 Keith Wetherby1
Keith Wetherby1 Joseph Chiao1
Joseph Chiao1 Emily Faulkner1
Emily Faulkner1 Yongjian Guo2
Yongjian Guo2 Shaobin Hou1J. Joanna Yu1Jinguo Chen1Lili Wan1,3H. Henry Li1,3*
Shaobin Hou1J. Joanna Yu1Jinguo Chen1Lili Wan1,3H. Henry Li1,3*
- 1Virant Diagnostics, Inc., Wheaton, MD, United States
- 2ScitechLink, LLC, Rockville, MD, United States
- 3Institute for Asthma and Allergy, Chevy Chase, MD, United States
Background: Hereditary angioedema (HAE) is an autosomal dominant genetic disorder caused by mutations in the C1 esterase inhibitor gene, SERPING1, leading to overproduction of bradykinin and debilitating swelling attacks. Variants in the SERPING1 gene are typically detected in a clinical setting by DNA sequencing or multiplex ligation-dependent probe amplification (MLPA), with over 893 total variants identified. Approximately 5% of patients with C1-esterase inhibitor deficiencies do not have detectable SERPING1 pathogenic variants. We further investigated a family with laboratory-confirmed HAE type I despite previous negative genetic test results for SERPING1 mutations.
Methods: We consented and collected whole blood samples from three family members with clinical diagnoses of HAE. The samples underwent genomic DNA extraction and evaluation for purity prior to sequencing. The DNA samples were processed through a semi-automated whole exome library prep pipeline and sequenced. SERPING1 MLPA was performed to assess exon-level copy number variation (CNV) for exons 1 through 8. Additionally, we incorporated a well-established bioinformatics technique called soft clipping into our variant analysis pipeline to detect structural variants.
Results: Clinical variant analysis revealed two common benign variants of SERPING1 in the proband. NGS and MLPA did not detect any SERPING1 pathogenic variants or genomic rearrangements, but additional structural variant analysis identified a high rate of soft clipping in exon 6 of the SERPING1 gene. Sanger sequencing of exon 6 revealed a heterozygous 56-base-pair deletion [NC_000011.10: g.57606508-57606563del, NM_000062(SERPING1): c.990_1029 + 16del] spanning the 3’ exon-intron boundary in all three subjects.
Summary: Without additional techniques following NGS and MLPA, such as a soft clipping analysis method, many difficult-to-detect large insertions and deletions may go undetected. We propose that a systematic approach to undetected HAE-causing mutation analysis, incorporating soft clipping as part of an overall strategy, would be more effective in identifying a small percentage of causal variants in approximately 5% of C1-esterase inhibitor HAE cases where no mutation is found by standard laboratory procedures, especially when there are high clinical suspicions of a familiar disorder.
Introduction
Hereditary angioedema (HAE) is a rare, life-threatening autosomal dominant genetic disease, most commonly caused by mutations in the SERPING1 gene on chromosome 11q. These mutations disrupt the regulation of the kallikrein-kinin system (KKS), resulting in the overproduction of bradykinin. Without treatment, angioedema episodes can be severely debilitating and may result in fatal laryngeal asphyxiation. To date, more than 893 SERPING1 variants have been identified, primarily through genetic techniques such as next-generation sequencing (NGS) and multiplex ligation-dependent probe amplification (MLPA) (1). However, clinicians must be aware of the limitations of these methods, especially when negative genetic test results conflict with clear clinical presentations of HAE. This highlights the importance of thorough clinical evaluation and a general understanding of genetic testing methods to ensure accurate diagnosis and effective patient management.
The SERPING1 gene contains 17 Alu elements, making it susceptible to non-homologous recombination events and prone to large deletions, insertions, and duplications (2, 3). Alu-repeat-mediated variations account for approximately 10% of all HAE cases (4). Due to technical limitations, conventional NGS short-read sequencing may miss small and large heterozygous rearrangements (5). Therefore, other molecular techniques, such as MLPA, are needed to investigate these events.
Herein, we describe three family members with HAE type I who test negative for pathogenic variants and large genomic rearrangements in SERPING1, as determined by NGS and MLPA, respectively. Using a systematic approach to HAE-causing mutation analysis that incorporates soft clipping, we identified a novel heterozygous disease-causing deletion in SERPING1, responsible for the C1 inhibitor deficiency in affected family members.
Materials and methods
Ethics declarations
All subjects analyzed in this study gave written informed consent before participation. The study protocol was approved by the Salus IRB (IRB protocol number: Virant-A0001).
HAE subtypes
Mutations in the SERPING1 gene cause two types of HAE. HAE-C1INH type I is caused by a quantitative C1 esterase inhibitor protein deficiency mainly due to truncating mutations. HAE-C1INH type II is caused by the reduced function of the C1 esterase inhibitor protein mainly resulting from missense or in-frame variants at or near the reactive site P1-P1’ (Arg466-Tyr467) on the exposed reactive mobile loop in exon 8 (6). HAE type I and type II affect about 1 in 50,000 people, with 85% of HAE patients representing type I (7, 8). NGS is typically used for molecular confirmation of HAE type I and type II in medical genomics studies.
Patient demographics
The clinical history of recurrent chronic angioedema and diagnostic test results for three family members (35-year-old female proband, her 8-year-old son, and her 62-year-old mother) were suggestive of HAE type I, characterized by low levels of complement C4, C1 esterase inhibitor antigen, and C1 esterase inhibitor function. Previous genetic testing of SERPING1 for each family member, conducted at another genetics laboratory, did not identify any mutations.
Blood collection + genomic DNA extraction
Whole blood was collected from the patients in BD vacutainer K2 EDTA tubes (Beckton Dickenson) and then stored at 4°C until DNA extraction. Genomic DNA was extracted from the patient's blood on the Qiagen EZ2 Connect MDx using the EZ1&2 DNA Blood 350ul kit (Qiagen) following the manufacturer's instructions. The DNA concentration and purity were evaluated using the Qiaxpert system (Qiagen).
Next generation sequencing (NGS)
Genomic DNA samples were processed through a semi-automated library prep pipeline (Agilent SureSelect Human All Exon V8, Agilent Magnis) and sequenced using Illumina technology. Reads were aligned and mapped to GRCh38 using Agilent Reporter v1.3.1 software. Variant filtering was performed using cloud-based commercial software (Agilent Interpret v5.4.2). The impact of variants was evaluated using commercial (Alamut Visual Plus, Sophia Genetics) and publicly available computational tools. Read alignment was visualized using Integrative Genomics Viewer (Broad Institute). Variants were classified according to ACMG guidelines (9).
MLPA
Exon level copy number variation (CNV) for exons 1 through 8 in SERPING1 was performed using Salsa MLPA Probemix P243 (MRC Holland) following the manufacturer's instructions. The probe mix kit consists of 11 control probes, 8 SERPING1 probes, 13 FXII probes and 1 APLNR probe, a gene upstream of SERPING1. Four DNA controls, including 3 healthy reference samples and one positive sample for a SERPING1 exon 4 deletion, were run with patient samples on an ABI 3500xl Genetic Analyzer (Applied Biosystems). Data analysis was performed using Coffalyser software v.240129.1959. The location of MLPA probes in relation to each SERPING1 exon is shown in Figure 1.
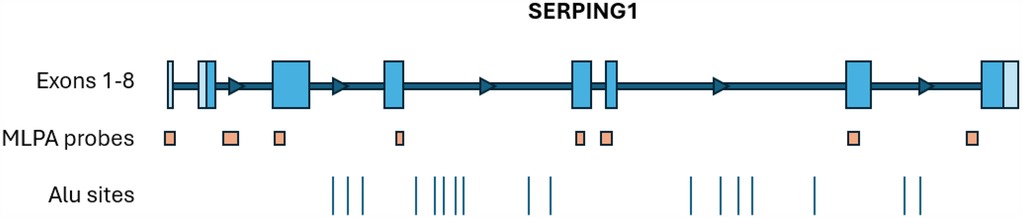
Figure 1. The approximate locations of SERPING1 exons, MLPA probes (MRC holland, probemix P243 SERPING1-F12), and Alu sites (2). The MLPA probe locations are as follows: ex1 probe covers the entire exon, ex2 probe is downstream of exon 2, ex3 probe is at the 5’ end of exon 3, ex4 probe is at the 3’ end of exon 4, ex5 probe is within exon 5, ex6 probe is on the 5’ intron-exon boundary, ex7 probe is at the 5’ end of the exon, and the ex8 probe is upstream of exon 8.
Sanger sequencing
Custom primers (forward, 5’-TCGGATCTCAATGTCCCTGC-3’ and reverse, 5’-TTGAGAATCCTGTTTCCAGCCT-3’) (Integrated DNA Technologies) were used to amplify a 459-base-pair product for SERPING1 exon 6 using the following PCR conditions: initial denaturation 98°C for 30 s followed by 35 cycles of denaturation at 98°C for 10 s, annealing at 60°C for 10 s and extension at 72°C for 90 s. The PCR product was cleaned up using Exo-SAP (Applied Biosystems) following the manufacturer's instructions. Bidirectional Sanger sequencing using Big Dye Terminator v3.1 (Applied Biosystems) and subsequent clean-up using Xterminator beads (Applied Biosystems) were performed following the manufacturer's instructions. Capillary electrophoresis was performed on a 3500xl Genetic Analyzer (Applied Biosystems). Analysis of the sequencing data was performed using Genecodes Sequencher (RRID:SCR_001528) version 5.4.6.
Bioinformatics tools
We implemented a well-established bioinformatics technique called soft clipping into our analysis pipeline, using BWA, BEDTOOLS, and BEDMAP, to detect potential genomic rearrangements in the SERPING1 gene (10–12). Soft clipping regions in NGS sequence alignment represent bases that do not align with the reference sequence without removing them from the read data. The clipping percentage is calculated by dividing the number of reads with soft clipping bases by the total number of reads. The clipping percentage for a given exon is then compared to other samples. An increase in percentage compared to the healthy samples suggests further investigation is needed. Low-quality MAPQ scores were filtered out of the analysis.
Results
NGS
Variant analysis for SERPING1 revealed two common benign variants (Table 1) in the proband but not in the mother or the son. Due to their high frequency in population databases (gnomAD, Broad Institute) and our lab, these variants were not given further consideration.
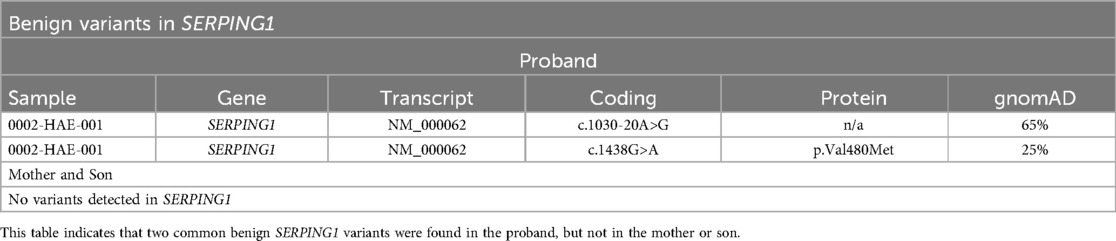
Table 1. Benign variants in SERPING1.
MLPA
Due to the presence of 17 Alu elements, MLPA is used as part of the systematic analysis of SERPING1 when no pathogenic variant is found during DNA sequencing. One positive control sample with a heterozygous exon 4 deletion in SERPING1 and three healthy reference samples were tested along with the proband and mother. The proband and mother were negative for copy number variations in exons 1–8. Therefore, we did not test the son. Negative MLPA results do not completely rule out structural variants >50 bp (13). Deletions or insertions occurring outside the MLPA probe locations will go undetected.
Soft clipping analysis
All three family members have low levels of C1-INH indicating a SERPING1 mutation, but no pathogenic mutations were found through NGS or MLPA. Soft clipping is a bioinformatics technique used in sequence alignment to mark bases that do not align with the reference sequence without removing them from the read data. We processed the NGS bam files for the proband, mother (son not tested) and two additional controls through our soft clipping bioinformatics workflow to investigate the SERPING1 gene for difficult-to-detect insertions and deletions (14). Of the two control samples, 0046 had a known heterozygous SERPING1 exon 4 deletion, and 0045 had a missense SERPING1 mutation in exon 3. A high rate of soft clipping was detected in SERPING1 exon 6 in this HAE family as shown in Table 2; Figure 2.
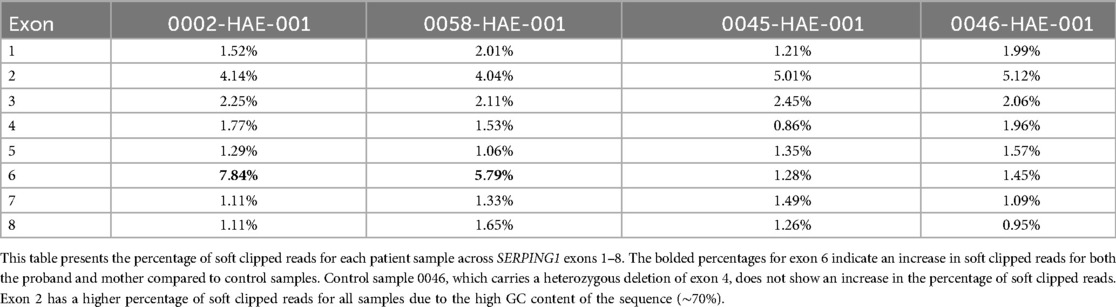
Table 2. A high rate of soft clipping in SERPING1 exon 6.
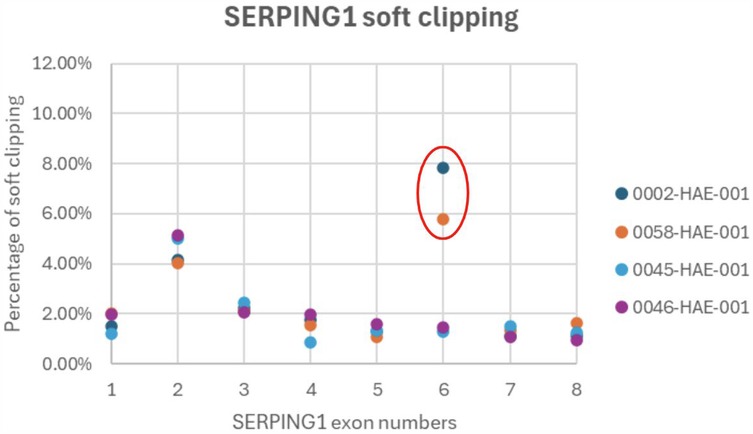
Figure 2. A high rate of soft clipping in SERPING1 exon 6. Soft clipping percentages graphed per patient for SERPING1 exons 1-8. Outliers are circled red and suggest further investigation.
Sanger sequencing
We performed Sanger sequencing for SERPING1 exon 6 for all three family members based on an increase of soft clipped reads compared to controls. A novel disease-causing heterozygous 56-base-pair deletion, [NM_000062 (SERPING1): c.990_1029+16del], spanning the 3’ exon-intron boundary was subsequently identified for all three family members. The deletion is predicted to cause a frameshift at coding position 332 resulting in the addition of 3 spurious amino acids before a premature translational stop codon (p.Val332Serfs*3) as shown in Table 3; Figures 3A–C.

Table 3. Pathogenic SERPING1 mutations.
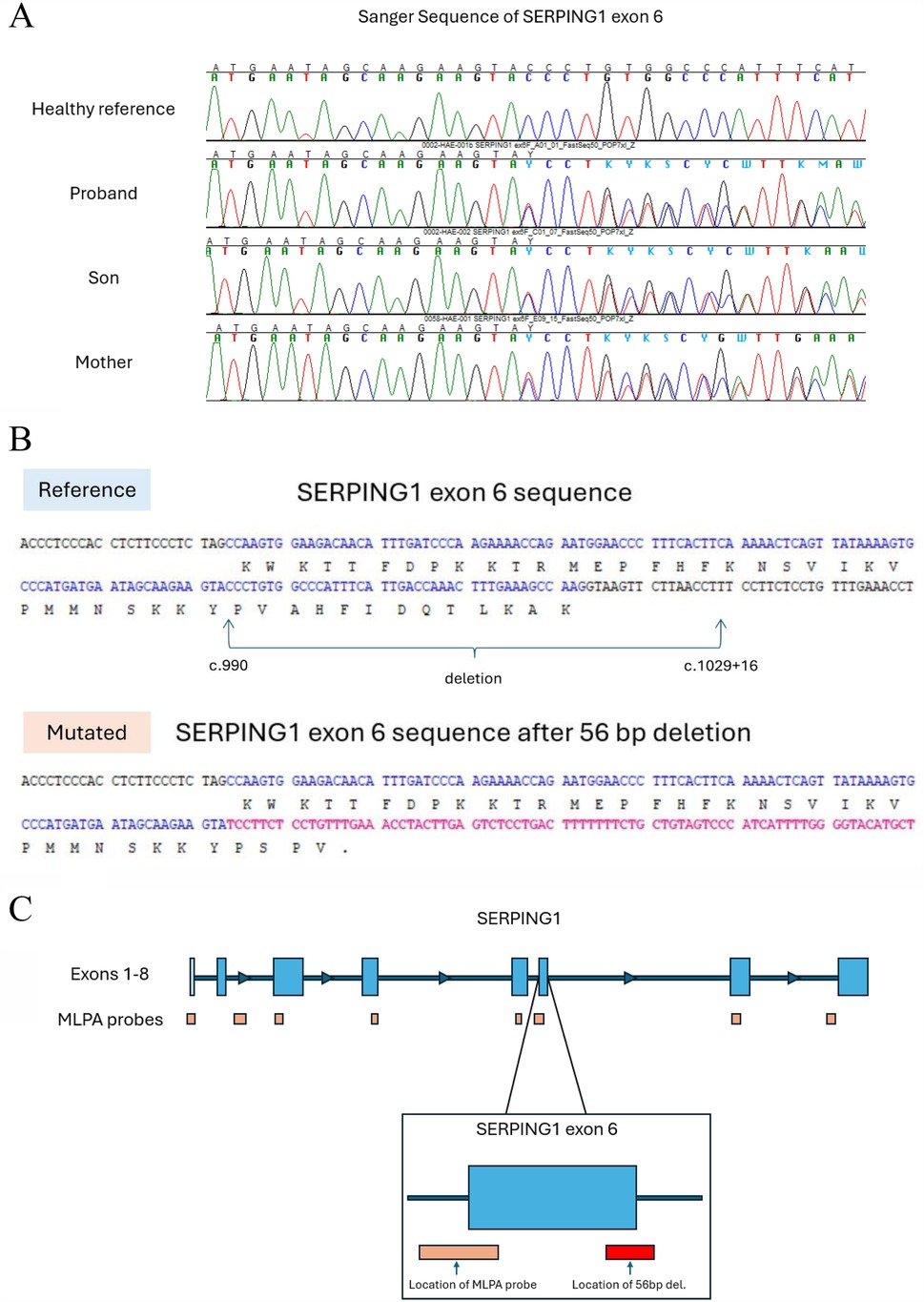
Figure 3. Sanger sequencing of exon 6 revealed a heterozygous 56-base-pair deletion. (A) Sanger electropherogram showing SERPING1 exon 6 for the three family members and a healthy control. (B) The reference nucleotide sequence of exon 6 (top) and the deleted sequence found in the HAE patients (bottom). The intron sequence is shown in black; the exon sequence is shown in blue with the corresponding amino acid code shown below; the new sequence after the deletion is shown in red. A new stop codon is introduced shortening the transcript to 335 total amino acids. (C) The approximate location of the MLPA probe and the 56-base-pair deletion show that a large structural variant >50 bp can be missed.
Discussion
We performed NGS sequencing and MLPA for three family members with chronic recurrent angioedema and abnormal C4 and C1-INH screening tests but found no obvious SERPING1 mutation. Prior analysis by another genetics laboratory also failed to detect SERPING1 mutations. Due to the high clinical suspicion, the allergist requested further investigations by our laboratory to identify a potential genetic association in this family. In response, we developed a soft clipping bioinformatics workflow to analyze NGS data for difficult-to-detect insertions and deletions. Results indicated a high percentage of soft clipping in SERPING1 exon 6 and Sanger sequencing identified a novel 56-base-pair deletion. The SERPING1 c.990_1029+16del variant likely creates a new premature stop codon, subjecting the transcript to nonsense-mediated mRNA decay (NMD).
The NMD quality control system ensures that potentially toxic polypeptide fragments do not accumulate while simultaneously coping with possible translational logjams that might arise from the inherent inefficiency of premature translation termination (15). The early stop codon and NMD explain the C4 and C1-INH deficiency in the three affected family members diagnosed with HAE type I.
We believe that a systematic approach to undetected HAE analysis, using soft clipping as part of an overall strategy, would be effective in determining a small percentage of causal variants in approximately 5% of HAE-C1INH cases where no mutation is found by standard clinical NGS procedures (16–18). Vatsiou 2020 used splice analysis to discover deep intronic cryptic splice sites. Splice site analysis is also an effective tool for finding potential pathogenic variants and SpliceAI (Broad Institute) is included in our overall strategy for undetected HAE analysis. SpliceAI is a deep neural network that models mRNA splicing and is used to predict cryptic splice sites in noncoding genomic sequences (19). Deep intronic variants are seldom covered in routine HAE clinical sequencing, making it easy for these variants to go undetected.
Identification of genetic variants, particularly those involving SERPING1, is critical for several reasons. Confirming a genetic mutation supports the patient's symptoms and clinical diagnosis of HAE, particularly in patients with overlapping or atypical symptoms. Determining the underlying genetic cause can guide targeted therapeutic interventions, ensuring the patient receives the most effective treatment for their disease subtype. In addition to receiving personalized treatment, it documents proof of the patient's disorder for insurance coverage of potentially expensive and chronic therapies for HAE. A clinical genetic diagnosis also enables family screening and early identification of at-risk relatives, allowing for possible life-saving preventative measures or early treatment interventions.
Soft clipping bioinformatics analysis reduces the need for exploratory genetic testing and facilitates molecular diagnosis through a more targeted approach. This method enables high-throughput screening and provides a more comprehensive analysis of NGS data compared to visual inspection of sequencing reads alone. Retrospective analysis of previously sequenced samples from undetected HAE patients can be performed, conserving valuable time and resources that would otherwise be allocated to exploratory sequencing efforts.
The use of soft clipping in genetic testing represents an important step in addressing diagnostic challenges in rare diseases like HAE. This approach marks areas for additional review that standard clinical analysis may miss, providing clinicians and patients with a more comprehensive diagnostic toolset. These findings can inform future research efforts and contribute to the development of novel therapeutic approaches.
Clinicians and genetic testing laboratories should collaborate closely to ensure systematic and thorough evaluation of suspected HAE cases, particularly in those with strong clinical suspicion and negative initial genetic testing results. The limitations of genetic methodologies should be acknowledged and discussed. It should be noted that NGS sequencing only detects pathogenic mutations in approximately 80%–85% of HAE cases, including single nucleotide variations such as missense, nonsense, and small insertions and deletions < 15 base pairs. Standard variant analysis commonly covers the exon and flanking sequence, so deep intronic splice variants, if present, are often excluded. The incorporation of MLPA to detect large deletions, insertions, and duplications, accounts for another 10% of HAE cases. Nevertheless, similar to the case of this reported family, large deletions may sometimes go undetected. The technical limitations of NGS and MLPA in detecting structural variants can be overcome with long-range sequencers, such as Pacific Biosciences’ single-molecule real-time sequencing (PacBIO) and Oxford Nanopore Technologies nanopore sequencing (ONT). However, these long-read sequencing technologies present their own limitations in base calling accuracy, with a single-pass accuracy of 85%–87% (20). Although Sanger sequencing is still considered the gold standard in sequencing and helped us detect this family's mutation, its low throughput limitations have led to it being largely replaced by high-throughput, cost-effective NGS in most clinical genetics laboratories.
To enhance the diagnostic accuracy of hereditary angioedema (HAE) caused by SERPING1 mutations, we recommend adhering to the general testing algorithm outlined by the World Allergy Organization and the European Academy of Allergy and Clinical Immunology (21). Clinicians ordering genetic tests should be informed about the laboratory's methodology and sequencing technology, particularly for rare diseases such as HAE, where multiple types of genetic variations exist within SERPING1. Most genetic laboratories have transitioned from Sanger sequencing—traditionally the preferred method for SERPING1 analysis—to next-generation sequencing (NGS) for greater efficiency. However, when NGS and multiplex ligation-dependent probe amplification (MLPA) yield negative results, soft clipping analysis should be considered. This analysis must be performed alongside other samples in the same NGS run, as variations in sequencing runs, laboratory pipelines, and instruments can introduce inconsistencies that affect reliability. Selecting a genetics laboratory with the capability to conduct additional NGS bioinformatics analyses is essential for improving mutation detection in SERPING1 and optimizing HAE diagnosis.
The Leiden Open Variation Database (LOVD) contains five SERPING1 deletion variants greater than 50 bases but less than an exon, as of the date of this publication. These variants (SERPING1 NM_000062: c.138_207del69, c.250_414del165, c.726_777del51, c.1391_1445del55, and c.*101_*254del153) were all identified by Sanger sequencing (16, 22–25). Generally, NGS SERPING1 studies report variations of small frame shifts and large gene rearrangements. To emphasize the importance of NGS limitations, the creation of an intermediate frame shift category, defined as greater than 50 bp and less than an exon, should be considered. The limitations of any laboratory's sequencing technology should be clearly stated on genetic test reports, as required by clinical regulatory agencies. It is suggested that the undetected 5% of HAE mutations may be found in intronic or untranslated regions of the gene. While we do not refute this stance, we want to expand this suggestion to cover technological limitations as well.
As commercial genetic laboratories increasingly adopt NGS as their primary method, replacing Sanger sequencing to improve overall efficiency, the integration of more techniques—such as soft clipping and splice site analysis—should be prioritized to further close diagnostic gaps and optimize patient care, despite the additional costs. Clinicians ordering genetic tests must be well-informed and vigilant, particularly when familial clinical symptoms suggest a genetic etiology that is not evident in negative test results. Enhanced awareness of these limitations can prompt reanalysis or alternative testing strategies. Furthermore, interdisciplinary collaboration to integrate these advanced techniques into routine NGS workflows holds the potential to improve diagnostic yields, facilitate timely access to effective treatments, and advance equitable healthcare for patients with rare and complex diseases.
Data availability statement
Public database of variant discussed in the article was sent to ClinVar and acknowledged on March 12, 2025,Y2xpbnZhckBuY2JpLm5sbS5uaWguZ292LA== File name(s): Submission ID: SUB15167532, Organization ID: 509915.
Ethics statement
The studies involving humans were approved by Salus IRB, Versiti Clinical Trials. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
KW: Conceptualization, Validation, Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Visualization. JC: Conceptualization, Formal analysis, Supervision, Validation, Writing – original draft, Writing – review & editing. EF: Methodology, Writing – original draft, Writing – review & editing. YG: Writing – original draft, Writing – review & editing. SH: Writing – original draft, Writing – review & editing. JY: Writing – original draft, Writing – review & editing. JC: Writing – original draft, Writing – review & editing. LW: Writing – original draft, Writing – review & editing. HL: Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
KW, JC, EF, SH, JY, JC, LW, HH were employed by Virant Diagnostics, Inc. YG was employed by ScitechLink, LLC.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Drouet C, López-Lera A, Ghannam A, López-Trascasa M, Cichon S, Ponard D, et al. SERPING1 variants and C1-INH biological function: a close relationship with C1-INH-HAE. Frontiers in Allergy. (2022) 3:835503. doi: 10.3389/falgy.2022.835503
2. Germenis AE, Speletas M. Genetics of hereditary angioedema revisited. Clin Rev Allergy Immunol. (2016) 51(2):170–82. doi: 10.1007/s12016-016-8543-x
3. Carter PE, Duponchel C, Tosi M, Fothergill JE. Complete nucleotide sequence of the gene for human C1 inhibitor with an unusually high density of alu elements. Eur J Biochem. (1991) 197(2):301–8. doi: 10.1111/j.1432-1033.1991.tb15911.x
4. Nicolicht P, Faria DOS, Martins-Silva L, Maia LSM, Moreno AS, Arruda LK, et al. Gene mapping strategy for alu elements rearrangements: detection of new large deletions in the SERPING1 gene causing hereditary angioedema in Brazilian families. Gene. (2019) 685:179–85. doi: 10.1016/j.gene.2018.10.084
5. Lincoln SE, Hambuch T, Zook JM, Bristow SL, Hatchell K, Truty R, et al. One in seven pathogenic variants can be challenging to detect by NGS: an analysis of 450,000 patients with implications for clinical sensitivity and genetic test implementation. Genet Med. (2021) 23(9):1673–80. doi: 10.1038/s41436-021-01187-w
6. Zuraw BL. The pathophysiology of hereditary angioedema. World Allergy Organ J. (2010) 3(9 Suppl):S25–28. doi: 10.1097/WOX.0b013e3181f3f21c
7. Sinnathamby ES, Issa PP, Roberts L, Norwood H, Malone K, Vemulapalli H, et al. Hereditary angioedema: diagnosis, clinical implications, and pathophysiology. Adv Ther. (2023) 40(3):814–27. doi: 10.1007/s12325-022-02401-0
8. Bowen B, Hawk JJ, Sibunka S, Hovick S, Weiler JM. A review of the reported defects in the human C1 esterase inhibitor gene producing hereditary angioedema including four new mutations. Clin Immunol. (2001) 98(2):157–63. doi: 10.1006/clim.2000.4947
9. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
10. Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. (2010) 26(5):589–95. doi: 10.1093/bioinformatics/btp698
11. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England). (2010) 26(6):841–42. doi: 10.1093/bioinformatics/btq033
12. Neph S, Scott Kuehn M, Reynolds AP, Haugen E, Thurman RE, Johnson AK, et al. BEDOPS: high-performance genomic feature operations. Bioinformatics. (2012) 28(14):1919–20. doi: 10.1093/bioinformatics/bts277
13. Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. (2011) 12(5):363–76. doi: 10.1038/nrg2958
14. Corominas J, Smeekens SP, Nelen MR, Yntema HG, Kamsteeg E-J, Pfundt R, et al. Clinical exome sequencing-mistakes and caveats. Hum Mutat. (2022) 43(8):1041–55. doi: 10.1002/humu.24360
15. Amrani N, Sachs MS, Jacobson A. Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol. (2006) 7(6):415–25. doi: 10.1038/nrm1942
16. Grombirikova H, Bily V, Soucek P, Kramarek M, Hakl R, Ballonova L, et al. Systematic approach revealed SERPING1 splicing-affecting variants to be highly represented in the Czech national HAE cohort. J Clin Immunol. (2023) 43(8):1974–91. doi: 10.1007/s10875-023-01565-w
17. Demidov G, Laurie S, Torella A, Piluso G, Scala M, Morleo M, et al. Structural variant calling and clinical interpretation in 6224 unsolved rare disease exomes. Eur J Human Genet. (2024) 32(8):998–1004. doi: 10.1038/s41431-024-01637-4
18. Vatsiou S, Zamanakou M, Loules G, Psarros F, Parsopoulou F, Csuka D, et al. A novel deep intronic SERPING1 variant as a cause of hereditary angioedema due to C1-inhibitor deficiency. Allergol Int. (2020) 69(3):443–49. doi: 10.1016/j.alit.2019.12.009
19. Jaganathan K, Panagiotopoulou SK, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. (2019) 176(3):535–548.e24. doi: 10.1016/j.cell.2018.12.015
20. Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. (2020) 21(1):30. doi: 10.1186/s13059-020-1935-5
21. Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui I, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-the 2021 revision and update. Allergy. (2022) 77(7):1961–90. doi: 10.1111/all.15214
22. Iwamoto K, Tanaka A, Hiragun M, Kawai M, Mihara S, Takenaka M, et al. Novel and recurrent C1 inhibitor gene mutations in nine Japanese patients with hereditary angioedema. J Dermatol Sci. (2012) 68(1):68–70. doi: 10.1016/j.jdermsci.2012.06.012
23. Bos IG, Lubbers YT, Roem D, Abrahams JP, Hack CE, Eldering E. The functional integrity of the serpin domain of C1-inhibitor depends on the unique N-terminal domain, as revealed by a pathological mutant. J Biol Chem. (2003) 278(32):29463–70. doi: 10.1074/jbc.M302977200
24. Qu L, Wei B, Liu M, Zhang L, Xiao T, Chen HD, et al. A novel mutation in exon 8 of C1 inhibitor (C1INH) gene leads to abolish its physiological stop codon in a large Chinese family with hereditary angioedema type I. Exp Dermatol. (2012) 21(10):788–91. doi: 10.1111/j.1600-0625.2012.01563.x
Keywords: SERPING1, hereditary angioedema (HAE), multiplex ligation-dependent probe amplification (MLPA), c1-inhibitor (C1INH), soft clipping, next generation sequencing (NGS)
Citation: Wetherby K, Chiao J, Faulkner E, Guo Y, Hou S, Yu JJ, Chen J, Wan L and Henry Li H (2025) Identification of an elusive SERPING1 deletion in a family with hereditary angioedema type I utilizing soft clipping. Front. Allergy 6:1565283. doi: 10.3389/falgy.2025.1565283
Received: 22 January 2025; Accepted: 31 March 2025;
Published: 17 April 2025.
Edited by:
Xin Wang, Brigham and Women's Hospital and Harvard Medical School, United StatesReviewed by:
Anastasios E. Germenis, University of Thessaly, GreeceJinyuan Ma, Henry Ford Health System, United States
Copyright: © 2025 Wetherby, Chiao, Faulkner, Guo, Hou, Yu, Chen, Wan and Henry Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: H. Henry Li, aGVucnlsaUBhbGxlcmd5YXN0aG1hLnVz