Neuroscience Programme, International School for Advanced Studies, Trieste, Italy
Early in postnatal life correlated GABAergic activity in the hippocampus is thought to play a crucial role in synaptogenesis and in the development of adult neuronal networks. Unlike adulthood, at this developmental stage, mossy fibers (MF) which are the axons of granule cells, release GABA into CA3 principal cells and interneurons. Here, we tested the hypothesis that at MF-CA3 connections, tonic activation of GABAB autoreceptors by GABA is responsible for the low probability of release and synapse silencing. Blocking GABAB receptors with CGP55845 enhanced the probability of GABA release and switched on silent synapses while the opposite was observed with baclofen. Both these effects were presynaptic and were associated with changes in paired-pulse ratio and coefficient of variation. In addition, enhancing the extracellular GABA concentration by repetitive stimulation of MF or by blocking the GABA transporter GAT-1, switched off active synapses, an effect that was prevented by CGP55845. In the presence of CGP55845, stimulation of MF-induced synaptic potentiation. The shift of EGABA from the depolarizing to the hyperpolarizing direction with bumetanide, a blocker of the cation-chloride co-transporter NKCC1, prevented synaptic potentiation and caused synaptic depression, suggesting that the depolarizing action of GABA observed in the presence of CGP55845 is responsible for the potentiating effect. It is proposed that, activation of GABAB receptors by spillover of GABA from MF terminals reduces the probability of release and contributes to synapses silencing. This would act as a filter to prevent excessive activation of the auto-associative CA3 network and the emergence of seizures.
In the developing hippocampus, GABAergic signaling plays a crucial role in generating primitive patterns of network activity known as giant depolarizing potentials or GDPs (Ben-Ari et al., 2007
). The higher intracellular chloride concentration present in immature neurons (Rivera et al., 1999
) leads to an efflux of chloride and to a membrane depolarization which triggers sodium spikes and excites postsynaptic cells (Ben-Ari et al., 1989
). GABA-induced membrane depolarization activates N-methyl-D-aspartate receptors and voltage-dependent calcium channels with consequent calcium rise inside the cells and activation of intracellular signaling cascades. Such mechanisms are central to the well-known trophic action of GABA which regulates several key developmental steps including DNA synthesis, cell migration, cell growth and synapse formation (Owens and Kriegstein, 2002
). In addition, evidence has been recently provided that the principal neurotransmitter released from mossy fibers (MF) during the first week of postnatal life is GABA (Safiulina et al., 2006
). In adulthood, these fibers are glutamatergic and are endowed with a number of receptor types which up or down-regulate the transmitter release. These include metabotropic glutamate receptors (Kamiya et al., 1996
), GABAA and GABAB receptors (Min et al., 1998
; Vogt and Nicoll, 1999
), adenosine receptors (Thompson et al., 1992
), kainate receptors (Castillo et al., 1997
; Vignes and Collingridge, 1997
) and peptides (Weisskopf et al., 1993
). GABAB receptors which are coupled to Gi and Go proteins are localized on both pre and postsynaptic membranes (Bettler et al., 2004
; Misgeld et al., 1995
). Activation of postsynaptic GABAB receptors causes the opening of inwardly rectifying potassium channels (Lüscher et al., 1997
) whereas activation of presynaptic GABAB receptors causes a reduction in transmitter release mainly via inhibition of P/Q and N types of voltage-dependent calcium channels (Poncer et al., 1997
). While postsynaptic GABAB receptors are developmentally regulated (Cherubini et al., 1998
; Gaiarsa et al., 1995
), presynaptic GABAB receptors are present and functional already at birth. It has been shown that they control network activity (McLean et al., 1996
) and modulate the amplitude of glutamate- or GABA-mediated postsynaptic currents in CA3 principal cells (Gaiarsa et al., 1995
). However, these studies failed in identifying where GABAA-mediated synaptic currents originated from.
Here, we addressed whether, immediately after birth, MF-induced GABAergic responses in CA3 principal cells could be modulated by GABAB autoreceptors. We report that blocking GABAB receptors with CGP55845 enhanced synaptic efficacy and converted silent synapses into active ones. In addition, by enhancing the concentration of “ambient” GABA with repetitive stimulation of MF or by blocking GABA transporters contributed to the switch-off of active synapses, an effect that was prevented by the GABAB receptor antagonist CGP55845. It is proposed that at early developmental stages, spillover of GABA from MF terminals activates GABAB receptors and reduces synaptic efficacy probably by interfering with the probability of GABA release. This would prevent excessive activation of the CA3 hippocampal subfield and the development of seizures.
Slice Preparation
Experiments were performed on hippocampal slices obtained from postnatal (P) day P1–P6 Wistar rats as previously described (Gasparini et al., 2000
). All experiments were carried out in accordance with the European Community Council Directive of 24 November 1986 (86/609EEC) and were approved by the local authority veterinary service. Briefly, animals were decapitated after being anaesthetized with an i.p. injection of urethane (2 g/kg). The brain was quickly removed from the skull and placed in ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl 130, KCl 3.5, NaH2PO4 1.2, NaHCO3 25, MgCl2 1.3, CaCl2 2, glucose 25, saturated with 95% O2 and 5% CO2 (pH 7.3–7.4). Transverse hippocampal slices (500 μm thick) were cut with a vibratome and stored at room temperature in a holding bath containing the same solution as above. After a recovery period of at least 1-h, an individual slice was transferred to the recording chamber where it was continuously superfused with oxygenated ACSF at a rate of 2–3 ml/min at 33–34°C. Experiments with NO-711 were performed either at 33–34°C or at room temperature (∼24°C).
Electrophysiological Recordings
Whole cell patch-clamp recordings were obtained from visually identified CA3 pyramidal cells in voltage-clamp configuration using an Axopatch 1D amplifier (Axon Instruments, Foster City, CA, USA). Pyramidal cells exhibited strong fire adaptation in response to long (400 ms) depolarizing current pulses. Ten cells injected with 3–4% biocytin (Nε-biotinyl-L-lysine, Sigma) for later morphological identification revealed the classical shape of pyramidal neurons (data not shown).
Patch electrodes were pulled from borosilicate glass capillaries (Hingelberg, Malsfeld, Germany). They had a resistance of 5–7 MΩ when filled with an intracellular solution containing (in mM): KCl 140, HEPES 10, EGTA 1, MgATP 2, MgCl2 1 (pH 7.3 with NaOH). In experiments with baclofen, to avoid concomitant activation of postsynaptic GABAB receptors, in the internal solution KCl was replaced with CsCl. The whole cell capacitance was fully compensated and the series resistance (10–20 MΩ) was compensated at 75–80%. The stability of the patch was checked by repetitively monitoring the input and series resistance during the experiment. Cells exhibiting >15 changes in series resistance were excluded from the analysis. On average, the series resistance value was 11.2 ± 1.4 MΩ.
Bipolar twisted NiCr-insulated electrodes localized into stratum granulosum of the dentate gyrus were used to evoke synaptic responses in CA3 principal cells. We used minimal stimulation of granule cells at the frequency of 0.05 Hz in order to activate only one or few presynaptic fibers. According to the technique described by Jonas et al. (1993) and Allen and Stevens (1994)
the stimulation intensity was decreased until only a single axon was activated. This was achieved when the mean amplitude of the postsynaptic currents and failure probability remained constant over a range of stimulus intensities near the threshold for detecting a response (Safiulina et al., 2006
). An abrupt increase in the mean peak amplitude of synaptic currents was observed when the stimulus intensity was increased above 1 μA (Figure 1
A). This all-or-none behavior suggests that only a single granule cell was stimulated. In addition, the latency, shape and rise time of the individual synaptic responses remained constant when DNQX (20 μM) and D-APV (50 μM) were added to the external solution, indicating that fast ionotropic glutamate receptors were not involved (on average the rise time and latency of MF-evoked synaptic events were 3.4 ± 0.2 and 3.1 ± 0.6 ms before and 3.4 ± 0.2 and 3.03 ± 0.6 ms after DNQX, respectively; n = 5, p > 0.05; Figure 1
). Therefore, although not otherwise stated, in the following experiments cells treated and non-treated with DNQX and D-APV were pooled together.
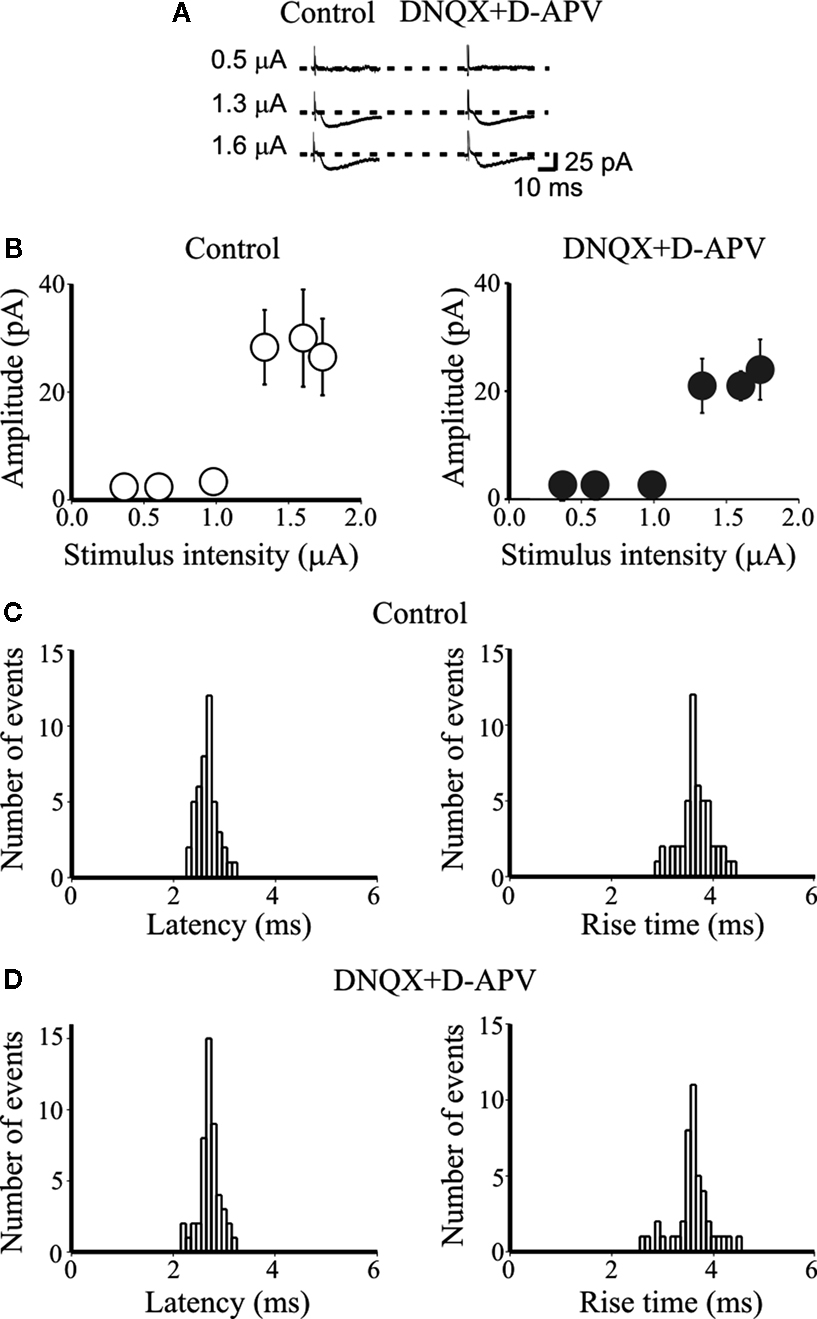
Figure 1. DNQX and D-APV do not modify the latency and rise time of unitary synaptic currents evoked in principal cells by minimal stimulation of granule cells in the dentate gyrus. (A) Unitary synaptic currents evoked
in a CA3 pyramidal cell at P4 with different stimulation intensities before
(left, control) and during application of DNQX (20 μM) and D-APV (50 μM, right) from a holding potential −70 mV. Each trace is the average of 15–20 responses (including failures). (B) Peak amplitudes of synaptic currents represented in (A) are plotted as a function of stimulus intensities. Bars are SEM. Latency and rise time distributions of individual currents evoked in a CA3 pyramidal cell by minimal stimulation of granule cells in the dentate gyrus in the absence (C) and in the presence (D) of DNQX and D-APV. Note that in the presence of DNQX and D-APV the unimodal distribution of latencies and rise times of individual responses did not change.
As already reported (Safiulina et al., 2006
), the monosynaptic nature of synaptic currents was supported by the unimodal and narrow latencies and rise time distributions which remained constant when the extracellular Ca2+/Mg2+ concentration ratio was reduced from 2:1.3 to 1:3 (data not shown). When the stimulation intensity was turned down, the probability of failure in synaptic transmission was near 1. Failures were usually estimated by visual discrimination. In most cases paired stimuli were applied at 50 ms interval. In a set of experiments, in order to control the adequacy of the visual selection, we used the methods described by Nicholls and Wallace (1978)
consisting in doubling the responses with positive amplitude. A similarity and a high correlation between the two methods were obtained (r = 0.95, p < 0.0001). Synapses were considered “presynaptically” silent if they did not respond to the first stimulus in 30–60 consecutive trials but exhibited occasional responses to a second pulse (following the first one with 50 ms delay).
Mossy fibers inputs were identified on the basis of their strong paired-pulse facilitation, of their sensitivity to group III mGluR agonist 2-amino-4-phosphonobutyric acid (L-AP4; Gutierrez et al., 2003
; Kasyanov et al., 2004
), and short-term frequency-dependent facilitation (Safiulina et al., 2006
). MF-induced synaptic responses were completely and reversibly blocked by bicuculline methiodide (20 μM) or picrotoxin (100 μM), confirming their GABAergic origin. In contrast to MF inputs, GABAergic inputs from interneurons were insensitive to L-AP4 (Safiulina et al., 2006
; Walker et al., 2001
). In some experiments, MF were activated with brief trains of stimuli delivered at the frequency of 0.1, 3 and 10 Hz in the presence of DNQX (20 μM) and D-APV (50 μM). To prevent potential long-term changes in synaptic strength only 20–30 events were collected for each stimulation frequency. Usually, after 10 stimuli MF-mediated synaptic responses reached a steady state. The last 10 events were averaged and compared with controls.
Drugs used were: D(−)-2-amino-5-phosphonopentanoic acid (D-AP5), bumetanide, 6,7-dinitroquinoxaline-2,3-dione (DNQX), picrotoxin, bicuculline methiodide, CGP 55845, L-AP4, all purchased from Tocris Cookson Ltd (Bristol, UK); (±)-baclofen and NO-711 purchased from Sigma (Milan, Italy). All drugs were dissolved in ACSF except CGP 55845, bumetanide and DNQX that were dissolved in DMSO and picrotoxin that was dissolved in ethanol. The final concentration of DMSO in the bathing solution was 0.1%. At this concentration, DMSO alone did not modify the shape or the kinetics of synaptic currents. Drugs were applied in the bath via a three-way tap system, by changing the superfusion solution to one differing only in its drug(s) content). The ratio of flow rate to bath volume ensured complete exchange within 2 min.
Data Acquisition and Analysis
Data were stored on a magnetic tape and transferred to a computer after digitization with an A/D converter (Digidata 1200; Axon Instruments, Foster City, CA, USA). Acquisition and analysis of evoked responses were performed with Clampfit 9 (Axon Instruments, Foster City, CA, USA). Data were sampled at 20 kHz and filtered with a cut off frequency of 1 kHz. The mean EPSC amplitude was obtained by averaging successes and failures. Responses contaminated with polysynaptic activity were excluded from this study. The paired-pulse ratio (PPR) was calculated as the mean amplitude of the synaptic response evoked by the second stimulus over that evoked by the first one. The coefficient of variation was calculated as the ratio between the standard deviation of synaptic currents amplitude and the mean. In some experiments, the decaying phase of GABAA-mediated synaptic currents was fitted with exponential functions in the form:
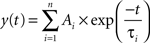
where τi and Ai are the time constants and relative fractions of the respective components. The concentration-response curve for baclofen was fitted with the Hill equation:
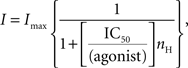
where Imax is the maximum response, IC50 is the concentration of the agonist producing a half-maximal response and nH is the Hill coefficient.
If not otherwise stated, data are expressed as mean ± SEM. Statistical comparisons were made with the use of paired t-test. A value of p < 0.05 was taken as significant.
Early in postnatal development, the probability (P) of obtaining successes at MF-CA3 synapses is quite low (on average 0.3 ± 0.04; see also Kasyanov et al., 2004
). These synapses include also presynaptically silent ones (see Materials and Methods for their identification). Thus, in several cases (12/52) stimulation of granule cells in the dentate gyrus failed to produce any synaptic response over 30 consecutive trials. However, occasional responses to the second stimulus occurred, suggesting that these synapses were “presynaptically” silent (Gasparini et al., 2000
; Kasyanov et al., 2004
). To distinguish between low P and presynaptically silent cells, throughout the text we will refer to low P non-silent and low P silent neurons. In accordance with their MF origin and GABAergic nature (Gutierrez, 2005
; Gutierrez et al., 2003
; Kasyanov et al., 2004
; Safiulina et al., 2006
), bath application of L-AP4 (10 μM) significantly reduced the amplitude of synaptic events (34 ± 5% of control; n = 8, p < 0.01), which were completely blocked by picrotoxin (100 μM; Figures 2
A,B). Depolarizing the cell to positive potentials (+40 mV) produced outward currents with latency, onset and deactivation kinetics similar to those obtained at −70 mV, further indicating that they were mediated by GABAA receptors.
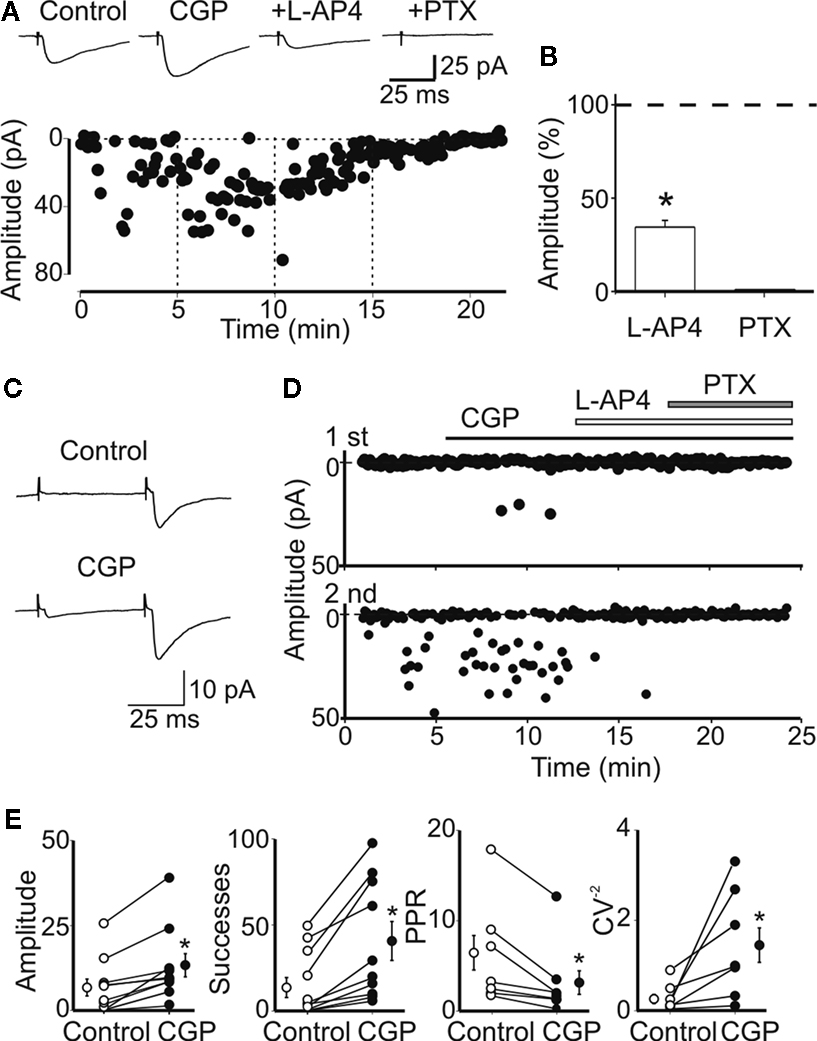
Figure 2. Block of GABAB receptors with CGP55845 enhances the probability of GABA release from mossy fiber terminals. (A) Peak amplitude of MF-evoked synaptic currents in control, during bath application of CGP55845 (1 μM), CGP plus L-AP4 (10 μM) and CGP plus L-AP4 and picrotoxin (100 μM, vertical dashed lines). Note the reduction in the number of failures in CGP. Current responses were reduced in amplitude by L-AP4 and were blocked by picrotoxin. Insets above the graph represent averaged traces (20 individual samples including failures). (B) Each column in the graph represents the mean amplitude of synaptic currents (expressed as percentage of controls) evoked during bath application of L-AP4 and picrotoxin (*p < 0.05). (C) A presynaptically silent neuron before (upper trace) and during bath application of CGP55845 (1 μM, lower trace). Note the appearance of a synaptic current in CGP (each trace is the average of 20 individual samples). (D) Peak amplitude of MF-evoked synaptic currents for the cell shown in (C) before and during bath application of CGP, L-AP4 and PTX (closed and open bars). The upper graph refers to the first response, the lower graph to the second one. (E) Summary plots of mean amplitude, success rate, paired-pulse ratio and inverse squared of CV in control (open circles) and during bath application of CGP (closed circles). In this and in the following figures, on the left and on the right of individual responses are represented averaged values; *p < 0.05. Amplitude and successes refer to three silent and seven non-silent neurons. PPR and CV−2 refer only to non-silent cells.
We used the selective GABAB receptor antagonist CGP55845 to examine whether activation of presynaptic GABAB receptors by endogenously released GABA could be responsible for the low P observed at immature MF-CA3 connections. Previous studies from the immature hippocampus have shown that GABAB receptors are developmentally regulated, being expressed during the first two postnatal weeks only at presynaptic level (Cherubini et al., 1998
; Gaiarsa et al., 1995
; Lei and McBain, 2003
; Nurse and Lacaille, 1999
).
Presynaptic GABAB Receptors Reduce the Probability of GABA Release From MF Terminals
The examples of Figure 2
illustrate the effects of CGP55845 on a low P non-silent (Figure 2
A) and a low P silent neuron (Figures 2
C,D). While in the first case bath application of CGP55845 (1 μM) enhanced the amplitude of single fiber-evoked synaptic current (in response to the first pulse), in the second case, this drug induced the appearance of synaptic responses. As already mentioned, the addition of L-AP4 (10 μM) to the external solution substantially reduced the amplitude of MF-evoked responses that were completely abolished by picrotoxin (100 μM; Figure 2
A). Overall, in 10 neurons (three low P silent and seven low P non-silent), CGP55845 significantly increased the amplitude and the number of successes to both the first and the second response (the amplitude of the first response changed from 6.6 ± 2.5 to 13.4 ± 3.5 pA, p < 0.05; the amplitude of the second response from 79.4 ± 30.3 to 87.8 ± 43.0 pA; the success rate of the first response changed from 14.5 ± 5.7% to 41.5 ± 11.2%, p < 0.05; the success rate of the second response from 56.9 ± 9.3% to 75.7 ± 9.1%; Figure 2
E). To test whether CGP55845-induced reduction in failure rate was due to an increase in transmitter release we measured the PPR and the CV−2 which are largely used to evaluate changes in P (Malinow and Tsien, 1990
; McAllister and Stevens, 2000
; Zucker and Regehr, 2002
). The PPR and the CV−2 values refer only to non-silent low P synapses (n = 7), because calculated ratios are infinitely large when the mean amplitude of the first response is close to 0 (compatible with a lack of quantal release, in these cases also the CV−2 was close to 0). On average, in non-silent low P synapses, the PPR was 6.4 ± 1.9 and 3.1 ± 1.3 before and during CGP55845, respectively while, in the presence of CGP55845, CV−2 changed from 0.3 ± 0.1 to 1.5 ± 0.4 (Figure 2
E). No changes in holding current or in membrane input resistance were observed during CGP55845 application. The mean holding current was 9.1 ± 1.6 and 10.5 ± 2.6 pA and the mean input resistance was 548 ± 56 and 637 ± 69 MΩ (n = 19, p > 0.05) in control and in the presence of CGP55845, respectively.
These results support the hypothesis that, in normal conditions, activation of GABAB autoreceptors by endogenously released GABA down-regulates GABA release and this may contribute in making functional synapses silent.
Baclofen Mimics the Effects of Endogenous GABA on Presynaptic GABAB Receptors
To see whether a direct activation of presynaptic GABAB receptors can mimic the effect of endogenous GABA, baclofen, a selective GABAB agonist (Misgeld et al., 1995
) was tested on MF-evoked synaptic currents (recorded in CA3 principal cells with patch pipettes containing CsCl to elude possible activation of postsynaptic GABAB receptors). To this purpose, baclofen was applied only to low P non-silent cells. Baclofen did not modify the holding current or the membrane resistance of the recorded neurons (mean input resistance was 558 ± 86 and 492 ± 96 MΩ (n = 12, p > 0.05) in control and in the presence of baclofen, respectively). These values were not significantly different form those observed in cells patched with KCl containing electrodes. This is probably due to the fact that immature neurons are rather compact (the dendritic arborization is still very poor, Tyzio et al., 1999
) and synaptic currents are generated close to the soma.
Exposure of individual slices to increasing concentrations of baclofen (from 10 nM to 3 μM) progressively reduced the amplitude of synaptic responses. As shown in Figure 3
G, a cumulative dose-response curve was constructed. Fitting the experimental points with the Hill equation gave an IC50 value of 400 nM and a Hill coefficient of 1.78, suggesting that two molecules of baclofen interact with each presynaptic GABAB receptor to produce the observed response. Baclofen-induced reduction in amplitude of the synaptic currents was mediated by GABAB receptors since it was prevented by the selective GABAB receptor antagonist CGP55845 (open circle in the graph of Figure 3
G). Baclofen (3 μM) reversibly and completely blocked the first MF-induced synaptic response and drastically reduced the amplitude of the second one (Figure 3
A). A lower concentration of the agonist, closer to the IC50 value (100 nM), significantly reduced the amplitude of both the first and the second response. In accordance with a presynaptic site of action on P, the reduction in amplitude of the first response was accompanied by an increase in the amplitude of the second response (Figure 3
B). Overall, in 12 cells, baclofen (100 nM) induced a significant reduction in amplitude (from 23.2 ± 3.9 to 8.0 ± 2.1 pA, p < 0.001; Figure 3
C) and in success rate (from 52.9 ± 3.5 to 28.7 ± 3.4, p < 0.001; Figure 2
D) of synaptic currents. These effects were associated with a significant increase in PPR (from 3.9 ± 0.5 to 9.3 ± 2.4, p < 0.05; Figure 3
E) and a decrease in the CV−2 (from 0.72 ± 0.09 to 0.32 ± 0.07, p < 0.001; Figure 3
F). These data further indicate that activation of GABAB receptors present on MF terminals is responsible for the reduction of P and for synapse silencing.
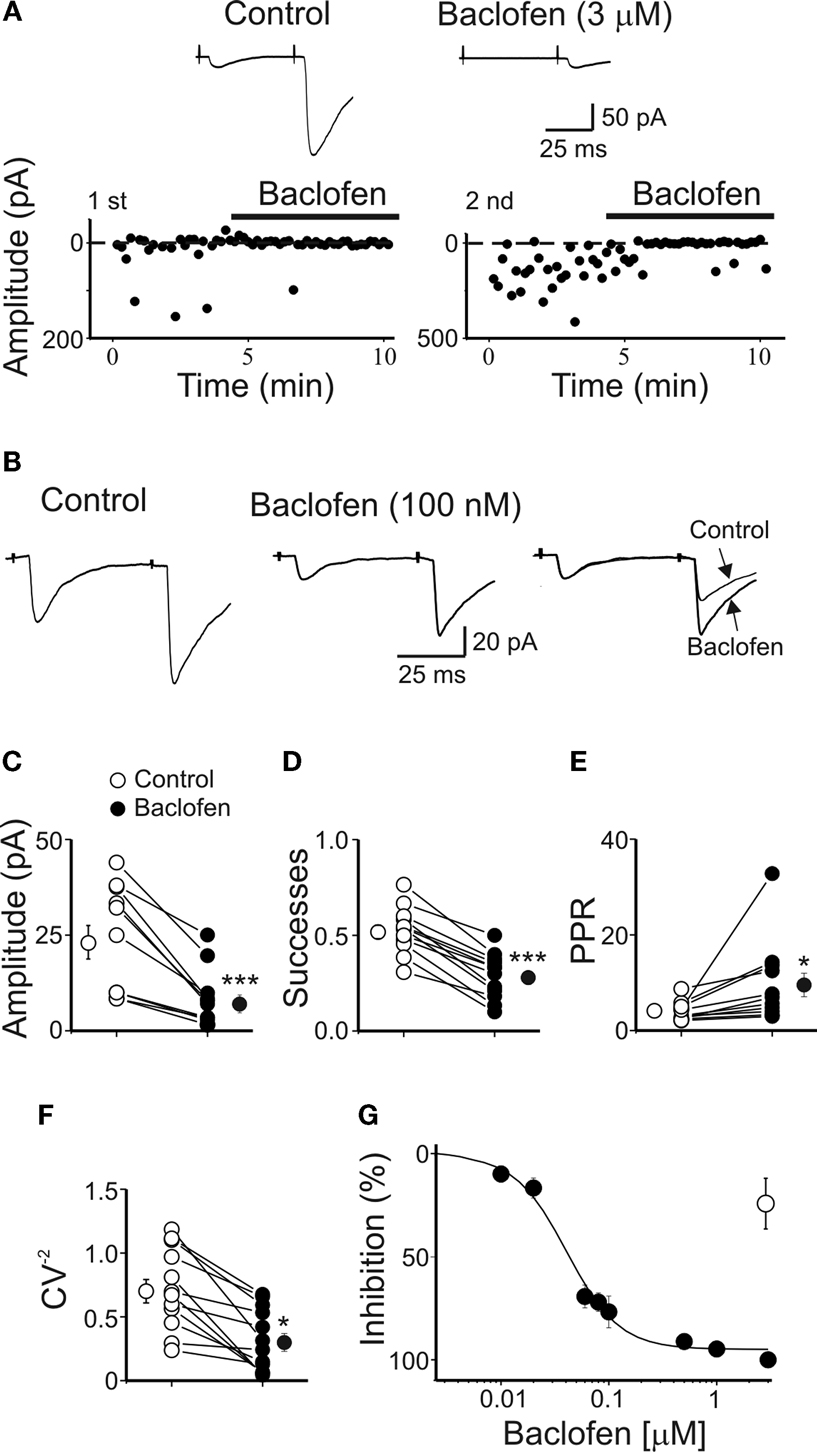
Figure 3. Baclofen reduces the amplitude of MF-evoked GABAergic currents in CA3 principal cells. (A) Peak amplitude of MF-evoked synaptic currents (in response to a first and a second stimulus, at P4) in control and during bath application of baclofen (3 μM, bars). Note that baclofen completely abolished the response to the first stimulus and strongly depressed the response to the second one. Insets above the graphs represent averaged traces (20 individual samples including failures) in control (left) and during application of baclofen (right). (B) A lower concentration of baclofen (100 nM) reduced the peak amplitude of both first and second responses (each trace is the average of 20 individual samples including failures). On the right, the two traces (on the left and on the middle) are normalized to the first response and superimposed to show baclofen-induced changes in PPR. Summary plots of mean amplitude (C), success rate (D), paired-pulse ratio (E) and inverse squared of CV (F) for 12 cells recorded in control (open symbols) and during bath application of baclofen (100 nM, closed symbols). *p < 0.05, ***p < 0.001. (G) Baclofen depressed in P1–P6 pyramidal cells (closed circles) the amplitude of MF-elicited synaptic currents in a dose-dependent way. Each point represents the mean value of three to five individual experiments. Bars are the SEM (they are often within the symbols). Data points were fitted with the Hill equation (see Materials and Methods; the IC50 value was 0.04 μM). The effect of baclofen (3 μM) was partially prevented by CGP55845 (1 μM, open circle) (n = 5).
Frequency-Dependent Modulation of Presynaptic GABAB Receptors
The low initial P found at MF-CA3 connections may account for the short-term frequency-dependent facilitation observed at these synapses (Henze et al., 2000
; McBain, 2008
; Nicoll and Schmitz, 2005
; Salin et al., 1996
). Synapses with higher P as those made by MF on interneurons express, in response to brief trains of stimuli, either a mild facilitation or a depression (Lei and McBain, 2003
; McBain, 2008
; Safiulina et al., 2006
). Synapses facilitation or depression depends on several factors including calcium dynamics in presynaptic nerve terminals, expression of presynaptic voltage-dependent calcium channels, activation of autoreceptors, desensitization of postsynaptic GABAA receptors, etc. In a previous study from immature GABAergic MF-CA3 synapses (Safiulina et al., 2006
), a marked frequency-dependent facilitation was induced by changes in stimulation frequency from 0.05 to 0.3 Hz. Whether frequency-dependent short-term depression may also occur at these synapses has not been investigated, yet. Therefore, in the following experiments, brief trains of stimuli (at frequencies of 3 and 10 Hz) were applied to granule cells in the dentate gyrus and the possible contribution of GABAB receptors to synaptic depression was assessed by comparing synaptic events obtained in the absence or in the presence of CGP55845. The rationale behind this is that an increase in GABA release, following repetitive stimulations of presynaptic terminals, may cause a delay in GABA clearance which in turn may facilitate spillover and activation of presynaptic GABAB receptors localized on MF terminals. Repetitive activation of presynaptic fibers at 3 Hz produced in seven out of twelve cells a depression of MF-evoked synaptic responses. As shown in the illustrative example of Figures 4
A,B, changing the stimulation frequency from 0.1 to 3 Hz induced a reduction in amplitude of MF responses that was partially rescued by CGP55845. In seven cells the reduction in amplitude (33 ± 13%) and in successes rate (26 ± 9%) of MF-evoked GPSCs was associated with an increase in PPR (142 ± 19%) and a decrease in the CV−2 (27 ± 17%) suggesting a presynaptic site of expression (Figure 4
C). These effects were partially blocked by CGP55845 (in comparison to controls, in CGP the amplitude of synaptic responses was 81 ± 17%, the success rate 87 ± 10%, the PPR 61 ± 19% and the CV−2 216 ± 64%; Figure 4
C).
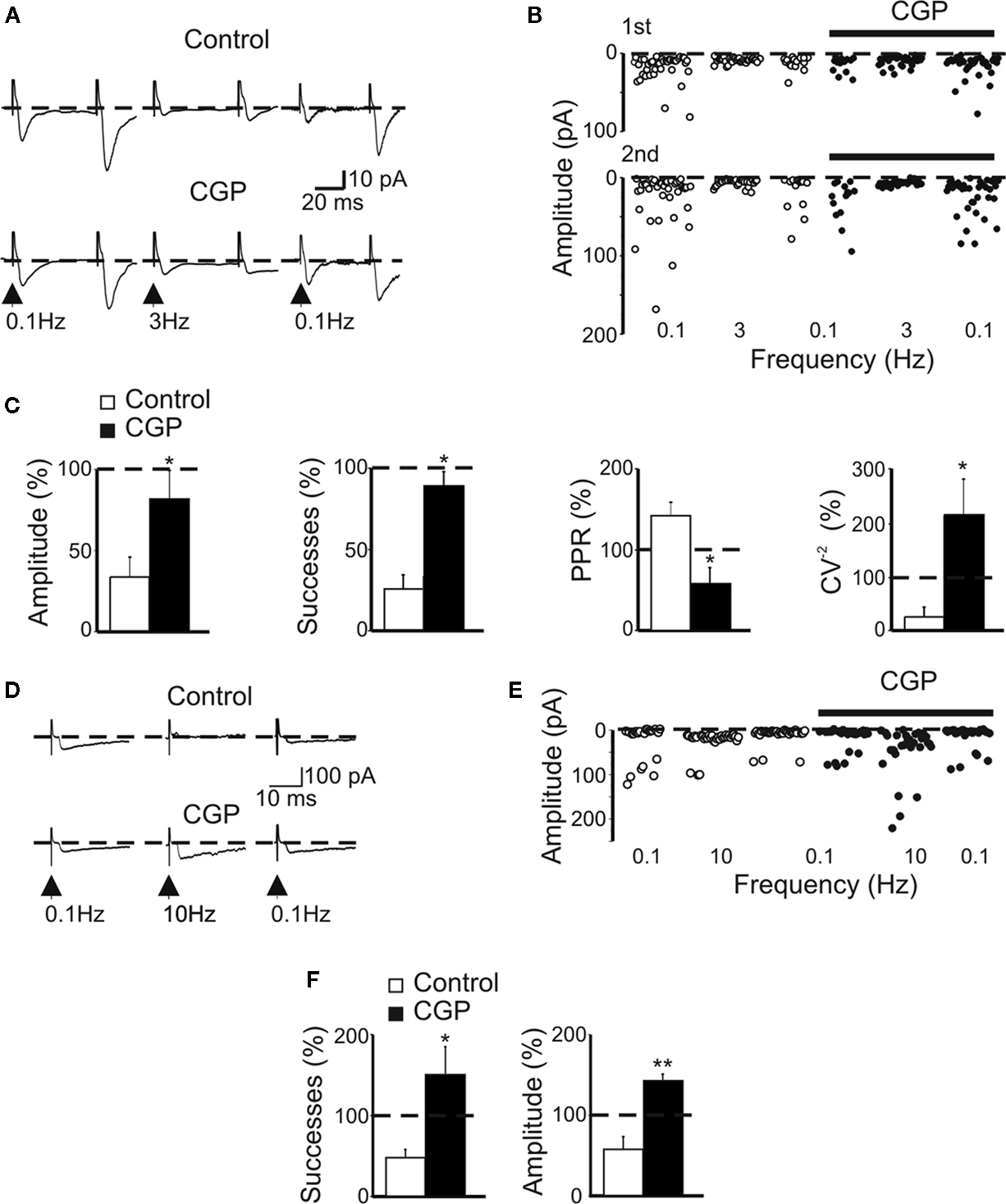
Figure 4. CGP 55845 partially prevents frequency-dependent depression of MF-evoked GABAergic currents in CA3 principal cells. (A) Synaptic currents evoked in control (upper traces) and during application of CGP 55845 (lower traces) by stimulation of granule cells in the dentate gyrus at 0.1 Hz (left) at 3 Hz (middle) and again at 0.1 Hz (right). Each trace is the average of 20 individual responses. (B) The peak amplitude of synaptic currents [shown in (A)] in control (open circles) and during CGP (bar, closed circles) are plotted as a function of different frequencies (shown below). (C) Summary data (n = 12) representing changes in amplitude, success rate, PPR and CV−2 at 3 Hz and normalized to those obtained at 0.1 Hz in control (white columns) and during CGP (black columns); *p < 0.05. (D) Synaptic currents evoked in control (upper traces) and during application of CGP 55845 (lower traces) by stimulation of granule cells in the dentate gyrus at 0.1 Hz (left), at 10 Hz (middle) and again at 0.1 Hz (right). Each trace is the average of 20 individual samples. (E) Peak amplitudes of synaptic responses [same cell shown in (D)] in control (open circles) and during CGP (bar, closed circles) plotted as a function of different frequencies (shown below). (F) Summary data (n = 7) representing changes in amplitude and successes in control and during CGP at 10 Hz (normalized to synaptic currents obtained at 0.1 Hz, dashed line). *p < 0.05, **p < 0.01.
In the remaining five cells, stimulating MF at 3 Hz caused a potentiation of GPSCs (208 ± 9%) which was depressed by bath application of CGP (the amplitude of GPSCs was 58 ± 4% of that obtained at 0.1 Hz; data not shown). Although the mechanisms underlying synaptic potentiation are still unclear, it is likely that the direction of short-term plasticity largely depends on the initial P, as demonstrated for glutamatergic MF-interneurons synapses (Toth et al., 2000
).
Brief trains of stimuli at 10 Hz produced in 6/13 a reduction in amplitude of the synaptic responses (58 ± 13%, p < 0.05, n = 6) and in the number of successes (49 ± 8%, p < 0.05, n = 6; Figures 5
D–F). Bath application of CGP55845 did fully prevent synaptic depression and induced synaptic potentiation (Figures 5
D–F). In the remaining seven cells changing stimulation frequency from 0.1 to 10 Hz induced synaptic potentiation (152 ± 26%). It should be stressed that two of the potentiated responses were low P silent cells.
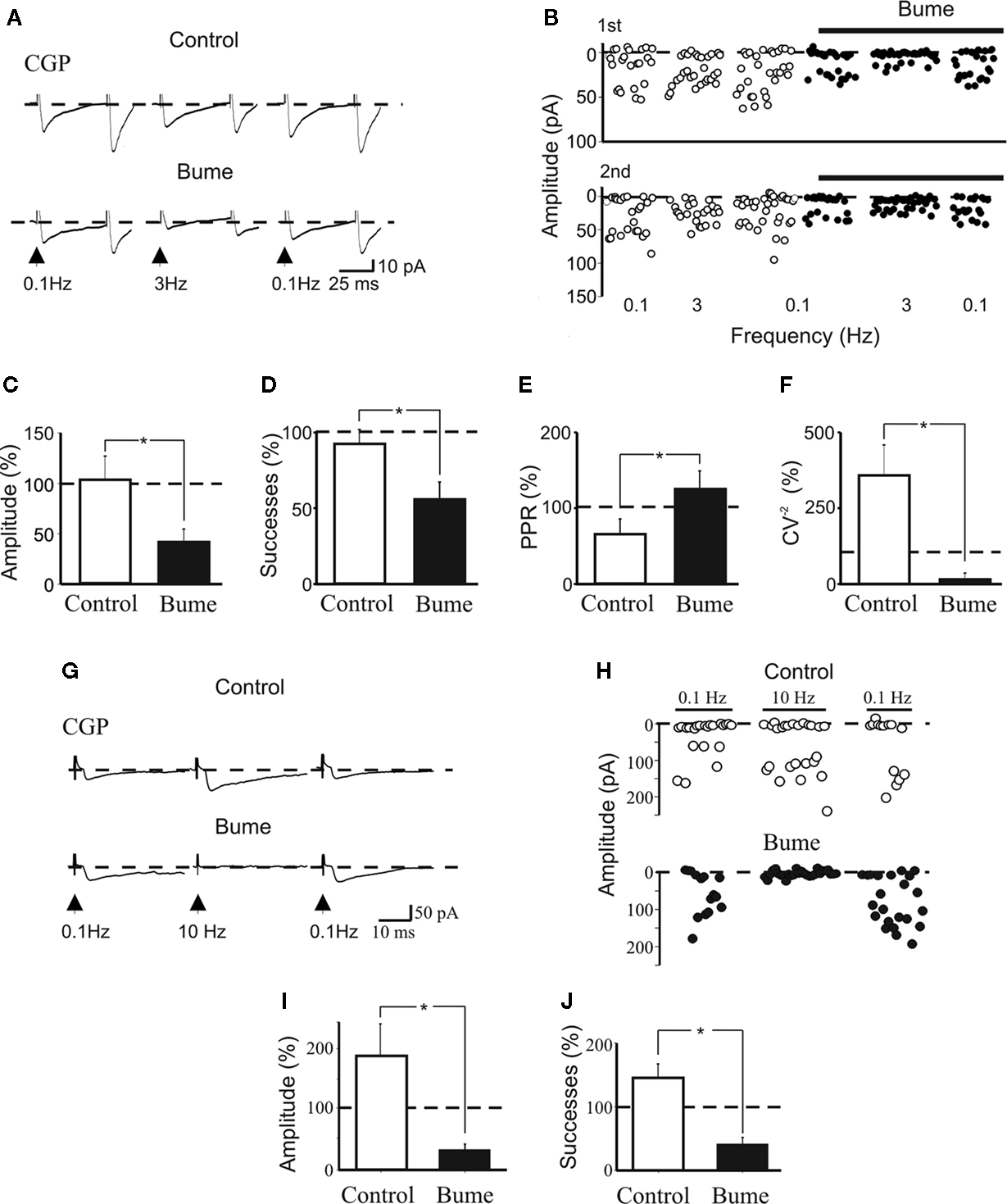
Figure 5. Prevention of frequency-dependent depression by CGP 55845 requires the depolarizing action of GABA on MF terminals. (A) Synaptic currents evoked in the presence of CGP 55845 (1 μM) in control conditions (upper traces) and after 30 min perfusion with bumetanide (10 μM, lower traces) at 0.1 Hz (left), at 3 Hz (middle) and again at 0.1 Hz (right). Each trace is the average of 10–15 individual responses. Note synaptic depression at 3 Hz in the presence of bumetanide. (B) The peak amplitude of synaptic currents shown in (A) in control (open circles) and during bumetanide (closed circles) are plotted as a function of different frequencies (shown below). (C–F) Summary data (n = 5) representing changes in amplitude (C), success rate (D), PPR (E) and CV−2 (F) obtained at 3 Hz (normalized to the responses obtained at 0.1 Hz) in control (white columns) and in bumetanide (black columns); *p < 0.05. (G) Synaptic currents evoked in the presence of CGP 55845 (1 μM) in control conditions (upper traces) and after 30 min perfusion with bumetanide (10 μM, lower traces) at 0.1 Hz (left), at 10 Hz (middle) and again at 0.1 Hz (right). Each trace is the average of 20 individual samples. Note synaptic depression at 10 Hz in the presence of bumetanide. (H) The peak amplitude of synaptic currents shown in (G) in control (open circles) and during bumetanide (closed circles) are plotted as a function of different frequencies (shown above). (I,J) Summary data (n = 5) representing changes in amplitude (I) and success rate (J) obtained at 10 Hz (normalized to the responses obtained at 0.1 Hz) in control (white columns) and in bumetanide (black columns); *p < 0.05.
The shift from synaptic depression to synaptic potentiation (at 10 Hz stimulation) with CGP55845 is intriguing. One possibility is that, the enhanced release of GABA, induced by blocking GABAB autoreceptors with CGP, may activate GABAA receptors present on MF boutons (Alle and Geiger, 2007
). Activation of these receptors is known to depolarize presynaptic terminals and facilitate GABA release (Khirug et al., 2008
; Nakamura et al., 2007
).
To test whether this hypothesis was correct, we applied bumetanide (10 μM for 20–30 min in the presence of CGP55845, DNQX and D-AP5), a blocker of the cation-chloride co-transporter NKCC1, responsible for the accumulation of chloride inside the cell and for the depolarizing action of GABA (Dzhala et al., 2005
; Safiulina et al., 2008
; Sipilä et al., 2006
). In a previous study, using gramicidin-perforated patch to preserve the anionic conditions of the recorded cell, we found that bumetanide, slightly hyperpolarized the neurons (3–6 mV) and shifted GABA reversal potential from −47.6 ± 3.3 to −71.5 ± 3.09 mV (Sivakumaran et al., 2009
). As shown in the example of Figure 5
, while in control (in the presence of CGP55845) repetitive stimulation of presynaptic fibers at 3 Hz, did not significantly (p > 0.05) modify the amplitude of GPSCs or their success rate, in the presence of bumetanide it significantly reduced both amplitude and success rate (n = 5; Figures 5
A–D). These effects were associated with a significant increase and decrease of PPR and CV−2, respectively (Figures 5
E,F), suggesting a presynaptic type of action. Changes in amplitude of GPSCs were not related to modifications in EGABA in the recorded cell, since these experiments were performed using symmetric chloride solutions. The figure shows also that bumetanide significantly reduced frequency-dependent potentiation of GPSCs induced by stimulating MF at 10 Hz (in the presence of CGP; n = 5; Figures 5
G–J). These data strongly suggest that the depolarizing action of GABA on GABAA receptors localized on MF terminals account for CGP-induced synaptic facilitation.
Presynaptic GABAB Receptors on MF are Tonically Activated by Ambient GABA
To further validate the hypothesis that the enhanced concentration of ambient GABA and the delayed clearance of this neurotransmitter from the cleft are responsible for the activation of GABAB autoreceptors and synaptic silencing, in the next series of experiments on low P non-silent cells, we increased the concentration of GABA in the synaptic cleft with NO711, a selective blocker of the glial and the neuronal GABA transporter GAT-1 (Borden, 1996
). Firstly, we performed these experiments at room temperature (22–24°C) because it is well-known that the action of GAT-1 is strongly temperature-dependent (Binda et al., 2002
). In the example of Figures 6
A–C, bath application of NO711 (3 μM) strongly reduced the amplitude of synaptic responses, an effect that was partially rescued by CGP55845, suggesting the involvement of GABAB autoreceptors. Summary data from seven out of eight cells are shown in Figures 6
D–G. NO711 induced a significant reduction in amplitude of synaptic currents (from 23.4 ± 2.3 to 9.1 ± 2.3 pA, p < 0.05) and in the number of successes (from 37.6 ± 6.7% to 21.9 ± 3.2%, p < 0.05). These effects were associated with a significant increase in PPR (from 1.6 ± 0.2 to 2.4 ± 0.3, p < 0.05) and a decrease in the CV−2 (from 0.9 ± 0.2 to 0.5 ± 0.1, p < 0.05). The PPR and the CV−2 were calculated only in six cells, because one was silenced by NO711. As expected (Marchionni et al., 2007
; Safiulina et al., 2006
), in the presence of NO711 synaptic currents exhibited a slowdown of their deactivation kinetics (the mean time constant increased from 14.7 ± 3.4 to 26 ± 3.3 ms, n = 4, p < 0.05; Figure 6
B).
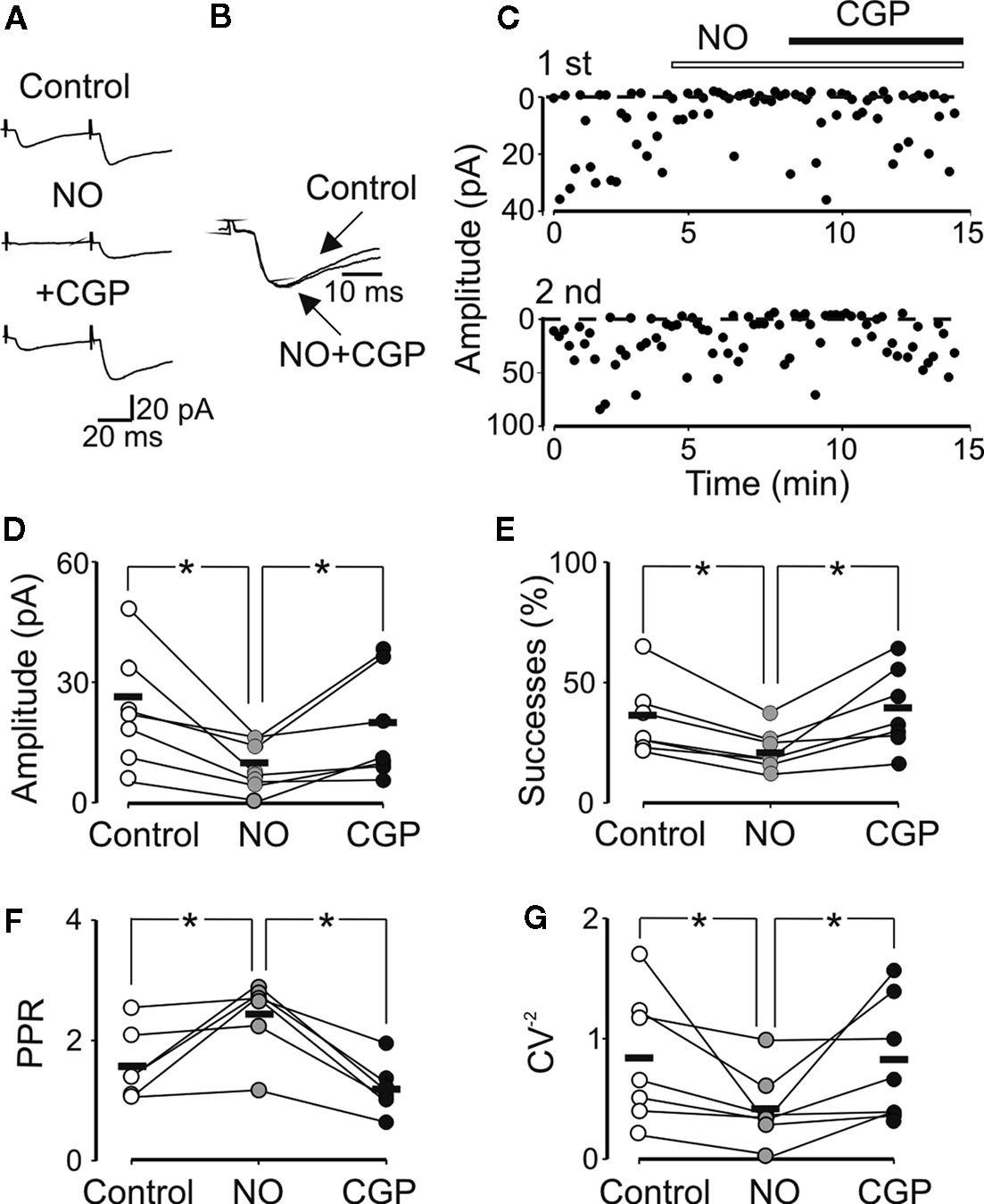
Figure 6. The GAT-1 transporter blocker NO711 enhances the level of ambient GABA and reduces GABA release through the activation of presynaptic GABAB receptors. (A) Average of 20 individual traces (including failures) recorded at room temperature from a CA3 pyramidal cell in control, during NO711 (3 μM) and NO711 plus CGP55845 (1 μM). Note the disappearance of the first response in NO711 and its reappearance after addition of CGP55845. (B) Synaptic responses obtained in control and in the presence of CGP55845 are normalized and superimposed to show NO7211-induced changes in decay kinetics. (C) Peak amplitudes of MF-evoked synaptic currents [shown in (A)] are plotted against time before and during bath application of NO711 (open bar) and NO711 plus CGP55845 (closed bar). Summary plots of the mean amplitude (D), success rate (E), paired-pulse ratio (F) and inverse squared of CV (G) for six to seven individual cells recorded in control (open symbols) and during bath application of NO711 (gray circles) and NO711 plus CGP (black circles). Horizontal bars represent the mean. *p < 0.05.
In one cell (out of eight), NO711 slightly increased the amplitude of the synaptic response from 34.7 to 44.5 pA. Also in this case the effect was presynaptic because it was associated with a reduction in success rate (from 60% to 53%) and with a modification in the PPR (from 1.09 to 0.77). Near physiological temperatures (33–34°C), NO711 was able to reduce the amplitude of synaptic responses in 16 out of 27 cells without silencing them (the amplitude of MF responses was 24.9 ± 3.4 and 15.7 ± 3.1 pA before and after NO711, respectively; p < 0.001; data not shown). Also in this case a reduction in the number of successes (from 62.7 ± 5.9 to 41.1 ± 6.6) was accompanied with an increase in PPR (from 1.8 ± 0.5 to 3.4 ± 0.9, p < 0.05) and a decrease in CV−2 (from 0.9 ± 0.2 to 0.6 ± 0.2, p < 0.05). In the remaining 11 cells, NO711 induced a potentiation of synaptic currents (from 36.1 ± 4.1 to 54.7 ± 5.9 pA). This effect was associated with an increase in the success rate (from 45.3 ± 4.8 to 69.6 ± 4.5, p < 0.05) and with a reduction of the PPR (from 1.9 ± 0.5 to 0.9 ± 0.1, p < 0.05) and CV−2 (from 0.7 ± 0.1 to 2.1 ± 0.6, p < 0.05), suggesting a presynaptic site of action. Although we do not know the cause of the NO711-induced potentiation of synaptic currents, it is likely that in particular circumstances spillover of GABA from the synaptic cleft may activate presynaptic GABAA receptors and enhance transmitter release via depolarization of MF terminals.
We showed that early in postnatal development, GABA released from MF terminals is under the powerful control of presynaptic GABAB receptors which contribute to synapses silencing.
In a previous study (Safiulina et al., 2006
), we provided evidence that at P0–P6, the principal neurotransmitter released from MF terminals into CA3 principal cells and interneurons is GABA. MF-evoked glutamatergic currents were rarely observed before P3 and, if present, they did not exhibit frequency-dependent facilitation before P6 (Marchal and Mulle, 2004
). In our case, GABAA-mediated synaptic responses exhibited all the characteristics of MF (Salin et al., 1996
) including low probability of release, frequency-dependent facilitation and sensitivity to L-AP4 (Safiulina et al., 2006
; see also Gutierrez et al., 2003
; Walker et al., 2001
). In a recent report (Uchigashima et al., 2007
), GABAergic responses evoked in CA3 principal neurons of developing mice by minimal stimulation of granule cells were attributed to GABA released from low threshold GABAergic interneurons. However, unlike MF responses described here, those reported by Uchigashima et al. (2007)
exhibited minimal frequency-dependent facilitation and higher probability of release. Markers of glutamatergic and GABAergic phenotypes have been shown to coexist in developing granule cells (Gutierrez, 2005
) which transiently express both mRNA coding for GAD 67 and GAD 67 protein (Dupuy and Houser, 1997
; Frahm and Draguhn, 2001
; Schwarzer and Sperk, 1995
; Sloviter et al., 1996
) as well as the vesicular GABA transporter VGAT in MF terminals (Lamas et al., 2001
; Safiulina et al., 2006
). It is unclear however, whether GABAA-mediated synaptic currents are generated by glutamatergic cells synthesizing GABA that would be down-regulated in adulthood (Frahm and Draguhn, 2001
) or by GAD positive interneurons migrating to the upper and middle portions of the granule layer cells (Dupuy and Houser, 1997
). Whatever is the source of GABA, it is clear from the present experiments that, under resting conditions, MF-evoked GABAergic responses exhibited a very low probability of release (they were often “presynaptically” silent; Gasparini et al., 2000
) and a robust paired-pulse facilitation. In the adult hippocampus, this short-term form of synaptic plasticity relies on specialized intrinsic properties of “giant” boutons present on MF terminals and on the tonic activation of presynaptic receptors by endogenous ligands present in the extracellular space (Bischofberger et al., 2006
; Nicoll and Schmitz, 2005
). Interestingly, while GABAB receptors are widely distributed at pre and postsynaptic sites (López-Bendito et al., 2004
), only the presynaptic ones are functional at birth (Gaiarsa et al., 1995
). Postsynaptic GABAB-mediated inhibition starts developing toward the end of the first–second postnatal week (Cherubini et al., 1998
; Gaiarsa et al., 1995
; Lei and McBain, 2003
). This was confirmed also in the present experiments in which no changes in membrane potential or input resistance were detected during application of CGP55845. Our results clearly demonstrate that at immature MF-CA3 synapses presynaptic GABAB receptors exert a powerful control on transmitter release: (i) CGP55845 significantly increased the probability of successes and enhanced the PPR, considered an index of presynaptic release probability (Zucker, 1989
). This effect was particularly evident in those synapses that appeared silent to the first pulse. (ii) Conversely, baclofen at very low concentrations induced a powerful depression of MF-evoked synaptic responses, and in several occasions made them silent. The depressing effect of baclofen was associated with a significant increase in transmitter failures and an increase in the PPR, indicating a presynaptic type of action. Interestingly, the IC50 value for baclofen found here was at least one order of magnitude lower than that found at GABAergic synapses in CA3 stratum radiatum interneurons (Lei and McBain, 2003
). This may be related to differences in GABAB receptors expression, distribution, and/or affinity among different cell types and suggests a target-specific modulation of GABAergic transmission by GABAB autoreceptors. (iii) Increasing the concentration of “ambient” GABA by repetitive stimulation of MF resulted, in the majority of cases, in synaptic depression that involved GABAB autoreceptors as demonstrated by the possibility to prevent this effect with CGP55845. The enhanced GABA release may cause also desensitization of postsynaptic GABAA receptors (Overstreet et al., 2000
). However, in the present experiments this is unlikely, since synaptic currents almost completely regained their control level in the presence of CGP55845. The frequency-dependent shift from synaptic depression to synaptic potentiation in the presence of CGP55845 observed at 10 Hz may involve the activation of different receptor subtypes. We can exclude the involvement of presynaptic kainate receptors known to exert a facilitatory effect on GABA release (Lerma, 2003
, 2006
), since these experiments were performed in the presence of DNQX. Other receptor types which may contribute to this phenomenon are GABAA receptors which have been shown to be localized on MF boutons (Alle and Geiger, 2007
). Activation of these receptors by GABA present in the extracellular space would depolarize presynaptic terminals by an outward flux of chloride (Khirug et al., 2008
; Nakamura et al., 2007
). This would lead to a larger flux of calcium in MF terminal through voltage-dependent calcium channels. In favor of this hypothesis are the experiments with bumetanide, in which the shift of EGABA from the depolarizing to the hyperpolarizing direction fully prevented the potentiating effect induced by high frequency stimulation of afferent inputs. It is likely that bumetanide exerted its effects on GABAA receptors localized on MF terminals as suggested by changes in PPR and CV−2 of GPSCs. (iv) Increasing the concentration of “ambient” GABA with NO711, a selective blocker of the GABA transporter GAT-1, induced synaptic depression that was rescued by CGP55845 indicating the involvement of GABAB receptors. This effect was particularly robust at room temperature, indicating that accumulation of GABA at synapses depends not only by the geometry, location, density and kinetics of the transporters but also by the temperature (Binda et al., 2002
). Also in this case, the effect was presynaptic since it was associated with changes in PPR and CV−2. Near physiological temperature (34°C), NO711 caused either a depression or a facilitation of MF-evoked synaptic currents. Although both changes were presynaptic, the precise mechanism underlying the potentiating effect remains unclear.
Unlike MF-CA3 responses, synaptic currents evoked in CA3 principal cells by activation of GABAergic interneurons in the hilus were unaffected by presynaptic GABAB receptors (Caillard et al., 1998
). Although surprising, this effect could be explained by a lower concentration of GABA at interneuron-CA3 synapses, insufficient to activate GABAB receptors present on interneuron terminals. The concentration of GABA in the synaptic cleft and its clearance by GABA transporters, GABAB receptors density, affinity and localization more or less close to the release sites, receptor coupling to downstream effectors, are all critical factors controlling the probability of transmitter release at both GABAergic and glutamatergic synapses. According to Moore et al. (2003)
at adult MF-CA3 connections, the basal release probability of glutamate is maintained at very low levels by the tonic action of extracellular adenosine on inhibitory A1 receptor subtypes. Although these results have been recently questioned (Kukley et al., 2005
), we can exclude a similar effect at immature GABAergic MF-CA3 connections because adenosine did not affect spontaneous GABAA-mediated synaptic events recorded from CA3 principal cells (Safiulina et al., 2005
).
In conclusion, our results show that early in postnatal life under basic synaptic transmission, at MF terminals, endogenously released GABA acting on presynaptic GABAB receptors exerts a powerful control on its own release and contributes to synapses silencing. This would reduce the possibility that, due to the depolarizing and excitatory action of GABA (Ben-Ari et al., 2007
), a single MF input generates action potentials in pyramidal cells and would prevent excessive activation of the auto-associative CA3 network and the emergence of seizures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Manuela Lough for carefully reading the manuscript. This work was supported by a grant from Ministero Istruzione Universita’ e Ricerca (MIUR) to EC.