heterocyclic amine transformation activities of reuterin. Sci. Rep. 6, 36246. doi: 10.1038/srep36246
Eren A. M., Esen Ö.C., Quince C., Vineis J. H., Morrison H. G., Sogin M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319. doi: 10.7717/peerj.1319
Fekry M. I., Engels C., Zhang J., Schwab C., Lacroix C., Sturla S. J., et al. (2016). The strict anaerobic gut microbe Eubacterium hallii transforms the carcinogenic dietary heterocyclic amine 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Environ. Microbiol. Rep. 8, 201–209. doi: 10.1111/1758-2229.12369
Felton J. S., Jägerstad M., Knize M. G., Skog K., Wakabayashi K. (2000) Contents in foods, beverages and tobacco (John Wiley & Sons Ltd). Available at: https://www.cabdirect.org/cabdirect/abstract/20001417463 (Accessed January 18, 2023).
Huber D. H., Ramirez-Garcia A., Chavarria-Palma J. E., Espinosa-Solares T., Lhilhi Noundou V., Montenegro-Garcia N. A., et al. (2020). Stress induced by crude glycerol in a thermophilic digester: microbial community divergence and resilience, but slow process recovery. Appl. Microbiol. Biotechnol. 104, 10769–10781. doi: 10.1007/s00253-020-10965-6
Hyatt D., Chen G.-L., LoCascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
IARC Working Group, Risks, on the E @ of C, Group, W, Risks, on the E @ of C (1993) PhIP (2-AMINO-1-METHYL-6-PHENYLIMIDAZO[4,5-b]PYRIDINE). Available at: https://www.ncbi.nlm.nih.gov/books/NBK513600/ (Accessed January 18, 2023).
Kang H. J., Lee S. Y., Lee D. Y., Kang J. H., Kim J. H., Kim H. W., et al. (2022). Main mechanisms for carcinogenic heterocyclic amine reduction in cooked meat by natural materials. Meat. Sci. 183, 108663. doi: 10.1016/J.MEATSCI.2021.108663
Kang D. D., Li F., Kirton E., Thomas A., Egan R., An H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359. doi: 10.7717/peerj.7359
Li H., Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li W., Godzik A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Masumura K., Matsui M., Yamada M., Horiguchi M., Ishida K., Watanabe M., et al. (1999). Mutagenicity of 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) in the new gpt delta transgenic mouse. Cancer Lett. 143, 241–244. doi: 10.1016/S0304-3835(99)00132-9
McMurdie P. J., Holmes S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217. doi: 10.1371/journal.pone.0061217
Milanese A., Mende D. R., Paoli L., Salazar G., Ruscheweyh H.-J., Cuenca M., et al. (2019). Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 10, 1014. doi: 10.1038/s41467-019-08844-4
Morita H., Toh H., Fukuda S., Horikawa H., Oshima K., Suzuki T., et al. (2008). Comparative genome analysis of Lactobacillus reuteri and Lactobacillus fermentum reveal a genomic island for reuterin and cobalamin production. DNA Res. 15, 151–161. doi: 10.1093/dnares/dsn009
Muñoz-Tamayo R., Laroche B., Walter E., Doré J., Duncan S. H., Flint H. J., et al. (2011). Kinetic modelling of lactate utilization and butyrate production by key human colonic bacterial species. FEMS Microbiol. Ecol. 76, 615–624. doi: 10.1111/j.1574-6941.2011.01085.x
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Ramirez Garcia A., Hurley K., Marastoni G., Diard M., Hofer S., Greppi A., et al. (2022). Pathogenic and commensal gut bacteria harboring Glycerol/Diol dehydratase metabolize glycerol and produce DNA-reactive Acrolein. Chem. Res. Toxicol. 1840–1850. doi: 10.1021/acs.chemrestox.2c00137
Ramirez Garcia A., Zhang J., Greppi A., Constancias F., Wortmann E., Wandres M., et al. (2021). Impact of manipulation of glycerol/diol dehydratase activity on intestinal microbiota ecology and metabolism. Environ. Microbiol. 23, 1765–1779. doi: 10.1111/1462-2920.15431
Santos F., Vera J. L., van der Heijden R., Valdez G., de Vos W. M., Sesma F., et al. (2008). The complete coenzyme B12 biosynthesis gene cluster of Lactobacillus reuteri CRL1098. Microbiology 154, 81–93. doi: 10.1099/mic.0.2007/011569-0
Scott K. P., Martin J. C., Duncan S. H., Flint H. J. (2014). Prebiotic stimulation of human colonic butyrate-producing bacteria and bifidobacteria, in vitro. FEMS Microbiol. Ecol. 87, 30–40. doi: 10.1111/1574-6941.12186
Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shetty S. A., Zuffa S., Bui T. P. N., Aalvink S., Smidt H., De Vos W. M. (2018). Reclassification of Eubacterium hallii as Anaerobutyricum hallii gen. nov., comb. nov., and description of Anaerobutyricum soehngenii sp. nov., a butyrate and propionate-producing bacterium from infant faeces. Int. J. Syst. Evol. Microbiol. 68, 3741–3746. doi: 10.1099/ijsem.0.003041
Sriramulu D. D., Liang M., Hernandez-Romero D., Raux-Deery E., Lünsdorf H., Parsons J. B., et al. (2008). Lactobacillus reuteri DSM 20016 produces cobalamin-dependent diol dehydratase in metabolosomes and metabolizes 1,2-propanediol by disproportionation. J. Bacteriol. 190, 4559–4567. doi: 10.1128/JB.01535-07
Stevens J. F., Maier C. S. (2008). Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 52, 7–25. doi: 10.1002/mnfr.200700412
Tanaka T., Suzuki R., Kohno H., Sugie S., Takahashi M., Wakabayashi K. (2005). Colonic adenocarcinomas rapidly induced by the combined treatment with 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and dextran sodium sulfate in male ICR mice possess beta-catenin gene mutations and increases immunoreactivity for beta-catenin, cyclooxygenase-2 and inducible nitric oxide synthase. Carcinogenesis 26, 229–238. doi: 10.1093/carcin/bgh292
Ursell L. K., Metcalf J. L., Wegener Parfrey L., Knight R. (2012). Defining the human microbiome. Nutr. Rev. 70, s38–s34. doi: 10.1111/j.1753-4887.2012.00493.x
Vollenweider S., Lacroix C. (2004). 3-hydroxypropionaldehyde: applications and perspectives of biotechnological production. Appl. Microbiol. Biotechnol. 64, 16–27. doi: 10.1007/s00253-003-1497-y
Zhang J., Empl M. T., Schneider M., Schröder B., Stadnicka-Michalak J., Breves G., et al. (2019a). Gut microbial transformation of the dietary mutagen MeIQx may reduce exposure levels without altering intestinal transport. Toxicol. Vitr. 59, 238–245. doi: 10.1016/J.TIV.2019.04.004
Zhang J., Lacroix C., Wortmann E., Ruscheweyh H.-J., Sunagawa S., Sturla S. J., et al. (2019b). Gut microbial beta-glucuronidase and glycerol/diol dehydratase activity contribute to dietary heterocyclic amine biotransformation. BMC Microbiol. 19, 99. doi: 10.1186/s12866-019-1483-x
Zhang J., Schwab C. (2022). Is acrolein a reuterin-borne chemical hazard in biopreserved foods? Food Chem. Adv. 1, 100044. doi: 10.1016/J.FOCHA.2022.100044
Keywords: dietary carcinogens, PHIP, glycerol/diol dehydratase, reuterin, Anaerobutyricum hallii
Citation: Ramirez Garcia A, Greppi A, Constancias F, Ruscheweyh H-J, Gasser J, Hurley K, Sturla SJ, Schwab C and Lacroix C (2023) Anaerobutyricum hallii promotes the functional depletion of a food carcinogen in diverse healthy fecal microbiota. Front. Microbiomes 2:1194516. doi: 10.3389/frmbi.2023.1194516
Received: 27 March 2023; Accepted: 17 August 2023;
Published: 18 September 2023.
Edited by:
Nancy D Turner, Michigan State University, United StatesReviewed by:
Zehra Esra Ilhan, INRAE Centre Jouy-en-Josas, FranceMichael Gänzle, University of Alberta, Canada
Copyright © 2023 Ramirez Garcia, Greppi, Constancias, Ruscheweyh, Gasser, Hurley, Sturla, Schwab and Lacroix. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Lacroix, Y2hyaXN0b3BoZS5sYWNyb2l4QGhlc3QuZXRoei5jaA==
†These authors have contributed equally to this work