Abstract
Objectives/hypothesis:
Chronic rhinosinusitis (CRS) may be triggered by environmental insults. We hypothesized that CRS results from epigenetic modifications of host DNA from external insults, leading to downstream RNA/DNA gene expression changes and immuno-mechanical disruptions. We therefore performed a multi-omics study integrating epigenetic (DNA methylation), transcriptomic (mRNA), and proteomic (cytokine) data of CRS sinonasal tissue to visualize interactions amongst these modalities to study our hypothesis.
Methods:
Sinonasal tissue was collected from 14 prospectively enrolled CRS and control subjects. Cytokine, mRNA transcriptome, and DNA methylome analysis were performed. Multi-omics analysis via joint dimensional reduction (JDR) was conducted.
Results:
Multi-omics unsupervised clustering separated subjects into two distinct groups: one cluster of 9 CRS subjects and another with 3 controls and 2 non-eosinophilic CRSsNP subjects. DNA methylation, followed by mRNA expression, contributed most to cluster assignment. DNA methylation was the most significant data modality contributing to total variance on JDR. Cytokines critical in CRS (IL-5, IL-13, IL-10, IFNγ, IL-6) associated with hundreds of differentially methylated regions (DMRs) and mRNA. On conjoint analyses, common upstream DMRs and mRNAs were linked to cytokines IL-5 and IL-13, cytokines IL-10 and IFNγ, and cytokines IFNγ and IL-6, respectively.
Conclusions:
Our results support the hypothesis that environmental insults may be significant drivers of CRS pathogenesis through epigenetic mechanisms that result in dysregulated mRNA transcription and cytokine expression. The most novel part of this study is our multi-omics approach that used integration of epigenetic (DNA methylation), transcriptomic (mRNA), and proteomic (cytokine) data to uncover insights into CRS pathogenesis; this is the first of its kind in CRS etiopathogenesis. The multi-omics analysis clearly separated clusters of control and CRS subjects, demonstrating its validity in future research. The study also identified interactions of methylated DNA, mRNA, and cytokines in CRS pathogenesis, highlighting novel molecules and pathways that may be potential therapeutic targets.
1 Introduction
A complex interaction of unfavorable environmental insults in the susceptible host has been postulated to disrupt normal homeostatic mechanisms in chronic rhinosinusitis (CRS) (1). Proposed external stressors (“the environment”) include microbial pathogens, microbiome dysbiosis, exposure to allergens, and air pollution (1). In addition, genetic susceptibility may be one of several host factors that result in disease. Even though familial clustering has long been reported, it is unclear whether familial clustering results from shared genes or shared environments, as identifiable monogenic alterations have not been identified in most CRS patients (2–5). In a large population-based study from Utah, U.S.A., 1,638 CRS with nasal polyposis (CRSwNP) and 24,200 CRS sans NP (CRSsNP) subjects were matched to random controls; 1st and 2nd-degree relatives were found to have a 4.1-fold and 3.3-fold elevated risk for CRSwNP, respectively. For CRSsNP, 1st and 2nd-degree relatives had a 2.4-fold and 1.4-fold risk, respectively (2). Interestingly, spouses of CRSsNP patients were also found to have a 2-fold increased risk of CRSsNP (2). In Sweden, Bohman, et al. (6) found that the prevalence of CRSwNP in relatives was 13.4% vs. 2.7% in controls; a relative risk of 4.9 in the first-degree relatives. These studies generate questions about the pathogenic roles of genes, shared environments, or both.
Epigenetics is the study of environmental influences on gene expression. Epigenetic studies are particularly helpful in disease states such as CRS, where multiple host or environmental factors may influence disease pathogenesis (7). External impact is modulated through mechanisms such as DNA methylation, histone modifications, non-coding RNAs, and alternative polyadenylation (APA) (8, 9). Epigenetic changes can notably persist and be passed to the progeny for 2–3 generations. Epigenetics may help explain both familial clustering and the increase in prevalence. Early studies on CRS epigenetics have shown several differences in DNA methylation between CRS and control tissue (10, 11). However, most of these are based in Asia, with only two of our previous studies being conducted in the United States. In this current study, our goal was to investigate the association, if any, of epigenetic modifications and mRNA transcriptomic and proteomic changes characterizing CRS. Transcriptomics analyzes RNA molecules, such as messenger RNA (mRNA), to understand gene expression (4, 12, 13) and has helped identify mechanistic pathways. However, most CRS transcriptomics studies have been conducted in Asia (14–17), with three studies incorporating non-Asian subjects (18–20) this is a relatively novel approach in North America, where population genetics and the environment differ (21, 22). When connecting transcriptomics with proteomics, studies have been divergent, reporting a correlation between CRS mRNA expression (transcriptome) and the proteome (17, 23, 24), as well as discordance (21) highlighting the need for further research using a multi-omics approach. Multiomics studies incorporate multiple modalities with large data sets through bioinformatics tools to help uncover complex relationships and interactions of biological processes at various levels, and can also help identify pathogenetic pathways that may not be apparent when studying each “omics” field individually. However, multiomics analyses involving the epigenome, transcriptome, and proteome (cytokine) have previously not been studied in CRS.
We hypothesize that external insults cause epigenetic modifications of host DNA, resulting in unfavorable DNA and associated RNA and protein expressions, which result in immuno-mechanical disruptions associated with CRS pathogenesis. We tested our hypothesis by performing multi-omics analyses integrating epigenetic (DNA methylation), transcriptomic (mRNA), and proteomic (cytokine) data of CRS sinonasal tissue. Our secondary goal was to visualize interactions amongst these modalities to uncover novel molecules and pathways with potential roles in CRS pathogenesis.
2 Methods
This study was conducted at a tertiary-level hospital in Arizona after approval from the institutional review board (IRB ID: 16-008609). Subjects were classified into controls and CRS based on nasal endoscopy and sinus CT according to 2015 consensus guidelines from the American Academy of Otolaryngology-Head and Neck Surgery (25). CRS subjects were further classified into CRSwNP and CRSsNP. Patients on systemic corticosteroids, biological therapy, and systemic or topical antibiotics in the last 4 weeks were excluded, so as not to affect the baseline cytokine profile of sinonasal tissue. Control subjects were undergoing transsphenoidal endoscopic resection of pituitary adenoma and were negative on CT and endoscopy for sinusitis and had no nasal history suggestive of allergic rhinitis. Prospective data was collected on demographics, clinical diagnoses, and disease severity [patient reported 22-item sinonasal outcome test (SNOT-22) scores (26) and Lund Mackay (27) Sinus CT scores].
STATA BE/18.0 was used to assess any differences in age and sex distribution between the cohorts. The Mann–Whitney U-test was used to compare the difference in age distribution, and Fisher's exact test was used to compare the gender distribution between the cohorts. A p-value of <0.05 was chosen as the criterion of statistical significance. Sinonasal tissue samples for all multi-omics analyses were obtained at a single time point under direct endoscopic guidance for 11 CRS and 3 control subjects. Since sinonasal mucosal samples were obtained during surgery, standard surgical aseptic precautions were used. Specimens were stored at −80°C until analysis. Samples were placed into sterile 7 ml polycarbonate tubes (Sarstedt 71.9923.610) and frozen within 15 min in a −90°C bath of Novec-engineered fluid (3M HFE-7000) cooled in a HistoChill freezing bath (SP Scientific HC80A0). Ethmoidal tissue was used for DNA methylation and cytokine studies. RNA sequencing was performed on ethmoidal tissue in CRS patients and inferior turbinate tissue in controls per IRB approval. A part of the ethmoidal tissue was sent in formalin at the time of surgery for structured histopathology analysis as described by Snidvongs et al. (28) Subjects with tissue eosinophils ≥10 eos/hpf were classified as eosinophilic CRS (eCRS) and those with <10 eos/hpf as non-eosinophilic CRS (neCRS).
2.1 DNA methylation
DNA extraction was done using the QIAamp DNA Mini kit by Qiagen (Reference no. 51306). Reduced Representation Bisulfite Sequencing (RRBS) Library prep and Sequencing were done on Illumina's HiSeq4000. RRBS data were analyzed using a streamlined analysis and annotation pipeline (SAAP) for RRBS, SAAP-RRBS (29). Cytosine followed by a guanine nucleotide (CpG) loci were called differentially methylated CpGs (DMCs) when p ≤ 0.05 and the mean methylation difference for the CpG loci between groups was at least 5% (delta ≥5%). A requirement of having at least four CpG loci within a candidate differentially methylated region (DMR) was set. Further details are included in the Supplementary Section.
2.2 RNA-Sequencing
RNA samples underwent library prep using Illumina TruSeq® RNA Exome Library Prep kit (San Diego, CA). Libraries were sequenced in 2 pools per lane on an Illumina HiSeq 4,000 (100 × 2 paired-end reads) and base-calling using Illumina's RTA v2.7.7. Paired-end RNA sequencing reads were processed through the RNA-Seq bioinformatics pipeline, MAP-RSeq v3.1.4 (30). Differentially expressed genes (DEG) were identified from raw gene counts using edgeR 2.6.2 (31). DEGs were reported with log2 fold change and False Discovery Rate (FDR <5%). Canonical pathway analysis using Ingenuity Pathway Analysis (IPA) software (Ingenuity® Systems) identified significant pathways (p-value <5%). Further details are included in the Supplementary Section.
2.3 Cytokine analysis
Frozen specimens were weighed, thawed, mixed with phosphate-buffered saline (PBS) and protease inhibitors (Millipore Sigma, Burlington, MA), and homogenized. Supernatants were collected after centrifugation. Cytokine and chemokine levels (48-plex) were measured using a Millipore multiplex kit (Billerica, MA) on a Bio-Rad MAGPIX multiplex reader (Hercules, CA). The concentrations of cytokines were normalized to the concentration of total protein in each sample. Total protein was analyzed by using a BCA Protein Assay Kit (Thermo Fisher Scientific). The values of cytokines were divided by the values of total protein. Samples below the minimum detectable concentration (MinDC) were assigned half the MinDC, and values above the standard curve limit were assigned the highest standard. Cytokines and chemokines detected in <10% of samples (17) were excluded. Eosinophil peroxidase (EPX) levels were assessed using an in-house sandwich enzyme-linked immunosorbent assay (ELISA), like that described by Ochkur et al. (32).
2.4 Multi-omics analysis
2.4.1 Preprocessing
Batch effects were corrected using ComBat (33). Modality-specific normalization was followed by transformation and filtering to facilitate an equal contribution to the JDR model. Normalized-FPKM RNA-seq counts were log10 + 1e−4 transformed to achieve a Gaussian distribution. To balance feature counts across modalities, cytokines (with the fewest features) were filtered along with methylation and RNA features, which were significantly different between CRS and controls (FDR <5%).
2.4.2 Modeling
Joint dimensionality reduction (JDR) was performed using the multi-omics factor analysis (MOFA) methodology to integrate data modalities and extract variability dimensions, called factors (34). The contribution of each modality to the variance explained by each factor was quantified. To determine the number of viable factors, a randomized dataset was used, and factors where this dataset contributed the most variation were disregarded. Default settings were used, with modifications to remove scaling between data modalities and to pre-scale the value ranges to ensure a more accurate comparison of feature loading weights between modalities.
2.4.3 Analysis
Hierarchical all-against-all (HAllA) clustering was used to link quantitative and categorical clinical variables to factors, identifying features driving factor-sample distributions. Joint pathway analysis or kinase enrichment inference was used to analyze contributing modalities and features after identifying the associating factor to the clinical variable of interest.
3 Results
Table 1 details the clinical characteristics of the subjects. No statistically significant differences were found in the age and sex distributions between CRS cases and control subjects.
Table 1
| Characteristic | CRSa (n = 11) N (%) or Mean (SDb) |
Controls (n = 3) N (%) or Mean (SDb) |
|---|---|---|
| Age | 52.5 (13.0) | 46.3 (20.8) p = 0.63 |
| Gender (M/F) | 2 (18.2%)/9 (81.8%) | 2 (66.7%)/1 (33.3%) p = 0.17 |
| Clinical diagnosis | Non-CRS (pituitary adenoma) | |
|
3 (27.2%) | |
|
8 (72.7%) | |
| SNOT-22e | 34.5 (31.0); missing in one subject | 6 (2) |
| Lund-Mackay score | 12.1 (3.9) | 0.7 (0.6) |
| Previous sinus surgery | 7 (73.7%) | 0 |
| Absolute serum eosinophils (×109) | 0.36 (0.15) | 0.32 (0.13) |
| Tissue Eosinophil/HPFf | ||
|
5 (45.5%) | 3 (100%) |
|
4 (36.4%) | |
|
2 (18.2%) | |
| Asthma | ||
|
7 (73.7%) | 0 |
|
4 (36.3%) | 3 (100%) |
| Allergic rhinitis | ||
|
9 (81.9%) | 0 |
|
2 (18.1%) | 1 (33.3%) |
|
0 | 2 (66.7%) |
| AERDg | ||
|
1 (9%) | 0 |
|
10 (91%) | 3 (100%) |
| Smoking history | ||
|
0 (18.1%) | 1 (33.3%) |
|
8 (72.7%) | 2 (66.7%) |
|
3 (27.2%) | 0 |
| Current nasal steroid spray | ||
|
2 (18.1%) | 0 |
|
5 (45.4%) | 0 |
|
4 (36.3%) | 0 |
Clinical characteristics of subjects.
CRS: Chronic rhinosinusitis.
SD: Standard deviation.
CRSwNP: CRS with nasal polyposis.
CRSsNP: CRS sans nasal polyposis.
SNOT-22: 22-item sinonasal outcome test.
HPF: high power field.
AERD: Aspirin Exacerbated Respiratory Disease.
3.1 The multi-omics approach was successful in separating CRS subjects from controls
Multi-omics unsupervised clustering separated CRS from Controls; DNA methylation modality most contributed to cluster assignment, followed by RNA transcripts.
Multi-omics unsupervised clustering revealed two distinct groups with clear separation (Figure 1A). Figure 1B depicts the clinical diagnosis of cluster constituents. Cluster 1 was found to be entirely constituted by CRS subjects (3 CRSwNP, 6 CRSsNP), and Cluster 2 included all 3 controls and 2 non-eosinophilic CRSsNP subjects. Figure 1C depicts cluster constituents based on tissue eosinophil status. Where all Cluster 2 constituents had <10 eos/hpf, 6 CRS subjects in Cluster 1 had high tissue eosinophilia (2 with >100 eos/hpf, and 4 with tissue eosinophils between 10 and 100 eos/hpf), and 2 had non-eosinophilic tissue. Next, we examined each cluster to identify associated pathways (Figure 1D, E). The known functions of the top pathways associated with Cluster 1 and Cluster 2 are depicted in Tables 2A, B, respectively. The modality that most contributed to cluster assignment was DNA methylation, followed by RNA transcripts (Figure 1F). DMRs and DE RNAs associated with both clusters were identified, the top 50 of which are listed in Tables 3A, B, respectively. Supplementary Tables S1A and S1B present the known functions of these genes.
Figure 1

(A) Multiomics clustering of samples considering all three data modalities, (B) constituents of each cluster by diagnosis (control, CRSwNP, CRSsNP), (C) constituents of each cluster by tissue eosinophil count, (D) significant pathways associated with cluster 1 and (E) cluster 2, (F) number of features from each data modality contributing to cluster assignment.
Table 2A
| Pathway | Considerations | Referencea |
|---|---|---|
| STAT3 | a The protein encoded by STAT3 gene is a member of the STAT protein family. In response to cytokines and growth factors, STAT family members are phosphorylated by the receptor associated kinases, and then form homo- or heterodimers that translocate to the cell nucleus where they function as transcription activators. STAT3 protein is activated through phosphorylation in response to various cytokines and growth factors including IFNs, EGF, IL5, and IL6. This protein mediates expression of a variety of genes in response to cell stimuli, and thus plays a key role in many cellular processes such as cell growth and apoptosis. PIAS3 protein is a specific inhibitor of this protein. This gene also plays a role in regulating host response to viral and bacterial infections. Mutations in this gene are associated with infantile-onset multisystem autoimmune disease and hyper-immunoglobulin E syndrome. This gene participates in immune response or antiviral activity. | Liu 2021 (A1) |
| SNARE Signaling | SNARE proteins are critical in granule fusion events | Lacy 2011 (A2) |
| Phagosome maturation | Phagosome maturation is the process by which bacteria and other ingested particles are degraded. This pathway is regulated by p38 mitogen-activated protein kinase (MAPK), which is activated by toll-like receptors (TLRs). | Blander 2004 (A3) |
| Gustation | In CRSwNP, downregulated genes were enriched for gustatory sensory perception and tissue homeostasis, | Wang 2022 (A4) |
| GNRH signaling | Preprogonadotropin-releasing hormone-like protein (GnRH) is overexpressed in COVID-19 convalescent subjects | Huoman 2022 (A5) |
| Choline degradation I | Toll-like receptor (TLR) activation enhances choline uptake by macrophages through induction of choline transporter CTL1 | Sanchez-Lopez 2019 (A6) |
| Cardiac B-adrenergic signaling | Reactive oxygen species formation regulates beta2-adrenergic receptor signal transduction | Michaeloudes 2022 (A7) |
| Cardiac hypertrophy signaling/enhanced signaling | Cellular metabolism, proliferation, non-coding RNAs, immune responses, translational regulation, and epigenetic modifications, positively or negatively regulate cardiac hypertrophy | Nakamura 2018 (A8) |
| CD27 Signaling in Lymphocytes | Is a costimulatory molecule of tumor necrosis factor receptor (TNFR) family, strongly expressed on activated CD4(+) and CD8(+) T lymphocytes | Behrendt 2010 (A9) |
Pathways associated with cluster 1 (9 CRS subjects).
Sources: National Institutes of Health National Library of Medicine, https://www.ncbi.nlm.nih.gov/gene National Human Genome Research Institute https://www.genome.gov/genetics-glossary Last accessed August 16, 2024.
Table 2B
| Pathway | Considerations | Reference |
|---|---|---|
| Zymosterol biosynthesis | Essential for membrane fluidity and function; important 2nd messenger lipids involved in developmental signaling | Germann 2005 (A10) |
| Superpathway of cholesterol biosynthesis; Cholesterol I, II (via 24,25-dihydrolanosterol); III (via desmosterol) | Cholesterol and cholesterol derivatives shape plasma membrane fluidity and lipid raft dynamics, affecting the formation of the immunological synapse and its downstream signalling events, modulating T-cell activation and function | Cardoso 2021 (A11) |
| Gustation pathway | In subjects diagnosed with CRSwNP, downregulated genes were predominantly enriched for gustatory sensory perception, tissue homeostasis, and muscle system process | Wang 2022 (A12) |
| CREB signaling in neurons | DNA-binding transcriptional regulator CREB is an intracellular protein that regulates expression of genes important in dopaminergic neurons | Wang 2018 (A13) |
| Role of JAK2 in hormone-like cytokine signaling | a Janus Kinase 2 (JAK2) protein has an N-terminal domain that is required for erythropoietin receptor association, an SH2 domain that binds STAT transcription factors, a pseudokinase domain and a C-terminal tyrosine kinase domain. Cytokine binding induces autophosphorylation and activation of this kinase. This kinase then recruits and phosphorylates signal transducer and activator of transcription (STAT) proteins. Growth factors like TGF-beta 1 also induce phosphorylation and activation of this kinase and translocation of downstream STAT proteins to the nucleus where they influence gene transcription. Mutations in this gene are associated with numerous inflammatory diseases and malignancies. This gene is a downstream target of the pleiotropic cytokine IL6 that is produced by B cells, T cells, dendritic cells, and macrophages to produce an immune response or inflammation. A nonsynonymous mutation in the pseudokinase domain of this gene disrupts the domains inhibitory effect and results in constitutive tyrosine phosphorylation activity and hypersensitivity to cytokine signalling. This gene and the IL6/JAK2/STAT3 signalling pathway is a therapeutic target for the treatment of excessive inflammatory responses to viral infections. | Wang 2016 (A14) |
| Role of hypercytokinemia/hyperchemokinemia in the pathogenesis of influenza | Hypercytokinemia/hyperchemokinemia precedes acute respiratory distress syndrome (ARDS) during influenza infection | Wei 2022 (A15) |
| Interferon signaling | Type I and II interferons and IL-27 inhibit ILC2 functions through the activation of STAT1 | Duer 2016 (A16) |
Pathways Associated with Cluster 2 (All 3 control and 2 CRS <10 eos/hpf subjects)
Sources: National Institutes of Health National Library of Medicine, https://www.ncbi.nlm.nih.gov/gene National Human Genome Research Institute https://www.genome.gov/genetics-glossary Last accessed August 16, 2024.
Table 3A
| Gene name | Gene description | Gene type |
|---|---|---|
| C8orf31 | Chromosome 8 open reading frame 31 | Non-coding RNA |
| EDN2 | Endothelin 2 | protein-coding |
| MIR320E | MicroRNA 320e | Non-coding RNA |
| SP6 | Sp6 transcription factor | protein-coding |
| NANS | N-acetylneuraminate synthase | protein-coding |
| COG1 | Component of oligomeric Golgi complex 1 | protein-coding |
| HEYL | HES related family bHLH transcription factor with YRPW motif like | protein-coding |
| ST3GAL4 | ST3 beta-galactoside alpha-2,3-sialyltransferase 4 | protein-coding |
| EXT1 | Exostosin glycosyltransferase 1 | protein-coding |
| SMAD3 | SMAD family member 3 | protein-coding |
| C1QTNF5 | C1q and TNF related 5 | protein-coding |
| MYCL | MYCL proto-oncogene, bHLH transcription factor | protein-coding |
| DNAJB6 | DNA heat shock protein family (Hsp40) member B6 | protein-coding |
| TULP1 | TUB like protein 1 | protein-coding |
| SLC44A2.2 | Solute carrier family 44, member 2 | protein-coding |
| SOX15 | SRY-box transcription factor 15 | protein-coding |
| SLC44A2 | Solute carrier family 44, member 2 | protein-coding |
| ZBTB16 | Zinc finger and BTB domain containing 16 | protein-coding |
| SEMA6C | semaphorin 6C | protein-coding |
| PEBP4 | Phosphatidylethanolamine binding protein 4 | protein-coding |
| EPHB3 | EPH receptor B3 | protein-coding |
| SPAG6 | Sperm associated antigen 6 | protein-coding |
| LINC00963 | Long intergenic non-protein coding RNA 963 | Non-coding RNA |
| SLC44A2.1 | Solute carrier family 44, member 2 | protein-coding |
| QRFP | Pyroglutamylated RFamide peptide | protein-coding |
| LINC00265 | Long intergenic non-protein coding RNA 265 | Non-coding RNA |
| SHISAL1 | Shisa like 1 | protein-coding |
| CACNA1H | Calcium voltage-gated channel subunit alpha1 H | protein-coding |
| PTPN21 | Protein tyrosine phosphatase non-receptor type 21 | protein-coding |
| GADD45B | Growth arrest and DNA damage inducible beta | protein-coding |
| FBLN1 | Fibulin 1 | protein-coding |
| ANKRD65 | Ankyrin repeat domain 65 | protein-coding |
| OSBPL5 | Oxysterol binding protein like 5 | protein-coding |
| KLK5 | Kallikrein related peptidase 5 | protein-coding |
| TFDP1 | Transcription factor Dp-1 | protein-coding |
| AGAP2-AS1 | AGAP2 antisense RNA 1 | Non-coding RNA |
| WFIKKN2 | WAP, follistatin, immunoglobulin, kunitz, netrin domain containing 2 | protein-coding |
| LINC01338 | Long intergenic non-protein coding RNA 1338 | Non-coding RNA |
| STMND1 | Stathmin domain containing 1 | protein-coding |
| SHANK2 | SH3 and multiple ankyrin repeat domains 2 | protein-coding |
| CD37 | CD37 molecule | protein-coding |
| PRR25 | Proline rich 25 | protein-coding |
| SLURP2 | Secreted LY6/PLAUR domain containing 2 | protein-coding |
| CACNA1C-IT3 | CACNA1C intronic transcript 3 | Non-coding RNA |
| LINC01814 | Long intergenic non-protein coding RNA 1814 | Non-coding RNA |
| IL31RA | Interleukin 31 receptor A | protein-coding |
| FBLN7 | Fibulin 7 | protein-coding |
| FAM78A | Family with sequence similarity 78 member A | protein-coding |
| PRMT7 | Protein arginine methyltransferase 7 | protein-coding |
| PLA2G4C | Phospholipase A2 group IVC | protein-coding |
The Top 50 differentially methylated regions in DNA between cluster 1 and 2.
Sources: National Institutes of Health National Library of Medicine, https://www.ncbi.nlm.nih.gov/gene National Human Genome Research Institute https://www.genome.gov/genetics-glossary Last accessed August 16, 2024.
Table 3B
| Gene name | Gene description | Gene type |
|---|---|---|
| ATP2A3 | ATP2A3 (ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporting 3) also known as SERCA3 | Protein coding |
| PNCK | Pregnancy Up-Regulated Nonubiquitous CaM Kinase | Protein coding |
| CCDC88B | CCDC88B coiled-coil domain containing 88B [Homo sapiens (human)] | Protein coding |
| CST4 | cystatin S | Protein coding |
| ARHGAP40 | Rho GTPase activating protein 40 | Protein coding |
| CST1 | cystatin SN | Protein coding |
| LDLRAD2 | low density lipoprotein receptor class A domain containing 2 | Protein coding |
| PRB1 | proline rich protein BstNI subfamily 1 | Protein coding |
| CCDC183 | coiled-coil domain containing 183 | Protein coding |
| RILP | Rab interacting lysosomal protein | Protein coding |
| MAPK8IP3 | mitogen-activated protein kinase 8 interacting protein 3 | Protein Coding |
| PLXNB3 | plexin B3 | Protein Coding |
| SRPK3 | SRSF protein kinase 3 | Protein coding |
| ARHGF16 | Rho guanine nucleotide exchange factor 16 | Protein coding |
| CARD14 | caspase recruitment domain family member 14 | Protein coding |
| CHDH | choline dehydrogenase | Protein coding |
| JSRP1 | junctional sarcoplasmic reticulum protein 1 | Protein coding |
| PTPRH | protein tyrosine phosphatase receptor type H | Protein coding |
| ATP10B | ATPase phospholipid transporting 10B | Protein coding |
| FUT3 | Also known as CD174/fucosyltransferase 3 (Lewis blood group) | Protein coding |
| EGLN3 | egl-9 family hypoxia inducible factor 3 | Protein coding |
| TMEM200A | transmembrane protein 200A | Protein coding |
| HBA2 | hemoglobin subunit alpha 2 | Protein coding |
| PRLR | prolactin receptor | Protein coding |
| LRFN5 | leucine rich repeat and fibronectin type III domain containing 5 | Protein coding |
| FRMD6 | FERM domain containing 6 | Protein coding |
| LUM | lumican | Protein coding |
| JAM2 | junctional adhesion molecule 2 | Protein coding |
| CNTN1 | Contactin 1 | Protein coding |
| SNHG14 | Small nucleolar RNA host gene 14 | Noncoding RNA |
| STAC | SH3 and cysteine rich domain | Protein coding |
| WNT5A | Wnt family member 5A | Protein coding |
| NELL2 | Neural EGFL like 2 | Protein coding |
| HMGN1P36 | High mobility group nucleosome binding domain 1 pseudogene 36 | Pseudogene |
| PTN | Pleiotrophin | Protein coding |
| SCN2B | Sodium voltage-gated channel beta subunit 2 | Protein coding |
| AR | Androgen receptor | Protein coding |
| SNORD116-18 | Small Nucleolar RNA, C/D Box 116-18 | snoRNA |
| SNORD 116-25 | Small Nucleolar RNA, C/D Box 116-25 | snoRNA |
| TCP1 | T-complex 1 | Protein coding |
| SNORD62A | Small nucleolar RNA, C/D box 62A | snoRNA |
| SNORD62B | Small nucleolar RNA, C/D box 62B | snoRNA |
| GPC3 | Glypican 3 | Protein coding |
| CCDC36 | Coiled-coil domain containing 36 | Protein coding |
| CSMD3 | CUB and Sushi multiple domains 3 | Protein coding |
| SLIT2 | Slit guidance ligand 2 | Protein coding |
| ZNF660 | Zinc finger protein 660 | Protein coding |
| DPP6 | Dipeptidyl peptidase like 6 | Protein coding |
| NPNT | Nephronectin | Protein coding |
The Top 50 Differentially Expressed mRNA between Clusters 1 and 2
snoRNA: Small nucleolar RNAs (snoRNAs), a class of small RNA molecules that primarily guide chemical modifications of other RNAs. Sources: National Institutes of Health National Library of Medicine, https://www.ncbi.nlm.nih.gov/gene National Human Genome Research Institute https://www.genome.gov/genetics-glossary Last accessed August 16, 2024.
3.2 DNA methylation was the most significant data modality contributing to total variance
Tissue samples from 14 subjects demonstrated several unique features across DNA methylation, transcriptomic, and cytokine data. Figure 2 depicts the filtering and transformation strategy for each data modality. DNA Methylation was the most significant data modality contributing to Total Variance.
Figure 2

Filtering and transformation strategy for each data modality through volcano plots and histogram representations. Data value distributions were transformed to be as close to Gaussian as possible.
JDR resulted in five dimensions of variation (“factors”), which captured the most significant patterns of information (Figure 3B). Methylation was the most significant data modality contributing to total variance (Figure 3C). Factor 1 is mostly driven by methylation. Factors 4 and 5 have similar contributions from methylation and RNA expression. Factor 3 is mostly correlated with methylation and less with RNA expression. Factor 2's important contribution comes from cytokines; however, it was also moderately correlated to DNA methylation, and weakly to RNA expression (Figure 3B). Table 4 details significant pathways associated with each factor as referenced by studies of inflammatory mechanisms.
Figure 3

Total variance contribution from each data modality (methylation, RNA, cytokines) is shown: (A) total number of data points for each modality, (B) joint dimension reduction resulted in five dimensions of variation (“Factors”). The darker the color, the more the contribution to the individual factor from the modality (methylation, RNA, cytokine), (C) Modality contributing to the total variance.
Table 4
| Pathways—factor | Considerations | Reference |
|---|---|---|
| Pathways associated with Factor 1 | ||
| Transcriptional regulatory network in embryonic stem cells | Specifies gene expression and imparts distinct cellular phenotypes | Chan 2011 (A17) |
| Signaling by Rho family GTPases | Precise regulation of actin cytoskeletal dynamics as well as other immunological functions of leukocytes | Dipankar 2021 (A18) |
| Role of JAK1 and JAK3 in gamma-c cytokine signaling | Janus kinases are associated with intracellular domains of IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 receptors; JAK3 binds to γ chain and JAK1 to the other chain | Haan 2011 (A19) |
| RHOA signaling | Ras homolog family member A (RHOA) is a molecular switch that is activated in response to binding of chemokines, cytokines, and growth factors, and regulates activation of cytoskeletal proteins and other factors | Bros 2019 (A20) |
| Myelination signaling | Signaling of Wnt/β-catenin, PI3K/AKT/mTOR, and ERK/MAPK oligodendrocyte precursor cell differentiation and myelination/remyelination regulation | Gaesser 2016 (A21) |
| Mouse embryonic stem cell pluripotency | Noncoding RNAs and regulation of chromatin packing dynamics by histone modifications and DNA methylation play a vital role in pluripotency maintenance | Chen 2016 (A22) |
| Human embryonic stem cell pluripotency | Combination of intrinsic and extrinsic signaling pathways that regulates self-renewal of human embryonic stem cell | Mohammadi 2020 (A23) |
| Chronic myeloid leukemia signaling | Proliferation, self-renewal, and survival of normal and malignant stem cells | Moradi 2019 (A24) |
| Caudal-related homeobox transcription (CDX) gastrointestinal cancer signaling | Intestine-specific nuclear transcription factor, strongly implicated in multiple tumorigenesis | Yu 2019 (A25) |
| Axonal guidance signaling | Axonal guidance signaling-associated pathways (including NGF and semaphorin 3A) are suppressed in CRSwNP | Wu 2018 (A26) |
| Pathways associated with Factor 2 | ||
| Role of JAK2 in hormone-like cytokine signaling | JAK2 is essential for signaling through hormone-like cytokines and growth factors such as interleukin-3 (IL-3), IL-5, granulocyte macrophage-colony stimulating factor (GM-CSF), erythropoietin (EPO), and thrombopoietin | Wang 2016 (A27) |
| Osteoarthritis | Related to pathological signaling pathways, such as Wnt/β-catenin, NF-κB, focal adhesion, hypoxia inducible factor (HIFs), TGFβ, and other pathways and the key regulators AMPK, mTOR, and RUNX2 | Yao 2023 (A28) |
| Myelination signaling | Signaling of Wnt/β-catenin, PI3K/AKT/mTOR regulators, and ERK/MAPK | Gaesser 2016 (A29) |
| Human embryonic stem cell pluripotency | Intrinsic and extrinsic signaling pathways that regulate self-renewal of human embryonic stem cell | Mohammadi 2020 (A30) |
| Hepatic fibrosis/Hepatic stellate cell activation | IL-17 directly induces production of collagen type I in hepatic stellate cells by activating signal transducer and activator of transcription 3 (STAT3) signaling pathway | Meng 2012 (A31) |
| Gustation | In CRSwNP, downregulated genes are predominantly enriched for gustatory sensory perception, tissue homeostasis, and muscle system process | Wang 2022 (A32) |
| Cardiac hypertrophy signaling (enhanced) | Cellular metabolism, proliferation, non-coding RNAs, immune responses, translational regulation, and epigenetic modifications, regulating cardiac hypertrophy | Nakamura 2018 (A33) |
| CREB signaling in neurons | Intracellular protein that regulates the expression of genes that are important in dopaminergic neurons | Wang 2018 (A34) |
| CMP-N-acetyl-neuraminate biosynthesis I | CMP-N-acetylneuraminate synthetase (CMAS) is a key enzyme in sialic acid incorporation pathway, and is crucial in the virulence and survival of several pathogenic bacteria | Bose 2019 (A35) |
| Androgen biosynthesis | Hyperandrogenism can activate mononuclear cells (MNC) in the fasting state, increasing MNC sensitivity to glucose | Gonzalez 2011 (A36) |
| Pathways associated with Factor 3 | ||
| TREM1 signaling | Triggering receptor expressed on myeloid cells- 1 (TREM1). Neutrophil Activation Pathway is suppressed in eosinophilic nasal polyps | Wu 2018 (A37) |
| Superpathway of cholesterol biosynthesis; Cholesterol I, II (via 24,25-dihydrolanosterol); III (via desmosterol) | Cholesterol and cholesterol derivatives shape plasma membrane fluidity and lipid raft dynamics, affecting the formation of the immunological synapse and its downstream signalling events, modulating T-cell activation and function | Cardoso 2021 (A38) |
| Role of cytokines in mediating communication between immune cells | In Th2-polarized environment of allergic asthma, high IL-4 levels produced by locally infiltrating innate lymphoid cells and helper T cells promote an alternatively activated M2a phenotype in macrophages, affecting local immune response and airway structure | Ewan 2021 (A39) |
| Intrinsic prothrombin activation pathway; Extrinsic prothrombin activation pathway; Coagulation system | Activation of the coagulation pathway, including increased thrombin-antithrombin and D-dimer, has been demonstrated in chronic urticaria | Kim 2015 (A40) |
| Airway inflammation asthma | Type 2 inflammation pathways link the pathogenesis of asthma and CRSwNP | Laidlaw 2020 (A41) |
| Pathways associated with Factor 4 | ||
| Wound healing signaling pathway | Involvement of JAK/STAT signaling in chronic wounds | Jere 2017 (A42) |
| Role of osteoclasts in rheumatoid arthritis signaling pathway | IL-1β, IL-6, TNF-α, IL-17 and hypoxia-inducible factor-1α (HIF-1α) are produced that could mediate bone loss | Hu 2022 (A43) |
| Phagosome maturation | Regulated by p38 mitogen-activated protein kinase (MAPK), which is activated by TLRs | Blander 2004 (A44) |
| Mineralocorticoid biosynthesis | Mineralocorticoid receptor activation result in increased tissue oxidative stress and vascular inflammation | Young 2008 (A45) |
| Microautophagy signaling pathway | During lysosomal inhibition, MyD88 is accumulated, and overabundant MyD88 autoactivates downstream signaling or enhance TLR/IL-1R-mediated signaling | Into 2017 (A46) |
| Huntington disease signaling | RhoA regulation and downstream cellular functions, and signaling in neurodegenerative diseases | Schmidt 2022 (A47) |
| Hepatic fibrosis/Hepatic stellate cell activation | IL-17 directly induced production of collagen type I in hepatic stellate cells by activating the signal transducer and activator of transcription 3 (STAT3) signaling pathway | Meng 2012 (A48) |
| Glucocorticoid biosynthesis | Elevated IL-17A level promotes pyroptosis in hNECs through the ERK-NLRP3/caspase-1 signaling pathway and contributes to glucocorticoid resistance by affecting glucocorticoid receptor homeostasis in CRSwNP | Li 2022 (A49) |
| CSDE1 signaling pathway | Cold shock domain-containing E1 is an RNA-binding protein that can directly interrupt transcription and translation of proteins and has been shown to prevent neurogenesis in human embryonic stem cells | Lee 2017 (A50) |
| Assembly of RNA polymerase I complex | RNA polymerase I and RNAPIII are protein complexes specializing in transcription of highly abundant non-coding RNAs, such as ribosomal RNA and transfer RNA | Turowski 2021 (A51) |
| Pathways associated with Factor 5 | ||
| Wound healing signaling pathway | JAK/STAT signaling and PBM in chronic wounds | Jere 2017 (A52) |
| Role of osteoclasts in rheumatoid arthritis (RA) signaling path | IL-1β, IL-6, TNF-α, IL-17 and hypoxia-inducible factor-1α (HIF-1α) are produced that could mediate bone loss | Hu 2022 (A53) |
| Role of osteoblasts in RA signaling pathway | Involves proinflammatory cytokines Tumor Necrosis factor-α, Interleukin-1 | Hu 2022 (A53) |
| Pulmonary fibrosis idiopathic signaling pathway | Primary human fibroblast cultures signaling leads to IL-6R overexpression. The IL-6/STAT3/Smad3 axis facilitates cellular responses and fibrotic disease | Shochet 2020 (A54) |
| Pathogen induced cytokine storm signaling pathway | Toll-like receptor-4 (TLR4) signaling activates diverse transcription factors and induces proinflammatory cytokine expression | Kobayashi 2013 (A55) |
| Neutrophil extracellular trap signaling pathway | Triggered by innate immune receptors through downstream intracellular signaling, which activate myeloperoxidase, neutrophil elastase, and protein-arginine deiminase type 4 to promote chromatin decondensation | Papayannopoulos 2018 (A56) |
| Microautophagy signaling pathway | During lysosomal inhibition, MyD88 is accumulated, and overabundant MyD88 autoactivates downstream signaling or enhance TLR/IL-1R-mediated signaling | Into 2017 (A57) |
| Iron homeostasis signaling pathway | IL-33 is associated with erythrocytes and heme to promote the generation of mature splenic red pulp macrophages through activation of the MyD88 adaptor protein and ERK1/2 kinases downstream of IL-33 receptor, IL1RL1 | Lu 2020 (A58) |
| Hepatic fibrosis/Hepatic stellate cell activation | IL-17 directly induced production of collagen type I in hepatic stellate cells by activating the signal transducer and activator of transcription 3 (STAT3) signaling pathway | Meng 2012 (A59) |
| GP6 signaling pathway | GP6 is a collagen and fibrin receptor for tissue repair, wound healing, general inflammation, and innate immunity | Nurden 2019 (A60) |
Significant pathways associated with each of the five factors shown in figure 2B.
3.3 Correlation of factor variation with clinical features
The five factors of variation were correlated with clinical features using HAllA (Figure 4). Significant correlations for Factor 1 were with allergic rhinitis, absolute blood eosinophil count, tissue eosinophil counts, and clinical diagnosis. For Factor 2, the most significant correlations were allergic rhinitis and immune deficiency. Factor 3's correlations were tissue eosinophil counts, smoking, allergic rhinitis, and gender. Factor 4's most significant correlations were tissue eosinophil counts, allergic rhinitis, and clinical diagnosis. Factor 5's most significant correlations were allergic rhinitis, clinical diagnosis, tissue eosinophil counts, and pre-operative SNOT-22. Tissue eosinophil counts mostly correlated with Factors 4 and 3, and absolute blood eosinophil counts only significantly correlated with Factor 1. Allergic rhinitis strongly correlated with Factors 1, 2, 4, and 5, and moderately correlated with Factor 3. Age, previous sinus surgery, asthma history, total IgE, CT score, steroid nasal spray use, and AERD presented with weak correlations.
Figure 4

Hierarchical-all-against-all clustering used to represent the correlation between factor distribution and clinical metrics of samples. Similarly, behaving factors and clinical metrics are binned into clusters. Correlation is shown from high (blue) to low (beige).
3.4 Examination of sample distribution across factors: tissue eosinophilia was able to better cluster subjects in two distinct groups compared to phenotypic status
We examined sample distribution across all five factors identified with JDR. Figure 5 (A, B, C) shows the sample distribution colored by diagnosis, tissue eosinophils/hpf, and SNOT-22 scores, respectively. Whereas SNOT-22 seemed to lead to a random distribution, both clinical diagnosis (CRSwNP, CRSsNP vs. control) and tissue eosinophils were able to better cluster subjects in two distinct groups. This was especially evident for factor 4, where all 3 controls were separated from CRSwNP and/or CRS ≥10 eos/hpf.
Figure 5
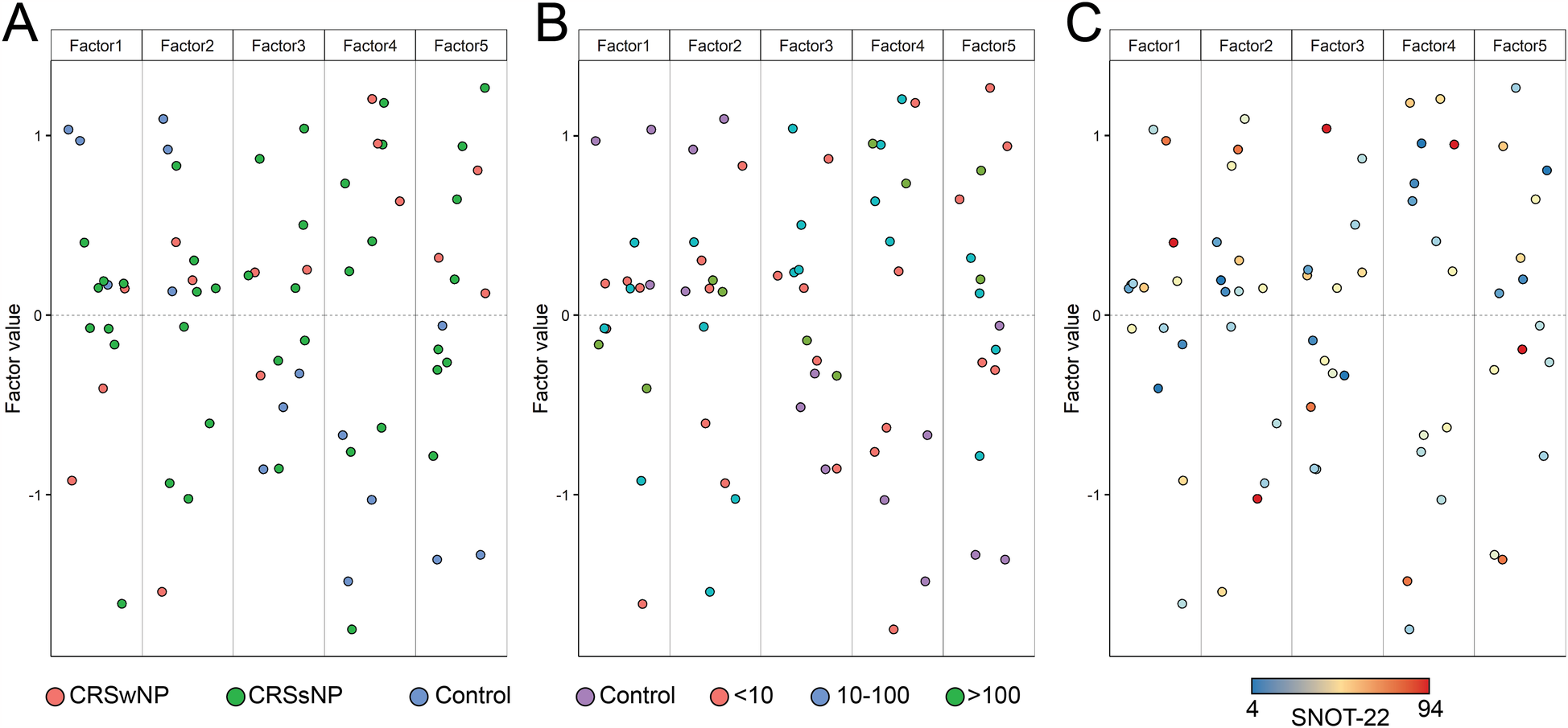
Sample distribution across all five factors colored by (A) diagnosis/polyp status, (B) tissue eosinophil numbers/hpf, and (C) SNOT-22 scores.
3.5 DNA methylation and mRNA heatmaps failed to cluster CRSwNP and CRSsNP separately
The association of each cytokine with DNA methylation and mRNA expression in each subject was examined. DNA methylation (Figure 6A) and mRNA heatmaps (Figure 6B) showed all 3 control samples clustered together. There was no clear clustering observed for phenotypical subtypes of CRS by methylation and mRNA expression status, likely exposing the limitations of classifying only by polyp status.
Figure 6

Heatmaps of the association between each subject (x-axis) with (A) DNA methylation and (B) RNA expression on the y-axis. Color key for Subjects: CRSwNP is orange, CRSsNP is purple, and Controls are green. Within the heatmap, red represents higher hypermethylation and mRNA expression.
3.6 Correlation between DNA, RNA, and cytokine expression: Two distinct clusters of cytokines were noted, with opposed positive, neutral, and negative correlations for cytokines
Next, we investigated the correlation between DNA, RNA, and cytokine expression and identified two distinct clusters with opposed positive, neutral, and negative correlations for the cytokine-methylation analysis (Figure 7A). The first cluster included MCSF, FLT3l, GROa, RANTES, VEGFa, FGF2, EGF, sCD40l, PDGFAA, IP10, MIG, IL-18, MCP1, and IL-12p40. The second cluster included IL-4, IL-13, IL-5, IL-1RA, IL-8, IL-10, INFγ, IL-6, G-CSF, Eotaxin, MIP-1b, MIP-1a, Fractalkine, MDC, EPX, MCP3, and TGFα. Two distinct clusters of cytokines were noted with opposed positive, neutral, and negative correlations for cytokines-RNA expression analysis as well (Figure 7B). The first cluster included MCP3, MIP-1a, IP10, IL-18, TGFα, GROa, RANTES, FGF2, VEGFa, sCD40l, and PDGFAA. The second cluster included EGF, IL-4, IL-1RA, MDC, EPX, IL-5, IL-13, MIG, Fractalkine, MCP1, IL-12p40, MCSF, FLT3l, Eotaxin, IL-8, IL-6, GCSF, MIP-1b, IFNγ, and IL-10.
Figure 7

Heatmap of (A) differential DNA methylation and (B) differential RNA expression to show correlations to cytokines. The y-axis represents the genes associated with the DMRs/ mRNAs, and the x-axis represents individual cytokines. Red: strong correlation; blue: weak correlation; white: no correlation.
3.7 Associations of individual cytokines with upstream DNA methylation and RNA expression were found
Isolated cytokine analysis was used next to study associations between cytokine, DNA, and RNA. The analysis revealed that IL-5 was associated with 720 differentially expressed (DE) RNAs and 172 differentially methylated regions (DMRs) on the DNA. IL-13 associated with 49 DE-RNAs and 180 DMRs, IL-10 to 54 DE-RNAs and 82 DMRs, IFNγ to 71 DE-RNAs and 123 DMRs, and IL-6 to 236 DE-RNAs and 178 DMRs. IL-4 and TGF did not significantly correlate with the other data modalities. Figures 8A,B illustrate the top 50 differentially methylated genes and differentially expressed mRNAs, respectively, for each of the 30 cytokines. Table 5 presents the top 10 DMRs and the differentially expressed mRNA identified in our study as related to many of these cytokines.
Figure 8

Heatmap correlating each cytokine (x-axis) with their top 50 genes with (A) differential DNA methylation & (B) differentially expressed RNA (y-axis).
Table 5
| Cytokines | Differentially methylated DNA | Differentially expressed mRNA |
|---|---|---|
| IL-1RA | SHANK2, STMND1, CD37, ANKRD65, EXT1, DNAJB6, SOX15, PRR25, TULP1, C1QTNF5 | FUT3, ATP10B, PTPRH, ATP2A3, CHDH, PLXNB3, PRB1, LDLRAD2, EGLN3, SRPK3 |
| IL-4 | OSBPL5, TFDP1, AGAP2-AS1, ANKRD65, SHISAL1, LINC00265 CD37, STMND1, EXT1, QRFP | CST4, ARHGAP40, ARHGEF16, CARD14, FUT3, EGLN3, PLXNB3, PTPRH, ATP10B, CST1 |
| IL-5 | CD37, CACNA1H, LINC00265 SHISAL1, PTPN21, LINC00963, GADD45B, SLC44A2.1, QRFP, OSBPL5 | PRB1, CARD14, RILP, ARHGEF16, CHDH, CCDC183, MAPK8IP3, SRPK3, ARHGAP40, PNCK |
| IL-6 | SHANK2, STMND1, TULP1, SLC44A2.2, C1QTNF5, ANKRD65, FBLN1, ZBTB16, MIR320E, EPHB3 | JSRP1, PTPRH, ATP10B, FUT3, EGLN3, FRMD6, LFRN5, ATP2A3, PNCK, CCDC88B |
| IL-8 | PEBP4, EPHB3, SOX15, TULP1, QRFP, STMND1, GADD45B, PTPN21, LINC00265, SPAG6 | ATP10B, EGLN3, PTPRH, FUT13, PRLR, CHDH, JSRP1, SRPK3, ARHGAP40, PNCK |
| IL-10 | C8ORF31, EDN2, MIR320E, LINC01338, KLK5, FBLN1, ZBTB16, COG1, SOX15, HEYL | JSRP1, PTPRH, EGLN3, ATP10B, ATP2A3, PNCK, CCDC88B, CST4, ARHGAP40, CST1 |
| IL-12p40 | PEBP4, PRR25, EPHB3, SPAG6, SLC44A2.1, QRFP, LINC00265, C8ORF31, MIR320E, CD37 | EGLN3, FUT3, CHDH, MAPK8IP3, CCSC183, LDLRAD2, CST1, ARHGEF16, PNCK, CCDC88B |
| IL-13 | OSBPL5, SHISAL1, DNAJB6, CD37, SMAD3, ST3GAL4, WFIKKN2, TFDP1, QRFP, AGAP2-AS1 | CARD14, ARHGEF16, RILP, PRB1, ARHGAP40, ATP2A3, CST4, CST1, SRPK3, CHDH |
| EPX | CD37, SHISAL1, SLC44A2.1, SPAG6, LINC00963, CACNA1H, PTPN21, GADD45B, WFIKKN2, AGAP2-AS1 | CCDC88B, PNCK, PRB1, SRPK3, CHDH, CARD14, CST4, RILP, CCDC183, CST1 |
| INF-γ | TULP1, SLC44A2.2, SOX15, SLC44A2, ZBTB16, SEMA6C, PEBP4, MIR320E, EPHB3, NANS | JSRP1, PTPRH, ATP10B, FUT3, EGLN3, LRFN5, JAM2, PRLR, HBA2, FRMD6 |
| TGF-alpha | FAM78A, IL31RA, ANKRD65, EXT1, SOX15, SP6, DNAJB6, CACNA1C-IT3, SHANK2, SLC44A2 | HBA2, NELL2, HMGN1P36, SNORD116-18, TCP1, SNORD116-25, ATP10B, PTPRH, FUT3, SNORD62A |
Top 10 differentially methylated DNA as well as differentially expressed mRNA molecules.
3.8 Conjoint cytokine analyses identified common upstream DNA methylation and RNA expression for some cytokines
Next, conjoint cytokine analysis was performed (Figure 9) to identify commonly shared genes. The conjoint analysis showed that cytokines IL-5 and IL-13 were similarly correlated with RNA expression of TMEM74B and CPNE7, and with DNA methylation of DICER1 and SHISAL1. IL-10 and IFNγ were correlated to RNA expression of CYP27C1 and SOX18, and with DNA methylation of CASZ1, SYNRG, SNORD149, NTF4, and CTBP2. IFNγ and IL-6 were similarly correlated to RNA expression of CD79B and GFBP3, and with DNA methylation of EGFL7, HOXA2, PEBP4, WNT7B, CTBP2, and INTS1 (Figure 9). Table 6 enlists the function of genes identified on conjoint cytokine analysis.
Figure 9
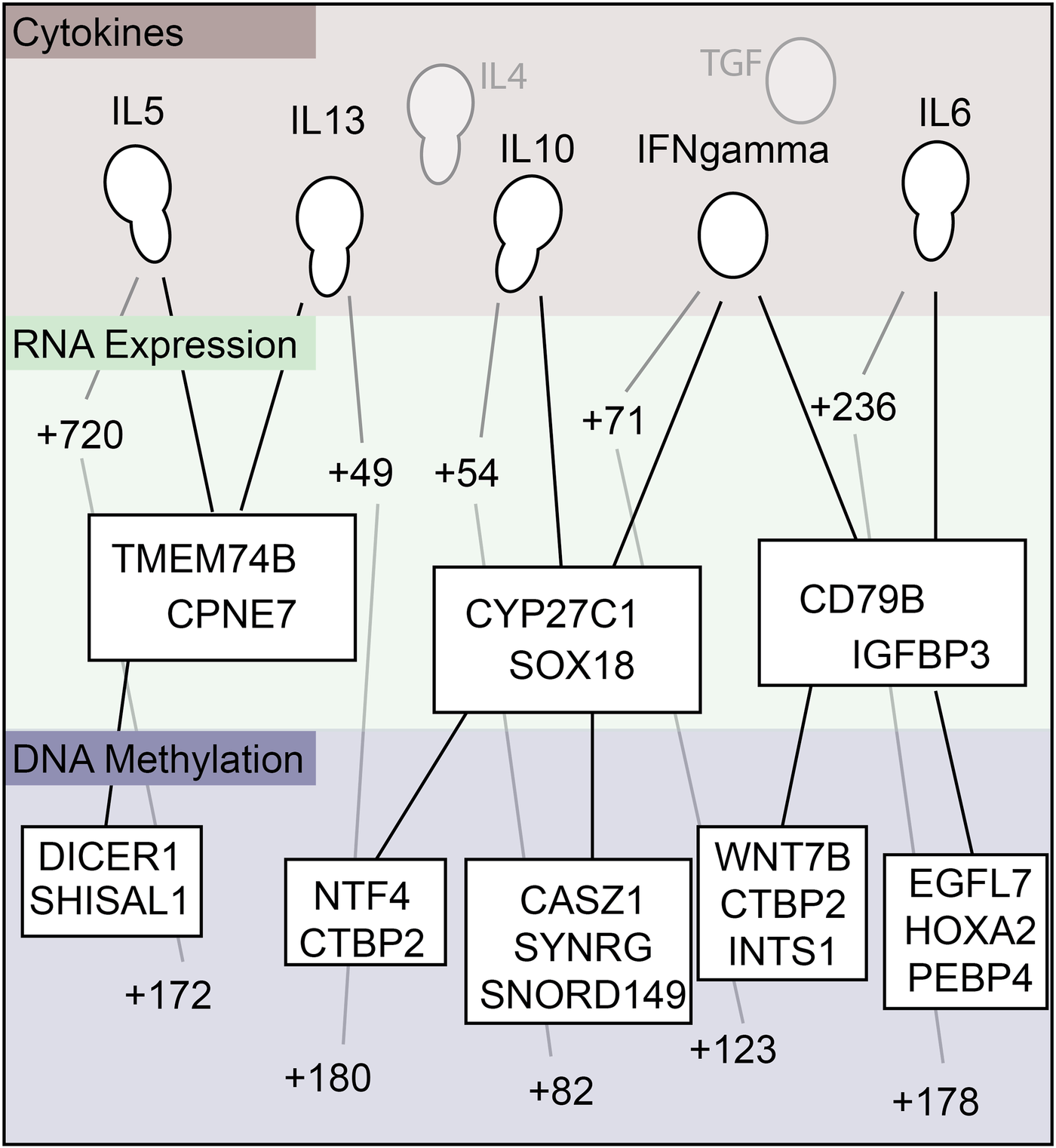
Summary map relating cytokine to RNA expression, and RNA expression to DNA methylation.
Table 6
| Gene | Name | Function | Cytokine |
|---|---|---|---|
| Differentially Methylated Genes | |||
| DICER1 | Dicer 1, ribonuclease III |
The encoded protein functions as a ribonuclease, and is a strong antiviral agent active against RNA viruses, including Zika and SARS-CoV-2 viruses | IL-5 and IL-13 |
| SHISAL1 | Shisa like 1 | Predicted to be integral component of membrane | IL-5 and IL-13 |
| NTF4 | Neurotrophin factor 4 | Neurotrophins control survival and differentiation of mammalian neurons | IL-10 and IFNγ |
| CTBP2 | C-terminal binding protein 2 | This gene can encode two distinct proteins: one isoform is a transcriptional repressor, while the other is a component of specialized synapses known as synaptic ribbons. Both proteins contain a NAD + binding domain similar to NAD + -dependent 2-hydroxyacid dehydrogenases. | IL-10 and IFNγ as well as IFNγ and IL-6 |
| CASZ1 | Castor zinc finger 1 | The protein encoded by this gene is a zinc finger transcription factor and may function as a tumor suppressor | IL-10 and IFNγ |
| SYNRG | Synergin gamma | Encodes a protein that interacts with the gamma subunit of AP1 clathrin-adaptor complex | IL-10 and IFNγ |
| SNORD 149 | Small Nucleolar RNA, 149 | *snoRNA (non-protein-coding); Small nucleolar RNAs (snoRNAs) are a class of small RNA that assist in chemical modifications of other RNAs, such as ribosomal RNAs | IL-10 and IFNγ |
| WNT7B | Wnt family member 7b | Member of the WNT gene family, which consists of structurally related genes encoding secreted signaling proteins regulating cell fate and patterning | IFNγ and IL-6 |
| INTS1 | Integrator complex subunit 1 | Is a subunit of the Integrator complex, which associates with the RNA polymerase II large subunit and mediates processing of small nuclear RNAs U1 and U2. | IFNγ and IL-6 |
| EGFL7 | Epidermal Growth Factor like domain multiple 7 | Encodes a secreted endothelial cell protein that contains two epidermal growth factor-like domains. The encoded protein may play a role in regulating vasculogenesis, and growth and proliferation of tumor cells. | IFNγ and IL-6 |
| HOXA2 | Homeobox A2 | Encodes a DNA-binding transcription factor regulating gene expression, morphogenesis, and differentiation | IFNγ and IL-6 |
| PEBP4 | Phosphatidylethanolamine binding protein 4 | Phosphatidylethanolamine (PE)-binding proteins, including PEBP4, are an evolutionarily conserved family of proteins with pivotal biologic functions, such as lipid binding and inhibition of serine proteases | IFNγ and IL-6 |
| Differentially Expressed mRNA | |||
| TMEM74B | Transmembrane protein 74B | Predicted to be integral component of membrane. | IL-5 and IL-13 |
| CPNE7 | Castor zinc finger 1 | Encodes a zinc finger transcription factor. The encoded protein may function as a tumor suppressor gene. | IL-5 and IL-13 |
| CYP27C1 | Cytochrome P450 family 27 subfamily C member 1 | Encodes a member of the cytochrome P450 superfamily of enzymes which are monooxygenases catalyzing many reactions involved in drug metabolism and synthesis of cholesterol, steroids, and other lipids. | IL-5 and IL-13 |
| SOX18 | SRY-box transcription factor 18 | This gene encodes a member of the SOX (SRY-related HMG-box) family of transcription factors. The encoded protein may function as a transcriptional regulator and play a role in blood vessel and lymphatic vessel development. | IL-10 and IFNγ |
| CD79B | CD79b molecule | Gene encodes Ig-beta protein of B-cell antigen component. It associates with Ig-alpha and Ig-beta, necessary for expression and function of B-cell antigen receptor | IFNγ and IL-6 |
| IGFBP3 | Insulin-like growth factor binding protein 3; this gene is a member of the IGFBP family. It prolongs half-life of IGFs and alters their interaction with cell surface receptors. | IFNγ and IL-6 | |
Function of genes identified on conjoint cytokine analysis.
Sources: National Institutes of Health National Library of Medicine, National Center for Biotechnology Information https://www.ncbi.nlm.nih.gov/gene National Human Genome Research Institute https://www.genome.gov/genetics-glossary/Pseudogene Last accessed August 16, 2024.
*snoRNA: small nucleolar RNA.
4 Discussion
The results of our study support our hypothesis that environmental insults may be significant in CRS pathogenesis through epigenetic mechanisms that result in dysregulated mRNA transcription and cytokine production downstream. Chronic dysregulated immune responses may continue long past the initial external insult through the induction of epigenetic changes, as seen in CRS (5).
Although the changes that occur at the histopathological and cytokine/protein levels have recently become better characterized in subjects with CRS (35–37), the genetic mechanism associated with such changes has not been fully characterized (3). In a sparse area of research, this study provides the first multi-omics analysis of CRS tissue from the United States, validating the association of epigenetic changes with transcriptomic and proteomic signatures seen in CRS. Furthermore, multi-omics analysis using DNA methylation, mRNA expression, and cytokine expression datasets successfully separated clusters of control and CRS subjects, demonstrating the utility of multi-omics analysis as a valuable tool in studying CRS. Our study is novel in using a multi-omics integration of DNA, RNA, and cytokine data to study CRS. Only two prior multi-omics studies have investigated CRS, but neither studied DNA data, and both were conducted outside of North America (18, 38). Miyata et al. (38), isolated eosinophils from six nasal polyp patients and performed multi-omics analysis using lipidomics, proteomics, and transcriptomics. Hoggard et al. (18), investigated temporal changes in polyp tissue in CRS in response to systemic corticosteroids in three males with CRSwNP subjects who underwent surgery, assessing natural variability over time and local response to systemic corticosteroid therapy. The authors found that the most highly abundant transcripts and proteins were associated with pathways involved in inflammation, FAS, cadherin, integrin, Wnt, apoptosis, cytoskeletal signaling, coagulation, and B- and T-cell activation. Given that DNA methylation was the most significant data modality contributing to the total variance between CRS and control subjects, epigenetic modifications are critical for further study in CRS for mechanistic and therapeutic targets. In addition, epigenetic mechanisms help explain shifts in the dominant CRS inflammatory pattern from non-type 2 to type 2, as is being noted in Asian regions as they undergo industrialization (36, 37, 39).
Our study further identified several known and potential mechanistic pathways and proteins involved in immunity and structural integrity, which may have roles in CRS pathogenesis. These are related to cytokine signal transduction, granule fusion events, phagosome maturation, toll-like receptors (TLRs) activation, reactive oxygen species formation, cellular metabolism, translational regulation, etc. The identification of JAK signaling also highlights the potential therapeutic role of JAK inhibitors in recalcitrant CRS, like current trials for asthma therapy (40). Many novel DMRs and DE mRNA (Tables 4A, B) were identified, including genes involved in membrane stability, homeostasis, as well as the gustation pathway, which are targets for further research (Supplementary Table S1).
Novel findings on conjoint cytokine analysis (Figure 9) showed that the cytokines IL-5 and IL-13 shared genes with RNA expression of TMEM74B and CPNE7, and with DNA methylation of DICER1 and SHISAL1. We also similarly noted shared genes for IL-10 and IFNγ, as well as IFNγ and IL-6. Table 6 details the functions of these genes. Both IL-5 and IL-13 are well recognized for their roles in the type-2 inflammatory process predominantly associated with CRSwNP, and hence, their association in differentially regulated upstream DNAs and RNAs is understandable, further reinforcing the utility of the multiomics approach. IFNγ is detected at lower levels in CRS tissue, reducing the antiviral immune response, which could result in or exacerbate the CRS following a viral infection (41). The role of IL-10 and IL-6 is reported in the literature (42, 43), and their association with IFNγ is interesting. DMRs and DE RNAs identified in association with key inflammatory cytokines involved in CRS pathogenesis, like IL-5, IL-13, IL-10, IFNγ, and IL-6 (Table 5), could be potentially important future therapeutic targets.
4.1 Limitations
This is a small study, albeit with the largest number of subjects published for CRS multiomics. While the multiomics approach distinguished two clusters, one of which was composed entirely of CRS patients, the other grouped three controls and two non-eosinophilic CRSsNP subjects, and perhaps these may have been clustered differently in a larger sample size. Genomic assays and multiomics analysis are prohibitively expensive, complex, and require technical expertise in integration, statistics, and systems biology. However, we hope that with reduced cost of genomic assays and multiomics analysis and support from extramural funding, larger prospective sampling can be performed for future studies.
Prospective, longitudinal studies with sample collection at multiple time points are needed to study CRS disease evolution. RNA expression is transient and may not correlate with protein level unless analyzed concurrently, which was mitigated by collecting tissue for histopathology, DNA methylation, and cytokine assay simultaneously in this study.
The multi-omics approach may also allow for focused upstream gene profiling of targeted cytokines of interest, such as IL-4, IL-5, IL-13, and others, in addition to an unsupervised approach that was used in this study. We anticipate that the use of single-cell RNA sequencing may be necessary for this approach rather than the bulk tissue sample that was used for this study.
Technical limitations in the study include bulk tissue RNA sequencing, which can only provide an average gene expression profile for the entire sample, but is cheaper than single-cell RNA sequencing (scRNA-seq) while identifying global differences in gene expression between disease and control states. Additionally, of the multiplex assay performed for cytokines, only 30 could be included per study methodology for multiomics analysis. Quantifying tissue eosinophilia with histopathology is imperfect, as degranulated eosinophils are difficult to measure. More sensitive novel assays, such as those from NanoString Technology (https://nanostring.com/), are planned for future study.
5 Conclusions
The study supports the hypothesis that environmental insults may be significant drivers of CRS pathogenesis through epigenetic mechanisms that result in dysregulated mRNA transcription and cytokine expression. The most novel part of this study is the integration of epigenetic (DNA methylation), transcriptomic (mRNA), and proteomic (cytokine) data to uncover novel insights into the pathogenesis of CRS. This multi-omics approach is the first of its kind to study environment-host interactions in CRS etiopathogenesis. The multi-omics analysis clearly separated clusters of control and CRS subjects, demonstrating its validity in future research. DNA methylation also contributed most to total variance, underscoring the role of environmental factors in CRS. Key cytokines like IL-5, IL-13, IL-10, IFNγ, and IL-6 were associated with hundreds of differentially methylated regions (DMRs) and differentially expressed mRNAs, providing future targets for study. IL-5 and IL-13, IL-10 and IFNγ, and IFNγ and IL-6 were associated with common upstream genes. The study further identified interactions of methylated DNA, mRNA, and cytokines in CRS pathogenesis, highlighting novel molecules and pathways that may be potential therapeutic targets.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Institutional review Board, Mayo Clinic, Arizona, US. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
DL: Project administration, Writing – original draft, Methodology, Resources, Visualization, Conceptualization, Validation, Writing – review & editing, Funding acquisition, Supervision. TB: Writing – original draft, Writing – review & editing. CM: Formal analysis, Data curation, Software, Investigation, Writing – review & editing. EJ: Investigation, Software, Writing – review & editing, Data curation, Formal analysis. NK: Writing – review & editing. PL: Writing – review & editing, Writing – original draft. MM: Writing – review & editing. AM: Writing – review & editing. HK: Conceptualization, Supervision, Writing – review & editing, Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was partly funded by an unrestricted grant through the FlinnFoundation, and intramural grants from Mayo Clinic in Arizona.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/falgy.2025.1606255/full#supplementary-material
References
1.
Kato A Schleimer RP Bleier BS . Mechanisms and pathogenesis of chronic rhinosinusitis. J Allergy Clin Immunol. (2022) 149(5):1491–503. 10.1016/j.jaci.2022.02.016
2.
Oakley G Curtin K Orb Q Schaefer C Orlandi R Alt J . Familial risk of chronic rhinosinusitis with and without nasal polyposis: genetics or environment. Int Forum Allergy Rhinol. (2015) 5(4):276–82. 10.1002/alr.21469
3.
Brar T Marks L Lal D . Insights into the epigenetics of chronic rhinosinusitis with and without nasal polyps: a systematic review. Front Allergy. (2023) 4:1165271. 10.3389/falgy.2023.1165271
4.
Lal D Brar T Ramkumar SP Li J Kato A Zhang L . Genetics and epigenetics of chronic rhinosinusitis. J Allergy Clin Immunol. (2023) 151(4):848–68. 10.1016/j.jaci.2023.01.004
5.
Brar T Marino MJ Lal D . Unified airway disease: genetics and epigenetics. Otolaryngol Clin North Am. (2023) 56(1):23–38. 10.1016/j.otc.2022.09.002
6.
Bohman A Juodakis J Oscarsson M Bacelis J Bende M Naluai ÅT . A family-based genome-wide association study of chronic rhinosinusitis with nasal polyps implicates several genes in the disease pathogenesis. PLoS One. (2017) 12(12):e0185244. 10.1371/JOURNAL.PONE.0185244
7.
Rakyan VK Down TA Balding DJ Beck S . Epigenome-wide association studies for common human diseases. Nat Rev Genet. (2011) 12(8):529–41. 10.1038/nrg3000
8.
Martin MJ Garcia-Sanchez A Estravis M Gil-Melcón M Isidoro-Garcia M Sanz C et al Genetics and epigenetics of nasal polyposis: a systematic review. J Investig Allergol Clin Immunol. (2021) 31(3):196–211. 10.18176/jiaci.0673
9.
Allis CD Jenuwein T . The molecular hallmarks of epigenetic control. Nat Rev Genet. (2016) 17(8):487–500. 10.1038/NRG.2016.59
10.
Brar T Baheti S Marino MJ Kita H Lal D . Genome-wide epigenetic study of chronic rhinosinusitis tissues reveals dysregulated inflammatory, immunologic and remodeling pathways. Am J Rhinol Allergy. (2023) 37:692–704. 10.1177/19458924231193526
11.
Brar T Baheti S Bhagwate AV Kita H Marino MJ Lal D . Genomewide epigenetic study shows significant DNA hypermethylation in chronic rhinosinusitis versus control ethmoidal tissue. Int Forum Allergy Rhinol. (2023) 13:2235–9. 10.1002/alr.23205
12.
Wang Z Gerstein M Snyder M . RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. (2009) 10(1):57–63. 10.1038/NRG2484
13.
Poole A Urbanek C Eng C Schageman J Jacobson S O'Connor BP et al Dissecting childhood asthma with nasal transcriptomics distinguishes subphenotypes of disease. J Allergy Clin Immunol. (2014) 133(3):670–8. 10.1016/J.JACI.2013.11.025
14.
Wang W Gao Z Wang H Li T He W Lv W et al Transcriptome analysis reveals distinct gene expression profiles in eosinophilic and noneosinophilic chronic rhinosinusitis with nasal polyps. Sci Rep. (2016) 6(May):1–14. 10.1038/srep26604
15.
Gill AS Pulsipher A Sumsion JS Oakley GM Leclair LW Howe H et al Transcriptional changes in chronic rhinosinusitis with asthma favor a type 2 molecular endotype independent of polyp Status. J Asthma Allergy. (2021) 14:405–13. 10.2147/JAA.S301825
16.
Plager DA Kahl JC Asmann YW Nilson AE Pallanch JF Friedman O et al Gene transcription changes in asthmatic chronic rhinosinusitis with nasal polyps and comparison to those in atopic dermatitis. PLoS One. (2010) 5(7):e11450. 10.1371/JOURNAL.PONE.0011450
17.
Hao Y Zhao Y Wang P Du K Li Y Yang Z et al Transcriptomic signatures and functional network analysis of chronic rhinosinusitis with nasal polyps. Front Genet. (2021) 12:609754. 10.3389/fgene.2021.609754
18.
Hoggard M Jacob B Wheeler D Zoing M Chang K Biswas K et al Multiomic analysis identifies natural intrapatient temporal variability and changes in response to systemic corticosteroid therapy in chronic rhinosinusitis. Immun Inflamm Dis. (2021) 9(1):90–107. 10.1002/iid3.349
19.
Kenney HM Rangel-Moreno J Peng Y Chen KL Bruno J Embong A et al Multi-omics analysis identifies IgG2b class-switching with ALCAM-CD6 co-stimulation in joint-draining lymph nodes during advanced inflammatory-erosive arthritis. Front Immunol. (2023) 14:1237498. 10.3389/fimmu.2023.1237498
20.
Xu Z Huang Y Meese T Van Nevel S Holtappels G Vanhee S et al The multi-omics single-cell landscape of sinus mucosa in uncontrolled severe chronic rhinosinusitis with nasal polyps. Clin Immunol. (2023) 256:109791. 10.1016/j.clim.2023.109791
21.
Workman AD Nocera AL Mueller SK Otu HH Libermann TA Bleier BS . Translating transcription: proteomics in chronic rhinosinusitis with nasal polyps reveals significant discordance with messenger RNA expression. Int Forum Allergy Rhinol. (2019) 9(7):776–86. 10.1002/alr.22315
22.
Brar T McCabe C Miglani A Marino M Lal D . Tissue eosinophilia is superior to an analysis by polyp Status for the chronic rhinosinusitis transcriptome: an RNA study. Laryngoscope. (2023) 133:2480–9. 10.1002/lary.30544
23.
Liu Z Kim J Sypek JP Wang IM Horton H Oppenheim FG et al Gene expression profiles in human nasal polyp tissues studied by means of DNA microarray. J Allergy Clin Immunol. (2004) 114(4):783–90. 10.1016/J.JACI.2004.04.052
24.
Stankovic KM Goldsztein H Reh DD Platt MP Metson R . Gene expression profiling of nasal polyps associated with chronic sinusitis and aspirin-sensitive asthma. Laryngoscope. (2008) 118(5):881–9. 10.1097/MLG.0B013E31816B4B6F
25.
Rosenfeld RM Piccirillo JF Chandrasekhar SS Brook I Ashok Kumar K Kramper M et al Clinical practice guideline (update): adult sinusitis. Otolaryngol Head Neck Surg. (2015) 152(2 suppl):S1–S39. 10.1177/0194599815572097
26.
Hopkins C Gillett S Slack R Lund VJ Browne JP . Psychometric validity of the 22-item sinonasal outcome test. Clin Otolaryngol. (2009) 34(5):447–54. 10.1111/J.1749-4486.2009.01995.X
27.
Lund VJ Kennedy DW . Quantification for staging sinusitis. The staging and therapy group. Ann Otol Rhinol Laryngol Suppl. (1995) 167:17–21. 10.1177/000348949510410s02
28.
Snidvongs K Lam M Sacks R Earls P Kalish L Phillips PS et al Structured histopathology profiling of chronic rhinosinusitis in routine practice. Int Forum Allergy Rhinol. (2012) 2(5):376–85. 10.1002/alr.21032
29.
Sun Z Baheti S Middha S Kanwar R Zhang Y Li X et al SAAP-RRBS: streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing. Bioinformatics. (2012) 28(16):2180–1. 10.1093/BIOINFORMATICS/BTS337
30.
Kalari KR Nair AA Bhavsar JD O'Brien DR Davila JI Bockol MA et al MAP-RSeq: mayo analysis pipeline for RNA sequencing. BMC Bioinformatics. (2014) 15:224. 10.1186/1471-2105-15-224
31.
Robinson MD McCarthy DJ Smyth GK . Edger: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. (2010) 26(1):139–40. 10.1093/BIOINFORMATICS/BTP616
32.
Ochkur SI Kim JD Protheroe CA Colbert D Condjella RM Bersoux S et al A sensitive high throughput ELISA for human eosinophil peroxidase: a specific assay to quantify eosinophil degranulation from patient-derived sources. J Immunol Methods. (2012) 384(1-2):10–20. 10.1016/j.jim.2012.06.011
33.
Johnson WE Li C Rabinovic A . Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. (2007) 8(1):118–27. 10.1093/biostatistics/kxj037
34.
Argelaguet R Arnol D Bredikhin D Deloro Y Velten B Marioni JC et al MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. (2020) 21(1):111. 10.1186/s13059-020-02015-1
35.
Bachert C Zhang N Hellings PW Bousquet J . Clinical reviews in allergy and immunology endotype-driven care pathways in patients with chronic rhinosinusitis. J Allergy Clin Immunol. (2019) 141(5):1543–51. 10.1016/j.jaci.2018.03.004
36.
Karin J Tim D Gabriele H Cardell LO Marit W Claus B . Type 2 inflammatory shift in chronic rhinosinusitis during 2007–2018 in Belgium. Laryngoscope. (2021) 131(5):E1408–14. 10.1002/lary.29128
37.
Wang X Zhang N Bo M Holtappels G Zheng M Lou H et al Diversity of TH cytokine profiles in patients with chronic rhinosinusitis: a multicenter study in Europe, Asia, and oceania. J Allergy Clin Immunol. (2016) 138(5):1344–53. 10.1016/j.jaci.2016.05.041
38.
Miyata J Fukunaga K Kawashima Y Watanabe T Saitoh A Hirosaki T et al Dysregulated fatty acid metabolism in nasal polyp-derived eosinophils from patients with chronic rhinosinusitis. Allergy. (2019) 74(6):1113–24. 10.1111/ALL.13726
39.
Katotomichelakis M Tantilipikorn P Holtappels G De Ruyck N Feng L Van Zele T et al Inflammatory patterns in upper airway disease in the same geographical area may change over time. Am J Rhinol Allergy. (2013) 27(5):354–60. 10.2500/ajra.2013.27.3922
40.
Georas SN Donohue P Connolly M Wechsler ME . JAK Inhibitors for asthma. J Allergy Clin Immunol. (2021) 148(4):953–63. 10.1016/j.jaci.2021.08.013
41.
Hwang JW Lee KJ Choi IH Han HM Kim TH Lee SH . Decreased expression of type I (IFN-β) and type III (IFN-λ) interferons and interferon-stimulated genes in patients with chronic rhinosinusitis with and without nasal polyps. J Allergy Clin Immunol. (2019) 144(6):1551–65.e2. 10.1016/j.jaci.2019.08.010
42.
Kubota K Takeno S Taruya T Sasaki A Ishino T Hirakawa K . IL-5 and IL-6 are increased in the frontal recess of eosinophilic chronic rhinosinusitis patients. J Otolaryngol Head Neck Surg. (2017) 46(1):36. 10.1186/s40463-017-0214-2
43.
Xuan L Zhang N Wang X Zhang L Bachert C . IL-10 family cytokines in chronic rhinosinusitis with nasal polyps: from experiments to the clinic. Front Immunol. (2022) 13:947983. 10.3389/fimmu.2022.947983
Summary
Keywords
epigenetics, chronic rhinosinusitis, transcriptomics, proteomics, multiomics, cytokines, differentially methylated DNA, differentially expressed mRNA
Citation
Lal D, Brar T, McCabe C, Jessen E, Kumar N, Lança Gomes P, Marino MJ, Miglani A and Kita H (2025) Epigenetic modifications are associated with mRNA and cytokine expression changes in chronic rhinosinusitis: a multiomics study from the United States. Front. Allergy 6:1606255. doi: 10.3389/falgy.2025.1606255
Received
04 April 2025
Accepted
12 May 2025
Published
05 June 2025
Volume
6 - 2025
Edited by
Diego Marcelo Conti, KU Leuven, Belgium
Reviewed by
Matija Rijavec, University Clinic of Pulmonary and Allergic Diseases Golnik, Slovenia
Eduardo Javier Correa, Nuevo Hospital Comarcal de La Linea de La Concepción, Spain
Updates
Copyright
© 2025 Lal, Brar, McCabe, Jessen, Kumar, Lança Gomes, Marino, Miglani and Kita.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Devyani Lal lal.devyani@mayo.edu
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.