 Yi Fang
Yi Fang Ting Gao
Ting Gao Baorong Zhang
Baorong Zhang Jiali Pu
Jiali Pu- Department of Neurology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Alzheimer’s disease (AD) is an irreversible neurodegenerative disorder that destroys cognitive functions. Recently, a number of high-profile clinical trials based on the amyloid cascade hypothesis have encountered disappointing results. The failure of these trials indicates the necessity for novel therapeutic strategies and disease models. In this review, we will describe how recent advances in stem cell technology have shed light on a novel treatment strategy and revolutionized the mechanistic investigation of AD pathogenesis. Current advances in promoting endogenous neurogenesis and transplanting exogenous stem cells from both bench research and clinical translation perspectives will be thoroughly summarized. In addition, reprogramming technology-based disease modeling, which has shown improved efficacy in recapitulating pathological features in human patients, will be discussed.
Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder characterized by progressive cognitive decline. AD affects 5–7% of older adults globally (Prince et al., 2013), and the expected number of affected patients is expected to grow continuously as the population ages in most countries. Currently, however, there is no cure for this condition. The Food and Drug Administration approved and actively marketed drugs for AD, including cholinesterase inhibitors and N-Methyl-D-Aspartate antagonists, whose effects improve daily functions to a certain degree (Rogers and Friedhoff, 1996; Tariot et al., 2004), yet they are not capable of altering disease progression. Tremendous efforts have been made to develop novel therapeutics to potentially reverse disease progression. Among the ongoing clinical trials designed to modify AD, a majority of them are intended to ameliorate Aβ, including β-secretase inhibitors, immunotherapies, and anti-aggregation agents (Cummings et al., 2017). Recently, several pioneering spotlighted trials targeting Aβ have met with dissatisfying results in terms of improved cognitive function (Doody et al., 2013; Salloway et al., 2014). One cannot jump to the conclusion that these negative clinical outcomes refute the prevailing amyloid cascade hypothesis, yet lessons should be learned from these dissatisfying results. Interestingly, successful elimination of amyloid in animal models, which typically overexpress APP or presenilin (PS1, PS2) genes, does not guarantee successful cognitive restoration in human patients. On the one hand, AD is a complex disease involving multiple cell types and cellular processes; therefore, targets other than amyloid should be considered and tested. On the other hand, developing solid models that better mimic disease pathologies in terms of NFTs, neuronal loss, and cellular interactions will undoubtedly benefit drug screening and mechanistic investigations. In this review, we will discuss how current advances in stem cell technology might address these unmet needs.
Efforts to Promote Endogenous Neurogenesis
Substantial neuronal loss is observed even in mild AD patients (Gómez-Isla et al., 1996). Intuitively, increasing the number of neurons or replacing lost neurons are potential therapeutic strategies for AD. Stem cells are capable of renewing themselves continuously and differentiating into specialized cells, including neurons. The human CNS was long considered as incapable of neural regeneration. Approximately two decades ago, the fact that neurons regenerate continuously throughout life was recognized and gradually became widely accepted (Eriksson et al., 1998). The process of generating new fate-specified, functional neurons from neural progenitor cells, which are functionally incorporated into a neural circuit, is defined as neurogenesis (Ming and Song, 2005). Across different species, neural regeneration mainly takes place at the DG of the hippocampus and the SVZ along the lateral ventricle (Kuhn et al., 1996; Alvarez-Buylla and Garcia-Verdugo, 2002). Notably, the DG, which plays a crucial role in memory formation processes [e.g., pattern separation (Leutgeb et al., 2007)], is related to early memory loss in AD (Ohm, 2007).
Neurogenesis decline accompanies normal aging (Klempin and Kempermann, 2007). Patients with neurodegenerative disorders continuously lose neurons while neurogenesis is insufficient. For AD, accumulating evidence suggests that impaired neurogenesis plays a role in its pathogenesis (Hollands et al., 2016). Multiple molecules involved in AD pathogenesis [such as ApoE (Yang et al., 2011), PS1 (Gadadhar et al., 2011), and APP (Ghosal et al., 2010)] were recognized to take part in neurogenesis modulation. Conversely, inhibition of NSCs results in deterioration of cognitive processes, such as hippocampal-dependent memory (Imayoshi et al., 2008). Therefore, understanding the mechanism of neurogenesis dysfunction and intervening with neurogenesis represents an alternative AD therapeutic strategy. Neurotrophic factors and transcription factors involved in signaling pathways, the vascular and immune systems, metabolic factors, and epigenetic regulation are recognized to participate in regulating neurogenesis (Horgusluoglu et al., 2017).
Generally, neurogenesis can be modulated by multiple factors that are related to lifestyle, including learning (Gould et al., 1999), exercise (van Praag et al., 1999), social interaction (Stranahan et al., 2006), caloric restriction (Bondolfi et al., 2004), blood oxygen level (Lange et al., 2016), and even microbial colonization (Ogbonnaya et al., 2015). In this regard, advocating a healthy lifestyle exerts at least a mild effect on preventing or controlling AD in the long run. Future animal and epidemiological studies need to elucidate the best feasible individualized strategy for lifestyle modification combination that takes a patient’s genetic background into account. Apart from lifestyle modification, which exerts mild effects, several pioneering studies identified key molecules or drugs that rescue or reverse NSC dysfunction in elderly animal models.
Proneurogenic Effects of Medications Available in the Clinic
Several drugs that are extensively used in the clinic have demonstrated robust proneurogenic effects in animals. Erythropoietin (EPO) is a cytokine that promotes hematopoiesis. Clinically, recombinant EPO is indicated for anemia. Its non-hematopoietic functions are being explored. During midgestation, EPO receptors are localized to regions of the neural tube that are responsible for neurogenesis (Alnaeeli et al., 2012). In the adult mouse brain, EPO receptors were detected primarily in the hippocampus, capsula interna, cortex, and midbrain (Digicaylioglu et al., 1995). Therefore, enhancing EPO receptor expression in the brain and increasing EPO is a potential strategy to enhance neurogenesis. In healthy young mice, 3 weeks of EPO administration significantly elevated the number of pyramidal neurons and oligodendrocytes (Hassouna et al., 2016). Furthermore, in an intracerebroventricular-streptozotocin rat model of sporadic AD, 2 weeks of EPO administration successfully prevented the memory deficit and the hippocampal neuronal loss induced by streptozotocin (Cevik et al., 2017).
Similarly, granulocyte colony-stimulating factor, a hematopoietic growth factor that stimulates proliferation and differentiation of neutrophil precursors, has been linked to enhanced neurogenesis (Schneider et al., 2005; Jung et al., 2006). In animal models of AD, restored memory was also observed (Tsai et al., 2007). If proven to be effective and safe in patients with AD, administration of these hematopoietic growth factors might be alternative options to modify symptoms.
Additionally, antidepressant drugs (primarily selective 5-HT reuptake inhibitors) have been established to play a crucial role in upregulating neurogenesis and achieving satisfying treatment response in patients with depression (Taupin, 2006; Hanson et al., 2011). Future studies need to determine whether antidepressants are efficacious for symptom reduction in patients with AD without comorbid depression. Further, additional research is needed to understand the relative contribution of enhanced neurogenesis and serotonin signaling, because previous studies have attributed reduced Aβ level and plaque formation in an aged APP/PS1 mouse model after EPO treatment to serotonin signaling regulation (Cirrito et al., 2011; Sheline et al., 2014).
Correcting Aberrant Metabolism to Modulate Neurogenesis
Lipids are a major component of the brain. Aberrant lipid metabolism is highly associated with AD (Di Paolo and Kim, 2011), yet the exact mechanism remains to be fully uncovered. Recent evidence indicates its role in neurogenesis defect. Lipid droplets selectively accumulated in the SVZ were found to distort NSCs and their daughter neuroblasts before amyloid accumulation in a 2-month-old 3xTg AD mouse model (Hamilton et al., 2015). Using an imaging mass spectrometry-based lipidomics strategy to identify the lipid responsible, oleic acid accumulation within the SVZ was observed (Hamilton et al., 2015). Microanalysis of microdissected SVZs demonstrated local aberrant lipid metabolism in the 3xTg brain, including increased expression of stearoyl-CoA-desaturase, the rate-limiting enzyme of oleic acid synthesis. Strikingly, when oleic acid synthesis was inhibited by intracerebroventricular infusion with a stearoyl-CoA-desaturase inhibitor, NSC proliferation reduction in the DG and SVZ was rescued in 2-month-old 3xTg mice (Hamilton et al., 2015). Current knowledge on the interaction between lipid metabolism and NSCs in both physiological and pathological conditions is inadequate. With advances in cutting-edge technology to trace lipid metabolism in the brain, future research needs to unravel the roles played by diverse lipid metabolites and metabolic processes in NSC dysfunction and AD.
Mature neurons are highly dependent on the mitochondrial electron transport chain and oxidative phosphorylation to meet the high energy demand. Using pharmacological and genetic approaches, the metabolic switch from glycolysis to mitochondrial electron transport chain and oxidative phosphorylation was reported to be required for NSCs to give rise to intermediate progenitor cells in adult neurogenesis (Beckervordersandforth et al., 2017). Moreover, eliminating the function of mitochondrial transcription factor A (Tfam) replicated age-related neurogenesis decline in young mice. The most exciting part of this study is that short-term treatment with piracetam, a drug that improves mitochondrial function through a number of pathways, was sufficient to double the number of proliferating cells in an aged animal model. Numerous lines of evidence have indicated the involvement of mitochondrial dysfunction in the pathogenesis of AD (Swerdlow et al., 2014); therefore, additional studies are needed to analyze the role of NSC’s metabolic switch in AD pathogenesis.
Recent studies revealed that NSC behavior is regulated by multiple metabolism-related processes, such as oxygen consumption, ATP production, and reactive oxygen species signaling (Almeida and Vieira, 2017). The metabolic drug metformin (Wang et al., 2012; Fatt et al., 2015) was recognized to take part in neurogenesis modulation, suggesting that the complex nature of metabolism and neurogenesis remains to be fully investigated.
Young Blood: ‘Resetting the Aging Clock’
Heterochronic parabiosis is an experimental method whereby the circulatory systems of young and elderly animals are joined and shared. Though controversial, there have been studies on the relationship between heterochronic parabiosis and rejuvenation for over a century (Conboy et al., 2013). The rationale behind these attempts is that there are signals from both the CNS itself and the body system outside the CNS that instruct neurogenesis in an age-related pattern. As NSCs in the CNS are exposed to blood vessels and cerebrospinal fluid, it is believed that by interfering with the blood carrying these signals, age-related neurodegeneration might be rescued. Administration of young plasma improves synaptic plasticity in the DG and elevated the cognitive function in elderly mice with the involvement of the cyclic AMP response element binding protein (Creb) signaling pathway (Villeda et al., 2014). Hunting for youth-promoting factors has attracted much research interest. Growth and differentiation factor 11 (GDF11) is one of the candidates that have shown promising potential regarding skeletal muscle, heart, and CNS rejuvenation (Jamaiyar et al., 2017). After systematic GDF11 administration, NSC proliferation in the SVZ was significantly elevated (Katsimpardi et al., 2014).
Conversely, chemokine CCL11, major histocompatibility complex component β2-microglobulin (β2-M), and transforming growth factor β (TGF-β) have been recognized as aging-promoting factors, which are elevated in the blood of the elderly and contribute to decreased neurogenesis and learning and memory (Villeda et al., 2011; Smith et al., 2015; Yousef et al., 2015). Furthermore, neurogenesis and cognitive functions can be mitigated in aged mice by reducing β2-M expression (Smith et al., 2015). In addition, inhibition of TGF-β1 signaling enhanced neurogenesis, as well as normalizing the β2-M level (Yousef et al., 2015).
It should be noted that enhancing neurogenesis does not equate to rejuvenating the brain. Rather than neurogenesis, elevated synaptic plasticity and hippocampal-dependent cognition were observed after intravenous administration of human cord plasma in aged mice (Castellano et al., 2017). Stem cell dysfunction is only one of the nine hallmarks of aging (López-Otín et al., 2013); therefore, future studies need to decipher the relative contribution of various modulators.
Shared mechanisms [e.g., synaptic plasticity (Hatanpää et al., 1999)] do exist between healthy aging and AD conditions; however, whether the interventions described above can sufficiently rescue cognitive decline in AD animal models and patients remains to be explored. The first clinical trial on transfusion of plasma from young donors to aged patients with AD is ongoing (identifier NCT02256306 on ClinicalTrials.gov). As previous studies have attributed better cognitive test results after plasma exchange to enhanced peripheral clearance of Aβ (Boada et al., 2009; Liu et al., 2015), plasma exchange studies need to use multiple outcome measures to evaluate neurogenesis elevation and increased amyloid clearance. Extra caution must be taken in clinical practice because of complications of plasma exchange (e.g., anaphylactic reaction). As more youth-promoting and aging-promoting factors are likely to be identified, it might be a prudent strategy to administer cocktail therapy comprising key modulators with known side effects.
Looking Forward
A large number of current neurogenesis studies based on animal models are not fully applicable to humans. However, postmortem studies are limited by their retrospective study design, tissue damage after death, and incomplete patient history information. Although attempts have been made to identify key metabolic biomarkers (Manganas et al., 2007; Spalding et al., 2013), current non-invasive macroscopic neuroimaging measures in the brain, such as magnetic resonance imaging and positron emission tomography are not sufficiently specific, precise, or sensitive to detect neurogenesis (for a review, see Ho et al., 2013). Collaboration between different academic domains is urgently needed to develop precise in vivo neurogenesis detection techniques.
Limited by the available research techniques, current understanding of neurogenesis dysfunction in AD is lacking. Numerous studies have pointed out that neurogenesis alterations start relatively early in the course of AD progression (Mu and Gage, 2011; Unger et al., 2016), making it a promising target for early intervention or prophylaxis. Therefore, a mechanistic insight into when and how early neurogenesis alteration is triggered is required. Furthermore, with advances in neurogenesis detection techniques in adult humans, neurogenesis alteration might be an early marker of AD. By contrast, during AD progression, how the classic pathology (e.g., amyloid plaques) interacts with neurogenesis is not well understood. Interventions that promote the function of newly generated neurons in the context of AD pathology remains to be investigated.
To summarize, accumulating evidence suggests the promising potential of intervening with endogenous NSC dysfunction and deteriorated neurogenesis to improve AD-related cognitive decline. More molecules involved in neurogenesis are likely to exist, and identifying these molecules and their underlying mechanisms might pave the way for novel AD therapeutics.
Transplanting Stem Cells to Support Neurons
Transplanting exogenous stem cells into CNS is an alternative strategy that has attracted much research interest. Significant effort has been made to engraft stem cells into degenerated neural tissue. However, the number of stem cells transplanted into brain decreased over time (Khoo et al., 2011). The efficacy of stem cell transdifferentiation into grafted tissue is low (Phinney and Prockop, 2007).
Accumulating evidence suggests that stem cells exert neurotrophic effects after transplantation (Lu et al., 2003; Martino and Pluchino, 2006). Transplanted stem cells elevate the levels of various factors, including BDNF (Blurton-Jones et al., 2009), glial cell line-derived neurotrophic factor (GDNF) (Kim S. et al., 2012), insulin-like growth factor 1 (IGF-1), Glucagon-like peptide-1 (GLP-1) (Klinge et al., 2011), vascular endothelial growth factor (VEGF) (Garcia et al., 2014), to exert a paracrine effect. Recent research comprising transplanting stem cells in animal models are listed in Table 1. Stem cells have been recognized to improve various cellular functions in animal models of AD, including synaptic strength (Blurton-Jones et al., 2009), neurogenesis (Kim S. et al., 2012; Kim D.H. et al., 2015), microglial activity (Lee et al., 2009a; Yang et al., 2013), angiogenesis (Garcia et al., 2014), mitochondrial function (Zhang et al., 2015), autophagy (Shin et al., 2014), and apoptosis (Lee et al., 2010). Stem cell transplantation influences AD via multiple mechanisms; therefore, it is promising compared with conventional treatments that target a single pathology.
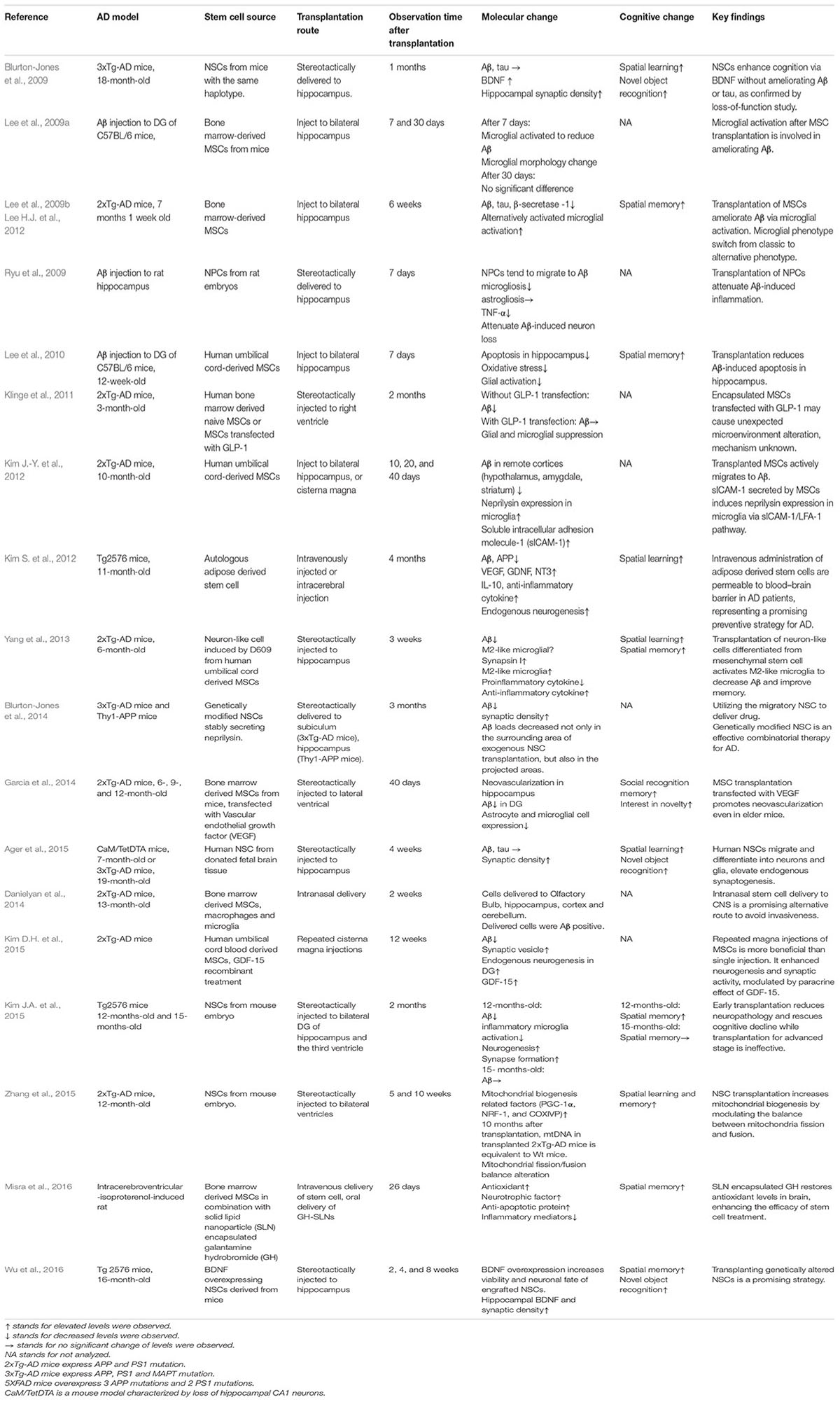
TABLE 1. Stem cell transplantation studies on animal models of Alzheimer’s disease.
Transplanting Genetically Altered Neural Stem Cells
Most NSC transplantation studies successfully rescued cognitive dysfunction in animal models of AD, yet failed to ameliorate Aβ deposition (Blurton-Jones et al., 2009; Zhang et al., 2014; Ager et al., 2015). To maximize the efficacy of transplantation, a strategy that harnesses NSCs to deliver key disease-modulating proteins has been proposed. Overexpression of neprilysin, the key Aβ degrading enzyme, in transplanted MSCs significantly reduces synaptic loss and the Aβ level (Blurton-Jones et al., 2014). Other cellular functions that are of significant importance to AD are modulated by transfecting NSCs with other factors. BDNF-overexpressing NSCs induced a better recovery of the hippocampal BDNF level, synaptic density and stronger cognitive function (Wu et al., 2016). IGF-1 is another trophic factor that promotes differentiation toward neuronal cells and is essential for neural proliferation and survival (Russo et al., 2005). An in vitro study overexpressing of IGF-1 in cortical neurons demonstrated increased GABAergic neuron differentiation, increased VEGF production, and elevated survival of the transplanted cells (McGinley et al., 2016). Despite showing promising potential, this strategy faces major challenges. The safety and efficacy of transplanting genetically altered cells in humans has not yet been validated. Furthermore, this strategy requires stem cell genome alteration, which could face stricter regulatory restrictions in clinical translation.
Inflammatory Responses Modulated by Mesenchymal Stem Cell Transplantation
Apart from NSCs, the most widely used source of stem cells for transplantation are MSCs. MSCs are cells that reside around blood vessels in bone marrow, supporting hematopoiesis and cartilage regeneration, and complementing the differentiated osteoblasts and adipocytes (Bianco et al., 2013). Not only do they differentiate into adipocytes, myocytes, osteoblasts, chondrocytes, and cardiovascular, and neurogenic cell types, but also tend to reside at sites of injury and inflammation (Karp and Leng Teo, 2009). Studies have confirmed modulation of inflammation after MSC transplantation.
Inflammation plays a critical role in AD pathogenesis (Heppner et al., 2015). Inflammatory responses in the CNS reflect endogenous efforts to clear pathological deposits. Microglia are the resident immune cells in the brain, which are involved in both neural protection and death. A number of studies have confirmed that MSC transplantation modulates microglial activity in the CNS to ameliorate Aβ (Lee et al., 2009a; Lee H.J. et al., 2012). Moreover, there are two opposite microglial phenotypes in the CNS: M1 and M2. M1 microglia releases pro-inflammatory cytokines such as TNF-α, IL-1β, and reactive oxygen species. M2 microglia, however, are anti-inflammatory. M2 microglia are induced by IL-4, IL-13, apoptotic cells, or other anti-inflammatory cytokines (Tang and Le, 2016). Several previous trials on mice confirmed that M2 microglia are involved in ameliorating Aβ after transplantation (Lee et al., 2009b; Ma et al., 2013; Yang et al., 2013). In this regard, targeting the M1/M2 microglia balance is a potential strategy to ameliorate inflammation in AD. CCL5 secreted by transplanted MSCs, for instance, has been recognized to activate M2 microglia (Lee J.K. et al., 2012).
The active homing mechanism of MSCs makes their systemic administration (e.g., intravenous injection) possible, which would possibly avoid direct invasion of the brain. Besides, they are convenient to access, lack ethical concerns, and have low immunogenicity. MSCs hold significant potential in clinical use. However, our current understanding of the MSC trafficking mechanism is lacking (Karp and Leng Teo, 2009). One recent study reported a low efficacy of MSC homing to lesion sites in an aged AD mouse model (Fabian et al., 2017). With increasing insights into the homing mechanism, manipulation to enhance the efficacy of transplanted MSCs that home specifically to the brain might benefit MSC transplantation.
Extracellular Vesicles Derived From Stem Cells
Besides secreting soluble molecules such as BDNF (the classic paracrine effect), recent studies have explored the therapeutic potential of stem cell-derived extracellular vesicles (Katsuda et al., 2013a). Exosomes are cell-derived membrane vesicles containing lipids, proteins, mRNAs, and microRNAs. Recently, they have been recognized to be one of the key mediators of cell-to-cell communication. In addition to functional proteins, they transfer genetic information to recipient cells to regulate physiological or pathological processes (Valadi et al., 2007; Record et al., 2011). For instance, one study confirmed that injection of exosomes secreted by self-derived dendritic cells achieved 60% mRNA and protein knockdown of β-secretase 1 and 55% Aβ reduction using short interfering RNAs in wild-type mice (Alvarez-Erviti et al., 2011). From this perspective, it is likely that stem cells transmit tissue repair or regeneration signals to lesions via exosomes. Current research has revealed the potential of stem cell-derived exosomes in the treatment of stroke (Xin et al., 2013), myocardial ischemia (Lai et al., 2010), and liver fibrosis (Li et al., 2012). For AD, one recent study suggested that adipose tissue-derived MSCs secret exosomes that contain enzymatically active neprilysin when co-cultured with Aβ (Katsuda et al., 2013b). Looking forward, harnessing stem cells to either deliver designed drugs or secret a combination of molecules and RNAs that represent the body’s response to the pathological microenvironment with spatial precision is a promising strategy (El Andaloussi et al., 2013). Furthermore, although there is a long way to go, administration of exosomes derived from stem cells represents an alternative therapy for AD to circumvent relatively unsafe cell transplantation.
Clinical Translation
There has been growing interest in exploring the potential of treating patients with AD using stem cell transplantation. Ongoing clinical trials intended to transplant stem cells into patients with AD are listed in Table 2. Various sources of MSCs, including human umbilical cord blood, placental tissue, autologous adipose tissue, and ischemia-tolerant MSCs, are being tested in clinical trials. Accessibility, invasiveness, potential tetratomic induction, proliferation rate, cost, and efficacy should be thoroughly evaluated and compared. The following paragraphs summarize several concerns and advances regarding transformation from the bench to the bedside.
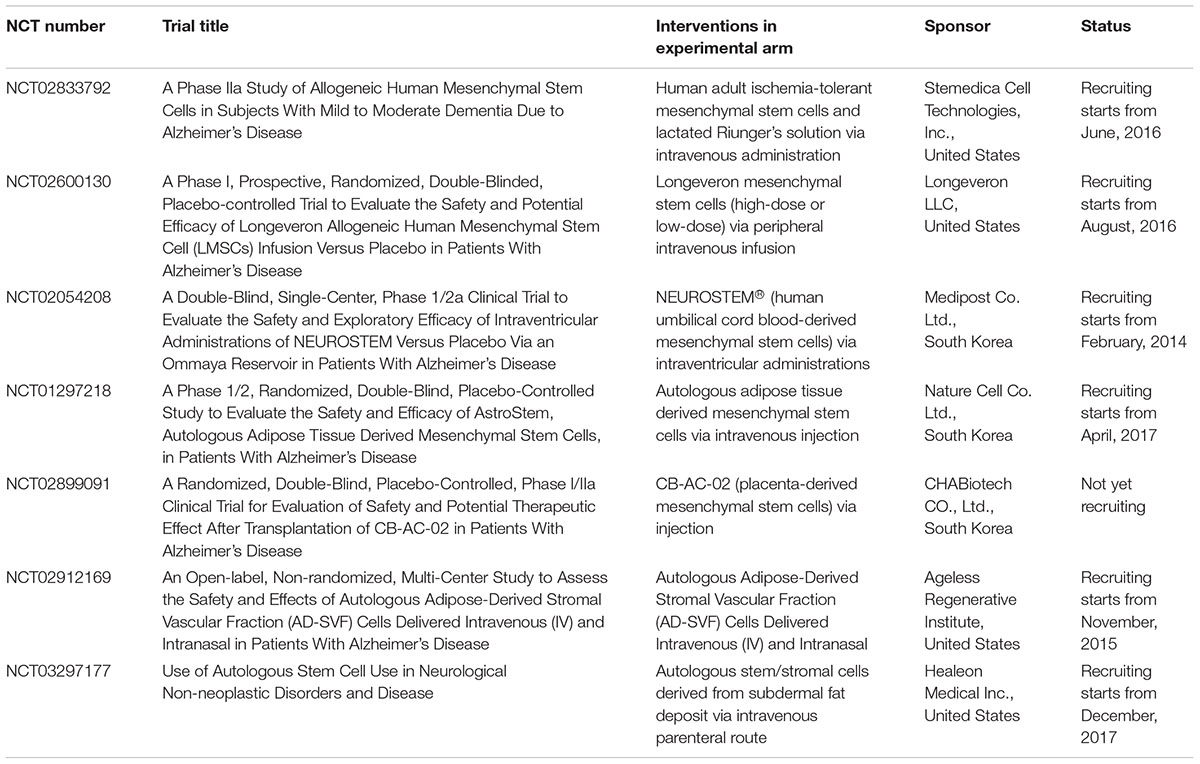
TABLE 2. Selected clinical trials registered at ClinTrials.gov on stem cell therapy for Alzheimer’s disease as of October, 2017.
First, the efficacious time frame for AD treatment is not unknown. Multiple studies demonstrated increased synaptic strength in animal models after NSC transplantation (Bae et al., 2007; Blurton-Jones et al., 2009; Kim D.H. et al., 2015). Previous studies have pointed out that synaptic dysfunction occurs before plaque formation (Selkoe, 2002), and loss of synapses in the neocortex and hippocampus is the predominant factor that correlates with cognitive impairment in AD (Terry et al., 1991). Although not carefully tested in clinical trials, it is intriguing to think that NSC transplantation might protect patients with AD at an early stage. By contrast, AD is a progressive chronic disease that typically lasts several years after initial diagnosis; therefore, the appropriate time window for stem cell transplantation in the course of AD progression requires further exploration. A large number of preclinical studies used mouse models at a relatively young age or at an early-stage of disease progression, yet the observation time was not long enough. One study using the Tg2576 mouse model, which develops age-related cognitive defects, demonstrated that transplantation recovered cognition and ameliorated neuropathology in 12-month-old mouse, while transplantation failed to recover either cognition or neuropathology in a 15-month-old mouse (Kim J.A. et al., 2015). Future research needs to elucidate whether stem cell transplantation is efficacious for patients with AD in an advanced stage, and whether stem cell transplantation is efficacious and necessary for prophylactic purposes.
As transplantation research transforms from the laboratory to the clinic, large-scale stem cell transplantation requires proper quality control protocol. Recent preclinical studies on AD (Marsh et al., 2017) and cervical spinal cord injury (Anderson et al., 2017) demonstrated that clinical-grade stem cell transplantation might not be as effective as research-grade cell transplantation. To prioritize the efficacy and safety of transplantation for human patients, longer-term observation on multiple animal models after transplantation and more comparability tests on large-scale stem cell manufacturing are needed.
Invasion of the brain might be a major concern for elderly and weak patients with AD. Several studies reported novel methods to circumvent invasive surgery. Intranasal and intravenous routes are being explored (Kim S. et al., 2012; Danielyan et al., 2014; Kanamaru et al., 2015). Recent advances in brain imaging allow magnetic resonance imaging-guided focused ultrasound to target specific structures, involving transient disruption of the blood–brain barrier to deliver therapeutic stem cells from blood to the parenchyma (Burgess et al., 2011). Novel sources of stem cell are also being tested. For instance, dental pulp cells are cranial neural crest-derived multipotent cells that present neurotrophic properties (Mead et al., 2017). They are being tested as a potential stem cell source for transplantation in an AD model (Apel et al., 2009; Ahmed Nel et al., 2016).
Another concern in current clinical practice is transplantation rejection. To lower the risk of a serious immune response, researchers are exploring the potential of either autogenic stem cells (e.g., adipose tissue-derived MSCs or bone marrow-derived MSCs) or allogenic cells with hypo-immunogenic properties [e.g., umbilical cord-derived MSCs (Weiss et al., 2008)]. Alternatively, to help the engrafted cells avoid possible immune rejection, cell encapsulation techniques have been applied in several studies. With a polymeric semi-permeable membrane that allows the exchange of essential factors for cell metabolism, the encapsulated cells are protected from immune attack for long-term stable delivery of therapeutic agents. Several studies have used encapsulated somatic cells to deliver various growth factors to treat AD in animal models (Garcia et al., 2010; Spuch et al., 2010) and humans (Eriksdotter-Jönhagen et al., 2012; Wahlberg et al., 2012). One study demonstrated suppression of microglia and astrocytes using encapsulated MSCs transfected with GLP-1 (Klinge et al., 2011).
Last but not least, as has been described previously, the number of transplanted stem cell is prone to decrease over time. Increasing the survival rate of transplanted stem cells and lowering negative responses in the body after their death is crucial for sustaining a long-term therapeutic effect. These concerns need to be resolved before stem cell transplantation goes into clinical practice. Regulation and oversight should be strengthened to ensure that the tremendous potential of stem cells is fully realized.
Investigating Alzheimer’S Disease by Reprogramming Techniques
It is difficult to obtain tissue samples from the human CNS to model disease; therefore, previous insights into AD relied heavily on post-mortem autopsy, which represents the pathology at the end of the disease, or in transgenic mice expressing or overexpressing APP or PS mutations. These animal models were developed on the basis of the prevailing amyloid cascade hypothesis, which holds that it is the deposition of APP cleavage products that causes the pathological changes. However, they are not capable of replicating the full spectrum of AD pathology observed in human patients, such as tau pathology, mutations in non-coding regions of the genome, and neurodegeneration. Furthermore, approximately 3–32% of patients clinically diagnosed with AD are amyloid-negative on positron emission tomography imaging (Ossenkoppele et al., 2015). The huge heterogeneity in AD patients requires them to be divided into subgroups or to be considered as individuals in terms of mechanistic studies and drug screening. The lack of proper disease models might be one of the reasons why drugs proven to efficiently ameliorate Aβ in animal models do not perform well in the human brain where much more complex pathologies are involved.
Induced pluripotent stem cells (iPSCs) are created using a technique that reprograms somatic cells back to the pluripotent-state by the overexpression of key transcription factors (Takahashi et al., 2007). The introduction of iPSCs has revolutionized neurological disease modeling. In 2011, the first AD model using iPSCs was reported (Yagi et al., 2011). Pluripotent stem cells were induced using five transcription factors (OCT4, SOX2, KLF4, LIN28, and NANOG) from fibroblasts of patients with familial AD. These iPSCs were then induced into neurons, which demonstrated typical pathology. Huge heterogeneity exists in patients with AD; therefore, the iPSC technique offers unique opportunities to study patients by subgroups and screen drugs in a patient-specific manner. The following paragraphs will review the phenotypes and drug reactions presented in recent iPSC-based AD models generated from a variety of patients.
Familial Alzheimer’s Disease
Familial AD (fAD) affects about 0.5% of all patients with AD, the majority of which are autosomal dominant with full penetrance that typically presents before 65 years old. To generate amyloid-β peptides, two sequential cleavages of APP occur, cleavage by β-secretase in the extracellular space, and then by γ-secretase within the membrane. Mutations in PS1, PS2, and APP genes are the major causes of fAD.
The presenilin protein is an essential component of γ-secretase. Gamma-secretase cleaves at multiple sites; therefore, Aβ varies in amino acid length (36–43 residues). Aβ40 is the most abundant type and Aβ42 is most prone to self-aggregation (Haass and Selkoe, 2007). iPSC-based studies observed elevated Aβ42/Aβ40 (Yagi et al., 2011; Koch et al., 2012; Sproul et al., 2014). Furthermore, a γ-secretase inhibitor effectively reduced Aβ secretion (Yagi et al., 2011; Koch et al., 2012). NSCs from iPSCs with the PS1 L166P mutation were generated (Koch et al., 2012). That study demonstrated selectively decreased Aβ40 secretion and an elevated Aβ42/Aβ40 ratio. The Aβ42 level did not differ from the control; therefore, the authors concluded that partial dysfunction of γ-secretase occurs in the PS1 L166P mutation, while other γ-secretase functions remain intact. Sproul et al. (2014) studied neural progenitor cells derived from iPSCs carrying the PS1 A246E or M146L mutations. Compared with the control, molecular profiling identified 14 genes with altered expression in the PS1 mutation lines. Among them, five genes were differentially expressed in late-onset AD. This study shed light on identifying genetic expression alterations, which will facilitate further studies on fAD pathogenesis.
Dissatisfying clinical outcomes cast doubt on the amyloid cascade hypothesis; therefore, its validity should be thoroughly tested using various models. Cells from patients with fAD carrying APP mutations provide models to study the relationship between Aβ and tau. Israel et al. (2012) studied two iPSC lines generated from patients with fAD who carried a duplication of the APP gene. Elevated Aβ40, active GSK3β (the kinase that phosphorylates tau at Thr231), phosphorylated tau at Thr231 and total tau was observed. To test if there was a direct causative relationship between the APP processing product and phosphorylated tau and active GSK3β, β-secretase and γ-secretase inhibitors were added. Notably, only the β-secretase inhibitor treatment partially reduced phosphorylated tau and active GSK3β levels, indicating that APP processing (products other than Aβ) is responsible for tau Thr231 phosphorylation. This study also confirmed the assumption that early endosomes are present in iPSC-iNs, implying that these early endosomes take part in modulating APP processing.
Muratore et al. (2014) explored the relationship between APP processing and tau in the APP V717I mutation. The APP V717I mutation alters the initial cleavage site of γ-secretase, causing altered APP cleavage by both β-secretase and γ-secretase. The iNs demonstrated increased levels of both Aβ42 and Aβ38, and increased total and phosphorylated tau. Early Aβ antibody treatment reverses tau, suggesting a partially causal relationship between altered APP processing (Aβ) and tau formation.
Moore et al. (2015) studied the relationship between APP processing and tau in different patients with AD of different genetic backgrounds. iPSC lines which altered the APP dosage (APP duplication) or ε-cleavage site (APP V717I) demonstrated elevated total or phosphorylated tau levels, while PS1 mutants (Y115C and intron 4), which elevated the Aβ42/Aβ40 ratio, did not. Furthermore, a β-secretase inhibitor, which prevents the β-C-terminal fragment (CTF) from forming, reduced the intracellular tau level. Meanwhile, a γ-secretase inhibitor that promotes β-CTF aggregation elevated the intracellular tau level. Therefore, Moore et al. (2015) proposed an intriguing hypothesis that the APP cleavage product (β-CTF) is involved in regulating tau pathology. Whether or not β-CTF is related to advanced tau/NFT pathology remains to be investigated.
Kondo et al. (2013) studied the phenotype of the APP-E693Δ mutation, a rare autosomal recessive mutation, using iPSCs. Intriguingly, rather than extracellular Aβ aggregation, intracellular accumulation of Aβ oligomers was observed in this APP-E693Δ line. In addition, intracellular Aβ aggregation leads to a cellular stress response causing endoplasmic reticulum and oxidative stress. One of two sporadic patient lines demonstrated the same phenotype as APP-E693Δ mutation, while the APP-V717L mutation did not. Furthermore, only the lines with intracellular Aβ are responsive to Docosahexaenoic acid treatment. The authors proposed dividing patients with AD into the intracellular Aβ type and extracellular Aβ type to achieve personalized treatment.
Sporadic Alzheimer’s Disease
The majority of patients with AD suffer from sporadic AD (sAD). As revealed by several iPSC studies, large heterogeneity exists among the phenotypes and drug responsiveness of patients with sAD (Israel et al., 2012; Kondo et al., 2013). Large-scale genome-wide association studies have identified numerous susceptible genetic variations in patients with late-onset sAD (Lambert et al., 2013), demonstrating the complex genetic nature of sAD. However, the biological functions of those key genes associated with the pathogenesis of AD have not been well studied. Furthermore, altered levels of Aβ were not observed in a large proportion of patients with sAD (Toledo et al., 2013), and tau predicts dementia symptoms better than Aβ (Brier et al., 2016). Thus, conventional animal models, which do not involve tau pathology, are not appropriate to study sAD. Several recent studies using iPSC-based modeling gained insights into the function of these genes and provided examples of how to study sporadic diseases.
Apolipoprotein E (APOE), the gene encoding the key protein for lipid catabolism has been recognized to play a key role in the pathogenesis of sAD. Sortilin-related receptor, L [DLR class] (SORL1) is a neuronal APOE receptor that is predominantly expressed in the CNS. Young et al. (2015) reported the function of SORL1 single nucleotide polymorphisms in non-coding regions in an iPSC-based model. When treated with BDNF, protective phenotypes (carrying one or two protective alleles) responded with significantly higher SORL1 expression and Aβ reduction compared with those of the risk phenotypes (carrying two risk alleles). Furthermore, SORL1 knockdown confirmed that BDNF-induced Aβ reduction is dependent on SORL1 expression. This study indicated that neurotrophic factors such as BDNF are potentially regulated by disease risk-related genetic mutations. Furthermore, this study implied the necessity of detailed stratification of patients with sAD to tackle the pathogenetic mechanisms.
Hossini et al. (2015) studied genetic expression by transcriptome analysis and demonstrated the first protein interaction network from one patient with sAD. By analyzing the transcriptome, the authors reported the upregulation and downregulation of several genes that differed from fAD-associated genes. Moreover, the authors observed ubiquitin-proteasome system dysfunction in this patient with sAD compared with an age-matched control. Protein interaction network analysis revealed the involvement of APP and GSK3β.
Three-Dimensional Modeling and Chimeric Modeling
In humans, hyperphosphorylated tau aggregates in dendrites and axons to form dystrophic neurites, and aggregates in cell bodies to form NFTs. As shown in Table 3, conventional iPSC-based modeling did not demonstrate robust NFTs. Choi et al. (2014) developed a three-dimensional human NSC-derived in vitro model of AD. Matrigel containing extracellular matrix proteins was used as the support. By limiting Aβ diffusion, this three-dimensional culture promoted Aβ aggregation and its downstream cascade. In addition, it closely mimics the real cell environment with regard to vertical cell growth, the synaptic distance between cells, and cell maturation (D’Avanzo et al., 2015). In Choi et al.’s study, iPSC lines carrying APP and PS1 mutations in three-dimensional culture successfully demonstrated robust extracellular Aβ plaques and NFTs. Furthermore, β-secretase and γ-secretase inhibitors reduced the Aβ level as well as the tau level, supporting the amyloid cascade hypothesis.
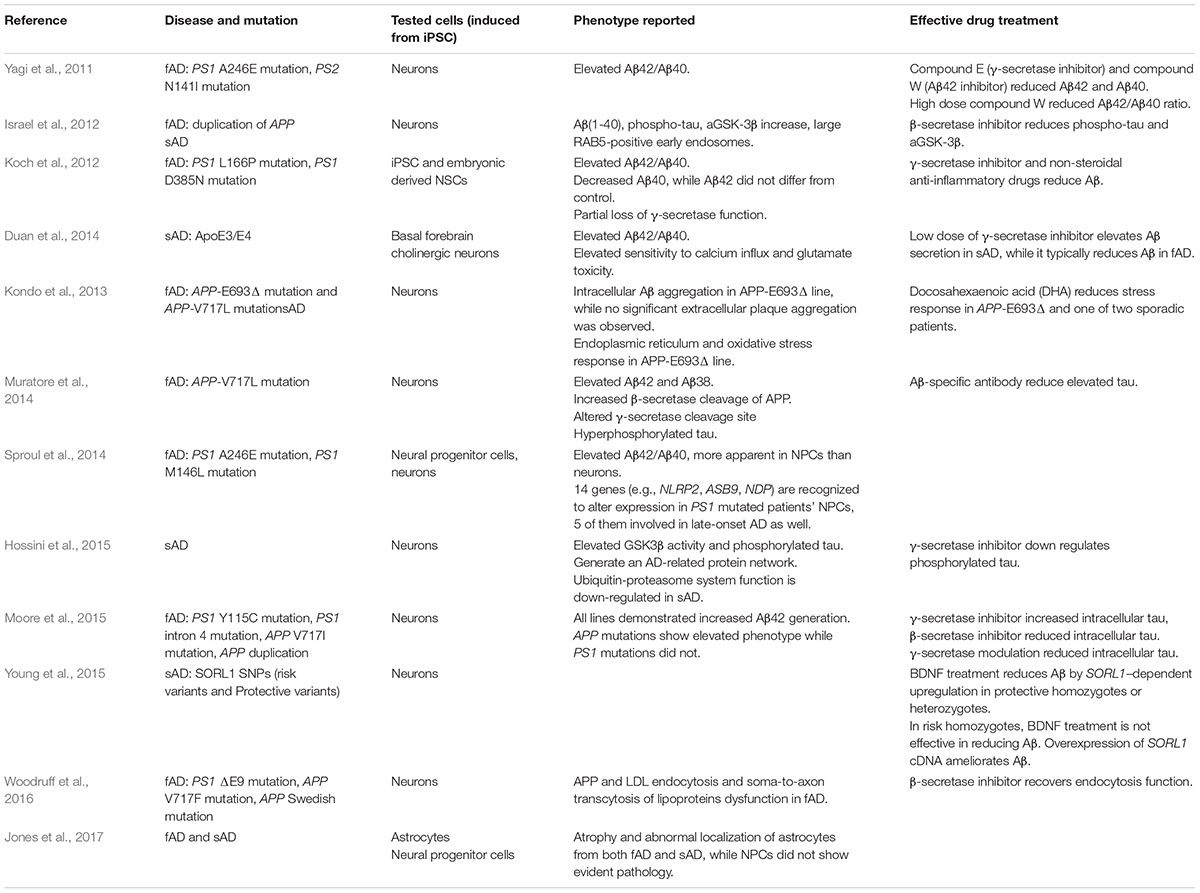
TABLE 3. iPSC modeled Alzheimer’s disease.
Another type of three-dimensional culture system, termed organoids or spheroids, is a scaffold-free self-organizing structure (Lancaster et al., 2013). Raja et al. (2016) developed brain organoids from iPSCs derived from patients with fAD. The organoid successfully recapitulated Aβ aggregation, hyperphosphorylated tau, and endosome abnormalities. Treatment with β-secretase and γ-secretase inhibitors reduced the Aβ and tau pathology. Lee et al. (2016) generated a three-dimensional neuro-spheroid culture from blood cell-derived iPSCs from patients with sAD. Similar to Raja et al.’s findings, the three-dimensional neuro-spheroids demonstrated Aβ aggregation, and hyperphosphorylated tau. Notably, the authors reported reduced Aβ-ameliorating efficacy of the β-secretase and γ-secretase inhibitors in three-dimensional model compared with a two-dimensional model. More research is needed to determine the cause of the diminished drug efficacy in the three-dimensional model. This type of three-dimensional modeling offers a great opportunity to study the role of cellular interactions (e.g., astrocytes and microglia involvement and neural cytoskeletal malfunction) in AD progression.
Chimeric modeling is another strategy to mimic the complex nature of multiple cellular interactions in the human brain. Espuny-Camacho et al. (2017) reported grafting healthy human neural precursor cells derived from iPSCs into the frontal cortices of immunodeficient newborn transgenic APP/PS1-21 mice. When exposed to Aβ, microglia, and astrocytes, the engrafted healthy human neurons developed significant degeneration, as observed by decreased synaptic density and dystrophic neurites, which has not been fully recapitulated in previous animal models. In comparison, the transplanted mouse cells did not develop striking neurodegeneration, indicating that it is crucial to use human neurons rather than murine neurons. Six months after transplantation, there was substantial human cell loss. Furthermore, the transplanted human cells demonstrated necrosis, 3R to 4R tau expression switching, and hyperphosphorylated tau accumulation. Intriguingly, the transplanted neurons died in the absence of NFT, leaving future studies to understand mechanism that generates NFTs and the cause of neuronal death. This novel chimeric model demonstrated promising potential to study human-specific AD pathogenesis.
Mimicking Aging in Reprogramming
Interestingly, aging is a major risk factor for AD (Lindsay et al., 2002). However, the iPSC technique resets age-associated traits (e.g., cellular senescence, telomere shortening, and mitochondrial dysfunction) back to a fetal stage (Mahmoudi and Brunet, 2012), which potentially contribute to AD pathogenesis (Moreira et al., 2010). This might lower the validity of iPSC modeling. Stress exposure (e.g., free radicals, hypoxemia), triggering progeria syndrome pathways has been suggested to accelerate aging in iPSCs (Vera and Studer, 2015; Soria-Valles and López-Otín, 2016).
Apart from resetting somatic cells to a primitive pluripotent state, the reprogramming technique allows iNs to be directly generated from somatic cells, without an embryonic state. Activating the transcription factor combination BAM (Brn2, Asc11, and Myt11) using exogenous ectopic expression of a transgene has been proven to be sufficient to generate functional iNs from mouse fibroblasts (Vierbuchen et al., 2010). With the addition of factor NeuroD1, human fibroblasts can be transdifferentiated into functional mature neurons (Pang et al., 2011). The combination of a micro-RNA (miR-124) and two transcription factors (Myt11 and Brn2) achieved successful conversion of human fibroblasts into functional iNs (Ambasudhan et al., 2011). Non-viral reprogramming methods, which are less invasive to the genome, are being explored. Black et al. (2016) reported that endogenous BAM transcription factors expression could be engineered using the CRISPR/Cas9 system to convert fibroblasts into neuronal cells. Small molecule cocktails have also been reported to directly induce neurons. By adding a cocktail of seven small chemicals that regulate neuronal transcription factor expression, Hu et al. (2015) converted fibroblasts from patients with fAD carrying APP or PS1 mutations into neuronal cells, and observed amyloid and tau phenotypes, showing the promising potential of using iNs to model AD. A large percentage of iNs induced by this cocktail are glutamatergic cells. Although iNs are considered to retain aging-related genetic and epigenetic characteristics that better mimic AD, the fate of iNs is not under precise control. Further work is needed to decipher the molecular mechanisms that govern the differentiation toward a specific neuronal subtype, for example, in AD, cholinergic neurons in the basal forebrain.
The paragraph above summarized several methods to convert fibroblasts into functional iNs in vitro, which might shed light on AD modeling and disease mechanism. Several studies have explored the therapeutic potential of converting glial cells to neural cells in injured brain tissue (Guo et al., 2014; Su et al., 2014). Guo et al. (2014) reported the successful reprogramming of reactive glial cells into functional neurons in an AD model. Overexpression of one transcription factor, NeuroD1, turned astrocytes into glutamatergic neurons, whereas it turned NG2 cells (oligodendrocyte precursor cells) into glutamatergic and GABAergic neurons. This study exploited the pathological gliosis that inhibits neural regeneration and survival in the AD brain as a source of direct reprogramming. Further work is needed to explore whether these neurons induced under pathological conditions are beneficial to improve pathological responses.
Looking Forward
Tremendous variations in the genetic background and life experience (epigenetic change) exist in patient-derived iPSC lines. Current advances in genome editing techniques (e.g., CRISPR/Cas9, zinc finger nuclease, helper-dependent adenovirus) allow us to observe different phenotypes caused by mutations of interest, while keeping the genetic background constant (Liu et al., 2011; Soldner et al., 2011; Kwart et al., 2017). For instance, Fong et al. (2013) exploited genetically engineered iPSCs to study the tauopathy phenotype. Using a zinc finger nuclease, the authors created isogenic iPSC lines carry wild-type TAU, and heterozygous or homozygous TAU-A152T mutation. Mutation dose-dependent neurodegeneration and axonal degeneration were observed.
Efforts have been made to transplant cells derived from iPSCs. Apart from the human-mice chimeric model (Espuny-Camacho et al., 2017), human iPSC-derived dopaminergic neurons were transplanted into a primate Parkinson’s disease model (Kikuchi et al., 2017). In Kikuchi et al.’s study, high grafted cell survival rate and no tumor were observed in 2 years, demonstrating the bright future of transplanting iPSC-based cells in terms of low tumorigenicity and high survival rate. Mandai et al. (2017) reported the first case of transplanting a retina derived from autologous iPSCs into a patient with age-related macular degeneration. Creating iPSCs for each patient is time-consuming, laborious, and expensive; therefore, efforts have been made to establish iPSC banks that are presumed to be sufficient to find at least partial HLA matched donor cells in Japan, the United States, and Europe. The first clinical trial of allogenic iPSC transplantation has been initiated by the same group in Japan to transplant retinas into patients with age-related macular degeneration. AD typically occurs in the elderly, and autologous cells are prone to contain accumulated genetic abnormalities that are potentially harmful; therefore, transplanting cells derived from HLA-matched iPSCs is a strategy worthy of further exploration.
Conclusion
Alzheimer’s disease is a complex neurodegenerative disorder that involves multiple cell types and a large variety of cellular activities. Identifying key molecules involved in the modulation of endogenous neurogenesis and intervening with them might be a preliminary, but promising, strategy to prevent or even reverse AD. Although several pioneering studies have demonstrated elevated neurogenesis in terms of metabolism and plasma exchange in animal models, future studies need to test the efficacy of these manipulations in human patients.
Transplanting stem cells to substitute for lost neurons is another intuitively feasible strategy. However, studies have confirmed that the main benefit of stem cell transplantation is a neurosecretory effect. Various neurotrophic factors involved in modulating multiple cellular functions that promote the amelioration of pathological features and cognition in animal models have been recognized. There has been increasing commercial interest to transform current advances in transplantation into clinical practice on human patients. Various stem cell sources and transplantation routes are being studied to promote the efficacy and safety of transplantation. Regulatory rules from governments should catch up with the growing enthusiasm for stem cell transplantation (see the summaries in Figure 1).
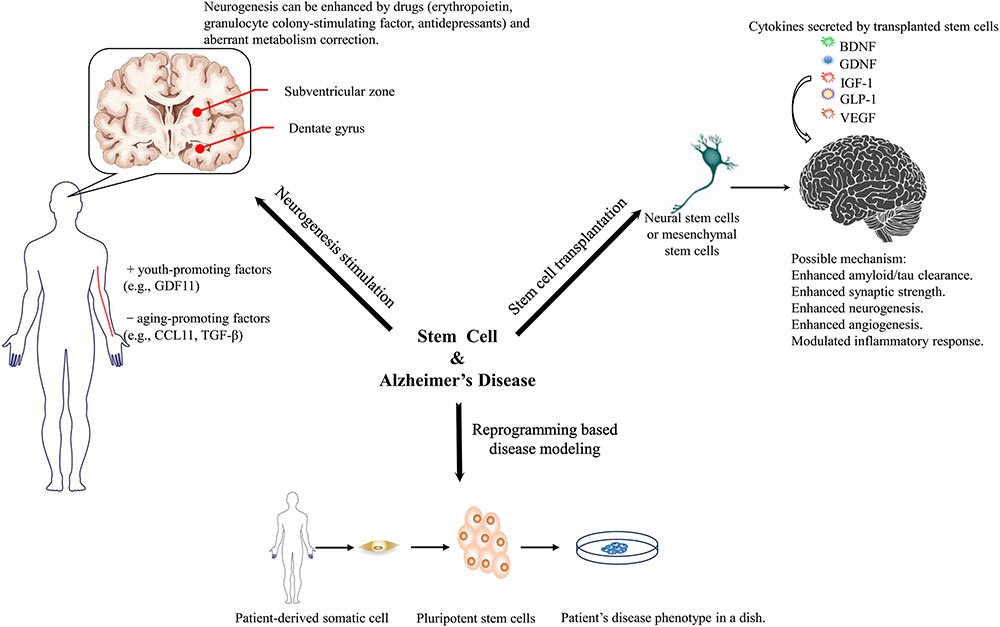
FIGURE 1. Three aspects that stem cell technology might benefit Alzheimer’s disease research and therapeutics.
One of the major hurdles in developing therapeutics for AD and studying its pathogenesis is the lack of animal models that fully recapitulate the pathological features observed in humans. iPSCs have revolutionized AD modeling because they make it possible to generate neuronal cells directly from patients. A substantial amount of research has proven their potency in modeling diseases and screening drugs. Long-term controversies over the disease mechanism (e.g., the challenged amyloid hypothesis) can be tested in iPSC-based models. Three-dimensional modeling and chimeric modeling have been proposed because they aggregate amyloid potency or/and better mimic various cellular interactions that take place in the patient’s brain. Direct reprogramming techniques circumvent the intermediate embryonic state; thus, aging-related features that potentially contribute to AD pathogenesis are maintained. Genome editing techniques allow isogenic comparison of various mutations while keeping the genetic background constant. The phenotypes and drug reactions of different iPSC lines from various patients have been accumulating; therefore, future research might develop detailed patient stratification rules to provide patients with personalized drug regimens. Combined with high-throughput drug screening, future translational studies will be easier.
Author Contributions
YF gathered the initial case information and drafted the initial manuscript. TG participated in the case gathering and drew the figure. BZ reviewed and revised the manuscript. JP reviewed and revised the manuscript and supervised the gathering of case information. All authors approved the manuscript as submitted and agreed to be accountable for all aspects of the work.
Funding
This study was financially supported by the grants from the National Natural Science Foundation of China [81771216 and 81520108010] and the Natural Science Foundation of Zhejiang Province [LY17H090005].
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
Aβ, amyloid beta; AD, Alzheimer’s disease; ApoE, apolipoprotein E; APP, amyloid precursor protein; BDNF, brain-derived neurotrophic factor; CNS, central nervous system; DG, dentate gyrus; fAD, familial Alzheimer’s Disease; iNs, induced neurons; iPSC, induced pluripotent stem cell; MSC, mesenchymal stem cell; NFTs, neurofibrillary tangles; NSC, neural stem cell; PS1, presenilin 1; PS2, presenilin 2; SVZ, subventricular zone.
References
Ager, R. R., Davis, J. L., Agazaryan, A., Benavente, F., Poon, W. W., LaFerla, F. M., et al. (2015). Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 25, 813–826. doi: 10.1002/hipo.22405
Ahmed N.-M., Murakami, M., Hirose, Y., and Nakashima, M. (2016). Therapeutic potential of dental pulp stem cell secretome for Alzheimer’s disease treatment: an in vitro study. Stem Cells Int. 2016:8102478. doi: 10.1155/2016/8102478
Almeida, A. S., and Vieira, H. L. A. (2017). Role of cell metabolism and mitochondrial function during adult neurogenesis. Neurochem. Res. 42, 1787–1794. doi: 10.1007/s11064-016-2150-3
Alnaeeli, M., Wang, L., Piknova, B., Rogers, H., Li, X., and Noguchi, C. T. (2012). Erythropoietin in brain development and beyond. Anat. Res. Int. 2012:953264. doi: 10.1155/2012/953264
Alvarez-Buylla, A., and Garcia-Verdugo, J. M. (2002). Neurogenesis in adult subventricular zone. J. Neurosci. 22, 629–634.
Alvarez-Erviti, L., Seow, Y., Yin, H., Betts, C., Lakhal, S., and Wood, M. J. (2011). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345. doi: 10.1038/nbt.1807
Ambasudhan, R., Talantova, M., Coleman, R., Yuan, X., Zhu, S., Lipton, S. A., et al. (2011). Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 9, 113–118. doi: 10.1016/j.stem.2011.07.002
Anderson, A. J., Piltti, K. M., Hooshmand, M. J., Nishi, R. A., and Cummings, B. J. (2017). Preclinical efficacy failure of human CNS-derived stem cells for use in the pathway study of cervical spinal cord injury. Stem Cell Rep. 8, 249–263. doi: 10.1016/j.stemcr.2016.12.018
Apel, C., Forlenza, O. V., de Paula, V. J. R., Talib, L. L., Denecke, B., Eduardo, C. P., et al. (2009). The neuroprotective effect of dental pulp cells in models of Alzheimer’s and Parkinson’s disease. J. Neural Transm. 116, 71–78. doi: 10.1007/s00702-008-0135-3
Bae, J.-S., Han, H. S., Youn, D.-H., Carter, J. E., Modo, M., Schuchman, E. H., et al. (2007). Bone marrow-derived mesenchymal stem cells promote neuronal networks with functional synaptic transmission after transplantation into mice with neurodegeneration. Stem Cells 25, 1307–1316. doi: 10.1634/stemcells.2006-0561
Beckervordersandforth, R., Ebert, B., Schäffner, I., Moss, J., Fiebig, C., Shin, J., et al. (2017). Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron 93, 560.e6–573.e6. doi: 10.1016/j.neuron.2016.12.017
Bianco, P., Cao, X., Frenette, P. S., Mao, J. J., Robey, P. G., Simmons, P. J., et al. (2013). The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat. Med. 19, 35–42. doi: 10.1038/nm.3028
Black, J. B., Adler, A. F., Wang, H.-G., D’Ippolito, A. M., Hutchinson, H. A., Reddy, T. E., et al. (2016). Targeted epigenetic remodeling of endogenous loci by CRISPR/Cas9-based transcriptional activators directly converts fibroblasts to neuronal cells. Cell Stem Cell 19, 406–414. doi: 10.1016/j.stem.2016.07.001
Blurton-Jones, M., Kitazawa, M., Martinez-Coria, H., Castello, N. A., Müller, F.-J., Loring, J. F., et al. (2009). Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13594–13599. doi: 10.1073/pnas.0901402106
Blurton-Jones, M., Spencer, B., Michael, S., Castello, N. A., Agazaryan, A. A., Davis, J. L., et al. (2014). Neural stem cells genetically-modified to express neprilysin reduce pathology in Alzheimer transgenic models. Stem Cell Res. Ther. 5:46. doi: 10.1186/scrt440
Boada, M., Ortiz, P., Anaya, F., and Hernández, I. (2009). Amyloid-targeted therapeutics in Alzheimer’s disease: use of human albumin in plasma exchange as a novel approach for abeta mobilization. Drug News Perspect. 22, 325–339. doi: 10.1358/dnp.2009.22.6.1395256
Bondolfi, L., Ermini, F., Long, J. M., Ingram, D. K., and Jucker, M. (2004). Impact of age and caloric restriction on neurogenesis in the dentate gyrus of C57BL/6 mice. Neurobiol. Aging 25, 333–340. doi: 10.1016/S0197-4580(03)00083-6
Brier, M. R., Gordon, B., Friedrichsen, K., McCarthy, J., Stern, A., Christensen, J., et al. (2016). Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med. 8:338ra66. doi: 10.1126/scitranslmed.aaf2362
Burgess, A., Ayala-Grosso, C. A., Ganguly, M., Jordão, J. F., Aubert, I., and Hynynen, K. (2011). Targeted delivery of neural stem cells to the brain using MRI-guided focused ultrasound to disrupt the blood-brain barrier. PLoS One 6:e27877. doi: 10.1371/journal.pone.0027877
Castellano, J. M., Mosher, K. I., Abbey, R. J., McBride, A. A., James, M. L., Berdnik, D., et al. (2017). Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 544, 488–492. doi: 10.1038/nature22067
Cevik, B., Solmaz, V., Yigitturk, G., Cavusoðlu, T., Peker, G., and Erbas, O. (2017). Neuroprotective effects of erythropoietin on Alzheimer’s dementia model in rats. Adv. Clin. Exp. Med. 26, 23–29. doi: 10.17219/acem/61044
Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D’Avanzo, C., et al. (2014). A three-dimensional human neural cell culture model of Alzheimer/’s disease. Nature 515, 274–278. doi: 10.1038/nature13800
Cirrito, J. R., Disabato, B. M., Restivo, J. L., Verges, D. K., Goebel, W. D., Sathyan, A., et al. (2011). Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans. Proc. Natl. Acad. Sci. U.S.A. 108, 14968–14973. doi: 10.1073/pnas.1107411108
Conboy, M. J., Conboy, I. M., and Rando, T. A. (2013). Heterochronic parabiosis: historical perspective and methodological considerations for studies of aging and longevity. Aging Cell 12, 525–530. doi: 10.1111/acel.12065
Cummings, J., Lee, G., Mortsdorf, T., Ritter, A., and Zhong, K. (2017). Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement. Transl. Res. Clin. Interv. 3, 367–384. doi: 10.1016/j.trci.2017.05.002
Danielyan, L., Beer-Hammer, S., Stolzing, A., Schäfer, R., Siegel, G., Fabian, C., et al. (2014). Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer’s and Parkinson’s disease. Cell Transplant. 23, 123–139. doi: 10.3727/096368914X684970
D’Avanzo, C., Aronson, J., Kim, Y. H., Choi, S. H., Tanzi, R. E., and Kim, D. Y. (2015). Alzheimer’s in 3D culture: challenges and perspectives: Alzheimer’s in 3D culture. Bioessays 37, 1139–1148. doi: 10.1002/bies.201500063
Di Paolo, G., and Kim, T.-W. (2011). Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat. Rev. Neurosci. 12, 284–296. doi: 10.1038/nrn3012
Digicaylioglu, M., Bichet, S., Marti, H. H., Wenger, R. H., Rivas, L. A., Bauer, C., et al. (1995). Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc. Natl. Acad. Sci. U.S.A. 92, 3717–3720. doi: 10.1073/pnas.92.9.3717
Doody, R. S., Raman, R., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 369, 341–350. doi: 10.1056/NEJMoa1210951
Duan, L., Bhattacharyya, B. J., Belmadani, A., Pan, L., Miller, R. J., and Kessler, J. A. (2014). Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol. Neurodegener. 9:3. doi: 10.1186/1750-1326-9-3
El Andaloussi, S., Mäger, I., Breakefield, X. O., and Wood, M. J. A. (2013). Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 12, 347–357. doi: 10.1038/nrd3978
Eriksdotter-Jönhagen, M., Linderoth, B., Lind, G., Aladellie, L., Almkvist, O., Andreasen, N., et al. (2012). Encapsulated cell biodelivery of nerve growth factor to the basal forebrain in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 33, 18–28. doi: 10.1159/000336051
Eriksson, P. S., Perfilieva, E., Björk-Eriksson, T., Alborn, A.-M., Nordborg, C., Peterson, D. A., et al. (1998). Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317. doi: 10.1038/3305
Espuny-Camacho, I., Arranz, A. M., Fiers, M., Snellinx, A., Ando, K., Munck, S., et al. (2017). Hallmarks of Alzheimer’s disease in stem-cell-derived human neurons transplanted into mouse brain. Neuron 93, 1066.e8–1081.e8. doi: 10.1016/j.neuron.2017.02.001
Fabian, C., Naaldijk, Y., Leovsky, C., Johnson, A. A., Rudolph, L., Jaeger, C., et al. (2017). Distribution pattern following systemic mesenchymal stem cell injection depends on the age of the recipient and neuronal health. Stem Cell Res. Ther. 8:85. doi: 10.1186/s13287-017-0533-2
Fatt, M., Hsu, K., He, L., Wondisford, F., Miller, F. D., Kaplan, D. R., et al. (2015). Metformin acts on two different molecular pathways to enhance adult neural precursor proliferation/self-renewal and differentiation. Stem Cell Rep. 5, 988–995. doi: 10.1016/j.stemcr.2015.10.014
Fong, H., Wang, C., Knoferle, J., Walker, D., Balestra, M. E., Tong, L. M., et al. (2013). Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep. 1, 226–234. doi: 10.1016/j.stemcr.2013.08.001
Gadadhar, A., Marr, R., and Lazarov, O. (2011). Presenilin-1 regulates neural progenitor cell differentiation in the adult brain. J. Neurosci. 31, 2615–2623. doi: 10.1523/JNEUROSCI.4767-10.2011
Garcia, K. O., Ornellas, F. L. M., Martin, P. K. M., Patti, C. L., Mello, L. E., Frussa-Filho, R., et al. (2014). Therapeutic effects of the transplantation of VEGF overexpressing bone marrow mesenchymal stem cells in the hippocampus of murine model of Alzheimer’s disease. Front. Aging Neurosci. 6:30 doi: 10.3389/fnagi.2014.00030
Garcia, P., Youssef, I., Utvik, J. K., Florent-Béchard, S., Barthélémy, V., Malaplate-Armand, C., et al. (2010). Ciliary neurotrophic factor cell-based delivery prevents synaptic impairment and improves memory in mouse models of Alzheimer’s disease. J. Neurosci. 30, 7516–7527. doi: 10.1523/JNEUROSCI.4182-09.2010
Ghosal, K., Stathopoulos, A., and Pimplikar, S. W. (2010). APP intracellular domain impairs adult neurogenesis in transgenic mice by inducing neuroinflammation. PLoS One 5:e11866. doi: 10.1371/journal.pone.0011866
Gómez-Isla, T., Price, J. L., McKeel, D. W., Morris, J. C., Growdon, J. H., and Hyman, B. T. (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 16, 4491–4500.
Gould, E., Beylin, A., Tanapat, P., Reeves, A., and Shors, T. J. (1999). Learning enhances adult neurogenesis in the hippocampal formation. Nat. Neurosci. 2, 260–265. doi: 10.1038/6365
Guo, Z., Zhang, L., Wu, Z., Chen, Y., Wang, F., and Chen, G. (2014). In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell 14, 188–202. doi: 10.1016/j.stem.2013.12.001
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hamilton, L. K., Dufresne, M., Joppé, S. E., Petryszyn, S., Aumont, A., Calon, F., et al. (2015). Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimer’s disease. Cell Stem Cell 17, 397–411. doi: 10.1016/j.stem.2015.08.001
Hanson, N. D., Owens, M. J., and Nemeroff, C. B. (2011). Depression, antidepressants, and neurogenesis: a critical reappraisal. Neuropsychopharmacology 36, 2589–2602. doi: 10.1038/npp.2011.220
Hassouna, I., Ott, C., Wüstefeld, L., Offen, N., Neher, R. A., Mitkovski, M., et al. (2016). Revisiting adult neurogenesis and the role of erythropoietin for neuronal and oligodendroglial differentiation in the hippocampus. Mol. Psychiatry 21, 1752–1767. doi: 10.1038/mp.2015.212
Hatanpää, K., Isaacs, K. R., Shirao, T., Brady, D. R., and Rapoport, S. I. (1999). Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 58, 637–643. doi: 10.1097/00005072-199906000-00008
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Ho, N., Hooker, J., Sahay, A., Holt, D., and Roffman, J. (2013). In vivo imaging of adult human hippocampal neurogenesis: progress, pitfalls and promise. Mol. Psychiatry 18, 404–416. doi: 10.1038/mp.2013.8
Hollands, C., Bartolotti, N., and Lazarov, O. (2016). Alzheimer’s disease and hippocampal adult neurogenesis; exploring shared mechanisms. Front. Neurosci. 10:178. doi: 10.3389/fnins.2016.00178
Horgusluoglu, E., Nudelman, K., Nho, K., and Saykin, A. J. (2017). Adult neurogenesis and neurodegenerative diseases: a systems biology perspective. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 93–112. doi: 10.1002/ajmg.b.32429
Hossini, A. M., Megges, M., Prigione, A., Lichtner, B., Toliat, M. R., Wruck, W., et al. (2015). Induced pluripotent stem cell-derived neuronal cells from a sporadic Alzheimer’s disease donor as a model for investigating AD-associated gene regulatory networks. BMC Genomics 16:84. doi: 10.1186/s12864-015-1262-5
Hu, W., Qiu, B., Guan, W., Wang, Q., Wang, M., Li, W., et al. (2015). Direct conversion of normal and Alzheimer’s disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell 17, 204–212. doi: 10.1016/j.stem.2015.07.006
Imayoshi, I., Sakamoto, M., Ohtsuka, T., Takao, K., Miyakawa, T., Yamaguchi, M., et al. (2008). Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 11, 1153–1161. doi: 10.1038/nn.2185
Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., et al. (2012). Probing sporadic and familial Alzheimer/’s disease using induced pluripotent stem cells. Nature 482, 216–220. doi: 10.1038/nature10821
Jamaiyar, A., Wan, W., Janota, D. M., Enrick, M. K., Chilian, W. M., and Yin, L. (2017). The versatility and paradox of GDF 11. Pharmacol. Ther. 175, 28–34. doi: 10.1016/j.pharmthera.2017.02.032
Jones, V. C., Atkinson-Dell, R., Verkhratsky, A., and Mohamet, L. (2017). Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. 8:e2696. doi: 10.1038/cddis.2017.89
Jung, K.-H., Chu, K., Lee, S.-T., Kim, S.-J., Sinn, D.-I., Kim, S. U., et al. (2006). Granulocyte colony-stimulating factor stimulates neurogenesis via vascular endothelial growth factor with STAT activation. Brain Res. 107, 190–201. doi: 10.1016/j.brainres.2005.12.037
Kanamaru, T., Kamimura, N., Yokota, T., Nishimaki, K., Iuchi, K., Lee, H., et al. (2015). Intravenous transplantation of bone marrow-derived mononuclear cells prevents memory impairment in transgenic mouse models of Alzheimer’s disease. Brain Res. 1605, 49–58. doi: 10.1016/j.brainres.2015.02.011
Karp, J. M., and Leng Teo, G. S. (2009). Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell 4, 206–216. doi: 10.1016/j.stem.2009.02.001
Katsimpardi, L., Litterman, N. K., Schein, P. A., Miller, C. M., Loffredo, F. S., Wojtkiewicz, G. R., et al. (2014). Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 344, 630–634. doi: 10.1126/science.1251141
Katsuda, T., Kosaka, N., Takeshita, F., and Ochiya, T. (2013a). The therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Proteomics 13, 1637–1653. doi: 10.1002/pmic.201200373
Katsuda, T., Tsuchiya, R., Kosaka, N., Yoshioka, Y., Takagaki, K., Oki, K., et al. (2013b). Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 3:1197. doi: 10.1038/srep01197
Khoo, M. L. M., Tao, H., Meedeniya, A. C. B., Mackay-Sim, A., and Ma, D. D. F. (2011). Transplantation of neuronal-primed human bone marrow mesenchymal stem cells in hemiparkinsonian rodents. PLoS One 6:e19025. doi: 10.1371/journal.pone.0019025
Kikuchi, T., Morizane, A., Doi, D., Magotani, H., Onoe, H., Hayashi, T., et al. (2017). Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 548, 592–596. doi: 10.1038/nature23664
Kim, D. H., Lee, D., Chang, E. H., Kim, J. H., Hwang, J. W., Kim, J.-Y., et al. (2015). GDF-15 secreted from human umbilical cord blood mesenchymal stem cells delivered through the cerebrospinal fluid promotes hippocampal neurogenesisnd synaptic activity in an Alzheimer’s disease model. Stem Cells Dev. 24, 2378–2390. doi: 10.1089/scd.2014.0487
Kim, J. A., Ha, S., Shin, K. Y., Kim, S., Lee, K. J., Chong, Y. H., et al. (2015). Neural stem cell transplantation at critical period improves learning and memory through restoring synaptic impairment in Alzheimer’s disease mouse model. Cell Death Dis. 6:e1789. doi: 10.1038/cddis.2015.138
Kim, J.-Y., Kim, D. H., Kim, J. H., Lee, D., Jeon, H. B., Kwon, S.-J., et al. (2012). Soluble intracellular adhesion molecule-1 secreted by human umbilical cord blood-derived mesenchymal stem cell reduces amyloid-β plaques. Cell Death Differ. 19, 680–691. doi: 10.1038/cdd.2011.140
Kim, S., Chang, K.-A., Kim, J. A., Park, H.-G., Ra, J. C., Kim, H.-S., et al. (2012). The preventive and therapeutic effects of intravenous human adipose-derived stem cells in Alzheimer’s disease mice. PLoS One 7:e45757. doi: 10.1371/journal.pone.0045757
Klempin, F., and Kempermann, G. (2007). Adult hippocampal neurogenesis and aging. Eur. Arch. Psychiatry Clin. Neurosci. 257, 271–280. doi: 10.1007/s00406-007-0731-5
Klinge, P. M., Harmening, K., Miller, M. C., Heile, A., Wallrapp, C., Geigle, P., et al. (2011). Encapsulated native and glucagon-like peptide-1 transfected human mesenchymal stem cells in a transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 497, 6–10. doi: 10.1016/j.neulet.2011.03.092
Koch, P., Tamboli, I. Y., Mertens, J., Wunderlich, P., Ladewig, J., Stüber, K., et al. (2012). Presenilin-1 L166P mutant human pluripotent stem cell–derived neurons exhibit partial loss of γ-secretase activity in endogenous amyloid-β generation. Am. J. Pathol. 180, 2404–2416. doi: 10.1016/j.ajpath.2012.02.012
Kondo, T., Asai, M., Tsukita, K., Kutoku, Y., Ohsawa, Y., Sunada, Y., et al. (2013). Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell 12, 487–496. doi: 10.1016/j.stem.2013.01.009
Kuhn, H. G., Dickinson-Anson, H., and Gage, F. H. (1996). Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 16, 2027–2033.
Kwart, D., Paquet, D., Teo, S., and Tessier-Lavigne, M. (2017). Precise and efficient scarless genome editing in stem cells using CORRECT. Nat. Protoc. 12, 329–354. doi: 10.1038/nprot.2016.171
Lai, R. C., Arslan, F., Lee, M. M., Sze, N. S. K., Choo, A., Chen, T. S., et al. (2010). Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 4, 214–222. doi: 10.1016/j.scr.2009.12.003
Lambert, J.-C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lancaster, M. A., Renner, M., Martin, C.-A., Wenzel, D., Bicknell, L. S., Hurles, M. E., et al. (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. doi: 10.1038/nature12517
Lange, C., Garcia, M. T., Decimo, I., Bifari, F., Eelen, G., Quaegebeur, A., et al. (2016). Relief of hypoxia by angiogenesis promotes neural stem cell differentiation by targeting glycolysis. EMBO J. 35, 924–941. doi: 10.15252/embj.201592372
Lee, H. J., Lee, J. K., Lee, H., Carter, J. E., Chang, J. W., Oh, W., et al. (2012). Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 33, 588–602. doi: 10.1016/j.neurobiolaging.2010.03.024
Lee, H. J., Lee, J. K., Lee, H., Shin, J., Carter, J. E., Sakamoto, T., et al. (2010). The therapeutic potential of human umbilical cord blood-derived mesenchymal stem cells in Alzheimer’s disease. Neurosci. Lett. 481, 30–35. doi: 10.1016/j.neulet.2010.06.045
Lee, H.-K., Sanchez, C. V., Chen, M., Morin, P. J., Wells, J. M., Hanlon, E. B., et al. (2016). Three dimensional human neuro-spheroid model of Alzheimer’s disease based on differentiated induced pluripotent stem cells. PLoS One 11:e0163072. doi: 10.1371/journal.pone.0163072
Lee, J. K., Jin, H. K., and Bae, J. (2009a). Bone marrow-derived mesenchymal stem cells reduce brain amyloid-β deposition and accelerate the activation of microglia in an acutely induced Alzheimer’s disease mouse model. Neurosci. Lett. 450, 136–141. doi: 10.1016/j.neulet.2008.11.059
Lee, J. K., Jin, H. K., Endo, S., Schuchman, E. H., Carter, J. E., and Bae, J. (2009b). Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-beta deposition and rescues memory deficits in Alzheimer’s disease mice by modulation of immune responses. Stem Cells 28, 329–343. doi: 10.1002/stem.277
Lee, J. K., Schuchman, E. H., Jin, H. K., and Bae, J.-S. (2012). Soluble CCL5 derived from bone marrow-derived mesenchymal stem cells and activated by amyloid β ameliorates alzheimer’s disease in mice by recruiting bone marrow-induced microglia immune responses. Stem Cells 30, 1544–1555. doi: 10.1002/stem.1125
Leutgeb, J. K., Leutgeb, S., Moser, M.-B., and Moser, E. I. (2007). Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science 315, 961–966. doi: 10.1126/science.1135801
Li, T., Yan, Y., Wang, B., Qian, H., Zhang, X., Shen, L., et al. (2012). Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 22, 845–854. doi: 10.1089/scd.2012.0395
Lindsay, J., Laurin, D., Verreault, R., Hébert, R., Helliwell, B., Hill, G. B., et al. (2002). Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am. J. Epidemiol. 156, 445–453. doi: 10.1093/aje/kwf074
Liu, G.-H., Suzuki, K., Qu, J., Sancho-Martinez, I., Yi, F., Li, M., et al. (2011). Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 8, 688–694. doi: 10.1016/j.stem.2011.04.019
Liu, Y.-H., Wang, Y.-R., Xiang, Y., Zhou, H.-D., Giunta, B., Mañucat-Tan, N. B., et al. (2015). Clearance of amyloid-beta in Alzheimer’s disease: shifting the action site from center to periphery. Mol. Neurobiol. 51, 1–7. doi: 10.1007/s12035-014-8694-9
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Lu, P., Jones, L., Snyder, E., and Tuszynski, M. (2003). Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp. Neurol. 181, 115–129. doi: 10.1016/S0014-4886(03)00037-2
Ma, T., Gong, K., Ao, Q., Yan, Y., Song, B., Huang, H., et al. (2013). Intracerebral transplantation of adipose-derived mesenchymal stem cells alternatively activates microglia and ameliorates neuropathological deficits in Alzheimer’s disease mice. Cell Transplant. 22(Suppl. 1), S113–S126. doi: 10.3727/096368913X672181
Mahmoudi, S., and Brunet, A. (2012). Aging and reprogramming: a two-way street. Curr. Opin. Cell Biol. 24, 744–756. doi: 10.1016/j.ceb.2012.10.004
Mandai, M., Watanabe, A., Kurimoto, Y., Hirami, Y., Morinaga, C., Daimon, T., et al. (2017). Autologous induced stem-cell–derived retinal cells for macular degeneration. N. Engl. J. Med. 376, 1038–1046. doi: 10.1056/NEJMoa1608368
Manganas, L. N., Zhang, X., Li, Y., Hazel, R. D., Smith, S. D., Wagshul, M. E., et al. (2007). Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain. Science 318, 980–985. doi: 10.1126/science.1147851
Marsh, S. E., Yeung, S. T., Torres, M., Lau, L., Davis, J. L., Monuki, E. S., et al. (2017). HuCNS-SC human NSCs fail to differentiate, form ectopic clusters, and provide no cognitive benefits in a transgenic model of Alzheimer’s disease. Stem Cell Rep. 8, 235–248. doi: 10.1016/j.stemcr.2016.12.019
Martino, G., and Pluchino, S. (2006). The therapeutic potential of neural stem cells. Nat. Rev. Neurosci. 7, 395–406. doi: 10.1038/nrn1908
McGinley, L. M., Sims, E., Lunn, J. S., Kashlan, O. N., Chen, K. S., Bruno, E. S., et al. (2016). Human cortical neural stem cells expressing insulin-like growth factor-I: a novel cellular therapy for Alzheimer’s disease: neural stem cells and induced IGF-I expression. Stem Cells Transl. Med. 5, 379–391. doi: 10.5966/sctm.2015-0103
Mead, B., Logan, A., Berry, M., Leadbeater, W., and Scheven, B. A. (2017). Concise review: dental pulp stem cells: a novel cell therapy for retinal and central nervous system repair. Stem Cells 35, 61–67. doi: 10.1002/stem.2398
Ming, G., and Song, H. (2005). Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci. 28, 223–250. doi: 10.1146/annurev.neuro.28.051804.101459
Misra, S., Chopra, K., Saikia, U. N., Sinha, V. R., Sehgal, R., Modi, M., et al. (2016). Effect of mesenchymal stem cells and galantamine nanoparticles in rat model of Alzheimer’s disease. Regen. Med. 11, 629–646. doi: 10.2217/rme-2016-0032
Moore, S., Evans, L. D. B., Andersson, T., Portelius, E., Smith, J., Dias, T. B., et al. (2015). APP metabolism regulates Tau proteostasis in human cerebral cortex neurons. Cell Rep. 11, 689–696. doi: 10.1016/j.celrep.2015.03.068
Moreira, P. I., Carvalho, C., Zhu, X., Smith, M. A., and Perry, G. (2010). Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 1802, 2–10. doi: 10.1016/j.bbadis.2009.10.006
Mu, Y., and Gage, F. H. (2011). Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 6:85. doi: 10.1186/1750-1326-6-85
Muratore, C. R., Rice, H. C., Srikanth, P., Callahan, D. G., Shin, T., Benjamin, L. N. P., et al. (2014). The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 23, 3523–3536. doi: 10.1093/hmg/ddu064
Ogbonnaya, E. S., Clarke, G., Shanahan, F., Dinan, T. G., Cryan, J. F., and O’Leary, O. F. (2015). Adult hippocampal neurogenesis is regulated by the microbiome. Biol. Psychiatry 78, e7–e9. doi: 10.1016/j.biopsych.2014.12.023
Ohm, T. G. (2007). “The dentate gyrus in Alzheimer’s disease,” in Progress in Brain Research The Dentate Gyrus: A Comprehensive Guide to Structure, Function, and Clinical Implications, ed. H. E. Scharfman (Amsterdam: Elsevier), 723–740. doi: 10.1016/S0079-6123(07)63039-8
Ossenkoppele, R., Jansen, W. J., Rabinovici, G. D., Knol, D. L., van der Flier, W. M., van Berckel, B. N. M., et al. (2015). Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 313:1939. doi: 10.1001/jama.2015.4669
Pang, Z. P., Yang, N., Vierbuchen, T., Ostermeier, A., Fuentes, D. R., Yang, T. Q., et al. (2011). Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223. doi: 10.1038/nature10202
Phinney, D. G., and Prockop, D. J. (2007). Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair-current views. Stem Cells 25, 2896–2902. doi: 10.1634/stemcells.2007-0637
Prince, M., Bryce, R., Albanese, E., Wimo, A., Ribeiro, W., and Ferri, C. P. (2013). The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 9, 63.e2–75.e2. doi: 10.1016/j.jalz.2012.11.007
Raja, W. K., Mungenast, A. E., Lin, Y.-T., Ko, T., Abdurrob, F., Seo, J., et al. (2016). Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS One 11:e0161969. doi: 10.1371/journal.pone.0161969
Record, M., Subra, C., Silvente-Poirot, S., and Poirot, M. (2011). Exosomes as intercellular signalosomes and pharmacological effectors. Biochem. Pharmacol. 81, 1171–1182. doi: 10.1016/j.bcp.2011.02.011
Rogers, S. L., and Friedhoff, L. T. (1996). The efficacy and safety of donepezil in patients with Alzheimer’s disease: results of a US multicentre, randomized, double-blind, placebo-controlled trial. Dement. Geriatr. Cogn. Disord. 7, 293–303. doi: 10.1159/000106895
Russo, V. C., Gluckman, P. D., Feldman, E. L., and Werther, G. A. (2005). The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev. 26, 916–943. doi: 10.1210/er.2004-0024
Ryu, J. K., Cho, T., Wang, Y. T., and McLarnon, J. G. (2009). Neural progenitor cells attenuate inflammatory reactivity and neuronal loss in an animal model of inflamed AD brain. J. Neuroinflammation 6:39. doi: 10.1186/1742-2094-6-39
Salloway, S., Sperling, R., Fox, N. C., Blennow, K., Klunk, W., Raskind, M., et al. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333. doi: 10.1056/NEJMoa1304839
Schneider, A., Krüger, C., Steigleder, T., Weber, D., Pitzer, C., Laage, R., et al. (2005). The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J. Clin. Invest. 115, 2083–2098. doi: 10.1172/JCI23559
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Sheline, Y. I., West, T., Yarasheski, K., Swarm, R., Jasielec, M. S., Fisher, J. R., et al. (2014). An antidepressant decreases CSF Aβ production in healthy individuals and in transgenic AD mice. Sci. Transl. Med. 6:236re4. doi: 10.1126/scitranslmed.3008169
Shin, J. Y., Park, H. J., Kim, H. N., Oh, S. H., Bae, J.-S., Ha, H.-J., et al. (2014). Mesenchymal stem cells enhance autophagy and increase β-amyloid clearance in Alzheimer disease models. Autophagy 10, 32–44. doi: 10.4161/auto.26508
Smith, L. K., He, Y., Park, J.-S., Bieri, G., Snethlage, C. E., Lin, K., et al. (2015). β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 21, 932–937. doi: 10.1038/nm.3898
Soldner, F., Laganière, J., Cheng, A. W., Hockemeyer, D., Gao, Q., Alagappan, R., et al. (2011). Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 146, 318–331. doi: 10.1016/j.cell.2011.06.019
Soria-Valles, C., and López-Otín, C. (2016). iPSCs: on the road to reprogramming aging. Trends Mol. Med. 22, 713–724. doi: 10.1016/j.molmed.2016.05.010
Spalding, K. L., Bergmann, O., Alkass, K., Bernard, S., Salehpour, M., Huttner, H. B., et al. (2013). Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227. doi: 10.1016/j.cell.2013.05.002
Sproul, A. A., Jacob, S., Pre, D., Kim, S. H., Nestor, M. W., Navarro-Sobrino, M., et al. (2014). Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS One 9:e84547. doi: 10.1371/journal.pone.0084547
Spuch, C., Antequera, D., Portero, A., Orive, G., Hernández, R. M., Molina, J. A., et al. (2010). The effect of encapsulated VEGF-secreting cells on brain amyloid load and behavioral impairment in a mouse model of Alzheimer’s disease. Biomaterials 31, 5608–5618. doi: 10.1016/j.biomaterials.2010.03.042
Stranahan, A. M., Khalil, D., and Gould, E. (2006). Social isolation delays the positive effects of running on adult neurogenesis. Nat. Neurosci. 9, 526–533. doi: 10.1038/nn1668
Su, Z., Niu, W., Liu, M.-L., Zou, Y., and Zhang, C.-L. (2014). In vivo conversion of astrocytes to neurons in the injured adult spinal cord. Nat. Commun. 5:3338. doi: 10.1038/ncomms4338
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2014). The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231. doi: 10.1016/j.bbadis.2013.09.010
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Tang, Y., and Le, W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Tariot, P. N., Farlow, M. R., Grossberg, G. T., Graham, S. M., McDonald, S., Gergel, I., et al. (2004). Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 291, 317–324. doi: 10.1001/jama.291.3.317
Taupin, P. (2006). Neurogenesis and the effect of antidepressants. Drug Target Insights 1, 13–17. doi: 10.1177/117739280600100005
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Toledo, J. B., Shaw, L. M., and Trojanowski, J. Q. (2013). Plasma amyloid beta measurements - a desired but elusive Alzheimer’s disease biomarker. Alzheimers Res. Ther. 5:8. doi: 10.1186/alzrt162
Tsai, K.-J., Tsai, Y.-C., and Shen, C.-K. J. (2007). G-CSF rescues the memory impairment of animal models of Alzheimer’s disease. J. Exp. Med. 204, 1273–1280. doi: 10.1084/jem.20062481
Unger, M. S., Marschallinger, J., Kaindl, J., Höfling, C., Rossner, S., Heneka, M. T., et al. (2016). Early changes in hippocampal neurogenesis in transgenic mouse models for Alzheimer’s disease. Mol. Neurobiol. 53, 5796–5806. doi: 10.1007/s12035-016-0018-9
Valadi, H., Ekström, K., Bossios, A., Sjöstrand, M., Lee, J. J., and Lötvall, J. O. (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659. doi: 10.1038/ncb1596
van Praag, H., Christie, B. R., Sejnowski, T. J., and Gage, F. H. (1999). Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci. U.S.A. 96, 13427–13431. doi: 10.1073/pnas.96.23.13427
Vera, E., and Studer, L. (2015). When rejuvenation is a problem: challenges of modeling late-onset neurodegenerative disease. Development 142, 3085–3089. doi: 10.1242/dev.120667
Vierbuchen, T., Ostermeier, A., Pang, Z. P., Kokubu, Y., Südhof, T. C., and Wernig, M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. doi: 10.1038/nature08797
Villeda, S. A., Luo, J., Mosher, K. I., Zou, B., Britschgi, M., Bieri, G., et al. (2011). The aging systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94. doi: 10.1038/nature10357
Villeda, S. A., Plambeck, K. E., Middeldorp, J., Castellano, J. M., Mosher, K. I., Luo, J., et al. (2014). Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 20, 659–663. doi: 10.1038/nm.3569
Wahlberg, L. U., Lind, G., Almqvist, P. M., Kusk, P., Tornøe, J., Juliusson, B., et al. (2012). Targeted delivery of nerve growth factor via encapsulated cell biodelivery in Alzheimer disease: a technology platform for restorative neurosurgery. J. Neurosurg. 117, 340–347. doi: 10.3171/2012.2.JNS11714
Wang, J., Gallagher, D., DeVito, L. M., Cancino, G. I., Tsui, D., He, L., et al. (2012). Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell 11, 23–35. doi: 10.1016/j.stem.2012.03.016
Weiss, M. L., Anderson, C., Medicetty, S., Seshareddy, K. B., Weiss, R. J., VanderWerff, I., et al. (2008). Immune properties of human umbilical cord Wharton’s Jelly-derived cells. Stem Cells 26, 2865–2874. doi: 10.1634/stemcells.2007-1028
Woodruff, G., Reyna, S. M., Dunlap, M., Van Der Kant, R., Callender, J. A., Young, J. E., et al. (2016). Defective transcytosis of APP and lipoproteins in human iPSC-derived neurons with familial Alzheimer’s disease mutations. Cell Rep. 17, 759–773. doi: 10.1016/j.celrep.2016.09.034
Wu, C.-C., Lien, C.-C., Hou, W.-H., Chiang, P.-M., and Tsai, K.-J. (2016). Gain of BDNF function in engrafted neural stem cells promotes the therapeutic potential for Alzheimer’s disease. Sci. Rep. 6:27358. doi: 10.1038/srep27358
Xin, H., Li, Y., Liu, Z., Wang, X., Shang, X., Cui, Y., et al. (2013). MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells 31, 2737–2746. doi: 10.1002/stem.1409
Yagi, T., Ito, D., Okada, Y., Akamatsu, W., Nihei, Y., Yoshizaki, T., et al. (2011). Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 20, 4530–4539. doi: 10.1093/hmg/ddr394
Yang, C.-P., Gilley, J. A., Zhang, G., and Kernie, S. G. (2011). ApoE is required for maintenance of the dentate gyrus neural progenitor pool. Development 138, 4351–4362. doi: 10.1242/dev.065540
Yang, H., Xie, Z. H., Wei, L. F., Yang, H. N., Yang, S. N., Zhu, Z. Y., et al. (2013). Human umbilical cord mesenchymal stem cell-derived neuron-like cells rescue memory deficits and reduce amyloid-beta deposition in an AβPP/PS1 transgenic mouse model. Stem Cell Res. Ther. 4:76. doi: 10.1186/scrt227
Young, J. E., Boulanger-Weill, J., Williams, D. A., Woodruff, G., Buen, F., Revilla, A. C., et al. (2015). Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 16, 373–385. doi: 10.1016/j.stem.2015.02.004
Yousef, H., Conboy, M. J., Morgenthaler, A., Schlesinger, C., Bugaj, L., Paliwal, P., et al. (2015). Systemic attenuation of the TGF-β pathway by a single drug simultaneously rejuvenates hippocampal neurogenesis and myogenesis in the same old mammal. Oncotarget 6, 11959–11978. doi: 10.18632/oncotarget.3851
Zhang, W., Gu, G.-J., Shen, X., Zhang, Q., Wang, G.-M., and Wang, P.-J. (2015). Neural stem cell transplantation enhances mitochondrial biogenesis in a transgenic mouse model of Alzheimer’s disease–like pathology. Neurobiol. Aging 36, 1282–1292. doi: 10.1016/j.neurobiolaging.2014.10.040
Keywords: Alzheimer’s disease, neurogenesis, stem cell transplantation, induced pluripotent stem cell, disease modeling
Citation: Fang Y, Gao T, Zhang B and Pu J (2018) Recent Advances: Decoding Alzheimer’s Disease With Stem Cells. Front. Aging Neurosci. 10:77. doi: 10.3389/fnagi.2018.00077
Received: 14 October 2017; Accepted: 07 March 2018;
Published: 22 March 2018.
Edited by:
Catarina Oliveira, University of Coimbra, PortugalReviewed by:
Charles Harrington, University of Aberdeen, United KingdomMariagrazia Grilli, Università degli Studi del Piemonte Orientale, Italy
Copyright © 2018 Fang, Gao, Zhang and Pu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiali Pu, amlhbGlwdUB6anUuZWR1LmNu; Y2FycmllXzExMDVAMTYzLmNvbQ==