 Yifan Zhou
Yifan Zhou Xiaojun Zhang
Xiaojun Zhang- College of Animal Science and Technology, Yangzhou University, Yangzhou, China
Non-O1/O139 Vibrio cholerae is a highly virulent pathogen that causes mass mortalities of various aquatic animals. In the present study, we sequenced the whole genome of non-O1/O139 V. cholerae GXFL1-4, isolated from Macrobrachium rosenbergii, to reveal the pathogenicity and antibiotic resistance. The result showed its genome contained two circular chromosomes and one plasmid with a total size of 4,282,243 bp, which harbored 3,869 coding genes. Among them, 3,047, 2,659, and 3,661 genes were annotated in the Clusters of Orthologous Genes (COG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG), respectively. In addition, 372 potential virulence genes were predicted based on the Virulence Factor Database (VFDB) database, such as type II, III, IV, and VI secretion systems related genes, flagella genes, and pilus formation or motility-related genes. Blast results in the Comprehensive Antibiotic Resistance Database (CARD) database showed that the strain contained 148 antibiotic resistance-related genes belonging to 27 categories, such as efflux pump complex antibiotic resistance genes and antibiotic resistance gene cluster genes. The Pathogen-Host Interaction (PHI) database annotated 320 genes related to pathogen-host interaction, such as T3SS, virulence regulatory factors, transcriptional regulators, and two-component response regulator related genes. The whole-genome analysis suggested that the pathogenic non-O1/O139 V. cholerae strain GXFL1-4 might have a complex molecular mechanism of pathogenicity and antibiotic resistance. This study provides a wealth of information about non-O1/O139 V. cholerae genes related to its pathogenicity and drug resistance and will facilitate the understanding of its pathogenesis as well as the development of prevention and treatment strategies for the pathogen.
Introduction
Vibrio cholerae is a Gram-negative bacterium with more than 200 serogroups, which do not possess the cholera toxin, and the toxin-coregulated pilus virulence factors are commonly designated as non-O1/O139 V. cholerae. Non-O1/O139 V. cholerae is an important pathogen of zoonosis that is ubiquitously distributed in aquatic environments (1). With the intensification of aquaculture, non-O1/O139 V. cholerae has become one of the most serious problems to endanger sustainable aquaculture development (2). The non-O1/O139 V. cholerae caused high mortality and great economic loss in a variety of aquatic animals, such as Penaeus vannamei, Macrobrachium nipponense, Cyprinus carpio, and Carassius auratus (3–5). For the great losses caused by non-O1/O139 V. cholerae to the aquaculture industry, our previous studies revealed that non-O1/O139 V. cholerae GXFL1-4 is a highly virulent strain isolated from M. rosenbergii that causes mass mortalities in M. rosenbergii (6). Despite these prior studies, the pathogenic mechanism of non-O1/O139 V. cholerae during infection in aquatic animals is poorly understood.
Recent vigoros complete genome analysis has been performed to unravel putative strain-specific virulence determinants. Complete genome analysis of pathogenic bacteria provides a powerful tool for better understanding the mechanisms of bacterial pathogenicity and their drug-resistant properties, which provide the new genetic and molecular approaches for controlling the disease (7). Whole-genome sequencing has been increasingly employed to analyze the relationship among the aquatic animals, the environment, and the virulence of strains (8, 9). At present, there are some complete genome sequences of V. cholerae from humans and environments in the Genbank genome database. However, the genetic features and evolutionary strategies of non-O1/O139 V. cholerae from aquatic animals remain largely unknown.
In this study, the complete genome of highly pathogenic non-O1/O139 V. cholerae strain GXFL1-4, isolated from the diseased M. rosenbergii, was sequenced, and the genes related to pathogenicity, drug resistance, and host–pathogens interaction were identified. This research will provide valuable insights into the prevention and treatment of diseases caused by non-O1/O139 V. cholerae.
Materials and Methods
Bacteria, Library Construction, and Sequencing
A non-O1/O139 V. cholerae GXFL1-4 was isolated from diseased M. rosenbergii at a commercial aquaculture farm in Gaoyou County, Jiangsu Province, China. Challenges showed that bath immersion of strain GXFL1-4 caused red body syndrome and mass mortalities of M. rosenbergii (6). The strain was inoculated into 100 ml of LB broth medium and incubated at 28°C with shaking at 200 rpm for 18 h. The culture was then centrifuged at 10,000 rpm for 2 min to pellet the cells. The collected cells were then washed and re-suspended in sterile 1 × phosphate-buffered saline (PBS). From this, 1 ml aliquots were collected in microcentrifuge tubes and used for further studies.
Lim et al. used the sodium dodecyl sulfate (SDS) method to extract genomic DNA (10). The harvested DNA was detected by the agarose gel electrophoresis and quantified by the Qubit® 2.0 Fluorometer (Thermo Scientific). A total amount of 1 μg DNA per sample was used as input material for the DNA sample preparations. Sequencing libraries were generated using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer's recommendations and index codes were added to attribute sequences to each sample. The whole genome of GXFL1-4 was sequenced using Illumina NovaSeq PE150 at the Beijing Novogene Bioinformatics Technology Co., Ltd.
Genome Assembly
Clean data are assembled with Short Oligonucleotide Alignment Program SOAP denovo software (11), SPAdes software (12), and Assembly By Short Sequences (Abyss) software (13). The assembly results of the three software were integrated with CISA software and the assembly result with the least scaffolds was selected. The GapCloser software was used to fill the gap in the preliminary assembly results. The same lane pollution by filtering the reads with low sequencing depth was removed to obtain the whole genome.
Genome Component Prediction
For bacteria, we used the GeneMarkS (Version 4.17) program to retrieve the related coding gene (14). The interspersed repetitive sequences were predicted using the RepeatMasker (Version open-4.0.5) (15). Transfer RNA (tRNA) genes were predicted by the tRNAscan-SE (Version 1.3.1) (16, 17). Ribosome RNA (rRNA) genes were analyzed by the rRNAmmer (Version 1.2). Small nuclear RNAs (snRNA) were predicted by BLAST against the Rfam database (Version 1.1rc4) (18, 19). The IslandPath-DIOMB program was used to predict the Genomics Islan (Version 0.2ds) (20).
Gene Function
We used different databases to predict gene functions, which were Gene Ontology (GO) (http://www.geneontology.org) (21), Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg) (22), and Clusters of Orthologous Groups (COG) (http://www.ncbi.nlm.nih.gov/COG/) (23), respectively. The prediction of Type I–VII proteins secreted by the pathogenic bacteria was based on the Effective T3 software (Version 1.0.1). For the pathogenicity and antibiotic resistance of pathogenic bacteria, we used the Pathogen Host Interactions (PHI) (http://www.phi-base.org/index.jsp) (24), Virulence Factors of Pathogenic Bacteria (VFDB) (http://www.mgc.ac.cn/VFs) (25), and Comprehensive Antibiotic Research Database (CARD) (http://arpcard.mcmaster.ca) (26) to perform the above analyses. A whole-genome BLAST search (E-value less than 1e-5, minimal alignment length percentage larger than 40%) was conducted against the above databases.
Antibiotic-Resistant Test
The sensitivity of strain GXFL1-4 to 36 antibiotics was evaluated by disc diffusion method (27) on Muller-Hinton agar using commercial antibiotic discs. All the tests were performed in triplicate. The antibiotic sensitivity of the strains as resistant, intermediate, or sensitive was measured by the inhibition zone diameter according to standards suggested by Hangzhou Tianhe Microorganism Reagent Co.
Results
Genome Structure and General Features
The genome of non-O1/O139 V. cholerae strain GXFL1-4 was sequenced using Nanopore, and the genome characteristics are summarized in Table 1. After filtering, all reads were assembled into two circular chromosomes and one circular plasmid (Figure 1). Chromosome I and II are 1,154,802 and 3,079,090 bp, with a G + C content of 45.99 and 47.97%, respectively (NCBI accession number CP090386 and CP090387). The plasmid is 48,351 bp, with a G+C content of 41.3% (NCBI accession number CP090388). Table 1 and Figure 1 show the annotation information of general features.
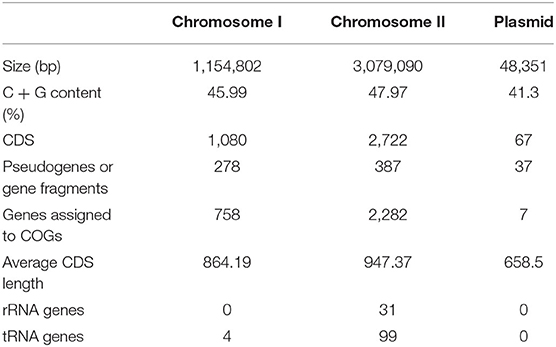
Table 1. Genomic information for non-O1/O139 Vibrio cholerae strain GXFL1-4.

Figure 1. Circular genome map of non-O1/O139 Vibrio cholerae GXFL1-4.
COG Analysis
Clusters of Orthologous Groups annotation results showed that 3,047 genes were annotated into 24 classes of genes, accounting for 78.75% of total genes in non-O1/O139 V. cholerae strain GXFL1-4. The number of each class of gene was as follows: RNA processing and modification gene (1), chromatin structure and dynamics gene (1), energy production and conversion genes (183), cell cycle control, cell division, and chromosome partitioning genes (43), amino acid transport and metabolism genes (284), nucleotide transport and metabolism genes (82), carbohydrate transport and metabolism genes (202), coenzyme transport and metabolism genes (172), lipid transport and metabolism genes (108), translation, ribosomal structure, and biogenesis genes (252), transcription genes (252), replication, recombination, and repair genes (145), cell wall/membrane/envelope biogenesis genes (196), cell motility genes (130), posttranslational modification, protein turnover, chaperones genes (159), inorganic ion transport and metabolism genes (173), secondary metabolites biosynthesis, transport, and catabolism genes (67), general function prediction only genes (253), function unknown genes (200), signal transduction mechanisms genes (308), intracellular trafficking, secretion, and vesicular transport genes (84), defense mechanisms genes (92), extracellular structures genes (28), and mobilome: prophages and transposons genes (63) (Figure 2). Supplementary Table 1 lists all COG functional annotation information.
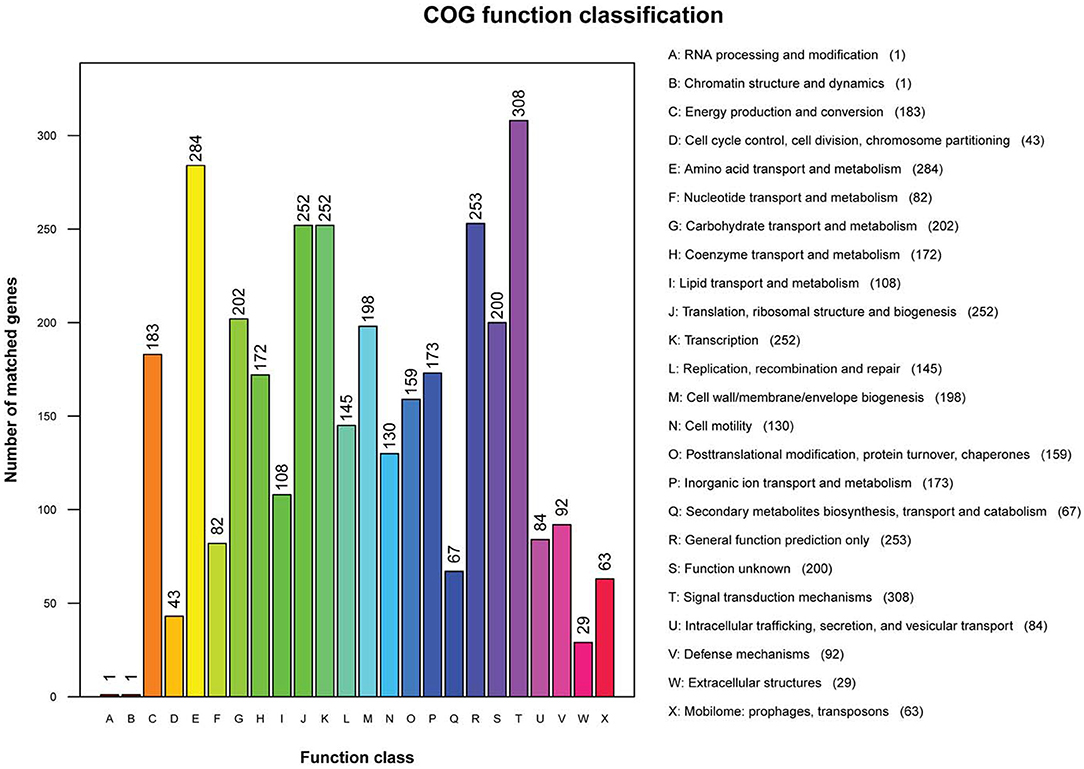
Figure 2. The Clusters of Orthologous Genes (COG) functional annotation in the whole genome of non-O1/O139 V. cholerae strain GXFL1-4.
GO Analysis
The functional annotation results in the GO database showed that 2,659 genes were annotated into three classes of genes, which accounted for 68.72% of total genes of non-O1/O139 V. cholerae strain GXFL1-4. Among them, 1,041 genes were related to the cellular component, 2,142 genes were related to the molecular function, and 2069 genes were related to the biological process. The highest numbers of genes in the classification of the biological process were biological adhesion process (1,507) and biological regulation (1,491). The highest numbers of genes in the classification of cellular components were cell (1,023), cell part(1,023), extracellular region (213), and extracellular region part (193). The highest numbers of genes in the classification of molecular function were antioxidant activity (1,429) and binding (1,273). More information is shown in Figure 3. Supplementary Table 2 lists all GO functional annotation information.
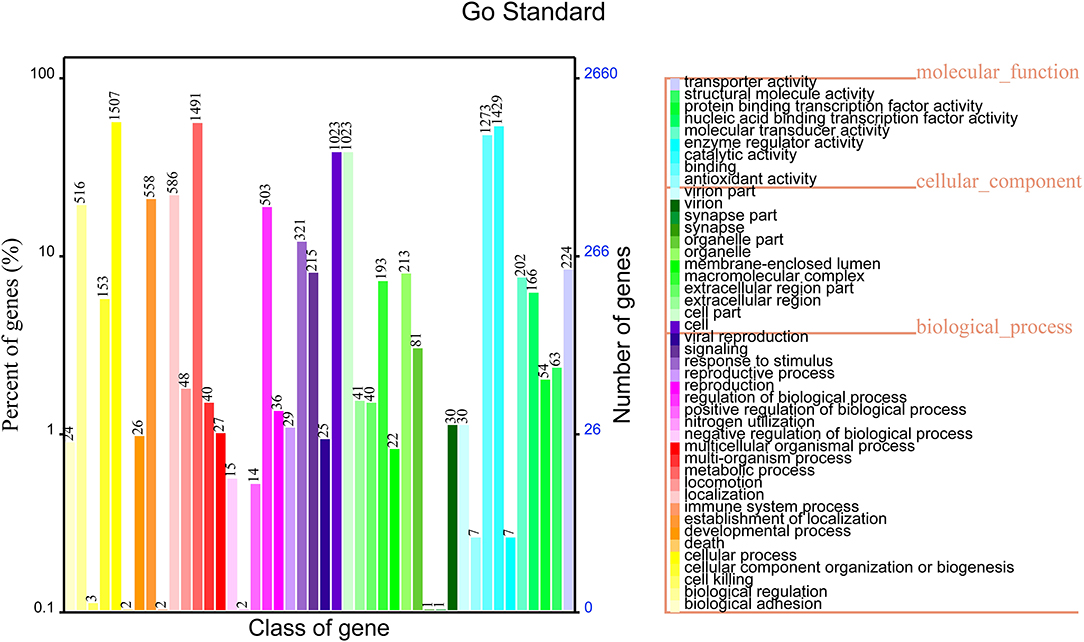
Figure 3. Gene Oncology (GO) functional annotation in the whole genome of non-O1/O139 V. cholerae strain GXFL1-4.
KEGG Analysis
The results of the KEGG pathway analysis showed that 3,661 genes were annotated into 189 known metabolic pathways. Among them, the largest number of genes was ABC transporters (103), followed by the two-component system (101), biofilm formation (83), ribosome (54), and quorum sensing (48). Cluster analysis showed that 196 metabolic pathways were categorized into six classifications of cellular processes, metabolism, human diseases, genetic information processing, organismal systems, and environmental information processing, and the numbers of genes in these six classifications were 215, 681, 78, 184, 37, and 275, respectively (Figure 4). The 215 genes in the classification of cellular processes could be divided into four categories, and most of them were clustered into the cellular community—prokaryotes (173) and cellular community-prokaryotes (52). The 681 genes in the classification of metabolism were divided into 11 categories. The categories included carbohydrate metabolism (287), amino acid metabolism (205), metabolism of cofactors and vitamins (159), and energy metabolism (127). The 78 genes in the classification of human diseases were clustered into 7 categories, and the categories of the largest gene number were antibiotic resistance: drug resistance (44), infectious diseases (22), and neurodegenerative diseases (14). The 184 genes in the classification of genetic information processing were clustered into three categories. The categories of the largest gene numbers were transcription (85), replication and repair (81), and folding, sorting, and degradation (49). The 37 genes in the classification of organismal systems were divided into eight categories. Among them, the top three categories with the largest gene numbers were the endocrine system (11), aging (7), immune system (6), and nervous system (5). The environmental information was divided into two categories, such as membrane transport (162) and signal transduction (115). Supplementary Table 3 shows all KEGG pathway annotation information.
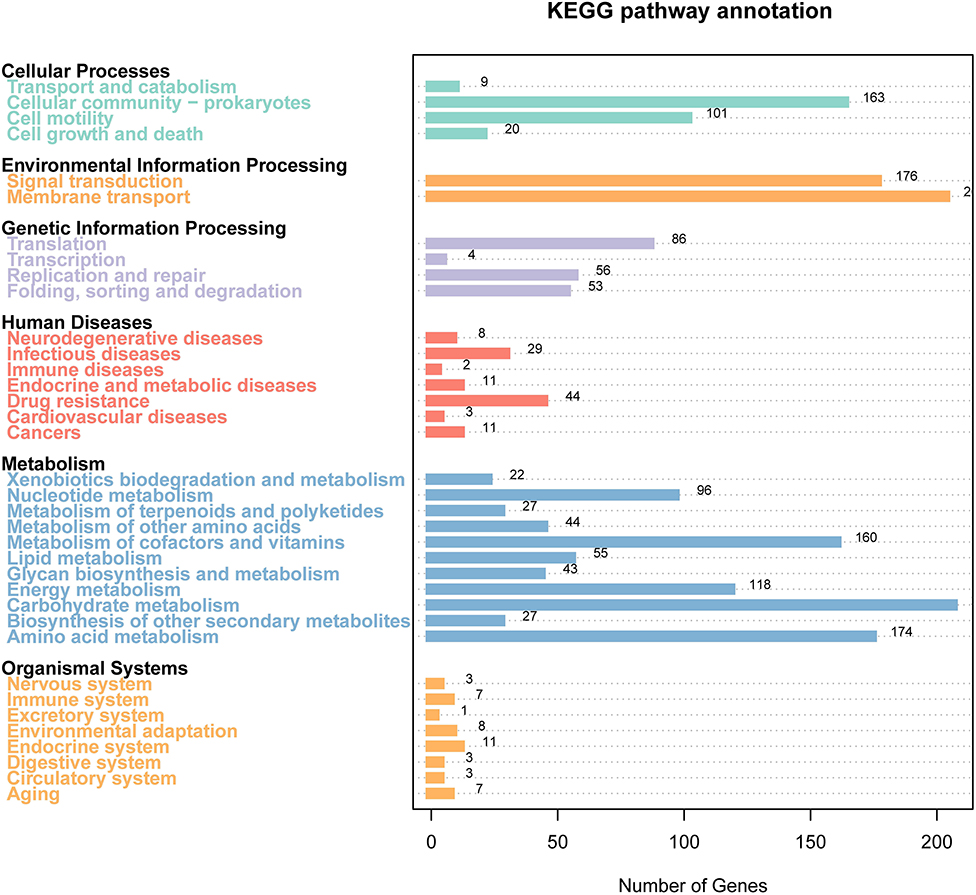
Figure 4. Gene distribution based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) classification of non-O1/O139 V. cholerae GXFL1-4.
Prediction of Virulence Genes of the Strains GXFL1-4
A total of 372 potential virulence genes were predicted in the whole genome of the strain GXFL1-4. Moreover, 174 genes had annotation information in VFDB database that included 143 offensive virulence genes, 22 defensive virulence genes, 6 virulence-associated regulation genes, and 3 non-specific virulence genes (Table 2). Supplementary Table 4 shows detailed VFDB annotation information.
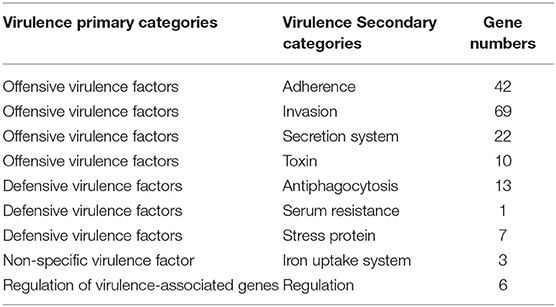
Table 2. The annotation of virulence factors of the strain GXFL1-4 in VFDB databases.
Among the offensive virulence genes, there were 42 genes related to adherence, including pilus formation or motility genes, such as mshB/C/D, mshE/F/G, membrane protein ompU, and multivalent adhesion molecule MAM7. There were 69 genes related to invasion, including flagella formation or motility exercise-related genes, such as flaA, fleQ, flgB, flaG, flhA, fliG, fliM, motA, transcriptional regulator fleQ, flrA, chemotaxis protein, cheA, and cheR.
Analysis of Antibiotic Resistance
A total of 36 antibiotics belonging to 12 categories were selected to test the antimicrobial phenotype of strain GXFL1-4 through the Kirby-Bauer disk diffusion method (Table 3). It was sensitive to cefoxitin, streptomycin, tobramycin, erythromycin, spectinomycin, chloramphenicol, amikacin, tetracycline, polymyxin B, and minocycline.
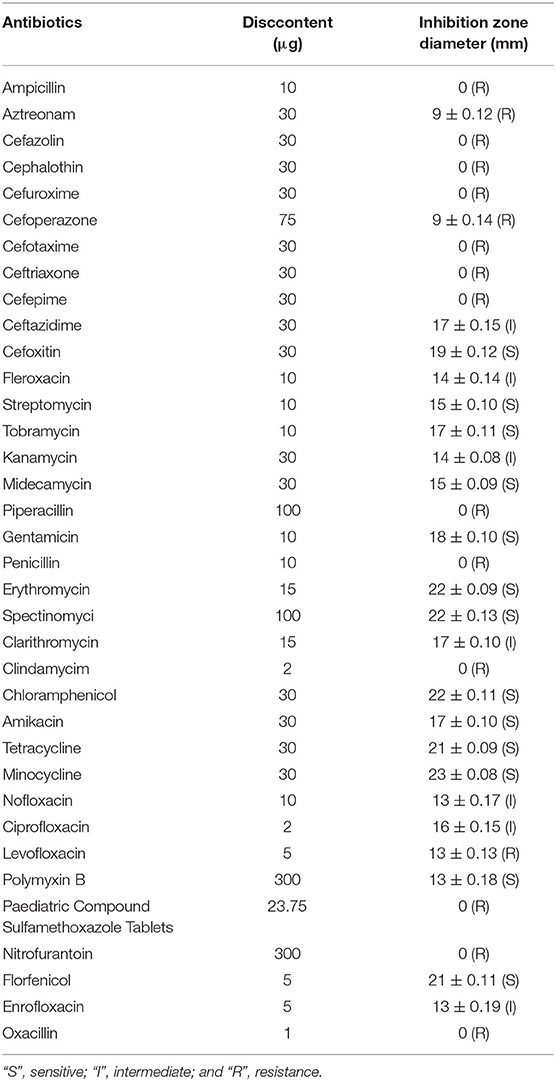
Table 3. Antibiotic resistance of non-O1/O139 V. cholera GXFL1-4.
The results of the CARD database showed that 148 antibiotic resistance genes belonging to 27 categories were found in the whole genome of the strain GXFL1-4 (Table 4). In terms of antibiotic resistance phenotype and genotype correlation, the strain GXFL1-4 contained the penam-resistant genes (evgS, adeL, CRP, NmcR, mecC, and smeR), cephalosporin-resistant genes (NmcR, CTX-M-99, and smeR), monobactam-resistant genes (OpmB), quinolone-resistant genes (evgS, adeL, patA, arlR, mfd, MexF/V/I, CRP, cdeA, QnrVC5, emrB, and hmrM), sulfonamide-resistant genes (sul2/3), and lincosamide-resistant genes [msrB, RlmA(II), and clbB]. The phenotype was consistent with the genotype. In addition, multidrug efflux pump systems and two-component regulatory systems were also found in the strain. Supplementary Table 5 shows antibiotic resistance genes additional information.
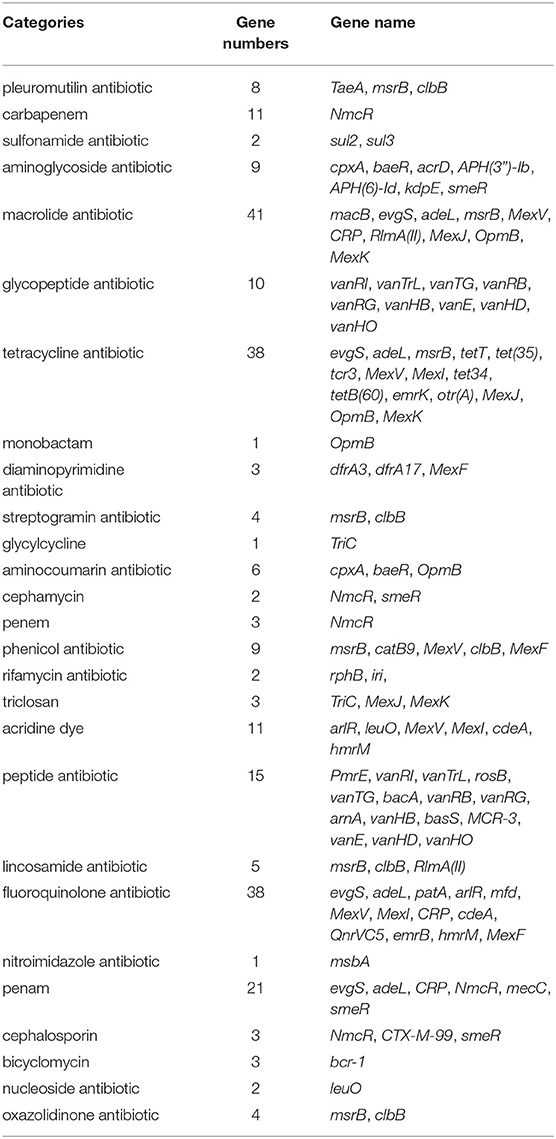
Table 4. The antibiotic resistance genes of the strain GXFL1-4 annotated in CARD.
Genomic Islands Analysis
Through the IslandPath-DIOMB online system, 9 genomic islands were predicted to be contained in the whole genome of the strain GXFL1-4 (Figure 5). The longest genomic island was 81,279 bp, and the shortest one was 6,594 bp. Supplementary Table 6 illustrates their detailed information.
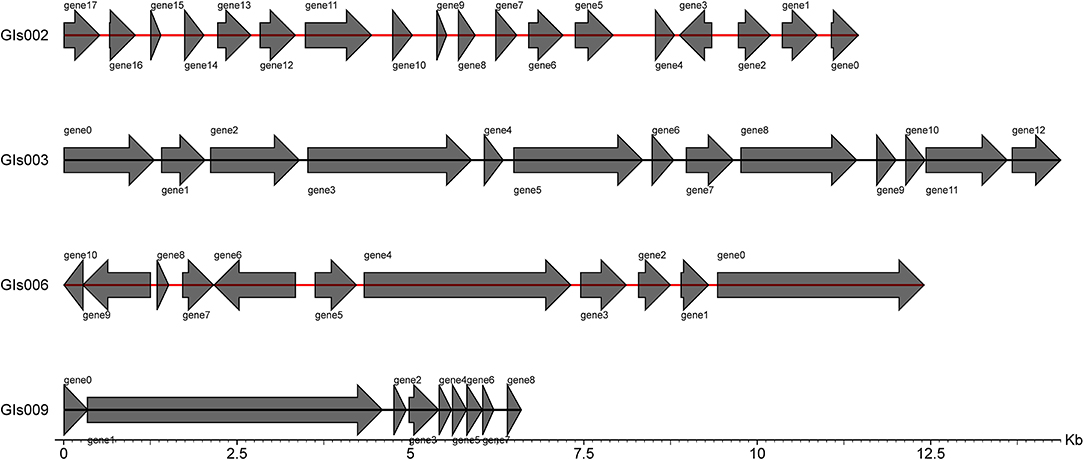
Figure 5. Statistical map of gene distribution in genomic islands.
Predictive Analysis of Pathogen-Host Interaction Between the Strain GXFL1-4 and Host
According to the annotation results of the PHI database (Figure 6), the strain GXFL1-4 contained 320 genes related to PHI. Among them, there were 201 genes related to reduced virulence, 27 genes related to loss of pathogenicity, 14 genes related to hypervirulence, 6 genes related to lethal factors, 5 genes related to effectors, 2 genes related to chemical resistance, 2 genes related to chemical sensitivity, and 61 genes related to unaffected pathogenicity. The key genes associated with high pathogenicity were hypervirulence and lethal genes. Among them, such as T3SS function genes: Ndk, GroEL, Rv2220, and fabG1; virulence regulatory factors: raxQ, raxP, and kdpE; transcriptional regulators, aphB and ohrR; and two-component response regulator, LuxO, arcA, and CpxR.
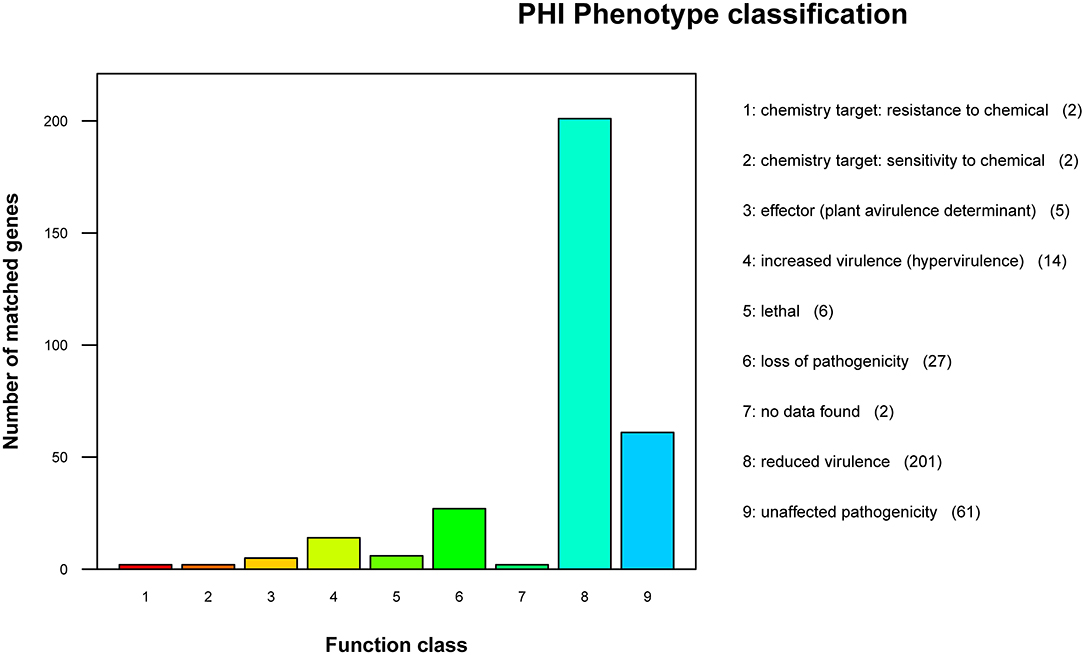
Figure 6. Pathogen-Host Interaction (PHI) phenotype mutation type distribution map.
Secretory System Analysis
In this study, type II, type III, type IV, and type VI secretory systems were represented by genes in the GXFL1-4 genome. Moreover, 13 genes were assigned to types II, 155 genes were assigned to type III, two genes were assigned to type VI, and one gene was assigned to type VI. The genes assigned to the type II secretory systems were all present on chromosome II. The genes assigned to the type IV secretory systems were present on plasmid I. The genes assigned to the type VI secretory systems were present on chromosome I. In non-O1/O139 V. cholerae strain GXFL1-4, type III secretory system related genes were encoded on both chromosomes I and II, as well as on the plasmid.
Discussions
In this study, we found that the whole genome of pathogenic non-O1/O139 V. cholerae strain GXFL1-4 was 4,282,243 bp in length, including two circular chromosomes and one plasmid, which was consistent with the genomes of V. cholera (29). In the gene annotation analysis, a large number of genes were annotated in the KEGG pathways, which are related to human diseases. Previous studies also showed that non-O1/O139 V. cholerae can cause liver disease, lung disease, gastroenteritis, and so on (28, 30, 31), which demonstrates the potential threat of non-O1/O139 V. cholerae strain GXFL1-4 to humans.
Extracellular products play an important role in the pathogenesis of various aquatic pathogens. In this study, the prediction of virulence-related genes showed that non-O1/O139 V. cholerae GXFL1-4 contains caseinase-related genes (Vsp), lipase-related genes (YplA), lecithinase–related genes (VvPlpA), and hemolysin-related genes (HlyA) in the whole genome. Previous studies have confirmed that the above extracellular products can cause harm to aquatic animals (6, 32). In addition, a number of genes coding motility and synergistic factors have been annotated, such as the flagella-related genes (flaA, fliD, flgB), chemotaxis factors (cheA, cheB, cheY), pili-related genes (pilE, fimT, mshB), and outer membrane proteins-related genes (OmpU). These genes were responsible for colonizing the host through movement and adhesion, finally causing the host disease (33, 34). These results indicated that non-O1/O139 V. cholerae carries many virulence genes, which might be important for pathogenicity.
It is generally believed that proteins and toxins of pathogenic bacteria are related to secretory systems. In this study, the type II, type III, type IV, and type VI secretory systems-related genes were predicted in the non-O1/O139 V. cholerae GXFL1-4 genome. In the genome data, the type II pathway components are encoded by 12 extracellular protein secretion (eps) genes and one mannose-sensitive hemagglutinin (MSHA) pili gene. These genes support proteins and toxins to cross the outer membrane, its process including translocation across the cytoplasmic membrane and the outer membrane (35). Often, the type II secretory system works along with other secretion systems to achieve full virulence. The type III secretory system promotes the transfer of bacterial effector proteins to the cytoplasm or the plasma membrane of target eukaryotic cells. In type III secretory system, a large number of flagellin genes, flagellar-related genes, and chemotaxis protein-related genes are annotated, which promote bacterial invasion and colonization, subvert host cells (36). The type IV secretory system is the most ubiquitous secretion system in nature, which annotated gene TraK can mediate the translocation of DNA or effector molecules into bacterial and eukaryotic target cells (37). The type VI secretory system is a cell envelope spanning machine in which the related gene VgrG translocates toxic effector proteins into eukaryotic and prokaryotic cells and has a pivotal role in pathogenesis and bacterial competition (38).
Multiple antibiotic resistance genes were identified in our predictive analysis of antibiotic resistance genes. Among them, activation of the CpxA system stimulates mar transcription, indicating that it can enhance the multidrug resistance cascade (39). Functional studies have shown that MsbA also is a polyspecific transporter capable of recognizing and transporting a wide spectrum of drug molecules (40). Moreover, multidrug efflux pumps are thought to be involved in mediating multidrug resistance in pathogenic bacteria. OpmB gene products showed functional cooperation with a membrane fusion protein of the MexVW-OprM multidrug efflux complex, MexV, a channel that is formed through both the inner and outer membrane, which can mediate the removal of xenobiotics, such as antibiotics, responding for resistance to antimicrobial agents (41). These results showed that the resistance mechanism of the strain GXFL1-4 might be complex.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
YZho, SG, JL, PJ, and YZha contributed to conception and design of the study. YZho organized the database. CW performed the statistical analysis. QJ wrote the first draft of the manuscript. XG and XZ wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by the National Natural Science Fund (31972830), the earmarked fund for Jiangsu Agricultural Industry Technology System (JATS [2021] 501, JATS [2021] 505), the National Key Research and Development Project (2019YFD0900305), and Revitalizing of Seed Industry—the Open Competition Mechanism to Select the Best Candidates Projects (JBGS [2021] 120). This work was generously supported by a studentship from the Yangzhou University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Novogene for sequencing, genome annotation, and helpful suggestions for the analysis of this data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.882885/full#supplementary-material
References
1. Li F, Du P, Li B, Ke C, Chen A, Chen J, et al. Distribution of virulence-associated genes and genetic relationships in non-O1/O139 Vibrio cholerae aquatic isolates from China. Appl Environ Microbiol. (2014) 80:4987–92. doi: 10.1128/AEM.01021-14
2. Zhang XJ, Yao DR, Yan BL, Bi KR, Liang LG, Qin GM. Identification of Vibrio cholerae as a causative bacterium for an ulcer disease of cultured loach Misgurnus anguillicaudatus in China. Afr J Microbiol Res. (2012) 6:2060–70. doi: 10.5897/AJMR11.1344
3. Cao H, An J, Zheng W, He S. Vibrio cholerae pathogen from the freshwater-cultured white leg shrimp Penaeus vannamei and control with Bdellovibrio bacteriovorus. J Invertebr Pathol. (2015) 130:13–20. doi: 10.1016/j.jip.2015.06.002
4. Li XX, Yang H, Gao XJ, Zhang HH, Chen N, Miao Z, et al. The pathogenicity characterization of non-O1 Vibrio cholerae and its activation on immune system in freshwater shrimp Macrobrachium nipponense. Fish Shellfish Immunol. (2019) 87:507–14. doi: 10.1016/j.fsi.2019.01.050
5. Raja ST, Gaurav R, Neeraj S, Singh LW. Detection of rtxA gene and other virulence genes in non-O1 and non-O139 Vibrio cholerae isolated from ornamental fishes in India. Indian J Anim Res. (2011) 78:787–9. doi: 10.1051/gse:2008013
6. Gao XJ, Miao Z, Li X, Chen N, Gu WW, Liu XD, et al. Pathogenicity of non-O1/ O139 Vibrio cholerae and its induced immune response in Macrobrachium rosenbergii. Fish Shellfish Immunol. (2019) 92:300–307. doi: 10.1016/j.fsi.2019.06.032
7. Zhang S, Huang J, Di J, Du H, Xu Q, Qiong Z, Congiu L, Wei Q. The genome sequence of a new strain of Mycobacterium ulcerans ecovar Liflandii, emerging as a sturgeon pathogen. Aquac. (2018) 489:141–7. doi: 10.1016/j.aquaculture.2018.02.001
8. Fu S, Ni P, Yang Q, Hu H, Liu Y. Delineating the key virulence factors and intra-species divergence of Vibrio harveyi by the use of whole genome sequencing. Can J Microbiol. (2020) 67:231–48. doi: 10.1139/cjm-2020-0079
9. Zhang XJ, Chen CZ, Yan BL, Fang H, Xu J. Phenotypic and molecular characterization of pathogenic Vibrio parahaemolyticus isolated from Penaeus vannamei. Oceanol Limnol Sin. (2009) 40:654–62. doi: 10.3321/j.issn:0029-814X.2009.05.021
10. Lim HJ, Lee EH, Yoon Y, Chua B, Son A. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J Appl Microbiol. (2016) 120:379–87. doi: 10.1111/jam.13011
11. Li R, Li Y, Kristiansen K, Jun W. Sequence analysis SOAP: short oligonucleotide alignment program. Bioinformatics. (2013) 24:713–4. doi: 10.1093/bioinformatics/btn025
12. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
13. Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. (2009) 19:1117–23. doi: 10.1101/gr.089532.108
14. John B, Alexandre L, Mark B. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. (2001) 29:2607–18. doi: 10.1093/nar/29.12.2607
15. Saha S, Bridges S, Magbanua ZV, Peterson DG. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. (2008) 36:2284–94. doi: 10.1093/nar/gkn064
16. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. (1997) 25:955–64. doi: 10.1093/nar/25.5.955
17. Karin L, Peter H, Andreas RE, Henrik SH, Torbjørn R, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. (2007) 35:3100–8. doi: 10.1093/nar/gkm160
18. Gardner PP, Jennifer D, Tate JG, Nawrocki EP, Kolbe DL, Stinus L, et al. Rfam: updates to the RNA families database. Nucleic Acids Res. (2009) 37:136–40. doi: 10.1093/nar/gkn766
19. Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. (2009) 25:1335–37. doi: 10.1093/bioinformatics/btp157
20. William H, Wan I, Jones SJ, Brinkman FSL. IslandPath: aiding detection of genomic islands in prokaryotes. Bioinform. (2003) 19:418–20. doi: 10.1093/bioinformatics/btg004
21. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. (2000) 25:25–9. doi: 10.1038/75556
22. Minoru K, Susumu G, Shuichi K, Yasushi O, Masahiro H. The KEGG resource for deciphering the genome. Nucleic Acids Res. (2004) 32:D277–80. doi: 10.1093/nar/gkh063
23. Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. (2015) 43:261–9. doi: 10.1093/nar/gku1223
24. Martin U, Rashmi P, Arathi R, Irvine AG, Helder P, Hammond-Kosack KE. The Pathogen-Host Interactions database (PHI-base): additions and future developments. Nucleic Acids Res. (2015) 36:572–76. doi: 10.1093/nar/gku1165
25. Chen L, Xiong Z, Sun L, Yang J, Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. (2012) 40:641–5. doi: 10.1093/nar/gkr989
26. Jia B, Raphenya AR, Alcock B, Waglechner N, McArthur AG. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. (2017) 45:566–73. doi: 10.1093/nar/gkw1004
27. Igbinosa IH, Chigor VN, Igbinosa EO, Obi LC, Okoh AI. Antibiogram. Adhesive characteristics, and incidence of class 1 integron in aeromonas species isolated from two south african rivers. Biomed Res Int. (2013) 13:2127570. doi: 10.1155/2013/127570
28. Wei Y, Yu Y, Yang H. Non-O1/Non-O139 Vibrio cholera bacteremia in a patient with autoimmune liver disease. J Emerg Crit Care Med. (2018) 2:1–2. doi: 10.21037/jeccm.2018.10.13
29. Kumar S, Lindquist IE, Sundararajan A. Genome sequence of non-O1 Vibrio cholerae PS15. Genome Announc. (2013) 1:e00227-12. doi: 10.1128/genomeA.00227-12
30. Shannon JD, Kimbrough RC. Pulmonary cholera due to infection with a non-O1 Vibrio cholerae strain. J Clin Microbiol. (2006) 9:3459–60. doi: 10.1128/JCM.02343-05
31. Namdari H, Klaips CR, Hughes JL. A cytotoxin-producing strain of Vibrio cholerae non-O1, non-O139 as a cause of cholera and bacteremia after consumption of raw clams. J Clin Microbiol. (2000) 9:3518. doi: 10.1128/JCM.38.9.3518-3519.2000
32. Jiang ZY, Gao XJ, Jiang Q, Zhu XH, Zhou YF, Zhang ZR, et al. Genomic characterization and pathogenicity analysis of the Vibrio mimicus Y4 causing red body disease in Macrobrachium nipponense. Aquaculture. (2021) 15:548–56.
33. Sharma A, Chaturvedi AN. Prevalence of virulence genes (ctxA, stn, OmpW and tcpA) among non-O1 Vibrio cholerae isolated from fresh water environment. Int J Hyg Environ Health. (2006) 209:521–6. doi: 10.1016/j.ijheh.2006.06.005
34. Girón JA, Torres AG, Freer E, James BK. The flagella of enteropathogenic Escherichia coli mediate adherence to epithelial cells. Mol Microbiol. (2002) 44:361–79. doi: 10.1046/j.1365-2958.2002.02899.x
35. Sandkvist M. Type II secretion and pathogenesis. Infect Immun. (2001) 69:3523–35. doi: 10.1128/IAI.69.6.3523-3535.2001
36. Gerlach RG, Hensel M. Protein secretion systems and adhesins: the molecular armory of Gram-negative pathogens. Int J Med Microbiol. (2007) 297:401–15. doi: 10.1016/j.ijmm.2007.03.017
37. Harris RL, Hombs V, Silverman PM. Evidence that F-plasmid proteins TraV, TraK and TraB assemble into an envelope-spanning structure in Escherichia coli. Mol Microbiol. (2010) 42:757–66. doi: 10.1046/j.1365-2958.2001.02667.x
38. Hachani A, Lossi NS, Hamilton A, Sophie B, David AJ, Alain F. Type VI secretion system in Pseudomonas aeruginosa: secretion and multimerization of VgrG proteins. J Biol Chem. (2011) 286:12317–27. doi: 10.1074/jbc.M110.193045
39. Griffin NW, Yang D, Kong W, Hua Z, Shi Y. The CpxR/CpxA two-component regulatory system up-regulates the multidrug resistance cascade to facilitate Escherichia coli resistance to a model antimicrobial peptide. J Biochem Chem. (2014) 47:32571–82. doi: 10.1074/jbc.M114.565762
40. Ward A, Reyes CL, Yu J, Roth CB, Geoffrey C. Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc Natl Acad Sci. (2009) 48:19005–10. doi: 10.1073/pnas.0709388104
Keywords: non-O1/O139 Vibrio cholerae, genomic characterization, virulence-related genes, antibiotic resistance-related genes, secretion systems
Citation: Zhou Y, Gu S, Li J, Ji P, Zhang Y, Wu C, Jiang Q, Gao X and Zhang X (2022) Complete Genome Analysis of Highly Pathogenic Non-O1/O139 Vibrio cholerae Isolated From Macrobrachium rosenbergii Reveals Pathogenicity and Antibiotic Resistance-Related Genes. Front. Vet. Sci. 9:882885. doi: 10.3389/fvets.2022.882885
Received: 24 February 2022; Accepted: 04 April 2022;
Published: 18 May 2022.
Edited by:
Lixing Huang, Jimei University, ChinaCopyright © 2022 Zhou, Gu, Li, Ji, Zhang, Wu, Jiang, Gao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojun Zhang, zxj9307@163.com